
Abstract
Oxidative stress is caused by an imbalance of reactive oxygen species (ROS)/reactive nitrogen species (RNS) and the antioxidative stress defense systems in cells. ROS/RNS or carcinogen metabolites can attack intracellular proteins, lipids, and nucleic acids, which can result in genetic mutations, carcinogenesis, and other diseases. Nrf2 plays a critical role in the regulation of many antioxidative stress/antioxidant and detoxification enzyme genes, such as glutathione S-transferases (GSTs), NAD(P)H:quinone oxidoreductase 1 (NQO1), UDP-glucuronyl transferases (UGTs), and heme oxygenase-1 (HO-1), directly via the antioxidant response element (ARE). Recently, many studies have shown that dietary phytochemicals possess cancer chemopreventive potential through the induction of Nrf2-mediated antioxidant/detoxification enzymes and anti-inflammatory signaling pathways to protect organisms against cellular damage caused by oxidative stress. In addition, carcinogenesis can be caused by epigenetic alterations such as DNA methylation and histone modifications in tumor–suppressor genes and oncogenes. Interestingly, recent studies have shown that several naturally occurring dietary phytochemicals can epigenetically modify the chromatin, including reactivating Nrf2 via demethylation of CpG islands and the inhibition of histone deacetylases (HDACs) and/or histone acetyltransferases (HATs). The advancement and development of dietary phytochemicals in cancer chemoprevention research requires the integration of the known, and as-yet-unknown, compounds with the Nrf2-mediated antioxidant, detoxification, and anti-inflammatory systems and their in vitro and in vivo epigenetic mechanisms; human clinical efficacy studies must also be performed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
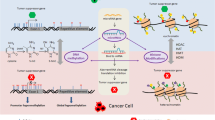
1 Introduction
Cancer chemoprevention is a major cancer preventive strategy that utilizes naturally occurring dietary phytochemicals or therapeutic drugs with relatively low toxicity. Phytochemicals, along with physical activity and mental relaxation, can inhibit, retard, or reverse carcinogenesis. With the advent of modern technology and instrumentation, many studies on dietary phytochemicals have been performed, including studies on their chemistry, biological activities, and mechanisms of action at the cellular level, in in vivo animal model systems, and in clinical trials. Carcinogenic species, such as environmental pollutants, dietary mutagens and radiation, may result in the production of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS), which further react with cellular molecules such as proteins, lipids, and DNA to induce carcinogenesis. Dietary phytochemicals not only directly scavenge ROS/RNS but also indirectly remove carcinogenic reactive intermediates via the transcription factor Nrf2 [nuclear factor erythroid 2 p45 (NF-E2)-related factor 2] antioxidant and detoxification system. When Nrf2 is released from Kelch-like ECH associated protein 1 (Keap1) and translocates to the nucleus, Nrf2 binds to antioxidant responsive elements (AREs) in the promoter/enhancer region of phase II detoxification and antioxidant enzyme genes with the Maf protein. Recent research has also shown that the reactivation of Nrf2 might be regulated by dietary phytochemicals through epigenetic modifications such as DNA methylation and histone modification. In this review we will summarize the correlations among oxidative stress, Nrf2 and cancer. The cancer chemopreventive effects of dietary phytochemicals on the activation of Nrf2-mediated antioxidant, detoxification and anti-inflammatory systems through Nrf2–Keap 1 and epigenetic pathways will also be discussed with regard to their roles in blocking the initiation of carcinogenesis.
2 Oxidative Stress and Cancer
2.1 Oxidative Stress
Free radicals are molecules or molecular fragments containing one or more unpaired electrons. The human body is under attack from free radicals, including superoxide (\( {\text{O}}_2^{ - }\bullet \)), nitric oxide (NO) and hydroxyl ions (\( {\text{OH}}\bullet \)) [1]. Hydrogen peroxide, superoxide, and hydroxyl radicals are more generally known as ROS generated as byproducts of the metabolism of oxygen, whereas nitrite, nitrate, and peroxynitrite, referred to as RNS, are generated as the products of NO metabolism [2]. ROS/RNS are generated through various processes, including mitochondria-catalyzed electron transport reactions, UV irradiation, X-rays and gamma rays, inflammatory processes, lipid peroxidation (LPO), and environmental pollutants [3].
Oxidative stress is an imbalance between the generation of ROS/RNS and the antioxidative stress defense systems [4, 5]. Cumulatively produced ROS/RNS in the body induce a cellular redox imbalance and subsequent biomolecular damage. Oxidative stress is a common pathogenic mechanism in aging and the development of various types of cancers and neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [6, 7].
2.2 Oxidative Stress and Cancer
Reactive species are well recognized for playing a dual role as both deleterious and beneficial species. ROS/RNS are important intracellular signaling molecules that play key roles in various physiological processes, including apoptosis [8]. ROS/RNS can regulate Bcl-2 expression levels, thereby impacting the function of Bcl-2 to induce cell death through the necrotic or apoptotic pathway [8]. Apoptotic regulation involves receptor activation, a change in the expression levels of the Bcl-2 family of proteins, caspase activation, and mitochondrial dysfunction [9]. C-Jun N-terminal kinase (JNK), or stress-activated protein kinase (SAPK), members of the mitogen-activated protein kinase superfamily (MAPK), are also involved in ROS/RNS-mediated cell death [10]. When at low to moderate concentrations, ROS may induce cellular senescence and apoptosis and play a beneficial physiological role as antitumorigenic species [11, 12]. However, ROS act as second messengers in signal-transduction pathways [13] and are considered to be important mediators of damage to cell structures, including lipids and membranes, proteins, and DNA [14].
Increased levels of reactive species are associated with oncogenic stimulation, and oxidative stress can be considered an important class of carcinogen [11]. Chronic inflammation is associated with an increased risk of various types of human cancers, and inflammation is associated with the induction of oxidative/nitrosative stress and LPO, which generate excess ROS/RNS and DNA-reactive aldehydes [15]. Cancer development is characterized by the cumulative action of multiple events in a single cell with initiation, promotion, and progression stages; the ROS are involved in all stages [16].
The initiation stage involves a non-lethal mutation in DNA [17]. Both ROS and RNS have been shown to be involved in DNA damage [18, 19]. The DNA mutations caused by reactive species include point mutations, deletions, insertions, chromosomal translocations, crosslinks, and other modifications. An early study demonstrated that DNA alterations by oxidative stress through 8-hydroxyguanine (8-OH-G) mutations, which may arise from the formation of 8-OH-dG, involve the GC → TA transversion [17]. This type of modified DNA is relatively easily formed, is mutagenic and carcinogenic, and can be used as a potential biomarker of carcinogenesis [20]. Direct DNA damage or genomic instability coupled with altered gene expression and changes in protein conformation occur simultaneously in cancer development [12].
The promotion stage is characterized by the clonal expansion of initiated cells by the induction of cell proliferation and the failure to induce cell death. A high level of oxidative stress is cytotoxic and induces cell apoptosis or necrosis. However, if the oxidative stress is present continuously at a relatively low level, cell division and subsequent tumor growth is stimulated [21]. Progression is an irreversible stage of the carcinogenic process. Further genetic damage and the disruption of chromosome integrity occur at this stage, corresponding to a cell transition from benign to malignant [21, 22].
2.3 The Antioxidant Defense System in Carcinogenesis
Antioxidants may be characterized as acting either through the inhibition of ROS generation or through the direct scavenging of free radicals [12, 23]. In living organisms the effects of ROS/RNS are balanced by the antioxidant action, which is composed of both enzymatic and nonenzymatic antioxidants. Antioxidants directly remove free radicals and maintain the intracellular redox status [24].
The nonenzymatic antioxidants include vitamin C (l-ascorbate), vitamin E, carotenoids, selenium, flavonoids, and thiol antioxidants such as glutathione, thioredoxin (Txn), and lipoic acid. [11, 17, 25]. Vitamin C is a water-soluble antioxidant and an enzyme cofactor present in plants and some animals. Humans must obtain vitamin C through the diet because of the inability to synthesize this nutrient endogenously. There are two chemical forms of vitamin C: the reduced form (ascorbic acid, AA) and the oxidized form (dehydroascorbic acid, DHA). Reduced AA is the more predominant chemical structure in the human body, and it is a potent antioxidant that efficiently quenches damaging free radicals. Many in vivo studies have shown a beneficial role of vitamin C in cancer prevention and treatment [26]. However, at high concentrations, vitamin C also serves as a pro-oxidant promoting ROS levels [26]. Vitamin C can also cooperate with vitamin E to regenerate alpha-tocopherol radicals in membranes and lipoproteins [27]. Vitamin E is a fat-soluble vitamin that exists in eight different forms, and this vitamin also serves as both an anti- and a pro-oxidant via different mechanisms [26].
The enzymatic antioxidants include superoxide dismutases (SODs), catalase, and glutathione peroxidases (GPxs) [27]. SODs are the major antioxidant defense systems against \( {\text{O}}_2^{ - }\bullet \) and consist of three isoforms in mammals: SOD1 (the cytoplasmic Cu/ZnSOD), SOD2 (the mitochondrial MnSOD), and SOD3 (the extracellular Cu/ZnSOD). All of the SOD isoforms require a catalytic metal (Cu or Mn) for activation [28]. Catalase is an enzyme that degrades hydrogen peroxide, reducing H2O2 to water and oxidizing it to molecular oxygen [29]. Glutathione S-transferases (GSTs) and GPxs are important in the defense against free-radical-induced oxidative damage [30, 31].
The thiol-containing small molecules, such as glutathione (GSH), are major intracellular antioxidants. γ-Glutamyl cysteine synthase (γGCS), including the glutamate cysteine ligase (Gcl), catalytic (Gclc), and modifier (Gclm) subunits, is essential for the biosynthesis of GSH. Some small thiol-containing compounds, such as Txn, glutaredoxins, and periredoxins, undergo rapid oxidization and regeneration and serve as substrates for antioxidant enzymes [24]. In addition to the above-described antioxidant enzymes (SODs, catalase, and GPxs), which inactivate ROS/RNS directly, the antioxidant system also includes enzymes such as glutathione reductase (GSR), NAD(P)H:quinone oxidoreductase 1 (NQO1), UDP-glucuronyl transferases (UGTs), and thioredoxin reductase (Txnd), sulfiredoxin (Srx), and GSTs, which recycle thiols or facilitate the excretion of oxidized and reactive secondary metabolites (e.g., quinones, epoxides, aldehydes, and peroxides) through reduction/conjugation reactions. In antioxidant systems there are other stress response proteins, such as heme oxygenase-1 (HO-1) and -2 (HO-2), metallothionines, and heat shock proteins that also provide cellular protection against various oxidant or pro-oxidant insults [24].
2.4 Antioxidant Gene Regulation and the Antioxidant Response Element
Most of the antioxidant genes listed above contain cis-acting antioxidant response elements (AREs) with a functional consensus sequence of 5′-RTGAYnnnGCR-3′ (where R = A or G and Y = C or T) [32]. The AREs have been widely used to screen for potential inducers of antioxidant enzymes [12, 32]. At the transcription level, the antioxidant enzymes are largely regulated by the binding of a particular transcription factor known as nuclear factor erythroid 2p45 (NF-E2)-related factor 2 (Nrf2) to the ARE [33, 34]. Nrf2 was first isolated in 1994 from a hemin-induced K562 erythroid cell line belonging to the basic leucine zipper nuclear transcription factor family, which share regions of homology with that of the Drosophila cap “n” collar (CNC) protein [35, 36]. The human Nrf2 showed a high sequence homology to the known p45 subunit of nuclear factor erythroid 2 (NF-E2) [35, 36]. The importance of Nrf2 was demonstrated with Nrf2-knockout mice, which were found to contain lower levels of detoxifying enzymes than wild-type mice and were susceptible to xenobiotics and environmental poisons [37, 38].
Nrf2 activity is mainly regulated by Kelch-like ECH-associated protein 1 (Keap1), a homolog of the Drosophila actin-binding protein Kelch, which binds to the actin cytoskeleton. Under homeostatic conditions, Nrf2 is mainly retained in the cytosol by the Keap1 protein [39]. Upon a challenge by oxidative or chemical stress, Nrf2 can be released from the Keap 1 sequestration and translocates to the nucleus [39, 40]. In the nucleus, Nrf2 selectively heterodimerizes with Maf, activation transcription factor (ATF), and/or members of the AP-1 family of leucine zipper proteins to trigger the transcription of its target genes [41, 42].
2.5 The Regulation of Nrf2 Activation
The MAPKs include extracellular signal-regulated kinases (ERKs), JNK, and protein 38 (p38). The MAPK cascade, protein kinase C (PKC), and phosphatidylinositol 3-kinase (PI3K) are involved in the activation of Nrf2–Keap1 with significant cross talk. Numerous studies have revealed that ERK and JNK have a positive effect on ARE-mediated activities [12, 43, 44] and that the phosphorylation of Nrf2 by p38 may inhibit Nrf2 activation by increasing Keap1/Nrf2 binding [45]. Nrf2 can be directly phosphorylated by PKC at serine 40 [46–49], and PI3K signaling facilitates Nrf2 nuclear translocation [50–53]. The direct phosphorylation of Nrf2 by MAPKs, however, has only a slight effect on Nrf2 translocation and activity [54]. However, recent evidence suggests that oxidative stress-mediated post-transcriptional control of Nrf2 activation may also play a role in the regulation of Nrf2 activation [23, 55].
2.6 Cancer Chemoprevention by Dietary Phytochemicals
Phytochemicals from dietary plants and medicinal herbs are becoming increasingly important factors in cancer chemoprevention or adjuvant chemotherapy because many of these plants exhibit effects on cell death and intracellular redox status modulation [40]. Many flavonoids and polyphenolic antioxidants, such as catechins, epigallocatechin gallate (EGCG), and curcumin, exert their anti-inflammatory and antioxidative effects through phase II detoxification/antioxidant enzymes that are mediated by integrated Nrf2 [12, 25, 56, 57]. One phytochemical compound may act on multiple pathways. For example, curcumin has an anti-inflammatory effect by inhibiting NF-κB by blocking IκB degradation. Curcumin has also been shown to regulate the antioxidant response by inhibiting the phosphorylation of Akt and ERK [58, 59]. In addition, curcumin regulates cell death by decreasing the expression levels of tumor necrosis factor-α and endogenous Bcl-2 and Bcl-xL [60, 61]. EGCG has been shown to have multiple effects on the cell cycle and on anti-inflammatory and anticancer regulation through the modulation of NF-κB, COX-2, DNA methyl transferase 1 (DNMT1), ERK-1/2, p38, and matrix metalloproteinase-2 (MMP2) [62–64].
3 Nrf2-Mediated Antioxidant and Detoxification Systems and Anti-inflammation and Cancer Prevention
Oxidative stress results in various pathological conditions and diseases such as inflammation and cancer because oxidative stress causes biochemical alterations in cellular components such as proteins, nucleic acids, and lipids [14]. Oxidative stress is caused by the imbalance between ROS formation and cellular antioxidant capacity. The antioxidant system in cells mitigates the toxic attack and ROS potential. Thiol-containing small molecules, such as GSH and Txn, which belong to the nonenzymatic antioxidant system, can eliminate ROS directly [65]. Enzymes such as catalase, GPx, and peroxiredoxins (Prdx) can remove ROS via catalytic reactions accompanied by GSH or Txn [66, 67].
Xenobiotics come from various drugs, carcinogens and environmental chemicals, and they are typically converted into intermediate molecules that may contain nucleophilic or electrophilic groups through the catalytic action of phase I enzymes such as cytochrome P450 enzymes [68, 69]. Some xenobiotic metabolites may possess toxic or carcinogenesis potentials, and the induction of oxidative stress may be one of the inducible phenomena. However, most if not all hydrophobic xenobiotic metabolites are eliminated after conjugation with hydrophilic molecules such as GSH and glucuronic acid by phase II detoxification and antioxidant enzymes [70].
Nrf2 is a crucial regulator in the induction of the phase II antioxidant and detoxification enzyme genes, which protect cells from damage resulting from oxidative and electrophilic attack [71, 72]. Therefore, dietary phytochemicals will be indirect antioxidants that improve cellular antioxidant capacity by enhancing the gene expression of phase II antioxidant and detoxification enzymes via the Nrf2 pathway.
3.1 Nrf2 and the Antioxidant and Detoxification Systems
The principal phase II antioxidant and detoxification enzymes include the classical conjugating enzymes such as GSTs and UGTs, reduction enzymes such as NQOs, and stress response enzymes such as HO-1 [67, 73]. Many phase II antioxidant and detoxification genes are regulated through the ARE in the promoter [74]. Nrf2 has been demonstrated in extensive studies to be an essential transcription factor for the regulation of the ARE [42, 75–77]. Nrf2 that has translocated from the cytoplasm to the nucleus interacts with other bZIP transcription factor partners, such as small Maf proteins (Maf F, Maf G, and Maf K) and ATF4, and transactivates AREs [78–81]. Many chemicals induce the expression of ARE-driven genes through the translocation of Nrf2, including phenolic antioxidants, such as BHA and tert-butyl hydroxyquinone (tBHQ); isothiocyanates, such as sulforaphane (SFN) and PEITC; and synthetic triterpenoids, such as oleanane [82–88].
GSTs have seven distinct classes based on amino-acid sequences, the physical structure of the genes and immunological cross-reactivity; these classes include alpha (α), mu (μ), omega (ω), pi (π), sigma (σ), theta (θ), and zeta (ζ) [89]. GSTs scavenge endogenous and exogenous electrophiles, such as epoxides, aldehydes, and peroxides, in cells [89]. A number of studies have demonstrated that Nrf2 plays a crucial role in the regulation of GSTs. Nrf2 induces significant changes in the mRNA expression levels of many subtypes of mouse hepatic GSTs [75]. GST mRNA and protein expression levels are decreased in Nrf2-KO mice compared with wild-type mice, and elevated Nrf2 activation in the liver resulted in a marked increase of GST mRNA expression in Keap1-knockdown mice [75, 90]. Chemopreventive synthetic antioxidants, such as butylated hydroxyanisole (BHA) and ethoxyquin, increased the expression of GSTs in the mouse liver through Nrf2 induction [91]. In addition, lithocholic acid, the most toxic bile acid, has been shown to increase hepatic glutathione and GST activity in wild-type mice compared with Nrf2-KO mice [92].
UGTs are important enzymes for the excretion of water-soluble glucuronides transformed from toxic exogenous (such as drugs, pesticides, and carcinogens) and endogenous (such as bilirubin, steroids, and hormones) compounds through a conjugation reaction [93]. UGTs play a critical protective role against environmental chemicals and carcinogens. For example, UGT-deficient cultured rat skin fibroblast is more susceptible to B[a]P carcinogenesis [94]. The reduction of DMBA–DNA adduct formation was found in breast cancer cells with elevated UGT1A1 [95]. It has also been found that tBHQ induces the UGT1A1 mRNA level and enzyme activity in the liver and intestine in UGT1A transgenic mice [96]. Lower basal mRNA expression levels of UGTs such as UGT1A6, UGT1A9, UGT2B34, UGT2B35, and UGT2B36 were observed in Nrf2-knockout mice compared with wild-type mice [86, 97, 98]. It has been demonstrated that Nrf2 up-regulates UGT activity and promotes a conjugation reaction of 4-aminobiphenyl (ABP) from tobacco smoke with glucuronic acid in the liver, which might protect the liver against ABP [99]. The GST activity was reduced in the liver and small intestine of Nrf2 KO mice, and oltipraz, a chemopreventive agent, does not affect the expression levels of these enzymes in Nrf2-KO mice compared with wild-type mice [100].
NQO1 is a cytosolic flavoprotein and facilitates the detoxification and excretion of endogenous and exogenous chemicals through a reduction reaction from quinones to hydroquinones [101, 102]. It has been reported that the disruption of NQO1 contributed to a higher susceptibility to B[a]P-induced skin carcinogenesis in mice [103]. Lower Nqo1 expression and activity were found in the liver, small intestine, and forestomach of Nrf2-KO mice [75, 99, 100]. Early carcinogenesis induced by cyclophosphamide, which causes oxidative stress in the rat liver, can be effectively inhibited by the powerful antioxidant astaxanthin accompanied by an increase in NQO-1 and HO-1 as mediated through the Nrf2-ARE pathway [104]. The lycopene metabolite apo-8′-lycopenal induced the accumulation of nuclear Nrf2, which resulted in an increase in HO-1 and NQO-1 expression levels in human hepatoma HepG2 cells [105]. In addition, NQO1 mRNA and protein expression levels can be increased by curcumin as mediated by restoring Nrf2 expression through DNA demethylation on Nrf2 promoter CpG islands [106].
HO-1 exhibits both antioxidative and anti-inflammatory capacities. HO-1 catalyzes the catabolism of the pro-oxidant heme to produce bilirubin and carbon monoxide, which have antioxidative and anti-inflammatory effects, respectively [107–109]. HO-1 mRNA and protein expression levels are induced when cells are exposed to oxidative stress that results in cellular injury [110], and Nrf2 is a critical transcription factor that regulates the induction of the HO-1 gene [111]. The administration of toxic paraquat and cadmium chloride induced the expression of HO-1 mRNA and protein in peritoneal macrophages of wild-type mice but not in Nrf2-KO mice [112]. Nordihydroguaiaretic acid (NDGA), a cancer chemopreventive agent, induced the protein expression of Nrf2 and HO-1 in kidney-derived LLC-PK1, in HEK293T cells, and in wild-type MEFs, but not in Nrf2-KO MEFs [113]. Berberine is an important active compound in the Chinese herb Rhizoma coptidis. Berberine promoted HO-1 mRNA and protein expression levels mediated by Nrf2 activation through the PI 3-kinase/AKT pathway in rat brain astrocytes [114].
3.2 Nrf2 and Anti-inflammation
In addition to oxidative stress, Nrf2 also participates in the protection against inflammation in cells [115–120]. It has been shown that lipopolysaccharide (LPS) increased NADPH oxidase-dependent ROS generation and the levels of TNF-alpha, IL-6 and chemokines (Mip2 and Mcp-1) in the peritoneal neutrophils from Nrf2-KO mice compared with wild-type mice [121]. Nrf2 is a crucial regulator that has been shown to modulate the innate immune response and survival during experimental sepsis using Nrf2-deficient mice and Nrf2-deficient mouse embryonic fibroblasts [122]. Some findings have suggested that there is cross-talk between Nrf2 and inflammation [123]. The Nrf2/ARE signaling pathway may be negatively regulated by proinflammatory signaling [124]. It was hypothesized that NF-κB/p65 could result in the inactivation of Nrf2 through the selective deprivation of the CREB binding protein (CBP) from Nrf2 [124]. NF-κB/p65 also promotes the interaction of HDAC3 with either CBP or MafK, which results in the repression of ARE [124].
It has been reported that Nrf2 mitigates chemical-induced pulmonary injury and inflammation [125, 126]. The genetic ablation of Nrf2 resulted in severe tobacco-smoke-induced emphysema, airway inflammation, and asthma in mice [127, 128]. The major reason for the expression of these phenotypes is that a disruption of Nrf2 caused lower antioxidant gene expression levels, enhanced the expression levels of the T helper type 2 cytokines interleukin (IL)-4 and IL-13 in bronchoalveolar lavage fluid and in splenocytes, and increased alveolar cell apoptosis after allergen challenge [127, 128]. The Nrf2-KO mice are also more susceptible to DSS-induced colitis. More severe colonic colitis was observed in Nrf2-KO mice, including the loss of colonic crypts, the massive infiltration of inflammatory cells, and anal bleeding, than in wild-type mice [117]. A lower induction of phase II antioxidant and detoxification enzymes, such as HO-1, NQO1, UGT1A1, and GSTM1, and a higher induction of proinflammatory biomarkers, such as interleukin (IL)-1β, IL-6, TNF-α, nitric oxide synthetase (iNOS), and cyclooxygenase 2 (COX2), were observed in Nrf2-KO mice [117]. It has also been shown that indirect antioxidants protected animals from inflammatory damage via Nrf2 activation, which may be a cancer-preventive mechanism [121, 129], and that Nrf2 is required for sulforaphane (SFN)-mediated anti-inflammatory response [130].
4 Cancer Prevention by Dietary Phytochemicals Via the Nrf2 Pathway
Chemoprevention involves the use of dietary compounds or synthetic chemicals to inhibit the development of invasive cancer. Chemoprevention can involve preventing carcinogens from reaching the target sites, from undergoing metabolic activation, or from subsequently interacting with crucial cellular macromolecules such as DNA, RNA, and proteins at the initiation stage. In addition, chemoprevention can inhibit the malignant transformation of initiated cells at either the promotion or the progression stage [71, 131, 132].
In this context, the induction of phase II detoxification and antioxidant enzymes is assumed to be one of the most effective ways to prevent carcinogenesis by both endogenous and exogenous carcinogens [133]. Thus, several dietary compounds that exhibit antioxidant activity and function as inducers and/or cell signals have been reported to increase phase II detoxification enzymes, and these compounds may act as chemopreventive agents [134, 135]. Most of these phase II detoxification enzymes are known to be induced by promoting the nuclear translocation of Nrf2 and its subsequent binding to the ARE sequence in those enzyme genes, leading to transcriptional activation [136]. Thus, Nrf2 is considered the major regulatory pathway of cytoprotective gene expression against oxidative and/or electrophilic stress [137].
Several studies have used in vitro and in vivo approaches involving natural dietary compounds to show that Nrf2 controls the expression of ARE-mediated gene expression and to demonstrate the role of Nrf2 in cancer chemoprevention [138, 139]. Some examples of Nrf2 inducers include curcumin from turmeric [106]; indole-3-carbinol (I3C), 3,3′-diindolylmethane (DIM), phenethyl isothiocyanate (PEITC), and sulforaphane (SFN) from cruciferous vegetables [56, 140]; epigallocatechin-3-gallate (EGCG) from green tea [141]; resveratrol from grapes [142], gamma-tocopherol-enriched mixed tocopherols from soybeans and corn oil [143]; and other compounds described in Table 1. To date, the Nrf2 downstream genes identified can be grouped into the following categories: intracellular redox-balancing proteins, which reduce the levels of ROS with enzymes such as glutamate cysteine ligase (GCL), GPx, Txn, Txnd, peroxiredoxin (Prx), and HO-1; phase II detoxifying enzymes, which metabolize xenobiotics into less toxic forms and/or catalyze conjugation reactions to increase the solubility of xenobiotics, thereby facilitating their elimination [133] with enzymes like HO-1, NQO1, GSTs, GSR, glutamate–cysteine ligase (the catalytic subunit, GCLC and the modifier subunit, GCLM), microsomal epoxide hydrolase 1 (mEH), and the UGT1 family polypeptide A6 (UGT1A6) [150]; and transporters, which control the uptake and efflux of endogenous substances and xenobiotics such as the multidrug resistance-associated protein (MRP) [112, 133]. Thus, this complicated crosstalk among various molecular targets and signaling pathways constitutes an elaborate network that responds coordinately to various xenobiotics, including carcinogens, drugs, and dietary bioactive compounds [134].
Interestingly, the Nrf2 pathway has also been connected to the inflammatory response by studies using the TRAMP mouse model of prostate carcinogenesis [154]. Mice lacking the Nrf2 pathway have proven to be more susceptible to experimentally induced colitis; as expected, these mice express low levels of phase II detoxification and antioxidant enzymes (i.e., HO1, NQO-1, UGST1A1, GST) and exhibit an increased expression of proinflammatory cytokines/mediators [i.e., cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor α (TNF-α)] [117]. In contrast, extracts from Chrysanthemum zawadskii (CZ) and licorice Glycyrrhiza uralensis (LE) have been shown (using in vitro and in vivo approaches) to possess a strong inhibitory effect against NF-κB-mediated inflammation and to have a strong activation of the Nrf2-ARE-antioxidative stress-signaling pathways [155].
Other studies have suggested Nrf2 involvement with MAPK pathways, including the ERK, JNK, and p38 pathways, in chemical-induced detoxifying enzyme regulation [148, 156]. For example, it has been demonstrated that blocking the ERK pathway attenuates the induction of ARE-mediated gene expression by tBHQ and SFN in human hepatoma HepG2 cells and in the murine hepatoma Hepa1c1c7 cells, whereas inhibition of the p38 pathway shows an opposite effect, implying the involvement of MAPKs in the modulation of ARE-mediated gene expression [157, 158]. These MAPKs, such as ERK, JNK, and p38, have also been activated by treatment with diallyl trisulfide (DATS), one of the three major organosulfur compounds of garlic. However, the inhibition of MAPKs did not affect DATS-induced ARE activity in HepG2-ARE-C8 cells (human hepatoma cells transfected with pARE-TI-luciferase) [148].
5 Epigenetic Alterations in Cancer
Cancer is caused by a series of genetic changes in tumor suppressor genes and oncogenes. However, a large amount of evidence has shown that epigenetic alterations such as DNA methylation and histone modifications can also contribute to carcinogenesis [159]. The term “epigenetics” was first defined as “the causal interactions between genes and their products, which bring the phenotype into being” by the developmental biologist Conrad H. Waddington in 1942 [160]. The concept of epigenetics has evolved as well. As Wolffe defined it, epigenetics became “the study of heritable changes in gene expression that occur without a change in DNA sequence” [161].
In cancer, hypermethylation of the promoter regions of certain tumor suppressor genes is thought to be the most relevant epigenetic change associated with malignant transformation. These heritable changes occur through the methylation of cytosine bases in the DNA and by post-transcriptional modifications of histones [162]. For example, hypermethylation of the CpG island located in the promoter region of tumor suppressor genes such as p16ink4a and BRCA1 results in gene silencing [163, 164]. Histones also play a pivotal role in epigenetic modification. Histone modification is known to regulate gene expression and chromatin structure, which are closely associated with DNA methylation [165].
Unlike genetic changes, epigenetic alterations are potentially reversible. Epigenetically modified genes can be restored, whereas genetic mutations are permanent. Transcriptionally repressed genes that are silenced by epigenetic alteration can be reactivated by epigenetic modification because these silenced genes are still intact. The removal of the methyl groups from the silenced tumor suppressor genes reverses the expression of these genes, leading to the recovery of function [166]. Therefore, the study of epigenetic targets and the mechanism of inhibition can be a novel approach to halt or delay carcinogenesis. The application of drugs to target epigenetic alterations represents a new and fascinating approach in the field of cancer prevention and therapy. With their relatively low toxicity levels and promising effects, dietary chemopreventive phytochemicals may provide a plausible avenue for epigenetic chemoprevention.
We present two important epigenetic mechanisms, DNA methylation and histone modification, that are of interest for cancer chemoprevention. Specific inhibitors of these epigenetic alterations and the dietary chemopreventive phytochemicals that have potential as epigenetic modifiers are also presented in this review.
5.1 DNA Methylation
DNA methylation is the most extensively studied epigenetic event. In mammalian cells, DNA methylation is the addition of a methyl group to the 5′ position of cytosine bases in CpG dinucleotides by DNA methyltransferases (DNMT) [167, 168]. The CpG dinucleotides are not distributed evenly throughout the genome but instead tend to group in regions known as CpG islands [168]. Approximately 60% of the human genome promoters are linked to CpG islands. Most CpG sites throughout the genome are known to be methylated. In contrast, the majority of CpG islands usually remain unmethylated in undifferentiated normal cells [168, 169]. These unmethylated CpG islands have an open structure and accord closely with the adjacent transcriptional promoter, leading the genes to remain transcriptionally active [170]. However, in cancer cells, the hypermethylation of CpG islands is known to cause gene silencing by preventing the recruitment of transcriptional protein from DNA [171]. In addition, DNA methylation can interact with various methyl-CpG binding domain proteins (MBDs), such as MBD1–MBD4 and methyl CpG binding protein 2 (MeCP2), by providing the binding site [172, 173]. These binding proteins can interact with a co-repressor complex, including histone deacetylases (HDACs), resulting in transcriptional repression [174, 175].
The primary goal of DNA methylation studies is to find DNMT inhibitors. However, other molecules are also involved in epigenetic mechanisms. Among the DNMT inhibitors, 5-azacytidine and 5-aza-2-deoxycytidine are the most widely studied epigenetic modifiers [176, 177]. However, there are many studies showing that DNA methylation is an essential function in normal mammalian cells [169]. In a mutant-DNMT mouse model, homozygous mouse embryos exhibited delayed development and did not survive past mid-gestation [178]. DNMT 3a and 3b are essential for de novo DNA methylation and mouse development. The inactivation of both genes by gene targeting blocks de novo methylation in embryonic stem cells and arrests embryonic development [179]. Thus, the genetic disruption of DNMTs in a mouse model shows that a balanced DNMT activity is important to maintaining cellular homeostasis. Accumulating evidence demonstrates that the DNA methylation of genes in most human cancers, similar to mutations and deletions, causes the transcriptional silencing of tumor suppressor genes [180].
5.2 Histone Modifications
Together with DNA methylation, histone modification plays an important role in gene expression and tumorigenesis by influencing chromatin structure [159, 181]. Chromatin is present in eukaryotic cells and is a densely packed macromolecular complex that is composed of DNA, histones, and non-histone proteins. The functional roles of chromatin are to package DNA into a small volume to fit within the nucleus and to influence gene expression and DNA replication. The nucleosome, the basic subunit of chromatin, is composed of a histone octamer that consists of an H3/H4 tetramer and two H2A/H2B dimers, and 146 bp of DNA is wrapped around this octamer. Higher-order structuring of nucleosomes results in a compact 30-nm fiber, which is then condensed to form chromosomes. The stability of these more highly folded structures is maintained by the addition of histones. The chromatin structure, which is closely involved in gene expression, is regulated by post-translational modifications of histones [182–184]. There are two different forms of chromatin structure: heterochromatin (condensed) and euchromatin (extended) [185]. In general, heterochromatin is a tightly packed structure, and it is difficult for transcription factors to access heterochromatin, which represses gene transcription. In contrast, euchromatin is loosely packed and more accessible to transcription factors, which enables active gene expression [186]. Histone proteins contain a globular C-terminal domain and an unsaturated N-terminal tail, which are amino-terminal residues protruding from nucleosomes [182]. Most histone modifications occur at the lysine, arginine, and serine residues of the N-terminal tails extending from the histone core by post-transcriptional modifications such as acetylation, methylation, phosphorylation, ubiquitination, and sumoylation [182, 187, 188]. The chromatin structure can be regulated through these modifications, which provide different levels of accessibility to transcription factors [189]. Various histone modifications are potentially reversible through the addition and removal of covalent alterations at the histone tail [181].
Interestingly, methylation on a lysine residue at histone H3 appears to induce two opposite structures, transcriptionally active chromatin or inactive chromatin, depending on which residue is methylated. Methylation at lysine 4 (Lys4) at the histone H3 tail is known to be associated with transcriptionally active chromatin, whereas methylation at lysine 9 (Lys9) in the same histone tail is reported to be related to transcriptionally repressed chromatin [185, 190, 191]. Moreover, important findings suggest that the methylation of H3 Lys9 might be required for DNA methylation [192, 193]. DNMT inhibitors, such as 5-azacytidine and 5-aza-2-deoxycytidine, trichostatin A and suberoylanilide hydroxamic acid (SAHA), are widely used as HDAC inhibitors in many studies [177, 194].
6 The Epigenomic Reactivation of Nrf2 by Dietary Phytochemicals
Epigenetic modification plays a prominent role in the development and differentiation of various cells in an organism. Defects in the epigenome have been implicated in many diseases and are known to be influenced, in whole or at least in part, by environmental factors. It is apparent that environmental factors, diet, and lifestyle have an impact on the development of various cancers in humans. Hence, minimizing exposure to environmental carcinogens, maintaining a healthier lifestyle, and consuming a healthy diet are thought to be reasonable approaches for cancer prevention. In addition to genetic mutations, epigenetic alterations play an important role in cancer development. It is believed that epigenetic changes arise before genetic alterations. The potential of dietary phytochemicals as cancer chemopreventive/anticancer agents through epigenetic modification has been demonstrated in many studies. In this chapter we will provide an overview of cancer epigenetics and discuss the potential for (and challenges of) using dietary phytochemicals as epigenetic modifiers for cancer chemoprevention.
The inclusion of epigenetics in the National Institutes of Health (NIH) research portfolio and roadmap in 2008 has indicated the urgent need for research in epigenetic mechanisms of diseases, including cancer. Unlike genetic mutations, changes in gene expression due to epigenetic regulation during carcinogenesis can be reversed or prevented by chemicals. Therefore, the pharmacological targeting of epigenetic events has emerged as a promising approach to treating or preventing cancers.
Several HDAC and DNMT inhibitors have been approved for the treatment of hematological malignancies and are currently at different phases of clinical trials [195, 196]. Similarly, the DNA-hypomethylating agents 5-azacitdine and 5-aza-2′-deoxycytidine (decitabine) have been tested in myelodysplastic syndrome (MDS), acute myelogenous leukemia (AML), and chronic myelogenous leukemia (CML) patients with some encouraging outcomes [197–200]. HDAC inhibitors, such as vorinostat (suberoylanilide hydroxamic acid or SAHA), belinostat, romidepsin, and panobinostat, have been used to treat hematological malignancies and solid tumors [201, 202]. The development of HDAC or DNMT inhibitors as anticancer drugs has been hindered by their adverse side effects [203]. Accumulating evidence suggests that some dietary phytochemicals may exert their cancer chemopreventive/anticancer effects via epigenetic modifications [204–206]. In this chapter, we focus on a few of the most widely studied dietary compounds as epigenetic modifiers.
6.1 Curcumin
Hailed as “Indian solid gold,” curcumin is a polyphenolic compound derived from the Curcuma longa plant. Despite its poor bioavailability, curcumin has been shown to be a strong anticancer agent against different types of cancers in animals and with in vitro cell culture systems [207]. At least 33 proteins have been identified as being targeted by curcumin. The potential of curcumin in targeting epigenetic modifications has recently been revealed [207].
6.1.1 Curcumin as a DNA Hypomethylation Agent
DNA methylation is a heritable epigenetic modification that modulates the transcriptional plasticity of the genome. The hypermethylation of promoter CpG islands, particularly at tumor suppressor genes, plays a causative role in carcinogenesis. In fact, recent findings suggest that epigenetic alterations may precede genetic mutations [159]. DNA methylation is regulated by DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) to transfer a methyl group from the methyl donor S-adenosyl-L-methionine (SAM) to cytosine residues at the C-5 position [208]. There are contradicting reports on the potential for curcumin as a DNMT inhibitor. Using a molecular docking approach, curcumin has been shown to bind covalently to the catalytic thiolate of C1226 of DNMT1, leading to its inhibitory effect [209]. In contrast, Medina-Franco et al. [210] found that curcumin has little or no pharmacologically relevant DNMT inhibitory activity. However, we have recently reported that curcumin can restore the expression of the Nrf2 and Neurog1 genes through DNA demethylation [57, 106]. Similarly, Jha et al. demonstrated that curcumin can reverse CpG hypermethylation, leading to the activation of the RARβ2 gene in cervical cancer cell lines [211]. However, in another report, demethoxycurcumin and bisdemethoxycurcumin, but not curcumin, were found to be able to demethylate the WIF-1 promoter region in A549 cells [212]. Further research is necessary to explain these discrepancies.
6.1.2 The Effect of Curcumin on Histone Modification
Post-translational histone modifications, including acetylation, methylation, phosphorylation, and ubiquitination, are important epigenetic events that regulate gene expression. Histone acetylation catalyzed by histone acetyltransferases (HATs) and HDACs is one of the most studied histone modifications. An accumulating body of evidence suggests that alterations in HAT and HDAC activity occur in cancer [213]. Curcumin has been reported to be a strong inhibitor of both HDACs and HATs. Curcumin is a specific inhibitor of the p300/CREB-binding protein (CBP) HAT activity but not of p300/CBP-associated factor, as demonstrated by Balasubramanyam et al. [214]. In agreement with this finding, Morimoto et al. found that the inhibition of p300 HAT activity by curcumin prevented heart failure in rats; Li et al. reported that curcumin possesses a protective effect against cardiac hypertrophy, inflammation, and fibrosis through the suppression of p300-HAT activity [215, 216]. The p300 and CBP proteins are transcriptional coactivators that function partially through their intrinsic HAT activities [217]. In addition to histones, p300 and CBP acetylate several non-histone proteins, including p53 [218]. Interestingly, curcumin was found to be able to inhibit p300-mediated acetylation of p53 in vivo [214]. In addition, Kang et al. reported that curcumin induces histone hypoacetylation in brain cancer cells, leading to the induction of apoptosis through a (PARP)- and caspase 3-mediated manner [219]. Mechanistically, Marcu et al. proposed that curcumin is a selective HAT inhibitor. The covalent binding of curcumin with p300 leads to a conformational change, resulting in a decreased binding efficiency of histones H3, H4, and acetyl CoA [220]. In addition to HAT, curcumin was found to be a strong inhibitor of HDACs. Chen et al. reported that curcumin significantly suppresses the expression of p300, HDAC1, and HDAC3 in Raji cells [221]. Similarly, Liu et al. reported the inhibitory effect of [222]. In a study by Bora-Tatar et al., curcumin was found to be the strongest HDAC inhibitor among 33 carboxylic acid derivatives tested [223]. Curcumin-induced HDAC4 inhibition in medulloblastoma was also recently reported [224].
6.2 The Isothiocyanates Sulforaphane and Phenethyl Isothiocyanate
Isothiocyanates (ITCs) are biologically active hydrolysis products of glucosinolates from cruciferous vegetables such as broccoli, brussels sprouts, cabbage, cauliflower, Chinese cabbage, and watercress. Studies have shown that PEITC and SFN, two examples of ITCs, are strong anticancer/cancer chemopreventive agents [225]. The induction of apoptosis, cell-cycle arrest, autophagy, phase II detoxifying/antioxidant genes and the inhibition of inflammation by blocking NFKb signaling pathways are reported to be possible mechanisms by which isothiocyanates exert their anticancer/cancer chemopreventive effect [225]. The role of isothiocyanates in modulating epigenetic changes has been recently reported.
6.2.1 The Effects of SFN/PEITC on DNA Methylation
The effects of SFN on DNA methylation were first reported by Meeran et al. These researchers found that SFN treatment exhibited a dose- and time-dependent [226] suppression of DNMT1 and DNMT3a. The suppression of DNMTs by SFN is associated with the site-specific CpG demethylation of the first exon of the hTERT gene. A subsequent ChIP assay revealed that SFN increased the level of the active chromatin markers acetyl-H3, acetyl-H3K9, and acetyl-H4 but suppressed the levels of the inactive chromatin markers trimethyl-H3K9 and trimethyl-H3K27. Wang et al. reported that PEITC demethylates the promoter and restores the expression of GSTP1 in both androgen-dependent and androgen-independent LNCaP cancer cells [227]. Interestingly, PEITC was found to be more effective than 5′-aza-2′-deoxycytidine in DNA methylation.
6.2.2 The Effects of SFN/PEITC on Histone Modification
SFN is known to be a dietary HDAC inhibitor, as demonstrated in in vitro and in vivo studies [228–230]. SFN was found to suppress HDAC activity without altering protein expression levels in the human embryonic kidney 293 cells and the human colorectal cancer cell HCT116 [228]. SFN and its glutathione conjugate (SFN–GSH) were found to be less effective than the two major metabolites of SFN, SFN-cysteine and SFN-N-acetylcysteine, as HDAC inhibitors in vitro. A similar HDAC inhibitory effect of SFN was also observed in BPH-1, LnCaP, and PC-3 prostate epithelial cells [231]. In addition, SFN as an HDAC inhibitor is being investigated in vivo in mice and in human subjects. HDAC activity was significantly inhibited as early as 6 h after a single oral dose of 10 μmol SFN with a concomitant increase in acetylated histones H3 and H4 in the colonic mucosa [232]. More importantly, SFN was found to suppress intestinal carcinogenesis in Apc(min) mice through histone modification, as demonstrated by an increase in acetylated histones in the polyps. SFN can also suppress the growth of PC-3 xenografts by inhibiting HDAC activity [233]. In humans, a single dose of SFN-rich broccoli sprouts is sufficient to inhibit significantly HDAC activity in peripheral blood mononuclear cells (PBMCs) 3 and 6 h after consumption [233]. Like SFN, PEITC inhibits HDAC. PEITC was reported to inhibit HDAC activity and expression levels in LNCaP cells, leading to the re-expression of GSTP1 [227]. Furthermore, PEITC increases the methylation of lysine 4 of histone H3 but decreases the level of trimethylated lysine 9 of H3. Similarly, PEITC restored p21 expression through HDAC inhibition in LNCaP cells [234].
6.3 Tea Polyphenols
There is a large body of evidence indicating that bioactive polyphenolic compounds in tea (Camellia sinensis, Theaceae) may reduce the risk of chronic diseases, including cancers. Catechins, which include (−)-epicatechin (EC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC), and (−)-epigallocatechin-3-gallate (EGCG), are the most abundant compounds present in tea [235]. Among these catechins, EGCG has been identified as one of the most effective compounds. Antioxidative stress, detoxification, antiproliferation, anti-inflammation, antiangiogenesis, and the induction of apoptosis have been proposed to be the mechanisms by which EGCG exerts its cancer chemopreventive effects [236]. The role of EGCG as an epigenetic modifier for cancer treatment and chemoprevention has received recent attention [205, 237].
6.3.1 The Effects of EGCG on DNA Methylation
One of the earliest reports to demonstrate the effect of EGCG on DNA methylation was the study by Fang et al. in 2003 [238]. EGCG inhibited DNMT activity, leading to a concentration-dependent and time-dependent reversal of the hypermethylation of p16 (INK4a), retinoic acid receptor beta (RARbeta), O(6)-methylguanine methyltransferase (MGMT), and human mutL homolog 1 (hMLH1) genes in human esophageal KYSE 510 cells. Similarly, Kato et al. found that treatment of oral cancer cells with EGCG partially reversed the hypermethylation status of the RECK gene and significantly enhanced the expression levels of RECK mRNA [239]. A dose-dependent inhibition of DNMT activity was observed in LNCaP cells after a 7-day exposure of cells to different doses of EGCG, leading to the re-expression of the GSTP1 gene [240]. In another study, EGCG treatment was found to decrease the global DNA methylation levels in A431 human skin cancer cells in a dose-dependent manner. EGCG decreased the levels of 5-methylcytosine, DNMT activity, and the mRNA and protein levels of DNMT1, DNMT3a, and DNMT3b [241]. In addition to the direct inhibitory effect on DNMT, EGCG was also found to inhibit indirectly DNMT activity by decreasing the availability of SAM [205, 242]. In contrast to the findings from in vitro studies, the in vivo hypomethylation effect of EGCG has been controversial. The oral administration of 0.3% green tea polyphenols (GTPs) to wild-type and transgenic adenocarcinomas of mouse prostate (TRAMP) mice showed decreased levels of 5-methyl-deoxycytidine (5mdC) in the liver at 12 weeks but did not alter the levels of 5mdC in the prostate, gut, and liver from WT mice at either 12 or 24 weeks of age [243]. However, EGCG treatment resulted in a significant inhibition of the UVB-induced global DNA hypomethylation pattern in the SKH-1 hairless mouse [244].
6.3.2 The Effects of EGCG on Histone Modification
In addition to its DNMT inhibitory effect, EGCG modulates gene expression via histone modification. EGCG was found to abrogate p300-induced p65 acetylation in vitro and in vivo, increase the level of cytosolic IkappaB alpha, and suppress tumor necrosis factor alpha (TNFα)-induced NF-κB activation. Despite a strong specificity for the majority of HAT enzymes, EGCG did not demonstrate activity toward HDAC, SIRT1, or HMTase [245]. However, EGCG was found to decrease HDAC activity and increase levels of acetylated lysine 9 and 14 on histone H3 (H3-Lys 9 and 14) and acetylated lysine 5, 12, and 16 on histone H4, but EGCG decreased levels of methylated H3-Lys 9 in A431 human skin cancer cells [241]. EGCG was also reported to inhibit HDAC1-3 expression and increase the levels of acetylated histone H3 (LysH9/18) and H4 levels in LNCaP cells [240]. The in vivo effect of EGCG on histone modification remains to be determined.
6.4 Genistein
Genistein is a natural isoflavone and phytoestrogen found in soy products. The antitumor properties of genistein have been extensively studied using cell culture systems and preclinical models. Epidemiological studies suggest that dietary intake of genistein is linked with a decreased risk of breast and prostate cancer [246, 247]. It has been reported that genistein can regulate gene transcription through the modulation of DNA methylation and histone modification.
6.4.1 The Effects of Genistein on DNA Methylation
The DNA hypomethylation effect of genistein on different cell lines has been previously reported. Genistein and 5aza-C treatment significantly decreased the promoter methylation of B-cell translocation gene 3 (BTG3), leading to its re-expression [248] in prostate cancer cell lines. Similarly, treatment of a squamous cervical cancer cell line, SiHa, with genistein resulted in promoter demethylation and the reactivation of the RARβ2 gene [211]. A similar promoter demethylation effect of genistein on different target genes was also observed in renal and breast cancer cell lines [248, 249]. It is believed that genistein modulates promoter demethylation through the direct inhibition of DNMTs and the methyl-CpG-binding domain 2.
6.4.2 The Effects of Genistein on Histone Modification
In addition to DNA methylation, genistein modulates gene expression through histone modification. Genistein was reported to increase acetylated histones 3, 4, and H3/K4 at the p21 and p16 transcription start sites, leading to the reactivation of the genes in human prostate cancer cells [250]. Genistein was also found to activate tumor suppressor genes, such as PTEN and CYLD, via the demethylation and acetylation of H3-K9 of the promoter region of the genes [251]. Interestingly, the suppression effect of genistein on SIRT-1 led to the acetylation of H3-K9 at the p53 and FOXO3a promoters [251].
7 Conclusions
Various toxins, such as carcinogens, environmental pollutants, solar radiation, and dietary mutagens, cause oxidative stress and inflammation and are the major drivers of cancer. Dietary phytochemicals and/or relatively nontoxic therapeutic drugs, such as cancer chemopreventive agents, are administered to inhibit, retard, or reverse the initiation and progression stages of carcinogenesis over time. The induction of the Nrf2-related antioxidant, detoxification, and anti-inflammation systems play an important role in blocking carcinogenesis. In addition to the Nrf2–Keap1 signaling pathway, epigenetic modifications are key mechanisms for the regulation of Nrf2-mediated antioxidant and detoxification genes. Therefore, a promising approach to cancer chemoprevention is the use of dietary phytochemicals to increase the expression of Nrf2 and Nrf2 downstream antioxidant and detoxification enzymes. The results from research investigating this approach may provide clinical benefits to human health.
References
Brüne B, Zhou J, von Knethen A (2003) Kidney Int Suppl:S22
Darley-Usmar V, Halliwell B (1996) Pharm Res 13:649
Yu BP (1994) Physiol Rev 74:139
Beckman KB, Ames BN (1998) Physiol Rev 78:547
Halliwell B, Gutteridge J (1999) Free radicals in biology and medicine. Oxford University Press, Oxford
Emerit J, Edeas M, Bricaire F (2004) Biomed Pharmacother 58:39
Cataldi A (2010) Curr Pharm Des 16:1387
Azad N, Iyer A, Vallyathan V, Wang L, Castranova V, Stehlik C, Rojanasakul Y (2010) Ann N Y Acad Sci 1203:1
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM (2007) Antioxid Redox Signal 9:49
Shen HM, Liu ZG (2006) Free Radic Biol Med 40:928
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006) Chem Biol Interact 160:1
Tan AC, Konczak I, Sze DM, Ramzan I (2011) Nutr Cancer 63:495
Zamocky M, Furtmuller PG, Obinger C (2008) Antioxid Redox Signal 10:1527
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Int J Biochem Cell Biol 39:44
Bartsch H, Nair J (2006) Langenbecks Arch Surg 391:499
Klaunig JE, Kamendulis LM (2004) Annu Rev Pharmacol Toxicol 44:239
Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J (2004) Mol Cell Biochem 266:37
Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H (2002) Free Radic Biol Med 32:1102
Brown GC, Borutaite V (2001) IUBMB Life 52:189
Shigenaga MK, Gimeno CJ, Ames BN (1989) Proc Natl Acad Sci USA 86:9697
Dreher D, Junod AF (1996) Eur J Cancer 32A:30
Carmeliet P (2000) Nat Med 6:389
He X, Ma Q (2009) Mol Pharmacol 76:1265
Reddy SP (2008) Curr Mol Med 8:376
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Mol Cell Biochem 345:91
Mamede AC, Tavares SD, Abrantes AM, Trindade J, Maia JM, Botelho MF (2011) Nutr Cancer 63:479
Kojo S (2004) Curr Med Chem 11:1041
Fukai T, Ushio-Fukai M (2011) Antioxid Redox Signal 15:1583
Nishikawa M, Hashida M, Takakura Y (2009) Adv Drug Deliv Rev 61:319
Limon-Pacheco JH, Gonsebatt ME (2010) Cent Nerv Syst Agents Med Chem 10:287
Jung KA, Kwak MK (2010) Molecules 15:7266
Rushmore TH, Morton MR, Pickett CB (1991) J Biol Chem 266:11632
Shen G, Kong AN (2009) Biopharm Drug Dispos 30:345
Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M (2011) Trends Mol Med 17:363
Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Proc Natl Acad Sci USA 91:9926
Chan JY, Han XL, Kan YW (1993) Proc Natl Acad Sci USA 90:11366
Chan K, Kan YW (1999) Proc Natl Acad Sci USA 96:12731
Slocum SL, Kensler TW (2011) Arch Toxicol 85:273
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Genes Dev 13:76
Shu L, Cheung KL, Khor TO, Chen C, Kong AN (2010) Cancer Metastasis Rev 29:483
Chinenov Y, Kerppola TK (2001) Oncogene 20:2438
Kobayashi M, Yamamoto M (2005) Antioxid Redox Signal 7:385
Kong AN, Yu R, Hebbar V, Chen C, Owuor E, Hu R, Ee R, Mandlekar S (2001) Mutat Res 480–481:231
Hu R, Shen G, Yerramilli UR, Lin W, Xu C, Nair S, Kong AN (2006) Arch Pharm Res 29:911
Keum YS, Yu S, Chang PP, Yuan X, Kim JH, Xu C, Han J, Agarwal A, Kong AN (2006) Cancer Res 66:8804
Huang HC, Nguyen T, Pickett CB (2000) Proc Natl Acad Sci USA 97:12475
Huang HC, Nguyen T, Pickett CB (2002) J Biol Chem 277:42769
Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T (2003) Am J Physiol Cell Physiol 285:C334
Bloom DA, Jaiswal AK (2003) J Biol Chem 278:44675
Kang KW, Lee SJ, Park JW, Kim SG (2002) Mol Pharmacol 62:1001
Lee JM, Hanson JM, Chu WA, Johnson JA (2001) J Biol Chem 276:20011
Kang KW, Choi SH, Kim SG (2002) Nitric Oxide 7:244
Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K (2003) FEBS Lett 546:181
Sun Z, Huang Z, Zhang DD (2009) PLoS One 4:e6588
Li W, Thakor N, Xu EY, Huang Y, Chen C, Yu R, Holcik M, Kong AN (2010) Nucleic Acids Res 38:778
Saw CL, Cintron M, Wu TY, Guo Y, Huang Y, Jeong WS, Kong AN (2011) Biopharm Drug Dispos 32:289
Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, Saw CL, Cheung KL, Kong AN (2011) AAPS J 13:606
Chun KS, Keum YS, Han SS, Song YS, Kim SH, Surh YJ (2003) Carcinogenesis 24:1515
Reuter S, Eifes S, Dicato M, Aggarwal BB, Diederich M (2008) Biochem Pharmacol 76:1340
Aggarwal BB, Kumar A, Bharti AC (2003) Anticancer Res 23:363
Deeb D, Xu YX, Jiang H, Gao X, Janakiraman N, Chapman RA, Gautam SC (2003) Mol Cancer Ther 2:95
Pandey M, Gupta S (2009) Front Biosci (Elite Ed) 1:13
Jeong WS, Kim IW, Hu R, Kong AN (2004) Pharm Res 21:649
Jagtap S, Meganathan K, Wagh V, Winkler J, Hescheler J, Sachinidis A (2009) Curr Med Chem 16:1451
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P (2008) Antioxid Redox Signal 10:1343
Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ (2005) Antioxid Redox Signal 7:42
Dinkova-Kostova AT, Talalay P (2008) Mol Nutr Food Res 52(Suppl 1):S128
Conney AH (2003) Annu Rev Pharmacol Toxicol 43:1
Jakoby WB, Ziegler DM (1990) J Biol Chem 265:20715
Jana S, Mandlekar S (2009) Curr Drug Metab 10:595
Chen C, Kong AN (2004) Free Radic Biol Med 36:1505
Kundu JK, Surh YJ (2010) Pharm Res 27:999
Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT (2010) Antioxid Redox Signal 13:1713
Wasserman WW, Fahl WE (1997) Proc Natl Acad Sci USA 94:5361
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y (1997) Biochem Biophys Res Commun 236:313
Kwak MK, Egner PA, Dolan PM, Ramos-Gomez M, Groopman JD, Itoh K, Yamamoto M, Kensler TW (2001) Mutat Res 480–481:305
Venugopal R, Jaiswal AK (1996) Proc Natl Acad Sci USA 93:14960
He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J (2001) J Biol Chem 276:20858
Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M (2005) Mol Cell Biol 25:8044
Igarashi K, Kataoka K, Itoh K, Hayashi N, Nishizawa M, Yamamoto M (1994) Nature 367:568
Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M (2002) Gene 294:1
Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, Lin W, Reddy B, Chan JY, Kong AN (2006) Cancer Lett 243:170
Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, Lin W, Reddy B, Chan JY, Kong AN (2006) Life Sci 79:1944
Kwak MK, Kensler TW (2010) Toxicol Appl Pharmacol 244:66
Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, Jain MR, Lin W, Keum YS, Liew C, Chan JY, Kong AN (2006) Pharm Res 23:2621
Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S (2002) Cancer Res 62:5196
Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, Silkworth JB, Taguchi K, Yamamoto M, Williams CR, Liby KT, Sporn MB, Sutter TR, Kensler TW (2009) Carcinogenesis 30:1024
Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, Honda T, Gribble GW, Sporn MB, Kensler TW (2006) Cancer Res 66:2488
Hayes JD, Flanagan JU, Jowsey IR (2005) Annu Rev Pharmacol Toxicol 45:51
Reisman SA, Yeager RL, Yamamoto M, Klaassen CD (2009) Toxicol Sci 108:35
Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, Wolf CR, Yamamoto M (2000) Biochem Soc Trans 28:33
Tan KP, Wood GA, Yang M, Ito S (2010) Br J Pharmacol 161:1111
Tukey RH, Strassburg CP (2000) Annu Rev Pharmacol Toxicol 40:581
Vienneau DS, DeBoni U, Wells PG (1995) Cancer Res 55:1045
Leung HY, Wang Y, Leung LK (2007) Toxicology 242:153
Yueh MF, Tukey RH (2007) J Biol Chem 282:8749
Yeager RL, Reisman SA, Aleksunes LM, Klaassen CD (2009) Toxicol Sci 111:238
Buckley DB, Klaassen CD (2009) Drug Metab Dispos 37:847
Paonessa JD, Ding Y, Randall KL, Munday R, Argoti D, Vouros P, Zhang Y (2011) Cancer Res 71:3904
Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW (2001) Proc Natl Acad Sci USA 98:3410
Nioi P, Hayes JD (2004) Mutat Res 555:149
Ross D (2004) Drug Metab Rev 36:639
Long DJ 2nd, Waikel RL, Wang XJ, Perlaky L, Roop DR, Jaiswal AK (2000) Cancer Res 60:5913
Tripathi DN, Jena GB (2010) Mutat Res 696:69
Yang CM, Huang SM, Liu CL, Hu ML (2012) J Agric Food Chem 60:1576
Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN (2011) Biochem Pharmacol 82:1073
Loboda A, Jazwa A, Grochot-Przeczek A, Rutkowski AJ, Cisowski J, Agarwal A, Jozkowicz A, Dulak J (2008) Antioxid Redox Signal 10:1767
Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM (1995) Mol Med 1:827
Wunder C, Potter RF (2003) Curr Drug Targets 3:199
Guo X, Shin VY, Cho CH (2001) Life Sci 69:3113
Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL (1999) J Biol Chem 274:26071
Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M (2000) J Biol Chem 275:16023
Rojo AI, Medina-Campos ON, Rada P, Zuniga-Toala A, Lopez-Gazcon A, Espada S, Pedraza-Chaverri J, Cuadrado A (2012) Free Radic Biol Med 52:473
Chen JH, Huang SM, Tan TW, Lin HY, Chen PY, Yeh WL, Chou SC, Tsai CF, Wei IH, Lu DY (2012) Int Immunopharmacol 12:94
Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C (2006) Am J Physiol 290:H1862
Chen XL, Kunsch C (2004) Curr Pharm Des 10:879
Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN (2006) Cancer Res 66:11580
Li N, Nel AE (2006) Antioxid Redox Signal 8:88
Rahman I, Biswas SK, Kirkham PA (2006) Biochem Pharmacol 72:1439
Yates MS, Kensler TW (2007) Drug News Perspect 20:109
Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S (2006) Biochem Biophys Res Commun 351:883
Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S (2006) J Clin Invest 116:984
Yang H, Magilnick N, Ou X, Lu SC (2005) Biochem J 391:399
Liu GH, Qu J, Shen X (2008) Biochim Biophys Acta 1783:713
Cho HY, Reddy SP, Yamamoto M, Kleeberger SR (2004) FASEB J 18:1258
Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, Iizuka T, Hegab AE, Hosoya T, Nomura A, Sakamoto T, Yamamoto M, Sekizawa K (2005) J Immunol 175:6968
Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S (2004) J Clin Invest 114:1248
Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S (2005) J Exp Med 202:47
Khor TO, Yu S, Kong AN (2008) Planta Med 74:1540
Lin W, Wu RT, Wu T, Khor TO, Wang H, Kong AN (2008) Biochem Pharmacol 76:967
Wattenberg LW (1985) Cancer Res 45:1
Hu R, Saw CL, Yu R, Kong AN (2010) Antioxid Redox Signal 13:1679
Yu S, Kong AN (2007) Curr Cancer Drug Targets 7:416
Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD (2008) Pharmacol Res 58:262
Finley JW, Kong AN, Hintze KJ, Jeffery EH, Ji LL, Lei XG (2011) J Agric Food Chem 59:6837
Kim HJ, di Luccio E, Kong AN, Kim JS (2011) Biotechnol J 6:525
Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M (2006) Mol Cell Biol 26:221
Surh YJ, Kundu JK, Na HK, Lee JS (2005) J Nutr 135:2993S
Saw CL, Wu Q, Kong AN (2010) Chinese Med 5:37
Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, Conney AH, Kong AN (2006) Cancer Res 66:8293
Shen G, Xu C, Hu R, Jain MR, Nair S, Lin W, Yang CS, Chan JY, Kong AN (2005) Pharm Res 22:1805
Kang HJ, Hong YB, Kim HJ, Wang A, Bae I (2011) Toxicol Lett 209:154
Barve A, Khor TO, Nair S, Reuhl K, Suh N, Reddy B, Newmark H, Kong AN (2009) Int J Cancer 124:1693
Haridas V, Hanausek M, Nishimura G, Soehnge H, Gaikwad A, Narog M, Spears E, Zoltaszek R, Walaszek Z, Gutterman JU (2004) J Clin Invest 113:65
Higgins LG, Cavin C, Itoh K, Yamamoto M, Hayes JD (2008) Toxicol Appl Pharmacol 226:328
Chen CC, Chen HL, Hsieh CW, Yang YL, Wung BS (2011) Acta Pharmacol Sin 32:62
Feng R, Lu Y, Bowman LL, Qian Y, Castranova V, Ding M (2005) J Biol Chem 280:27888
Chen C, Pung D, Leong V, Hebbar V, Shen G, Nair S, Li W, Kong AN (2004) Free Radic Biol Med 37:1578
Wu TY, Saw CL, Khor TO, Pung D, Boyanapalli SS, Kong AN (2011) Mol Carcinog
Lian F, Wang XD (2008) Int J Cancer 123:1262
Wondrak GT, Cabello CM, Villeneuve NF, Zhang S, Ley S, Li Y, Sun Z, Zhang DD (2008) Free Radic Biol Med 45:385
Shin JW, Ohnishi K, Murakami A, Lee JS, Kundu JK, Na HK, Ohigashi H, Surh YJ (2011) Cancer Prev Res 4:860
Hou DX, Korenori Y, Tanigawa S, Yamada-Kato T, Nagai M, He X, He J (2011) J Agric Food Chem 59:11975
Swanson HI, Njar VC, Yu Z, Castro DJ, Gonzalez FJ, Williams DE, Huang Y, Kong AN, Doloff JC, Ma J, Waxman DJ, Scott EE (2010) Drug Metab Dispos 38:539
Wu TY, Khor TO, Saw CL, Loh SC, Chen AI, Lim SS, Park JH, Cai L, Kong AN (2011) AAPS J 13:1
Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, Kong AN (2000) J Biol Chem 275:39907
Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, Kong AN (1999) J Biol Chem 274:27545
Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AN (2000) J Biol Chem 275:2322
Esteller M (2008) N Engl J Med 358:1148
Waddington CH (1942) Endeavour 1:18
Wolffe AP, Matzke MA (1999) Science 286:481
Baylin SB, Herman JG (2000) Trends Genet 16:168
Esteller M (2005) Annu Rev Pharmacol Toxicol 45:629
Jones PA, Baylin SB (2002) Nat Rev Genet 3:415
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M (2005) Nat Genet 37:391
Issa JP (2002) J Nutr 132:2388S
Esteller M (2002) Oncogene 21:5427
Bird A (2002) Genes Dev 16:6
Suzuki MM, Bird A (2008) Nat Rev Genet 9:465
Esteller M (2007) Nat Rev Genet 8:286
Prendergast GC, Ziff EB (1991) Science 251:186
Hendrich B, Bird A (1998) Mol Cell Biol 18:6538
Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A (1992) Cell 69:905
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Nature 393:386
Feng Q, Zhang Y (2001) Genes Dev 15:827
Lee BH, Yegnasubramanian S, Lin X, Nelson WG (2005) J Biol Chem 280:40749
Yu SW, Khor TO, Cheung KL, Li WG, Wu TY, Huang Y, Foster BA, Kan YW, Kong AN (2010) PLoS One 5:e8579
Li E, Bestor TH, Jaenisch R (1992) Cell 69:915
Okano M, Bell DW, Haber DA, Li E (1999) Cell 99:247
Herman JG, Baylin SB (2003) N Engl J Med 349:2042
Ellis L, Atadja PW, Johnstone RW (2009) Mol Cancer Ther 8:1409
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Nature 389:251
Tremethick DJ (2007) Cell 128:651
Berlowitz L, Pallotta D (1972) Exp Cell Res 71:45
Jenuwein T, Allis CD (2001) Science 293:1074
Lund AH, van Lohuizen M (2004) Genes Dev 18:2315
Berger SL (2007) Nature 447:407
Kouzarides T (2007) Cell 128:693
Rodenhiser D, Mann M (2006) CMAJ 174:341
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Nature 410:120
Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Science 292:110
Jackson JP, Lindroth AM, Cao X, Jacobsen SE (2002) Nature 416:556
Tamaru H, Selker EU (2001) Nature 414:277
Kim SH, Kang HJ, Na H, Lee MO (2010) Breast Cancer Res 12:R22
Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, Anzalone L, Pezzani L, Di Giacomo AM, Fonsatti E, Colizzi F, Altomonte M, Calabro L, Maio M (2007) J Cell Physiol 212:330
Mai A, Altucci L (2009) Int J Biochem Cell Biol 41:199
Abdulhaq H, Rossetti JM (2007) Expert Opin Investig Drugs 16:1967
Fenaux P, Ades L (2009) Leuk Res 33(Suppl 2):S7
Griffiths EA, Gore SD (2008) Semin Hematol 45:23
Issa JP (2007) Clin Cancer Res 13:1634
Rasheed WK, Johnstone RW, Prince HM (2007) Expert Opin Investig Drugs 16:659
Prince HM, Bishton MJ, Harrison SJ (2009) Clin Cancer Res 15:3958
Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M (2008) Blood 112:981
Davis CD, Uthus EO (2004) Exp Biol Med (Maywood) 229:988
Fang M, Chen D, Yang CS (2007) J Nutr 137:223S
Yang CS, Fang M, Lambert JD, Yan P, Huang TH (2008) Nutr Rev 66(Suppl 1):S18
Ravindran J, Prasad S, Aggarwal BB (2009) AAPS J 11:495
Kanai Y (2008) Pathol Int 58:544
Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, Li PK, Lin J, Fuchs JR, Marcucci G, Li C, Chan KK (2009) Bioorg Med Chem Lett 19:706
Medina-Franco JL, Lopez-Vallejo F, Kuck D, Lyko F (2011) Mol Divers 15:293
Jha AK, Nikbakht M, Parashar G, Shrivastava A, Capalash N, Kaur J (2010) Folia Biol (Praha) 56:195
Liu YL, Yang HP, Gong L, Tang CL, Wang HJ (2011) Mol Med Rep 4:675
Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A (2001) Cell Mol Life Sci 58:728
Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK (2004) J Biol Chem 279:51163
Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, Komeda M, Fujita M, Shimatsu A, Kita T, Hasegawa K (2008) J Clin Invest 118:868
Li HL, Liu C, de Couto G, Ouzounian M, Sun M, Wang AB, Huang Y, He CW, Shi Y, Chen X, Nghiem MP, Liu Y, Chen M, Dawood F, Fukuoka M, Maekawa Y, Zhang L, Leask A, Ghosh AK, Kirshenbaum LA, Liu PP (2008) J Clin Invest 118:879
Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG (2000) Mol Cell 6:551
Brooks CL, Gu W (2003) Curr Opin Cell Biol 15:164
Kang SK, Cha SH, Jeon HG (2006) Stem Cells Dev 15:165
Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, Neckers L (2006) Med Chem 2:169
Chen Y, Shu W, Chen W, Wu Q, Liu H, Cui G (2007) Basic Clin Pharmacol Toxicol 101:427
Liu HL, Chen Y, Cui GH, Zhou JF (2005) Acta Pharmacol Sin 26:603
Bora-Tatar G, Dayangac-Erden D, Demir AS, Dalkara S, Yelekci K, Erdem-Yurter H (2009) Bioorg Med Chem 17:5219
Lee SJ, Krauthauser C, Maduskuie V, Fawcett PT, Olson JM, Rajasekaran SA (2011) BMC Cancer 11:144
Cheung KL, Kong AN (2010) AAPS J 12:87
Meeran SM, Patel SN, Tollefsbol TO (2010) PLoS One 5:e11457
Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW (2007) Mol Carcinog 46:24
Myzak MC, Karplus PA, Chung FL, Dashwood RH (2004) Cancer Res 64:5767
Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E (2011) Mol Nutr Food Res 55:999
Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Ho E, Dashwood RH (2011) Mol Cancer 10:68
Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E (2006) Carcinogenesis 27:811
Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH (2006) FASEB J 20:506
Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E (2007) Exp Biol Med (Maywood) 232:227
Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, Feng J, Liu D, Chiao JW (2008) Int J Oncol 33:375
Graham HN (1992) Prev Med 21:334
Yang CS, Wang H (2011) Mol Nutr Food Res 55:819
Li Y, Tollefsbol TO (2010) Curr Med Chem 17:2141
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS (2003) Cancer Res 63:7563
Kato K, Long NK, Makita H, Toida M, Yamashita T, Hatakeyama D, Hara A, Mori H, Shibata T (2008) Br J Cancer 99:647
Pandey M, Shukla S, Gupta S (2010) Int J Cancer 126:2520
Nandakumar V, Vaid M, Katiyar SK (2011) Carcinogenesis 32:537
Lee WJ, Zhu BT (2006) Carcinogenesis 27:269
Morey Kinney SR, Zhang W, Pascual M, Greally JM, Gillard BM, Karasik E, Foster BA, Karpf AR (2009) Cancer Prev Res (Phila) 2:1065
Mittal A, Piyathilake C, Hara Y, Katiyar SK (2003) Neoplasia 5:555
Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, Kim MJ, Cha JH, Kim YJ, Jun WJ, Lee JM, Yoon HG (2009) Cancer Res 69:583
Adlercreutz H (2002) Lancet Oncol 3:364
Wu AH, Ziegler RG, Nomura AM, West DW, Kolonel LN, Horn-Ross PL, Hoover RN, Pike MC (1998) Am J Clin Nutr 68:1437S
Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, Saini S, Tanaka Y, Dahiya AV, Khatri G, Dahiya R (2009) Carcinogenesis 30:662
King-Batoon A, Leszczynska JM, Klein CB (2008) Environ Mol Mutagen 49:36
Majid S, Kikuno N, Nelles J, Noonan E, Tanaka Y, Kawamoto K, Hirata H, Li LC, Zhao H, Okino ST, Place RF, Pookot D, Dahiya R (2008) Cancer Res 68:2736
Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, Majid S, Igawa M, Dahiya R (2008) Int J Cancer 123:552
Acknowledgments
This work is supported in part by Institutional Funds and by RO1-CA073674, RO1-CA094828, R01-CA118947, and R01-CA152826 awarded to Dr. Ah-Ng Tony Kong from the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Su, ZY., Shu, L., Khor, T.O., Lee, J.H., Fuentes, F., Kong, AN.T. (2012). A Perspective on Dietary Phytochemicals and Cancer Chemoprevention: Oxidative Stress, Nrf2, and Epigenomics. In: Pezzuto, J., Suh, N. (eds) Natural Products in Cancer Prevention and Therapy. Topics in Current Chemistry, vol 329. Springer, Berlin, Heidelberg. https://doi.org/10.1007/128_2012_340
Download citation
DOI: https://doi.org/10.1007/128_2012_340
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-34574-6
Online ISBN: 978-3-642-34575-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)