
Abstract
Many different types of organisms synthesize hydrocarbons in nature, but for all their ubiquity, the biochemical and genetic bases for how these compounds are synthesized are not well understood. Several biochemical mechanisms have been proposed for non-isoprenoid hydrocarbon biosynthesis, most notably the head-to-head condensation and elongation-decarboxylation pathways from fatty acid precursors, but definitive characterization of these and other possible mechanisms have largely remained elusive. This review explores the possible metabolic pathways that various plant, algal, and microbial species use to synthesize linear hydrocarbons and the genetic factors that are involved in regulating those pathways.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Fatty Acid Synthesis
- Very Long Chain Fatty Acid
- Precursor Fatty Acid
- Botryococcus Braunii
- Synechococcus Elongatus
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
It has long been known that hydrocarbons from plants, algal, and microbial organisms are present in crude oil deposits [7, 23]. This was originally recognized for the possibility of using these molecules as evolutionary indicators, since evolutionary processes can be deduced from the biochemistry of distinct compounds [11]. While the substantial portion of these deposits is the result of the breakdown of biomass over time, these original hydrocarbons maintain their integrity due to their extremely stable nature [6]. In some cases, these hydrocarbon markers are still viable in deposits that are over three billion years old [3]. Recently though, interest in the biosynthesis of hydrocarbons has grown substantially due to the prospect of using hydrocarbon producing organisms as a source of renewable fuels.
Long-chain hydrocarbons, consisting of alkanes and alkenes, are promising targets for biofuels as they are already the major constituents of petroleum-based fuels that are in use today. Synthesis of these compounds in nature is accomplished by an assortment of different types of organisms and play a variety of roles, as insect epicuticular waxes and pheromones [15, 30], protective coatings on fungal spores [35], epicuticular waxes covering stems and leaves in higher plants [18, 26], and various other purposes in many microorganisms, including blue-green and green algae, cyanobacteria, and yeasts, among others [11, 16].
2 Hydrocarbon Biosynthesis
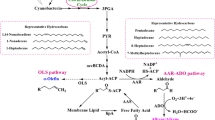
It has been well documented that fatty acid metabolites are precursors to long-chain hydrocarbons. This has been demonstrated through radiocarbon labeling studies in a multitude of species [17, 33]. While many details are not well characterized, there appears to be two main metabolic pathways for the synthesis of hydrocarbons from fatty acids (Fig. 1).

Possible pathways for long-chain hydrocarbon biosynthesis from a fatty acid precursor
The first, primarily found in bacteria, is head-to-head condensation of two fatty acids [2, 4, 22]. In this putative pathway, a bond is formed between the carboxyl carbon of one fatty acid and the α-carbon of a second fatty acid. The resulting compound is then decarboxylated to form a hydrocarbon that is one carbon less than the total carbons from the two fatty acids [1]. The ensuing hydrocarbon chain length would be odd assuming the number of carbons in the fatty acid groups was even, as is typically the case.
The second pathway, evidence of which has been found in many plant and algal species [10, 20, 25], is the elongation-decarboxylation mechanism. It is characterized by the elongation of the precursor fatty acid (primarily C16 or C18) to a very long-chain fatty acid (VLCFA, longer than C22) in a process analogous to de novo fatty acid synthesis. There are several possible mechanisms from which the hydrocarbon could then be formed from the VLCFA. The first is decarboxylation, in which the carboxylic end of the VLCFA is cleaved to form a hydrocarbon that is one carbon shorter than the VLCFA. A second possibility is reduction of the carboxylic end of the VLCFA to an aldehyde, with subsequent decarbonylation to a hydrocarbon identical to that formed from decarboxylation. The final mechanism is reduction of the aldehyde to a primary alcohol, which could then be dehydrated to the resulting hydrocarbon without the loss of the terminal carbon. These pathways are discussed in more detail in the ensuing sections.
2.1 Head-to-Head Condensation
Early investigations of the head-to-head condensation mechanism of hydrocarbon biosynthesis took place in the late 1960s. Incorporation of 14C-labeled fatty acids in the tobacco plant [17] and bacterium Sarcina lutea [1] suggested that two fatty acids were being incorporated into the final hydrocarbon. Additionally, cell extracts have shown that the carboxyl group for only one of the fatty acids was being lost as carbon dioxide while the other was retained in the hydrocarbon. Together, these results were consistent with head-to-head condensation over alternative mechanisms such as elongation-decarboxylation, but the underlying biochemistry and genetics were not revealed with any clarity. Only recently have further studies shed light on the underlying basis for this chemistry.
One such study looked at hydrocarbon synthesis in the bacterium Micrococcus luteus, a close relative to the previously studied S. lutea, for which a genome sequence was available. A search for homologs of the KAS enzymes responsible for condensation of fatty acyl groups in de novo fatty acid synthesis resulted in a single target gene (Mlut_13230) that showed promise in heterologous expression in E. coli. Due to its sequence similarity to KAS III, the enzyme responsible for catalyzing the initial condensation reaction in fatty acid synthesis, a mechanism for head-to-head condensation was proposed that is very similar to the fatty acid synthesis reaction (Fig. 2). One fatty acid is initially oxidized to a β-ketoacyl-CoA (similar to malonyl-CoA), followed by a decarboxylative Claisen condensation with the second fatty acid in the form of fatty acyl-CoA, catalyzed by Mlut_13230. The condensed product is theorized to be a diketone, with three subsequent reductions and two dehydrations needed to form the monounsaturated hydrocarbon, reactions similar to the reduction and dehydration reactions that take place in fatty acid synthesis. In vitro studies with the purified Mlut_13230 protein, tetradecanoyl-CoA, and E. coli lysate resulted in unsaturated C27 monoketones, a possible intermediate between the diketone and the final hydrocarbon in Fig. 2 [4]. Enzymes catalyzing the initial oxidation steps, as well as the reduction and dehydration steps at the end of the possible pathway were not found, however, leaving many of the proposed mechanisms unsupported. Furthermore, two additional M. luteus genes (Mlut_13240 and Mlut_13250) were found to be required for successful heterologous expression in E. coli, yet no clear function for them was found.

Possible head-to-head condensation mechanism, where R1 and R2 are alkyl groups of the precursor fatty acid. The monoketones observed in vitro were hypothesized to occur as an intermediate in the reduction and dehydration steps between the diketone and the hydrocarbon [4]
Soon after the preceding pathway was put forth, a separate investigation came out with substantially different conclusions as to the underlying mechanisms for head-to-head condensation. In that study, an oleABCD gene family was identified as necessary for head-to-head hydrocarbon biosynthesis. Of the 3,558 genomes surveyed, only 69 were qualified as potential candidates for having hydrocarbon producing ole genes. All the positive results were from bacterial genomes, while none of the more than 2,100 Eukaryota and 84 Archaea genomes showed strong evidence for containing the oleABCD gene family [31]. Of the four ole genes identified as being integral to head-to-head condensation, oleA was the primary candidate for the catalysis of the condensing reaction, as it was homologous to the thiolase superfamily of condensing enzymes. Additionally, the Shewanella oneidensis oleA gene was found to have 31% sequence similarity to the corresponding Mlut_13230 condensing enzyme in M. luteus [32]. Up to six different gene arrangements were found for oleABCD, the highest population consisted of an unbroken cluster of all four genes arranged continuously, while second was the fusion of the oleBC genes into one gene within the larger cluster (a configuration that M. luteus was shown to exhibit, hence the three genes found as in the previous study). Various other cluster arrangements and dispersion of individual genes throughout the genome were observed as well, but with much less species exhibiting those characteristics. Despite the large difference in populations, each of the six arrangements of genes had at least one bacterial species that produced hydrocarbons. While different compounds were created, all were greater in length than C23 and had at least one unsaturation, consistent with head-to-head synthesis. Among the 14 different types of bacteria tested, 10 had a single product with a carbon length of 31 with nine double bonds known as 3, 6, 9, 12, 15, 19, 22, 25, 28-hentriaconanonaene. This is consistent with the condensation of two molecules of the same hexadecatetraenoic acid, a compound known to be an intermediate in the biosynthesis of long-chain polyunsaturated fatty acids (PUFAs) that are produced in various Shewanella strains that were a part of a parallel study [32]. This link was confirmed when the PUFA synthesis pathway was blocked in a mutant strain and no hydrocarbons could be detected, while restoration of the blocked gene restored hydrocarbon biosynthesis.
The fact that many of the bacteria produced just a single hydrocarbon compound from a relatively non-abundant fatty acid precursor points to a probable substrate specificity for the oleA protein. A series of heterologous expression experiments of Stenotrophomona maltophilias oleA in Shewanella oneidensis showed that not only was oleA selective, but the entire gene cluster was as well. Additionally, the S. maltophilia oleA showed specificity for saturated and monounsaturated fatty acids by synthesizing mono and di-unsaturated ketones. The corresponding oleBCD gene set was not present to metabolize the intermediates, so the ketones were the final product of the heterologous expression. The native oleA was only able to produce the C31 ketone with eight double bonds when a mutant S. oneidensis was created without oleBCD genes. When present, the final product was the hentriaconanonaene [31].
While the previously presented mechanism proposed a decarboxylative Claisen condensation due to homology of the oleA gene to KAS III, the thiolase superfamily of genes catalyzes both decarboxylative and nondecarboxylative condensation reactions [14]. An alternative mechanism consisting of the nondecarboxylative reaction was therefore offered (Fig. 3). Based on the experimental data, it is believed that oleA catalyzes the first reaction where the two fatty acyl groups are condensed to the β-keto thioester intermediate. Absence of the oleBCD genes could allow the β-keto intermediate to undergo hydrolysis to an unstable β-keto acid, which can spontaneously decarboxylate to the ketones observed in the heterologous expression experiments discussed previously [32]. The presence of the appropriate oleBCD genes would preclude this branch mechanism, channeling the β-keto thioester intermediate to the hydrocarbon instead. There is some indication of the possible underlying mechanisms based on the known gene families of the oleBCD enzymes, but these individual steps have not been described at this point.

A second possible fatty acyl head-to-head condensation mechanism. The nondecarboxylative Claisen condensation takes place first through the action of oleA, producing the β-keto thioester. OleBCD presumably catalyze the reactions that lead to the hydrocarbon, while in their absence the β-keto thioester could undergo hydrolysis and then spontaneously decarboxylate to the ketone (adapted from Sukovich et al. (22))
2.2 Elongation
Investigation of the so-called “elongation decarboxylation” pathway has basically followed the same course as that of head-to-head condensation. Early studies indicating this mechanism were first accomplished in the 1960s, with incorporation of radiocarbon-labeled fatty acids or acetates used as the main tools for distinguishing between the condensation and elongation mechanisms [2, 18, 19]. This mechanism is characterized by elongation of precursor fatty acids via the fatty acid elongation pathway to a very long chain fatty acid (VLCFA). An initial fatty acid, such as palmitate (16:0) or oleate (18:1) is elongated in the same manner as de novo fatty acid synthesis, with a couple of exceptions [27]. First, malonyl-CoA units are not converted to malonyl-ACP prior to condensation with the acyl-CoA. Secondly, instead of being located in the cytoplasm, as in eukaryotes and bacteria or in the plastids for photosynthetic organisms for de novo fatty acid synthase, the VLCFA synthase is located in the membrane of the endoplasmic reticulum (ER). The mechanism for elongation behaves mostly in the same way as de novo synthesis though, with repeated malonyl-CoA units added to the growing acyl-CoA chain until the desired length is reached, typically 24–34 carbons long [28]. In order to make the hydrocarbon from the resulting VLCFA, the carboxylic end must be replaced. There are several possible mechanisms for which this can happen.
2.2.1 Decarboxylation
Until the 1980s, elongation-decarboxylation was the only mechanism considered for converting VLCFAs to hydrocarbons. While it was known that the carboxylic carbon was lost in hydrocarbon synthesis, the direct cleavage of the carboxyl end was not considered feasible without an electron withdrawing group to stabilize the negative charge on the leaving CO2 [9]. It was thought that such a group would exist in the immediate precursor to the decarboxylic reaction, but this has never been detected [21]. In the 1970s, experimental results started to show that perhaps an aldehyde was a precursor to the hydrocarbon instead [5], but the mechanism by which that would happen was unknown. It was not until the mid 1980s that a possible reduction-decarbonylation mechanism was first suggested [8].
2.2.2 Reduction-Decarbonylation
Inhibition of alkane synthesis in young leaves of the plant Pisum sativum was the first indication that aldehydes were an intermediate to hydrocarbon synthesis, though this was not recognized at the time [5]. Addition of metal ion chelators inhibited the normal synthesis of the alkane hentriacontane, with a C32 aldehyde accumulating instead. Since decarboxylation was the consensus mechanism at the time, this was thought to be a side reaction of an intermediate in that pathway. Subsequent experiments in P. sativum have shown that the aldehyde is likely the immediate precursor to the hydrocarbon. In cell-free preparations, the microsomal fraction (inclusive of ER remnants) was found to convert octadecanoyl-CoA to the aldehyde octadecanal along with some hydrocarbon synthesis activity. The heavy particulate fraction (inclusive of cell wall, cutin, and other membranes) did not have any activity with the acyl-CoA, but readily converted exogenous aldehyde to alkanes [8]. In concert with hydrocarbon synthesis activity, CO was measured as a byproduct in nearly a 1:1 mol ratio with produced hydrocarbon while no CO2 was indicated, further demonstrating a decarbonylation mechanism over decarboxylation.
Similar enzymatic activity was observed in the algae Botryococcus braunii. A purified aldehyde decarbonylase was shown to produce CO in nearly a 1:1 stoichiometry with the alkane formed. Crude preparations, however, produced mainly CO2 instead of CO, something not observed in the previous P. sativum (pea) experiments. It was theorized that the algae had evolved a CO oxidizing mechanism to circumvent the toxic effects of CO, while the probable location of the decarbonylase in the cuticle of peas would allow for a direct venting to atmosphere and no need for the oxidizing action [9].
Several other organisms have been studied for the presence of a decarbonylase as well, including the cyanobacteria Synechococcus elongatus and plant Arabidopsis thaliana. In the latter, a cer1 wax deficient mutant was found to have an increase in aldehydes and a drop in alkanes from the wild-type [12]. While one would expect this behavior if an aldehyde decarbonylase was deficient in that mutant, the same relationship could be seen if the aldehydes were a side product of the main pathway [28]. Subsequent analysis of the wax production in the cer1 mutant showed that the cer1 deletion could not have been simply a block of the decarbonylation reaction [13]. In the cyanobacteria S. elongates, two candidate genes for alkane biosynthesis, PCC7942_orf1593 and orf1594 were identified as the aldehyde decarbonylase and acyl-CoA reductase, respectively [29]. Heterologous expression of the proposed reductase _orf1594 in E. coli produced even-chained fatty aldehydes and alcohols, while expression of the two together resulted in odd-chained alkanes and alkenes. As would be expected when no precursor was present, expression of _orf1593 alone was indistinguishable from the negative control. Expression of _orf1593 genes from 15 other species of cyanobacteria with _orf1594 from S. elongatus produced the same combination of results [29].
While the ability to make alkanes makes the elongation-decarbonylation mechanism unique, alkenes are also a possible hydrocarbon product from this pathway. However, a potential mechanism for their synthesis is often ignored. Does VLCFA elongation start with an unsaturated fatty acid or is the VLCFA desaturated in the process of being reduced and decarbonylated? A recent study looking at wax composition in maize silks suggests that both possibilities may be true. When the double bond was located in the elongated portion (that which was added in VLCFA synthesis) of the hydrocarbon, it was determined that the precursor fatty acid was elongated first and then desaturated. Alternatively, if the double bond was in the original fatty acid portion of the molecule, that position was consistent with the particular unsaturated fatty acid that had entered VLCFA elongation [25].
2.2.3 Reduction-Dehydration
In 2004, a new reduction pathway was proposed that does not result in the loss of the carboxylic end of a VLCFA [24]. Moreover, the bacterium being studied, Vibrio furnisii M1, produced even-chained hydrocarbons, a rarity from the elongation pathway. C14-labeling studies at the 1-position of the precursor hexadecanoic acid showed incorporation of label in hexadecanal, hexadecanol, as well as the hydrocarbon hexadecane in cell extracts. This indicates that a consecutive reduction from the fatty acid to an aldehyde to a primary alcohol is possible, with dehydration of the alcohol forming the hydrocarbon.
A follow-up study was done in 2007 by a different group that seemed to contradict the earlier findings. Several different strains of V. furnissii were tested for hydrocarbon synthesis in both cell extracts and in intact cells. However, none produced hydrocarbons, much less in the unique manner described previously. Additionally, the complete genome from V. furnissii was sequenced yet no alkane biosynthetic or degradation genes were identified [34].
3 Conclusion
The mechanisms by which organisms synthesize hydrocarbons have been studied for nearly five decades. Two main synthesis pathways have been identified as the most prominent, with head-to-head condensation of fatty acids exhibited primarily in prokaryote and elongation-decarbonylation existing throughout the animal domains, but principally in eukarya. Recent genetic approaches have shed light on the mechanisms by which hydrocarbons are produced that traditional techniques could not. However, until the key enzymes involved in these pathways are cloned and fully characterized, the mechanistic details of how fatty acids are converted to hydrocarbons will remain unresolved.
References
Albro PW, Dittmer JC (1969) Biochemistry of long-chain nonisoprenoid hydrocarbons.3. metabolic relationship of long-chain fatty acids and hydrocarbons and other aspects of hydrocarbon metabolism in Sarcina lutea. Biochemistry 8:1913
Albro PW, Dittmer JC (1969) The biochemistry of long-chain, nonisoprenoid hydrocarbons. I. Characterization of the hydrocarbons of Sarcina lutea and the isolation of possible intermediates of biosynthesis. Biochemistry 8:394–404
Barghoorn ES (1971) The oldest fossils. Sci Am 224:30
Beller HR, Goh EB, Keasling JD (2009) Genes involved in long-chain alkene biosynthesis in micrococcus luteus. Appl Environ Microbiol 76:1212–1223
Buckner JS, Kolattukudy PE (1973) Specific inhibition of alkane synthesis with accumulation of very long-chain compounds by dithioerythritol, dithiothreitol, and mercaptoethanol in Pisum-Sativum. Arch Biochem Biophys 156:34–45
Calvin M (1969) Chemical evolution; molecular evolution towards the origin of living systems on the earth and elsewhere. Oxford University Press, New York
Cane RF (1969) Coorongite and the genesis of oil shale. Geochim Cosmochim Acta 33:257
Cheesbrough TM, Kolattukudy PE (1984) Alkane biosynthesis by decarbonylation of aldehydes catalyzed by a particulate preparation from Pisum-Sativum. In: Proceedings of the national academy of sciences of the United States of America-biological sciences, vol 81. pp 6613–6617
Dennis M, Kolattukudy PE (1992) A Cobalt-Porphyrin enzyme converts a fatty aldehyde to a Hydrocarbon and Co. Proc Natl Acad Sci USA 89:5306–5310
Dennis MW, Kolattukudy PE (1991) Alkane biosynthesis by decarbonylation of aldehyde catalyzed by a microsomal preparation from Botryococcus braunii. Arch Biochem Biophys 287:268
Han J, Calvin M (1969) Hydrocarbon distribution of algae and bacteria, and microbiological activity in sediments. Proc Natl Acad Sci USA 64:436–443
Hannoufa A, Mcnevin J, Lemieux B (1993) Epicuticular Waxes of Eceriferum Mutants of Arabidopsis-Thaliana. Phytochemistry 33:851–855
Hansen JD, Pyee J, Xia Y, Wen TJ, Robertson DS, Kolattukudy PE, Nikolau BJ, Schnable PS (1997) The glossy1 locus of maize and an epidermis-specific cDNA from Kleinia odora define a class of receptor-like proteins required for the normal accumulation of cuticular waxes. Plant Physiol 113:1091–1100
Heath RJ, Rock CO (2002) The Claisen condensation in biology. Nat Prod Rep 19:581–596
Howard R, Howard RW (2005) Ecological, behavioral, and biochemical aspects of insect hydrocarbons. Annu Rev Entomol 50:371
Kalacheva GS, Zhila NO, Volova TG, Gladyshev MI (2002) The effect of temperature on the lipid composition of Botryococcus. Mikrobiologiia 71:336–344
Kaneda T (1968) Biosynthesis of long-chain hydrocarbons. 2. Studies on biosynthetic pathway in tobacco. Biochemistry 7:1194–1202
Kolattukudy PE (1966) Biosynthesis of wax in Brassica oleracea. Relation of fatty acids to wax. Biochemistry 5:2265–2275
Kolattukudy PE (1967) Mechanisms of synthesis of waxy esters in Broccoli (Brassica Oleracea). Biochemistry 6:2705–2717
Kolattukudy PE (1968) Tests whether a head to head condensation mechanism occurs in the biosynthesis of n-hentriacontane, the paraffin of spinach and pea leaves. Plant Physiol 43:1466
Ladygina N, Dedyukhina E, Vainshtein M (2006) A review on microbial synthesis of hydrocarbons. Process Biochem 41:1001–1014
Markey SP, Tornabene TG (1971) Characterization of branched monounsaturated hydrocarbons of Sarcina lutea and Sarcina flava. Lipids 6:190–195
Meinschein WG (1959) Origin of petroleum. The American association of petroleum geologists bulletin 43:925
Park MO (2005) New pathway for long-chain n-alkane synthesis via 1-alcohol in Vibrio furnissii M1. Biochem J 187:1426–1429 Feb 2005
Perera MADN, Qin WM, Yandeau-Nelson M, Fan L, Dixon P, Nikolau BJ (2010) Biological origins of normal-chain hydrocarbons: a pathway model based on cuticular wax analyses of maize silks. Plant J 64:618–632
Post-Beittenmiller D (1996) Biochemistry and molecular biology of wax production in plants. Annu Rev Plant Physiol Plant Mol Biol 47:405–430
Riezman H (2007) The long and short of fatty acid synthesis. Cell 130:587–588
Samuels L, Kunst L, Jetter R (2008) Sealing plant surfaces: cuticular wax formation by epidermal cells. Annu Rev Plant Biol 59:683–707
Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB (2010) Microbial Biosynthesis of Alkanes. Science 329:559–562
Singer T, Singer TL (1998) Roles of hydrocarbons in the recognition systems of insects. Am Zool 38:394
Sukovich DJ, Seffernick JL, Richman JE, Gralnick JA, Wackett LP (2010) Widespread head-to-head hydrocarbon biosynthesis in bacteria and role of OleA. Appl Environ Microbiol 76:3850–3862
Sukovich DJ, Seffernick JL, Richman JE, Hunt KA, Gralnick JA, Wackett LP (2010) Structure, function, and insights into the biosynthesis of a head-to-head hydrocarbon in Shewanella oneidensis strain MR-1. Appl Environ Microbiol 76:3842–3849
Tornabene TG, Oro J (1967) 14c incorporation into fatty acids and aliphatic hydrocarbons of Sarcina lutea. J Bacteriol 94:349–358
Wackett LP, Frias JA, Seffernick JL, Sukovich DJ, Cameron SM (2007) Genomic and biochemical studies demonstrating the absence of an alkane-producing phenotype in Vibrio furnissii M1. Appl Environ Microbiol 73:7192–7198
Weete J, Weete JD (1972) Aliphatic hydrocarbons of the fungi. Phytochemistry 11:1201
Acknowledgments
This work was supported in part by National Science Foundation Grant EFRI-0938157. M. Brown is an Iowa State University Plant Sciences Institute Fellow.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag London Limited
About this chapter
Cite this chapter
Brown, M., Shanks, J. (2012). Linear Hydrocarbon Producing Pathways in Plants, Algae and Microbes. In: Gopalakrishnan, K., van Leeuwen, J., Brown, R. (eds) Sustainable Bioenergy and Bioproducts. Green Energy and Technology. Springer, London. https://doi.org/10.1007/978-1-4471-2324-8_1
Download citation
DOI: https://doi.org/10.1007/978-1-4471-2324-8_1
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-2323-1
Online ISBN: 978-1-4471-2324-8
eBook Packages: EngineeringEngineering (R0)