
Abstract
Induction of enzymes that enhance the detoxication of chemical carcinogens has been a broadly effective strategy for chemoprevention of experimental carcinogenesis in rodent models. Several inducing agents are now in clinical trials to evaluate utility for prevention of cancers associated with unavoidable high exposures to environmental carcinogens. The successes of these pre-clinical and clinical interventions lead to studies to define the molecular basis for protection by these agents, which now include phenolic antioxidants, dithiolethiones, isothiocyanates, and triterpenoids. In the mid-1990s, the NF-E2-related factor 2 (Nrf2) transcription factor was identified as a key regulator of the inducible expression of enzymes such as glutathione S-transferases and NAD(P)H: quinone oxidoreductase in catalyzing the detoxication of reactive electrophiles and oxidants that contribute to the formation of mutations and ultimately cancers. Nrf2 is now recognized to regulate a broad cytoprotective, transcriptional response leading to prevention of damage to DNA, proteins and lipids; recognition, repair and removal of macromolecular damage; and tissue renewal following toxic assaults. Highlighting the importance of this pathway as a determinant of susceptibility to carcinogenesis, multiple studies now demonstrate enhanced incidence, multiplicity, and/or tumor burden in Nrf2-disrupted mice compared to wild-type in models of inflammation and colon cancer, bladder cancer, lung disease and cancer, stomach cancer, mammary cancer, skin cancer, and hepatocarcinogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The NF-E2-related factor 2 (Nrf2) transcription factor-signaling pathway has been targeted for prevention of chemical carcinogenesis, both in animal models and in clinical trials well before its initial molecular characterization in the late 1990s. In the early 1970s, Wattenberg and colleagues established that phenolic antioxidants such as butylated hydroxyanisole and butylated hydroxytoluene were effective anticarcinogens in rodents, especially when administered prior to carcinogen challenge (Wattenberg 1972). Early mechanistic studies focused on the possibility that elevation of cellular glutathione levels or of glutathione-utilizing enzymes such as glutathione S-transferases (GSTs) by these antioxidants would lead to protection against chemical carcinogenesis. GSTs, now known to be regulated in part through Nrf2, were known at that time to detoxify the electrophilic intermediates of some carcinogens. In particular, Benson et al. (1978) showed that liver cytosols from butylated hydroxyanisole-fed mice exhibited much higher GST activities than controls and that cytosols prepared from the livers of these rodents eliminated the mutagenic activity in urine from mice treated with the carcinogen benzo[a]pyrene (Benson et al. 1978). Subsequently, the concerted induction of multiple carcinogen detoxication enzymes was found to occur in many tissues of the mouse leading to the hypothesis that a broad-based approach to chemical protection against carcinogenesis, mutagenesis, and other forms of toxicity would be the modulation of enzymes involved in the metabolism and disposition of the reactive intermediates of toxicants, namely, electrophiles and free radicals (Bueding et al. 1982; Benson et al. 1978). Substantial experimental evidence has been developed to support the view that induction of such cytoprotective enzymes is a critical and sufficient mechanism to engender protection against carcinogenesis provoked by environmental (i.e., dietary and airborne carcinogens) and endogenous (i.e., inflammatory states) factors. Since the validation of enzyme induction as a successful means for prevention in scores of animal models of chemical carcinogenesis, there has been an explosion of knowledge on several fronts. Foremost has been the molecular dissection of the pathway by which the initial classes of enzyme inducers acted. These studies have been abetted by the utilization of genetically engineered mice in which components of the Keap1-Nrf2-signaling pathway have been disrupted, either globally or by tissue-specific means. A general theme that emerges is that mice in which Nrf2 has been genetically disrupted are more sensitive to chemical carcinogenesis. Moreover, the chemopreventive activities of several classes of enzyme inducers, including phenolic antioxidants, dithiolethiones, isothiocyanates, and triterpenoids, are lost in these pathway-deficient mice. Additionally, comparisons of the pharmacodynamic actions of these inducers, especially at low concentrations, to those of genetic activation of the pathway highlight a strong convergence of qualitative if not quantitative action. Pleiotropic effects of these inducers, such as induction of apoptosis, are seen at higher concentrations. Emerging insights into the dark side of the Nrf2 pathway, namely, its propensity to be usurped through mutation and epigenetic marks by cancer cells, wherein a pro-survival, chemoresistant phenotype is facilitated, also point to a key role for the pathway as a multifaceted determinant of susceptibility to carcinogenesis.
Keap1-Nrf2 signaling
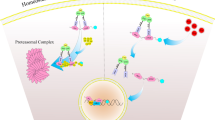
The Keap1-Nrf2-signaling axis provides a broad-based cytoprotective response toward disruption of cellular homeostasis by extrinsic and intrinsic stresses. The current model of Keap1-Nrf2 interactions, as depicted in Fig. 1, and as addressed in recent reviews (Dinkova-Kostova et al. 2005; Hayes and McMahon 2009), involves the Kelch domains of a Keap1 homodimer functionally interacting with two different sites within the Neh2 domain of Nrf2, the ETGE, or high affinity ‘hinge’ site, and the DLG, the lower affinity ‘latch’ site. Under normal cellular conditions, Tong et al. (2006) propose that Nrf2 first interacts with the Keap1 dimer through the ETGE hinge interaction, tethering Nrf2 to the Keap1 homodimer, and subsequently the Cul3-Rbx1 complex which, following the stable interaction of Nrf2 to Keap1 through the DLG latch motif, leads to the appropriate orientation of proteins to facilitate the ubiquitination and subsequent proteasomal targeting as well as destruction of Nrf2. Upon cellular stress or pharmacologic induction, the ability of Keap1 to maintain both points of contact, the hinge and the latch, is thought to be disrupted by the alteration of the tertiary or quaternary structure of the Keap1 homodimer, accomplished via alterations of the many reactive cysteines within Keap1 through oxidation or covalent modification. The disruption of this efficient turnover of Nrf2 allows for the accumulation of the protein and permits Nrf2 to translocate into the nucleus. Once within the nucleus, Nrf2 forms heterodimers with small Maf proteins, and drives the transcription of genes with a functional Antioxidant Response Element (ARE) within their promoters (Nioi et al. 2003; Malhotra et al. 2010). These genes include, but are not limited to, conjugation/detoxication proteins, antioxidative enzymes, anti-inflammation proteins, the proteasome, and cellular chaperones, creating a general cytoprotective response following pathway activation. Recently, the response of Nrf2 has been broadened in scope, with research documenting interactions between Nrf2 and Notch signaling (Wakabayashi et al. 2010a), p53/p21 (Chen et al. 2009), p62-based autophagy (Komatsu et al. 2010; Lau et al. 2010), aryl hydrocarbon receptor signaling (Shin et al. 2007; Miao et al. 2005), and other processes (Wakabayashi et al. 2010b). These interactions provide the means to elicit the broad-based cell survival responses that now typify the pathway.

General schematic for the Keap1-Nrf2 signaling axis. Under homeostatic conditions, Nrf2 is bound by Keap1 through the ‘hinge’ (ETGE) and ‘latch’ (DLG) domains of Nrf2. Following binding, Nrf2 is ubiquitinated by the Cul3/Rbx1/E2 ubiquitin ligase complex, marking it for proteasomal degradation. Induction of Nrf2 signaling by pharmacologic agents or endogenous cellular stress (see Table 1) leads to a disruption of the interaction between the ‘latch’ motif of Nrf2 and Keap1. This interaction is disrupted via tertiary or quaternary structural changes in the Keap1 dimer, likely by alteration of reactive cysteines within Keap1. This disruption leads to an inefficient ubiquitination of Nrf2, allowing for accumulated Nrf2 to translocate into the nucleus where Nrf2 interacts with small Maf and co-activator proteins, binds to AREs in promoters of target genes, and drives transcription of cytoprotective genes.
Genetic mouse models
The genetic manipulations of the Keap1-Nrf2 pathway have produced a number of transgenic mice, with one of the most useful varieties being the knock out of Nrf2. This mouse has no gross phenotypes within the laboratory setting (Chan et al. 1996); however, it displays impaired homeostasis of reactive oxygen species (ROS), a lupus-like autoimmune syndrome (Ma et al. 2006) and is more sensitive to toxic insult. The sensitivity to toxic insult has been shown in a variety of circumstances, including demonstrations of acute toxicity as well as prolonged exposure to both toxic compounds and carcinogens including acetaminophen (Enomoto et al. 2001; Chan et al. 2001), ovalbumin (Rangasamy et al. 2005), cigarette smoke (Rangasamy et al. 2004; Iizuka et al. 2005), pentachlorophenol (Umemura et al. 2006), 4-vinylcyclohexene diepoxide (Hu et al. 2006), benzo[a]pyrene (Ramos-Gomez et al. 2001), diesel exhaust (Aoki et al. 2001) and N-nitrosobutyl (4-hydroxybutyl) amine (Iida et al. 2004). In order to examine the situation where Nrf2 signaling is upregulated, a Keap1 knock-out mouse was created; however, this genetic construct is lethal at approximately 3 weeks post-birth, due to hyperkeratosis of the esophagus and forestomach. Prior to death, however, these mice demonstrate a high level of Nrf2 signaling and within the liver show an upregulation of detoxication enzymes including GSTs as well as Nqo1 (Wakabayashi et al. 2003). In order to circumvent this consequence of upregulated Nrf2 signaling, a hepatocyte-specific Keap1 knock-out mouse was created, utilizing flanking loxP sites and a Cre recombinase driven under the albumin promoter. This mouse, hereby referred to as a CKO mouse (conditional Keap1 knock-out), demonstrates no overt phenotypic effects of the upregulated Nrf2 signaling within the liver; however, the upregulation of signaling confers enhanced expression of cytoprotective enzymes and demonstrated protection against acetaminophen poisoning (Okawa et al. 2006). These mice do exhibit a shortened lifespan, indicating that sustained, constitutive activation of Nrf2 is disadvantageous to long-term survival (Taguchi et al. 2010). This transgenic mouse allows for a genetic upregulation of the Keap1-Nrf2-signaling axis, giving another angle of inquiry into the consequences of the alteration of Nrf2 signaling.
The general cytoprotective response of activation of the Nrf2 pathway has made it an attractive target for pharmacological intervention, with a major driving force behind the research being that of chemoprevention. Numerous classes of compounds are known to activate Nrf2 signaling through interaction with Keap1, with the more prominent molecules including dithiolethiones (e.g., olitpraz Egner et al. 1994), isothiocyanates (e.g., sulforaphane Dinkova-Kostova et al. 2007), and triterpenoids (Bensasson et al. 2010) (e.g., CDDO-Im Yates et al. 2007; Liby et al. 2005), with a detailed list in Table 1. These three examples demonstrate a dynamic range of activating potential with a 1,000-fold difference in Nrf2 induction as measured by the induction of ARE-driven genes (Kensler and Wakabayashi 2010). This difference in potency may reflect mechanism of interaction with sensor cysteines in Keap1 (Kobayashi et al. 2009; Dinkova-Kostova et al. 2010). Of these prototypic agents, oltipraz and sulforaphane have either undergone or are actively undergoing clinical trials as chemoprevention agents. Triterpenoids are just entering clinical development as therapeutic agents, but also have a huge potential for chemoprevention.
These drugs are often utilized in conjunction with Nrf2−/− mice in order to demonstrate that the effects shown in a drug trial against carcinogens are due to upregulation of the Keap1-Nrf2-signaling axis. With that said, of the compounds utilized to induce Nrf2 signaling, many are relatively unspecific, with the exact mode of action being unknown. The use of pharmacologic induction in conjunction with Nrf2−/− mice provides solid evidence of the role of Nrf2 signaling within the experimental context being explored. Additionally, a comparison of gene expression patterns by microarray between the effects of pharmacologic dosing with CDDO-Im with that of genetic alteration via Keap1 disruption in the CKO mouse allow for the assertion that the primary mode of action of CDDO-Im at low doses is that of a specific activator of Nrf2 (Yates et al. 2009). Higher concentrations affect additional targets (Liby et al. 2007), which may in fact also contribute to their protective actions. Sulforaphane, for example, has been shown to cause apoptosis (Gamet-Payrastre et al. 2000; Pledgie-Tracy et al. 2007) and inhibition of proliferation, angiogenesis, and metastasis (Zhang and Tang 2007). Pharmacologic induction of the pathway, however, allows for pulsed induction rather than permanent induction of the Keap1-Nrf2-signaling axis, which may reduce any untoward effects of constant pathway activation.
The role of the Keap1-Nrf2-signaling axis on carcinogenesis
Inflammation and colon cancer
The role of Nrf2 in protection against numerous acute toxicities and carcinogenesis has been investigated, within the contexts of genetic ablation of Nrf2 as well as pharmacologic interventions. For example, Nrf2 is an effective modifier of inflammatory-based promotion of carcinogenesis within the setting of colorectal cancer. Osburn et al. (2007) as well as Khor et al. (2006) have examined the effect of Nrf2 genotype on the susceptibility of mice to azoxymethane (AOM), and dextran sulfate sodium (DSS) induced colorectal cancer. Within this model of colon cancer, AOM is utilized as a potent initiator and is followed up by exposure to DSS as a promoter via induction of inflammation within the colon. Khor et al. (2006) found that following 1 week of AOM/DSS treatment of both wild-type and Nrf2−/− mice, Nrf2−/− mice demonstrate reduced length of colon as well as an increased severity of colitis. In an additional study, Khor et al. (2008) show that following 20 weeks of AOM/DSS treatment, Nrf2−/− mice show a 40% higher incidence of colonic tumors, with 80% of those tumors being adenocarcinomas versus adenomas. Osburn et al. (2007) also demonstrate that following dosage with the regimen of AOM/DSS, Nrf2−/− mice demonstrate significantly higher levels of inflammatory cytokine mRNAs, polymorphonuclear leukocyte invasion within the colon, and a higher level of protein oxidation when compared with wild-type mice. Within this experimental setting, Nrf2 was shown to attenuate inflammatory damage within the tissue by counteracting the generation of ROS and subsequent macromolecular damage, thereby removing the need to create a microniche of pro-growth within the tissue to compensate for any damage. The activity of Nrf2 thereby reduces ROS generation, a source of potential mutagens, and minimizes the generation of a tumor-promoting microniche within the colonic crypts.
Bladder cancer and interactions with p53
Iida et al. (2004) have shown that Nrf2 plays a role in the detoxification of N-nitrosobutyl(4-hydroxybutyl)amine (BBN), a carcinogen derived metabolically from an N-nitroso compound found in cigarette smoke which acts, when given orally to mice, as a urinary bladder carcinogen. Within the study, it was shown that bladder cancer incidence, following treatment with BBN, was highest within Nrf2−/− mice at 65% incidence, followed by wild-type at 36%, and the lowest incidence of cancer was present within wild-type mice dosed with oltipraz at 14%. Iida et al. then show that UDP-glucuronosyltransferase (UGT) is upregulated within the wild-type and oltipraz-treated mice when compared with Nrf2−/− mice, shunting the carcinogen through a known metabolic pathway leading to detoxication and excretion when Nrf2 is activated. This upregulation is also seen when examining cellular expression of GSTπ. Additionally, Iida et al. show through HPLC that within the urine of Nrf2−/− mice, N-nitrosobutyl(3-carboxypropyl)amine, a carcinogenic metabolite of BBN, is present in significantly higher quantities when compared to wild-type mice in either the oltipraz treated or untreated groups. This reduction was not seen, however, in Nrf2−/− mice treated with oltipraz, demonstrating the efficacy of the Nrf2 pathway both alone and in combination with pharmacologic activation in reducing the carcinogenic potential of BBN. The carcinogenic potential of BBN in bladder cancer has also been shown to be modulated by Nrf2 through a synergistic effect with p53 (Iida et al. 2007). Iida et al. demonstrated that compound transgenic mice (Nrf2−/− genotype together with a mutation in a single p53 allele) dosed with BBN have a significantly higher incidence of bladder cancer than that of solely an Nrf2−/− mouse or a p53+/− mouse. The Nrf2−/−::p53+/− mice also demonstrate a significantly higher rate of muscle-invasive cancer when compared with the Nrf2−/−, p53+/− or wild-type mice. This demonstrates the cooperative nature of Nrf2 and p53 in defense against carcinogenic insults, with a two-pronged defensive mechanism. Nrf2 acts to detoxify compounds, and p53 initiates cell cycle checkpoints to either allow for DNA and cellular repair or begins the induction of cellular apoptosis in order to remove potentially harmful cells. Lines of direct interactions between these two pathways are beginning to emerge (Wakabayashi et al. 2010b; You et al. 2011).
Lung disease and cancer
Nrf2 has also been shown to be involved in the response of the lung to numerous toxicological insults including exposure to diesel exhaust and cigarette smoke (Iizuka et al. 2005; Sussan et al. 2009). Aoki et al. (2001) demonstrated that following exposure to diesel exhaust, Nrf2−/− mice have significantly higher levels of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-OHdG) DNA adducts as well as a higher extent of hyperplasia when compared with Nrf2+/−, a genotype that is comparable with wild-type mice in expression of cytoprotective genes. Nrf2 has also been shown to modulate the effects of cigarette smoke exposure within the lungs. Lizuka et al. (2005) as well as Rangasamy et al. (2004) have shown that Nrf2−/− mice are significantly more susceptible to cigarette smoke-induced emphysema and lung damage when compared with wild-type mice. Iizuka et al. demonstrated this vulnerability following both an 8- and 16-week exposure, as shown by levels of inflammatory signals, macrophage, and neutrophil counts. This phenotype is accounted for by a reduction in the ability of Nrf2−/− mice to increase levels of antioxidant genes following exposure to smoke, as well as an impairment of macrophages to phagocytose neutrophils leading to an increase in elastase activity. Rangasamy et al. (2004) demonstrated an earlier onset, and increase in severity of cigarette smoke induced emphysema in Nrf2−/− mice versus their wild-type littermates. The effect of pharmacological activation of the Nrf2 pathway on the effects of cigarette smoking has also been examined, where mice were dosed with CDDO-Im at 90 mg/kg of diet during a 6-month (5-day/week; 5-h/day) exposure to cigarette smoke. The CDDO-Im-treated mice showed a significant reduction in alveolar destruction, cigarette smoke induced apoptosis and oxidative stress, and DNA damage as shown by examination of 8-OHdG adducts (Sussan et al. 2009). This reduction in damage was attributed to an increase in antioxidant gene expression caused by the upregulation of Nrf2 signaling via pharmacologic induction.
Stomach cancer
Nrf2 has also been shown to be involved in the detoxication of benzo[a]pyrene, a potent pro-carcinogen formed by the incomplete combustion of carbon. The impact of Nrf2 on benzo[a]pyrene carcinogenicity has been examined both in Nrf2−/− mice as well as pharmacologically induced mice with different activators of the Nrf2 pathway, oltipraz (Ramos-Gomez et al. 2001, 2003) and sulforaphane (Fahey et al. 2002) via exposure to Nrf2-activating compound either prior to or concurrent with exposure to benzo[a]pyrene. The activation of the Nrf2 pathway leads to an increase in the activity of conjugating enzymes, allowing for increased conjugation and excretion of benzo[a]pyrene, thereby reducing the toxicological impact (Fig. 2). These induced changes allow for a reduction in tumor incidence within the forestomach that is not seen within the Nrf2−/− genotype, indicating that the pharmacological activation of Nrf2 is responsible for the diversion of benzo[a]pyrene through a less harmful metabolic pathway. Moreover, wild-type mice dosed with vehicle show a lower multiplicity of forestomach tumors than Nrf2−/− mice, further reinforcing the role of Nrf2 as a cytoprotective transcription factor.

The role of Nrf2 in the metabolism of B[a]P. a Nrf2 serves to upregulate various enzymes involved in the detoxication of B[a]P (Uppstad et al. 2010). b The effect of Nrf2 genotype on benzo[a]pyrene metabolism through DNA adducts or tumorigenesis (left and right panel, respectively). B[a]P adducts were measured as total tetrols released from DNA isolated from mouse forestomach 24-h post-exposure. Tumorigenesis was evaluated 30 weeks after the first of four weekly exposures to B[a]P. Oltipraz or vehicle was administered 48 h prior to B[a]P exposure (Ramos-Gomez et al. 2001, 2003)
Skin cancer
The effect of Nrf2 activation has also been examined within the context of the carcinogen 7,12-dimethylbenz(a)anthracene (DMBA) as an initiator of skin cancer. Xu et al. (2006) examined the consequences of both the pharmacological induction of Nrf2 by topical application of sulforaphane as well as the genetic disruption of Nrf2 on the carcinogenic potential of DMBA followed by promotion via the repetitive application of 12-O-teradecanoylphorbol-13-acetate (TPA). Within this experimental context, Nrf2−/− mice developed significantly more skin tumors than did wild-type mice, 95 to 45%, respectively. The induction of the Nrf2 pathway by treatment of the skin with sulforaphane prior to application of DMBA led to a threefold reduction in tumor burden in the wild-type mice, from 60 to 20%, but was ineffective in the Nrf2−/− mouse, demonstrating an Nrf2-dependent protection from tumorigenesis following DMBA/TPA treatment.
Mammary cancer
The role of Nrf2 has also been examined in mammary gland carcinogenesis. Becks et al. (2010) induced female mice with medroxyprogesterone acetate, followed by treatment with DMBA and examined the influence of both Nrf2 genotype, as well as the potential induction of Nrf2 via the citrus coumarin auraptene. The Nrf2−/− mice, when compared to the wild-type controls, had the same occurrence of tumors; however, the masses of the tumors in the Nrf2−/− mice were significantly larger than those within the wild-type. This suggests a potential role for Nrf2 in regulating the malignant progression of these mammary tumors. Activation of antioxidant signaling by auraptene treatment, as shown by GST activity, had no effect on carcinogenesis within this study.
Endogenous ROS
Nrf2 has also been shown to play a role in the modulation of endogenous stresses, specifically in the case of an increase in intracellular ROS when Nrf2 expression is lowered or knocked out. Frohlich et al. (2008) demonstrated this phenomenon within constructs of prostate cancer cell lines, as well as in Nrf2−/− cells. With a reduction or elimination of Nrf2 expression, intracellular ROS is increased due to a decrease in the expression of antioxidative enzymes. This creates an increase in oxidative damage within the cell, which manifests in increased DNA damage as shown by comet assay. It is easy to concede that an increase in oxidative damage to DNA has the potential to create mutagenic lesions, which if located in the correct area of the genome, can lead to harmful accrued mutations and subsequently, initiation and promotion of cancer (Yu and Kensler 2005; Kwak et al. 2004).
Hepatocarcinogenesis
Nrf2 has also been shown to play a role in mediating hepatocarcinogenesis following exposure to multiple toxins in the mouse. Umemura et al. (2006) have shown that Nrf2−/− mice are more susceptible to pentachlorophenol (PCP) toxicity than the wild-type controls within the study. Following exposure to 1,200 ppm doses of PCP for 4 weeks, Nrf2−/− mice showed significantly higher levels of 8-OHdG within the liver, as well as higher levels of fatty acid oxidation. Additionally, Nrf2−/− mice demonstrate significantly higher levels of cellular proliferation following exposure to PCP as measured through BrdU-staining. Umemura, and colleagues observed a reduction in both Nqo1 as well as UDP-glucuronosyltransferase expression in the Nrf2−/− mice, demonstrating a reduction in detoxication enzymes that contribute to the increase in cellular insult by PCP. Additionally, Kitamura et al. (2007) have shown that Nrf2−/− mice demonstrate an increased susceptibility to hepatocarcinogenesis following exposure to 2-amino-3-methylimidazo[4,5-f-quinoline (IQ). Kitamura and colleagues show that Nrf2−/− mice display significantly higher levels of hepatocellular adenomas and hepatocellular carcinomas following a 52-week exposure of IQ in the diet. A second experiment within the study demonstrates a significantly higher incidence of preneoplastic and neoplastic lesions within the forestomach of Nrf2−/− mice following a 4-week exposure to IQ. This study also demonstrated a depression in UGT and GST activity within the liver of Nrf2−/− mice when compared to control following exposure to IQ. This result, in conjunction with a demonstration of increased fatty acid oxidation, a higher level of cellular proliferation, and higher levels of DNA adduct formation within the livers of Nrf2−/− mice following IQ treatment, show that Nrf2 deficiency leaves these mice in a compromised position of hepatocellular integrity. Within all of these examples, Nrf2−/− mice show an increased sensitivity to cellular stressors and carcinogens, demonstrating the importance of functional Nrf2 in combating toxicological insults through alterations of the cellular defense mechanism, and an increase in detoxification enzymes leads to a reduction in exposure of toxin, leading to lowered risks of mutations and subsequent disease.
The mycotoxin aflatoxin B1, produced by the fungus Aspergillus, has been shown to be a potent hepatic carcinogen in humans (Ross et al. 1992; Sun et al. 1999), rats (Wogan et al. 1974), and infant mice (Vesselinovitch et al. 1972) among many species. This carcinogen is present within staple foods around the world, especially corn and peanuts. This widespread contamination makes aflatoxin an important target for chemopreventive efforts, as while the contamination in foods can be minimized, it is nearly impossible to eradicate. Aflatoxin, once ingested, is metabolically activated to the AFB1-8-9-exo epoxide by Cyp450, which then rapidly binds to DNA, forming an N7-guanine adduct, which in turn is rapidly depurinated from the DNA-yielding abasic sites which hold the potential to lead to mutations within the cell. Nrf2 has been shown to modulate this metabolic pathway. Yates et al. (2006) have shown that CDDO-Im confers protection against aflatoxin-induced tumorigenesis in the rat. Graded dosing with CDDO-Im resulted in reductions in the hepatic burden of GSTπ-positive foci, a presumptive marker of precancerous lesions, within the liver from 39% up to a complete elimination when compared to control. In order to implicate the Keap1-Nrf2-signaling axis into the mechanism of cancer control in the rat, analysis of numerous Nrf2 target genes was examined; expression of Nrf2 targets Nqo1, Gstα, aflatoxin aldehyde reductase (AFAR) and Hmox1 expression were all elevated. Due to the lack of an Nrf2−/− rat model, the pharmacologic effects of CDDO-Im were investigated in both wild-type and Nrf2−/− mice by microarray, which implicated Nrf2 in the CDDO-Im-based induction of many of these cytoprotective enzymes. It is likely that Nrf2 activation by CDDO-Im is at least partially responsible for the reduction in aflatoxin-based carcinogenesis within the rat; however, the field of aflatoxin research would be well served with an Nrf2−/− rat in order to further investigate the role of Nrf2 in the resistance of aflatoxin toxicity and carcinogenesis.
In an effort to examine the effects of the upregulation of AFAR on the processing of AFB1, one of the consequences of CDDO-Im intervention, a transgenic rat overexpressing AFAR was created. When this rat was challenged with AFB1, higher levels of the AFB1 dialcohol metabolites were excreted, with a concurrent reduction in AFB1-lysine adducts which are formed from the same precursor aflatoxin-dialdehyde metabolite; however, this alteration in metabolism did not significantly impact either the acute toxic or tumorigenic effects of AFB1 dosage as measured by bile duct proliferation or GSTπ positive foci, respectively (Roebuck et al. 2009).
A complication in the examination of the role of the Keap1-Nrf2-signaling axis in aflatoxin-induced carcinogenesis within the mouse is, with the exception of the infant mouse (Vesselinovitch et al. 1972), the wild-type mouse is refractory to the effects of aflatoxin due to high levels of expression of the mouse isoform of glutathione S-transferase A3 (Gsta3), an Nrf2-regulated gene that functions to detoxify the mutagenic aflatoxin-epoxide metabolites (Jowsey et al. 2003). Llic et al. (2010) have developed a Gsta3 knock-out mouse, which has been shown to be sensitive to the acute cytotoxic and genotoxic effects of AFB1; however, as of yet, this mouse model has not been examined in its ability to develop AFB1-induced tumors.
Dark side of Nrf2
Recently, Nrf2 has been shown to be upregulated in numerous types of cancer, indicating a potential role in the promotion of malignancy. A variety of tumor types, including primary head and neck cancers (Stacy et al. 2006) lung tumors (Shibata et al. 2008b; Ohta et al. 2008; Singh et al. 2006), skin (Kim et al. 2010), gallbladder (Shibata et al. 2008a), breast (Nioi and Nguyen 2007), prostate (Zhang et al. 2010), and endometrial cancers (Jiang et al. 2010) have been shown to have upregulated Nrf2 signaling. Mutations in Keap1 and Nrf2, principally in sequences affecting the interaction domains between these two proteins, are observed in these settings.
It is hypothesized that this upregulation in Nrf2 signaling, and subsequent increase in cellular defensive enzymes, gives tumors an advantage, not only over a challenging tumor microenvironment, but leads to drug resistance, as many standard chemotherapeutics agents are cytotoxic. With the increased levels of protective enzymes as well as drug exporters, many of the cytotoxic effects of chemotherapeutics can be abrogated by the tumor cells by upregulation of Nrf2 signaling, thereby yielding a tumor more suited to aggressive growth and expansion and more capable of escaping anticancer therapy.
Nrf2 may also play into the phenotype of more aggressive growth directly through interactions with Notch1, a cell fate decision molecule, of which Nrf2 has been shown to directly contribute to its expression (Wakabayashi et al. 2010a). An upregulation of Nrf2 thereby has the potential to increase the expression levels of Notch1 within cells, leading to an increase in proliferation rate and a more malignant phenotype. Additionally, Nrf2 has been observed to interact with the p53 pathway via contribution to the basal expression of Mdm2, a direct inhibitor of p53 (You et al. 2011). An increase in the expression levels of Nrf2 could thereby increase the inhibition of p53, as well as confounding any ROS-based apoptotic signals through detoxication, enhancing cellular survival in the face of the challenging tumor microenvironment (Wakabayashi et al. 2010b). Any upregulation in Nrf2 signaling, therefore, has the potential to lead to a more aggressive growth phenotype of tumor in the face of both inherent (e.g., ROS) and external (chemotherapeutics) challenges.
Nrf2 has been shown to facilitate the drug-resistant tumor phenotype in numerous manners, with one instance being that of Nrf2 regulating the expression of the multidrug-resistant protein-3 (MRP3) in both human bronchial epithelial and non-small-cell lung carcinoma (Mahaffey et al. 2009). MRP3 is a member of the multidrug resistance protein family, which, when combined with an upregulation of detoxication enzymes, many of which are direct targets of Nrf2 signaling including GSTs, can lead to the increased hydrophilicity, and excretion of a variety of cytotoxic agents utilized in chemotherapy including chlorambucil, cisplatin, etoposide, and doxorubicin (Wang et al. 2008; Meijerman et al. 2008). The ability of Nrf2 to influence both of these actions, the increase in detoxication enzymes as well as the expression level of MRPs, demonstrates a powerful combination of alterations allowing for an increase in tumor cell survival in the face of cytotoxic agents.
The ability of Nrf2 to modulate the effects of chemotherapeutic drugs has also been shown within the context of pancreatic cancer cell lines by Hong et al. (2010). Within this study, upregulation of Nrf2 protein levels is shown in pancreatic cancer cell lines when compared with normal pancreatic cell lines indicating the phenotype of increased Nrf2 signaling promoting a pro-cell survival phenotype. This is reinforced by the examination of the protein levels of numerous direct downstream targets of Nrf2 including HMOX1, NQO1, and GCLC. Additionally, increasing the levels of Nrf2 through transfection in pancreatic cell lines conferred resistance to the cytotoxic agents cisplatin and camptothecin, while the reduction in Nrf2 signaling through the transfection of a dominant negative construct of Nrf2 leads to a diminuation of cell survival rates in the face of such cytotoxic agents.
Upregulation of Nrf2 signaling within the tumor environment has also been observed within lung cancer. Shibata et al. (2008b) observed a number of mutations within the Keap1-Nrf2-signaling axis serving to upregulate Nrf2 signaling, both within lung tumors, and lung cancer cell lines. Shibata et al. also discovered reduced levels of expression of Keap1 within both primary lung cancer tissues and in lung cancer cell lines, indicated that Keap1 activity may be abrogated not only by introduced mutations, but by epigenetic or other methods of control of protein expression levels, in order to ensure the phenotype of increased Nrf2 signaling, and the benefits therein. Also, within the vein of lung cancer research, Singh et al. (2010) have shown that a gain of Nrf2 function confers radioresistance within non-small-cell lung cancer cells. Singh and colleagues hypothesize that this gain in radioresistance is acquired through the increased levels of antioxidative and cytoprotective enzymes that exist within the battery of Nrf2 target genes. This idea is supported by the observation that the disruption of Nrf2 signaling supplemented with the application of exogenous antioxidants prior to exposure to radiation leads to an abrogation of radiation-induced cell death. The utilization of Keap1−/− cells, mimicking the mutations found naturally within tumors, also confers radioresistance to cells, demonstrating an additional consequence of the upregulation of Nrf2 within tumors.
Disruption of Keap1 either through genetic mutation or deletion has also been found to occur in biliary tract cancer, with Shibata et al. (2008a) finding somatic mutations within 11.6% of tumors analyzed. These mutations, when introduced into gallbladder cancer cell lines, were found to abrogate the ability of Keap1 to catalyze the ubiquitination and subsequent degradation of Nrf2, allowing for elevated activity of Nrf2 signaling. The upregulation of Nrf2-based cytoprotective signaling leads to a resistance to 5-fluorouracil (5-FU), via neutralization of 5-FU-generated mitochondrial ROS and subsequent apoptotic signaling. The opposite of this effect is true in cell lines with a reduction in Nrf2 signaling demonstrating a survival advantage conferred by an increased Nrf2 signaling.
Nrf2, a multitiered response signaling protein
Nrf2 may protect against carcinogenesis by means other than the detoxication of xenobiotics and cellular toxicants. Nrf2 has been shown to interact with numerous other cellular pathways, including the pro-growth cell differentiation factor Notch1 (Wakabayashi et al. 2010a). In the recent examination of this interaction, Wakabayashi and colleagues demonstrate that Nrf2 is at least partially responsible for the expression of Notch1 in adult mice, as demonstrated though the rescue of the delay in lever regeneration following partial hepatectomy in Nrf2−/− mice by expression of a liver-specific Notch1 intracellular domain (NICD) construct. This interaction demonstrates that Nrf2 not only plays a role in the immediate damage control response to cellular toxic via enzyme induction, but that Nrf2 plays a role in a damaged tissue recovery process. This specific example is not isolated, as Nrf2 has recently been described to interact with a host of cellular signaling networks and processes including p62-mediated autophagy (Komatsu et al. 2010; Lau et al. 2010), NF-κB signaling (Liu et al. 2008), and p21/p53-based signaling (Chen et al. 2009; Wakabayashi et al. 2010b). These interactions show that Nrf2 responds to toxicological insults in a multitiered manner (Table 2). The primary response involves damage prevention through detoxication, conjugation, and excretion of compounds to eliminate reactive intermediates of oxidants and electrophiles. A secondary response entails damage control through increases in DNA repair (Yates et al. 2009), proteasomal activity (Kwak et al. 2003) as well as autophagy (Komatsu et al. 2010; Lau et al. 2010) in order to facilitate recognition, repair and removal of damaged macromolecules from the cell. A tertiary response facilitates cell renewal through interactions with factors such as Notch1 in order to allow for the regrowth of damaged tissues (Wakabayashi et al. 2010a).
Nrf2 functions as a double-edged sword in cellular defense signaling. Under a disruption of cellular homeostasis within a normal, functioning cell, Nrf2 catalyzes the general cytoprotective response to any number of insults that may affect a cell. In the case of a malfunctioning Keap1-Nrf2-signaling axis, however, unfettered Nrf2 signaling allows for protection of cells against numerous chemotherapeutic and cytotoxic agents. The Keap1-Nrf2-signaling axis, thereby, allows two different potential outcomes to pharmacologic interventions. First, the transient upregulation of Nrf2 signaling within the context of chemoprevention against exposure to harmful agents, in order to heighten their detoxification and excretion, and second, the interruption of Nrf2 signaling allowing for the sensitization of chemoresistant tumors to chemotherapeutic and cytotoxic agents. In this setting, inhibitors of the Nrf2 pathway may exhibit therapeutic utility (Ren et al. 2011). Greater understanding of the impacts of diminished and amplified Nrf2 signaling in normal and malignant tissues will be required to safely and effectively utilize small molecule activators and inhibitors as modifiers of multiple stages of carcinogenesis.
References
Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M (2001) Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol 173(3):154–160
Becks L, Prince M, Burson H, Christophe C, Broadway M, Itoh K, Yamamoto M, Mathis M, Orchard E, Shi R, McLarty J, Pruitt K, Zhang S, Kleiner-Hancock HE (2010) Aggressive mammary carcinoma progression in Nrf2 knockout mice treated with 7, 12-dimethylbenz[a]anthracene. BMC Cancer 10:540
Bensasson RV, Zoete V, Berthier G, Talalay P, Dinkova-Kostova AT (2010) Potency ranking of triterpenoids as inducers of a cytoprotective enzyme and as inhibitors of a cellular inflammatory response via their electron affinity and their electrophilicity index. Chem Biol Interact 186(2):118–126
Benson AM, Batzinger RP, Ou SY, Bueding E, Cha YN, Talalay P (1978) Elevation of hepatic glutathione S-transferase activities and protection against mutagenic metabolites of benzo(a)pyrene by dietary antioxidants. Cancer Res 38(12):4486–4495
Bueding E, Dolan P, Leroy JP (1982) The antischistosomal activity of oltipraz. Res Commun Chem Pathol Pharmacol 37(2):293–303
Chan K, Lu R, Chang JC, Kan YW (1996) NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA 93(24):13943–13948
Chan K, Han XD, Kan YW (2001) An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA 98(8):4611–4616
Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD (2009) Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell 34(6):663–673
Dinkova-Kostova AT, Holtzclaw WD, Kensler TW (2005) The role of Keap1 in cellular protective responses. Chem Res Toxicol 18(12):1779–1791
Dinkova-Kostova AT, Fahey JW, Wade KL, Jenkins SN, Shapiro TA, Fuchs EJ, Kerns ML, Talalay P (2007) Induction of the phase 2 response in mouse and human skin by sulforaphane-containing broccoli sprout extracts. Cancer Epidemiol Biomarkers Prev 16(4):847–851
Dinkova-Kostova AT, Talalay P, Sharkey J, Zhang Y, Holtzclaw WD, Wang XJ, David E, Schiavoni KH, Finlayson S, Mierke DF, Honda T (2010) An exceptionally potent inducer of cytoprotective enzymes: elucidation of the structural features that determine inducer potency and reactivity with Keap1. J Biol Chem 285(44):33747–33755
Egner PA, Kensler TW, Prestera T, Talalay P, Libby AH, Joyner HH, Curphey TJ (1994) Regulation of phase 2 enzyme induction by oltipraz and other dithiolethiones. Carcinogenesis 15(2):177–181
Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, Harada T, Yamamoto M (2001) High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 59(1):169–177
Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, Talalay P, Lozniewski A (2002) Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci USA 99(11):7610–7615
Frohlich DA, McCabe MT, Arnold RS, Day ML (2008) The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 27(31):4353–4362
Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, Gasc N, Tulliez J, Terce F (2000) Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res 60(5):1426–1433
Hayes JD, McMahon M (2009) NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 34(4):176–188
Hong YB, Kang HJ, Kwon SY, Kim HJ, Kwon KY, Cho CH, Lee JM, Kallakury BV, Bae I (2010) Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas 39(4):463–472
Hu X, Roberts JR, Apopa PL, Kan YW, Ma Q (2006) Accelerated ovarian failure induced by 4-vinyl cyclohexene diepoxide in Nrf2 null mice. Mol Cell Biol 26(3):940–954
Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, Shimazui T, Akaza H, Yamamoto M (2004) Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 64(18):6424–6431
Iida K, Itoh K, Maher JM, Kumagai Y, Oyasu R, Mori Y, Shimazui T, Akaza H, Yamamoto M (2007) Nrf2 and p53 cooperatively protect against BBN-induced urinary bladder carcinogenesis. Carcinogenesis 28(11):2398–2403
Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, Morishima Y, Hegab AE, Homma S, Nomura A, Sakamoto T, Shimura M, Yoshida A, Yamamoto M, Sekizawa K (2005) Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10(12):1113–1125
Ilic Z, Crawford D, Vakharia D, Egner PA, Sell S (2010) Glutathione-S-transferase A3 knockout mice are sensitive to acute cytotoxic and genotoxic effects of aflatoxin B1. Toxicol Appl Pharmacol 242(3):241–246
Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, Zhang DD (2010) High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res 70(13):5486–5496
Jowsey IR, Jiang Q, Itoh K, Yamamoto M, Hayes JD (2003) Expression of the aflatoxin B1–8, 9-epoxide-metabolizing murine glutathione S-transferase A3 subunit is regulated by the Nrf2 transcription factor through an antioxidant response element. Mol Pharmacol 64(5):1018–1028
Kensler TW, Wakabayashi N (2010) Nrf2: friend or foe for chemoprevention? Carcinogenesis 31(1):90–99
Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN (2006) Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res 66(24):11580–11584
Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, Cheung WK, Chan JY, Reddy BS, Yang CS, Kong AN (2008) Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila Pa) 1(3):187–191
Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, Eom HS, Yoo NJ, Lee SH (2010) Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol 220(4):446–451
Kitamura Y, Umemura T, Kanki K, Kodama Y, Kitamoto S, Saito K, Itoh K, Yamamoto M, Masegi T, Nishikawa A, Hirose M (2007) Increased susceptibility to hepatocarcinogenicity of Nrf2-deficient mice exposed to 2-amino-3-methylimidazo[4, 5-f]quinoline. Cancer Sci 98(1):19–24
Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, Yamamoto M (2009) The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol 29(2):493–502
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–223
Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW (2003) Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem 278(10):8135–8145
Kwak MK, Wakabayashi N, Kensler TW (2004) Chemoprevention through the Keap1-Nrf2 signaling pathway by phase 2 enzyme inducers. Mutat Res 555(1–2):133–148
Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30(13):3275–3285
Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB (2005) The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 65(11):4789–4798
Liby KT, Yore MM, Sporn MB (2007) Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer 7(5):357–369
Liu GH, Qu J, Shen X (2008) NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta 1783(5):713–727
Ma Q, Battelli L, Hubbs AF (2006) Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol 168(6):1960–1974
Mahaffey CM, Zhang H, Rinna A, Holland W, Mack PC, Forman HJ (2009) Multidrug-resistant protein-3 gene regulation by the transcription factor Nrf2 in human bronchial epithelial and non-small-cell lung carcinoma. Free Radic Biol Med 46(12):1650–1657
Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, Biswal S (2010) Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 38(17):5718–5734
Meijerman I, Beijnen JH, Schellens JH (2008) Combined action and regulation of phase II enzymes and multidrug resistance proteins in multidrug resistance in cancer. Cancer Treat Rev 34(6):505–520
Miao W, Hu L, Scrivens PJ, Batist G (2005) Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem 280(21):20340–20348
Nioi P, Nguyen T (2007) A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun 362(4):816–821
Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD (2003) Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J 374(Pt 2):337–348
Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M, Hirohashi S (2008) Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res 68(5):1303–1309
Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M (2006) Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun 339(1):79–88
Osburn WO, Karim B, Dolan PM, Liu G, Yamamoto M, Huso DL, Kensler TW (2007) Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer 121(9):1883–1891
Pledgie-Tracy A, Sobolewski MD, Davidson NE (2007) Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther 6(3):1013–1021
Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW (2001) Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA 98(6):3410–3415
Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW (2003) Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 24(3):461–467
Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114(9):1248–1259
Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S (2005) Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202(1):47–59
Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, Zhang DD (2011) Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci USA 108(4):1433–1438
Roebuck BD, Johnson DN, Sutter CH, Egner PA, Scholl PF, Friesen MD, Baumgartner KJ, Ware NM, Bodreddigari S, Groopman JD, Kensler TW, Sutter TR (2009) Transgenic expression of aflatoxin aldehyde reductase (AKR7A1) modulates aflatoxin B1 metabolism but not hepatic carcinogenesis in the rat. Toxicol Sci 109(1):41–49
Ross RK, Yuan JM, Yu MC, Wogan GN, Qian GS, Tu JT, Groopman JD, Gao YT, Henderson BE (1992) Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet 339(8799):943–946
Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, Hirohashi S (2008a) Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135(4):1358–1368, 1368 e1351–e1354
Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S (2008b) Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA 105(36):13568–13573
Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, Kensler TW (2007) NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol 27(20):7188–7197
Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S (2006) Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med 3(10):e420
Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S (2010) Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid Redox Signal 13(11):1627–1637
Stacy DR, Ely K, Massion PP, Yarbrough WG, Hallahan DE, Sekhar KR, Freeman ML (2006) Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck 28(9):813–818
Sun Z, Lu P, Gail MH, Pee D, Zhang Q, Ming L, Wang J, Wu Y, Liu G, Wu Y, Zhu Y (1999) Increased risk of hepatocellular carcinoma in male hepatitis B surface antigen carriers with chronic hepatitis who have detectable urinary aflatoxin metabolite M1. Hepatology 30(2):379–383
Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D, Yates MS, Kombairaju P, Yamamoto M, Liby KT, Sporn MB, Gabrielson KL, Champion HC, Tuder RM, Kensler TW, Biswal S (2009) Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA 106(1):250–255
Taguchi K, Maher JM, Suzuki T, Kawatani Y, Motohashi H, Yamamoto M (2010) Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol 30(12):3016–3026
Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M (2006) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 26(8):2887–2900
Umemura T, Kuroiwa Y, Kitamura Y, Ishii Y, Kanki K, Kodama Y, Itoh K, Yamamoto M, Nishikawa A, Hirose M (2006) A crucial role of Nrf2 in in vivo defense against oxidative damage by an environmental pollutant, pentachlorophenol. Toxicol Sci 90(1):111–119
Uppstad H, Ovrebo S, Haugen A, Mollerup S (2010) Importance of CYP1A1 and CYP1B1 in bioactivation of benzo[a]pyrene in human lung cell lines. Toxicol Lett 192(2):221–228
Vesselinovitch SD, Mihailovich N, Wogan GN, Lombard LS, Rao KV (1972) Aflatoxin B 1, a hepatocarcinogen in the infant mouse. Cancer Res 32(11):2289–2291
Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35(3):238–245
Wakabayashi N, Shin S, Slocum SL, Agoston ES, Wakabayashi J, Kwak MK, Misra V, Biswal S, Yamamoto M, Kensler TW (2010a) Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci Signal 3(130):ra52
Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW (2010b) When NRF2 talks, who’s listening? Antioxid Redox Signal 13(11):1649–1663
Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD (2008) Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29(6):1235–1243
Wattenberg LW (1972) Inhibition of carcinogenic and toxic effects of polycyclic hydrocarbons by phenolic antioxidants and ethoxyquin. J Natl Cancer Inst 48(5):1425–1430
Wogan GN, Paglialunga S, Newberne PM (1974) Carcinogenic effects of low dietary levels of aflatoxin B1 in rats. Food Cosmet Toxicol 12(5–6):681–685
Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, Conney AH, Kong AN (2006) Inhibition of 7, 12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 66(16):8293–8296
Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, Honda T, Gribble GW, Sporn MB, Kensler TW (2006) Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-, 12-dioxooleana-1, 9(11)-dien-28-oyl]imidazole. Cancer Res 66(4):2488–2494
Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB, Kensler TW (2007) Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 6(1):154–162
Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, Silkworth JB, Taguchi K, Yamamoto M, Williams CR, Liby KT, Sporn MB, Sutter TR, Kensler TW (2009) Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30(6):1024–1031
You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW, Kwak MK (2011) Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Arch Biochem Biophys 507(2):356–364
Yu X, Kensler T (2005) Nrf2 as a target for cancer chemoprevention. Mutat Res 591(1–2):93–102
Zhang Y, Tang L (2007) Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacol Sin 28(9):1343–1354
Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG, Biswal S (2010) Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther 9(2):336–346
Acknowledgments
We would like to thank the National Institutes of Health for grant support: CA39416, CA94076, ES003819, ES006052. SLS is supported by T32 ES07141.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Slocum, S.L., Kensler, T.W. Nrf2: control of sensitivity to carcinogens. Arch Toxicol 85, 273–284 (2011). https://doi.org/10.1007/s00204-011-0675-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-011-0675-4