
Abstract
Despite the several advances in the last few years into treatment of advanced lung cancer, the 5-year survival remains extremely low. New therapeutic strategies are currently under investigation, and immunotherapy seems to offer a promising treatment alternative. In the last decade, therapeutic cancer vaccines in lung cancer have been rather disappointing, mainly due to the lack of efficient predictive biomarkers. A better refinement of the patient population that might respond to treatment might finally lead to a success story. For the first time, the immune checkpoint inhibitors are demonstrating sustained antitumor response and improved survival and they may be the first immunotherapeutics available for patients with lung cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction—NSCLC and Immunotherapy
Lung cancer is the most common cause of cancer mortality globally, accounting for 1.2 million deaths per year (Ferlay et al. 2010). Non-small cell lung cancer (NSCLC) accounts for 80–85 % of all cases and small cell lung cancer accounts for the remaining 15–20 % (Peters et al. 2012). Within NSCLC, the squamous cell carcinoma accounts for approximately 30 % and the non-squamous NSCLC for the 70 %: the latter group is mainly formed by adenocarcinomas, but it can also include large cell carcinomas and less well-differentiated variants (Goldstraw et al. 2011) (Fig. 1).

Fig. 1 Cellular classification of lung cancer
The majority of lung cancer patients present with advanced disease (stage IIIb/IV), and despite targeted therapy has increased the treatment options in the latest years, the overall 5-year survival rate is less than 5 % (Detterbeck et al. 2009).
In first line, the standard of care for stage IV patients consists in the combination of cytotoxic chemotherapy (carboplatin or cisplatin in combination with paclitaxel, gemcitabine, docetaxel, vinorelbine, irinotecan, and pemetrexed) (National Comprehensive Cancer Network (NCCN) 2014).
More recently, targeted therapies have increased the treatment options for patients with certain genetic mutations, opening the path for a more personalized treatment (Blackhall et al. 2013). For instance, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, and afatinib offer a therapeutic window in patients with identified genetic alterations of a key oncogenic driver, the EGFR (Peters et al. 2012). The EML4-ALK fusion gene, resulting from an inversion in chromosome 2, has also been identified as an oncogenic driver in NSCLC (Kwak et al. 2010). It is encountered more frequently in never-smokers, the adenocarcinoma subtype and in younger patients, representing probably ∼5 % of adenocarcinoma (Shaw et al. 2009). Patients with ALK mutation can be efficiently treated by the TKI crizotinib (Shaw et al. 2013).
While targeted therapies against driver oncoproteins such as EGFR and ALK have increased the treatment options for patients with non-squamous NSCLC, there are few therapeutic alternatives against tumors without known-driver mutations, and chemotherapy (carboplatin + paclitaxel or pemetrexed) remains the standard of care for squamous NSCLC patients.
The development and implementation of new therapeutic strategies is therefore essential to improve prognosis in lung cancer, and immunotherapy may offer a promising treatment alternative.
Immunotherapy using therapeutic cancer vaccines has shown promise in early clinical trials and has advanced to late-phase development with disappointing results. These include the therapeutic vaccines that target MAGE-3 and MUC1. A promising approach currently in clinical evaluation in NSCLC is the use of immune checkpoint modulators. By blocking inhibitory molecules or, alternatively, activating stimulatory molecules, these treatments are designed to unleash and/or enhance preexisting anticancer immune responses.
2 The Role of the Immune System in NSCLC
The immune system has a complex interaction with the tumor, as it can have either a tumor-promoting or tumor-inhibitory role, depending on the tissue localization, the cell types of the tumor immune infiltrate and the cytokines they secrete. The nature of the immune cells within the tissue can influence tumor progression and has prognostic significance.
An increased tumor infiltration with CD4+ and CD8+ T-helper (Th) 1 cells has been considered a strong favorable prognostic predictor independently associated with improved survival in lung cancer (Kawai et al. 2008). Similarly, a Th1-enriched gene signature in the tumor microenvironment may favor the presence of immune effector cells in the tumor of patients who responded to the MAGE-3 cancer vaccine (Ulloa-Montoya et al. 2013). Conversely, the IL4 gene pathway and other genes associated with a Th2 signature are significantly enriched in the blood of NSCLC patients in tumor progression (Chen et al. 2013). In NSCLC, higher number of cytotoxic T cells (CTL) and Th1 type cells have been associated with survival benefit regardless of the disease subtype (Bremnes 2011; Dieu-Nosjean 2008), whereas a low CTL/Treg ratio and Th2 type cells are a predictor of recurrence and shorter survival (Petersen et al. 2006).
Like the majority of other tumors, also NSCLC uses different strategies to evade the immune system and prevent destruction by effector T cells. Firstly by down-regulating key molecules such as MHC class I molecules and tumor-associated antigens to avoid immune recognition, and secondly preventing T-cell activation by disabling T-cell function or inducing T-cell apoptosis (Schreiber et al. 2011).
In surgically resected specimens, 25–94 % of NSCLCs have down-regulated HLA class I expression (So et al. 2005) and abnormal expression of the β2-microglobulin (Baba et al. 2007), hampering an efficient antigen presentation of tumor-associated epitopes to T cells. Lung cancer cells also express the programmed death ligand-1 (PD-L1) which has been shown to suppress immune responses through engagement with the negative regulator PD-1 expressed on activated T cells and B cells (Konishi et al. 2004; Topalian et al. 2012).
Alternatively, tumor escape may result from an immunosuppressive tumor microenvironment via the production of vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), or indoleamine 2,3-dioxygenase (IDO) and/or the recruitment of regulatory immune cells that function as the effectors of immunosuppression (Schreiber et al. 2011).
Several cellular and soluble suppressive mechanisms have been described in NSCLC. The increased number of M2 macrophages, which secrete IL-8 and IL-10 and inhibit Th1 immune response, is associated with poor prognosis and disease recurrence in NSCLC (Suzuki et al. 2011). Similarly, a tumor accumulation of myeloid-derived suppressor cells (MDSC) and regulatory T cells (Tregs) is associated with unfavorable prognosis in NSCLC patients (Diaz-Montero et al. 2009; Woo et al. 2001).
MDSC can strongly suppress T-cell function via up-regulation of reactive oxygen species (ROS) production (Huang et al. 2013). Tumor-infiltrating Foxp3+ Tregs were positively correlated with intratumoral cyclooxygenase-2 (COX-2) expression and associated with a worse prognosis in resected NSCLC (Shimizu et al. 2010; Hanagiri et al. 2013). Both the inhibition of ROS and COX2 might offer a therapeutic option to counterbalance the effects of these two suppressive regulators.
The immune system plays an active role not only in adenocarcinoma but also in squamous cell carcinoma. For instance, a higher number of infiltrating CD8+ CTL and a significantly lower number of Tregs have been found in squamous than adenocarcinoma (Black 2013; Stinchcombe 2014). However, in squamous cell carcinoma, the favorable CTL/Treg ratio does not correlate with a survival advantage, possibly due to an immunosuppressive tumor environment and other immune-evading strategies (Suzuki et al. 2011; Stinchcombe 2014).
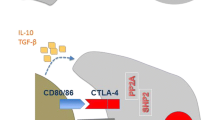
A major component of the adaptive immune response relies on the specific recognition of T-cell antigens. This starts with the T-cell receptor recognizing the antigenic peptide presented by the major histocompatibility complex (MHC) on the antigen-presenting cells (APCs), leading to T-cell activation. Engagement of the co-receptor CD28 with B7-1 (CD80) and B7-2 (CD86) ligands on the APCs provides a stimulatory signal for T cells, sustaining T-cell activation (Chambers and Allison 1997). Subsequent engagement of the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) with the same ligands on the APCs results in attenuation of the response (Walunas et al. 1994; Chambers and Allison 1997). Tregs are also known to up-regulate CTLA-4, further contributing to suppressed activation and expansion of CTL (Peggs et al. 2009). Another negative regulator expressed on activated T cells is programmed death 1 (PD1), whose binding with its ligands PD-L1 and PD-L2 on APCs and tumor cells results in down-regulation of T-cell activation (Dong et al. 2002). Tumors are able to up-regulate the PD-L1 ligand expression in order to bind to the PD1 expressed on T cells and thus down-regulating the immune response and promoting T-cell apoptosis (Dong et al. 2002). Blocking the negative T-cell regulators has proven to sustain the T-cell immune response and enhanced antitumor immunity (Leach et al.1996; Brahmer 2014). In NSCLC , elevated PD-L1 levels were positively correlated with increased TILs and associated with better outcome in lung carcinomas (Velcheti et al. 2014). Higher PD-L1 levels were detected in squamous cell carcinomas compared to adenocarcinoma (56.7 % vs. 27.5 %; P = 0.009) despite having comparable lymphocytic infiltrates (Velcheti et al. 2014).
The potential to identify subsets of NSCLC patients with an inflammatory tumor microenvironment that predicts for longer survival may also predict for response to immunotherapy and checkpoint inhibitor blockade.
Understanding of the immune evasion mechanisms regulated by tumor cells is necessary for developing more effective immunotherapeutic approaches to lung cancer. Ultimately, it is likely that the success of immune therapy in lung cancer will depend on a particular immune biomarker signature and the integration of strategies that aims to boost the immune response while down-regulating the cancer-induced immune suppression.
3 Vaccines
Vaccines investigated in NSCLC are usually antigen specific and are based on peptides, viral vectors, cell lines, or anti-idiotypic antibodies. Ideally, antigens selected for the immunological approaches should be highly expressed by the tumor but not by healthy tissues and should be immunogenic (Bradbury and Shepherd 2008).
This article will focus on vaccines in advanced stage of clinical development. Table 1 provides a list of vaccines which are currently investigated in phase III clinical trials.
3.1 Melanoma-Associated Antigen 3 (MAGE-A3, GSK1572932A)
The MAGE-A3 antigen is expressed by various tumors. It is not expressed in normal tissue except testis and placenta; however, testis and placenta lack the presentation by HLA molecules (De Plaen et al. 1994). MAGE-A3 is expressed in 35–50 % of NSCLC tumors (Van den Eynde BJ 1997; Sienel 2004; Vansteenkiste et al. 2007), and its expression seems to be a prognostic factor for the clinical outcome in NSCLC (Brichard 2007).
Consequently, MAGE-A3 is a tumor specific antigen which makes it unique for the development of an antigen-specific cancer immunotherapy (ASCI). The MAGE-A3 immunotherapeutic GSK1572932A consists of the MAGE-A3 peptide in combination with an adjuvant. The peptide used in newer clinical trials is produced by recombinant technology (Tyagi and Mirakhur 2009). In earlier clinical trials, the adjuvant AS02B was used, including the phase II study in NSCLC. However, compared to AS02B, the adjuvant AS15 resulted in an increased clinical activity as demonstrated in a melanoma trial (Kruit 2008). Therefore, the AS15 immunostimulant was chosen for all further trials (MAGRIT).
Phase II trial in adjuvant NSCLC
This was a randomized, double-blind, placebo-controlled trial in completely resected MAGE-A3 positive-stage IB or II NSCLC patients (Vansteenkiste et al. 2013). 33 % of the 1,089 screened patients were MAGE-A3 positive, 183 patients were enrolled, and 122 patients were assigned to MAGE-A3, 60 to placebo.
After a median observation period of 44 months, the hazard ratio for the primary endpoint disease-free interval (DFI) was 0.75 (95 % CI, 0.46–1.23; p = 0.254) in favor of the MAGE-A3 treatment. This effect was not statistically significant. Similar results have been observed for the secondary endpoints disease-free survival (DFS) and overall survival (OS). After 70 weeks of median observation, the effect for the primary endpoint DFI was almost the same with HR = 0.78 (95 % CI, 0.49–1.24; p = 0.259). In addition, this effect was consistent for all stratification factors (disease stage, histology, and resection technique). However, after 70 months of median observation, the treatment effect on the secondary endpoint DFS was weaker and did even no longer exist for OS.
A total of 117 of 119 MAGE-A3-treated patients who were eligible for immune response assessment developed anti-MAGE-A3 IgG antibodies after four doses of the vaccine. The remaining two patients developed an immune response after six and nine doses. No induction of immune response was observed in 57 placebo-treated patients. There was no correlation between immune response and DFI.
MAGE-A3 immunotherapeutic treatment was safe with no relevant difference in terms of grade 3/4 AES compared to placebo. Local reactions were more frequently observed in the MAGE-A3 arm and were all of mild-to-moderate intensity.
Gene signature (GS) was investigated in tumor samples in a phase II trial in melanoma (Kruit et al. 2013). 84 genes, mainly immune related, were identified as potentially predictive for the efficacy of MAGE-A3. This GS was then prospectively investigated in the phase II trial in NSCLC (Ulloa-Montoya et al. 2013). 61 (39 %) of 157 tested NSCLC patients were GS positive. Comparing the effect of the vaccine with placebo for the primary endpoint DFI, hazard ratio was 0.42 (95 % CI, 0.17–1.03; p = 0.06) for the GS-positive patients and hazard ratio was 1.17 (95 % CI, 0.59–2.31; p = 0.65) for the GS-negative patients. Also, for OS, where no clinical effect was seen after 70 months of median observation in the whole population, hazard ratio was 0.63 (95 % CI, 0.22–1.78; p = 0.81) in favor of MAGE-A3 vaccination in patients with positive GS.
Phase III trial in NSCLC: MAGRIT
A randomized, double-blind phase III trial in patients with resected stage IB/II/IIIA NSCLC was initiated based on the phase II data. Patients were eligible if they were MAGE-A3 positive, underwent surgery with or without standard adjuvant chemotherapy. 13 intramuscular injections are scheduled over 27 months. The primary endpoint was DFS, and the secondary endpoint was prospective validation of GS (Tyagi and Mirakhur 2009).
MAGRIT started in October 2007 and aimed to recruit 2,270 patients from around 400 sites in 33 countries (clinicaltrials.gov). On April 2, 2014 GlaxoSmithKline announced its decision to stop the MAGRIT trial, after establishing that it will not be possible to identify a subpopulation of gene signature-positive NSCLC patients that may benefit from the treatment (GSK press release). Data showed that MARGIT did not meet its first or second co-primary endpoints as it did not significantly extend disease-free survival when compared to placebo in either the overall MAGE-A3-positive population or in those MAGE-A3-positive patients who did not receive chemotherapy.
In summary, although the phase II trial indicated a clinical benefit of MAGE-A3 immunotherapy in patients with completely resected stage IB/II NSCLC, an effect which was even more pronounced in patients with positive GS, the phase III trial MARGIT failed to demonstrate a clinical benefit for the patients.
3.2 L-BLP25 (Tecemotide, Formerly Stimuvax)
The mucinous glycoprotein MUC1, a member of a family of mucins, is an integral membrane protein with extracellular, transmembrane, and cytoplasmic domains. MUC1 has a broad distribution in a variety of normal tissues and tumor tissues (Zotter et al. 1988; Ho et al. 1993; Kufe 2009). MUC1 has oncogenic potential and is able to confer resistance to genotoxic agents (Agrawal et al. 1998). Further, MUC1 seems to play a role in tumor progression, because it can stimulate cell proliferation through growth factor receptor, β-catenin, and ERα, and also suppresses apoptosis through the regulation of JNK, NF-kB, HSP90, and extrinsic apoptotic pathways (Bafna et al. 2010). MUC1 also alters various signaling pathways (Zhao et al. 2009; Raijna et al. 2011). In contrast to normal tissue, MUC1 is aberrantly glycosylated in tumors which makes it a unique target for cancer treatment. Furthermore, underglycosylated MUC1 primes class I-restricted CTL more efficiently than glycosylated MUC1 (Hiltbold 1999). MUC1 is expressed in >90 % of early-stage NSCLC independent of histology. MUC1 expression is generally maintained in paired primary/nodal tumor samples (Mitchell et al. 2013).
Tecemotide is a peptide-based vaccine consisting of BLP25 lipopeptide, immunoadjuvant monophosphoryl lipid A (MPL), and three lipids forming a liposomal product (Butts et al. 2005). Three days after a low-dose intravenous cyclophosphamide administration, tecemotide is administered subcutaneously weekly over 8 weeks, followed by administration every 6 weeks until disease progression.
Phase II trials in stage III/IV NSCLC
Tecemotide was investigated as maintenance treatment in a randomized phase IIB trial in stage IIIB/IV NSCLC (Butts et al. 2005). Patients were eligible if they did not progress after initial therapy, i.e. chemoradiotherapy or chemotherapy. They were randomly assigned to tecemotide plus best supportive care (BSC) or BSC alone. The primary endpoint was OS. 171 patients were accrued, 65 with stage III disease, and 106 with stage IV disease. The median OS time was 17.4 months for patients in the tecemotide arm and 13 months in the BSC arm with a hazard ratio of 0.739 (95 % CI, 0.509–1.073; p = 0.112) in favor of tecemotide. This effect was even more pronounced in the subgroup of patients with stage IIIB locoregional disease; the hazard ratio was 0.524 (95 % CI, 0.261–1.052; p = 0.069). An updated survival analysis revealed that patients with stage IIIB locoregional disease had a median OS of 30.6 months in the tecemotide arm versus 13.3 months in the BSC arm with a 45 % reduced risk to die [HR 0.548, 95 % CI 0.301–0.999 (Butts et al. 2011)].
Comparable survival data, observed in a single-arm phase II trial in stage III locoregional NSCLC patients (n = 22), provided some consistency (Butts et al. 2010). Long-term observation data of this trial indicated a median OS of 51.9 months (Butts et al. 2012).
Phase III trial START
Encouraged by the phase II data, a randomized, double-blind phase III trial was conducted in stage III locoregional NSCLC (Butts et al. 2013, 2014). 1,513 patients that did not progress after chemoradiotherapy were randomized to tecemotide or placebo in a 2:1 manner. Chemoradiotherapy was given concurrently or sequentially and consisted of a platinum-based chemotherapy and at least 50 Gy of radiotherapy. Caused by a clinical hold, the primary analysis cohort consisted of 1,239 patients. Median OS was 25.6 months in the tecemotide arm versus 22.3 months in the placebo arm (HR 0.88, 95 % CI, 0.75–1.03, p = 0.123). A subgroup analysis for the strata of START revealed a more pronounced treatment effect in patients with prior concurrent chemoradiotherapy (n = 806). The median OS in this subgroup was 30.8 months for patients in the tecemotide arm and 20.6 months for patients in the placebo arm (HR 0.78, 95 % CI, 0.64–0.95, p = 0.016). Tecemotide was well tolerated with no safety concerns.
Based on the subgroup finding of START, the START2 trial was initiated. START2 is a phase III, multicenter, 1:1 randomized, double-blind, placebo-controlled clinical trial designed to assess the efficacy, safety, and tolerability of tecemotide in patients suffering from unresectable, locally advanced (stage III) NSCLC who have had a response or stable disease after at least two cycles of platinum-based concurrent chemoradiotherapy (CRT). The study is expected to recruit about 1,000 patients. The study’s primary endpoint is OS. Secondary endpoints include time to symptom progression, progression-free survival, and time to progression (Merck KGaA press release, clinicaltrials.gov NCT02049151). The trial has been stopped in September 2014 (press release Merck KGaA).
In summary, the phase III trial could not demonstrate a survival benefit for patients treated with tecemotide. However, subgroup analyses indicate that patients with prior concurrent chemoradiotherapy may benefit from tecemotide treatment. The value of this subgroup finding is currently investigated in another phase III trial.
3.3 GV1001 (Telomerase Vaccine, Tertomotide)
The reverse transcriptase subunit of telomerase (hTERT) is overexpressed in the majority of tumors and, under normal conditions, by embryonic cells and bone marrow stem cells (Kim et al. 1994). It is absent in most adult tissues. Therefore, it is an attractive target for antigen-specific cancer immunotherapy.
GV1001 is a 16-mer peptide vaccine which is injected intradermally 10–15 min after the administration of the adjuvant GM-CSF. It induces both, CD4+ and CD8+, responses and may cause epitope spreading (Inderberg-Suso EM et al. 2012). The administration schedule consists of 3 injections in the first week, followed by injections in weeks 2, 3, 4, 6, 10, and then booster injections every 4 weeks. GV1001 was investigated in several tumor entities such as breast and prostate cancer, melanoma, and other solid tumors; however, most data were generated in pancreatic cancer and NSCLC. Based on phI/II data in a trial in patients with pancreatic cancer (Bernhardt et al. 2006; Kyte et al. 2009), two phase III trials have been initiated in this indication. One was stopped early because a lack of efficacy (press release), the second failed to demonstrate evidence for efficacy (Middleton et al. 2013).
Phase I/II trials in NSCLC
In the trial CTN-2006, 23 unresectable stage III NSCLC patients were vaccinated after the initial treatment with radiotherapy plus weekly docetaxel. Specific T-cell response was shown in 16 of 20 eligible patients. Survival was longer in patients with immune response. The treatment was well tolerated (Brunsvig et al. 2011).
A phase I/II trial, CTN 2000, investigated the vaccination in 26 patients, most of them in stage IV disease. Immune responses were detected in 11 out of 24 eligible patients (Brunsvig et al. 2006). An 8-year update of this trial revealed that OS was significantly increased in immune responders compared to non-responders with a median survival of 19 and 3.5 months, respectively (Brunsvig et al. 2011). Four patients were long-term survivors, two of them received vaccinations still 9 years after start of vaccination. All four long-term survivors exhibited strong and durable T-cell responses.
Phase III trial LucaVax
A phase III trial was planned investigating the effect of GV1001 in stage III NSCLC . Patients with unresectable disease who got chemoradiotherapy with curative intent are eligible. Chemoradiotherapy has to consist of platinum-based doublets which must not contain gemcitabine and radiotherapy with a dose of up to 66 Gy. Patients are randomized in a double-blind manner to either GV1001 or placebo. The trial was planned to start in April 2012 but is not recruiting, yet (clinical trials.gov, NCT01579188).
In summary, there is some evidence for immunological responses after vaccination of patients with GV1001. GV1001 seems to be well tolerated and showed some efficacy in early trials. However, ongoing phase III trials in pancreatic cancer are failed, while the phase III trial in NSCLC did not start recruitment. It is open if the development of GV1001 will be continued.
3.4 Belagenpumatucel-L (TGF-β Antisense Gene-Modified Allogeneic Tumor Cell Vaccine, LucanixTM)
Belagenpumatucel-L is a cell-based vaccine which consists of four human NSCLC cell lines: two adenocarcinomas, one squamous cell carcinoma, and one large cell carcinoma. All cell lines were transfected with an antisense gene for TGF-β (Nemunaitis et al. 2006). Using defined human cancer cell lines presenting antigens for vaccination of NSCLC patients is a reasonable approach, because data from gene expression profiling in adenocarcinomas indicate that the majority of tumor-related genes are co-expressed by different lung tumor subtypes (Hayes et al. 2006). In addition, this approach has the advantage that the vaccine can be used “off the shelf.”
TGF-β has a broad variety of effects on tumors, which may be even controversial (Katz et al. 2013). TGF-β signaling can result in tumor promotion by several modes of action. Immune evasion is one of them, and blocking it may be a useful tool to increase the effect of a vaccine. There are other direct effects of TGF-β which promote tumors, for example promotion of invasion and angiogenesis and development of metastases.
Belagenpumatucel-L is administered intradermally (2.5 × 107 cells in a volume of 0.4 mL) monthly for 18 months and then once at 21 and 24 months in the absence of disease progression (clinical trial.gov, NCT00676507).
Phase II trials
Belagenpumatucel-L was investigated in an open-label, three-arm phase II trial (Nemunaitis et al. 2006). Three doses of the vaccine were investigated: 1.25, 2.5, and 5 × 107 cell/injection (cohorts 1, 2, and 3, respectively). Patients were eligible if they had a confirmed stage II (n = 2), IIIA (n = 12), IIIB (n = 15) or IV (n = 46) NSCLC . Treatment with belagenpumatucel-L was started after at least 30 days following the initial cytotoxic therapy.
The treatment was safe with no difference in serious adverse events across dose cohorts. All but two-grade 3/4 adverse events were attributed to disease progression.
Dose-related survival results were observed. Cohorts 2 and 3 were significant different compared to cohort 1 (p = 0.0069). Also 1- and 2-year survival probability was higher for cohorts 2 and 3 compared to cohort 1: 68 and 52 % compared to 39 and 20 %, respectively. The median survival time was 581 days in dose cohorts 2 and 3, i.e., significantly higher than in cohort 1 (252 days, p = 0.0186).
In PBMC of all patients, an increased intracellular cytokine production was measured at week 8 compared to base line. Further, patients with stable disease (SD) or better had higher frequencies of IFN-γ, IL-6 and IL4. Seven of nine patients with negative ELISPOT at baseline developed a response at week 12. The majority of stage IIIB/IV NSCLC patients with SD or better produced a markedly elevated ELISPOT response. With regard to the humoral immune response, a correlation of positive response and clinical outcome was described.
The clinical data were confirmed in a smaller phase II study in 21 patients with advanced stage NSCLC (Nemunaitis et al. 2009). In addition, further immunological tests were performed in late-stage NSCLC patients of phase II patients (Fakhari et al. 2009). According to these data, immune responders survived 32.5 months compared to 11.6 months for non-responders.
Phase III trial
An international multicenter, randomized, double-blind, placebo-controlled study of Lucanix™ maintenance therapy for stages III/IV NSCLC subjects who have responded to or have stable disease following one regimen of front-line, platinum-based combination chemotherapy was initiated in 2008 (STOP; Clinicaltrials.gov, NCT00676507). Primary endpoint was overall survival.
STOP did not meet its predefined primary endpoint in the entire patient population (Giaccone 2013); median OS was 20.3 months with the vaccine compared to 17.8 months with placebo (HR 0.94; p = 0.594).
However, prognostic factors for improved outcome were identified in subgroup analyses. The OS was shown to be significantly impacted by the time interval between randomization and the end of frontline chemotherapy (p = 0.002). The OS was improved by 7.3 months with belagenpumatucel-L in 305 stage IIIB/IV patients who were randomised within 12 weeks of chemotherapy completion. In this cohort, the median OS was 20.7 months with belagenpumatucel-L compared to 13.4 months with placebo (HR 0.75; p = 0.083).
Other factors prognostic of better outcome were disease stage, whether prior radiation was received and histology. The patients with confirmed pretreatment radiation showed optimally improved median OS of 29.8 months difference between the treatments; patients receiving belagenpumatucel-L following radiation showed median OS of 40.1 months compared to 10.3 months with placebo (HR 0.45; p = 0.014).
In summary, despite encouraging data from early clinical trials, the phase III trial failed to demonstrate efficacy of belagenpumatucel-L for the intent to treat population. However, subgroup analyses revealed that defined patient populations may benefit from the vaccination. Further studies are warranted to confirm this data.
3.5 Racotumomab (Anti-idiotypic NeuGc-GM3 mAb, 1E10 Antibody)
NGcGM3 ganglioside is expressed by many tumors and seems to be practically undetectable in healthy human tissue (Fernandez et al. 2010; Gomez and Ardigo 2012). GM3 is expressed in >90 % of NSCLC tumors and is involved in tumor induced dentritic cell suppression (van Cruijsen et al. 2009).
Racotumomab is an IgG1 anti-idiotypic monoclonal antibody developed by the Center of Molecular Immunology, Havana, Cuba (Vázquez et al. 2012). Racotumomab is administered intradermally (1 mg/ml) five times every 2 weeks during the induction period followed by monthly vaccinations (clinicaltrials.gov, NCT01460472).
Phase I trials were performed in different tumor types including melanoma, breast cancer, SCLC, and NSCLC (Vázquez et al. 2012). It was found that the administration is safe and that it resulted in a strong and specific immune response against NeuGc containing gangliosides (Hernández et al. 2008).
Phase II trials
A multicenter, randomized, placebo-controlled, double-blind trial in patients with advanced NSCLC (stage IIIB/IV) who had achieved stable disease or better after initial onco-specific treatment was conducted (Macías et al. 2012). 176 patients were randomized to the vaccine or placebo. Moderate-to-mild local injection site reaction has been observed in the vaccination arm; otherwise, no difference was seen between both arms. The median OS was 8.3 months in the racotumomab arm and 6.3 months in the placebo arm. The OS rate at 1 and 2 years was 38 and 17 % in the racotumomab arm and 24 and 7 % in the placebo arm, respectively. For patients who received >5 vaccinations/placebo administrations (n = 135), this effect was more pronounced.
In another open, non-randomized phase II trial, patients with advanced NSCLC (stage IIIB/IV) who did progress following initial onco-specific treatment were treated with racotumomab (Gomez et al. 2013). Most of the patients had received 4–6 cycles of cisplatin plus vinblastine. 180 patients were accrued. Median survival was 8.06 months, and the OS rate at 2 years was 21 %. Compared to the above-mentioned trial, a comparable OS was observed for patients who received the vaccine.
Phase III trial
A prospective, randomized multicenter, open-label phase III study of racotumomab plus BSC versus BSC in patients with advanced NSCLC was initiated in 2010 (clinicaltrials.gov, NCT01460472). The primary endpoint is OS, secondary endpoints are safety, PFS and immunology. 1,082 patients will be enrolled, and data are expected in September 2015. Patients are eligible if they suffer from stage III or IV NSCLC and did not progress after standard one line treatment. The trial is conducted in Argentina, Brazil, Cuba and Singapore.
In summary, racotumomab was well tolerated and showed efficacy immunological response in phase II trials and no details on data have been published so far. Obviously, the ganglioside seems to be an interesting target and the phase III trial may provide more information on this vaccine.
3.6 TG4010 (MVA-MUC1-IL2)
TG4010 is an antigen-specific vaccine targeting MUC1 (for MUC1, see also chapter about tecemotide). TG4010 is based on a viral vector, a modified vaccinia of Ankara (MVA), which, in addition to MUC1, expresses interleukin 2. TG4010 is administered subcutaneously at a dose of 108 pfu weekly for 6 weeks and then every 3 weeks (Limacher and Quoix 2012).
Two small phase I trials in 13 patients with MUC1 positive metastatic solid tumors were conducted (Rochlitz et al. 2003). The trials showed that the administration of TG4010 is well tolerated with injection site reactions and flulike symptoms only. It was further observed, that four patients had disease stabilization. One patient with NSCLC , who initially progressed, developed a long lasting response. In addition, some T-cell proliferative immune responses were observed.
Phase II studies
A phase II study was conducted in 65 patients randomized to either chemotherapy (cisplatin plus vinorelbine) plus TG4010 (arm 1) or to TG 4010 alone (arm 2) for first-line therapy of stage IIIB/IV NSCLC (Ramlau et al. 2008a). All patients had MUC1-positive tumors as assessed by IHC. The stronger clinical effect was observed in 44 patients randomized to arm 1. 33 of these 44 patients were evaluable for response, 13 achieved a partial response with a response rate of 35.1 %.12 other patents (27.3 %) achieved stabilization of disease. Immunological data (T-cell proliferation test and ELISPOT) were not conclusive. Immunological responses were seen as well before (baseline) as after vaccination. Actually, positive ELISPOTs at any time during the trial correlated with a better survival.
Based on these data, another randomized, open-label phase IIb trial investigated the effect of TG4010 in first-line therapy for stage IV NSCLC which was tested MUC1 positive (Quoix et al. 2011; Ramlau et al. 2008b). 148 patients were recruited, 74 patients were randomized to the combination therapy group (cisplatin plus gemcitabine plus TG4010), and the remaining patients were randomized to the chemotherapy alone group. Progression-free survival (PFS) at 6 months was the chosen primary endpoint. In the combination group, 43.2 (95 % CI 33.4–53.5) were progression free at 6 months, while only 35.1 % (95 % CI 25.9–45.3) were progression free at 6 months in the chemotherapy group. Objective response rate was 41.9 (95 % CI 30.5–53.9) and 28.4 (95 % CI 18.5–40.1) for combination therapy and chemotherapy, respectively. There was no difference in overall survival. A pre-specified analyses revealed that the percentage of activated natural killer cells (CD16+, CD56+, and CD69+) had predictive value. Patient with normal percentage of activated natural killer cells (73.2 % of patients) benefited more from TG4010 treatment, and this included also median OS which was 17.1 months for the combination group and 11.3 months for the chemotherapy group. The interaction between pretreatment percentage of activated natural killer cells and treatment group was significant for OS (p = 0.0023). Overall TG4010 was well tolerated with fever, abdominal pain, and injection site pain as site effects which occurred more frequently in the combination group. While the incidence of serious adverse events was similar in both treatment groups of the ITT population, there was a significantly higher incidence for these events in the combination group for the subpopulation with higher percentage of activated natural killer cells.
Phase IIb/III trial TIME
This is a double-blind trial comparing the combination of first-line chemotherapy with TG4010 or placebo in stage IV NSCLC with MUC1 expressing tumor (Quoix et al. 2012). The phase IIb part (n = 210) aimed at prospectively validating the level of activated NK-cells, the so-called triple-positive activated lymphocyte levels (CD16+, CD56+, CD69+; TrPAL), as predictive marker with PFS as primary endpoint. In contract to the originally planned approach, recently communicated data indicated that a PFS analysis using a quartile approach for the TrPAL lead to predictive threshold for the phase III part of the trial (press release 2014). The quartile analysis showed that in the 75 % of patients having the lower baseline level of TrPAL and who received TG4010, there was a clinically meaningful improvement in PFS, as indicated by a >25 % reduction in the risk of progression or death compared to placebo. Conversely, in the 25 % of patients with the higher level of TrPAL (highest quartile) and who received TG4010, there was no improvement in PFS. Additionally, in subgroup analyses using the quartile approach, an even larger improvement in PFS was obtained in patients with non-squamous tumors not treated with bevacizumab (73 % of initial study population). The design of the phase III part is now under discussion with regulatory authorities. The study started in 2012 with an estimated primary completion date end of 2015 (clinicaltrials.gov, NCT01383148).
In summary, TG4010 is well tolerated and showed clinical efficacy in combination with chemotherapy in metastatic NSCLC in a randomized phase II trial. A biomarker was identified (activated NK-cells, TrPAL). Data of the phase IIb part of the registration trial will be discussed with regulatory authorities in order to design the phase III part.
3.7 EGF Vaccine (CimaVax)
The epidermal growth factor (EGF) plays an important role in tumor growth, mitosis, and metastasis. The EGF receptor (EGFR) is expressed in NSCLC (Salomon et al. 1995), and two types of anti-EGF receptor approaches have been investigated in NSCLC: tyrosine kinase inhibitors and monoclonal antibodies (Ciardello and Tortora 2008; Pirker 2013).
An EGF vaccine was developed in Cuba with recombinant human EGF linked to a carrier protein (P64k Neisseria Meningitidis recombinant protein). A pilot trial in 10 patients with solid tumors indicated that this approach is immunologically effective and tolerable (González et al. 1998).
Two pilot trials were performed in 40 stage IIIB/IV NSCLC patients after onco-specific treatment in order to investigate the effectiveness of two different adjuvants in combination with the vaccine. Based on the antibody responses measured, Montanide ISA 51 was chosen for future clinical trials (Gonzáles et al. 2003). Another analysis using pooled data of the pilot trials showed clinical efficacy, in particular in seroconverted patients (Gonzáles et al. 2007).
Phase II trial
80 patients with stage IIIB/IV NSCLC who received first-line treatment were randomized to the vaccine plus supportive care versus supportive care (Neninger Vinageras et al. 2008; García et al. 2008). About 30 % of these patients had progressive disease at the time of randomization. In the treatment arm, patients received a low-dose cyclophosphamide administration 3 days prior to vaccination, then 4 weekly intra-muscular injections of 50 µg EGF followed by monthly injections.
The median OS was 6.47 months in the vaccine arm and 5.33 months in the control arm (p = 0.098). This effect was more pronounced in patients younger than 60 years with a median OS of 11.57 versus 5.33 months in the vaccination and control arm, respectively. Antigen responses and EGF concentration were measured in 42 patients (26 vaccinated and 16 controls) only. Data showed that patients with good antibody response had the better survival benefit with a median OS of 11.7 months compared to 3.6 months in patients with poor antibody response. Further, an inversed correlation between EGF concentration and survival was demonstrated. Vaccination was safe with mild-to-moderate-related adverse events only.
Phase III trial
In 2006, a phase III trial was initiated at 18 sites in Cuba which is still ongoing. 579 patients are randomized 2:1 to vaccine or control (Rodríguez et al. 2010). The therapeutic schedule for the vaccine is the same as in the phase II study; however, the vaccine is administered in four injection sites resulting in a four times higher dose. Patients are stratified for age: >60 years versus ≤60 years. Preliminary results of 160 patients show a trend toward a delayed separation of the survival curves in favor of the vaccine arm (Rodríguez et al. 2010).
A total of 40 patients of this phase III trial who received the high-dose regimen were compared to 40 patients of the phase II trial who received the lower-dose regimen (Rodríguez et al. 2011). The patient groups from both trials were balanced in terms of baseline and tumor characteristics. Both regimens were well tolerated with only grade 1- and grade 2-related adverse events. The humoral response was more pronounced in the high-dose group with an antibody titer of 1:7328 (geometric mean) compared to 1:3160 in the control group. In the high-dose group, the rate of good antibody responders was 54.8 % and the rate of “super good responders” was 30.8 %. In the phase II trial, the respective rates are 52.8 and 10 %. The median OS in high-dosed patients was 13.57 months compared to 6.47 months in low-dosed patients.
Another phase III open-label, randomized trial in stage III/IV NSCLC patients is ongoing and finished recruitment. Patients in the age of 20–65 years were eligible and randomized to the vaccine or BSC as first-line therapy. According to changes in clinicaltrials.gov (March 2014), the trial has been terminated in order to initiate a new phase III design including biomarker to enrich the patient population and to further strengthen OS benefit (clinicaltrials.gov, NCT01444118). The level of circulating EGF is used as biomarker and a minimum threshold level has been set as inclusion criterion for the trial.
In summary, phase II data on safety, efficacy, and immunology are encouraging. First data of an ongoing phase III trial suggest that an increased dose of the vaccine may have a stronger effect in terms of immunology and efficacy without an increasing risk for the patients. The phase III trial was terminated, and the outcome has not been made public so far. CimaVax was licensed in Cuba for the treatment of adult patients with stage IIIB/IV NSCLC (Rodríguez et al. 2010). Bioven has licensed in the rights on the EGF vaccine developed in Cuba. Bioven names this product EGF Pathway-Targeted Immunization, PTI, instead of vaccine in order to make clear distinction between classical vaccines targeting tumor antigens such as MUC1, MAGE 3, L-BLP-25 presented here, and the PTI approach.
3.8 Talactoferrin Alfa
Talactoferrin alfa is a recombinant human lactoferrin (Kelly and Giaccone 2010). It is structurally and functionally similar to native human lactoferrin as it can be purified from human milk. Lactoferrin has multiple known biological effects such as anti-inflammatory effects or anti-tumor effects. In addition, there are different immune modulatory effects. After oral administration, talactoferrin recruits dentritic cells in the Peyer’s patches of the intestine. These dentritic cells present (tumor associated) antigens and initiate an immune stimulatory cascade in the gut associated lymphoid tissue (GALT).
The anti-tumor effect of talactoferrin was demonstrated in mouse models. It was shown that oral talactoferrin induces mucosal IFN-gamma production as well as expansion of CD8+ T lymphocytes and NK-cells and enhancement of CD8+ cytotoxicity (Spadaro et al. 2007).
A phase I trial was conducted in patients with solid tumors. Ten patients who had failed conventional chemotherapy were recruited in the first part of the trial which was intended to escalate the dose and to identify the maximum-tolerated dose (MTD) (Hayes et al. 2006b). Talactoferrin was well tolerated, an MTD was not defined. Talactoferrin induced an increase in circulating IL18. Five patients had stable disease after 2 months of treatment, seven patients showed a reduction in tumor growth rate. In the second part, further 26 patients were recruited that received the two highest daily doses tested in part one (4.5 or 9 g/day) (Hayes and Falchook 2010). 12 of the total 36 patients suffered from NSCLC. 11 of them have received prior CTX or radiochemotherapy. Out of the 12 patients, seven had SD. The median PFS was 4.3 months, the median OS was 8.8 months. The substance was well tolerated.
Phase II trials
Two randomized, double-blind, placebo-controlled phase II studies have been initiated: one in stage IV NSCLC patients that progressed after CTX and one in previously untreated stage IV NSCLC patients.
In the first trial, 100 patients were included of which 81 patients received at least one dose (evaluable population) (Parikh et al. 2011). The majority of patients received one prior therapy, about 1/4 received two or more prior therapies. Talactoferrin was dosed 1.5 mg twice daily for 12 weeks followed by 2 weeks off for a maximum of three cycles until progression. The median OS was 6.1 months in the talactoferrin arm and 3.1 months in the placebo arm and the primary endpoint was met (p = 0.05). The 1-year survival rates were 29 and 16 %, respectively.
The second trial recruited 110 stage IV NSCLC patients who did not receive prior therapy for NSCLC (Digumarti et al. 2011). Patients were randomized to CTX plus talactoferrin or CTX plus placebo. Patients in the talactoferrin arm received 1.5 mg twice daily for three 6-week cycles (5 weeks on, 1 week off the drug) until progression. Primary endpoint was response rate (RR). In the evaluable population, RR was 47 % in the talactoferrin arm and 29 % in the placebo arm (p = 0.05). Median PFS was 7.0 and 4.2 months (HR = 0.85; p = 0.24), and median OS was 10.4 and 8.5 months (HR = 0.87; p = 0.26), respectively.
In both phase II trials, talactoferrin was well tolerated with grade 3 and 4 events occurring slightly more frequent in the placebo arms.
Phase III trials Based on the phase II data, two phase III trials were initiated: FORTIS-M and FORTIS-C.
FORTIS-M was a randomized, double-blind, placebo-controlled trial which compared the clinical effect of talactoferrin-alfa plus BSC with placebo plus BSC in patients with stage IV NSCLC who have failed two or more prior treatment regimens (clinicaltrials.gov, NCT00707304). The study did not meet the primary endpoint, i.e., to demonstrate a significant survival benefit for talactoferrin compared to placebo (aggenix press release). 742 patients were enrolled globally at 160 sites. Patients in the talactoferrin arm had a median OS of 7.5 months compared with 7.7 months for the placebo arm (HR = 1.04, p = 0.66).
FORTIS-C was a randomized, placebo-controlled study of talactoferrin-alfa in combination with carboplatin and paclitaxel as first-line therapy in patients with stage IV NSCLC. Co-primary endpoints were OS and PFS (clintrials.gov NCT00706862). This trial was stopped early because of the negative results from FORTIS-M (press release). At this time, 94 of the planned 1,100 patients were enrolled in the US. For the 94 enrolled patients, median PFS was 5.8 months in the talactoferrin arm and 5.6 months in the placebo arm (HR = 0.97; p = 0.89). Median OS was 11.4 versus 12.7 months (HR = 1.25; p = 0.36), respectively.
In summary, despite encouraging phase II data, talactoferrin-alfa failed to demonstrate clinical efficacy in late-stage NSCLC. The question was raised if this very late stage of disease, i.e., stage IV NSCLC with several previous lines of chemotherapy, is the right candidate for immunotherapy (Madan et al. 2013).
3.9 Tergenpumatucel-L (HyperAcute)
The α(1,3)galactosyl (αGal) gene is functional in mammalian species but inactive in humans (Joziasse and Orial 1999). Due to continuous presentation of the antigen αGal by intestinal and pulmonal bacteria, the human immune system develops specific antibodies recognizing αGal. These antibodies are responsible for a hyperacute reaction to cells expressing αGal, e.g., after xenotransplantation.
Already more than a decade ago, the approach of eliciting hyperacute xenograft response to treat cancer was described (Link et al. 1998). In animal models, it was shown that immunity against αGal can induce antitumor activity (Rossi et al. 2008).
Tergenpumatucel-L is a cell-based vaccine consisting of genetically modified allogeneic NSCLC cells bearing αGal moieties (Morris et al. 2013). Tergenpumatucel is administered intradermally.
Phase II trial
28 patients with metastatic or recurrent NSCLC were recruited into a single-arm phase II trial prior to systemic therapies (Morris et al. 2012, 2013). Patients received eight doses of 300 × 106 cells every 2 weeks. The median OS was 11.3 months. Eight patients had stable disease over more than 16 weeks. Nine out of 16 patients who received follow-up chemotherapy developed a response according to RECIST criteria. The vaccine was well tolerated. All patients showed increased anti-αGal antibody levels and 61 % of patients showed increased interferon-gamma levels (ELISPOT, no details described). These patients were characterized by a longer median OS of 21.9 months compared to 5.5 months for patients with lower IFN-gamma levels (Morris et al. 2012). The authors concluded that, compared to historical controls, the observed survival duration is encouraging. Further, due to the responses seen in the follow-up chemotherapies, the authors concluded that the vaccination has a chemosensitizing effect.
Phase IIb/III trial
Based on the phase II data, an open-label, randomized, multi-institutional adaptive design phase IIb/III trial was initiated in January 2013 (clintrials.gov, NCT01774578). Patients are eligible if they have stage IIIB/IV NCSLC and received prior line(s) of chemotherapy. In the phase IIb part, two dosing schedules will be investigated either 300 × 106 cells weekly for 11 weeks and then every 2 months for five additional doses or 300 × 106 cells every 2 weeks for six doses followed by additional 10 monthly doses. In both groups, a total of 16 immunizations is planned. The phase III part will then further investigate the clinical effect of Tergenpumatucel-L administering the selected dose from phase IIb part. 240 patients will be enrolled and randomized to either the vaccine or to chemotherapy. Primary endpoint is overall survival; results are expected in July 2015.
In summary, data from the phase II study indicate some clinical efficacy, although this trial was not randomized and no details have been published so far.
3.10 Summary—Vaccines
Several vaccines have been/are in phase III clinical development in NSCLC. To date, there is no evidence from phase III trials that vaccines can cause a clinically meaningful benefit in patients suffering from NSCLC. All finished trials did not meet the primary endpoint: MAGRIT (MAGE-A3), START (tecemotide), STOP (Lucanix), FORTIS M and FORTIS C (talactoferrin alpha) and probably also the CIMAVax trial (EGF vaccine). In addition, the LucaVax trial (GV1001) is not followed up and the START2 (tecemotide) follow-up trial was discontinued due to disappointing phase II data from a Japanese study in the same setting. Overall, the results of vaccinations in NSCLC are disappointing so far.
However, there are trials ongoing which can tell us more about the value of vaccinations in NSCLC. TIME (TG4010) as well as the phase III trials with racotumomab and tergenpumatucel-L will generate further data over the next years. Also, new phase III trials with belagenpumatucel-L (Lucanix) and CIMAVax may be initiated based on further analyses of the data.
Outlook: In addition to the development of vaccines as single immunological treatment approach, the combination of vaccines with non-specific immune stimulators, in particular checkpoint inhibitors , seems to be a promising approach which may strengthen the specific immunological effects of vaccines (Rangachari and Brahmer 2013). We are still in an early phase of investigating vaccines. Further understanding of the mode of action and in particular combination therapies may help to make vaccines more successful in NSCLC.
4 Immune Checkpoint Blockers
Since the mechanism of action of immune checkpoint blockers is not dependent on the expression of specific antigens in contrast to the vaccination approach, a broad application in different tumor types is conceivable. The strategy to augment antitumor responses through the blockade of immune checkpoint pathways has only recently been started to be explored for lung cancer, especially in NSCLC, and therefore, this part is not reporting the very limited experience in SCLC which has recently been summarized by Spiegel and Socinski (2013). Two pathways, the CTLA-4:B7-1/-2 and the PD-1:PD-L1/-L2 axis, are clinically investigated, and the current status is described below.
4.1 CTLA-4 Blockade
Positive phase III results in metastatic melanoma (Hodi et al. 2010; Robert et al. 2011) have stimulated the exploration of the anti-CTLA-4 monoclonal antibody ipilimumab in lung cancer. Thus, ipilimumab was evaluated in combination with paclitaxel and carboplatin (PC) in a randomized, double-blind, phase II, first-line clinical study in patients with locally advanced or metastatic NSCLC , or extended SCLC (phase II study for previously untreated subjects with non-small cell lung cancer (NSCLC) or small cell lung cancer (SCLC) (clinicaltrials.gov NCT00527735). Since the optimal sequence of chemo- and immunotherapy is challenging, two schedules, i.e., concurrent or phased, were explored. The study met its primary endpoint (defined as significant improvement in immune-related (ir)PFS) for phased treatment versus control (HR 0.72; p = 0.05), but not for concurrent treatment (HR 0.81; p = 0.13). The median irPFS/PFS/OS for phased, concurrent, and control treatments (i.e., PC only) were 5.7/5.1/12.2; 5.5/4.1/9.7 and 4.6/4.2/8.3 months, respectively (Lynch et al. 2010). Observed incidences of grade 3/4 treatment-related adverse events were similar: 39, 41, and 37 % for patients in the phased, concurrent and control groups, respectively. However, the rate of grade 3/4 immune-related AEs were differing as could be expected: 15 % in the phased-group, 20 % in the concurrent-group, and 6 % in the control-group patients. In addition, a trend for greater clinical activity was observed in patients with squamous histology (Lynch et al. 2010). Overall, these results triggered the initiation of a phase III trial in patients with stage IV squamous cell NSCLC to investigate ipilimumab plus PC versus placebo plus PC in August 2011 (clinicaltrials.gov NCT01285609).
Tremelimumab, another monoclonal anti-CTLA-4 antibody was explored in phase II in the maintenance setting after first-line chemotherapy for advanced NSCLC, where it did not improve PFS (Brahmer 2013). Up to date, no further trials have been initiated.
4.2 PD-1/PD-L1 Blockade
While CTLA-4 primarily regulates early stages of T-cell activation at their initial response to antigen as a signal dampener, the role of PD-1 is to limit the activity of T cells in peripheral tissues, especially in inflammatory situations (Pardoll 2012).
The first trial using a blocker of the PD-1/PD-L1 pathway, i.e., nivolumab/BMS-936558 an anti-PD-1 antibody, was a first-in-man single-agent dose-escalation trial. In this trial, one durable complete response (CR) (colorectal carcinoma, CRC) and two partial responses (PR) (melanoma and renal cell carcinoma, RCC) were observed in 39 patients. Two additional patients (melanoma, NSCLC) had significant lesional tumor regressions not meeting PR criteria. The initial safety profile was favorable in comparison with ipilimumab (Brahmer et al. 2010). These promising results boosted the clinical development activities on this specific immune checkpoint . The consequent multiple-dose-escalation trial with nivolumab containing several expansion cohorts recruited 296 patients overall (Topalian et al. 2012). Remarkably, 14 of 76 (18 %) advanced NSCLC patients evaluable for efficacy displayed an objective response (or) and five additional patients a stable disease (SD) for more than 24 weeks. The RR was higher in the squamous (6/18, 33 %) compared to the non-squamous subtype (7/56, 12 %). Again, the safety profile in the overall study population was favorable with a 14 % rate of grade 3/4 treatment-related adverse events, while 3 deaths from pulmonary toxicity were reported. Nivolumab monotherapy follow-up data presented at ASCO 2013 reported an overall RR of 17 % (22 responses in 129 patients; squamous vs. non-squamous: 17 vs. 18 %), a median PFS of 2.3 months, and a median OS of 9.6 months (Brahmer et al. 2013). This cohort was also investigated for the association of tumor PD-L1 expression with clinical activity. The tumors were defined as PD-L1 (PD-L1+) positive when ≥5 % of the tumor cells had membrane staining at any intensity. Using this cutoff, 5 of 31 patients defined as PD-L1+ had an OR, while 4 of 32 patients defined as PD-L1− had an OR as well (Antonia et al. 2013). Hence, further evaluation of PD-L1 as a molecular marker of nivolumab therapy is required.
First preliminary data in NSCLC have also been reported recently for lambrolizumab/MK-3475, which is another anti-PD-1 antibody. Thirty-eight patients (19/38) previously treated with two systemic regimens had been enrolled. The ORR (confirmed and unconfirmed using RECIST 1.1) was 21 % applying an independent central images review and 24 % using investigator-assessed irRC. The median duration of response by irRC had not been reached, with a median follow-up of 9 months (minimum 6 months). Pretreatment tumor PD-L1 expression was a statistically significant predictor of response. In patients with evaluable tumor PD-L1 expression, all confirmed responses by RECIST v 1.1 (and irRC) occurred in patients with tumors strongly positive for PD-L1. Fifty percent of the patients had drug-related adverse events, and there was only one case of a grade 3 (pulmonary edema), but no higher drug-related adverse events (Garon et al. 2013). Follow-up data presented at AACR 2014 reported a median PFS of 9 weeks, and a median OS of 51 weeks. PD-L1 IHC score was above a potential cut point in nine patients and below a potential cut point in 22 patients (seven patients could not be evaluated), and significant associations between tumor PD-L1 expression and ORR (57 vs. 5 %) were observed (Gandhi et al. 2014).
BMS-936559, an anti-PD-L1 antibody was explored in a multiple-dose-escalation phase I trial in 207 patients covering 49 advanced NSCLC patients evaluable for efficacy (Brahmer et al. 2012). RR for squamous and non-squamous subtypes were similar (1/13, 8 vs. 4/36, 11 %; all patients 5/49, 10 %) and not that impressive. However, as for nivolumab, a dose dependency in NSCLC patients could clearly be observed showing activity at 3 and 10 mg/kg. Grade 3/4 treatment-related adverse events were observed in only 9 % of the overall trial population.
Another anti-PD-L1 antibody, MPDL3280A, is also explored in phase I (Spiegel and Socinski 2013). The NSCLC expansion cohort (locally advanced or metastatic disease) was reported to display an impressive overall RR of 24 % (9 of 37 patients with both squamous and non-squamous histology). The incidence of grade 3/4 treatment-emergent adverse events in the NSCLC safety cohort was 34 %. Interestingly, no grade 3–5 pneumonitis or diarrhea was reported. Biomarker analyses from archival tumor showed a correlation between PD-L1 status and efficacy. Latest analyses revealed that patients with PD-L1-positive tumors showed an ORR of 100 % (4/4), while patients who were PD-L1 tumor status negative had an ORR of 15 % (4/26). Further, it was concluded that MPDL3280A is probably the first targeted agent showing more activity in smoking patients than in never-smokers. Moreover, the 24-week PFS was reported to be 46 % (Soria et al. 2013).
Several trials have recently been initiated with nivolumab in NSCLC: two open-label randomized phase III trials comparing nivolumab versus docetaxel in previously treated advanced or metastatic NSCLC , one trial in squamous and the other trial in non-squamous histology (clinicaltrials.gov NCT01642004, NCT01673867). An open-label phase III safety trial of nivolumab in subjects with advanced or metastatic NSCLC who have progressed during or after receiving at least one prior systemic regimen to estimate the incidence and characterize the outcome of high-grade, select adverse events has also been recently initiated (clinicaltrials.gov NCT02066636). Additional phase II trials are actively recruiting or ongoing (clinicaltrials.gov NCT02041533, NCT01721759).
A phase I trial in stage IIIB/IV NSCLC patients is exploring different combinations of nivolumab with (a) gemcitabine/cisplatin, (b) pemetrexed/cisplatin, (c) carboplatin/paclitaxel, (d) erlotinib, (e) ipilimumab, (f) bevacizumab maintenance, (g) switch maintenance, or (h) as monotherapy in first-line patients with brain metastases (clinicaltrials.gov NCT01454102). In addition, a randomized phase II trial in subjects with recurrent metastatic NSCLC exploring epigenetic priming with azacitidine and entinostat or oral azacitidine alone prior to nivolumab treatment has been initiated (clinicaltrials.gov NCT01928576). Priming with these methylation blockers holds promise as DNA demethylation may contribute to PD-1 overexpression. Another anti-PD-1 antibody, lambrolizumab/MK-3475, is also put forward to phase II/III: A randomized trial is exploring its efficacy and safety versus docetaxel in previously treated subjects with NSCLC (clinicaltrials.gov NCT01905657). A phase I study of lambrolizumab is investigating the combination with cisplatin/pemetrexed or carboplatin/paclitaxel in patients with advanced NSCLC (clinicaltrials.gov NCT01840579). Further, phase I and II trials with MK-3475 in NSCLC in monotherapeutic setting and in combination settings with paclitaxel, carboplatin, pemetrexed, bevacizumab, ipilimumab, and erlotinib have been initiated (clinicaltrials.gov NCT02039674). The anti-PD-L1 antibody MPDL3280A is also further explored in one phase III and three phase II trials in advanced NSCLC (clinicaltrials.gov NCT02008227, NCT01846416, NCT02031458,). Moreover, a phase III trial with the anti-PD-L1 antibody MEDI4736 as sequential therapy in patients with locally advanced, unresectable NSCLC (stage III) who have not progressed following definitive, platinum-based, concurrent chemoradiation therapy has been initiated (clinicaltrials.gov NCT02087423).
5 Conclusion
Lung cancer has for a long time not been considered to be a very immunogenic tumor type such as melanoma or renal cancer. This perception has nowadays changed. Several vaccines are in phase III clinical development in NSCLC . Only few data are available so far, and no evidence for the clinical efficacy of vaccines could be demonstrated, yet. Recently, a phase III trial of talactoferrin alfa did not meet the primary endpoint (not described here because the development was stopped). Also, for tecemotide and Lucanix, a clinical benefit in terms of OS could not be demonstrated in large phase III trials. The appropriate patient selection, either with gene signature or using the optimal combination treatment, may help yield the survival improvement with vaccines. Over the next few years, we will get a clearer picture about the role of vaccines in the treatment of NSCLC. Currently, the most promising results in NSCLC have been observed in early clinical trials using immune checkpoint inhibitors which led to an accelerated clinical development. If the promises of the initial results prove true, the first approval of an immune checkpoint blocker for NSCLC can be expected around 2016.
References
Agrawal B et al (1998) The biological role of mucins in cellular interactions and immune regulation: prospects for cancer immunotherapy. Mol Med Today 4:397–403
Fernandez LE et al (2010) NGcGM3 ganglioside: a privileged target for cancer vaccines. Clin Dev Immunol, Article ID 814397
Soria JC et al (2013) Clinical activity, safety and biomarkers of PD-L1 blockade in non-small cell lung cancer (NSCLC): additional analyses from a clinical study of the engineered antibody MPDL3280A (anti-PDL1). European cancer congress 2013 (ECCO-ESMO-ESTRO), Abstract 3408. http://eccamsterdam2013.ecco-org.eu/Scientific-Programme/Abstract-search.aspx#
Gandhi L et al (2014) MK-3475 (anti-PD-1 monoclonal antibody) for non-small cell lung cancer (NSCLC): antitumor activity and association with tumor PD-L1 expression. Abstract CT105. AACR annual meeting
Antonia SJ et al (2013) Association of PD-L1 tumor expression and immune biomarkers with clinical activity in patients with non-small cell lung cancer (NSCLC) treated with nivolumab (anti-PD-1; BMS-936558;Ono-4538). J Thorac Oncol 8(Suppl 2):S907–S908
Baba T et al (2007) Lack and restoration of sensitivity of lung cancer cells to cellular attack with special reference to expression of human leukocyte antigen class I and/or major histocompatibility complex class I chain related molecules A/B. Cancer Sci 98:1795–1802
Bafna S et al (2010) Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 29:2893–2904
Bernhardt SL et al (2006) Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer 95:1474–1482
Black CC et al (2013) Adenocarcinoma contains more immune tolerance regulatory T-cell lymphocytes (versus squamous carcinoma) in non-small-cell lung cancer. Lung 191(3):265–270
Blackhall F et al (2013) The impact on the multidisciplinary teams of molecular profiling for personalized therapy in non-small cell lung cancer. Lung Cancer 79:101–103
Bradbury PA, Shepherd F (2008) Immunotherapy for lung cancer. J Thorac Oncol 3(Suppl 2):S164–S170
Brahmer J (2013) Harnessing the immune system for the treatment of non-small-cell-lung cancer. J Clin Oncol 31:1021–1028
Brahmer JR (2014) Immune checkpoint blockade: the hope for immunotherapy as a treatment of lung cancer? Semin Oncol 41:126–132
Brahmer J et al (2010) Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 28:3167–3175
Brahmer J et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465
Brahmer J et al (2013) Survival and long-term follow-up of the phase I trial of nivolumab (Anti-PD-1; BMS-936558; ONO-4538) in patients (pts) with previously treated advanced non-small cell lung cancer (NSCLC). J Clin Oncol 31(Suppl):8030
Bremnes RM et al (2011) The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: emphasis on non-small cell lung cancer. J. Thorac Oncol 6:824–833
Brichard VG (2007) GSK’s antigen-specific cancer immunotherapy program: pilot results leading to phase III clinical development. Vaccine 25(Suppl 2):B61–B71
Brunsvig P et al (2006) Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother 55:1553–1564
Brunsvig P et al (2011) Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res 17:6847–6857
Butts C et al (2005) Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small cell lung cancer. J Clin Oncol 23:6674–6681
Butts C et al (2010) A multicenter open-label study to assess the safety of a new formulation of BLP25 liposome vaccine in patients with unresectable stage III non-small-cell lung cancer. Clin Lung Cancer 11:391–395
Butts C et al (2011) Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposomal vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol 137:1337–1342
Butts C et al (2012) Long-term efficacy and safety of L-BLP25 vaccine in a multi-center open-label study of patients with unresectable stage III NSCLC. Annals of Oncol 23(Suppl 9, ix):395
Butts C et al (2013) START: a phase III study of L-BLP25 cancer immunotherapy for unresectable stage III non-small cell lung cancer. J Clin Oncol 31:7500
Butts C et al (2014) Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol 15(1):59–68
Chambers CA, Allison JP (1997) Co-stimulation in T cell responses. Curr Opin Immunol 9:396–404
Chen YC et al (2013) Peripheral immune cell gene expression changes in advanced non-small cell lung cancer patients treated with first line combination chemotherapy. PLoS One 8:e57053
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358:1160–1174
De Plaen E et al (1994) Structure, chromosomal location, and expression of 12 genes of the MAGE family. Immunogenetics 40:360
Detterbeck FC, Boffa DJ, Tanoue LT (2009) The new lung cancer staging system. Chest 136:260–271
Diaz-Montero CM et al (2009) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 58:49–59
Dieu-Nosjean MC et al (2008) Long term survival for patients with non-small cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 26:4410–4417
Digumarti R et al (2011) A randomized, double-blind, placebo-controlled, phase II study of oral talactoferrin in combination with carboplatin and paclitaxel in previously untreated locally advanced or metastatic non-small cell lung cancer. J Thorac Oncol 6:1098–1103
Dong H et al (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800 Erratum in: Nat Med 8(9):1039
Fakhari H et al (2009) Correlation of immune responses and survival in a phase II study of belagenpumatucel-L in non-small cell lung cancer. J Clin Oncol 27(Suppl):3013
Ferlay J et al (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127:2893–2917
García B et al (2008) Effective inhibition of the epidermal growth factor/epidermal growth factor receptor binding by anti-epidermal growth factor antibodies is related to better survival in advanced non-small cell lung cancer patients treated with epidermal growth factor cancer vaccine. Clin Cancer Res 14:840–846
Garon EB et al (2013) Preliminary clinical safety and activity of MK-3475 monotherapy for the treatment of previously treated patients with non-small cell lung cancer. J Thorac Oncol 8(Suppl 2):S364–S365
Giaccone G (2013) Late breaking abstract: a phase III study of belagenpumatucel-L therapeutic tumor cell vaccine for non-small cell lung cancer (NSCLC). Abstract search—European cancer congress 2013. http://eccamsterdam2013.ecco-org.eu/Scientific-Programme/Abstract-search.aspx?abstractid=8961
Goldstraw P et al (2011) Non-small-cell lung cancer. Lancet 378:1727–1740
Gomez RE, Ardigo ML (2012) Anti-idiotype antibodies in cancer treatment: the pharmaceutical industry perspective. Frontiers Oncol 2, Article 147
Gomez RE et al (2013) Active immunotherapy in patients with progressive disease (PD) after first line therapy: Racotumomab experience. J Clin Oncol 31(Suppl):3086
González G et al (1998) A novel cancer vaccine composed of human-recombinant epidermal growth factor linked to a carrier protein: report of a pilot clinical trial. Ann Oncol 9:431–435
González G et al (2003) Epidermal growth factor-based cancer vaccine for non-small cell lung cancer therapy. Ann Oncol 14:461–466
González G et al (2007) Therapeutic vaccination with epidermal growth factor (EGF) in advanced lung cancer. Human Vaccines 3:8–13
Hanagiri T et al (2013) Clinical significance of the frequency of regulatory T cells in regional lymph node lymphocytes as a prognostic factor for non-small-cell lung cancer. Lung Cancer 81:475–479
Hayes TG, Falchook GS (2010) Phase IB of oral talactoferrin in the treatment of patients with metastatic solid tumors. Invest New Drugs 28:156–162
Hayes DN et al (2006a) Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol 24:5079–5090
Hayes TG et al (2006b) Phase I trial of oral talactoferrin alfa in refractory solid tumors. Invest New Drugs 24:233–240
Hernández AM et al (2008) Characterization of the antibody response against NeuGcGM3 ganglioside elicited in non-small cell lung cancer patients immunized with an anti-idiotypic antibody. J Immunol 181:6625–6634
Hiltbold EM (1999) Presentation of MUC1 tumor antigen by class I MHC and CTL function correlate with the glycosylation state of the protein taken up by dendritic cells. Cell Immunol 194:143–149
Ho SB et al (1993) Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res 53:641–651
Hodi SF et al (2010) Ipilimumab plus dacarbacine for previously untreated metastatic melanoma. N Engl J Med 363:711–723
Huang A et al (2013) Increased CD14+HLA-DR-/low myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother 62:1439–1451
Inderberg-Suso E-M et al (2012) Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with single hTERT peptide GV1001. Oncoimmunology 1:670–686
Joziasse DH, Orial R (1999) Xenotransplantation: the importance of the Galalpha1,3Gal epitope in hyperacute vascular rejection. Biochim Biophys Acta 1455:403–418
Katz LH et al (2013) Targeting TGF-β signaling in cancer. Expert Opin Ther Targets 17:743–760
Kawai O et al (2008) Predominant infiltration of macrophages and CD8(+) T cells in cancer nests is a significant predictor of survival in stage IV non-small cell lung cancer. Cancer 113:1387–1395
Kelly RJ, Giaccone G (2010) The role of talactoferrin alfa in the treatment of non-small cell lung cancer. Expert Opin Biol Ther 10:1379–1386
Kim NW et al (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266:2011–2015
Konishi J et al (2004) B7-H1 expression on non small cell lung cancer cells and its relationship with tumor infiltrating lymphocytes and their PD-1 expression. Clin Cancer Res 10:5094–5100
Kruit WH (2008) Immunization with recombinant MAGE-A3 protein combined with adjuvant systems AS15 or AS 02B in patients with unresectable and progressive metastatic cutaneous melanoma: a randomized open-label Phase II study of the EORTC Melanoma Group (16032–18031). J Clin Oncol 26(Suppl):9065
Kruit WH et al (2013) Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organization for Research and Treatment of Cancer Melanoma Group in metastatic melanoma. J Clin Oncol 31:2413–2420
Kufe DW (2009) Mucins in cancer: function, prognosis and therapy. Nature Reviews 9:874–885
Kwak EL, Bang YJ, Camidge DR et al (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703
Kyte JA (2009) Cancer vaccination with telomerase peptide GV1001. Expert Opin Investig Drugs 18:687–694
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–1736
Limacher J-M, Quoix E (2012) TG4010. A therapeutic vaccine against MUC1 expressing tumors. Oncoimmunology 1:791–792
Link CR Jr et al (1998) Eliciting hyperacute xenograft response to treat human cancer: alpha (1,3) galactosyltransferase gene therapy. Anticancer Res 18:2301–2308
Lynch TJ et al (2010) Phase II trial of ipilimumab (IPI) and paclitaxel/carboplatin (P/C) in first line stage IIIb/IV non-small cell lung cancer (NSCLC). J Clin Oncol 28(Suppl):7531
Macías A et al (2012) Active specific immunotherapy with racotumomab in the treatment of advanced non small cell lung cancer (NSCLC). Ann Oncol 23(Suppl 9, ix):406
Madan RA et al (2013) Effect of talactoferrin alfa on the immune system in adults with non-small cell lung cancer. The Oncologist 18:821–822
Middleton GW et al (2013) A phase III randomized trial of chemoimmunotherapy comprising gemcitabine and capecitabine with or without telomerase vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer. J Clin Oncol 31(Suppl):LBA4004
Mitchell PL et al (2013) Mucin 1 (MUC1) expression in patients (pts) with early stage non-small cell lung cancer (NSCLC): relationship between immunohistochemistry (IHC), tumor characteristics, and survival. J Clin Oncol 31(Suppl):3011
Morris J-C et al (2012) Correlation of interferon-g (γ) response with survival in a phase II hyperacute (HAL) immunotherapy trial for non-small cell lung cancer (NSCLC). J Clin Oncol 30(Suppl):2571
Morris JC et al (2013) Potential chemo-sensitization effect of tergenpumatucel-L immunotherapy in treated patients with adjuvant non-small cell lung cancer (NSCLC). J Clin Oncol 31(Suppl):8094
National Comprehensive Cancer Network (NCCN) (2014) Clinical Practice Guidelines in Oncology. Non-small cell lung cancer, version 3.2014. Accesses 22 April 2014
Nemunaitis J et al (2006) Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small cell lung cancer. J Clin Oncol 24:4721–4730
Nemunaitis J et al (2009) Phase II trial of belagenpumatucel-L, a TGF-beta2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther 16:620–624
Neninger Vinageras E et al (2008) Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small cell lung cancer. J Clin Oncol 26:1452–1458
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264
Parikh PM et al (2011) Randomized, double-blind, placebo-controlled phase II study of single-agent oral talactoferrin in patients with locally advanced or metastatic non-small cell lung cancer that progressed after chemotherapy. J Clin Onc 29:4129–4136
Peggs KS et al (2009) Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 206(8):1717–1725
Peters S et al (2012) Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 23(Suppl 7):vii56–vii64
Petersen RP et al (2006) Tumor infiltrating FOXP3 regulators T cells associated with recurrence in pathologic stage I NSCLC patients. Cancer 107:2866–28762
Pirker R (2013) EGFR-directed monoclonal antibodies in non-small cell lung cancer. Targ Oncol 8:47–53
Quoix E et al (2011) Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small cell lung cancer: a controlled phase IIb trial. Lancet Oncol 12:1125–1133
Quoix E et al (2012) TIME: a phase IIb/III randomized, double-blind, placebo-controlled study comparing first-line therapy with or without TG4010 immunotherapy product inn patients with stage IV non-small cell lung cancer (NSCLC). J Clin Oncol 30(Suppl):TPS7610
Raijna D et al (2011) Dependence on the MUC1-C oncoprotein in non–small cell lung cancer cells. Mol Cancer Ther 10:806–816
Ramlau R et al (2008a) Randomized phase IIB trial evaluating the therapeutic vaccine TG4010 (MVA-MUC1-IL2) as an adjunct to chemotherapy in patients with advanced non-small cell lung cancer (NSCLC). J Clin Oncol 26(Suppl):8023
Ramlau R et al (2008b) A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV non-small cell lung cancer. J Thor Oncol 3:735–744
Rangachari D, Brahmer JR (2013) Targeting the immune system in the treatment of non-small cell lung cancer. Curr Treat Options Oncol 14(4):580–594
Robert C et al (2011) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 364:2517–2526
Rochlitz C et al (2003) Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med 5:690–699
Rodríguez PC et al (2010) Clinical development and perspectives of CIMAvax EGF, Cuban vaccine for non-small cell lung cancer therapy. MEDICC Review 12:17–23
Rodríguez PC et al (2011) Safety, immunogenicity and preliminary efficacy of multiple-site vaccination with an epidermal growth factor (EGF) based cancer vaccine in advanced non small cell lung cancer (NSCLC) patients. J Immune Based Ther Vaccines 9:1–6
Rossi G et al (2008) Allogeneic melanoma vaccine expressing aGal epitopes induced antitumor immunity to autologous antigen in mice without signs of toxicity. J Immunother 31:545–554
Salomon DS et al (1995) Epidermal growth factor-related peptides and their receptors in human malignancies. Cir Rev Oncol Hematol 19:183–232
Schreiber RD et al (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331:1565–1570
Shaw AT et al (2009) Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 27:4247–4253
Shaw AT et al (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368:2385–2394
Shimizu K et al (2010) Tumor infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol 5:585–590
Sienel W et al (2004) Melanoma associated antigen (MAGE)-A3 expression in stages I and II non-small cell lung cancer: results of a multicenter study. Eur J Cardiothorac Surg 25:131–134
So T et al (2005) Haplotype loss of HLA class I antigen as an escape mechanism from immune attack in lung cancer. Cancer Res 65:5945–5952
Spadaro M et al (2007) Requirement for INF-γ, CD8+ T Lymphocytes, and NKT cells in talactoferrin-induced inhibition of neu+ tumors. Cancer Res 67:6425–6432
Spiegel R, Socinski M (2013) Rationale for chemotherapy, immunotherapy, and checkpoint blockade in SCLC. J Thorac Oncol 8:587–589
Stinchcombe TE (2014) Unmet needs in squamous cell carcinoma of the lung: potential role for immunotherapy. Med Oncol 31:960
Suzuki K et al (2011) Prognostic immune markers in non-small cell lung cancer clin. Cancer Res 17:5247–5256
Topalian SL et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Tyagi P, Mirakhur B (2009) MAGRIT: the largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clin Lung Cancer 10:371–374
Ulloa-Montoya F et al (2013) Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 31:2388–2395
Van Cruijsen H et al (2009) Tissue micro array analysis of ganglioside N-glycolyl GM3 expression and signal transducer and activator of transcription (STAT)-3 activation in relation to dendritic cell infiltration and microvessel density in non-small cell lung cancer. BMC Cancer 9:180–189
Van den Eynde BJ et al (1997) T cell defined tumor antigens. Curr Opin Immunol 9:684–693
Vansteenkiste J et al (2007) Final results of a multi-center, double blind, randomized placebo controlled phase II study to assess the efficacy of MAGE-A3 immunotherapeutic as adjuvant therapy in stage IB/II non-small cell lung cancer (NSCLC). J Clin Oncol 25(Suppl):7554
Vansteenkiste J et al (2013) Adjuvant MAGE-A3 immunotherapy in resected non-small cell lung cancer: phase II randomized study results. J Clin Oncol 31:2396–2403
Vázquez AM et al (2012) Racotumomab: an anti-idiotype vaccine related to N-glycolyl-containing gangliosides—preclinical and clinical data. Frontiers Oncol 2:150
Velcheti V et al (2014) Programmed death ligand-1 expression in non-small cell lung cancer. Lab Investig 94:107–116
Walunas TL et al (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 5:405–413
Woo EY et al (2001) Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 61:4766–4772
Zhao Q et al (2009) Circulating galectin-3 promotes metastasis by modifying MUC1 localization on cancer cell surface. Cancer Res 69:6799–6806
Zotter S et al (1988) Tissue and tumor distribution of human polymorphic epithelial mucin. Cancer Rev 11–12:55–101
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Quaratino, S., Forssmann, U., Marschner, JP. (2014). New Approaches in Immunotherapy for the Treatment of Lung Cancer. In: Savelyeva, N., Ottensmeier, C. (eds) Cancer Vaccines. Current Topics in Microbiology and Immunology, vol 405. Springer, Cham. https://doi.org/10.1007/82_2014_428
Download citation
DOI: https://doi.org/10.1007/82_2014_428
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23909-5
Online ISBN: 978-3-319-23910-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)