
Abstract
Preeclampsia is a pregnancy-induced complex of multiple pathological changes. Numerous stresses during pregnancy, including hypoxia, immune activation, inflammatory cytokines, and oxidative stress were reported as contributing factors to the preeclamptic pathology. Seeking common sensors of various stressors in preeclampsia is of new interest and can potentially benefit in disease prevention and treatment. Recent studies have highlighted the role of the Gadd45a protein as a stress sensor in preeclampsia. In response to various pathophysiological stressors, notably hypoxia, inflammatory cytokines, and AT1-AAs, Gadd45a activates Mkk3-p38 and or JNK signaling. This, in turn, results in immunological and inflammatory changes as well as triggering the production of circulating factors such as sFlt-1, which are believed to account for many of the pathophysiological-related symptoms of preeclampsia. Activation of inflammatory/immune responses in preeclampsia may function in a feedback loop to maintain elevated expression of Gadd45a protein.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
7.1 Stress and Preeclampsia
Preeclampsia, which affects approximately 5–8 % of all pregnancies, is one of the leading causes of maternal and fetal morbidity and mortality (Turner 2010; MacKay et al. 2001). It is a pregnancy-induced complex of multiple pathological changes, which are manifested as elevated blood pressure, proteinuria, and edema in the mid-late term of gestation (ACOG 2002). Multiple stresses were found contributing to the preeclamptic condition (Hubel 1999; Benyo et al. 2001; Teran et al. 2001).
7.1.1 Hypoxia
Hypoxia (i.e., placental ischemia) is essential in the pathogenesis of preeclampsia and is caused through a variety of mechanisms involved with abnormal placentation. Inadequate trophoblast invasion that results in deficient remodeling of the uterine spiral arteries is regarded as a primary cause of placental ischemia (Conrad and Benyo 1997). Poor placentation impairs the development of the early placenta and the maternal blood supply (Redman and Sargent 2005). This process starts from the 6th week of gestation and is prolonged to the latter two trimesters, eventually resulting in typical clinical presentations of preeclampsia, including intrauterine growth retardation (IUGR) (Redman and Sargent 2005).
Hypoperfusion can be both a cause and a consequence of abnormal placental development. A causal connection between poor placental perfusion, abnormal placental development, and preeclampsia is supported by the following evidences: medical conditions associated with vascular insufficiency (e.g., hypertension, diabetes, systemic lupus erythematosus, renal disease, acquired and inherited thrombophilias) increase the risk of abnormal placentation and preeclampsia (ACOG 2002; Dekker 1999). Recent updates, on the other hand, showed that reducing uteroplacental blood flow in pregnant rats can reproduce characteristic preeclamptic manifestations (Li et al. 2012; Makris et al. 2007).
One remarkable consequence of hypoxia is the endothelial cell dysfunction, which subsequently increases circulating factors such as fms-like tyrosine kinase receptor-1 (sFlt-1) and soluble endoglin (sENG) from the placenta and triggers preeclamptic pathology (Maynard et al. 2003). Both sFlt-1 and sENG were found elevated in the serum of preeclamptic patients as well as in their placentas. sFlt-1 is a splicing variant of the VEGF receptor and acts as a VEGF antagonist due to the absence of transmembrane and cytoplasmic domains, resulting in vessel constriction and high blood pressure (Maynard et al. 2005). Injecting sFlt-1 into pregnant rats generated systemic preeclamptic changes such as hypertension, proteinuria, and renal pathology (Maynard et al. 2003). sENG, a soluble TGF-β co-receptor, induces vascular permeability and hypertension in vivo, correlated with disease severity. Injection of sFlt-1 in combination with sENG into pregnant rats produced nephrotic-range proteinuria, severe hypertension, and biochemical evidence of HELLP syndrome (Venkatesha et al. 2006).
7.1.2 Immune Activation
7.1.2.1 Multiple Factors Triggers Immune Activation in Preeclampsia
Paternal Antigen: Retrospective studies have shown that preeclampsia occurs mostly in the first pregnancy. Likewise, partner change is correlated with increased risks of preeclamptic or hypertension in pregnancy (Zhang and Patel 2007). The prevailing hypothesis is that after the first pregnancy, the maternal immune system has “recognized” the paternal antigens and could tolerate the same antigens in subsequent pregnancies. Changing partner introduces new paternal antigens and with it a new risk for preeclampsia. The maternal immune system, therefore, has to reestablish an immune tolerance (Zhang and Patel 2007). Failure of this tolerance to occur may contribute to preeclampsia.
HLA System: Human trophoblast has a limited expression of strong transplantation antigens. These include nonpolymorphic HLA-E, F, and G (without signal paternal specificity) and HLA-C, on extravillous cytotrophoblast in interface II (with signal paternal specificity). It is reported that this interface regresses in the second half of pregnancy (Choudhury and Knapp 2001a, b). Since it is devoid of HLA expression at the third trimester, alloantigen-provoked pathological change occurs in the first half of pregnancy with the clinical presentation of preeclampsia in the late second or third trimester of the pregnancy.
Autoimmune Antibodies: Autoimmune antibodies were highlighted recently by numerous researches of their role in preeclampsia. Agonistic angiotensin II type 1 (AT1) receptor autoantibodies (AT1-AAs) that share the same AT1 receptor with angiotensin II (Wallukat et al. 1999; Zhou et al. 2008) and were found exclusively in peripheral blood of preeclamptic patients (Wallukat et al. 1999) are stressors that elicit preeclamptic symptoms (hypertension, proteinuria, renal damage, and sFlt-1 elevation) in vivo (Zhou et al. 2008). Therefore, triggering AT1 receptor signaling by circulating autoimmune antibodies (AT1-AAs) is notable evidence of how immune activation is involved in preeclamptic pathology (Zhou et al. 2008). In addition, angiotensin II by itself was elevated in preeclamptic placentas and increases systemic sensitivity to angiotensin II in preeclampsia (Shah 2005).
7.1.3 Inflammatory Cytokines
Although normal pregnancy evokes systemic inflammatory including innate immune responses which mainly take place in the third trimester (Redman et al. 1999), preeclampsia is associated with a more extreme maternal systemic inflammatory response (Christopher and Sargent 2004).
Tumor necrosis factor (TNF-α) is a multifunctional pro-inflammatory cytokine. It is produced chiefly by activated macrophages (Carswell et al. 1975) and can also be produced by other cells/tissues including human placentas (Wang and Walsh 1996; Kirwan et al. 2002). The primary role of TNF-α is regulating immune cells. TNF, as an endogenous pyrogen, induces fever. It elicits apoptotic cell death, sepsis, cachexia, and inflammation and inhibits tumorigenesis and viral replication (Idriss and Naismith 2000). It was reported that TNF-α was abnormally elevated in the peripheral blood of preeclamptic patients (Wang et al. 1996). Chronic infusion of TNF-α into normal pregnant rats results in significant increases in arterial pressure and a decrease in renal hemodynamics (Babbette et al. 2007). TNF-α infusion in pregnant rats also triggered AT1-AAs production (LaMarca et al. 2008), suggesting that TNF-α can cause both inflammatory and immune activation in preeclampsia.
IL-1, including IL-1α and IL-1β, is also an important inflammatory and immune regulator. Both IL-1α and IL-1β are produced by macrophages, monocytes, fibroblasts, and dendritic cells (Dinarello 2011). They play an important role against infection. IL-1 is also an endogenous pyrogen and regulates hematopoiesis. Increased IL-1 levels were found in the peripheral blood of preeclamptic patients with other inflammatory cytokines (Greer et al. 1994). Intracisternal or intravenous infusion of IL-1 beta increases blood pressure in a prostaglandin-dependent manner in rats (Takahashi et al. 1992).
IL-6 is secreted by T cells and macrophages (Kishimoto 2010). It is one of the most important mediators of fever and the main regulator of acute-phase response. Increased IL-6 levels were found in the serum of severe preeclamptic patients (Greer et al. 1994). Chronic infusion of IL-6 into normal pregnant rats resulted in similar effect as TNF-α, causing significant increases in arterial pressure and a decrease in renal hemodynamics (LaMarca et al. 2007). However, TNF-α activates the endothelin system in placental, renal, and vascular tissues, whereas IL-6 stimulates the renin–angiotensin system. In addition, these inflammatory cytokines may activate the sympathetic nervous system. They may also play an important role in causing hypertension in response to chronic reductions in uterine perfusion during pregnancy, by activating multiple neurohumoral and endothelial factors (LaMarca et al. 2007).
7.1.4 Oxidative Stress
Free radicals are atoms with an unpaired number of electrons that can be formed when oxygen interacts with certain molecules. Once formed, these highly reactive radicals can start a chain reaction. They react with and thus damage cellular components such as DNA or the cell membrane. The most common physiological radical is the superoxide anion. Sources of superoxide under physiological conditions include the enzymes nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, 5 cytochrome P450, and other oxidoreductases (Muller et al. 2007).
Oxidative stress (i.e., NADPH oxidase) is generated substantially at the maternal–fetal interface during pregnancy, particularly in the early trimester. It functions in the normal development of the placenta and contributes to the pathophysiology of pregnancy complications such as miscarriage, preeclampsia, intrauterine growth restriction (IUGR), and premature rupture of the membranes (Burton and Jauniaux 2004; Jauniaux et al. 2006). Unlike in normal pregnancy, oxidative stress and the systemic inflammatory response are more critical in preeclampsia (Redman and Sargent 2007). Preeclampsia, particularly early-onset preeclampsia, was associated with placental oxidative stress including increased concentrations of protein carbonyls, lipid peroxides, nitrotyrosine residues, and DNA oxidation (Myatt and Cui 2004; Burton et al. 2009). Moreover, early-onset preeclampsia, which is frequently associated with intrauterine growth retardation (IUGR), was reported with high levels of ER stress in the placenta (Burton et al. 2009).
Autoantibodies AT1-AAs also trigger oxidative stress in preeclampsia. They stimulate NADPH oxidase, resulting in an increase in ROS production (Dechend et al. 2003).
Cellular response to oxidative stress is via the mitogen-activated protein kinases (MAPK) pathway. For examples, ROS-induced activation of extracellular-regulated kinases (ERK1/2) generally promotes cell survival and proliferation, whereas stimulation of p38 and stress-activated protein kinase–c-Jun amino terminal kinases (SAPK–JNK) mostly induces apoptosis (Trachootham et al. 2008; Liebermann and Hoffman 2008).
7.2 The Role of Gadd45 Stress Sensors in Preeclampsia
Evidence accumulating in recent years has highlighted the role of the growth arrest and DNA damage-inducible 45 (Gadd45) family of genes as important sensors of environmental and physiological stress, including genotoxic damage (UV, X-ray), hypoxia, oxidative stress, and pro-inflammatory cytokines (Fornace et al. 1992; Liebermann and Hoffman 2002). Gadd45 proteins are, in essence, signal transducers that convert environmental and physiological stresses into various cellular stress responses including inflammation (Gupta et al. 2006), innate immunity (Gupta et al. 2006; Lu et al. 2004), and autoimmune diseases (Salvador et al. 2005). Gadd45 proteins bind to and regulate the activity of several downstream stress-response proteins (Liebermann and Hoffman 2002) such as MTK1 (MEKK4), an upstream activator of MKK3 and MKK6 that ultimately mediates activation of both p38 and JNK stress-response kinases (Takekawa and Saito 1998; Gupta et al. 2005).
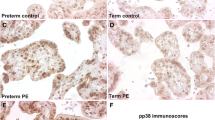
The first direct evidence showing Gadd45 as a stress sensor contributing to preeclampsia was via the placental examination. Placental tissues from both preeclamptic and normotensive (control) patients were examined for the mRNA levels of the Gadd45 family genes (a, b, and g). Although the expression of all three genes were elevated in preeclamptic placentas, the difference was statistically significant only for Gadd45a mRNA. In addition, Gadd45a protein was readily detectable only in preeclamptic placentas, and this elevation was independent of different BMI or race between the preeclamptic and control groups. Further, via immunohistochemical detection, Gadd45a protein was found localized in preeclamptic placentas, particularly in endothelial and trophoblast cells with the increased expression of Gadd45a downstream effector p38 protein. With dual immunofluorescence staining for both Gadd45a and sFlt-1(circulating factor and a key player in preeclampsia), the co-expression of these two proteins was targeted at the preeclamptic placental endothelial cells (Xiong et al. 2009).
7.2.1 Hypoxia and Gadd45a in Preeclampsia
As previously discussed, hypoxia is essential in the pathogenesis of preeclampsia. In vitro culture of both endothelial cells and placental explants showed that Gadd45a protein was induced with the downstream p38 protein phosphorylation under hypoxic circumstances. The activation of Gadd45a signaling caused elevation of sFlt-1 in the supernatant of cultured endothelial cells of placental explants. When Gadd45a expression was knocked down by specific Gadd45a RNAi, the elevation of sFlt-1 was depleted. The regulation of sFlt-1 secretion by Gaddd45a occurred via the p38 activation (Xiong et al. 2009, 2011).
7.2.2 AT1-AAs and Gadd45a in Preeclampsia
Angiotensin II is a vessel constrictor which causes increasing blood pressure and shares the same AT1 receptor with AT1-AAs. In order to study the interaction of Gadd45a and AT1-AAs in preeclampsia, angiotensin II was introduced to cultured placental explants. Treatment of placental explants with angiotensin II resulted in Gadd45a induction, p38 phosphorylation (i.e., activation), and elevation of sFlt-1 in the supernatant (Xiong et al. 2011). To establish a causal link between Gadd45a induction, p38 activation, and elevated secretion of sFlt-1, Gadd45a expression was knocked down with Gadd45a RNAi in the placental explants. RNAi-mediated knockdown of Gadd45a abolished angiotensin II-induced p38 activation and significantly reduced sFlt-1 levels in culture. Furthermore, blocking p38 activation with the specific chemical inhibitor also resulted in attenuated levels of sFlt-1 in the culture medium. On the other hand, blocking the activation of JNK, which is also a downstream effector of Gadd45a, did not attenuate sFlt-1 secretion (Xiong et al. 2011).
7.2.3 Inflammatory Cytokines and Gadd45a in Preeclampsia
Two important preeclampsia-associated inflammatory cytokines IL-6 and TNF-α were examined with Gadd45a stress-response cascade.
Incubation with IL-6 induced Gadd45a in placental explants is associated with activation of the downstream effectors p38 and phospho-JNK as well as elevated levels of sFlt-1 in the culture medium. RNAi-mediated knockdown of Gadd45a abolished p38 activation and significantly reduced sFlt-1 levels in the culture medium following IL-6 treatment. Blocking p38 also attenuated sFlt-1 secretion in the culture medium, whereas blocking JNK activation had no effect on sFlt-1 levels (Xiong et al. 2011).
Induction of Gadd45a in response to TNF-α was prompt (peak time at 10 or 20 min), compared to the other stressors discussed above. In addition, it was associated with both p38 and JNK activation and increased sFlt-1 levels in the culture medium. However, unlike other pre-inflammatory stressors, it was the inhibition of JNK activation, but not p38 activation, that attenuated sFlt-1 secretion (Xiong et al. 2011).
7.3 Conclusions
Gadd45a protein works as a stressor sensor in preeclampsia. In response to various pathophysiological stressors, notably hypoxia, inflammatory cytokines, and AT1-AAs, Gadd45a activates Mkk3-p38 and/or JNK signaling. This, in turn, results in immunological and inflammatory changes as well as triggering the production of circulating factors such as sFlt-1, which are believed to account for many of the pathophysiological-related symptoms of preeclampsia (Maynard et al. 2003). Inflammatory/immune activation in preeclampsia may function in a feedback loop to maintain elevated expression of Gadd45a protein (Fig. 7.1).

Fig. 7.1
References
ACOG Committee on Practice Bulletins – Obstetrics (2002) ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet Gynecol 99:159–167
Benyo DF, Smarason A, Redman CW, Sims C, Conrad KP (2001) Expression of inflammatory cytokines in placentas from women with preeclampsia. J Clin Endocrinol Metab 86(6):2505–2512
Burton GJ, Jauniaux E (2004) Placental oxidative stress; from miscarriage to preeclampsia. J Soc Gynecol Investig 11:342–352
Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS (2009) Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 30(Suppl A):S43–S48
Carswell EA, Old RL, Kassel S, Green N, Fiore WF, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumors (activated macrophage). Proc Natl Acad Sci USA 72(9):3666–3670
Choudhury SR, Knapp LA (2001a) Human reproductive failure I: immunological factors. Hum Reprod Update 7(2):113–134
Choudhury SR, Knapp LA (2001b) Human reproductive failure II: immunogenetic and interacting factors. Hum Reprod Update 7(2):135–160
Conrad KP, Benyo DF (1997) Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol 37(3):240–249
Dechend R, Viedt C, Müller DN, Ugele B, Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, Fiebeler A, Homuth V, Dietz R, Haller H, Kreuzer J, Luft FC (2003) AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 107(12):1632–1639
Dekker GA (1999) Risk factors for preeclampsia. Clin Obstet Gynecol 42:422
Dinarello CA (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117(14):3720–3732
Fornace AJ Jr, Jackman J, Hollander MC, Hoffman-Liebermann B, Liebermann DA (1992) Genotoxic-stress-response genes and growth-arrest genes. gadd, MyD, and other genes induced by treatments eliciting growth arrest. Ann N Y Acad Sci 663:139–153
Greer IA, Lyall F, Perera T, Boswell F, Macara LM (1994) Increased concentrations of cytokines interleukin-6 and interleukin-1 receptor antagonist in plasma of women with preeclampsia: a mechanism for endothelial dysfunction? Obstet Gynecol 84(6):937–940
Gupta M, Gupta SK, Balliet AG, Hollander MC, Fornace AJ, Hoffman B, Liebermann DA (2005) hematopoietic cells from Gadd45a- and Gadd45b-deficient mice are sensitized to genotoxic-stress-induced apoptosis. Oncogene 24:7170–7179
Gupta M, Gupta SK, Hoffman B, Liebermann DA (2006) Gadd45a and gadd45b protect hematopoietic cells from UV induced apoptosis via distinct signaling pathways including p38 activation and JNK inhibition. J Biol Chem 281:17552–17558
Hubel CA (1999) Oxidative stress in the pathogenesis of preeclampsia. Proc Soc Exp Biol Med 222(3):222–235
Idriss HT, Naismith JH (2000) TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech 50(3):184–195
Jauniaux E, Poston L, Burton GJ (2006) Placental-related diseases of pregnancy: involvement of oxidative stress and implications in human evolution. Hum Reprod Update 12:747–755
Kirwan JP, Hauguel-De Mouzon S, Lepercq J, Challier JC, Huston-Presley L, Friedman JE, Kalhan SC, Catalano PM (2002) TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 51(7):2207–2213
Kishimoto T (2010) IL-6: from its discovery to clinical applications. Int Immunol 22(5):347–352
LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP (2007) Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep 9(6):480–485
Li J, LaMarca B, Reckelhoff JF (2012) A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol 303(1):H1–H8
Liebermann DA, Hoffman B (2002) Myeloid differentiation (MyD)/growth arrest DNA damage (GADD) genes in tumor suppression, immunity& inflammation. Leukemia 16:527–541
Liebermann DA, Hoffman B (2008) Gadd45 in stress signaling. J Mol Signal 3:15
MacKay AP, Berg CJ, Atrash HK (2001) Pregnancy-related mortality from preeclampsia and eclampsia. Obstet Gynecol 97(4):533–538
Makris A, Thornton C, Thompson J et al (2007) Uteroplacental ischemia results in proteinuric hypertension and elevated sFLT-1. Kidney Int 71:977
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111(5):649–658
Maynard SE, Venkatesha S, Thadhani R, Karumanchi SA (2005) Soluble Fms-like tyrosine kinase 1 and endothelial dysfunction in the pathogenesis of preeclampsia. Pediatr Res 57(5 Pt 2):1R–7R
Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (2007) Trends in oxidative aging theories. Free Radic Biol Med 43(4):477–503
Myatt L, Cui X (2004) Oxidative stress in the placenta. Histochem Cell Biol 122:369–382
Redman CW, Sargent IL (2003) Pre-eclampsia, the placenta and the maternal systemic inflammatory response – a review. Placenta 24(Suppl A):S21–S27
Redman CW, Sargent IL (2005) Latest advances in understanding preeclampsia. Science 308(5728):1592–1594
Redman CW, Sargent IL (2009) Placental stress and pre-eclampsia: a revised view. Placenta 30(Suppl A):S38–S42
Redman CW, Sacks GP, Sargent IL (1999) Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol 180(2 Pt 1):499–506
Salvador JM, Mittelstadt PR, Belova GI, Fornace AJ Jr, Ashwell JD (2005) The autoimmune suppressor Gadd45alpha inhibits the T cell alternative p38 activation pathway. Nat Immunol 6:396–402
Shah DM (2005) Role of the renin-angiotensin system in the pathogenesis of preeclampsia. Am J Physiol Renal Physiol 288(4):F614–F625
Takahashi H, Nishimura M, Sakamoto M, Ikegaki I, Nakanishi T, Yoshimura M (1992) Effects of interleukin-1 beta on blood pressure, sympathetic nerve activity, and pituitary endocrine functions in anesthetized rats. Am J Hypertens 5(4 Pt 1):224–229
Takekawa M, Saito H (1998) A family of stress-inducible GADD45-like proteins mediates activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95:521–530
Teran E, Escudero C, Moya W, Flores M, Vallance P, Lopez-Jaramillo P (2001) Elevated C-reactive protein and pro-inflammatory cytokines in Andean women with pre-eclampsia. Int J Gynaecol Obstet 75(3):243–249
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P (2008) Redox regulation of cell survival. Antioxid Redox Signal 10(8):1343–1374
Turner JA (2010) Diagnosis and management of pre-eclampsia: an update. Int J Womens Health 2:327–337
Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D’Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA (2006) Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 12(6):642–649
Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC (1999) Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 3:945–952
Wang Y, Walsh SW (1996) TNF alpha concentrations and mRNA expression are increased in preeclamptic placentas. J Reprod Immunol 32(2):157–169
Zhang J, Patel G (2007) Partner change and perinatal outcomes: a systematic review. Paediatr Perinat Epidemiol 21(Suppl 1):46–57
Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y (2008) Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med 14:855–862
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Geifman-Holtzman, O., Xiong, Y., Holtzman, E.J. (2013). Gadd45 Stress Sensors in Preeclampsia. In: Liebermann, D., Hoffman, B. (eds) Gadd45 Stress Sensor Genes. Advances in Experimental Medicine and Biology, vol 793. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8289-5_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8289-5_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8288-8
Online ISBN: 978-1-4614-8289-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)