
Abstract
Uveitis encompasses a spectrum of diseases whose common feature is intraocular inflammation, which may be infectious or noninfectious in etiology (Nussenblatt and Whitcup 2010). Infectious causes of uveitis are typically treated with appropriate antimicrobial therapy and will not be discussed in this chapter. Noninfectious uveitides are thought have an autoimmune component to their etiology and are thus treated with anti-inflammatory agents.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Uveitis encompasses a spectrum of diseases whose common feature is intraocular inflammation, which may be infectious or noninfectious in etiology (Nussenblatt and Whitcup 2010). Infectious causes of uveitis are typically treated with appropriate antimicrobial therapy and will not be discussed in this chapter. Noninfectious uveitides are thought have an autoimmune component to their etiology and are thus treated with anti-inflammatory agents.
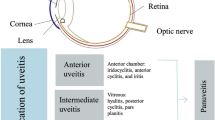
Uveitis may affect various sites within the eye, and the Standardization of Uveitis Nomenclature working group has recommended the following classifications: (1) Anterior uveitis in cases where the anterior chamber is the primary site of inflammation, (2) intermediate uveitis in cases where the vitreous is the primary site of inflammation, (3) posterior uveitis in cases where the retina or choroid are the primary sites of inflammation, and (4) panuveitis in cases where all of these sites are involved (Jabs et al. 2005). Depending on the primary sites of inflammation, different routes of anti-inflammatory therapy may be most appropriate. Below, we discuss the various anti-inflammatory agents and various routes of administration that may be employed in the pharmacologic treatment of noninfectious uveitis.
2 Corticosteroids
The first reports of corticosteroid use in the treatment of noninfectious uveitis were in the early 1950s (Woods 1950; Gordon et al. 1951). Corticosteroids remain a critical treatment modality for acute control of inflammation in noninfectious uveitis because of their rapid onset of action as well as their broad and robust anti-inflammatory effects. Corticosteroids function to activate anti-inflammatory genes while suppressing proinflammatory genes (Barnes 2011). Corticosteroids diffuse across the cell membrane, bind cytosolic glucocorticoid receptor (GR) alpha, and the corticosteroid/GR complex then translocates to the cell nucleus. In the nucleus, corticosteroid/GR complexes may act in several ways to promote anti-inflammatory and suppress proinflammatory responses. GR homodimers may bind glucocorticoid response elements (GREs) within promoter regions of corticosteroid-responsive genes, thereby activating anti-inflammatory gene transcription in a process known as trans-activation. Rarely, GR homodimers interact with GREs to suppress proinflammatory gene transcription in a process known as cis-repression. Corticosteroid/GR complexes may also interact with and suppress the signaling pathways activated by proinflammatory transcription factors, such as nuclear factor-kB (NF-kB), thereby blocking gene transcription of proinflammatory genes in a process known as trans-repression. The downstream effects of these gene interactions include inhibition of cytokine, chemokine, and adhesion molecule expression, which in turn leads to reduced chemotaxis and function of immune cells. In addition, corticosteroids have been shown to upregulate proteins involved in tight junction formation between retinal endothelial cells, suggesting a role in stabilizing the blood–retinal barrier and potentially reducing influx of inflammatory cells and molecules into the normally immune-privileged intraocular environment (Felinski et al. 2008; Keil et al. 2013). The full range of molecular mechanisms employed by corticosteroids in suppressing inflammation has yet to be elucidated and remains an active area of investigation.
Given the large number of genes and proteins affected by corticosteroids, it is not surprising that corticosteroid treatment is accompanied by a variety of side effects, several of which are potentially very serious. Systemic side effects include increased risk of infection, Cushing’s syndrome, osteoporosis, hypertension, dyslipidemia, adrenal insufficiency, insulin resistance, very rarely avascular necrosis of the joints, and growth retardation in children. Corticosteroid treatment also has specific ocular side effects, namely, development or progression of cataract and elevation of intraocular pressure (IOP) potentially leading to glaucoma, when administered locally in or around the eye. These adverse effects limit the long-term use of corticosteroids.
As mentioned above, corticosteroids may be administered systemically or locally to eye. Multiple formulations are available for both systemic and local ocular use (Table 1). In general, the location and severity of intraocular inflammation dictates the mode and specific corticosteroid used. The different corticosteroid formulations and routes of delivery are discussed in detail below.
2.1 Local Corticosteroids
Various options currently exist for local ocular corticosteroid administration, including topical formulations, preparations for injection in the sub-Tenon’s space, inferior orbit, or vitreous cavity, as well as sustained release intraocular implants placed in the posterior segment. Topical formulations are primarily used for treatment for anterior uveitis, although they may also be utilized for mild vitreous inflammation and uveitic macular edema. The goal of topical corticosteroid treatment of anterior uveitis is to eliminate the cellular and inflammatory protein (flare) responses in the anterior chamber to prevent irreversible inflammatory damage to the eye in addition to reducing patient symptoms of ocular pain and light sensitivity. Unfortunately, long-term use and higher corticosteroid potency often result in complications of cataract formation or progression and elevation of intraocular pressure (Becker and Mills 1963; Urban and Cotlier 1986). Systemic side effects are rare but have been reported with topical corticosteroid treatment (Sendrowski et al. 2008).
Many topical corticosteroid formulations are available, and they vary in their potency and dosing regimens. While much research has been conducted investigating bioavailability and relative anti-inflammatory effects for these medications, most of these studies focused on ocular surface disease (Sendrowski et al. 2008). For effective treatment of anterior uveitis, the corticosteroid must penetrate the cornea and reach therapeutic levels within the anterior chamber. The size and chemical composition of the corticosteroid molecule, as well as the topical formulation (e.g., solution, suspension, or emulsion), are factors that may affect corneal penetration and access to the anterior chamber. Studies investigating topical dexamethasone administration have shown that suspensions penetrate the anterior chamber better than solutions (Cagini et al. 2016) and that vitreous concentrations of dexamethasone after hourly dosing were negligible (Weijtens et al. 2002).
Few well-conducted clinical trials exist comparing the efficacy of different topical corticosteroid formulations and doses in noninfectious anterior uveitis. Prednisolone acetate ophthalmic suspension 1.0% (Pred Forte, Allergan, Irvine, California) was shown to have superior anti-inflammatory activity in terms of resolving anterior chamber cell and flare compared to loteprednol etabonate ophthalmic suspension 0.5% (Lotemax, Pharmos Corporation and Bausch and Lomb Pharmaceuticals, Tampa, Florida) in patients with acute anterior uveitis (The Loteprednol Etabonate US Uveitis Study Group 1999). Fewer patients experienced IOP elevations ≥10 mmHg in the loteprednol etabonate group compared to the prednisolone acetate group; however, statistics comparing the groups were not provided. Difluprednate is a high-potency difluorinated prednisolone corticosteroid. The 0.5% difluprednate ophthalmic emulsion (Durezol, Alcon Laboratories, Fort Worth, TX) has high glucocorticoid receptor affinity, tissue penetration, and bioavailability (Foster et al. 2010). Studies in patients with endogenous anterior uveitis have shown non-inferiority of difluprednate 0.05% dosed four times daily compared to prednisolone acetate 1% dosed eight times daily for 14 days (Foster et al. 2010; Sheppard et al. 2014). Clinically important IOP elevations were observed in 8.9–12% of eyes that received difluprednate compared to 3.7–5% of eyes that received prednisolone acetate 1%. However, other studies have reported IOP elevations ≥10 mmHg in 39–50% of eyes, with some eyes experiencing increases of over 30 mmHg (Birnbaum et al. 2011; Slabaugh et al. 2012). These studies also suggest that IOP elevation with topical difluprednate may be worse in the pediatric population.
As mentioned above, most topical corticosteroid formulations do not achieve sufficient concentrations in the posterior segment to be clinically useful for intermediate or posterior uveitis (Weijtens et al. 2002). The 0.5% difluprednate ophthalmic emulsion may be effective in treating some forms posterior uveitis (Onishi et al. 2015). In general, periocular or intravitreal injection of corticosteroids are required to achieve therapeutic levels in the posterior segment and are effective in the treatment of both active inflammation and macular edema (Sen et al. 2014). Clinically, these injections are most useful in cases of unilateral disease, in patients who are pseudophakic given the risk of cataract development, or in patients who are unable to tolerate systemic corticosteroids, such as poorly controlled diabetic patients.
Periocular treatments may be injected into the sub-Tenon’s space or transcutaneously into the orbital floor. Typically, 40 mg of methylprednisolone or triamcinolone is injected (Ferrante et al. 2004). The half-life of triamcinolone after a single posterior sub-Tenon’s injection in humans has been estimated at 25 days in the vitreous cavity (Shen et al. 2010). Injections can be repeated every 1–3 months as needed to control intraocular inflammation. In a retrospective review of 1,192 eyes that received at least one periocular corticosteroid injection, clinically meaningful cataract formation occurred in 20.2% of eyes (Sen et al. 2014). In the same study, intraocular pressure elevations to ≥24 and 30 mmHg occurred in 34% and 15% of eyes, respectively, and glaucoma surgery was required in 2.4% of eyes. Additional potential side effects of periocular corticosteroids injections include ptosis, orbital fat atrophy or prolapse, and inadvertent entrance into the globe (Lafranco Dafflon et al. 1999; Giles 1974; Dal Canto et al. 2005).
Intravitreal triamcinolone injection is another option for treating posterior segment inflammation or uveitic macular edema (Habot-Wilner et al. 2011). Typically, 2–4 mg of preservative-free triamcinolone is injected (Cunningham et al. 2008). Triamcinolone has been detected in the vitreous of non-vitrectomized eyes up to 2.75 months following a single 4 mg intravitreal injection (Mason et al. 2004). As with periorbital injections, intravitreal injections may be repeated for recurrent disease with close monitoring for side effects. In addition to development or progression of cataract and elevation of intraocular pressure, potential adverse effects of intravitreal steroid injections include vitreous hemorrhage, retinal detachment, as well as infectious or sterile endophthalmitis (Marticorena et al. 2012).
Two sustained release intravitreal corticosteroid implants have been approved by the US Food and Drug Administration (FDA) for the treatment of noninfectious uveitis. The dexamethasone intravitreal implant (Ozurdex, Allergan, Inc., Irvine, CA; 0.7 mg), which is administered in an office-based procedure, provides sustained release of dexamethasone via poly(lactic acid-co-glycolic acid) (PLGA) matrix material, which dissolves completely in vivo (Chang-Lin et al. 2011a). Studies in animal eyes have shown detection of dexamethasone in the vitreous and retina for up to 6 months, with peak concentrations in the first 2 months, without significant differences in concentrations between vitrectomized and non-vitrectomized eyes during the first month (Chang-Lin et al. 2011a, b). The 0.7 mg dexamethasone intravitreal implant was shown to significantly improve visual acuity and reduce vitreous haze scores in a multicenter randomized controlled clinical trial of patients with noninfectious intermediate or posterior uveitis compared to sham injection over 26 weeks (Lowder et al. 2011). 7.1% of eyes experienced intraocular pressure elevation ≥25 mmHg, and 15% of phakic eyes developed cataract. The median duration of therapeutic effect for first injections has been estimated to be 6 months, consistent with pharmacokinetic studies demonstrating detection of dexamethasone in the vitreous and retina for up to 6 months (Chang-Lin et al. 2011a; Tomkins-Netzer et al. 2014), and the mean time to second injections was estimated at 6.6 months (Zarranz-Ventura et al. 2014). Repeated insertions of the dexamethasone intravitreal implant have been reported without complication; (Querques et al. 2013) however, the number of applications that can be safely delivered to an eye remains unknown. Recently, results of a retrospective study showed the 0.7 mg dexamethasone intravitreal implant to be safe and effective in pediatric patients with noninfectious uveitis (Tomkins-Netzer et al. 2016).
The fluocinolone acetonide (FA) intravitreal implant (Retisert, 0.59 mg), which requires surgical implantation via sclerotomy in the operating room, provides sustained release of FA for approximately 30 months (Callanan et al. 2008). The Multicenter Uveitis Steroid Treatment (MUST) trial was designed to compare the efficacy of the FA intravitreal implant against systemic corticosteroid therapy, in addition to other systemic immunosuppressive medications when indicated, in patients with noninfectious intermediate uveitis, posterior uveitis, or panuveitis (Multicenter Uveitis Steroid Treatment Trial Research Group et al. 2010). Through 54 months of the study, visual acuity did not significantly differ between the groups at any time point; however, visual acuities were overall very good at the start of the trial, thereby limiting the potential for improvement with treatment (Multicenter Uveitis Steroid Treatment Trial Research group et al. 2015). While both treatments reduced the percentage of patients with active uveitis, the FA implant was significantly better at controlling inflammation at all time-points assessed. The FA implant was also significantly better at resolving macular edema through the first 2 years of treatment, after which systemic therapy showed equal efficacy. Cataract surgery was required significantly more often in the implant group through 54 months (87.7% vs 43% in the FA implant vs systemic treatment groups, respectively) with most surgeries occurring in the first 2 years (Multicenter Uveitis Steroid Treatment Trial Follow-up Study Research Group 2015). Elevation of intraocular pressure and IOP-lowering surgeries (31.1% vs 4.5% in the FA implant vs systemic treatment groups, respectively, through 2 years) were also significantly more common in the implant. However, systemic adverse events were not different between the groups.
2.2 Systemic Corticosteroids
For severe cases of intraocular inflammation, especially bilateral disease, systemic treatment with corticosteroids is often employed. Oral prednisone at a dose of 1 mg/kg/day is a common starting dose, with gradual taper as inflammation subsides (Jabs et al. 2000). For particularly severe cases of sight-threatening ocular inflammation, such as Behçet’s retinitis (Reed et al. 1998), corticosteroids may be administered intravenously (e.g., methylprednisolone dosed at 1,000 mg/day, which can be divided into four equal doses, for 3 days followed by high-dose oral prednisone with taper). Steroid-sparing immunosuppressive therapy (as discussed in following sections of this chapter) should be initiated if intraocular inflammation persists or recurs during steroid taper.
High doses of corticosteroids (e.g., 30 mg/day or more of prednisone) are associated with numerous adverse effects, and doses should be reduced as quickly yet safely as possible (Jabs et al. 2000). Adverse effects may occur anytime during corticosteroid treatment but are more common with higher doses and longer duration of use. Systemic side effects include, but are not limited to, osteoporosis, avascular necrosis, myopathy, hyperglycemia, weight gain, hypertension, hyperlipidemia, atherosclerosis, impaired wound healing, infection, psychological disturbance, and peptic ulcer disease. Supplementation with calcium and vitamin D should be prescribed to patients taking systemic corticosteroids, especially those on treatment for more than 3 months. Also, patients with a history of gastritis or gastroesophageal reflux disease or those who are concomitantly on non-steroidal anti-inflammatory drugs (NSAIDs) should be prescribed histamine-2 receptor blockers or proton pump inhibitors to reduce the risk of peptic ulcer disease.
3 T-Cell Inhibitors
Although corticosteroids remain a mainstay of treatment for uveitis, concern stemming from the side effects associated with their long-term use has prompted additional use of corticosteroid-sparing agents, such as the T-cell inhibitors cyclosporine and tacrolimus (Table 2). These agents decrease inflammation by interfering with signaling pathways involved in the function and proliferation of T cells (Knickelbein et al. 2015).
3.1 Cyclosporine
Cyclosporine was first isolated as an antifungal agent in 1970 by Borel and co-workers in Switzerland (De Smet and Nussenblatt 1993). Although as an antifungal agent it proved to be too narrow in its spectrum of activity, it was incidentally found to have potential as an immunosuppressant through its effects on T cells. When cyclosporine enters T cells, it binds to cyclophilin A, increasing its affinity for calcineurin and thereby preventing calcineurin’s ability to dephosphorylate proteins called nuclear factor of activated T cells (NFAT). Without dephosphorylation, NFAT proteins are unable to become activated and thus do not translocate to the nucleus, where they would normally influence the transcription of numerous inflammatory cytokines, including IL-2, IL-4, IL-10, IL-17, as well as tumor necrosis factor alpha (TNFα) and interferon gamma (IFN-γ) (Barbarino et al. 2013).
Initially, cyclosporine was primarily utilized to prevent and treat solid organ transplant rejections; however, in 1983, Nussenblatt et al. introduced a pilot study exhibiting the effectiveness of cyclosporine A in the treatment of ocular inflammation (Nussenblatt et al. 1983a), which was subsequently supported by several controlled and uncontrolled trials (Nussenblatt et al. 1983b, 1991; Masuda et al. 1989). More recently, a retrospective cohort study of 373 patients with noninfectious uveitis treated with cyclosporine in the Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study (SITE Study) found that 51.9% of patients had achieved complete control of inflammation by 12 months and that corticosteroid-sparing success was achieved by 36.1% by 12 months. 8.2% achieved control without the need for systemic corticosteroids by 12 months (Kacmaz et al. 2010).
Although cyclosporine has been established as a useful alternative to corticosteroid monotherapy, its use has been limited due to concern for side effects associated with long-term use. Nephrotoxicity is one of the primary concerns, and even low doses of cyclosporine over the long term can significantly impair renal function, with decreases in glomerular filtration rate and irreversible kidney damage assessed by biopsy (Isnard Bagnis et al. 2002). Therefore, regular monitoring of serum creatinine and urea is essential, and patients should avoid concomitant NSAID use. Hypertension is seen in 15–20% of patients, and while it can be treated with antihypertensives, potassium-sparing diuretics should be avoided as cyclosporine may induce hyperkalemia (Kashani and Mearza 2008). Hepatotoxicity also may occur; however, it is of less concern as perturbations in liver enzymes are transient and patients tend to be asymptomatic. It also is important to monitor for the development of infection or malignancy (De Smet and Nussenblatt 1993), although Kempen and colleagues found that calcineurin inhibitors do not increase cancer risk to a degree that outweighs the expected benefits of therapy (Kempen et al. 2008).
Adult patients are treated with 3–5 mg/kg by mouth, divided into twice daily equal doses (Knickelbein et al. 2015). The dose should be decreased if blood pressure or creatinine levels rise, and treatment should be discontinued if values fail to normalize following dose adjustment. Children are also dosed at 3–5 mg/kg/day divided into two equal doses.
Cyclosporine use during pregnancy has been associated with increased rates of prematurity and other complications including preeclampsia (Bung and Molitor 1991); however, it does not appear to be a major teratogen (Bar Oz et al. 2001). Contraindications include severe infection, uncontrolled hypertension, or current malignancies (Kashani and Mearza 2008).
3.2 Tacrolimus
Tacrolimus was isolated from Streptomyces tsukubaensis in 1984 and is used widely to prevent rejection in solid organ transplant recipients (Gul et al. 2013). Like cyclosporine it functions as a T-cell inhibitor; however, it binds to the immunophilin FK-binding protein 12 rather than cyclophilin A. The downstream effects are the same, resulting in decreased transcription of inflammatory cytokines associated with T-cell activation (Barbarino et al. 2013). Although the mechanism of immunosuppression is similar, tacrolimus is thought to be associated with fewer adverse effects as lower doses are possible due to its increased potency up to 100 times that of cyclosporine (Barbarino et al. 2013).
In 1988, Kawashima and colleagues found that the capacity of tacrolimus to prevent experimental autoimmune uveitis induction in rats was 10–30 times more intense than that of cyclosporine (Kawashima et al. 1988). A decade later, another study examined the effects of low-dose tacrolimus in a small cohort of patients with endogenous posterior uveitis who had failed cyclosporine therapy and found that visual improvement was achieved for 3 months or more with a mean maintenance dose of 0.06 ± 0.02 mg/kg/day without development of nephrotoxicity, the primary reason for discontinuing cyclosporine A therapy (Kilmartin et al. 1998a). More recently, a randomized trial of tacrolimus versus cyclosporine in the treatment of posterior and intermediate uveitis also demonstrated similar efficacy with improved safety profile in tacrolimus versus cyclosporine (Murphy et al. 2005). Despite its improved safety profile, tacrolimus is associated with several adverse effects similar to cyclosporine, including nephrotoxicity, neurotoxicity, hypertension, gastrointestinal disturbances, infections, and malignancy (Barbarino et al. 2013). Both tacrolimus and cyclosporine also have been associated with the development of new-onset diabetes mellitus in renal transplant recipients; however, several studies have demonstrated improved glucose metabolism with cyclosporine compared to tacrolimus (Ghisdal et al. 2008; Mora 2010; Ramos-Cebrian et al. 2007; Wyzgal et al. 2003). This finding was further supported by a recent study of 67 patients with new-onset diabetes after renal transplantation randomized to receive either continuation of tacrolimus or conversion to cyclosporine (Rathi et al. 2015). HbA1c levels improved significantly only in the cyclosporine group, and the decline in fasting plasma glucose and insulin requirement was more significant in subjects on cyclosporine.
Tacrolimus therapy is typically started at a dose of 0.05–0.15 mg/kg/day with a maximum dose of 0.3 mg/kg/day (Jabs et al. 2000). While 95% of tacrolimus metabolites are removed via the biliary tract, renal excretion accounts for 2% (Moller et al. 1999). Weekly laboratory assessments should include complete blood count and complete metabolic panel as well as assessment of blood pressure monthly at initiation of treatment and subsequently every 3 months after stable dosing has been achieved (Jabs et al. 2000).
Tacrolimus use during pregnancy has been associated with an increased risk of preterm birth and low birth weight; however, most studies involve its use in solid organ transplantations, and thus results may have been confounded by maternal condition (Nevers et al. 2014). According to the National Transplantation Pregnancy Registry, the incidence of major malformations associated with tacrolimus use was not much higher than in the general population (McKay and Josephson 2008). However, the available data are limited, and concerns have been raised about more subtle defects that may go unrecognized at birth, such as neurocognitive deficits. Calcineurin and FK-binding protein 12 are known to be increased in the fetal brain, and stimulation with tacrolimus may contribute to alterations in fetal cognitive development (Victor et al. 1995; Avramut et al. 2001).
4 Antimetabolites
Antimetabolites, including methotrexate, azathioprine, and mycophenolate mofetil, comprise another class of corticosteroid-sparing agents used in the treatment of ocular inflammation (Table 2). These drugs inhibit the proliferation of rapidly dividing cells, such as T and B lymphocytes, by antagonizing or competing with a metabolite needed for nucleotide synthesis (Kim and Foster 2006).
4.1 Methotrexate
Methotrexate was used initially as an antineoplastic agent in 1948 and for treatment of rheumatoid arthritis four decades later (Farber et al. 1948; Gangaputra et al. 2009). It was first implemented in the treatment of ocular inflammation in 1965 (Wong and Hersh 1965). Methotrexate functions as a potent inhibitor of dihydrofolate reductase, a key enzyme in the production of tetrahydrofolate, thereby decreasing the production of purines and pyrimidines required for DNA synthesis (Chan and Cronstein 2013). Just as it targets the rapidly proliferating cells of malignancy, its success in the treatment of uveitis is due to its ability to diminish the high turnover rate of inflammatory cells. In addition, methotrexate is thought to increase the rate of T-cell apoptosis and alter cytokine production (Wessels et al. 2008).
While methotrexate is most commonly administered orally, it can also be given via subcutaneous or intravitreal injections. When given orally, up to 35% of the dose is metabolized by intestinal flora prior to absorption; however, when parenterally administered, it is fully absorbed (Gangaputra et al. 2009; Larson et al. 2011). Gangaputra and colleagues retrospectively reviewed the records of 384 patients identified from the Systemic Immunosuppressive Therapy for Eye Diseases (SITE) Study and found no significant difference in the effectiveness of subcutaneous versus oral routes of administration (Gangaputra et al. 2009). Among patients with anterior uveitis, intermediate uveitis, posterior or panuveitis, complete suppression of inflammation sustained for ≥28 days was reached in 55.6%, 47.4% and 38.6%, respectively, and corticosteroid-sparing success was achieved in 6 months among 46.1%, 41.3%, and 20.7%, respectively. When considering scleritis, ocular mucous membrane pemphigoid, and other forms of ocular inflammation in addition to uveitis, the overall success within 12 months was 66% and 58.4% for sustained control and corticosteroid-sparing, respectively.
Intravitreal administration of methotrexate was first used in the treatment of uveitis in 2006 and was found to achieve a faster onset of action than systemic administration (Hardwig et al. 2006; Taylor et al. 2009), which typically takes up to 6 months to reach its full effect (Gangaputra et al. 2009). When given intravitreally, the mechanism of action is thought to be primarily mediated by the release of adenosine into the extracellular space, ultimately inhibiting the activity of neutrophils, macrophages, and T lymphocytes (Cronstein et al. 1993; Chan and Cronstein 2002; Bouma et al. 1994; Constantin et al. 1998). The largest series to date of intravitreal methotrexate reported improvement in vision and control of inflammation in 79% of 38 eyes from 30 patients (Taylor et al. 2013). Furthermore, 73% of those who responded to treatment achieved a period of extended remission for a median of 17 months after a single intravitreal injection of methotrexate. However, adverse effects, including elevated intraocular pressure (Taylor et al. 2013) and corneal epitheliopathy (Smith et al. 2002), have been reported, and intravitreal methotrexate is rarely used in the routine management of noninfectious uveitis.
Systemic administration of methotrexate has the potential to cause several serious side effects, including hepatotoxicity, bone marrow suppression, and interstitial pneumonia (Jabs et al. 2000). Concomitant administration of folic acid can mitigate these effects at the recommended dose of 1 mg by mouth daily, excluding the day that methotrexate is taken (Knickelbein et al. 2015). Patients should also be advised to abstain from alcohol use during treatment. In addition to the side effects mentioned above, others more commonly seen include gastrointestinal upset with associated anorexia, nausea and vomiting, as well as stomatitis, alopecia, and rash (Jabs et al. 2000; Durrani et al. 2011).
Prior to initiation of therapy, the following should be obtained: complete blood count, serum chemistry profile, hepatitis B surface antigen, and hepatitis C antibody. Regular monitoring should occur every 1–2 months and should include complete blood count and liver function tests (Jabs et al. 2000). If liver enzymes are elevated to ≥2-times the upper limit of normal on two separate occasions, the dose should be reduced. Liver biopsy is warranted if enzyme abnormalities continue despite discontinuation of the drug. Treatment should be terminated if the following conditions occur: WBC <2,500 μl, platelet count <75,000/μl, or liver enzymes ≥5 times the upper limit of normal (Knickelbein et al. 2015).
In adults, systemic methotrexate is typically started at 2.5–10 mg/week, which is then increased to an average of 15 mg/week (ranging from 7.5 to 25 mg) after several weeks if well tolerated (Jabs et al. 2000; Knickelbein et al. 2015). Intravitreal injections are dosed at 400 μg in 0.1 mL (Taylor et al. 2013). Guidelines for the dosage in children are less clear, but a recent systematic review and meta-analysis indicated 15 mg/m2 was the most common dose based on body surface area (Simonini et al. 2013).
Methotrexate is a known teratogenic and abortive agent, and thus pregnancy and lactation should be avoided at any dose. Both men and women should be advised to discontinue treatment at least 3 months before attempting to conceive (Knickelbein et al. 2015; Visser et al. 2009).
4.2 Azathioprine
Azathioprine was introduced in the 1960s as an antileukemic agent (Elion 1989) and was soon utilized in solid organ transplantation (Murray et al. 1963; Danovitch 1999) as well as rheumatologic diseases such as systemic lupus erythematosus (Abu-Shakra and Shoenfeld 2001) and psoriatic arthritis (Lee et al. 2001). Its use in ophthalmic disease was first in the treatment of corneal graft rejection (Polack 1967) and later for noninfectious ocular inflammatory conditions (Pasadhika et al. 2009) such as active chronic iridocyclitis (Mathews et al. 1969), Behçet’s disease (Yazici et al. 1990), and retinal vasculitis (Greenwood et al. 1998).
Azathioprine is a purine nucleoside analog that is metabolized to 6-mercaptopurine (6-MP), which after further metabolism can inhibit the first step in de novo purine-ring biosynthesis and ultimately become incorporated into replicating DNA and RNA, rendering it nonfunctional and thereby inhibiting the division and proliferation of inflammatory cells (Maltzman and Koretzky 2003). This mechanism targets lymphocytes due to their lack of a salvage pathway. Azathioprine is also thought to induce T-cell anergy or apoptosis through blockade of CD28 costimulation (Maltzman and Koretzky 2003; Elion 1993; Tiede et al. 2003).
Randomized clinical trials of azathioprine in ocular inflammation are limited and have largely focused on its use in Behçet’s disease (Yazici et al. 1990; Hamuryudan et al. 1997). The SITE Study retrospectively reviewed the records of 145 patients, 63% of whom had uveitis, and found that 62% of patients initially gained complete control of inflammation sustained over at least 28 days within 1 year of therapy and 47% were able to maintain control while tapering systemic corticosteroids to ≤10 mg/day (Pasadhika et al. 2009). Patients with intermediate uveitis had the greatest rate of success, with 90% achieving sustained inflammatory inactivity within 1 year. However, when compared to other corticosteroid-sparing treatments for noninfectious ocular inflammation, azathioprine was found to have a longer median time to treatment success compared to mycophenolate mofetil (MMF) and a higher rate of side effects compared to both MMF and methotrexate (Galor et al. 2008).
Side effects associated with azathioprine most commonly include gastrointestinal intolerance, myelosuppression, and, less often, liver toxicity (Jabs et al. 2000; Knickelbein et al. 2015; Clunie and Lennard 2004). Rarely, interstitial pneumonitis, pancreatitis, stomatitis, and alopecia have also been reported. Variations in metabolism of 6-MP can cause increased toxicity of the drug. For example, thiopurine methyltransferase (TPMT), a key enzyme in the methylation of 6-MP to an inactive metabolite, is controlled by a genetic polymorphism inherited as an autosomal codominant trait (Clunie and Lennard 2004; Weinshilboum and Sladek 1980). Decreased activity of TPMT leads to elevated cytotoxicity, possibly even within days of initiating azathioprine therapy (Clunie and Lennard 2004). Genetic testing or an assay of TPMT activity in red blood cells should be performed prior to starting treatment to allow for dose adjustment when necessary (Knickelbein et al. 2015; Durrani et al. 2011). Allopurinol, a strong xanthine oxidase (XO) and TPMT inhibitor, is known to interfere with the metabolism of azathioprine (Broekman et al. 2015). Consequently, the dose of azathioprine should be reduced by 25% for patients treated simultaneously with both drugs (Durrani et al. 2011).
Prior to initiating azathioprine, complete blood count with differential, serum creatinine, and liver enzymes should be obtained and repeated every 1–3 months throughout treatment (Jabs et al. 2000; Knickelbein et al. 2015; Durrani et al. 2011). In adults, 2 mg/kg/day is the most common dose, with ranges from 1 to 3 mg/kg/day. Patients are typically started at lower doses, and the dose is escalated if well tolerated. Doses can be given daily or twice daily when divided equally (Jabs et al. 2000; Knickelbein et al. 2015; Larson et al. 2011). Treatment should be discontinued if the following conditions occur: WBC ≤2,500/μl, platelet count <75,000/μl, liver enzymes ≥5 times the upper limit of normal, or an absolute neutrophil count below 1,000/μl (Knickelbein et al. 2015). If liver enzymes increase to ≥3 times the upper limit of normal, the dose should be reduced and liver enzymes should be retested 2 weeks later.
Azathioprine is pregnancy category D, meaning that the benefits of use during pregnancy may outweigh the potential teratogenic risks; however, most studies were based on its use in renal transplantation and inflammatory bowel disease (Caprilli et al. 2006; Ostensen and Förger 2013; Gerosa et al. 2014). A recent systematic review and meta-analysis on fetal outcomes after thiopurine use found that exposure in women was associated with preterm birth but not low birth weight or congenital abnormalities, and exposure in men at the time of conception was not associated with congenital abnormalities (Akbari et al. 2013). Nevertheless, due to the potential risk, contraception is important with the use of azathioprine and ideally, patients should not attempt to conceive for 3–4 months following the discontinuation of treatment (Knickelbein et al. 2015; Teruel et al. 2010).
4.3 Mycophenolate Mofetil
Mycophenolate mofetil (MMF) was initially introduced in 1946 as an antibiotic from Penicillium brevicompactum (Florey et al. 1946) and was first used as an immunosuppressant in the 1970s to treat psoriasis (Spatz et al. 1978). Two decades later it was utilized in solid organ transplant recipients as an alternative to other immunosuppressive agents associated with undesirable side effects due to their non-selective antiproliferative mechanism (Allison and Eugui 1993). MMF is the prodrug of mycophenolic acid, an inhibitor of the rate-limiting enzyme in de novo synthesis of guanosine nucleotides, inosine monophosphate dehydrogenase (Allison and Eugui 2000). MMF preferentially inhibits the type II isoform of this enzyme, allowing it to specifically target activated lymphocytes, which express this form, thereby inhibiting the division and proliferation of inflammatory cells. By decreasing guanosine nucleotides, MMF also suppresses the expression of vascular endothelial adhesion molecules, which decreases recruitment of lymphocytes and monocytes to sites of inflammation.
MMF use in ocular inflammation was first explored in animal models of experimental autoimmune uveoretinitis (Chanaud et al. 1995), leading to a number of studies supporting its use in refractory human inflammatory eye diseases, including noninfectious uveitis and scleritis (Kilmartin et al. 1998b; Larkin and Lightman 1999; Sen et al. 2003; Greiner et al. 2002; Lau et al. 2003; Baltatzis et al. 2003; Siepman et al. 2006; Thorne et al. 2005; Teoh et al. 2008). Doycheva and colleagues conducted a retrospective case series of 60 uveitis patients treated with MMF for at least 5 years and found that control of inflammation was achieved in 72% of patients after 1 year of treatment and in 82% after 2 years (Doycheva et al. 2011). Rates of long-term side effects were similar to those reported in studies of short-term use (Siepman et al. 2006; Thorne et al. 2005). In a retrospective cohort study, MMF was found to have a more rapid time to control of ocular inflammation than methotrexate and an improved side effect profile compared to azathioprine (Galor et al. 2008). A recent randomized clinical trial in patients with noninfectious intermediate uveitis, posterior uveitis, or panuveitis did not find a statistically significant difference in corticosteroid-sparing control of intraocular inflammation between patients receiving mycophenolate mofetil or methotrexate; however, there was a trend toward higher treatment success in the methotrexate group (Rathinam et al. 2014). There was no difference in the time to treatment effect between mycophenolate mofetil and methotrexate.
Side effects associated with MMF most commonly include gastrointestinal upset, malaise, fatigue, headaches, and infection (Jabs et al. 2000; Doycheva et al. 2011). Bone marrow suppression and liver toxicities are less common but routine laboratory monitoring is essential and patients should limit alcohol consumption (Jabs et al. 2000; Knickelbein et al. 2015). In 2008, the FDA warned of the potential association of progressive multifocal leukoencephalopathy (PML) with MMF use (FDA 2008). However, further studies, including a retrospective cohort study of 32,757 renal transplant recipients as well as the SITE study with over 200 patients with ocular inflammation treated with MMF, failed to support this potential association (Daniel et al. 2010; Neff et al. 2008).
Prior to initiating therapy, complete blood count, serum creatinine, and liver function tests should be obtained and subsequently repeated every 1–3 months during treatment (Knickelbein et al. 2015; Durrani et al. 2011). In adults, orally administered MMF is typically initiated at 500 mg twice daily for 1–2 weeks, which is increased to 1 g twice daily if well tolerated. Once control of inflammation is achieved, patients should continue therapy until they have been free of disease recurrences for 1–2 years. Treatment should be discontinued if the following conditions occur: WBC <2,500/μl, platelet count <75,000/μl, liver enzymes ≥5 times the upper limit of normal, or an absolute neutrophil count below 1,000/μl (Jabs et al. 2000). The dose should be reduced if liver function tests exceed ≥2–3-times the upper limit of normal or if there is a milder decrease in platelet count (Knickelbein et al. 2015). Patients should avoid simultaneous ingestion of antacids containing magnesium and aluminum hydroxide, as these reduce the bioavailability of MMF (Durrani et al. 2011).
MMF is a known teratogenic agent and has been found to decrease the effectiveness of oral contraceptives (Ostensen and Förger 2013; Gerosa et al. 2014; Sifontis et al. 2006). Therefore, two forms of contraception are needed, and male and female patients should avoid conception for at least the first 6 weeks but preferably 3–4 months after discontinuation of treatment (Knickelbein et al. 2015; Ostensen and Förger 2013).
5 Alkylating Agents
Alkylating agents, including cyclophosphamide and chlorambucil (Table 2), are derived from sulfur mustard or mustard gas, which was synthesized in 1860 and utilized in chemical warfare during the First World War (Frunzi 2007). In the 1940s, its ability to cause profound lymphopenia and myeloid suppression led to its introduction as a novel chemotherapeutic agent (Goodman et al. 1946). A decade later, the nitrogen mustard-derivative cyclophosphamide was first utilized in the treatment of ocular inflammation (Perez 1951), and another, chlorambucil, was added to the armamentarium in the 1970s (Patel et al. 2014). These agents inhibit the rapidly dividing cells of inflammation through disruption of DNA replication (Gallego-Pinazo et al. 2013). In general, their use has declined since the introduction of biologic agents.
5.1 Cyclophosphamide
Cyclophosphamide is comprised of a nitrogen mustard group attached to an oxazaphosphorine ring, which upon enzymatic activation functions as an alkylating agent to form DNA cross-links and DNA protein cross-links which inhibit DNA replication and lead to cell death (de Jonge et al. 2005). This results in a cytotoxic effect, particularly of the rapidly proliferating cells of malignancy as well as T and B lymphocytes involved in inflammation (Pujari et al. 2010). Thus, in addition to its use in chemotherapy, cyclophosphamide has also been utilized in the treatment of autoimmune diseases such as systemic lupus erythematosus and granulomatosis with polyarteritis (GPA, previously known as Wegener’s granulomatosis) (Jabs et al. 2000). It was first introduced in the treatment of ocular inflammation in 1951 (Perez 1951).
Although cyclophosphamide has been found to be effective in the treatment of ocular inflammation, its risk of associated toxicities has limited its use. The SITE Study retrospectively reviewed the records of 215 patients with ocular inflammation and found that 49.2% and 76% of patients achieved sustained control of inflammation for at least 28 days within 6 and 12 months, respectively (Pujari et al. 2010). However, the authors cautioned that given the substantial risk of serious side effects, use of the drug should be limited to the most severe sight-threatening cases.
The most common side effects include reversible bone marrow suppression, nausea, vomiting, alopecia, and gonadal damage (de Jonge et al. 2005; Kruh and Foster 2012). Bladder injury potentially leading to hemorrhagic cystitis or malignant transformation is another concern and is thought to be due to the formation of acrolein, a highly reactive aldehyde metabolite excreted in the urine (Yazici et al. 1990; de Jonge et al. 2005; Cox 1979). To minimize the risk of bladder injury, patients should hydrate with 3–4 L of fluid per day to promote frequent voiding throughout the day (Knickelbein et al. 2015). In addition, patients should be advised to take cyclophosphamide in the morning to limit retention of harmful metabolites in the urine overnight (Monach et al. 2010). Sodium 2-mercaptoethane sulphonate (Mesna) may also be prescribed, as it binds to acrolein to promote its safe excretion (Manz et al. 1985). Increased risk of infection due to leukopenia can be treated prophylactically with trimethoprim-sulfamethoxazole (Bactrim) if needed (Jabs et al. 2000; Knickelbein et al. 2015). Other side effects include hepatic injury and interstitial pneumonitis (de Jonge et al. 2005). Concern has also been raised over possible increased risk of cutaneous malignancy as well as myeloproliferative disorders (Jabs et al. 2000; Yazici et al. 1990). Cyclophosphamide relies on the CYP enzymes for its degradation. Therefore, genetic polymorphisms of CYPs or concomitant use of drugs that inhibit CYPs may result in increased bioavailability and toxicity of the drug (de Jonge et al. 2005).
Cyclophosphamide may be administered orally or intravenously, and several studies have investigated whether pulsed IV delivery could offer rapid control of inflammation while avoiding prolonged bladder exposure and neutropenia (Wakefield 2014). Results have been conflicting, but most studies have reported IV therapy to be less effective than oral (Jabs et al. 2000; Rosenbaum 1994; Ozyazgan et al. 1992), while a small number have concluded that IV pulse alone or in combination with low-dose corticosteroid treatment is as effective as oral with fewer side effects and decreased mortality (Khan et al. 2013; Suelves et al. 2013). The SITE Study demonstrated a trend for increased cancer-related mortality, leading to the authors’ suggestion that even though IV delivery may be less effective for inflammation control, it may be preferable in order to reduce the risk of malignancy (Kempen et al. 2008; Pujari et al. 2010; Martin et al. 1997).
The dosing of oral cyclophosphamide for ocular inflammation is typically 1–3 mg/kg/day (Jabs et al. 2000; Knickelbein et al. 2015; Larson et al. 2011; Yazici et al. 1990) and should be titrated for a target WBC of 3,000–4,000/mm3 (Knickelbein et al. 2015). IV pulse therapy may be dosed at 1 g/m2 body surface area every 3–4 weeks (Larson et al. 2011; Durrani et al. 2004). Upon initiation of treatment, complete blood count, platelet count, and urine analysis should be checked weekly and eventually monthly, once values have stabilized (Knickelbein et al. 2015). Treatment should be discontinued if WBC falls below 2,500/mm3 or if hematuria occurs, which should prompt a urology consult (Jabs et al. 2000; Knickelbein et al. 2015).
Cyclophosphamide is contraindicated in pregnancy as it is a known teratogen that has been associated with increased risk of skeletal and central nervous system abnormalities (Ostensen and Förger 2013). Lactation should also be avoided as the drug can be excreted in breast milk. Patients should be counseled on methods of fertility preservation as cyclophosphamide leads to infertility in both men and women due to disruption of oogenesis and spermatogenesis (Knickelbein et al. 2015). Simultaneous use of gonadotropin-releasing hormone treatment may increase the chance of continued fertility after completing treatment with cyclophosphamide (Knickelbein et al. 2015; Durrani et al. 2011; Blumenfeld and Haim 1997; Slater et al. 1999).
5.2 Chlorambucil
Chlorambucil is a nitrogen mustard alkylating agent introduced in 1953 as a more stable and less toxic derivative than cyclophosphamide (Miserocchi et al. 2002). Similar to cyclophosphamide, chlorambucil creates DNA cross-links that interfere with replication and transcription; however, its onset of action is slower (Larson et al. 2011). Although initially developed for treatment of malignancies, it was later utilized as an immunosuppressant to combat rheumatologic disorders. Since the 1970s, it has also been used to treat a variety of ocular inflammatory conditions such as Behçet’s disease and sympathetic ophthalmia (Patel et al. 2014; Goldstein et al. 2002; Tessler and Jennings 1990).
The use of chlorambucil has been limited due to its potential to cause serious side effects such as bone marrow suppression, infections, sterility, and malignancy (Miserocchi et al. 2002). Other less common side effects include skin rash, gastrointestinal upset, nausea, vomiting, anorexia, and alopecia (Tessler and Jennings 1990; Godfrey et al. 1974; Andrasch et al. 1978). Unlike cyclophosphamide, it is not associated with hemorrhagic cystitis or malignant transformation of the bladder epithelium (Goldstein et al. 2002).
Several studies have demonstrated an increased risk of malignancy, particularly acute leukemia, associated with cumulative dose and duration of treatment (Khan et al. 1979; Berk et al. 1981; Palmer et al. 1984). Khan and colleagues retrospectively reviewed the records of 2006 patients treated for chronic inflammatory rheumatic conditions and found that development of acute leukemia was uncommon when duration of therapy was fewer than 6 months or total cumulative dose was less than 1.0 g (Khan et al. 1979). More recently, several studies have found that high-dose, short-term therapy may offer sustained control of inflammation while minimizing the risk of associated side effects (Patel et al. 2014; Goldstein et al. 2002). At the low doses used in long-term therapy, chlorambucil acts as an inhibitor of protein synthesis, specifically of histones, while at high doses, it acts as a DNA alkylator leading to apoptosis (Sourlingas and Sekeri-Pataryas 1997). Disruption of histones causes structural instability and increased rate of mutations of the p53 gene leading to secondary malignancies (Sturm et al. 2003). Thus, short-term high-dose therapy may offer a way to circumvent the process of malignant transformation. Although further studies are needed, the use of chlorambucil may be warranted in patients with severe disease refractory to other forms of treatment.
For ocular inflammation, chlorambucil is typically dosed at 0.1–0.2 mg/kg/day and continued for 1 year following control of inflammation (Knickelbein et al. 2015). Alternatively, high-dose therapy may be offered, consisting of 2 mg per day for 1 week followed by 2 mg per day each week until quiescence is achieved, the WBC count decreases to 2,400 cells per microliter or the platelet count drops below 100,000 cells per microliter (Larson et al. 2011; Tessler and Jennings 1990; Mamo 1976). Upon initiation of treatment, complete blood count should be checked weekly and eventually monthly, once values have stabilized.
Chlorambucil is a known teratogen and is contraindicated in pregnancy. Patients should be counseled on fertility preservation as it has been associated with testicular hypotrophy and azoospermia in men and premature ovarian failure in women (Patel et al. 2014; Blumenfeld et al. 2000).
6 Biologic Agents
Antibodies and other proteins that target specific components of the immune cascade to downregulate the immune response have become an important treatment modality for ocular inflammatory disease (Table 2), especially in cases of refractory uveitis or patient intolerance to conventional immunomodulatory therapy. In fact, an expert panel advocates using biologics as first-line agents in vision-threatening ocular Behçet’s disease and second-line agents for many other types of chronic, vision-threatening ocular inflammatory disease (Levy-Clarke et al. 2014). Anti-tumor necrosis factor α (TNFα) agents are the most frequently used biologic agents for ocular inflammation, and other classes of biologic agents are emerging as potentially effective as well. The use of biologics for uveitis is considered off-label in the USA, given the lack of applicable randomized, controlled clinical trials. However, TNF inhibitors, for example, are approved for the treatment of uveitis in Japan and some European countries.
Since anti-inflammatory biologic agents downregulate the immune system, infections and an increased risk for malignancy are potential side effects. In addition to an increased frequency of upper respiratory infections, there is an increased risk for serious opportunistic infections and reactivation of latent infections. Current recommendations include screening for tuberculosis, hepatitis B, and hepatitis C prior to biologic administration (Selmi et al. 2015). Patients receiving biologic therapy should be vaccinated against influenza (inactivated vaccine), pneumococcal disease, and hepatitis B; these patients should not receive live vaccines. The biologic agents used in ocular inflammatory disease have been developed relatively recently, so the long-term safety of these medications is unclear, especially given the possibility of an increased risk for malignancy.
Side effects that may limit biologic agent tolerance include systemic infusion reaction, injection site reaction, sustained liver function test abnormality, severe neutropenia, and severe thrombocytopenia. Also, cost is a major limitation to the use of biologic agents. Analysis of a US claims database using data from 2007 to 2011 showed that the average yearly cost per patient treated with an anti-TNFα agent ranged from $17,767 to $24,273, depending on the agent used (Schabert et al. 2013). Insurance companies differ in their coverage of these medications, especially for off-label use as in uveitis. Also, many biologic agents are administered intravenously, which further increases costs and inconveniences the patient by requiring visits to an infusion center. Notable exceptions that may be administered subcutaneously by the patient or a family member in a more flexible setting are adalimumab, golimumab, and certolizumab.
6.1 TNFα Antagonists
TNFα is a potent proinflammatory cytokine implicated in the primary pathogenesis of uveitis. TNFα levels are increased in both serum and aqueous of patients with active uveitis (Santos Lacomba et al. 2001). Intravitreal injection of TNFα in rabbits was shown to cause ocular inflammation by disrupting the blood-ocular barrier (Rosenbaum et al. 1988). Systemic TNFα administration conferred susceptibility to ocular inflammation in an experimental autoimmune uveitis (EAU) mouse model (Nakamura et al. 1994), and blocking TNFα in an EAU model suppressed the ocular inflammation (Sartani et al. 1996).
Anti-TNFα agents are biologically derived products first approved for the treatment of rheumatoid arthritis (Woodrick and Ruderman 2011) and now are the most commonly used biologic agents for uveitis (Levy-Clarke et al. 2014). Among the TNFα antagonists, infliximab has the most evidence for efficacy in ocular inflammatory disease, followed by adalimumab. Golimumab and certolizumab are newer anti-TNFα agents that also are potentially useful for treating ocular inflammatory disease. However, etanercept, the first anti-TNFα agent developed, is not recommended in uveitis. Substituting agents within the anti-TNFα class even if there is lack of response to the initial anti-TNFα agent may be beneficial in uveitis: a meta-analysis of anti-TNFα agent use in pediatric chronic autoimmune uveitis showed that among children who did not maintain disease remission with the initially prescribed anti-TNFα agent, 75% responded to a second anti-TNFα agent (Simonini et al. 2014).
Known multiple sclerosis (MS) is a contraindication to anti-TNFα therapy. Both patients in a case series of two patients with rapidly progressive MS treated with anti-TNFα antibody infusions showed increased numbers of gadolinium-enhancing brain lesions (van Oosten et al. 1996), and a randomized controlled clinical trial of relapsing-remitting MS patients showed more frequent disease exacerbations in patients undergoing TNF blockade (Group TLMSSGaTUoBCMMA 1999).
Anti-TNFα therapy has been associated with other paradoxical autoimmune manifestations, including new cases of sarcoidosis with infliximab, adalimumab, etanercept, and certolizumab (Tong et al. 2012; Moisseiev and Shulman 2014). Psoriasiform rashes (Nguyen et al. 2013) and alopecia areata (Tauber et al. 2014) also have been reported with anti-TNFα therapy.
An early observational study suggested that the risk of lymphoma is higher in rheumatoid arthritis patients treated with anti-TNFα agents compared to conventional immunomodulatory therapy, but after subsequent analysis with an increased number of patients and follow-up duration the authors concluded that anti-TNFα agent use was not associated with an increased risk of lymphoma (Wolfe and Michaud 2007). Increased rates of lymphoma development have been found in other observational studies of TNFα antagonists compared to placebo (Geborek et al. 2005; Wong et al. 2012), but this finding is not consistent, and in all studies the number of lymphomas is small. Such analysis is further complicated by the increased risk of lymphoma in severe rheumatoid arthritis (Ekstrom et al. 2003). Although a theoretical risk for increased risk of various malignancies with biologic therapy persists, and further follow-up is indicated to definitively address this possibility, the studies to date do not support an increased risk for systemic malignancy.
The use of TNFα antagonists during pregnancy is generally avoided, but a prospective observational study showed no significant difference in the rate of congenital abnormalities in pregnancies with anti-TNFα exposure in the first trimester compared to disease-matched control pregnancies without anti-TNFα exposure and other pregnancies in normal controls (Diav-Citrin et al. 2014). TNFα antagonist exposure during early pregnancy may be less concerning since placental transfer of IgG antibodies is minimal during the first trimester, but placental transfer of IgG antibodies does become more efficient as pregnancy progresses, with case reports showing that infliximab is present in neonatal serum if the mother has been treated during the third trimester (Djokanovic et al. 2011).
Infliximab (Remicade, Janssen Biotech, Titusville, NJ, USA) is a chimeric (human-murine), monoclonal anti-TNFα IgG1k antibody. Multiple, relatively large case series show that infliximab is efficacious and well tolerated in Behçet’s disease-associated uveitis (Arida et al. 2011; Calvo-Rio et al. 2014a; Takeuchi et al. 2014; Vallet et al. 2015), leading to the recommendation that it be considered a first-line agent for ocular Behçet’s disease (Levy-Clarke et al. 2014).
Case series, observational studies, and open-label prospective clinical trials show that infliximab is efficacious for uveitis refractory to conventional immunosuppressive therapy in the context of scleritis, JIA-associated anterior uveitis, HLA-B27-associated anterior uveitis, ocular sarcoidosis, birdshot retinochoroidopathy, Vogt–Koyanagi–Harada disease (VKH), and idiopathic uveitis (Simonini et al. 2014; Murphy et al. 2004; Suhler et al. 2009; Sen et al. 2009; Ragam et al. 2014; Kruh et al. 2014). An open-label study examining the efficacy of infliximab for noninfectious uveitis refractory to at least one standard immunosuppressive medication showed a 77% response rate at 10 weeks and a 48% response rate at 50 weeks (Suhler et al. 2009).
Adalimumab (Humira; AbbVie, North Chicago, IL, USA) is a fully humanized monoclonal anti-TNFα IgG1 antibody. Multiple studies show that adalimumab is efficacious in Behçet’s-associated uveitis (Arida et al. 2011; Calvo-Rio et al. 2014a; Vallet et al. 2015; Díaz-Llopis et al. 2012), leading to the recommendation that it be considered a first-line agent for ocular Behçet’s disease (Levy-Clarke et al. 2014).
Case series, observational studies, and open-label prospective clinical trials show that adalimumab can be efficacious for uveitis refractory to conventional immunosuppressive therapy in the context of scleritis, JIA-associated anterior uveitis, HLA-B27-associated anterior uveitis, tubulointerstitial nephritis and uveitis syndrome (TINU), ocular sarcoidosis, birdshot retinochoroidopathy, VKH, and idiopathic uveitis (Simonini et al. 2014; Ragam et al. 2014; Díaz-Llopis et al. 2012; Restrepo and Molina 2010; Suhler et al. 2013). An open-label study examining the efficacy of adalimumab for noninfectious uveitis refractory to at least one standard immunosuppressive medication showed a 68% response rate at 10 weeks and a 39% response rate at 50 weeks (Suhler et al. 2013).
An open question in uveitis is whether infliximab and adalimumab are equivalent in terms of efficacy and safety. Comparisons of early studies suggested that infliximab may be more effective but have more serious side effects than adalimumab (Knickelbein et al. 2015), but these comparisons were limited by small sample size and often were indirect. For instance, the prospective, open-label study of infliximab in refractory uveitis showed a more favorable response rate and more toxicity compared to adalimumab (Suhler et al. 2013), but these studies were not performed concurrently and thus were not designed to be directly compared. A systemic review and meta-analysis did not find a significant difference in response to infliximab versus adalimumab in pediatric chronic noninfectious uveitis (Simonini et al. 2014). A recent patient series examining outcomes in Behçet’s disease showed no difference in efficacy or safety between infliximab and adalimumab (Vallet et al. 2015).
Golimumab (Simponi; Janssen Biotech, Titusville, NJ, USA) is a fully humanized monoclonal anti-TNFα antibody that first was reported to be useful for uveitis treatment in a series of two cases published in 2011 (Cordero-Coma et al. 2011). Additional case reports and retrospective case series show that golimumab can effectively control noninfectious intraocular inflammation (Faez et al. 2014; Miserocchi et al. 2014; Cordero-Coma et al. 2014; Calvo-Rio et al. 2014b). A recently published 3-year safety update shows that the safety profile of golimumab is similar to that of other TNFα antagonists (Kay et al. 2015).
Certolizumab pegol (Cimzia; UCB, Smyrna, GA, USA) is a humanized monoclonal anti-TNFα antibody Fab’ fragment that has been PEGylated to prolong its half-life. Of the available anti-TNFα agents, certolizumab has the least amount of published data to document its efficacy in uveitis, although the available data are promising. The first case reporting efficacy of certolizumab in uveitis was published in 2015 and showed a clinical response in HLA-B27 spondylarthropathy-associated anterior uveitis with certolizumab, after failing infliximab and adalimumab (Maiz Alonso et al. 2015). A recent case series showed that certolizumab had a response rate of 71.4% in cases of autoimmune uveitis that previously failed other anti-TNFα therapy (Llorenc et al. 2015).
Etanercept (Enbrel; Amgen, Thousand Oaks, CA, USA) is a recombinant fusion protein consisting of two copies of the soluble portion of the human TNF receptor fused to the human IgG1 Fc domain, and it downregulates TNFα signaling by binding free TNFα and preventing it from binding to cell surface TNFα receptors. Etanercept effectively treats inflammatory arthritis, but it appears to be less effective than other anti-TNFα agents in treating uveitis and scleritis (Smith et al. 2001, 2005; Doycheva et al. 2014; Galor et al. 2006). Etanercept use has been associated with new-onset uveitis and scleritis in challenge–dechallenge–rechallenge cases (Reddy and Backhouse 2003; Gaujoux-Viala et al. 2012), and a review of adverse drug events databases showed a significantly higher risk of uveitis with etanercept than with infliximab or adalimumab (Lim et al. 2007). As a result, etanercept use is avoided in uveitis patients, even in those with quiescent uveitis who may potentially benefit from its effects on inflammatory joint disease. This idea is evident in the prescribing patterns for juvenile idiopathic arthritis in the UK: the initially prescribed biologic agent in JIA patients with a history of chronic anterior uveitis is much more likely to be adalimumab or infliximab rather than etanercept, even though only etanercept and the interleukin-6 (IL-6) antagonist tocilizumab are approved for use in JIA by the National Institute for Health and Care Excellence (Kearsley-Fleet et al. 2016).
6.2 Anti-IL6 Agents
Elevated levels of IL-6, a proinflammatory cytokine, also have been demonstrated in serum (Kramer et al. 2007), aqueous (Murray et al. 1990), and vitreous (Perez et al. 2004) of uveitis patients. Yoshimura and colleagues have shown that IL-6 expression is necessary for ocular inflammation in an EAU mouse model and that IL-6 blockade ameliorates ocular inflammatory disease in that same EAU model (Yoshimura et al. 2009).
Tocilizumab (Actemra; Genentech, South San Francisco, CA, USA), a humanized monoclonal antibody that targets the IL-6 receptor, is becoming more widely used in noninfectious uveitis refractory to anti-TNFα treatment (Lin 2015), with case reports describing therapeutic success of tocilizumab in refractory birdshot chorioretinopathy (Muselier et al. 2011; Papo et al. 2014), idiopathic granulomatous panuveitis, juvenile idiopathic arthritis-associated uveitis (Tappeiner et al. 2012), Behçet’s disease (Deroux et al. 2015), Castleman’s disease-associated anterior uveitis and retinal vasculitis (Oshitari et al. 2012), and atypical Cogan’s syndrome-associated anterior uveitis (Shibuya et al. 2013). Tocilizumab also may be effective in treating uveitic macular edema. Case series show improvement in refractory uveitic macular edema with tocilizumab (Mesquida et al. 2014), even in the absence of obvious active uveitis (Muselier et al. 2011; Deuter et al. 2016).
Safety studies of tocilizumab have shown an increased risk of serious infections comparable to that of anti-TNFα agents (Nishimoto et al. 2009), relatively uncommon and usually mild transfusion reactions, neutropenia, thrombocytopenia, transaminase elevations, and serum lipid elevations. Initial clinical trials identified 18 cases of gastrointestinal perforation in rheumatoid arthritis patients treated with tocilizumab (Gout et al. 2011). Subsequent analysis showed that gastrointestinal perforation occurred in the setting of diverticulitis in the majority of these cases, and the risk of gastrointestinal perforation with tocilizumab was not significantly different than the risk with anti-TNFα agents and was significantly lower than the risk with corticosteroids. Postmarketing studies in rheumatoid arthritis and juvenile idiopathic arthritis patients confirm that tocilizumab is well tolerated and identify opportunistic infections and bone marrow suppression with varying degrees of neutropenia and/or thrombocytopenia as the most common adverse effects (Genovese et al. 2013; Koike et al. 2014; Yokota et al. 2015).
Other anti-IL6 agents include clazakizumab (Alder BioPharmaceuticals, Bothell, WA, USA), olokizumab (UCB, Brussels, Belgium, and R-Pharm, Moscow, Russia), sarilumab (Regeneron Pharmaceuticals, Tarrytown, NY, USA), siltuximab (Janssen Biotech, Titusville, NJ, USA), and sirukumab (Janssen Biotech, Titusville, NJ, USA). Ongoing clinical trials assess the effectiveness of tocilizumab in juvenile idiopathic arthritis-associated uveitis (phase I/II); tocilizumab in noninfectious intermediate, posterior, and panuveitis (phase I/II); and sarilumab in noninfectious intermediate, posterior, and panuveitis (phase II) (clinicaltrials.gov).
6.3 Anti-CD20 Agents
Rituximab (Rituxan; Genentech, South San Francisco, CA, USA) is a chimeric (human/murine) monoclonal antibody against CD20, a cell surface molecule found only on the surface of mature B lymphocytes. A single treatment series, given as two infusions separated by 2 weeks, depletes mature B cells for 4–6 months. Of note, plasma cells do not express CD20 and thus are not depleted by rituximab. The exact mechanism of action for rituximab in autoimmune disease is unclear but may include prevention of plasma cell formation, alteration of B-cell–T-cell interactions, and/or diversion of immune effector cells toward rituximab–B-cell complexes and away from disease-specific immune complexes within affected tissues (Taylor and Lindorfer 2007). In addition to the side effects shared by the other biologic agents discussed in this chapter, a rare but daunting potential side effect of rituximab treatment is progressive multifocal leukoencephalopathy (PML) in the absence of human immunodeficiency virus infection. This potentially fatal outcome of JC virus reactivation occurred at an estimated rate of 1 in 25,000 in a rheumatoid arthritis population (Clifford et al. 2011).
A prospective interventional trial of 12 patients (Suhler et al. 2014) and a retrospective case series of 15 patients (Cao et al. 2016) both showed that rituximab can effectively treat noninfectious scleritis refractory to other treatment modalities. A pilot study in Behçet’s disease showed that rituximab was more efficacious than cytotoxic therapy (Davatchi et al. 2010). A case series showed that rituximab may achieve long-term quiescence in severe JIA-associated uveitis refractory to anti-TNFα agents (Miserocchi et al. 2015). Additionally, there are case reports documenting efficacy of rituximab in recalcitrant VKH (Caso et al. 2015) and diffuse subretinal fibrosis uveitis syndrome (Cornish et al. 2015). Of note, with its more favorable side effect profile, rituximab has essentially replaced cyclophosphamide in the treatment of GPA (Lally and Spiera 2015).
6.4 Other Biologic Agents
Other biologic agents that show promise in the treatment of ocular inflammatory disease include the anti-IL1 agents anakinra, canakinumab, and gevokizumab as well as the anti-IL17 agent secukinumab. Both IL-1β (Wan et al. 2016) and IL-17 (Amadi-Obi et al. 2007) are thought to be involved in the primary pathogenesis of human uveitis and EAU, with IL-1β acting upstream of IL-17. Case studies have shown efficacy of the recombinant IL-1 receptor antagonist anakinra (Kineret; Sobi, Stockholm, Sweden), the human monoclonal anti-IL1β antibody canakinumab (Ilaris; Novartis, Basel, Switzerland), and the human monoclonal anti-IL1β antibody gevokizumab (XOMA, Berkeley, CA, USA) in Behçet’s disease; the potentially lower risk of tuberculosis reactivation with these medications compared to anti-TNFα agents suggests an advantage to using the anti-IL1 agents in tuberculosis-endemic areas (Cantarini et al. 2015). Studies of the human anti-IL17A monoclonal antibody secukinumab (Cosentyx; Novartis, Basel, Switzerland) in uveitis document both treatment success and treatment failure, perhaps because of differences in bioavailability based on administrative route. A recent randomized controlled trial of secukinumab in noninfectious active intermediate, posterior, or panuveitis showed an acceptable response rate of 72.7% and low relapse rates with high-dose intravenous administration; however, subcutaneous administration produced a response rate of only 33.3% (Letko et al. 2015).
7 Conclusions
Multiple therapeutic options currently exist for the treatment of noninfectious uveitis. Both local as well as systemic medications with various mechanisms of action may be utilized to combat intraocular inflammation. A common paradigm for treating chronic or recurrent noninfectious uveitis involves the “step-ladder” approach (Foster et al. 2016). This approach involves rapidly achieving disease quiescence with either local or systemic corticosteroids along with early initiation of traditional steroid-sparing therapy, such as mycophenolate, methotrexate, or cyclosporine. If these medications fail to control the disease, biologic agents are then added to the regimen. In especially recalcitrant cases, intraocular surgery in the form of pars plana vitrectomy may be indicated. As advancements are made in the understanding of inflammatory diseases, such as noninfectious uveitis, new therapeutic targets will be discovered and additional treatment options will become available.
References
The Loteprednol Etabonate US Uveitis Study Group (1999) Controlled evaluation of loteprednol etabonate and prednisolone acetate in the treatment of acute anterior uveitis. Loteprednol Etabonate US Uveitis Study Group. Am J Ophthalmol 127:537–544
Abu-Shakra M, Shoenfeld Y (2001) Azathioprine therapy for patients with systemic lupus erythematosus. Lupus 10:152–153
Akbari M, Shah S, Velayos FS, Mahadevan U, Cheifetz AS (2013) Systematic review and meta-analysis on the effects of thiopurines on birth outcomes from female and male patients with inflammatory bowel disease. Inflamm Bowel Dis 19:15–22
Allison AC, Eugui EM (1993) The design and development of an immunosuppressive drug, mycophenolate mofetil. Springer Semin Immunopathol 14:353–380
Allison AC, Eugui EM (2000) Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 47:85–118
Amadi-Obi A, Yu C-R, Liu X et al (2007) TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med 13:711–718
Andrasch RH, Pirofsky B, Burns RP (1978) Immunosuppressive therapy for severe chronic uveitis. Arch Ophthalmol 96:247–251
Arida A, Fragiadaki K, Giavri E, Sfikakis PP (2011) Anti-TNF agents for Behcet’s disease: analysis of published data on 369 patients. Semin Arthritis Rheum 41:61–70
Avramut M, Zeevi A, Achim CL (2001) The immunosuppressant drug FK506 is a potent trophic agent for human fetal neurons. Brain Res Dev Brain Res 132:151–157
Baltatzis S, Tufail F, Yu EN, Vredeveld CM, Foster CS (2003) Mycophenolate mofetil as an immunomodulatory agent in the treatment of chronic ocular inflammatory disorders. Ophthalmologe 110:1061–1065
Bar Oz B, Hackman R, Einarson T, Koren G (2001) Pregnancy outcome after cyclosporine therapy during pregnancy: a meta-analysis. Transplantation 71:1051–1055
Barbarino JM, Staatz CE, Venkataramanan R, Klein TE, Altman RB (2013) PharmGKB summary: cyclosporine and tacrolimus pathways. Pharmacogenet Genomics 23:563–585
Barnes PJ (2011) Glucocorticosteroids: current and future directions. Br J Pharmacol 163:29–43
Becker B, Mills DW (1963) Elevated intraocular pressure following corticosteroid eye drops. JAMA 185:884–886
Berk PD, Goldberg JD, Silverstein MN et al (1981) Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N Engl J Med 304:441–447
Birnbaum AD, Jiang Y, Tessler HH, Goldstein DA (2011) Elevation of intraocular pressure in patients with uveitis treated with topical difluprednate. Arch Ophthalmol 129:667–668
Blumenfeld Z, Haim N (1997) Prevention of gonadal damage during cytotoxic therapy. Ann Med 29:199–206
Blumenfeld Z, Shapiro D, Shteinberg M, Avivi I, Nahir M (2000) Preservation of fertility and ovarian function and minimizing gonadotoxicity in young women with systemic lupus erythematosus treated by chemotherapy. Lupus 9:401–405
Bouma MG, Stad RK, van den Wildenberg FA, Buurman WA (1994) Differential regulatory effects of adenosine on cytokine release by activated human monocytes. J Immunol 153:4159–4168
Broekman MTJ, Roelofs HMJ et al (2015) Allopurinol and 5-aminosalicylic acid influence thiopurine-induced hepatotoxicity in vitro. Cell Biol Toxicol 31:161–171
Bung P, Molitor D (1991) Pregnancy and postpartum after kidney transplantation and cyclosporine therapy -- review of the literature adding a new case. J Perinat Med 19:397–401
Cagini C, Cometa F, Torroni G, Pellegrino A, Pellegrino R, Cavallini GM (2016) Dexamethasone disodium phosphate penetration into the human aqueous humor after topical application. Curr Eye Res 41(7):897–899
Callanan DG, Jaffe GJ, Martin DF, Pearson PA, Comstock TL (2008) Treatment of posterior uveitis with a fluocinolone acetonide implant: three-year clinical trial results. Arch Ophthalmol 126:1191–1201
Calvo-Rio V, Blanco R, Beltran E et al (2014a) Anti-TNF-alpha therapy in patients with refractory uveitis due to Behcet’s disease: a 1-year follow-up study of 124 patients. Rheumatology 53:2223–2231
Calvo-Rio V, de la Hera D, Blanco R et al (2014b) Golimumab in uveitis previously treated with other anti-TNF-alpha drugs: a retrospective study of three cases from a single centre and literature review. Clin Exp Rheumatol 32:864–868
Cantarini L, Lopalco G, Caso F et al (2015) Effectiveness and tuberculosis-related safety profile of interleukin-1 blocking agents in the management of Behcet’s disease. Autoimmun Rev 14:1–9
Cao JH, Oray M, Cocho L, Stephen Foster C (2016) Rituximab in the treatment of refractory non-infectious scleritis. Am J Ophthalmol 164:22–28
Caprilli R, Gassull MA, Escher JC, European Crohn’s and Colitis Organisation et al (2006) European evidence based consensus on the diagnosis and management of Crohn’s disease: special situations. Gut 55:i36–i58
Caso F, Rigante D, Vitale A et al (2015) Long-lasting uveitis remission and hearing loss recovery after rituximab in Vogt-Koyanagi-Harada disease. Clin Rheumatol 34:1817–1820
Chan ES, Cronstein BN (2002) Molecular action of methotrexate in inflammatory diseases. Arthritis Res 4:266–273
Chan ES, Cronstein BN (2013) Mechanisms of action of methotrexate. Bull Hosp Joint Dis 71(Suppl 1):S5–S8
Chanaud NP 3rd, Vistica BP, Eugui E, Nussenblatt RB, Allison AC, Gery I (1995) Inhibition of experimental autoimmune uveoretinitis by mycophenolate mofetil, an inhibitor of purine metabolism. Exp Eye Res 61:429–434
Chang-Lin JE, Attar M, Acheampong AA et al (2011a) Pharmacokinetics and pharmacodynamics of a sustained-release dexamethasone intravitreal implant. Invest Ophthalmol Vis Sci 52:80–86
Chang-Lin JE, Burke JA, Peng Q et al (2011b) Pharmacokinetics of a sustained-release dexamethasone intravitreal implant in vitrectomized and nonvitrectomized eyes. Invest Ophthalmol Vis Sci 52:4605–4609
Clifford DB, Ances B, Costello C et al (2011) Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol 68:1156–1164
Clunie GPR, Lennard L (2004) Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology 43:13–18
Constantin A, Loubet-Lescoulie P, Lambert N et al (1998) Antiinflammatory and immunoregulatory action of methotrexate in the treatment of rheumatoid arthritis: evidence of increased interleukin-4 and interleukin-10 gene expression demonstrated in vitro by competitive reverse transcriptase-polymerase chain reaction. Arthritis Rheum 41:48–57
Cordero-Coma M, Salom D, Diaz-Llopis M, Lopez-Prats MJ, Calleja S (2011) Golimumab for uveitis. Ophthalmology 118:1892.e3-4
Cordero-Coma M, Calvo-Rio V, Adan A et al (2014) Golimumab as rescue therapy for refractory immune-mediated uveitis: a three-center experience. Mediators Inflamm 2014:717598
Cornish KS, Kuffova L, Forrester JV (2015) Treatment of diffuse subretinal fibrosis uveitis with rituximab. Br J Ophthalmol 99:153–154
Cox PJ (1979) Cyclophosphamide cystitis--identification of acrolein as the causative agent. Biochem Pharmacol 28:2045–2049
Cronstein BN, Naime D, Ostad E (1993) The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest 92:2675–2682
Cunningham MA, Edelman JL, Kaushal S (2008) Intravitreal steroids for macular edema: the past, the present, and the future. Surv Ophthalmol 53:139–149
Dal Canto AJ, Downs-Kelly E, Perry JD (2005) Ptosis and orbital fat prolapse after posterior sub-Tenon’s capsule triamcinolone injection. Ophthalmology 112:1092–1097
Daniel E, Thorne JE, Newcomb CW et al (2010) Mycophenolate mofetil for ocular inflammation. Am J Ophthalmol 149:423–32.e1-2
Danovitch GM (1999) Choice of immunosuppressive drugs and individualization of immunosuppressive therapy for kidney transplant patients. Transplant Proc 31:2S–6S
Davatchi F, Shams H, Rezaipoor M et al (2010) Rituximab in intractable ocular lesions of Behcet’s disease; randomized single-blind control study (pilot study). Int J Rheum Dis 13:246–252
de Jonge ME, Huitema AD, Rodenhuis S, Beijnen JH (2005) Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet 44:1135–1164
De Smet MD, Nussenblatt RB (1993) Clinical use of cyclosporine in ocular disease. Int Ophthalmol Clin 33:31–45
Deroux A, Chiquet C, Bouillet L (2015) Tocilizumab in severe and refractory Behcet’s disease: four cases and literature review. Semin Arthritis Rheum. doi:10.1016/j.semarthrit.2015.11.012
Deuter CM, Zierhut M, Igney-Oertel A et al (2016) Tocilizumab in uveitic macular edema refractory to previous immunomodulatory treatment. Ocul Immunol Inflamm 5:1–6 (Epub ahead of print)
Diav-Citrin O, Otcheretianski-Volodarsky A, Shechtman S, Ornoy A (2014) Pregnancy outcome following gestational exposure to TNF-alpha-inhibitors: a prospective, comparative, observational study. Reprod Toxicol 43:78–84
Díaz-Llopis M, Salom D, Garcia-de-Vicuña C et al (2012) Treatment of refractory uveitis with adalimumab: a prospective multicenter study of 131 patients. Ophthalmology 119:1575–1581
Djokanovic N, Klieger-Grossmann C, Pupco A, Koren G (2011) Safety of infliximab use during pregnancy. Reprod Toxicol 32:93–97
Doycheva D, Zierhut M, Blumenstock G, Stuebiger N, Deuter C (2011) Long-term results of therapy with mycophenolate mofetil in chronic non-infectious uveitis. Graefes Arch Clin Exp Ophthalmol 249:1235–1243
Doycheva D, Zierhut M, Blumenstock G et al (2014) Immunomodulatory therapy with tumour necrosis factor alpha inhibitors in children with antinuclear antibody-associated chronic anterior uveitis: long-term results. Br J Ophthalmol 98:523–528
Durrani K, Papaliodis GN, Foster CS (2004) Pulse IV cyclophosphamide in ocular inflammatory disease: efficacy and short-term safety. Ophthalmology 111:960–965
Durrani K, Zakka FR, Ahmed M, Memon M, Siddique SS, Foster CS (2011) Systemic therapy with conventional and novel immunomodulatory agents for ocular inflammatory disease. Surv Ophthalmol 56:474–510
Ekstrom K, Hjalgrim H, Brandt L et al (2003) Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 48:963–970
Elion GB (1989) The purine path to chemotherapy. Science 244:41–47
Elion GB (1993) The George Hitchings and Gertrude Elion lecture. The pharmacology of azathioprine. Ann N Y Acad Sci 685:400–407
Faez S, Lobo AM, Sobrin L, Papaliodis GN (2014) Treatment of seronegative spondyloarthropathy-associated uveitis with golimumab: retrospective case series. Clin Experiment Ophthalmol 42:392–395
Farber S, Diamond LK, Mercer RD et al (1948) Temporary remissions in acute leukemia in children produced by folic antagonist 4-aminopteroylglutamic acid (aminopterin). N Engl J Med 238:787–793
FDA (2008) Communication about an ongoing safety review of CellCept (mycophenolate mofetil) and Myfortic (mycophenolate acid). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm072438.htm
Felinski EA, Cox AE, Phillips BE, Antonetti DA (2008) Glucocorticoids induce transactivation of tight junction genes occludin and claudin-5 in retinal endothelial cells via a novel cis-element. Exp Eye Res 86:867–878
Ferrante P, Ramsey A, Bunce C, Lightman S (2004) Clinical trial to compare efficacy and side-effects of injection of posterior sub-Tenon triamcinolone versus orbital floor methylprednisolone in the management of posterior uveitis. Clin Experiment Ophthalmol 32:563–568
Florey HW, Jennings MA et al (1946) Mycophenolic acid; an antibiotic from Penicillium brevicompactum Dlerckx. Lancet 1:46–49
Polack FM (1967) Effect of azathioprine (Imuran) on corneal graft reaction. Am J Ophthalmol 64:233–244
Foster CS, Davanzo R, Flynn TE, McLeod K, Vogel R, Crockett RS (2010) Durezol (Difluprednate Ophthalmic Emulsion 0.05%) compared with Pred Forte 1% ophthalmic suspension in the treatment of endogenous anterior uveitis. J Ocul Pharmacol Ther 26:475–483
Foster CS, Kothari S, Anesi SD et al (2016) The Ocular Immunology and Uveitis Foundation preferred practice patterns of uveitis management. Surv Ophthalmol 61:1–17
Frunzi J (2007) From weapon to wonder drug. Hospitalist
Gallego-Pinazo R, Dolz-Marco R, Martinez-Castillo S, Arevalo JF, Diaz-Llopis M (2013) Update on the principles and novel local and systemic therapies for the treatment of non-infectious uveitis. Inflamm Allergy Drug Targets 12:38–45
Galor A, Perez VL, Hammel JP, Lowder CY (2006) Differential effectiveness of etanercept and infliximab in the treatment of ocular inflammation. Ophthalmology 113:2317–2323
Galor A, Jabs DA, Leder HA et al (2008) Comparison of antimetabolite drugs as corticosteroid-sparing therapy for noninfectious ocular inflammation. Ophthalmology 115:1826–1832
Gangaputra S, Newcomb CW, Liesegang TL et al (2009) Methotrexate for ocular inflammatory diseases. Ophthalmology 116:2188–2198.e1
Gaujoux-Viala C, Giampietro C, Gaujoux T et al (2012) Scleritis: a paradoxical effect of etanercept? Etanercept-associated inflammatory eye disease. J Rheumatol 39:233–239
Geborek P, Bladstrom A, Turesson C et al (2005) Tumour necrosis factor blockers do not increase overall tumour risk in patients with rheumatoid arthritis, but may be associated with an increased risk of lymphomas. Ann Rheum Dis 64:699–703
Genovese MC, Rubbert-Roth A, Smolen JS et al (2013) Longterm safety and efficacy of tocilizumab in patients with rheumatoid arthritis: a cumulative analysis of up to 4.6 years of exposure. J Rheumatol 40:768–780
Gerosa M, Meroni PL, Cimaz R (2014) Safety considerations when prescribing immunosuppression medication to pregnant women. Expert Opin Drug Saf 13:1591–1599
Ghisdal L, Bouchta NB, Broeders N et al (2008) Conversion from tacrolimus to cyclosporine A for new-onset diabetes after transplantation: a single-centre experience in renal transplanted patients and review of the literature. Transpl Int 21:146–151
Giles CL (1974) Bulbar perforation during periocular injection of corticosteroids. Am J Ophthalmol 77:438–441
Godfrey WA, Epstein WV, O’Connor GR et al (1974) The use of chlorambucil in intractable idiopathic uveitis. Am J Ophthalmol 78:415–428
Goldstein DA, Fontanilla FA, Kaul S, Sahin O, Tessler HH (2002) Long-term follow-up of patients treated with short-term high-dose chlorambucil for sight-threatening ocular inflammation. Ophthalmology 109:370–377
Goodman LS, Wintrobe MM et al (1946) Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. JAMA 132:126–132
Gordon DM, Mc LJ, Koteen H et al (1951) The use of ACTH and cortisone in ophthalmology. Am J Ophthalmol 34:1675–1686
Gout T, Ostor AJ, Nisar MK (2011) Lower gastrointestinal perforation in rheumatoid arthritis patients treated with conventional DMARDs or tocilizumab: a systematic literature review. Clin Rheumatol 30:1471–1474
Greenwood AJ, Stanford MR, Graham EM (1998) The role of azathioprine in the management of retinal vasculitis. Eye 12:783–788
Greiner K, Varikkara M, Santiago C, Forrester JV (2002) Efficiency of mycophenolate mofetil in the treatment of intermediate and posterior uveitis. Ophthalmologe 99:691–694
Group TLMSSGaTUoBCMMA (1999) TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 53:457–465
Gul FC, Turgut B, Dagli F, Ilhan N, Ozgen M (2013) The comparison of the impact of ghrelin and tacrolimus on vitreous cytokine levels in an experimental uveitis model. Graefes Arch Clin Exp Ophthalmol 251:1235–1241
Habot-Wilner Z, Sallam A, Pacheco PA, Do HH, McCluskey P, Lightman S (2011) Intravitreal triamcinolone acetonide as adjunctive treatment with systemic therapy for uveitic macular edema. Eur J Ophthalmol 21(Suppl 6):S56–S61
Hamuryudan V, Ozyazgan Y, Hizli N et al (1997) Azathioprine in Behçet’s syndrome: effects on long-term prognosis. Arthritis Rheum 40:769–774
Hardwig PW, Pulido JS, Erie JC, Baratz KH, Buettner H (2006) Intraocular methotrexate in ocular diseases other than primary central nervous system lymphoma. Am J Ophthalmol 142:883–885
Isnard Bagnis C, Tezenas du Montcel S, Beaufils H et al (2002) Long-term renal effects of low-dose cyclosporine in uveitis-treated patients: follow-up study. J Am Soc Nephrol 13:2962–2968
Jabs DA, Rosenbaum JT, Foster CS et al (2000) Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol 130:492–513
Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature Working Group (2005) Standardization of uveitis nomenclature for reporting clinical data. Results of the first international workshop. Am J Ophthalmol 140:509–516
Kacmaz RO, Kempen JH, Newcomb C et al (2010) Cyclosporine for ocular inflammatory diseases. Ophthalmology 117:576–584
Kashani S, Mearza AA (2008) Uses and safety profile of ciclosporin in ophthalmology. Expert Opin Drug Saf 7:79–89
Kawashima H, Fujino Y, Mochizuki M (1988) Effects of a new immunosuppressive agent, FK506, on experimental autoimmune uveoretinitis in rats. Invest Ophthalmol Vis Sci 29:1265–1271
Kay J, Fleischmann R, Keystone E et al (2015) Golimumab 3-year safety update: an analysis of pooled data from the long-term extensions of randomised, double-blind, placebo-controlled trials conducted in patients with rheumatoid arthritis, psoriatic arthritis or ankylosing spondylitis. Ann Rheum Dis 74:538–546
Kearsley-Fleet L, Davies R, Baildam E et al (2016) Factors associated with choice of biologic among children with Juvenile Idiopathic Arthritis: results from two UK paediatric biologic registers. Rheumatology (Oxford) 55(9):1556–1565
Keil JM, Liu X, Antonetti DA (2013) Glucocorticoid induction of occludin expression and endothelial barrier requires transcription factor p54 NONO. Invest Ophthalmol Vis Sci 54:4007–4015
Kempen JH, Gangaputra S, Daniel E, Levy-Clarke GA, Nussenblatt RB, Rosenbaum JT, Suhler EB, Thorne JE, Foster CS, Jabs DA, Helzlsouer KJ (2008) Long-term risk of malignancy among patients treated with immunosuppresive agents for ocular inflammation: a critical assessment of the evidence. Am J Ophthalmol 146:802–812
Khan MF, Arlet J, Bloch-Michel H et al (1979) Acute leukemias after treatment using cytotoxic agents for rheumatologic purpose. Nouv Presse Med 8:1393–1397
Khan IJ, Barry RJ, Amissah-Arthur KN et al (2013) Ten-year experience of pulsed intravenous cyclophosphamide and methylprednisolone protocol (PICM protocol) in severe ocular inflammatory disease. Br J Ophthalmol 97:1118–1122
Kilmartin DJ, Forrester JV, Dick AD (1998a) Tacrolimus (FK506) in failed cyclosporine A therapy in endogenous posterior uveitis. Ocul Immunol Inflamm 6:101–109
Kilmartin DJ, Forrester JV, Dick AD (1998b) Rescue therapy with mycophenolate mofetil in refractory uveitis. Lancet 352:35–36
Kim EC, Foster CS (2006) Immunomodulatory therapy for the treatment of ocular inflammatory disease: evidence-based medicine recommendations for use. Int Ophthalmol Clin 46:141–164
Knickelbein J, Jaworski L, Hasan J, Kaushal P, Sen HN, Nussenblatt RB (2015) Therapeutic options for the treatment of non-infectious uveitis. Expert Rev Ophthalmol 10:359–373
Koike T, Harigai M, Inokuma S et al (2014) Effectiveness and safety of tocilizumab: postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J Rheumatol 41:15–23
Kramer M, Monselise Y, Bahar I, Cohen Y, Weinberger D, Goldenberg-Cohen N (2007) Serum cytokine levels in active uveitis and remission. Curr Eye Res 32:669–675
Kruh J, Foster CS (2012) Corticosteroid-sparing agents: conventional systemic immunosuppressants. Dev Ophthalmol 51:29–46
Kruh JN, Yang P, Suelves AM, Foster CS (2014) Infliximab for the treatment of refractory noninfectious uveitis: a study of 88 patients with long-term follow-up. Ophthalmology 121:358–364
Lafranco Dafflon M, Tran VT, Guex-Crosier Y, Herbort CP (1999) Posterior sub-Tenon’s steroid injections for the treatment of posterior ocular inflammation: indications, efficacy and side effects. Graefes Arch Clin Exp Ophthalmol 237:289–295
Lally L, Spiera R (2015) Current therapies for ANCA-associated vasculitis. Annu Rev Med 66:227–240
Larkin G, Lightman S (1999) Mycophenolate mofetil. A useful immunosuppressive in inflammatory eye disease. Ophthalmology 106:370–374
Larson T, Nussenblatt RB, Sen HN (2011) Emerging drugs for uveitis. Expert Opin Emerg Drugs 16:309–322
Lau CH, Comer M, Lightman S (2003) Long-term efficacy of mycophenolate mofetil in the control of severe intraocular inflammation. Clin Experiment Ophthalmol 31:487–491
Lee JC, Gladman DD, Schentag CT, Cook RJ (2001) The longterm use of azathioprine in patients with psoriatic arthritis. J Clin Rheumatol 7:160–165
Letko E, Yeh S, Foster CS, Pleyer U, Brigell M, Grosskreutz CL (2015) Efficacy and safety of intravenous secukinumab in noninfectious uveitis requiring steroid-sparing immunosuppressive therapy. Ophthalmology 122:939–948
Levy-Clarke G, Jabs DA, Read RW, Rosenbaum JT, Vitale A, Van Gelder RN (2014) Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology 121:785–796.e3
Lim LL, Fraunfelder FW, Rosenbaum JT (2007) Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum 56:3248–3252
Lin P (2015) Targeting interleukin-6 for noninfectious uveitis. Clin Ophthalmol 9:1697–1702
Llorenc V, Mesquida M, Sainz de la Maza M et al (2015) Certolizumab Pegol, a new anti-TNF-alpha in the armamentarium against ocular inflammation. Ocul Immunol Inflamm 24:167–172
Lowder C, Belfort R Jr, Lightman S et al (2011) Dexamethasone intravitreal implant for noninfectious intermediate or posterior uveitis. Arch Ophthalmol 129:545–553
Maiz Alonso O, Blanco Esteban AC, Egues Dubuc CA, Martinez Zabalegui D (2015) Effectiveness of certolizumab pegol in chronic anterior uveitis associated to Crohn’s disease and ankylosing spondylitis. Reumatol Clin 11:189–190
Maltzman JS, Koretzky GA (2003) Azathioprine: old drug, new actions. J Clin Invest 111:1122–1124
Mamo JG (1976) Treatment of Behcet disease with chlorambucil. A follow-up report. Arch Ophthalmol 94:580–583
Manz I, Dietrich I, Przybylski M et al (1985) Identification and quantification of metabolite conjugates of activated cyclophosphamide and ifosfamide with mesna in urine by ion-pair extraction and fast atom bombardment mass spectrometry. Biomed Mass Spectrom 12:545–553
Marticorena J, Romano V, Gomez-Ulla F (2012) Sterile endophthalmitis after intravitreal injections. Mediators Inflamm 2012:928123
Martin F, Lauwerys B, Lefebvre C, Devogelaer JP, Houssiau FA (1997) Side-effects of intravenous cyclophosphamide pulse therapy. Lupus 6:254–257
Mason JO 3rd, Somaiya MD, Singh RJ (2004) Intravitreal concentration and clearance of triamcinolone acetonide in nonvitrectomized human eyes. Retina 24:900–904
Masuda K, Nakajima A, Urayama A et al (1989) Double masked trial of cyclosporin versus colchicine and long-term open study of cyclosporin in Behcet’s disease. Lancet 1:1093–1096
Mathews JD, Crawford BA, Bignell JL, Mackay IR (1969) Azathioprine in active chronic iridocyclitis. A double-blind controlled trial. Br J Ophthalmol 53:327–330
McKay DB, Josephson MA (2008) Pregnancy after kidney transplantation. Clin J Am Soc Nephrol 3(Suppl 2):S117–S125
Mesquida M, Molins B, Llorenc V, de la Maza MS, Adan A (2014) Long-term effects of tocilizumab therapy for refractory uveitis-related macular edema. Ophthalmology 121:2380–2386
Miserocchi E, Baltatzis S, Ekong A, Roque M, Foster CS (2002) Efficacy and safety of chlorambucil in intractable noninfectious uveitis: the Massachusetts Eye and Ear Infirmary experience. Ophthalmology 109:137–142
Miserocchi E, Modorati G, Pontikaki I, Meroni PL, Gerloni V (2014) Long-term treatment with golimumab for severe uveitis. Ocul Immunol Inflamm 22:90–95
Miserocchi E, Modorati G, Berchicci L, Pontikaki I, Meroni P, Gerloni V (2015) Long-term treatment with rituximab in severe juvenile idiopathic arthritis-associated uveitis. Br J Ophthalmol. doi:10.1136/bjophthalmol-2015-306790
Moisseiev E, Shulman S (2014) Certolizumab-induced uveitis: a case report and review of the literature. Case Rep Ophthalmol 5:54–59
Moller A, Iwasaki K, Kawamura A et al (1999) The disposition of 14C-labeled tacrolimus after intravenous and oral administration in healthy human subjects. Drug Metab Dispos 27:633–636
Monach PA, Arnold LM, Merkel PA (2010) Incidence and prevention of bladder toxicity from cyclophosphamide in the treatment of rheumatic diseases: a data-driven review. Arthritis Rheum 62:9–21
Mora PF (2010) New-onset diabetes after renal transplantation. J Invest Med 58:755–763
Multicenter Uveitis Steroid Treatment Trial Follow-up Study Research Group (2015) Quality of life and risks associated with systemic anti-inflammatory therapy versus fluocinolone acetonide intraocular implant for intermediate uveitis, posterior uveitis, or panuveitis: fifty-four-month results of the Multicenter Uveitis Steroid Treatment Trial and Follow-up Study. Ophthalmology 122:1976–1986
Multicenter Uveitis Steroid Treatment Trial Research Group, Kempen JH, Altaweel MM, Holbrook JT, Jabs DA, Sugar EA (2010) The multicenter uveitis steroid treatment trial: rationale, design, and baseline characteristics. Am J Ophthalmol 149:550–561.e10
Multicenter Uveitis Steroid Treatment Trial Research Group, Kempen JH, Altaweel MM et al (2015) Benefits of systemic anti-inflammatory therapy versus fluocinolone acetonide intraocular implant for intermediate uveitis, posterior uveitis, and panuveitis: fifty-four-month results of the Multicenter Uveitis Steroid Treatment (MUST) Trial and Follow-up Study. Ophthalmology 122:1967–1975
Murphy CC, Ayliffe WH, Booth A, Makanjuola D, Andrews PA, Jayne D (2004) Tumor necrosis factor alpha blockade with infliximab for refractory uveitis and scleritis. Ophthalmology 111:352–356
Murphy CC, Greiner K, Plskova J et al (2005) Cyclosporine vs tacrolimus therapy for posterior and intermediate uveitis. Arch Ophthalmol 123:634–641
Murray JE, Merrill JP, Harrison JH, Wilson RE, Dammin GJ (1963) Prolonged survival of human-kidney homografts by immunosuppressive drug therapy. N Engl J Med 268:1315–1323
Murray PI, Hoekzema R, van Haren MA, de Hon FD, Kijlstra A (1990) Aqueous humor interleukin-6 levels in uveitis. Invest Ophthalmol Vis Sci 31:917–920
Muselier A, Bielefeld P, Bidot S, Vinit J, Besancenot J-F, Bron A (2011) Efficacy of tocilizumab in two patients with anti-TNF-alpha refractory uveitis. Ocul Immunol Inflamm 19:382–383
Nakamura S, Yamakawa T, Sugita M et al (1994) The role of tumor necrosis factor-alpha in the induction of experimental autoimmune uveoretinitis in mice. Invest Ophthalmol Vis Sci 35:3884–3889
Neff RT, Hurst FP, Falta EM et al (2008) Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 86:1474–1478
Nevers W, Pupco A, Koren G, Bozzo P (2014) Safety of tacrolimus in pregnancy. Can Fam Physician 60:905–906
Nguyen K, Vleugels RA, Velez NF, Merola JF, Qureshi AA (2013) Psoriasiform reactions to anti-tumor necrosis factor alpha therapy. J Clin Rheumatol 19:377–381
Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J (2009) Long-term safety and efficacy of tocilizumab, an anti-IL-6 receptor monoclonal antibody, in monotherapy, in patients with rheumatoid arthritis (the STREAM study): evidence of safety and efficacy in a 5-year extension study. Ann Rheum Dis 68:1580–1584
Nussenblatt RB, Whitcup SM (2010) Uveitis: fundamentals and clinical practice, 4th edn. Mosby/Elsevier, Amsterdam/Edinburgh
Nussenblatt RB, Palestine AG, Rook AH, Scher I, Wacker W, Gery I (1983a) Treatment of intraocular inflammatory disease with Cyclosporine A. Lancet 2:235–238
Nussenblatt RB, Palestine AG, Chan CC (1983b) Cyclosporine A therapy in the treatment of intraocular inflammatory disease resistant to systemic corticosteroids and cytotoxic agents. Am J Ophthalmol 96:275–282
Nussenblatt RB, Palestine AG, Chan CC, Stevens G Jr, Mellow SD, Green SB (1991) Randomized, double-masked study of cyclosporine compared to prednisolone in the treatment of endogenous uveitis. Am J Ophthalmol 112:138–146
Onishi SM, Asahi MG, Chou C, Gallemore RP (2015) Topical difluprednate for the treatment of Harada’s disease. Clin Ophthalmol 9:157–167
Oshitari T, Kajita F, Tobe A et al (2012) Refractory uveitis in patient with castleman disease successfully treated with tocilizumab. Case Rep Ophthalmol Med 2012:968180
Ostensen M, Förger F (2013) How safe are anti-rheumatic drugs during pregnancy? Curr Opin Pharmacol 13:470–475
Ozyazgan Y, Yurdakul S, Yazici H et al (1992) Low dose cyclosporin A versus pulsed cyclophosphamide in Behcet’s syndrome: a single masked trial. Br J Ophthalmol 76:241–243
Palmer R, Doré CJ, Denman AM (1984) Chlorambucil-induced chromosome damage to human lymphocytes is dose-dependent and cumulative. Lancet 1:246–249
Papo M, Bielefeld P, Vallet H et al (2014) Tocilizumab in severe and refractory non-infectious uveitis. Clin Exp Rheumatol 32:S75–S79
Pasadhika S, Kempen JH, Newcomb CW et al (2009) Azathioprine for ocular inflammatory diseases. Am J Ophthalmol 148:500–509.e2
Patel SS, Dodds EM, Echandi LV et al (2014) Long-term, drug-free remission of sympathetic ophthalmia with high-dose, short-term chlorambucil therapy. Ophthalmology 121:596–602
Perez R (1951) Case of uveitis of unknown etiology treated with nitrogen mustards. Rev Clin Esp 41:265–267
Perez V, Papaliodis G, Chu D, Anzaar F, Christen W, Foster C (2004) Elevated levels of interleukin 6 in the vitreous fluid of patients with pars planitis and posterior uveitis: the Massachusetts eye & ear experience and review of previous studies. Ocul Immunol Inflamm 12:205–214
Pujari SS, Kempen H, Newcomb CW, Gangaputra S, Daniel E, Suhler EB, Thorne JE, Jabs DA, Levy-Clarke GA, Nussenblatt RB, Rosenbaum JT, Foster CS (2010) Cyclophosphamide for ocular inflammatory diseases. Ophthalmology 117:356–365
Querques L, Querques G, Lattanzio R et al (2013) Repeated intravitreal dexamethasone implant (Ozurdex(R)) for retinal vein occlusion. Ophthalmologica 229:21–25
Ragam A, Kolomeyer AM, Fang C, Xu Y, Chu DS (2014) Treatment of chronic, noninfectious, nonnecrotizing scleritis with tumor necrosis factor alpha inhibitors. Ocul Immunol Inflamm 22:469–477
Ramos-Cebrian M, Torregrosa JV, Gutierrez-Dalmau A, Oppenheimer F, Campistol JM (2007) Conversion from tacrolimus to cyclosporine could improve control of posttransplant diabetes mellitus after renal transplantation. Transplant Proc 39:2251–2253
Rathi M, Rajkumar V, Rao N et al (2015) Conversion from tacrolimus to cyclosporine in patients with new-onset diabetes after renal transplant: an open-label randomized prospective pilot study. Transplant Proc 47:1158–1161
Rathinam SR, Babu M, Thundikandy R et al (2014) A randomized clinical trial comparing methotrexate and mycophenolate mofetil for noninfectious uveitis. Ophthalmology 121:1863–1870
Reddy AR, Backhouse OC (2003) Does etanercept induce uveitis? Br J Ophthalmol 87:925
Reed JB, Morse LS, Schwab IR (1998) High-dose intravenous pulse methylprednisolone hemisuccinate in acute Behcet retinitis. Am J Ophthalmol 125:409–411
Restrepo JP, Molina MP (2010) Successful treatment of severe nodular scleritis with adalimumab. Clin Rheumatol 29:559–561
Rosenbaum JT (1994) Treatment of severe refractory uveitis with intravenous cyclophosphamide. J Rheumatol 21:123–125
Rosenbaum JT, Howes EL Jr, Rubin RM, Samples JR (1988) Ocular inflammatory effects of intravitreally-injected tumor necrosis factor. Am J Pathol 133:47–53
Santos Lacomba M, Marcos Martín C, Gallardo Galera JM et al (2001) Aqueous humor and serum tumor necrosis factor-α in clinical uveitis. Ophthalmic Res 33:251–255
Sartani G, Silver PB, Rizzo LV et al (1996) Anti-tumor necrosis factor alpha therapy suppresses the induction of experimental autoimmune uveoretinitis in mice by inhibiting antigen priming. Invest Ophthalmol Vis Sci 37:2211–2218
Schabert VF, Watson C, Joseph GJ, Iversen P, Burudpakdee C, Harrison DJ (2013) Costs of tumor necrosis factor blockers per treated patient using real-world drug data in a managed care population. J Manag Care Pharm 19:621–630
Selmi C, Ceribelli A, Naguwa SM, Cantarini L, Shoenfeld Y (2015) Safety issues and concerns of new immunomodulators in rheumatology. Expert Opin Drug Saf 14:389–399
Sen HN, Suhler EB, Al-Khatib SQ, Djalilian AR, Nussenblatt RB, Buggage RR (2003) Mycophenolate mofetil for the treatment of scleritis. Ophthalmology 110:1750–1755
Sen HN, Sangave A, Hammel K, Levy-Clarke G, Nussenblatt RB (2009) Infliximab for the treatment of active scleritis. Can J Ophthalmol 44:e9–e12
Sen HN, Vitale S, Gangaputra SS et al (2014) Periocular corticosteroid injections in uveitis: effects and complications. Ophthalmology 121:2275–2286
Sendrowski DP, Jaanus SD, Semes LP, Stern ME (2008) Anti-inflammatory drugs. In: Bartlett JD, Jaanus SD (eds) Clinical ocular pharmacology, 5th edn. Butterworh-Heinemann, St. Louis, pp 222–224
Shen L, You Y, Sun S, Chen Y, Qu J, Cheng L (2010) Intraocular and systemic pharmacokinetics of triamcinolone acetonide after a single 40-mg posterior subtenon application. Ophthalmology 117:2365–2371
Sheppard JD, Toyos MM, Kempen JH, Kaur P, Foster CS (2014) Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study. Invest Ophthalmol Vis Sci 55:2993–3002
Shibuya M, Fujio K, Morita K, Harada H, Kanda H, Yamamoto K (2013) Successful treatment with tocilizumab in a case of Cogan’s syndrome complicated with aortitis. Mod Rheumatol 23:577–581
Siepman K, Huber M, Stubiger N, Deuter C, Zierhut M (2006) Mycophenolate mofetil is a highly effective and safe immunosuppressive agent for the treatment of uveitis. Graefes Arch Clin Exp Ophthalmol 244:788–794
Sifontis NM, Coscia LA, Constantinescu S, Lavelanet AF, Moritz MJ, Armenti VT (2006) Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation 82:1698–1702
Simonini G, Paudyal P, Jones GT, Cimaz R, Macfarlane GJ (2013) Current evidence of methotrexate efficacy in childhood chronic uveitis: a systematic review and meta-analysis approach. Rheumatology 52:825–831
Simonini G, Katie D, Cimaz R, Macfarlane GJ, Jones GT (2014) Does switching anti-TNFalpha biologic agents represent an effective option in childhood chronic uveitis: the evidence from a systematic review and meta-analysis approach. Semin Arthritis Rheum 44:39–46
Slabaugh MA, Herlihy E, Ongchin S, van Gelder RN (2012) Efficacy and potential complications of difluprednate use for pediatric uveitis. Am J Ophthalmol 153:932–938
Slater CA, Liang MH, McCune JW, Christman GM, Laufer MR (1999) Preserving ovarian function in patients receiving cyclophosphamide. Lupus 8:3–10
Smith JR, Levinson RD, Holland GN et al (2001) Differential efficacy of tumor necrosis factor inhibition in the management of inflammatory eye disease and associated rheumatic disease. Arthritis Rheum 45:252–257
Smith JR, Rosenbaum JT, Wilson DJ et al (2002) Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvement. Ophthalmology 109:1709–1716
Smith JA, Thompson DJS, Whitcup SM et al (2005) A randomized, placebo-controlled, double-masked clinical trial of etanercept for the treatment of uveitis associated with juvenile idiopathic arthritis. Arthritis Care Res 53:18–23
Sourlingas TG, Sekeri-Pataryas KE (1997) S and G2 phase histone biosynthesis of HEp-2 cells after the influence of the bisalkylating agent, chlorambucil. Biochem Mol Biol Int 42:1103–1114
Spatz S, Rudnicka A, McDonald CJ (1978) Mycophenolic acid in psoriasis. Br J Dermatol 98:429–435
Sturm I, Bosanquet AG, Hermann S, Guner D, Dorken B, Daniel PT (2003) Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy. Cell Death Differ 10:477–484
Suelves AM, Arcinue CA, Gonzalez-Martin JM, Kruh JN, Foster CS (2013) Analysis of a novel protocol of pulsed intravenous cyclophosphamide for recalcitrant or severe ocular inflammatory disease. Ophthalmology 120:1201–1209
Suhler EB, Smith JR, Giles TR et al (2009) Infliximab therapy for refractory uveitis: 2-year results of a prospective trial. Arch Ophthalmol 127:819–822
Suhler EB, Lowder CY, Goldstein DA et al (2013) Adalimumab therapy for refractory uveitis: results of a multicentre, open-label, prospective trial. Br J Ophthalmol 97:481–486
Suhler EB, Lim LL, Beardsley RM et al (2014) Rituximab therapy for refractory scleritis: results of a phase I/II dose-ranging, randomized, clinical trial. Ophthalmology 121:1885–1891
Takeuchi M, Kezuka T, Sugita S et al (2014) Evaluation of the long-term efficacy and safety of infliximab treatment for uveitis in Behcet’s disease: a multicenter study. Ophthalmology 121:1877–1884
Tappeiner C, Heinz C, Ganser G, Heiligenhaus A (2012) Is tocilizumab an effective option for treatment of refractory uveitis associated with juvenile idiopathic arthritis? J Rheumatol 39:1294–1295
Tauber M, Buche S, Reygagne P et al (2014) Alopecia areata occurring during anti-TNF therapy: a national multicenter prospective study. J Am Acad Dermatol 70:1146–1149
Taylor RP, Lindorfer MA (2007) Drug insight: the mechanism of action of rituximab in autoimmune disease--the immune complex decoy hypothesis. Nat Clin Pract Rheumatol 3:86–95
Taylor SR, Habot-Wilner Z, Pacheco P, Lightman SL (2009) Intraocular methotrexate in the treatment of uveitis and uveitic cystoid macular edema. Ophthalmology 116:797–801
Taylor SR, Banker A, Schlaen A et al (2013) Intraocular methotrexate can induce extended remission in some patients in noninfectious uveitis. Retina 33:2149–2154
Teoh SC, Hogan AC, Dick AD, Lee RW (2008) Mycophenolate mofetil for the treatment of uveitis. Am J Ophthalmol 146:752–760, 760.e1–3
Teruel C, Lopez-San Roman A, Bermejo F et al (2010) Outcomes of pregnancies fathered by inflammatory bowel disease patients exposed to thiopurines. Am J Gastroenterol 105:2003–2008
Tessler HH, Jennings T (1990) High-dose short-term chlorambucil for intractable sympathetic ophthalmia and Behcet’s disease. Br J Ophthalmol 74:353–357
Thorne JE, Jabs DA, Qazi FA, Nguyen QD, Kempen JH, Dunn JP (2005) Mycophenolate mofetil therapy for inflammatory eye disease. Ophthalmology 112:1472–1477
Tiede I, Fritz G, Strand S et al (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest 111:1133–1145
Tomkins-Netzer O, Taylor SR, Bar A et al (2014) Treatment with repeat dexamethasone implants results in long-term disease control in eyes with noninfectious uveitis. Ophthalmology 121:1649–1654
Tomkins-Netzer O, Talat L, Seguin-Greenstein S, Bar A, Lightman S (2016) Outcome of treating pediatric uveitis with dexamethasone implants. Am J Ophthalmol 161:110–115.e2
Tong D, Manolios N, Howe G, Spencer D (2012) New onset sarcoid-like granulomatosis developing during anti-TNF therapy: an under-recognised complication. Intern Med J 42:89–94
Urban RC Jr, Cotlier E (1986) Corticosteroid-induced cataracts. Surv Ophthalmol 31:102–110
Vallet H, Riviere S, Sanna A et al (2015) Efficacy of anti-TNF alpha in severe and/or refractory Behcet’s disease: multicenter study of 124 patients. J Autoimmun 62:67–74
van Oosten BW, Barkhof F, Truyen L et al (1996) Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 47:1531–1534
Victor RG, Thomas GD, Marban E, O’Rourke B (1995) Presynaptic modulation of cortical synaptic activity by calcineurin. Proc Natl Acad Sci U S A 92:6269–6273
Visser K, Katchamart W, Loza E et al (2009) Multinational evidence-based recommendations for the use of methotrexate in rheumatic disorders with a focus on rheumatoid arthritis: integrating systematic literature research and expert opinion of a broad international panel of rheumatologists in the 3E Initiative. Ann Rheum Dis 68:1086–1093
Wakefield D (2014) Does cyclophosphamide still have a role in the treatment of severe inflammatory disease. Ocul Immunol Inflamm 22:306–310
Wan C-K, He C, Sun L, Egwuagu CE, Leonard WJ (2016) Cutting edge: IL-1 receptor signaling is critical for the development of autoimmune uveitis. J Immunol 196:543–546
Weijtens O, Schoemaker RC, Romijn FP, Cohen AF, Lentjes EG, van Meurs JC (2002) Intraocular penetration and systemic absorption after topical application of dexamethasone disodium phosphate. Ophthalmology 109:1887–1891
Weinshilboum RM, Sladek SL (1980) Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet 32:651–652
Wessels JA, Huizinga TW, Guchelaar HJ (2008) Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology 47:249–255
Wolfe F, Michaud K (2007) The effect of methotrexate and anti-tumor necrosis factor therapy on the risk of lymphoma in rheumatoid arthritis in 19,562 patients during 89,710 person-years of observation. Arthritis Rheum 56:1433–1439
Wong VG, Hersh EM (1965) Methotrexate in the therapy of cyclitis. Trans Am Acad Ophthalmol Otolaryngol 69:279–293
Wong AK, Kerkoutian S, Said J, Rashidi H, Pullarkat ST (2012) Risk of lymphoma in patients receiving antitumor necrosis factor therapy: a meta-analysis of published randomized controlled studies. Clin Rheumatol 31:631–636
Woodrick RS, Ruderman EM (2011) Safety of biologic therapy in rheumatoid arthritis. Nat Rev Rheumatol 7:639–652
Woods AC (1950) Clinical and experimental observation on the use of ACTH and cortisone in ocular inflammatory disease. Am J Ophthalmol 33:1325–1349
Wyzgal J, Oldakowska-Jedynak U, Paczek L et al (2003) Posttransplantation diabetus mellitus under calcineurin inhibitor. Transplant Proc 35:2216–2218
Yazici H, Pazarli H, Barnes CG et al (1990) A controlled trial of azathioprine in Behçet’s syndrome. N Engl J Med 322:281–285
Yokota S, Itoh Y, Morio T et al (2015) Tocilizumab in systemic juvenile idiopathic arthritis in a real-world clinical setting: results from 1 year of postmarketing surveillance follow-up of 417 patients in Japan. Ann Rheum Dis. doi:10.1136/annrheumdis-2015-207818
Yoshimura T, Sonoda KH, Ohguro N et al (2009) Involvement of Th17 cells and the effect of anti-IL-6 therapy in autoimmune uveitis. Rheumatology 48:347–354
Zarranz-Ventura J, Carreno E, Johnston RL et al (2014) Multicenter study of intravitreal dexamethasone implant in noninfectious uveitis: indications, outcomes, and reinjection frequency. Am J Ophthalmol 158:1136–1145.e5
Acknowledgment
This research was supported by the Intramural Research Program of the NIH, NEI.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 SpringerInternationalPublishingSwitzerland
About this chapter
Cite this chapter
Knickelbein, J.E., Armbrust, K.R., Kim, M., Sen, H.N., Nussenblatt, R.B. (2016). Pharmacologic Treatment of Noninfectious Uveitis. In: Whitcup, S., Azar, D. (eds) Pharmacologic Therapy of Ocular Disease. Handbook of Experimental Pharmacology, vol 242. Springer, Cham. https://doi.org/10.1007/164_2016_21
Download citation
DOI: https://doi.org/10.1007/164_2016_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58288-7
Online ISBN: 978-3-319-58290-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)