
Abstract
Obesity is associated with multiple metabolic disorders that drive cardiovascular disease, T2D and cancer. The doubling in the number of obese adults over the past 3 decades led to the recognition of obesity as a “disease”. With over 42 million children obese or overweight, this epidemic is rapidly growing worldwide. Obesity and T2D are both associated together and independently with an increased risk for cancer and a worse prognosis. Accumulating evidence from epidemiological studies revealed potential factors that may explain the association between obesity-linked metabolic disorders and cancer risk. Studies based on the insulin resistance MKR mice, highlighted the roe of the insulin receptor and its downstream signaling proteins in mediating hyperinsulinemia's mitogenic effects. Hypercholesterolemia was also shown to promote the formation of larger tumors and enhancement in metastasis. Furthermore, the conversion of cholesterol into 27-Hydroxycholesterol was found to link high fat diet-induced hypercholesterolemia with cancer pathophysiology. Alteration in circulating adipokines and cytokines are commonly found in obesity and T2D. Adipokines are involved in tumor growth through multiple mechanisms including mTOR, VEGF and cyclins. In addition, adipose tissues are known to recruit and alter macrophage phenotype; these macrophages can promote cancer progression by secreting inflammatory cytokines such as TNF-α and IL-6.
Better characterization on the above factors and their downstream effects is required in order to translate the current knowledge into the clinic, but more importantly is to understand which are the key factors that drive cancer in each patient. Until we reach this point, policies and activities toward healthy diets and physical activities remain the best medicine.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Growing evidence, over the past decade or more, has provided us with the knowledge that obesity and type 2 diabetes (T2D) are associated with an increased risk of many types of cancer and more importantly, perhaps, increased mortality in these individuals. The major emphasis today is focused on the epidemic of obesity and, secondarily, T2D and the metabolic derangements of these metabolic disorders. In addition, there are numerous ongoing studies attempting to understand the etiology of the vascular complications resulting from obesity and T2D, as well as potential preventative medications. On the other hand, given the increase in cases of obesity and T2D worldwide, focus should also be directed towards a major complication, namely, cancer risk and cancer mortality, which undoubtedly increases in parallel with the obesity/T2D epidemic.
In this review we present the epidemiological evidence for the association between these metabolic disorders and cancer risk, we will describe the studies identifying causative factors, and we will try to relate the metabolic aspects of both obesity and T2D and how they impact on cancer.
2 Epidemic of Obesity and T2D and the Increased Cancer Risk
The epidemic of obesity is now recognized as a worldwide problem. While the USA still leads the world in the percent of overweight and obese individuals, many westernized countries are fast catching up, and developing countries are increasingly being affected. According to the World Health Organization, in 2008, over 1.4 billion adults (11%) worldwide are overweight, of which ~300 million were obese (Haslam and James 2005), and the incidence is rising rapidly. The universally accepted definition of overweight and obesity is a body mass index (BMI) of >25 and >30 kg/m2, respectively. Using these criteria, more than 60% of adults in the USA are overweight and at least half of these are obese. In Southeast Asian countries, due to different body habitus, it is clearly more appropriate to define obesity using waist circumference, >40 inches in men and >35 inches in women. These measurements correlate better with visceral adiposity, and visceral adiposity closely tracks with elements of metabolic syndrome such as dyslipidemia, hypertension, and glucose intolerance.
The concern over the growing epidemic of obesity and the health ramifications has recently motivated the American Medical Association to designate obesity as a “disease,” whereas previously it was considered a “disorder,” a condition that carries less concern. The clear evidence that obesity predisposes to cardiovascular disease and cancer, for example, warrants this refocus on obesity as a disease entity.
This obesity epidemic is driving a similar trend in T2D, and the International Diabetes Federation (IDF) data suggests that ~8% of the world’s population has diabetes with levels as high as ~10% in the Middle East and North Africa. By the year 2030, this number will more than double, since many obese individuals are unaware they have diabetes.
2.1 Obesity and Cancer
The increased association of obesity and metabolic syndrome with cancer risk and cancer mortality has become evident over the past decade, from numerous epidemiological studies. Thus, in countries where obesity prevalence has increased rapidly, such as the USA, a significant proportion (~20%) of all new cancers may be attributable to obesity (Calle et al. 2003; Jemal et al. 2007). Specific examples of common cancers include breast, endometrial, colon, and prostate cancers. In one study of over 33,000 men, the presence of metabolic syndrome was associated with a 56% enhanced risk of cancer mortality over the following 14 years of follow-up (Jaggers et al. 2009; Pothiwala et al. 2009). While the Nurses’ Health Study suggested that central adiposity determined by waist circumference and waist to hip ratio was associated with an increased risk of postmenopausal breast cancer (Huang et al. 1999), more recent studies have claimed that premenopausal obesity is also a risk factor for breast cancer risk (Pierobon and Frankenfeld 2013; Robinson et al. 2014). The Million Women Study in the UK and the Cancer Prevention Study II in the USA similarly reported increased cancer mortality in obese individuals (Petrelli et al. 2002; Reeves et al. 2007).
Strong supporting evidence for the association of obesity and cancer risk and mortality was obtained from a number of bariatric surgery studies, which dramatically reverse obesity. In the Swedish Obesity Subjects (SOS) study, following more than 30% weight loss, a reduction of cancer in women of about 41% was recorded (Sjostrom et al. 2009). A similar effect was seen in the Utah obesity study (Adams et al. 2009). Furthermore, in the Women’s Intervention Nutrition Study (WINS), a 24% reduction in breast cancer was seen after only a 4% reduction in weight over 5 years (Prentice et al. 2006). The actual mechanisms involved in this effect are as yet undefined, but most likely reflect a correction of factors that are abnormal in cases of obesity and metabolic syndrome.
2.2 Children/Adolescents
The obesity epidemic has not spared children and adolescents. While definitions of obesity and metabolic syndrome are less well defined in children and adolescents, the increase is clear (Cook et al. 2003; Weiss et al. 2004). Furthermore, nonalcoholic fatty acid liver disease (NAFLD, another complication of obesity) is increased from 2.6% in normal weight children to 77% in overweight children (Franzese et al. 1997; Schwimmer et al. 2006). In line with this increase in NAFLD is the increase in adult hepatocellular carcinoma (HCC) in individuals who were obese during their childhood years, with a hazard ratio (HR) of 1.2–1.3 (Berentzen et al. 2014). Other studies have shown that BMI in the upper quartile in children from 2 to 14 years of age was associated with increased cancer risk in adulthood by 40% (Park et al. 2012), particularly colorectal, ovarian, cervical, and kidney cancer.
2.3 T2D Epidemic
The obesity epidemic is driving an epidemic of T2D worldwide. While there exists a group of healthy obese individuals, the majority demonstrate features of metabolic syndrome or prediabetes and, in genetically predisposed, progress to frank T2D (Cornier et al. 2008). Different manifestations of metabolic syndrome have different diagnostic criteria, but most include an increased waist circumference, dyslipidemia, hypertension, and even elevated fasting plasma glucose. Almost 70–80% of individuals with T2D are obese; moreover, several long-term prospective studies have shown a higher risk of T2D with increasing body weight. A National Health and Nutrition Examination Survey (NHANES) 25 years ago highlighted an association between being overweight and suffering from diabetes (Van Itallie 1985). Recently, it has been discovered that even apparently metabolically healthy overweight or obese men are still at significantly higher risk of developing T2D (Arnlov et al. 2011).
2.4 T2D and Cancer
Evidence is now emerging for a direct association between T2D and a higher risk of cancer mortality independent of the effects of obesity. (Coughlin et al. 2004; Verlato et al. 2003; Yancik et al. 2001a, b). It was found that hyperinsulinemia and insulin resistance, as shown by elevated levels of circulating C-peptide (a commonly used biomarker for insulin secretion in T2D), were significant risk factors for breast cancer (Bruning et al. 1992; Gunter et al. 2009; Verheus et al. 2006). A prospective study in Sweden of 80,000 women (average age 64.2 years) found that individuals with T2D had an increased incidence of breast cancer (Weiderpass et al. 1997). In another prospective study in 2003, an increased risk of estrogen receptor-positive breast cancer, postmenopausally, was found to be associated with T2D (Michels et al. 2003). Although T2D patients are known to be at high risk of developing pancreatic cancer (Coughlin et al. 2004; Pisani 2008; Rousseau et al. 2006), it has also been discovered that pancreatic cancer precedes diabetes (Isaksson et al. 2003; Permert et al. 1994). Colorectal cancer is also positively associated positively with T2D. A case-control study of around 10,000 adults in the UK showed that the risk of both colonic and rectal cancers is increased in both male and female diabetic patients (odds ratio = 1.42) (Yang et al. 2005), as was also shown in the Physicians’ Health Study, where the relative risk of colorectal cancer in men with T2D was 1.5 (Sturmer et al. 2006). Endometrial cancer risk has also been shown to be associated with high circulating insulin or C-peptide levels, and mortality risk is relatively high in diabetes compared to some other cancers (relative risk ≥2) (Folsom et al. 2004).
In contrast to most epithelial cancers, T2D has been found in several meta-analyses to be inversely associated with prostate cancer risk (Bonovas et al. 2004; Coughlin et al. 2004; Kasper and Giovannucci 2006). On the other hand, obese males with high levels of circulating C-peptide, who develop prostate cancer, are at higher risk of dying from the disease, suggesting a relationship between insulin and aggressive, high-grade prostatic tumors specifically (Ma et al. 2008).
3 Potential Mechanisms
Following the numerous observations that obesity and diabetes are clearly associated with an increased risk of cancer and cancer-related mortality, there has been increased interest in establishing the causal factors involved in this effect. Some of the possible factors are listed in Table 1. In-depth analyses of the observational studies have strongly suggested that insulin, insulin-like growth factor 1 (IGF-1) and insulin-like growth factor 1 receptor (IGF-1R), leptin, inflammatory cytokines, caloric intake, and lipids are examples of potential causal factors that may explain the association between metabolic disorders and cancer risk. To study these various factors, a number of groups have developed appropriate mouse models for these preclinical experiments.
4 Animal Models
4.1 Obesity
High-fat diet (HFD)-induced obesity has been used extensively in mice to study the ramifications on cancer growth. Using this model it was demonstrated that the growth of Lewis lung carcinoma and mouse colon 38 adenocarcinoma cell lines was increased. This was associated with insulin resistance, hyperinsulinemia, glucose intolerance, and hyperleptinemia. Male mice demonstrated more severe metabolic derangements compared to female mice, and the effect on tumor growth was more pronounced. Interestingly, when female obese mice underwent ovariectomy, they developed the same degree of metabolic derangement as the male obese mice, and the effect on tumor growth was markedly enhanced. This suggested that estradiol was protective against metabolic derangements in the face of HFD-induced obesity (Yakar et al. 2006). Similarly, in ovariectomized mice fed with a HFD or a calorie-restricted (CR) diet, HFD-induced obesity had the largest tumors whereas CR mice had the smallest (Rondini et al. 2011). In other models of diet-induced obesity and CR diet, it was demonstrated that prostate, pancreatic, and skin, in addition to colon and breast, cancers are affected by these manipulations of the metabolic changes (Blando et al. 2011; Ford et al. 2013; Lashinger et al. 2013; Moore et al. 2012; Olivo-Marston et al. 2014). The factors mediating these effects include hyperinsulinemia, leptin, inflammatory cytokines, and, as recently demonstrated, hypercholesterolemia.
4.2 Hypercholesterolemia
In mice fed with HFD to induce obesity, Nelson et al. showed that mammary tumor growth was increased and dependent on a metabolite of cholesterol, namely, 27-OH cholesterol, as well as the presence of the estrogen receptor. The conversion of cholesterol to its 27-OH metabolite occurred both in tumor-associated macrophages and tumor cells as well (Nelson et al. 2013). In a separate study, ApoE knockout (ApoE-/-) mice, which demonstrate hypercholesterolemia and hypertriglyceridemia, were shown to have enhanced mammary tumor growth and lung metastases (Alikhani et al. 2013). Interestingly, ApoE-/- mice are insulin sensitive and have normal glucose homeostasis, making them an excellent model to study the isolated effects of lipid abnormalities as their circulating insulin levels are also normal if somewhat lower than wild-type controls (Kawashima et al. 2009). These observations, along with studies in prostate cancer cells (Pelton et al. 2012), have identified cholesterol as tumor promoters and support the epidemiological studies that show an association of hyperlipidemia and cancer growth and mortality and the reduction of mortality in statin users (Nielsen et al. 2012).
4.3 Hyperinsulinemia
The MKR mice were developed by engineering muscle insulin resistance through a transgenic overexpression of a defective IGF-1R that interferes with insulin receptor (IR) as well, through hybrid receptors, resulting in severe insulin resistance. Male MKR mice are diabetic with severe hyperglycemia, hyperinsulinemia, and hyperlipidemia, but without being obese, since the diabetes is not brought about by HFD-induced obesity and diabetes. Female MKR mice, on the other hand, do not demonstrate hyperlipidemia and hyperglycemia but still have hyperinsulinemia; thus they are extremely useful for studying the effect of isolated hyperinsulinemia on cancer. Using multiple oncogenic-induced mouse tumor models, both by orthotopic injections of cancer cell lines and crossing with mammary tumor transgenic mice (Table 2), we were able to demonstrate that hyperinsulinemia was causative in the growth and progression of mammary tumors as well as metastases to the lungs. The role of hyperinsulinemia was demonstrated by reducing the circulating levels of insulin. This was achieved by the use of a β3-adrenergic receptor agonist (CL-316,243) previously shown to improve the insulin resistance and hyperinsulinemia in male MKR mice (Kim et al. 2006). The reduced insulin levels led to a marked reduction in breast tumor growth (Fierz et al. 2010a). Since insulin may affect tumor growth via IR or IGF-1R, we blocked the tyrosine kinase activity of these receptors using a small molecule tyrosine kinase inhibitor (BMS-536924). Receptor activity inhibition resulted in reduced tumor growth. A similar result was obtained with the use of low-dose picropodophyllin (PPP), a compound designed as an IGF-1R-specific inhibitor, but in our hands PPP inhibits both IR and IGF-1R (Rostoker et al. 2013).
Since mTOR is activated by insulin and is a central mediator of tumor progression (Fig. 1), we studied the impact of mTOR inhibition (by rapamycin) on mammary tumor progression and the metabolic state of the mice. Mammary tumor progression was studied in the double transgenic, MMTV-PyVmT/MKR, and two orthotopic models using the Met-1 and MCNeuA cells. In both the wild-type and MKR (insulin-resistant, hyperinsulinemic) mice, glucose intolerance and hypertriglyceridemia worsened significantly after rapamycin treatment. Nonetheless, tumor growth was inhibited in all three mammary tumor models, despite the worsening of insulin resistance and higher levels of circulating insulin (Fierz et al. 2010b). Inhibition of phosphatidylinositol 3-kinase (PI3K) alone, using the oral pan-class I PI3K inhibitor (NVP-BKM120), or together with mTOR, using NVP-BEZ235, inhibited tumor growth. However, in regard to the metabolic effects, inhibiting PI3K alone led to more severe metabolic derangements with increased insulin resistance, hyperinsulinemia, and hyperglycemia, in comparison to the dual inhibitor of PI3K/mTOR (Gallagher et al. 2012).

Insulin receptor/PI3K/mTOR signaling pathway. ERK extracellular signal-regulated kinase, Grb2 growth factor receptor-bound protein 2, IRS-1 insulin receptor substrate 1, MEK mitogen-activated protein kinase kinase, P phosphate, Raf rapidly accelerated fibrosarcoma kinase, Ras rat sarcoma protein
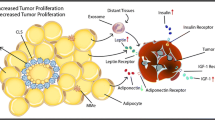
4.4 Adipokines
Circulating adipokines are commonly altered in obesity and T2D (Nalabolu et al. 2014). Leptin is classically elevated in these conditions, whereas adiponectin is reduced. Leptin has been shown to stimulate cancer cell growth in cell cultures, suggesting a role in the effect of obesity in enhancing cancer growth and prognosis (Park and Scherer 2011; Somasundar et al. 2003). The role of leptin in cancer progression has been studied in genetically induced obese mouse models. The Lep0b/Lep0b mice are obese, insulin resistant, and hyperinsulinemic, with low leptin levels. These competing features may explain the varying results when studying cancer in these mice. On the other hand, Lepdb/Lepdb mice, carrying a leptin receptor mutation that results in obesity with elevated leptin levels, show more marked cancer growth and metastases. Interestingly, serum from Lep0b/Lep0b induces a mesenchymal phenotype in the B16 melanoma cells that may explain the enhanced pulmonary metastases in the absence of increased primary tumor growth (Kushiro and Nunez 2011).
On the other hand, adiponectin has been shown to induce cancer cell apoptosis and may explain the effect of low adiponectin levels in obesity and T2D on cancer prognosis (Kelesidis et al. 2006; Körner et al. 2006). In a model of chemical (azoxymethane, AOM)-induced colon carcinogenesis and adiponectin and adiponectin receptor knockout mice, HFD increased the AOM effect (Fujisawa et al. 2008). Adiponectin administration inhibited tumor growth through multiple mechanisms, including inhibition of cell proliferation and inhibition of mTOR, VEGF, and cyclins (Moon et al. 2013).
Interestingly, there seems to be an interplay between the effect of adiponectin and cholesterol on tumor growth (Liu et al. 2013). In bigenic mice (MMTV-PyVmT and adiponectin deficient) fed with high-fat high-cholesterol diet, the resultant hypercholesterolemia and increased expression of the low density lipoprotein (LDL) receptor (LDLR) increased tumor cholesterol content and enhanced tumor growth. Adiponectin downregulated the LDLR in vitro and in vivo and demonstrated anti-cancer effects. Thus, the potential effects of adiponectin deficiency in obesity and T2D on both metabolism and enhanced cancer growth may be explained by a number of mechanisms. Insulin resistance associated with reduced adiponectin may secondarily lead to enhanced LDLR expression in cancer cells and increased uptake of cholesterol, leading to enhanced cancer growth. This may partially explain the increased LDLR expression seen in triple negative breast cancers and the worse prognosis (Antalis et al. 2011; Rudling et al. 1986).
4.5 Inflammatory Cytokines
Both obesity and T2D have been labeled “inflammatory disorders.” Adipose tissue from obese individuals shows a marked increase in macrophage infiltration and dysregulation of these macrophages in the tumor environment, suggesting that local tumor-associated macrophages (TAM) may regulate tumor progression (Fig. 2). TAMs are recruited by monocyte chemotactic protein-1 (MCP-1) found in tumor tissues and are generally of the M1 macrophage phenotype that is pro-inflammatory. The switch from M2 (anti-inflammatory) to M1 macrophages is commonly seen in obese individuals and rodents. These macrophages are also capable of secreting pro-inflammatory factors that affect the adipose tissue and cancer cells leading to cancer progression. Thus, in the case of breast cancer, obese women have increased circulating levels of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6). Elevated levels of these cytokines are associated with increased cancer progression. Moreover, local breast adipose tissue may similarly affect tumor growth via cytokines. Reversal of these effects was seen with low-calorie diets, low-fat diets, and weight loss. Similarly, cyclooxygenase (COX) and prostaglandin E2 (PGE2) inhibitors may prove to be useful as they inhibit the elevated levels of COX-2 and PGE2 found in white adipose tissue inflammation (Howe et al. 2013). These drugs have additional relevant effects, including activation of adenosine monophosphate-activated protein kinase (AMPK, which inhibits mTOR) and nuclear factor-κB (NF-κB) antagonism that may be important in inhibiting the inflammatory processes.

Adipocyte and macrophage dysregulation favor tumor development
5 Metformin, Insulin, and Cancer
Metformin is a biguanide that has multiple biological properties, many of which may be beneficial in reducing cancer risk or cancer progression. Metformin reduces the activity of complex I in the respiratory chain in hepatocytes, causing energetic stress, which in turn activates liver kinase B1 (LKB1)/AMPK pathway and inhibits gluconeogenesis (Shaw et al. 2005). Reduced hepatic gluconeogenesis lowers blood glucose levels and, secondarily, circulating insulin concentrations. Since hyperinsulinemia has been shown to be associated with cancer and cancer-related mortality in obese and pre-diabetic individuals, this may be one explanation for metformin’s reduction of cancer risk and mortality in these situations, as demonstrated by epidemiological studies (Wang et al. 2014; Zhang et al. 2013). Other effects of metformin on adipokines and inflammatory cytokines were recently revealed. These include resistin and adiponectin (Gomez-Diaz et al. 2012; Singh et al. 2012). What effect these may have on insulin resistance and secondarily on cancer remains to be determined.
Effects of metformin on cancer may also be via direct mechanisms. Otto Warburg demonstrated increased glycolysis in cancer cells, also known as the Warburg effect (Warburg 1956); however, a significant amount of ATP is derived from oxidative phosphorylation, the latter being affected by metformin-induced stress and AMPK activation. AMPK, in turn, inhibits cancer cell growth by inhibiting fatty acid synthesis and mTOR-induced protein translation (Algire et al. 2010; Larsson et al. 2012). p53 mutations in certain cancers may make them more sensitive to biguanides, due to increased oxidative phosphorylation. Finally, metformin may play a role in inhibiting tumor-initiating cells that are commonly resistant to therapy (Zhu et al. 2014).
6 Conclusions
While the epidemiological documentation of a relationship between obesity, metabolic syndrome, T2D, and cancer risk and prognosis is becoming clearer, the causal factors remain to be defined. Specific animal models and human studies have identified strong contenders, such as hyperinsulinemia, hyperlipidemia, IGF-1, and leptin levels; the relevance of each of these may vary depending on the model. Identifying, quantifying, and proving which factors are of greater importance are critical if investigators wish to target molecules to be used as adjunct chemotherapy, especially for chemoresistant- or radiation-resistant cancers.
References
Adams TD, Stroup AM, Gress RE, Adams KF, Calle EE, Smith SC, Halverson RC, Simper SC, Hopkins PN, Hunt SC (2009) Cancer incidence and mortality after gastric bypass surgery. Obesity (Silver Spring) 17:796–802
Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M (2010) Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer 17:351–360
Alikhani N, Ferguson RD, Novosyadlyy R, Gallagher EJ, Scheinman EJ, Yakar S, LeRoith D (2013) Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene 32:961–967
Antalis CJ, Uchida A, Buhman KK, Siddiqui RA (2011) Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis 28:733–741
Arnlov J, Sundstrom J, Ingelsson E, Lind L (2011) Impact of BMI and the metabolic syndrome on the risk of diabetes in middle-aged men. Diabetes Care 34:61–65
Berentzen TL, Gamborg M, Holst C, Sorensen TI, Baker JL (2014) Body mass index in childhood and adult risk of primary liver cancer. J Hepatol 60:325–330
Blando J, Moore T, Hursting S, Jiang G, Saha A, Beltran L, Shen J, Repass J, Strom S, DiGiovanni J (2011) Dietary energy balance modulates prostate cancer progression in Hi-Myc mice. Cancer Prev Res (Phila) 4:2002–2014
Bonovas S, Filioussi K, Tsantes A (2004) Diabetes mellitus and risk of prostate cancer: a meta-analysis. Diabetologia 47:1071–1078
Borowsky AD, Namba R, Young LJ, Hunter KW, Hodgson JG, Tepper CG, McGoldrick ET, Muller WJ, Cardiff RD, Gregg JP (2005) Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis 22:47–59
Bruning PF, Bonfrer JM, van Noord PA, Hart AA, de Jong-Bakker M, Nooijen WJ (1992) Insulin resistance and breast-cancer risk. Int J Cancer 52:511–516
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348:1625–1638
Campbell MJ, Wollish WS, Lobo M, Esserman LJ (2002) Epithelial and fibroblast cell lines derived from a spontaneous mammary carcinoma in a MMTV/neu transgenic mouse. In Vitro Cell Dev Biol Anim 38:326–333
Cook S, Weitzman M, Auinger P, Nguyen M, Dietz WH (2003) Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med 157:821–827
Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, Wang H, Eckel RH (2008) The metabolic syndrome. Endocr Rev 29:777–822
Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ (2004) Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol 159:1160–1167
Crawford ED (2009) Understanding the epidemiology, natural history, and key pathways involved in prostate cancer. Urology 73:S4–S10
Del Prete A, Allavena P, Santoro G, Fumarulo R, Corsi MM, Mantovani A (2011) Molecular pathways in cancer-related inflammation. Biochem Med (Zagreb) 21:264–275
Djiogue S, Nwabo Kamdje AH, Vecchio L, Kipanyula MJ, Farahna M, Aldebasi Y, Seke Etet PF (2013) Insulin resistance and cancer: the role of insulin and IGFs. Endocr Relat Cancer 20:R1–R17
Dunlap SM, Chiao LJ, Nogueira L, Usary J, Perou CM, Varticovski L, Hursting SD (2012) Dietary energy balance modulates epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prev Res (Phila) 5:930–942
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA (2013) Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13:759–771
Ewens A, Mihich E, Ehrke MJ (2005) Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res 25:3905–3915
Ferguson RD, Gallagher EJ, Scheinman EJ, Damouni R, LeRoith D (2013) The epidemiology and molecular mechanisms linking obesity, diabetes, and cancer. Vitam Horm 93:51–98
Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D (2010a) Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes 59:686–693
Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D (2010b) Mammalian target of rapamycin inhibition abrogates insulin-mediated mammary tumor progression in type 2 diabetes. Endocr Relat Cancer 17:941–951
Folsom AR, Anderson KE, Sweeney C, Jacobs DR Jr (2004) Diabetes as a risk factor for death following endometrial cancer. Gynecol Oncol 94:740–745
Ford NA, Nunez NP, Holcomb VB, Hursting SD (2013) IGF1 dependence of dietary energy balance effects on murine Met1 mammary tumor progression, epithelial-to-mesenchymal transition, and chemokine expression. Endocr Relat Cancer 20:39–51
Franzese A, Vajro P, Argenziano A, Puzziello A, Iannucci MP, Saviano MC, Brunetti F, Rubino A (1997) Liver involvement in obese children. Ultrasonography and liver enzyme levels at diagnosis and during follow-up in an Italian population. Dig Dis Sci 42:1428–1432
Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, Inamori M, Nakajima N, Watanabe M, Kubota N, Yamauchi T, Kadowaki T, Wada K, Nakagama H, Nakajima A (2008) Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut 57:1531–1538
Gallagher EJ, Fierz Y, Vijayakumar A, Haddad N, Yakar S, LeRoith D (2012) Inhibiting PI3K reduces mammary tumor growth and induces hyperglycemia in a mouse model of insulin resistance and hyperinsulinemia. Oncogene 31:3213–3222
Gomez-Diaz RA, Talavera JO, Pool EC, Ortiz-Navarrete FV, Solorzano-Santos F, Mondragon-Gonzalez R, Valladares-Salgado A, Cruz M, Aguilar-Salinas CA, Wacher NH (2012) Metformin decreases plasma resistin concentrations in pediatric patients with impaired glucose tolerance: a placebo-controlled randomized clinical trial. Metabolism 61:1247–1255
Grossmann ME, Cleary MP (2012) The balance between leptin and adiponectin in the control of carcinogenesis - focus on mammary tumorigenesis. Biochimie 94:2164–2171
Gunter MJ, Hoover DR, Yu H, Wassertheil-Smoller S, Rohan TE, Manson JE, Li J, Ho GY, Xue X, Anderson GL, Kaplan RC, Harris TG, Howard BV, Wylie-Rosett J, Burk RD, Strickler HD (2009) Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J Natl Cancer Inst 101:48–60
Gunther EJ, Belka GK, Wertheim GB, Wang J, Hartman JL, Boxer RB, Chodosh LA (2002) A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. FASEB J 16:283–292
Guy CT, Cardiff RD, Muller WJ (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12:954–961
Hadsell DL, Greenberg NM, Fligger JM, Baumrucker CR, Rosen JM (1996) Targeted expression of des(1-3) human insulin-like growth factor I in transgenic mice influences mammary gland development and IGF-binding protein expression. Endocrinology 137:321–330
Haslam DW, James WP (2005) Obesity. Lancet 366:1197–1209
Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ (2013) Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res 19:6074–6083
Huang Z, Willett WC, Colditz GA, Hunter DJ, Manson JE, Rosner B, Speizer FE, Hankinson SE (1999) Waist circumference, waist:hip ratio, and risk of breast cancer in the Nurses' Health Study. Am J Epidemiol 150:1316–1324
Imamura N, Yanagihara K, Kusunoki Y (1984) Tumor-associated antigen of spontaneous mammary tumor in rats. Oncology 41:206–209
Isaksson B, Strommer L, Friess H, Buchler MW, Herrington MK, Wang F, Zierath JR, Wallberg-Henriksson H, Larsson J, Permert J (2003) Impaired insulin action on phosphatidylinositol 3-kinase activity and glucose transport in skeletal muscle of pancreatic cancer patients. Pancreas 26:173–177
Jaggers JR, Sui X, Hooker SP, LaMonte MJ, Matthews CE, Hand GA, Blair SN (2009) Metabolic syndrome and risk of cancer mortality in men. Eur J Cancer 45:1831–1838
Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ (2007) Cancer statistics, 2007. CA Cancer J Clin 57:43–66
Kasper JS, Giovannucci E (2006) A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev 15:2056–2062
Kawashima Y, Chen J, Sun H, Lann D, Hajjar RJ, Yakar S, Leroith D (2009) Apolipoprotein E deficiency abrogates insulin resistance in a mouse model of type 2 diabetes mellitus. Diabetologia 52:1434–1441
Kelesidis I, Kelesidis T, Mantzoros CS (2006) Adiponectin and cancer: a systematic review. Br J Cancer 94:1221–1225
Kim H, Pennisi PA, Gavrilova O, Pack S, Jou W, Setser-Portas J, East-Palmer J, Tang Y, Manganiello VC, Leroith D (2006) Effect of adipocyte beta3-adrenergic receptor activation on the type 2 diabetic MKR mice. Am J Physiol Endocrinol Metab 290:E1227–E1236
Körner A, Kratzsch J, Kiess W (2006) Effect of adiponectin on proliferation of breast cancer cells. Exp Clin Endocrinol Diabetes 114:P03_043
Kushiro K, Nunez NP (2011) Ob/ob serum promotes a mesenchymal cell phenotype in B16BL6 melanoma cells. Clin Exp Metastasis 28:877–886
Lanari C, Lamb CA, Fabris VT, Helguero LA, Soldati R, Bottino MC, Giulianelli S, Cerliani JP, Wargon V, Molinolo A (2009) The MPA mouse breast cancer model: evidence for a role of progesterone receptors in breast cancer. Endocr Relat Cancer 16:333–350
Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014:149185
Larsson O, Morita M, Topisirovic I, Alain T, Blouin MJ, Pollak M, Sonenberg N (2012) Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc Natl Acad Sci U S A 109:8977–8982
Lashinger LM, Harrison LM, Rasmussen AJ, Logsdon CD, Fischer SM, McArthur MJ, Hursting SD (2013) Dietary energy balance modulation of Kras- and Ink4a/Arf+/–driven pancreatic cancer: the role of insulin-like growth factor-I. Cancer Prev Res (Phila) 6:1046–1055
Lee AV, Yee D (1995) Insulin-like growth factors and breast cancer. Biomed Pharmacother 49:415–421
Levine AJ, Feng Z, Mak TW, You H, Jin S (2006) Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev 20:267–275
Liu J, Xu A, Lam KS, Wong NS, Chen J, Shepherd PR, Wang Y (2013) Cholesterol-induced mammary tumorigenesis is enhanced by adiponectin deficiency: role of LDL receptor upregulation. Oncotarget 4:1804–1818
Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, Gaziano JM, Pollak M, Stampfer MJ (2008) Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol 9:1039–1047
McHenry PR, Sears JC, Herrick MP, Chang P, Heckman-Stoddard BM, Rybarczyk M, Chodosh LA, Gunther EJ, Hilsenbeck SG, Rosen JM, Vargo-Gogola T (2010) P190B RhoGAP has pro-tumorigenic functions during MMTV-Neu mammary tumorigenesis and metastasis. Breast Cancer Res 12:R73
McNamara KM, Sasano H (2015) The intracrinology of breast cancer. J Steroid Biochem Mol Biol. Ahead of print 145:172–178
Michels KB, Solomon CG, Hu FB, Rosner BA, Hankinson SE, Colditz GA, Manson JE, Nurses’ Health S (2003) Type 2 diabetes and subsequent incidence of breast cancer in the Nurses’ Health Study. Diabetes Care 26:1752–1758
Middleton PJ (1965) The histogenesis of mammary tumours induced in the rat by chemical carcinogens. Br J Cancer 19:830–839
Moon HS, Liu X, Nagel JM, Chamberland JP, Diakopoulos KN, Brinkoetter MT, Hatziapostolou M, Wu Y, Robson SC, Iliopoulos D, Mantzoros CS (2013) Salutary effects of adiponectin on colon cancer: in vivo and in vitro studies in mice. Gut 62:561–570
Moore T, Beltran L, Carbajal S, Hursting SD, DiGiovanni J (2012) Energy balance modulates mouse skin tumor promotion through altered IGF-1R and EGFR crosstalk. Cancer Prev Res (Phila) 5:1236–1246
Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P (1988) Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54:105–115
Nalabolu MR, Palasamudram K, Jamil K (2014) Adiponectin and leptin molecular actions and clinical significance in breast cancer. Int J Hematol Oncol Stem Cell Res 8:31–40
Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, Umetani M, Geradts J, McDonnell DP (2013) 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342:1094–1098
Nielsen SF, Nordestgaard BG, Bojesen SE (2012) Statin use and reduced cancer-related mortality. N Engl J Med 367:1792–1802
Noto H, Goto A, Tsujimoto T, Osame K, Noda M (2013) Latest insights into the risk of cancer in diabetes. J Diabetes Investig 4:225–232
Novosyadlyy R, LeRoith D (2010) Hyperinsulinemia and type 2 diabetes: impact on cancer. Cell Cycle 9:1449–1450
Ntikoudi E, Kiagia M, Boura P, Syrigos KN (2014) Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat Rev 40:22–30
Olivo-Marston SE, Hursting SD, Perkins SN, Schetter A, Khan M, Croce C, Harris CC, Lavigne J (2014) Effects of calorie restriction and diet-induced obesity on murine colon carcinogenesis, growth and inflammatory factors, and microRNA expression. PLoS One 9:e94765
Park J, Scherer PE (2011) Leptin and cancer: from cancer stem cells to metastasis. Endocr Relat Cancer 18:C25–C29
Park MH, Falconer C, Viner RM, Kinra S (2012) The impact of childhood obesity on morbidity and mortality in adulthood: a systematic review. Obes Rev 13:985–1000
Pei XF, Noble MS, Davoli MA, Rosfjord E, Tilli MT, Furth PA, Russell R, Johnson MD, Dickson RB (2004) Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In Vitro Cell Dev Biol Anim 40:14–21
Pelton K, Freeman MR, Solomon KR (2012) Cholesterol and prostate cancer. Curr Opin Pharmacol 12:751–759
Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Pour PM, Westermark P, Adrian TE (1994) Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med 330:313–318
Petrelli JM, Calle EE, Rodriguez C, Thun MJ (2002) Body mass index, height, and postmenopausal breast cancer mortality in a prospective cohort of US women. Cancer Causes Control 13:325–332
Pierobon M, Frankenfeld CL (2013) Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat 137:307–314
Pisani P (2008) Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem 114:63–70
Pothiwala P, Jain SK, Yaturu S (2009) Metabolic syndrome and cancer. Metab Syndr Relat Disord 7:279–288
Prentice RL, Caan B, Chlebowski RT, Patterson R, Kuller LH, Ockene JK, Margolis KL, Limacher MC, Manson JE, Parker LM, Paskett E, Phillips L, Robbins J, Rossouw JE, Sarto GE, Shikany JM, Stefanick ML, Thomson CA, Van Horn L, Vitolins MZ, Wactawski-Wende J, Wallace RB, Wassertheil-Smoller S, Whitlock E, Yano K, Adams-Campbell L, Anderson GL, Assaf AR, Beresford SA, Black HR, Brunner RL, Brzyski RG, Ford L, Gass M, Hays J, Heber D, Heiss G, Hendrix SL, Hsia J, Hubbell FA, Jackson RD, Johnson KC, Kotchen JM, LaCroix AZ, Lane DS, Langer RD, Lasser NL, Henderson MM (2006) Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 295:629–642
Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D, Million Women Study C (2007) Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ 335:1134
Rizner TL (2013) Estrogen biosynthesis, phase I and phase II metabolism, and action in endometrial cancer. Mol Cell Endocrinol 381:124–139
Robinson WR, Tse CK, Olshan AF, Troester MA (2014) Body size across the life course and risk of premenopausal and postmenopausal breast cancer in Black women, the Carolina Breast Cancer Study, 1993–2001. Cancer Causes Control. Ahead of print 25(9):1101–1117
Rondini EA, Harvey AE, Steibel JP, Hursting SD, Fenton JI (2011) Energy balance modulates colon tumor growth: Interactive roles of insulin and estrogen. Mol Carcinog 50:370–382
Rostoker R, Bitton-Worms K, Caspi A, Shen-Orr Z, LeRoith D (2013) Investigating new therapeutic strategies targeting hyperinsulinemia’s mitogenic effects in a female mouse breast cancer model. Endocrinology 154:1701–1710
Rousseau MC, Parent ME, Pollak MN, Siemiatycki J (2006) Diabetes mellitus and cancer risk in a population-based case-control study among men from Montreal, Canada. Int J Cancer 118:2105–2109
Rudling MJ, Stahle L, Peterson CO, Skoog L (1986) Content of low density lipoprotein receptors in breast cancer tissue related to survival of patients. Br Med J (Clin Res Ed) 292:580–582
Sandgren EP, Schroeder JA, Qui TH, Palmiter RD, Brinster RL, Lee DC (1995) Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res 55:3915–3927
Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C (2006) Prevalence of fatty liver in children and adolescents. Pediatrics 118:1388–1393
Sciacca L, Vigneri R, Tumminia A, Frasca F, Squatrito S, Frittitta L, Vigneri P (2013) Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis 23:808–815
Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646
Singh S, Akhtar N, Ahmad J (2012) Plasma adiponectin levels in women with polycystic ovary syndrome: impact of metformin treatment in a case-control study. Diabetes Metab Syndr 6:207–211
Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P (1987) Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49:465–475
Sirotnak FM, DeGraw JI, Schmid FA, Goutas LJ, Moccio DM (1984) New folate analogs of the 10-deaza-aminopterin series. Further evidence for markedly increased antitumor efficacy compared with methotrexate in ascitic and solid murine tumor models. Cancer Chemother Pharmacol 12:26–30
Sjostrom L, Gummesson A, Sjostrom CD, Narbro K, Peltonen M, Wedel H, Bengtsson C, Bouchard C, Carlsson B, Dahlgren S, Jacobson P, Karason K, Karlsson J, Larsson B, Lindroos AK, Lonroth H, Naslund I, Olbers T, Stenlof K, Torgerson J, Carlsson LM (2009) Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol 10:653–662
Somasundar P, Yu AK, Vona-Davis L, McFadden DW (2003) Differential effects of leptin on cancer in vitro. J Surg Res 113:50–55
Stewart TA, Pattengale PK, Leder P (1984) Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 38:627–637
Sturmer T, Buring JE, Lee IM, Gaziano JM, Glynn RJ (2006) Metabolic abnormalities and risk for colorectal cancer in the physicians’ health study. Cancer Epidemiol Biomarkers Prev 15:2391–2397
Tseng CH, Tseng FH (2014) Diabetes and gastric cancer: the potential links. World J Gastroenterol 20:1701–1711
Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE (1988) Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 55:619–625
Tzeng YJ, Guhl E, Graessmann M, Graessmann A (1993) Breast cancer formation in transgenic animals induced by the whey acidic protein SV40 T antigen (WAP-SV-T) hybrid gene. Oncogene 8:1965–1971
Uddin S, Hussain AR, Khan OS, Al-Kuraya KS (2014) Role of dysregulated expression of leptin and leptin receptors in colorectal carcinogenesis. Tumour Biol 35:871–879
Van Itallie TB (1985) Health implications of overweight and obesity in the United States. Ann Intern Med 103:983–988
Vansaun MN (2013) Molecular pathways: adiponectin and leptin signaling in cancer. Clin Cancer Res 19:1926–1932
Verheus M, Peeters PH, Rinaldi S, Dossus L, Biessy C, Olsen A, Tjonneland A, Overvad K, Jeppesen M, Clavel-Chapelon F, Tehard B, Nagel G, Linseisen J, Boeing H, Lahmann PH, Arvaniti A, Psaltopoulou T, Trichopoulou A, Palli D, Tumino R, Panico S, Sacerdote C, Sieri S, van Gils CH, Bueno-de-Mesquita BH, Gonzalez CA, Ardanaz E, Larranaga N, Garcia CM, Navarro C, Quiros JR, Key T, Allen N, Bingham S, Khaw KT, Slimani N, Riboli E, Kaaks R (2006) Serum C-peptide levels and breast cancer risk: results from the European Prospective Investigation into Cancer and Nutrition (EPIC). Int J Cancer 119:659–667
Verlato G, Zoppini G, Bonora E, Muggeo M (2003) Mortality from site-specific malignancies in type 2 diabetic patients from Verona. Diabetes Care 26:1047–1051
Wang L, Di LJ (2014) BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact? Int J Biol Sci 10:563–573
Wang Z, Lai ST, Xie L, Zhao JD, Ma NY, Zhu J, Ren ZG, Jiang GL (2014) Metformin is associated with reduced risk of pancreatic cancer in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract 106(1):19–26
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Weiderpass E, Gridley G, Persson I, Nyren O, Ekbom A, Adami HO (1997) Risk of endometrial and breast cancer in patients with diabetes mellitus. Int J Cancer 71:360–363
Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW, Allen K, Lopes M, Savoye M, Morrison J, Sherwin RS, Caprio S (2004) Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 350:2362–2374
Williams JC, Gusterson B, Humphreys J, Monaghan P, Coombes RC, Rudland P, Neville AM (1981) N-methyl-N-nitrosourea-induced rat mammary tumors. Hormone responsiveness but lack of spontaneous metastasis. J Natl Cancer Inst 66:147–155
Xu CX, Zhu HH, Zhu YM (2014) Diabetes and cancer: associations, mechanisms, and implications for medical practice. World J Diabetes 5:372–380
Yakar S, Nunez NP, Pennisi P, Brodt P, Sun H, Fallavollita L, Zhao H, Scavo L, Novosyadlyy R, Kurshan N, Stannard B, East-Palmer J, Smith NC, Perkins SN, Fuchs-Young R, Barrett JC, Hursting SD, LeRoith D (2006) Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology 147:5826–5834
Yancik R, Ganz PA, Varricchio CG, Conley B (2001a) Perspectives on comorbidity and cancer in older patients: approaches to expand the knowledge base. J Clin Oncol 19:1147–1151
Yancik R, Wesley MN, Ries LA, Havlik RJ, Edwards BK, Yates JW (2001b) Effect of age and comorbidity in postmenopausal breast cancer patients aged 55 years and older. JAMA 285:885–892
Yang YX, Hennessy S, Lewis JD (2005) Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol 3:587–594
Zhang P, Li H, Tan X, Chen L, Wang S (2013) Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol 37:207–218
Zhu P, Davis M, Blackwelder AJ, Bachman N, Liu B, Edgerton S, Williams LL, Thor AD, Yang X (2014) Metformin selectively targets tumor-initiating cells in ErbB2-overexpressing breast cancer models. Cancer Prev Res (Phila) 7:199–210
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ben-Shmuel, S., Rostoker, R., Scheinman, E.J., LeRoith, D. (2015). Metabolic Syndrome, Type 2 Diabetes, and Cancer: Epidemiology and Potential Mechanisms. In: Herzig, S. (eds) Metabolic Control. Handbook of Experimental Pharmacology, vol 233. Springer, Cham. https://doi.org/10.1007/164_2015_12
Download citation
DOI: https://doi.org/10.1007/164_2015_12
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-29804-7
Online ISBN: 978-3-319-29806-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)