
Abstract
Bacteriophages, or phages for short, are viruses that infect bacteria and are considered the main regulators of microbial balance on Earth. These viruses are extremely specific, and their long-term survivability and ability to reproduce quickly in suitable hosts play a major role in the preservation of a dynamic equilibrium amid the diverse variety of bacterial species in the Earth’s ecosystem.
The many advantages of bacteriophages make them valuable tools for the detection and identification of bacterial pathogens as well as potentially promising recognition elements in biosensors as they have distinct advantages over other biorecognition receptors. Among these advantages are the specificity of the interaction of this type of virus with its target host cell, its ability to lyse and kill its host, and its capacity to multiply during the infection process. In addition, they are ubiquitous in nature, harmless to humans, economically and easily produced, have a far longer shelf life as they withstand harsh environments, reducing the environmental limitations and enabling regeneration of the biosensor surface. They can also be immobilized onto transducing devices in much the same manner as antibodies or DNA probes. For these reasons, the present chapter will provide an overview of the techniques, methods, and assays that employ bacteriophages in biosensing applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Bacteriophages, or phages for short, are viruses that infect bacteria and are considered the main regulators of microbial balance on Earth as they total an estimated 1032 entities (Fig. 11.1) (Kutter and Sulakvelidze 2005). Bacteriophages are found in abundance in the soil, manure and feces, thermal vents, and water. These viruses are extremely specific, and their long-term survivability and ability to reproduce quickly in suitable hosts play a major role in the preservation of a dynamic equilibrium amid the diverse variety of bacterial species in the Earth’s ecosystem (Kutter and Sulakvelidze 2005).


Multiple bacteriophage attached to the outer surface of a bacterial cell (from http://www.Wikipedia.org)
The many advantages of bacteriophages make them valuable tools for the detection and identification of bacterial pathogens as well as potentially excellent vehicles for diagnostics and therapeutics of bacterial disease. Among these advantages are the specificity of the interaction of this type of virus with its target host cell, its ability to lyse and kill its host, and its capacity to multiply during the infection process.
Modern phage research is attributed to Frederick W. Twort who began serious bacteriological work in 1915. Twort was trying to grow Caccinia virus on agar media when he noticed the growth of many mucoid and glassy colonies of micrococci, which he identified as being bacterial contamination. However, when examined under the microscope, the colonies seemed to have disintegrated into small granules that could be dyed red with Giemsa stain (Twort 1915).
In 1917, Félix d’Herelle discovered microbes able to lyse bacteria and provoke their death, and thus called them “bacteriophages” (from Greek “to eat” bacteria) (D’Herelle 1917). D’Herelle later noted that phages were natural resistant agents to bacteria as phage titers increased in patients with infectious diseases when they started to convalesce (D’Herelle 1919a). In 1919 he went on to test phages as prophylaxis against the infection of chickens by Bacillus gallinarum. The results suggested that chickens treated with phages died less and recovered faster, and that subsequent infections by B. gallinarum were prevented (D’Herelle 1919b).
In 1954 Cherry et al. used a broad host range phage to detect the presence of Salmonella enterica and noted that the replication of phages in a plated bacterial culture resulted in the development of clear zones of cell lysis referred to as plaques (Fig. 11.2) (Cherry et al. 1954).


Bacteriophage plaques formed on a lawn of bacterial host cells (from http://www.Wikipedia.org)
Phages are parasites that use the bacterial machinery to direct their replication in the target host. Each phage particle, called a virion, encloses its genome in a protein or lipoprotein coat called a capsid. The capsid and the nucleic acid together form the nucleocapsid (Kutter and Sulakvelidze 2005). Phages are grouped in 12 families and multiple genera based on their morphology, nucleic acid homology and type, and serology (Mandeville et al. 2003). Moreover, phages can be further divided into two categories depending on their life cycles and means of propagation (Fig. 11.3) (Mandeville et al. 2003). The first group comprises the virulent (or lytic, or productive) phages that are only capable of lytic propagation consisting of infection of the host bacterium by the phage, replication of the phage genome, production of the phage structural components, and release of the newly assembled phages. More often than not, this process results in bacterial lysis and death. The second category comprises the temperate (or reductive) phages that are capable of lytic as well as lysogenic propagation where the genome will assume a quiescent state called a prophage, rendering the coexistence of the bacteriophage and host cell possible. This is achievable by incorporating the phage genome into the chromosome of the bacterium by a site-specific integration procedure (Mandeville et al. 2003). Advantages of temperate phages consist in shielding their hosts from infection by other phages, and inducing considerable changes in the properties of their host-bacteria, such as restriction systems and resistance to antibiotics (Kutter and Sulakvelidze 2005).


The lytic and lysogenic cycles of bacteriophage replication
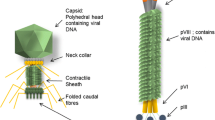
Tailed phages of the Caudovirales represent over 95% of the bacteriophages described in the literature. Half of the mass of the virion is made of double-stranded DNA while the other half is made of protein. The head is icosahedral and is constructed from multiple copies of one or two specific proteins (Kutter and Sulakvelidze 2005).
Tail morphology defines the three main families of tailed phages. The Siphoviridae family comprises approximately 60% of phages, the latter possessing long flexible tails. Twenty-five percent are Myoviridae with contractile tails, and the remaining 15% are Podoviridea with tiny, thick tails (Kutter and Sulakvelidze 2005). Archaephages, as their name suggests, infect Archae bacteria and have polymorphic shapes.
Tailless phages are divided into ten families differentiated by phage shape, the presence or absence of a lipid coat, the possession of single- or double-stranded DNA or RNA, whether or not they are released from their host cells by lysis, and whether or not they are segmented (Kutter and Sulakvelidze 2005).
Bacteriophages are promising recognition elements in biosensors as they have distinct advantages over antibodies as recognition receptors (Shabani et al. 2008, 2009; Balasubramanian et al. 2007). First, they are ubiquitous in nature and highly specific to bacteria while remaining harmless to humans. Second, phage production can be less complicated and less expensive than antibody production. Third, phages have a far longer shelf life as they withstand harsh environments, reducing the environmental limitations and enabling regeneration of the biosensor surface. Fourth, they can also be immobilized onto transducing devices in much the same manner as antibodies or DNA probes. For these reasons, the present chapter will review the developments of bacteriophages as Recognition Receptors in Biosensors.
2 Detection by Phage Amplification
Phages have stirred much attention in recent years as a means to detect bacteria in food and water and for disease surveillance. The reason remains that requirements for pathogenic bacterial detection are rapidity, sensitivity, specificity, and cost. Historically, conventional culture methods were and are still used for bacterial detection. These methods are, however, time consuming and require enrichment steps in order to visualize colonies on agar plates. Over the years, mass spectrometry and biochemical detection systems have been developed to ameliorate and increase the speed and sensitivity of culture methods but these methods retain negative aspects such as high cost and difficulty in handling.
Polymerase chain reaction (PCR) and immunoassays are other approaches designed to ameliorate the sensitivity of detection. However, PCR techniques detect the presence of nucleic acid and thus cannot differentiate living cells from dead cells. It is, however, possible to couple PCR and reverse transcription to detect mRNA, but high cost render RT-PCR difficult for regular use. Immunoassays such as ELISA are simple and rapid but they can have low sensitivity for detection of pathogens.
As mentioned above, bacteriophages bind specifically to their host and are optimized in a fashion that makes them ideal tools for the detection of bacterial pathogens. They are able to distinguish between live and dead cells, are robust, and easy to produce. Many detection techniques have been developed to exploit the specific binding ability of bacteriophages. These include the detection of intracellular components released from the bacterium after its lysis, the production of fluorescently labeled phages or fluorescently labeled antibodies directed against specifically adsorbing phage, inhibition of metabolism and growth, cell-wall recognition, and use of reporter phages. All of these methods will be discussed in this chapter.
3 Detection of Released Intracellular Components During Phage Lysis
The lysis of bacterial cells by bacteriophages culminates in the release of intracellular materials of the host bacterium and phage progeny. Two proteins are involved in the destruction of the bacterial cell wall. The first protein is a holin that perforates the cytoplasmic membrane by forming small pores in it. The second protein is an endolysin that will degrade the peptidoglycan provoking the release of a variety of detectable intracellular markers that can be easily measured (Schmelcher and Loessner 2008). The specificity of this detection method relies on the use of host-specific lytic phage.
3.1 Measurement of Adenosine Triphosphate Release
The most popular phage lysis method measures bacterial intracellular adenosine triphosphate (ATP). The amount of ATP in an average, actively metabolizing bacterial cell is quite consistent (approximately 10−15 g/cell) (Rees and Dodd 2006). The amount of ATP metabolized by a bacterial cell decreases rapidly after cell death. It is possible to determine the total number of viable cells by detecting the released intracellular ATP by the firefly luciferase/luciferin enzyme system after cell disrubtion using detergent-based agents (Stanley 1989). The amount of light produced can be determined by small luminometers and the quantity of ATP in the assay is directly proportional to the amount of light produced by the luciferase/luciferin reaction:
However, detection of specific pathogens is not possible using this method.
Specificity can be achieved by specific cell lysis, which can be accomplished by intact phage particles or purified recombinant phage endolysins (Schmelcher and Loessner 2008). Blasco et al. (1998), for example, developed the phage adenylate kinase (AK) assay. AK is an enzyme that directs the conversion of ADP to ATP and AMP:
When a cell bursts open after a phage infection event, AK is released as part of the cellular milieu. The Escherichia coli-specific phage NCIMB 10359 or the Salmonella-specific phage Newport was added to bacterial cultures, and, if suitable host cells were present, infection, lysis, and release of AK occurred. ADP was then added to drive the AK-mediated reaction toward the generation of ATP, and resulting ATP pools were detected using a commercially available firefly luciferase assay. Detection limits approached 104 cfu/mL of E. coli or Salmonella within an assay time of less than 2 h. These less than optimum detection limits were attributed to the high unspecific ATP background in such samples. Wu et al. (2001) later optimized assay incubation times and phage concentrations to increase detection limits to 103 cfu/mL. Squirrel et al. (2002) improved the sensitivity of the assay further by capturing and concentrating the pathogens of interest using immunomagnetic beads coated with antibodies before performing the phage-mediated bioluminescence AK assay.
Stewart et al. (1996) employed Ply 118 purified recombinant phage endolysins for specific lysing of Listeria monocytogenes and coupled cellular lysis to a bioluminescence-based ATP assay. Schuch et al. (2002) similarly detected Bacillus anthracis using the PlyG recombinant endolysins. They reported detection limits as low as 100 spores in 60 min (after incubation with germination solution) and 2.5 × 103 spores after 10 min.
3.2 Measurement of Enzymes and Other Cytoplasmic Markers
Other intracellular constituents can be used to monitor phage–cell lysis such as β-galactosidase and the endolysins that are produced as late gene products during phage replication to release progeny phage from the bacterial cells. Neufeld et al. (2003) used electrochemistry to measure amperometric changes in solution due to phage-mediated cell lysis. Infection of E. coli by a lytic version of phage lambda ultimately leads to the release of cellular components, such as the enzyme β-d-galactosidase. β-d-galactosidase can be measured amperometrically with a potentiostat via the addition of the substrate p-aminophenyl-β-d-galactopyranoside (β-PAPG) to yield the product p-aminophenol, which is oxidized at the carbon anode. E. coli could be detected within 6–8 h at a detection limit of 1 cfu/100 mL. Yemini et al. (2007) used the same principle to detect Bacillus cereus, where lysis by phage B1-7064 instigated cellular release of the enzyme α-glucosidase, as well as Mycobacterium smegmatis using phage D29 and the cellular release of β-glucosidase. Theoretically and advantageously, any phage/host combination can be detected using this method as long as the phage is lytic and an appropriate electrochemically detectable enzymatic marker is released by the target cell. However, a single phage would likely be insufficient to infect across the spectrum of target bacterial cells desired and false negative signals arising from cross-infections or naturally lysing cells would have to be accounted for. Neufeld et al. (2005) addressed some of these concerns in their assays using a phage-encoded alkaline phosphatase enzyme that had to be delivered to the host cell in order to be expressed. Thus, only after an active infection event would the enzyme be synthesized and then later released by the cell during lysis. This assay could detect a single E. coli cfu/mL in less than 3 h in both pure and mixed cultures.
3.3 Measurement of Phage Progeny
Since phage-infected cells ultimately synthesize and then release often times large numbers of progeny phage, a phage infection event can be assayed for simply by monitoring for a release and accumulation of progeny phage. The most basic format of these assays involves combining wild-type lytic phage with a bacterial culture. If bacterial hosts infectable by the phage are present, then progeny phage are produced and the resulting amplification in phage numbers serves as the signal for that infection occurrence. The technique was first demonstrated by Hirsh and Martin (1983) who used phage Felix-01 to detect Salmonella. Felix-01 was added to a sample, and if Salmonella were present, infection occurred and progeny phage was produced. The resulting amplification in phage numbers was detected by high-performance liquid chromatography (HPLC). Detection limits were fairly poor at 106 Salmonella per gram or mL sample. Stewart et al. (1998) demonstrated a more sensitive phage amplification assay again using phage Felix O-1 for detecting Salmonella as well as phages NCIMB 10116 and 10884 for detecting Pseudomonas aeruginosa. Phages were combined with bacterial cultures and incubated for up to 25 min to allow for infection to occur. Residual phages, i.e., those that had not participated in an active infection, were then destroyed via the addition of a virucide, in this case, pomegranate rind. After 3 min, virucidal activity was neutralized and “helper cells” were added to provide fresh hosts for the new progeny phage to infect. A standard top agar plaque plate was then prepared, where each plaque ideally represented a single phage/host infection event. By enumerating plaques, an estimate of the number of phages present could be calculated. Within 4 h, this assay could detect as few as 600 S. typhimurium or 40 P. aeruginosa cells/mL.
Favrin et al. (2001) used phage SJ2 for the detection of Salmonella Enteritidis. Salmonella cells were initially concentrated using immunomagnetic separation and then incubated with phage SJ2 for 10 min. The paramagnetic beads allowed the Salmonella cells to be easily washed and recovered while simultaneously removing free phage from the sample. Upon continued incubation, Salmonella cells infected with phage now lysed and released their phage progenies. Healthy Salmonella “signal-amplifying cells” (SACs) were then added to serve as new hosts for the progeny phage, and their subsequent infection could be monitored simply by measuring optical density, where a decrease in optical density indicated that signal-amplifying cell concentrations were declining due to infection and lysing by phage while an increase in optical density indicated an unaffected and growing population of signal-amplifying cells (Fig. 11.4). The assay was tested in artificially contaminated skim milk powder, chicken rinses, and ground beef, with an average detection limit of 3 cfu/g or mL within a total assay time of 20 h, inclusive of requisite preenrichment incubations. It has also been applied toward the detection of E. coli O157:H7 using phage LG1 and anti-E. coli paramagnetic beads, with a detection limit of 2 cfu/g ground beef within an assay time of 23 h (Favrin et al. 2003).


A phage amplification assay incorporating immunomagnetic separation. Salmonella cells present in the sample are captured with polyclonal antibody-coated paramagnetic beads. The addition of Salmonella-specific phage results in infection, cell lysis, and the amplified release of progeny phage. The addition of healthy signal-amplifying cells (SACs) provides new hosts for these phage, and subsequent infection (or noninfection if Salmonella was not initially present) is monitored by optical density. (With permission from Favrin et al. (2003), copyright 2008, American Society for Microbiology)
Jassim and Griffiths (2007) developed a phage amplification assay that used fluorochromic stains to monitor target host cell viability associated with phage infection and lysis. SYTO9 dye stains viable cells fluorescent green while propidium iodide stains dead cells fluorescent red. The propidium iodide penetrates only those cells with damaged cell membranes, as occurs when a host bacterium is infected with a phage. Therefore, after performing the phage amplification assay, each dye is added to the sample and the ratio of green-to-red fluorescent cells is used to indicate the occurrence of phage infection and, therefore, the presence of target bacterial cells. Using phage NCIMB 10116, target P. aeruginosa cells could be detected down to 10 cfu/mL in pure culture within 4 h with no preenrichment.
Ulitzur and Ulitzur (2006) used novel phage mutant repair mechanisms to identify endpoint plaque formation due to infected target bacteria. Phage carrying amber mutations (phage Felix-O1 for Salmonella), ultraviolet light-irradiated mutations (phage OE for E. coli), or temperature-sensitive mutations (phage AR1 for E. coli O157:H7) were constructed. These phages could not form plaques on their host cells unless their mutations were repaired by recombination or complementation, thereby negating the need to wash and/or centrifuge the assay samples to remove free phage. For example, two temperature-sensitive phage mutants were allowed to coinfect their E. coli O157:H7 targets at the permissive temperature (37°C), then incubated at their restrictive temperature (42°C) to prevent further infection cycles. Subsequent plaque formation was, therefore, only possible if the mutation had been repaired since any remaining mutant phage, due to their temperature sensitivity, could not plaque at 42°C, and the number of plaques thus reflected the number of E. coli host cells in the sample. Detection was achieved down to 1 cell/mL in a 3.5 h assay. Similar coinfection strategies with the other phage mutants yielded detection limits of 10 or less target cells/mL in 3–5 h assay formats.
One of the most applied phage amplification assays is for the clinical detection of Mycobacterium tuberculosis using the commercially available FAST-PlaqueTB™ kit (Mole and Maskell 2001). The phage, referred to as Actiphage™, is added to the sample for 1 h followed by the addition of a virucide (Virusol™). Since these tests are highly standardized for this specific phage/host combination, the virucidal approach works exceedingly well. After a 5-min incubation, the virucide is neutralized and a rapidly growing mycobacterial cell suspension is added (Sensor™ cells) to promote additional phage infection and amplification. Resulting phage are enumerated on plaque plates. Two large-scale studies verified the detection of 65–83% of confirmed M. tuberculosis infections in sputum samples within 2 days using this assay (Pai et al. 2005). Although direct microscopic methods can achieve results in 2 h with corresponding sensitivity, the simplicity of phage amplification, especially when packaged in kit form, and the ability to perform these assays with no additional costly equipment and minimal user training, offers significant benefits, particularly in economically disadvantaged countries and low resource environments. A FASTPlaque-Response™ kit is also available for establishing rifampicin resistant M. tuberculosis. The sample is preincubated in the presence or absence of rifampicin antibiotic and then subjected to the phage amplification assay. If the cells are resistant to rifampicin, the number of plaques enumerated will be similar in both samples. If the cells are sensitive to rifampicin, the number of plaques in the rifampicin-treated sample will be less than the rifampicin free sample. Susceptibility testing for various other antituberculosis drugs (isoniazid, ethambutol, streptomycin, pyrazinamide, ciprofloxacin) can additionally be achieved (Mole and Maskell 2001).
Microphage Inc., based in Colorado, USA, (http://www.microphage.com) developed a phage amplification assay for methicillin-resistant Staphylococcus aureus (MRSA). The clinical sample is incubated with MRSA-specific phages. The subsequent increase in yield of phages due to the presence of MRSA in the sample is detected via immunological identification of a phage-specific protein. Blood samples can be processed in as little as 5 h. Their detection assays can also be used for antibiotic susceptibility testing.
Several research groups have developed unique technologies to monitor the amplification of progeny phage after infection events. Guan et al. (2006), for example, combined phage amplification with a competitive enzyme-linked immunosorbent assay (cELISA) to detect Salmonella Typhimurium using phage BP1 and a biotinylated version of BP1. Salmonella cultures were incubated with wild-type BP1 phage and resulting phage supernatants added to ELISA microtiter plates coated with Salmonella Typhimurium smooth lipopolysaccharide (LPS) to which the phage attached. The biotinylated version of the phage was additionally added, which could be detected by the colorimetric substrate 3,3′,5,5′-tetramethylbenzidine peroxidase (TMB). If excess wild-type BP1 phage were present, due to the availability of suitable Salmonella host cells, then few biotinylated phage would attach and a weak colorimetric signal would be detected. If no target Salmonella were present, then BP1 replication would not occur and excess biotinylated phage would bind to the smooth LPS to yield an intense yellow color. In another technique, Madonna et al. (2003) used matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) to identify the molecular weight signature of the phage capsid protein. E. coli in pure culture was concentrated by immunomagnetic separation and then infected with phage MS2. Analysis of 1 µL of this sample by MALDI-MS was sufficient to detect the MS2 capsid protein, providing a detection limit of approximately 104 E. coli cells/mL in an assay time of 2 h.
4 Direct Detection Through Cell Wall Recognition
4.1 Phage Immobilization
Bacteriophage specificity can be used to capture bacterial targets, and by adhering bacteriophages directly to a transducer interface, fully standalone biosensors can be created. Bacteriophages have been immobilized onto transducer interfaces using various immobilization techniques including physical adsorption (Bennett et al. 1997; Sun et al. 2001; Singh et al. 2009; Gervais et al. 2007; Petrenko 2008; Balasubramanian et al. 2006; Nanduri et al. 2007a, b; Lakshmanan et al. 2007a, b; Petrenko 2008; Johnson et al. 2008; Wan et al. 2007; Li et al. 2009), Langmuir-Blodgett deposition (Olsen et al. 2006; Guntupalli et al. 2008), and covalent immobilization (Shabani et al. 2009; Zhu et al. 2008; Handa et al. 2008). Bennett et al. (1997) reported a method to separate and concentrate Salmonella from food materials by using a biosorbent coated with phages physically attached to a solid phase. In their approach, they soaked two different polystyrene surfaces, microplates and dipsticks, into a 5 × 1010 pfu/mL Sapphire-specific lytic phage solution, followed by washing in order to remove unbound phage, and blocking the remaining adsorption sites. These biosorbents were then incubated with Salmonella and mixed bacterial cultures, and after washing, the specific recovery of Salmonella cells was assessed either by PCR or by epifluorescence microscopy using acridine orange as a dye for labeling the bacterial nucleic acids. In both cases, Salmonella could be separated from mixed bacterial cultures, and 9 of 11 Salmonella strains gave positive signals. One of the strains not detected by the test, Salmonella enterica serovar Arizonae CRA 1568, is known to synthesize an incomplete lipopolysaccharide molecule that results in an inability of the phage to adsorb to this strain. However, the detection limit for this method was rather high. With a PCR detection step, a concentration of 105 cfu/mL was required to generate a positive signal, and a concentration of 107 cfu/mL was necessary in the initial culture to ensure that this amount of cells was captured by the biosorbent. This represents a capture efficiency of 1%, which is clearly not sufficient.
Sun et al. (2001) described specific immobilization of phages, exploiting the high affinity of biotin to streptavidin, as opposed to physical adsorption. In this work, phage SJ2 for Salmonella was biotinylated using sulfosuccinimido-biotin, which reacts with primary amino groups of the phage coat proteins. Afterward, the biotinylated phages were incubated with streptavidin-coated magnetic beads. This phage-based biosorbent was applied to capture target cells of S. enteritidis by magnetic separation, using magnetic beads coated with nonbiotinylated phage as a negative control. The capture efficiency was determined by using a recombinant bioluminescent Salmonella strain as a target organism and measuring relative light output (RLU) after capture. With this technique, approximately 20% of the target cells could be recovered when a culture of 2 × 106 cfu/mL was used, which was a significant improvement when compared to the physical adsorption method.
Gervais et al. (2007) demonstrated streptavidin-mediated attachment of bacteriophage genetically modified to display biotin on their capsid. The biotinylated phages were immobilized on streptavidin-coated gold surface electrodes fabricated in microtitre chips. This approach yielded a phage attachment density of 4.4 phages/µm2, representing a 15-fold increase over simple physisorption (Fig. 11.5). Impedance was used to monitor cell growth. The immobilized phage lyse their target cells and inhibit or slow cell growth in comparison to unmodified wells. The main limitation of this method is that the phage requires genetic modification which can be a time-consuming process.


T4 phage immobilized on a gold-coated surface. (Provided courtesy of Dr. S. Evoy-Alberta University)
Singh et al. (2009) reported the immobilization of wild-type T4 phage on gold surfaces through hydrogen bonding using different approaches. The gold surfaces were modified by hydrophilic interaction with sugars (dextrose and sucrose) and through thiol linkage with the amino acids histidine and cysteine. In addition, gold surfaces modified with amino acids where further activated with glutaraldehyde. The attachment density of phages with glutaraldehyde activation was 18 ± 0.15 phages/µm2. This represented a 4-fold improvement over the values obtained with biotinylated phages, and a 37-fold improvement compared with simple physisorbtion. Subsequently, the E. coli host bacterial capture density improved by 9-fold compared with physically adsorbed phages.
Jabrane et al. (2008, 2009) studied the effect of charged coated papers on the immobilization of phages. They precoated a low charge density cationic layer composed of PolyDADMAC onto the paper surface and showed maintenance of T4 phage bioactivity and increased phage spatial orientation perpendicular to the surface of the paper. They explained this by the fact that the capsid of the T4 phage carries a negative net charge while the tail fibers are slightly cationic.
Udit et al. (2008) expressed hexahistidine sequences from bacteriophage Qβ via coexpression of the wild-type and hexahistidine-modified coat proteins in E. coli. The resulting polyvalent hexahistidine moieties impart strong affinity for immobilized metal ions. SPR gold surfaces were modified to display Ni (II)-NTA moieties for immobilizing the expressed Qβ bacteriophage. A dissociation constant for binding to Ni–NTA of approximately 10 nM was measured by surface plasmon resonance under noncompetitive, physiological conditions. Affinity chromatography over immobilized metal columns was also demonstrated to purify the phages from both crude cell lysates and after chemical derivatization.
T4 bacteriophages were immobilized onto screen printed electrode (SPE) microarrays via 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) by formation of amide bonds between the phage and the electrochemically generated carboxylic groups at the carbon surface (Shabani et al. 2008, 2009). The attachment of the phage was investigated using time-of-flight secondary ion mass spectroscopy (TOF-SIMS). Surface chemical mapping (secondary ion spectra) was acquired from an area of 40 × 40 µm, using at least three different positions per electrode, to verify the immobilization process and confirm the attachment of phage at the electrode surface. Fig. 11.6 shows 40 × 40 μm2 intensity maps of negative and positive fragments. It is clear from the intensity map that CN− and CNO− fragments are present on the EDC- and T4-modified surface, showing gradually higher intensities. Also, the presence of K+ is a good indication of the presence of biological entities such as cells and viruses (Nygren et al. 2007), which is only observed after T4 immobilization. The relative intensity map for total ion reflects a homogenous distribution for each surface following the modification processes. This same research group immobilized gamma bacteriophage as probes to electrochemically functionalized SPE microarrays for the detection of Bacillus anthracis Sterne vegetative cells (Shabani et al. 2009). The carbon electrodes were initially functionalized through cyclic-voltammetric reduction of a nitroaryl diazonium moiety, followed by further reduction of nitro groups to amino groups, and finally activated with glutaraldehyde. Functionalization of the carbon electrodes and the binding of Gamma phage were verified by X-ray photoelectron spectroscopy and TOF-SIMS, respectively.


Approximately 40 × 40 μm2 intensity maps of various positive and negative ions from each surface during the modification process, bare (Surface 1), EDC (Surface 2), phage T4 (Surface 3). Ion intensity is scaled individually to show maximum counts as white and zero counts in black
4.2 Affinity Detection
4.2.1 Electrical
An electrooptical analyzer (ELUS EO) technique was used to characterize the biospecific binding between the bacterium E. coli XL-1 and the phage M13K07, a filamentous phage of the family Inoviridae (Bunin et al. 2004; Guliy et al. 2007). The operating principle of the analyzer is based on the polarizability of microorganisms, which depends strongly on their composition, morphology, and phenotype. The principle of analysis of the interaction of E. coli with phage M13K07 is based on recording changes of optical parameters of bacterial suspensions upon phage adsorption to cells, nucleic acid injection, phage amplification, and cell lysis. The interaction of E. coli with phage M13K07 induced a strong and specific electrooptical signal as a result of substantial changes in the electrooptical properties of the mixed bacterial suspension (E. coli K-12 and Azospirillum brasilense Sp7).
Kish et al. (2005) (Dobozi-King et al. 2005; Biard and Kish 2005) proposed a method called SEnsing of Phage-Triggered Ion Cascade (SEPTIC) for the rapid detection and identification of bacteria via the electrical field caused by the stochastic emission of ions during phage infection. The method is based on the massive transitory ion leakage that occurs at the moment of phage DNA injection into the host cell. The ion fluxes require only that the cells be physiologically viable (i.e., have energized membranes) and can occur within seconds after mixing the cells with sufficient concentrations of phage particles. These fluxes were detected using a nano-well, a lateral, micrometer-sized capacitor of titanium electrodes with a gap size of 150 nm, and used it to measure the electrical field fluctuations in microliter samples containing phage and bacteria. In mixtures where the analyte bacteria were sensitive to the phage, large stochastic waves with various time and amplitude scales were observed, with power spectra approximately following a 1/f 2 law from 1–10 Hz. The calculations indicated that the detection and identification of a single bacterium could be achieved with wild-type phages within a time window of 10 min. The advantage of this method is fast, immobilization-free detection but the cells must be physiologically viable.
The bacteriophage/host cell relationship can be used to not only detect target bacteria but to detect and identify bacteriophage as well. In the dairy fermentation industry, for example, the infection of bacterial starter cultures by contaminating bacteriophages results in significant economic losses. To detect these bacteriophages, the research group of Muñoz-Berbel et al. (2008) developed an impedimetric approach that monitored capacitive changes in a bacterial biofilm upon exposure to a bacteriophage. E. coli WG5 were immobilized on an electrode surface and PhiX174 bacteriophages were introduced to the sensor surface. Impedimetric changes occurring at the microelectrode surface due to bacteriophage infection and degradative lysis of the E. coli biofilm were measured over a 24-h period by nonfaradic impedance spectroscopy (IS) in urban sewage. The method was sensitive enough to detect phage concentrations of approximately 104 pfu/mL. Impedimetric detection was later adapted to an on-chip biosensor format and tested in milk samples (García-Aljaro et al. 2009). Detection of bacteriophages occurred down to 1 pfu/mL but responses were nonlinear, making this assay more of a presence/absence indicator than a quantitative test.
An impedance technique was employed for specific, direct and label-free detection of bacteria using bacteriophages as recognition receptors immobilized covalently onto functionalized SPE microarrays (Shabani et al. 2007; Shabani et al. 2008, 2009). T4 bacteriophages were immobilized onto SPE microarrays via 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) by formation of amide bonds between the phage and the electrochemically generated carboxylic groups at the carbon surface. The immobilized T4 phages were then used to specifically detect E. coli bacteria. Scanning electron microscopy (SEM) was used to verify the presence of surface-bound bacteria (Fig. 11.7). Impedance measurements (Nyquist plots) show shifts of the order of 104 Ω due to the binding of E. coli bacteria to the T4 phages (Fig. 11.8). No significant change in impedance was observed for control experiments using immobilized T4 phage in the presence of Salmonella. Concentration–response curves yielded a detection limit of 104 cfu/mL in 50 μL samples. Impedimetric techniques were also employed for the detection of Bacillus anthracis Sterne vegetative cells using Gamma phage as probes attached to electrochemically functionalized SPE microarrays (Shabani et al. 2009). Concentration–response curves yielded a detection limit of 103 cfu/mL in 40-µL samples.


SEM images of bacteria bound to phage-modified surface. (a) T4 phage immobilized to surface, (b) bacteria bound to immobilized T4 phage (high resolution), (c) bacteria bound to immobilized T4 phage (low resolution).


Nyquist plots for a T4-modified surface in the presence of E. coli at different concentrations
4.2.2 Optical Sensors
Bacteriophages as recognition receptors were employed in a number of optical biosensors including surface plasmon resonance sensors (García-Aljaro et al. 2008; Nanduri et al. 2007a, b; Balasubramanian et al. 2007), optical light microscope systems (Guntupalli et al. 2008), optical microresonators (Zhu et al. 2008), colorimetric microtiter plates (Knezevic and Petrovic 2008), and ELISA and atomic force microscopy (Handa et al. 2008).
Surface plasmon resonance (SPR) is a well-established technique for the measurement of molecules binding to surfaces and the quantification of binding constants between surface-immobilized ligand and analyte in solution. A two-channel microfluidic SPR sensor device was used to detect the presence of somatic coliphages, a group of bacteriophages that have been proposed as fecal pollution indicators in water, using their host, E. coli WG5, as a target for their selective detection (García-Aljaro et al. 2008). E. coli WG5 was immobilized on gold sensor chips using avidin–biotin and exposed to bacteriophages extracted from wastewater. SPR was able to monitor the binding of bacteriophages to their bacterial hosts, and demonstrated detection of as few as 1 PFU/mL of bacteriophage after an incubation period of 120 min, which equates to an approximate limit of detection of around 102 PFU/mL. The bacteriophage–bacterium interaction appeared to cause a structural change in the surface-bound bacteria, possibly due to collapse of the cell, which was observed as an increase in mass density on the sensor chip. Balasubramanian et al. (2006) employed an SPR-based SPREETA™ sensor for label-free detection of Staphylococcus aureus using lytic phage as highly specific and selective biorecognition elements. The lytic phage was physically adsorbed on the gold surface by flowing the phage solution overnight on the gold surface followed by washing. S. aureus bacteria were then exposed to the immobilized phages. The detection limit was determined to be 104 cfu/mL. Detection specificity was investigated by an inhibition assay while selectivity was examined with Salmonella typhimurium. The inhibition assay was performed by preincubating different concentrations of phages with a known concentration of S. aureus for 1 h to block the receptor sites from binding and the solutions were introduced through the sensor. It was found that the response of the sensor decreased as the concentration of the phage increased. This technique can be employed for rapid and label-free detection of different bacterial pathogens. SPR was employed for the detection of β-galactosidase (β-gal) using specific landscape phage 1G40 physically adsorbed on the gold surface of SPR SPREETA™ sensor chips (Nanduri et al. 2007a, b). Wild-type phage F8-5 was used in the reference channel as nonspecific to the β-gal phage. It was shown that concentrations of β-gal between 10−12 M and 10−7 M could be readily detected, with the linear part of the calibration curve between 10−9 M and 10−6 M (Fig. 11.9). When β-gal was preincubated with different concentrations of free 1G40 phage prior to exposure to the biosensor, concentration-dependent inhibition was observed, indicating the biosensor had high specificity toward β-gal. Batch mode and flow through modes were employed to study the binding of β-gal to immobilized phage on the SPR sensor surface. Experiments using a flow through mode provided more consistent results in the full dose range and showed higher sensitivity as opposed to the batch mode studies. The mean K d and binding valences for the flow through mode studies was 1.3 ± 0.001 nM and 1.5 ± 0.03, in comparison to 26 ± 0.003 nM and 2.4 ± 0.01 for the batch mode studies. The average thickness of phage 1G40 adlayer deposited through flow through and batch mode was 3 ± 0.002 nm and 0.66 ± 0.001 nm, respectively.


Dose–responses of a phage-immobilized SPR sensor in a flow through mode. Curve A represents responses obtained from the working phage, 1G40 immobilized on the SPR in one of the two channels of the sensor against graded concentrations of β-gal (0.0032–210 nM). B represents the responses obtained from the wild-type phage, F8-5 immobilized on the second channel. (Reproduced from Naduri et al. (2007) with permission of Elsevier)
S. aureus-specific bacteriophage were used as a probe for the detection of methicillin-resistant S. aureus (MRSA) in aqueous solution using a CytoViva™ optical light microscope system (Guntupalli et al. 2008). Biorecognition phage monolayers were transferred to glass substrates using Langmuir–Blodgett (LB) technique and were exposed individually to MRSA in solution at logarithmic concentrations ranging from 106 to 109 cfu/mL, and observed for real-time binding using a CytoViva™ optical light microscope system. Results indicate that LB monolayers possessed high levels of elasticity (K), measuring 22 and 29 mN/m for 109 and 1011 pfu/mL phage concentrations, respectively. Near-instantaneous MRSA–phage binding produced 33 ± 5%, 10 ± 1%, 1.1 ± 0.1%, and 0.09 ± 0.01% coverage of the substrate that directly correlated to a decrease in MRSA concentrations of 109, 108, 107, and 106 cfu/mL. The exclusive selectivity of phage monolayers was verified with Salmonella enterica subsp. enterica serovar typhimurium (S. typhimurium) and Bacillus subtilis.
An opto-fluidic ring resonator (OFRR) was used to monitor the binding of filamentous phage R5C2, displaying peptides to streptavidin specifically, and employed as a model receptor (Zhu et al. 2008). The experimental detection limit was approximately 100 pM streptavidin and the K d(apparent) was 25 pM. Specificity was verified using the RAP5 phage, which is not specific to streptavidin, as the negative control. Sensing surface regeneration results show that the phage maintained functionality after surface regeneration, which greatly improves the sensors’ reusability.
Colorimetric microtiter plates based on crystal violet staining and measurement of optical density were used for the selection of the most effective P. aeruginosa phages for inhibition of biofilm formation and its eradication (Knezevic and Petrovic 2008). Four different bacteriophages isolated from municipal water were tested in this study with P. aeruginosa PA-4u and ATCC 9027 as the bacterial hosts. Biofilm formation could be inhibited by over 50% under active growth conditions but relatively little effect was observed when mature biofilms were exposed to phage.
A monolayer of bacteriophage P22 whose tailspike proteins specifically recognize Salmonella serotypes was covalently bound to glass substrates through a bifunctional cross-linker 3-aminopropyltrimethoxysilane deposited using chemical vapor deposition. The amino-functionalized glass surface was activated with N-hydroxysulfosuccinimide (Sulfo-NHS) and 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) prior to phage immobilization (Handa et al. 2008). The specificity of the binding of S. typhimurium to the immobilized bacteriophage monolayer was studied by enzyme-linked immunosorbent assay (ELISA) and atomic force microscopy. E. coli and a Gram-positive Listeria monocytogenes bacterium were also studied as control bacteria. The durability of the P22 phage coating in ambient air was tested to show that 50% of immobilized phage particles were still capable of recognizing S. typhimurium after remaining in a dry state for 7 days.
4.2.3 Mass Sensitive Sensors
Bacteriophages as bioprobes were employed in a number of mass sensitive sensors based on quartz crystal microbalance (Nanduri et al. 2007a, b), surface acoustic wave (Olsen et al. 2006), magnetoelastic (Lakshmanan et al. 2007a, b; Petrenko 2008; Johnson et al. 2008; Wan et al. 2007), and magnetostrictive millimeter cantilever (Li et al. 2009) sensors.
Landscape phage 1G40 was immobilized by physical adsorption on the surface of a quartz crystal microbalance and used for detection of β-galactosidase from E. coli (Nanduri et al. 2007a, b). The sensor had a detection limit of a few nanomoles and a response time of ~100 s over the range of 0.003–210 nM. The binding dose–response curve had a typical sigmoid shape and the signal was saturated at a β-galactosidase concentration of about 200 nM. No response was observed in mixed solutions even when the concentration of BSA exceeded the concentration of β-galactosidase by a factor of approximately 2,000.
Surface acoustic wave biosensors were demonstrated for the detection of Salmonella typhimurium using filamentous phage physically adsorbed onto a piezoelectric transducer (Olsen et al. 2006). Quantitative deposition studies of the phages onto the transducer surface indicated that approximately 3 × 1010 phage particles/cm2 could be irreversibly adsorbed for 1 h at room temperature to prepare working biosensors. The biosensors possessed a rapid response time of <180 s, had a low-detection limit of 102 cells/mL, and were linear over a range of 101–107 cells/mL with a sensitivity of 10.9 Hz per order of magnitude of S. typhimurium concentration.
Millimeter magnetoelastic ribbon sensors were demonstrated in buffer for the detection of S. typhimurium (Lakshmanan et al. 2007a, b; Petrenko 2008) and Bacillus anthracis spores using specific filamentous bacteriophage. Phage were immobilized onto the surface of the sensors by physical adsorption. The phage-immobilized magnetoelastic sensors were exposed to S. typhimurium cultures at concentrations ranging from 5 × 101 to 5 × 108 cfu/mL, and the corresponding changes in resonance frequency were monitored. It was experimentally shown that the sensitivity of the magnetoelastic sensors was higher for sensors with smaller physical dimensions. An increase in sensitivity from 159 Hz/decade for a 2 mm sensor to 770 Hz/decade for a 1 mm sensor was observed. SEM was used to verify the frequency changes caused by S. typhimurium binding to phage immobilized on the sensor surface. A detection limit on the order of 103 cfu/mL was obtained for a sensor with dimensions of 1 × 0.2 × 0.015 mm (Lakshmanan et al. 2007a, b). A magnetoelastic biosensor immobilized with filamentous bacteriophage for the detection of Salmonella typhimurium in water and fat-free milk was also demonstrated using sensors with dimensions of 2 × 0.4 × 0.015 mm (Lakshmanan et al. 2007a, b). A detection limit of 5 × 103 cfu/mL, with a sensitivity of 159 Hz/decade, was obtained for the sensors tested in water samples, as compared to 118 Hz/decade in fat-free milk. The dissociation constant K d and the binding valencies for S. typhimurium spiked water samples was 136 and 2.4, respectively, while the K d and binding valencies in spiked fat-free milk samples was 149 and 2.5 cfu/mL, respectively.
Fabricated microscale magnetoelastic particles (500 × 100 × 4 μm) composed of an amorphous iron–boron binary alloy was demonstrated for B. anthracis spore detection using landscape affinity-selected filamentous phages physically adsorbed (Johnson et al. 2008; Wan et al. 2007; Petrenko 2008). The biosensors were tested in B. anthracis spore solutions with concentrations from 103 to 108 cfu/mL. A detection limit of 103 cfu/mL, with a sensitivity of 6.5 kHz/decade, was observed. SEM images were used for visual correlation of the sensors’ responses (Fig. 11.10). Specificity of the biosensor was measured after exposure to mixed spore solutions containing B. anthracis Sterne, Bacillus cereus, and Bacillus megaterium strains. Binding kinetics demonstrated that the K d and the binding valency for the sensor in B. anthracis spore solutions was 193 cfu/mL and 2.32, respectively (Wan et al. 2007).


SEM image of a gold-coated magnetoelastic sensor coated (a) with filamentous phage (b) attachment of the spores to the immobilized phage. (Reproduced from Johnson et al. (2008) with permission of Elsevier)
Magnetostrictive millimeter (length of 2.7 mm and width of 1 mm) cantilevers were recently employed for B. anthracis spore detection using physically adsorbed f8/8 landscape phage (Li et al. 2009). It was found that the resonance frequency changes with time continuously and eventually reaches its saturated value. Captured spore cells were observed using SEM. It was demonstrated that the shift in the resonance frequency was related to the total mass of spore cells on the sensor surface. The results showed good dose–response relationships between 106 and 108 cfu/mL. The reported detection limit was 105 cfu/mL.
4.3 Fluorescently Labeled Phages
Labeled phage relies on the initial attachment of the phage to its host cell to signal host cell presence. Goodridge et al. (1999a, b) demonstrated this technique by staining the DNA of the E. coli O157:H7 specific phage LG1 with the fluorescent dye YOYO-1. Upon attachment of this fluorescently labeled phage to a suitable E. coli O157:H7 host, a fluorescent halo could be visualized around the cell when viewed with an epifluorescent microscope (Fig. 11.11). When confronted with a mixed bacterial culture, immunomagnetic separation was first used to separate target E. coli cells from the mix, and then fluorescently labeled LG1 phage was added. Flow cytometry permitted rapid enumeration of phage attached E. coli cells. Detection limits in artificially contaminated ground beef and raw milk approached approximately 2 cfu/g after a 6 h enrichment and 10 cfu/mL after a 10-h enrichment, respectively. Kenzaka et al. (2006) demonstrated a similar procedure using DAPI (4′,6-diamidino-2-phenylindole)-labeled phage T4 to detect E. coli. Using epifluorescence microscopy, they successfully enumerated E. coli within 30 min in water samples obtained from a fecally contaminated canal in Thailand. Mosier-Boss et al. (2003) labeled P22 phage with a fluorescent SYBR gold dye. In this case, the labeled DNA was visualized after phage-mediated injection into target Salmonella Typhimurium host cells.


Escherichia coli O157:H7 cells labeled with fluorescent bacteriophages (With permission from Goodridge et al. (1999b), copyright 2008 Elsevier)
5 Indirect Detection
5.1 Detection Based on Inhibition of Metabolism and Growth
Generally, lytic phages infect host cells leading to inhibition of growth of cells and killing cells when phages are present at high enough concentrations. A number of studies was demonstrated for monitoring the metabolic activity and microbial growth of microbial populations including impedimetric (Okigbo et al. 1985; Andrews et al. 1997; Mclntyre and Griffiths 1997; Chang et al. 2002), conductivity (Pugh and Arnott 1987; Carminat and Neviani 1991; Sevensson 1994; Chang et al. 2002), and optically (Preer et al. 1971).
Chang et al. (2002), based on the knowledge that growth in a microbial culture could be monitored electrochemically by measuring changes in electrical parameters occurring as complex growth media substrates were broken down into smaller highly charged molecules such as acids, hypothesized that the presence of phage within a bacterial culture, provided that suitable host cells were present, would impede culture growth and, therefore, directly affect growth media composition. Thus, by comparing conductance measurements between phage-supplemented and phage-free samples or between phage-specific and nonphage-specific bacterial cultures, one could easily screen samples for the presence of phage-specific pathogens. Phage AR1 and its E. coli O157:H7 host were used to demonstrate the technique. Pure cultures of E. coli O157:H7 or non-O157:H7 cells at 106 cfu/mL with or without phage AR1 addition were placed in test tubes fitted with platinum electrodes and conductance measurements were taken every 6 min. Resulting conductance curves could discriminate between E. coli O157:H7 and non-O157:H7 cultures within a 24-h period.
6 Detection by Reporter Phages
Reporter phages are genetically endowed with reporter genes that transmit an easily visualized signal that correlates with productive phage attachment or infection events. Due to ease of measurement, signals are most often optical, taking the form of bioluminescence, fluorescence, or colorimetric via the luciferase (lux and luc), green fluorescent protein (gfp), and β-galactosidase (lacZ) reporter genes, respectively. More uncommon signals such as ice formation (inaW) have been applied as well. The reporter gene is placed within the phage’s genome, and upon infection of an appropriate bacterial host, is transferred to that host whereupon only then is it expressed. Interfacing the assay with an appropriate transducer capable of measuring the signal realizes the complete detection scheme, which can be carried out in formats that range from single sample test tubes to high-throughput microtiter plates to lab-on-a-chip devices.
6.1 Bioluminescent Reporter Phages (lux and luc)
Bioluminescence is the production and emission of light by a living organism and luciferase is the general term for the enzyme that mediates the light-emitting reaction. Luciferases derived from bacteria are denoted as lux while those derived from insects, primarily the firefly (Photinus pyralis), are denoted as luc (the firefly luciferase is sometimes also referred to as fflux). Both lux and luc have been very well-characterized and routinely applied in reporter gene assays (Daunert et al. 2000; Viviani 2002). Signal detection is typically achieved using photomultiplier tubes (PMTs), charge-coupled device (CCD) cameras, luminometers and microluminometers, or even something as simple as X-ray film or the naked eye.
6.1.1 lux Reporter Phages
Bioluminescent bacteria are the most abundant and widely distributed of the light-emitting organisms on Earth and can be found in both aquatic and terrestrial environments. The majority of these microorganisms are classified into three genera: Vibrio, Photobacterium, and Photorhabdus (Xenorhabdus). Their luciferase consists of two proteins, LuxA and LuxB, that cooperatively generate 490 nm light from the oxidation of a long chain fatty aldehyde in the presence of a reduced riboflavin phosphate (FMNH2) and oxygen. Three other proteins in the operon, LuxC, LuxD, and LuxE, regenerate the aldehyde substrate required for this reaction. Reporter systems can contain only the luxAB genes or the entire luxCDABE gene cassette. luxAB alone generates a bioluminescent signal only after addition of an aldehyde substrate, normally n-decanal, due to the absence of the luxCDE genes that would normally perform this function. Including the full luxCDABE operon permits autonomous generation of light independent of any substrate additions. Phage reporters so far developed utilize only the luxAB genes since incorporation of the entire lux operon is difficult due to its size and potential interference with headful packaging constraints of the phage. Ulitzur and Kuhn (Ulitzur and Kuhn 1987) first demonstrated luxAB reporter phage detection using phage λ Charon 30 to detect E. coli down to 10–100 cells/mL in artificially contaminated milk or urine within 1 h. Kodikara et al. (1991) later used this assay to detect E. coli in swab samples from slaughterhouse surfaces and swine carcasses at a detection limit of 10 cells/cm2 or g after a 4-h preenrichment. Waddell and Poppe (2000) inserted the luxAB genes into phage ΦV10 for the specific detection of E. coli O157:H7 within 1 h at concentrations of 106 cfu/mL.
Besides E. coli hosts, phage reporters have been designed for additional targets as well. Chen and Griffiths (1996) constructed several P22-based luxAB reporter phage specific for the A, B, and D1 serotypes of Salmonella enterica and directly detected 108 cfu/mL within 1–3 h. By adding a 6 h preincubation step, detection of as few as 10 cfu/mL were possible. Reporter phages were also injected directly into poultry eggs artificially contaminated with Salmonella at a lower inoculum of 63 cfu/mL. By imaging the entire egg with a CCD camera, points of bioluminescent light corresponding to phage-infected Salmonella cells could be visualized after n-decanal addition, permitting localized identification of infected areas within the unadulterated egg. Thouand et al. (2008) recently adapted these reporter phage to a more commercially viable kit-like format consisting of four steps; (1) addition of the luxAB reporter phage to a 12–14-h enriched culture, (2) a 1 h incubation sufficient to allow contact between phage and host Salmonella, (3) addition of fresh growth medium followed by a 2–4-h incubation, and (4) addition of the n-decanal reagent and measurement of light. They showed that bacterial concentrations enriched above 106 cfu/mL could be detected within 16 h in various artificially contaminated poultry feed, feces, and litter samples. Loessner et al. (1997) created an A511 luxAB reporter phage for Listeria monocytogenes. Phage A511 infects approximately 95% of the L. monocytogenes serovars responsible for human listeriosis. Cheeses, chocolate pudding, cabbage, lettuce, ground beef, liverwurst, milk, and shrimp were each inoculated with L. monocytogenes from 0.1 to 1,000 cfu/g and allowed to preincubate for 20 h. The reporter phages and n-decanal substrate were then added and detection limits of 1 cell/g were demonstrated within less than 24 h. Foods such as ground beef that contained more complex microflora yielded detection limits of approximately 10 cfu/g. A total of 348 naturally contaminated meats, poultry, dairy products, and other environmental samples were additionally assayed in parallel with standard plating techniques, and Listeria positive samples were found to correspond between the two methods. With standard plating requiring 72–96 h, the less than 24 h A511 luxAB reporter phage assay was shown to be highly efficient while generating statistically comparable results.
Using lux but in a different approach, Brigati et al. (2007) designed a phage reporter assay for the detection of E. coli O157:H7 that did not rely on the addition of n-decanal for signal generation. This allowed the assay to be fully autonomous and capable of continuous monitoring of target pathogens. Besides the luxCDABE genes discussed above, the lux operon also contains a luxI and luxR gene that are involved in a cell-to-cell communication network referred to as quorum sensing. The luxI gene product synthesizes the chemical N-3-(oxohexanoyl)-l-homoserine lactone (OHHL) that interacts with the luxR gene which, in turn, activates the luxCDABE gene cassette to generate bioluminescence. The luxI gene was inserted into phage PP01 and was expressed upon infection of an E. coli O157:H7 host, thereby obligating the E. coli cell to generate OHHL (Fig. 11.12). Within the assay there was also included a bioluminescent bioreporter cell containing the luxRCDABE genes and its exposure to the OHHL chemical instigated bioluminescent light emission. Thus, E. coli O157:H7 could be detected indirectly via its phage-derived synthesis of a signature chemical analyte. The assay was demonstrated in artificially contaminated apple juice and tap water with detection limits of 1 cfu/mL within 22 and 12.5 h, respectively, using a 96-well microtiter plate format. Monitoring of an artificially contaminated spinach leaf rinsate was performed with a CCD camera that allowed real-time, continuous visualization of the bioluminescent signal, permitting detection of 1 cfu/mL within 6 h after a 2-h preincubation (Ripp et al. 2008).


Bacteriophage-based bioluminescent assay format for the detection of Escherichia coli O157:H7. The PP01 reporter phage carries a luxI gene that, upon infection of its E. coli host, forces synthesis of the chemical OHHL. A bioluminescent bioreporter cell sensitive to OHHL subsequently yields a bioluminescent light response to denote the presence of phage infective E. coli O157:H7 cells
6.1.2 luc Reporter Phages
The luc firefly luciferase catalyzes the two-step conversion of d-luciferin to oxyluciferin to generate a 560-nm bioluminescent light signal. Analogous to the addition of n-decanal in luxAB reporter assays, the d-luciferin substrate must also be added exogenously during luc-based reporter assays. The use of luc in reporter phage has primarily been associated with Mycobacterium specific phages and the detection and assessment of drug susceptibility in M. tuberculosis (Carriere et al. 1997; Sarkis et al. 1995). Susceptibility to antimycobacterial drugs can be determined by comparing bioluminescent output kinetics from reporter phage added to antibiotic-free or antibiotic-amended Mycobacterium cultures. If the drug is effective, fewer host Mycobacterium cells are available for phage infection and less light is therefore emitted as compared to the antibiotic-free control. These assays can require several days to complete, but far surpass the several week time periods required using conventional antibiotic susceptibility assays. Bardarov et al. (2003) used luc reporter phage phAE142 to assay sputum samples for M. tuberculosis presence and establish antibiotic susceptibility profiles. M. tuberculosis could be detected within 7 days at concentrations greater than 104 cfu/mL, while antibiotic susceptibility profiles could be obtained within 3 days. This compares to conventional growth assays such as the Mycobacterial Growth Indicator Tube (MGIT) that requires 9 days until results and antibiotic resistant tests that require up to 12 days. The phage assay is inexpensive and simple to perform, and has been marketed as a diagnostic tool valuable to resource-poor countries. To demonstrate, Banaiee et al. (2008) provided the assay to laboratory technicians in a South African hospital setting and showed an approximate 98% agreement between it and conventional BACTEC methods. A phage, referred to as Che12, showing greater specificity toward M. tuberculosis has recently been described and genetically incorporated with the luc gene (Kumar et al. 2008)
6.2 Fluorescent Reporter Phages (gfp)
The green fluorescent protein (gfp) reporter gene emits a 508-nm fluorescent signal when activated by ultraviolet or blue light. Funatsu et al. (2002) first demonstrated gfp signaling in phage lambda for broad-based detection of E. coli. Reporter phage genomically incorporated with the gfp gene were combined with a mixed culture of E. coli and Mycobacterium smegmatis for 4 h and then observed under an epifluorescent microscope. E. coli cells that had been infected with the phage fluoresced while noninfectable M. smegmatis cells did not. Tanji et al. (2004) constructed another E. coli-specific gfp reporter phage using a phage T4 modified to inactivate its lytic activity. Thus, these phage were no longer able to kill their hosts, which is an advantage in an enumeration assay where destroying the target one wishes to detect is not altogether favorable. This reporter phage, referred to as T4e-/GFP, was added to a mixed culture of E. coli and P. aeruginosa where it was shown to differentiate between the two microbes when observed under the epifluorescent microscope (Fig. 11.13). Miyanaga et al. (2006) later used T4e−/GFP reporter phage to detect E. coli in sewage influent. Since the host range of T4e−/GFP was not inclusive of all E. coli present in the sewage samples, only approximately 8% of the total E. coli population was detected, which was not unexpected since the use of a single reporter phage severely confines target acquisitions. Namura et al. (2008) addressed this by isolating two additional E. coli-specific phage from sewage and genetically modifying them with gfp inserts. Together, these reporter phages demonstrated a host range covering nearly 50% of the E. coli sewage isolates.


(a) A microscopic image of an Escherichia coli/Pseudomonas aeruginosa bacterial mix, and (b) a same field-of-view fluorescent microscopic image showing fluorescent E. coli cells after addition of the E. coli-specific T4e−/GFP reporter phage. (With permission from Tanji et al. (2004), copyright 2008 Elsevier)
Oda et al. (2004) created a much narrower host range gfp reporter phage using the E. coli O157:H7-specific phage PP01 which was capable of discriminating between E. coli O157:H7 and E. coli K12 cells within 10 min based on fluorescence emission. Sensitivity was later improved by once again inactivating the lytic activity of the phage (Awais et al. 2006), providing clearer and sharper epifluorescent images that translated into easier and more direct identification of E. coli O157:H7 in mixed cultures. These assays additionally discriminated between healthy and stressed cells, where healthy cells emitted bright green fluorescence and metabolically stressed cells emitted faded fluorescent signals. This permitted simultaneous differentiation of healthy cells from viable but nonculturable (VBNC) cells, which cannot be accomplished with conventional plating methods without supplemental and time-consuming steps.
6.3 Colorimetric Reporter Phages (lacZ)
The lacZ gene encodes a β-galactosidase enzyme that catalyzes the hydrolysis of β-galactosides. With the addition of a fluorescent, luminescent, or chemiluminescent substrate, the reaction endpoint can be optically observed in a typical reporter gene fashion. Goodridge and Griffiths (2002) inserted the lacZ reporter gene into phage T4 for the detection of E. coli. After addition of a chemiluminescent substrate to phage-infected E. coli cells, they were able to achieve detection down to 100 cfu/mL in pure culture within 12 h. The lacZ reporter gene has, however, seen very little additional use in reporter phage assays since lux, luc, and gfp assays typically maintain greater sensitivities.
6.4 Ice Nucleation Reporter Phages (inaW)
A more unusual application of reporter phages involves the inaW ice nucleation gene. Its protein product, InaW, integrates within the bacterial outer cell membrane where it acts as a catalyst for ice crystal formation within a temperature range of −2 to −10°C. The signaling endpoint for inaW is, therefore, ice crystal formation at supercooled temperatures. Wolber and Green (1990) inserted the inaW gene into Salmonella phage P22 to create the BIND™ (Bacterial Ice Nucleation Diagnostic) assay. Upon infection of Salmonella by the reporter phage, the inaW gene is expressed and samples freeze faster when supercooled as compared to Salmonella or other cells not expressing inaW. To easily visualize ice formation, the BIND™ assay used an indicator dye that turned orange if freezing occurred and fluorescent green if it did not, thereby providing a simple colorimetric or fluorescent signaling endpoint. When tested in raw eggs and milk artificially contaminated with Salmonella, detection limits of less than 10 cells/mL were demonstrated. The BIND™ assay, although once sold as a commercial kit, is no longer marketed.
7 Other Detection Methods Using Phages
7.1 Phage-Conjugated Quantum Dots
Edgar et al. (2006) developed a unique phage-conjugated quantum dot method for the detection of E. coli. Quantum dots are fluorescent probes consisting of colloidal semiconductor nanocrystals that exhibit high quantum yield and excellent photostability. Phage T7 was engineered to display a biotinylation peptide on its major capsid protein. Upon infection, progeny phage synthesized within the E. coli cell became biotinylated. After lytic release from the cell, these biotinylated phage could be detected by a streptavidin functionalized quantum dot designed to attach to and label only biotinylated phage. Thus, if no suitable hosts were present, then no biotinylated phages were produced and the functionalized quantum dot, having nothing to attach to, would simply be washed away. Fluorescence microscopy permitted visualization of a single quantum dot conjugated phage. Typical detection limits in laboratory mixed culture approached 10 cells/mL within an assay time of 1 h. Testing of river water samples demonstrated detection of 20 E. coli cells/mL within 1 h.
8 Conclusions and Future Remarks
Biosensing relies on specific analyte recognition by an appropriate receptor. A good recognition receptor should be highly specific, stable, and versatile, and few receptors meet these prerequisites as well as bacteriophages do. Considering the global abundance of phage with which we share this planet, appropriate phage/host specificity ranges should theoretically exist for any bacterial host desired. Their ease of production, long shelf life, and conduciveness to immobilization further warrants their application in biosensing regimens. With applications ranging from the detection of food and waterborne pathogens to military and homeland defence-related biological threat identification to in vivo diagnostic surveillance of disease states, the innovative simplicity, specificity, and sensitivity of bacteriophage-mediated biosensors cannot go unnoticed.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- RNA:
-
Ribonucleic acid
- PCR:
-
Polymerase chain reaction
- mRNA:
-
Messenger RNA
- RT-PCR:
-
Real-time polymerase chain reaction
- ELISA:
-
Enzyme-linked immunosorbant assay
- ATP:
-
Adenosine triphosphate
- AMP:
-
Adenosine monophosphate
- PP1:
-
Protein phosphatase 1
- AK:
-
Adenylate kinase
- ADP:
-
Adenosine diphosphate
- b-PAPG:
-
p-aminophenyl-b-d-galactopyranoside
- HPLC:
-
High-performance liquid chromatography
- SACs:
-
Signal-amplifying cells
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- cELISA:
-
Competitive enzyme-linked immu nosorbent assay
- LPS:
-
Lipopolysaccharide
- TMB:
-
3,3′,5,5′-tetramethylbenzidine peroxidase
- MALDI-MS:
-
Matrix-assisted laser desorption/ionization mass spectrometry
- RLU:
-
Relative light output
- PolyDADMAC:
-
Polydiallyldimethylammonium chloride
- Ni (II):
-
Nickel (II)
- NTA:
-
Nitrilotriacetic acid
- SPR:
-
Surface plasmon resonance
- SPE:
-
Screen printed electrode
- EDC:
-
1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride
- TOF-SIMS:
-
Time-of-flight secondary ion mass spectroscopy
- SEPTIC:
-
SEnsing of phage-triggered ion cascade
- IS:
-
Impedance spectroscopy
- SEM:
-
Scanning electron microscopy
- LB:
-
Langmuir–Blodgett
- OFRR:
-
Opto-fluidic ring resonator
- pM:
-
Pico Molar
- Sulfo-NHS:
-
N-hydroxysulfosuccinimide
- BSA:
-
Bovine serum albumin
- DAPI:
-
4′,6-diamidino-2-phenylindole
- Gfp:
-
Green fluorescent protein
- PMTs:
-
Photomultiplier tubes (PMTs)
- CCD:
-
Charge-coupled device
- OHHL:
-
N-3-(oxohexanoyl)-l-homoserine lactone
- MGIT:
-
Mycobacterial growth indicator tube
- VBNC:
-
Viable but nonculturable
References
Andrews DMA, Gharbia SE, Shah HN (1997) Characterization of a novel bacteriophage in Fusobacterium varium, Clinical Infectious Diseases 25, Supplement 2. Proceedings of the 1996 Meeting of the Anaerobe Society of the Americas, Sep 1997, pp S287–S288
Awais R, Fukudomi H, Miyanaga K, Unno H, Tanji Y (2006) A recombinant bacteriophage-based assay for the discriminative detection of culturable and viable but nonculturable Escherichia coli O157:H7. Biotechnol Prog 22:853–859
Balasubramanian S, Sorokulova IB, Vodyanoy VJ, Simonian AL (2007) Lytic phage as a specific and selective probe for detection of Staphylococcus aureus – A surface plasmon resonance spectroscopic study. Biosens Bioelectron 22:948–955
Banaiee N, January V, Barthus C, Lambrick M, RoDiti D, Behr MA, Jacobs WR, Steyn LM (2008) Evaluation of a semi-automated reporter phage assay for susceptibility testing Myobacterium tuberculosis isolates in South Africa. Tuberculosis 88:64–68. doi:10.1016/j.tube.2007.08.006
Bardarov S, Dou H, Eisenach K, Banaiee N, Ya S, Chan J, Jacobs WR, Riska PF (2003) Detection and drug-susceptibility testing of M. tuberculosis from sputum samples using luciferase reporter phage: comparison with the Mycobacteria Growth Indicator Tube (MGIT) system. Diagn Microbiol Infect Dis 45:53–61
Bennet AR, Davids FG, Valhodimou S, Banks JG, Betts RP (1997) The use of bacteriophage-based systems for the separation and concentration of Salmonella. J App Microbiol 83:259–265
Blasco R, Murphy MJ, Sanders MF, Squirrell DJ (1998) Specific assays for bacteria using phage mediated release of adenylate kinase. J Appl Microbiol 84:661–666
Biard JR, Kish LB (2005) Enhancing the sensitivity of the septic bacterium detection method by concentrating the phage-infected bacteria via DC electrical current. Fluctuation Noise Lett 5:L153–L158
Brigati JR, Ripp S, Johnson CM, Jegier P, Sayler GS (2007) Bacteriophage-based bioluminescent bioreporter for the detection of Escherichia coli O157:H7. J Food Prot 70:1386–1392
Bunin VD, Ignatov OV, Guliy OI, Zaitseva IS, O’Neil D, Ivnitski D (2004) Electrooptical analysis of the Escherichia coli–phage interaction. Anal Biochem 328:181–186
Carminat D, Neviani E (1991) Application of the Conductance Measurement Technique for Detection of Streptococcus salivarius ssp. thermophilus Phages. J Dairy Sci 74:1472–1476
Carriere C, Riska PF, Zimhony O, Kriakov J, Bardarov S, Burns J, Chan J, Jacobs WR (1997) Conditionally replicating luciferase reporter phages: improved sensitivity for rapid detection and assessment of drug susceptibility of Mycobacterium tuberculosis. J Clin Microbiol 35:3232–3239
Chang TC, Ding HC, Chen S (2002) A conductance method for the identification of Escherichia coli O157:H7 using bacteriophage AR1. J Food Prot 65:12–17
Chen J, Griffiths MW (1996) Salmonella detection in eggs using lux + bacteriophages. J Food Prot 59:908–914
Cherry WB, Davis BR, Edwards PR, Hogan RB (1954) A simple procedure for the identification of the genus Salmonella by means of a specific bacteriophage. J Lab Clin Med 44:51–55
Corbitt AJ, Bennion N, Forsythe SJ (2000) Adenylate kinase amplification of ATP bioluminescence for hygiene monitoring in the food and beverage industry. Lett Appl Microbiol 30:443–447
Daunert S, Barrett G, Feliciano JS, Shetty RS, Shrestha S, Smith-Spencer W (2000) Genetically engineered whole-cell sensing systems: coupling biological recognition with reporter genes. Chem Rev 100:2705–2738
D'Herelle F (1917) Sur un microbe invisible antagoniste des bacilles dysentériques. Comptes Rend Acad Sci Paris 165:373–375
D'Herelle F (1919a) Du rôle du microbe filtrant bacteriophage dans la fièvre thyphoide. Compte Rend Acad Sci Paris 168:631–634
D'Herelle F (1919b) Sur une épizooric de thyphose aviaire. Comptes Rend Acad Sci Paris 169:817–819
Dobozi-King M, Seo S, Kim JU, Young R, Cheng M, Kish LB (2005) Rapid detection and identification of bacteria: SEnsing of Phage-Triggered Ion Cascade (SEPTIC). J Biol Phys Chem 5:3–7
Easter MC, Gibson DM (1985) Rapid and automated detection of Salmonella by electrical measurements. J Hyg (Lond) 94:245–262
Edgar R, McKinstry M, Hwang J, Oppenheim AB, Fekete RA, Giulian G, Merril C, Nagashima K, Adhya S (2006) High-sensitivity bacterial detection using biotin-tagged phage and quantum-dot nanocomplexes. Proc Natl Acad Sci USA 103:4841–4845
Favrin SJ, Jassim SA, Griffiths MW (2003) Application of a novel immunomagnetic separation-bacteriophage assay for the detection of Salmonella enteritidis and Escherichia coli O157: H7 in food. Int J Food Microbiol 85:63–71
Favrin SJ, Jassim SA, Griffiths MW (2001) Development and optimization of a novel immunomagnetic separation-bacteriophage assay for detection of Salmonella enterica serovar enteritidis in broth. Appl Environ Microbiol 67:217–224
Funatsu T, Taniyama T, Tajima T, Tadakuma H, Namiki H (2002) Rapid and sensitive detection method of a bacterium by using a GFP reporter phage. Microbiol Immunol 46:365–369
García-Aljaro C, Muñoz-Berbel X, Jenkins A, Blanch AR, Muñoz FX (2008) Surface plasmon resonance assay for real-time monitoring of somatic coliphages in wastewaters. Appl Environ Microbiol 74:4054–4058
García-Aljaro C, Muñoz-Berbel X, Muñoz FJ (2009) On-chip impedimetric detection of bacteriophages in dairy samples. Biosens Bioelectron 24:1712–1716
Gervais L, Gel M, Allain B, Tolba M, Brovko L, Zourob M, Mandeville R, Griffiths M, Evoy S (2007) Immobilization of biotinylated bacteriophages on biosensor surfaces. Sens Actuat B 12:615–621
Goodridge L, Chen J, Griffiths MW (1999a) Development and characterization of a fluorescent-bacteriophage assay for detection of Escherichia coli O157:H7. Appl Environ Microbiol 65:1397–1404
Goodridge L, Chen J, Griffiths MW (1999b) The use of fluorescent bacteriophage assay for detection of Escherichia coli O157:H7 in inoculated ground beef and raw milk. Int J Food Microbiol 47:43–50
Goodridge L, Griffiths MW (2002) Reporter bacteriophage assays as a means to detect foodborne pathogenic bacteria. Food Res Int 35:863–870
Griffiths M, Goodridge L, McIntyre L, Ilenchuk TT (2003) Diagnostic and therapeutic applications of lytic phages. Anal Lett 36:3241–3259
Guan JW, Chan M, Allain B, Mandeville R, Brooks BW (2006) Detection of multiple antibiotic-resistant Salmonella enterica serovar Typhimurium DT104 by phage replication-competitive enzyme-linked immunosorbent assay. J Food Protect 69:739–742
Guliy OI, Bunin VD, O’Neil D, Ivnitski D, Ignatov OV (2007) A new electro-optical approach to rapid assay of cell viability. Biosens Bioelectron 23:583–587
Guntupalli R, Sorokulova I, Krumnow A, Pustovyy O, Olsen E, Vodyanoy V (2008) Real-time optical detection of methicillin-resistant Staphylococcus aureus using lytic phage probes. Biosens Bioelectron 24:151–154
Handa H, Gurczynski S, Jackson MP, Auner G, Walker J, Mao G (2008) Recognition of Salmonella typhimurium by immobilized phage P22 monolayers. Surf Sci 602:1392–1400
Hirsh DC, Martin LD (1983) Rapid detection of Salmonella spp by using Felix-01 bacteriophage and high-performance liquid-chromatography. Appl Environ Microbiol 45:260–264
Jabrane T, Jeaidi J, Dubé M, Mangin PJ (2008) Gravure printing of enzymes and phages. Adv Print Media Technol 35:279–288
Jabrane T, Dubé M, Mangin PJ (2009) Bacteriophage immobilization on paper surface: effect of cationic pre-coat layer. In: Proceedings of Canadian PAPTAC 95th Annual Meeting 2009, Montreal, Canada, pp 31–314
Jassim SAA, Griffiths MW (2007) Evaluation of a rapid microbial detection method via phage lytic amplification assay coupled with live/dead fluorochromic stains. Lett Appl Microbiol 44:673–678
Johnson ML, Wan J, Huang S, Cheng Z, Petrenko VA, Kim D-J, Chen I-H, Barbaree JM, Hong JW, Chin BA (2008) A wireless biosensor using microfabricated phage-interfaced magnetoelastic particles. Sens Actuat A 144:38–47
Kenzaka T, Utrarachkij F, Suthienkul O, Nasu M (2006) Rapid monitoring of Escherichia coli in southeast Asian urban canals by fluorescent-bacteriophage assay. J Health Sci 52:666–671
Kish LB, Cheng M, Kim JU, Seo S, King MD, Young R, Der A, Sshmera G (2005) Estimation of detection limits of the phage-invasion based identification of bacteria. Fluctuat Noise Lett 5:L105–L108
Kodikara CP, Crew HH, Stewart GSAB (1991) Near on-line detection of enteric bacteria using lux recombinant bacteriophage. FEMS Microbiol Lett 83:261–266
Knezevic P, Petrovic O (2008) A colorimetric microtiter plate method for assessment of phage effect on Pseudomonas aeruginosa biofilm. J Microbiol Meth 74:114–118
Kumar V, Loganathan P, Sivaramakrishnan G, Kriakov J, Dusthakeer A, Subramanyam B, Chan J, Jacobs WR, Rama NP (2008) Characterization of temperate phage Che12 and construction of a new tool for diagnosis of tuberculosis. Tuberculosis 88:616–623. doi:10.1016/j.tube.2008.02.007
Kutter E, Sulakvelidze A (2005) Bacteriophages: biology and applications. CRC Press, Boca Raton, p 510
Lakshmanan RS, Guntupalli R, Hu J, Kim D-J, Petrenko VA, Barbaree JM, Chin BA (2007a) Phage immobilized magnetoelastic sensor for the detection of Salmonella typhimurium. J Microbiol Meth 71:55–60
Lakshmanan RS, Guntupalli R, Hu J, Petrenko VA, Barbaree JM, Chin BA (2007b) Detection of Salmonella typhimurium in fat free milk using a phage immobilized magnetoelastic sensor. Sens Actuat B 126:544–550
Li S, Fu L, Barbaree JM, Cheng Z-Y (2009) Resonance behavior of magnetostrictive micro/milli-cantilever and its application as a biosensor. Sens Actuat B 137:692–699
Loessner MJ, Rudolf M, Scherer S (1997) Evaluation of luciferase reporter bacteriophage A511::luxAB for detection of Listeria monocytogenes in contaminated foods. Appl Environ Microbiol 63:2961–2965
Mandeville R, Griffiths M, Goodridge L, McIntyre L, Ilenchuk TT (2003) Diagnostic and Therapeutic Applications of Lytic Phages. Anal Lett 3241–3259
Mclntyre L, Griffiths MW (1997) A bacteriophage-based impedemtric method for the detection of pathogens in diary products. J Diary Sci 80:107
Miyanaga K, Hijikata TF, Furukawa C, Unno H, Tanji Y (2006) Detection of Escherichia coli in the sewage influent by fluorescent labeled T4 phage. Biochem Eng J 29:119–124
Madonna AJ, Van Cuyk S, Voorhees KJ (2003) Detection of Escherichia coli using immunomagnetic separation and bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rap Commun Mass Sp 17:257–263
Mole RJ, Maskell WOC (2001) Phage as a diagnostic - the use of phage in TB diagnosis. J Chem Technol Biotechnol 76:683–688
Mosier-Boss PA, Lieberman SH, Andrews JM, Rohwer FL, Wegley LE, Breitbart M (2003) Use of fluorescently labeled phage in the detection and identification of bacterial species. Appl Spectrosc 57:1138–1144
Muñoz-Berbel X, García-Aljaro C, Muñoz FJ (2008) Impedimetric approach for monitoring the formation of biofilms on metallic surfaces and the subsequent application to the detection of bacteriophage. Electrochimica Acta 53(2008):5739–5744
Namura M, Hijikata T, Miyanaga K, Tanji Y (2008) Detection of Escherichia coli with fluorescent labeled phages that have a broad host range to E. coli in sewage water. Biotechnol Prog 24:481–486
Nanduri V, Balasubramanian S, Sista S, Vodyanoy VJ, Simonian AL (2007a) Highly sensitive phage-based biosensor for the detection of β-galactosidase. Anal Chim Acta 589:166–172
Nanduri V, Sorokulova IB, Samoylov AM, Simonian AL, Petrenko VA, Vodyanoy V (2007b) Phage as a molecular recognition element in biosensors immobilized by physical adsorption. Biosens Bioelectron 22:986–992
Neufeld T, Mittelman AS, Buchner V, Rishpon J (2005) Electrochemical phagemid assay for the specific detection of bacteria using Escherichia coli TG-1 and the M13KO7 phagemid in a model system. Anal Chem 77:652–657
Neufeld T, Schwartz-Mittelmann D, Biran EZ, Rishpon RJ (2003) Combined phage typing and amperometric detection of released enzymatic activity for the specific identification and quantification of bacteria. Anal Chem 75:580–585
Nygren H, Hagenhoff B, Malmberg P, Nilsson M, Richter K (2007) Bioimaging TOF-SIMS: high resolution 3D imaging of single cells. Microsc Res Tech 70:969–974
Oda M, Morita M, Unno H, Tanji Y (2004) Rapid detection of Escherichia coli O157:H7 by using green fluorescent protein-labeled PP01 bacteriophage. Appl Environ Microbiol 70:527–534
Olsen EV, Sorokulova IB, Petrenko VA, Chen I-H, Barbaree JM, Vodyanoy VJ (2006) Affinity-selected filamentous bacteriophage as a probe for acoustic wave biodetectors of Salmonella typhimurium. Biosens Bioelectron 21:1434–1442
Okigbo LM, Ooberg CJ, Richardson GH (1985) Lactic culture activity tests using ph and impedance instrumentation. J Dairy Sci 68:2521–2526
Pai M, Kalantri S, Pascopella L, Riley LW, Reingold AL (2005) Bacteriophage-based assays for the rapid detection of rifampicin resistance in Mycobacterium tuberculosis: a meta-analysis. J Infect 51:175–187
Petrenko VA (2008) Landscape phage as a molecular recognition interface for detection devices. Microelectronics J 39:202–207
Preer JR Jr, Preer LB, Rudman B, Jurand A (1971) Isolation and composition of bacteriophage-like particles from kappa of killer paramecia. Mol Gen Genet 111:202–208
Pugh SJ, Arnott ML (1987) Automated conductemetric detection of Salmonella in confectionary products. In rapid microbiological methods for foods, beverages & pharmaceuticals. Soc Appl Bacteriol Tech Series 185
Rees CE, Dodd CE (2006) Phage for rapid detection and control of bacterial pathogens in food. Adv Appl Microbiol 59:159–186
Ripp S, Jegier P, Johnson CM, Brigati J, Sayler GS (2008) Bacteriophage-amplified bioluminescent sensing of Escherichia coli O157:H7. Anal Bioanal Chem 391:507–514
Sarkis GJ, Jacobs WR, Hatfull GF (1995) L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol 15:1055–1067
Sanders MF (1995) A rapid bioluminescent technique for the detection and identification of Listeria monocytogenes in the presence of Listeria innocua. In: Campbell AK, Kricka LJ, Stanley PE (eds) Bioluminescence and chemiluminescence: fundamental and applied aspects. Wiley, Chichester, U.K., pp 454–457
Schmelcher M, Loessner MJ (2008) Chapter 27 page 727. In Zourob M, Elwary S, Turner A (eds) Principles of bacterial detection. Springer, New York, p 970
Schuch R, Nelson D, Fischetti VA (2002) A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889
Sevensson UK (1994) Conductimetric analyses of bacteriophage infection of two groups of bacteria in DL-Lactococcal starter cultures. J Dairy Sci 77:3524–3531
Silley P, Forsythe S (1996) Impedance microbiology–a rapid change for microbiologists. J Appl Bacteriol 80:233–243
Singh A, Glass N, Tolba M, Brovko L, Griffiths M, Evoy S (2009) Immobilization of bacteriophages on gold surfaces for the specific capture of pathogens. Biosens Bioelectron 24:3645–3651
Shabani A, Zourob M, Allain B, Lawrence M, Mandeville R (2007) Electrochemical detection of bacteria using bacteriophage. Signals Syst Electron 2:45–47
Shabani A, Zourob M, Allain B, Marquette CA, Lawrence MF, Mandeville R (2008) Bacteriophage-modified microarrays for the direct impedimetric detection of bacteria. Anal Chem 80:9475–9482
Shabani A, Zourob M, Marquette CA, Lawrence MF, Mandeville R (2009) Carbon microarrays for the direct impedimetric detection of bacillus anthracis using gamma phage as probe. International Journal of Environmental Analytical Chemistry (Submitted)
Squirrel DJ, Price RL, Murphy MJ (2002) Rapid and specific detection of bacteria using bioluminescence. Anal Chim Acta 457:109–114
Sun W, Brovko L and Griffiths M (2001) Use of bioluminescent Salmonella for assessing the efficiency of constructed phage-based biosorbent. J Ind Microbiol Biotechol 27:126–128
Stanley PE (1989) A review of bioluminescent ATP techniques in rapid microbiology. J Biolumin Chemilumin 4:375–380
Stewart GSAB, Jassim SAA, Denyer SP, Newby P, Linley K, Dhir VK (1998) The specific and sensitive detection of bacterial pathogens within 4 h using bacteriophage amplification. J Appl Microbiol 84:777–783
Stewart GSAB, Loessner MJ, Scherer S (1996) The bacterial lux gene bioluminescent biosensor revisited. ASM News 62:297–301
Tanji Y, Furukawa C, Na SH, Hijikata T, Miyanaga K, Unno H (2004) Escherichia coli detection by GFP-labeled lysozyme-inactivated T4 bacteriophage. J Biotechnol 114:11–20
Thouand G, Vachon P, Liu S, Dayre M, Griffiths MW (2008) Optimization and validation of a simple method using P22:: luxAB bacteriophage for rapid detection of Salmonella enterica serotypes A, B, and D in poultry samples. J Food Prot 71:380–385
Twort F (1915) An investigation on the nature of ultramicroscopic viruses. Lancet 11:1241
Udit AK, Brown S, Baksh MM, Finn MG (2008) Immobilization of bacteriophage Qβ on metal-derivatized surfaces via polyvalent display of hexahistidine tags. J Inorg Biochem 102:2142–2146
Ulitzur S, Kuhn J (1987) Introduction of lux genes into bacteria, a new approach for specific determination of bacteria and their antibiotic susceptibility. In: Scholmerich J et al (eds) Bioluminescence and chemiluminescence: new perspectives. Wiley, New York, pp 463–472
Ulitzur N, Ulitzur S (2006) New rapid and simple methods for detection of bacteria and determination of their antibiotic susceptibility by using phage mutants. Appl Environ Microbiol 72:7455–7459
Viviani VR (2002) The origin, diversity, and structure function relationships of insect luciferases. Cell Mol Life Sci 59:1833–1850
Wan J, Johnson ML, Guntupalli R, Petrenko VA, Chin BA (2007) Detection of Bacillus anthracis spores in liquid using phage-based magnetoelastic micro-resonators. Sens Actuat B 127:559–566
Waddell TE, Poppe C (2000) Construction of mini-Tn10luxABcam/Ptac-ATS and its use for developing a bacteriophage that transduces bioluminescence to Escherichia coli O157:H7. FEMS Microbiol Lett 182:285–289
Wolber PK, Green RL (1990) Detection of bacteria by transduction of ice nucleation genes. Trends Biotechnol 8:276–279
Wu Y, Brovko L, Griffiths MW (2001) Influence of phage population on the phage-mediated bioluminescent adenylate kinase (AK) assay for detection of bacteria. Lett Appl Microbiol 33:311–315
Yemini M, Levi Y, Yagil E, Rishpon J (2007) Specific electrochemical phage sensing for Bacillus cereus and Mycobacterium smegmatis. Bioelectrochemistry 70:180–184
Zhu H, White IM, Suter JD, Fan X (2008) Phage-based label-free biomolecule detection in an opto-fluidic ring resonator. Biosens Bioelectron 24:461–466
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Zourob, M., Ripp, S. (2010). Bacteriophage-Based Biosensors. In: Zourob, M. (eds) Recognition Receptors in Biosensors. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-0919-0_11
Download citation
DOI: https://doi.org/10.1007/978-1-4419-0919-0_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-0918-3
Online ISBN: 978-1-4419-0919-0
eBook Packages: EngineeringEngineering (R0)