
Abstract
Rapidly emerging countries like India and China are lifting millions of people out of poverty. This perpendicular growth in the number of people demanding access to reliable and affordable energy will drive energy demand in the decades to come. Much effort has been employed toward optimizing microbes and predominantly microalgae, to resourcefully produce compounds that can be substitute for fossil fuels. Oils acquired from algal feedstock are rich in triacylglycerols and could be converted into biodiesel via transesterification. Apart from the triacylglycerols and carbohydrates which are predominant in microalgae, there are several biomolecules like pigments and vitamins which play crucial role in pharmaceutical industries. There is an urgent need to understand which drives the production of such economic important biofuels or chemicals to improve the sustainability of the process. Integrative omics is a strong technique to know the complete system of microalgae and develop as microbial cell factories. Genomics and transcriptomics of microalgae have provided basic understanding toward lipid biosynthesis. Proteomics and metabolomics are now complementing “microalgal omics” and offer accurate functional insights into the attendant static and dynamic physiological contexts. Current chapter focuses on the application of omics approaches which considered powerful tools for a better understanding of algae cells metabolism. Then, the data would be used to develop sustainable strategies for biodiesel and by-products yield and quality improvement and a profitable microalgae industry.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The biological and biomedical scientific landscape has seen the escalation in the use and applications of “omics” technologies in the last decade. These technologies offer approaches that allow for a comprehensive description of nearly all components within the cell (Maghembe et al. 2020). Microalgae not only play a significant ecological role, but also of commercial importance. They have emerged as a promising group in the production of bioproducts and biofuel, as well as for the remediation of effluents. Indigenous inhabitants have used microalgae for centuries and the commercial application of microalgae has been extensively reviewed (Bleakley and Hayes 2017). The productivity of the microalgal production process depends on higher biomass, productivity, and yield and process robustness (Ummalyma 2020). These parameters mainly depend on the host microorganism. Natural screening, mutagenesis, bioprocess development, genetic and metabolic engineering strategies have been implemented to enhance the metabolic capabilities of the host microbes (Barh et al. 2013). The problems such as the accumulation of lethal intermediates or metabolic stress consequential decreased cellular fitness need to be solved. The inadequate knowledge about the regulatory mechanisms of key enzymes and the complex associations between genotype and phenotype are still obstacles to the development of efficient cell factories. The introduction of heterologous genes or deletion genes in specific metabolic pathways does not always consequence in the desired phenotype. Currently, remarkable innovations in platforms for omics-based study and application development have imparted crucial solutions to these problems. A combinatorial approach using numerous omics platforms and the incorporation of their outcomes is now an effective strategy for clarifying the molecular systems that are integral to improving algal productivity (Guarnieri and Pienkos 2015; Rai et al. 2016).
2 Proteomics and Molecular Examination of Microalgal Lipid Accumulation
Figure 11.1 shows the diagrammatic representation of identification of potential targets for enhancing the lipid accumulation in microalgae through the proteomic approach. Proteomics based studies are used in several fields such as identification of different diagnostic markers, pathogenicity mechanisms, and biomarkers for vaccine production as well as modification of expression patterns in response to altered signals and interpretation of protein pathways in several diseases (Aslam et al. 2017). Proteomics is basically complex because it contains the analysis as well as the categorization of total protein signatures of a genome. Mass spectrometry along with LC–MS and MALDI-TOF-TOF-MS being frequently used approach, is the main among current proteomics approaches. Though exploitation of proteomics facilities containing the software for equipment and databases as well as the need of skilled personnel substantially raises the costs, hence limit their wider use mainly in the developing world. Additionally, the proteome is an extremely dynamic intricate regulatory system that governs the expression levels of proteins. Microalgae are currently being explored as bio-production platforms for hydrocarbon and lipid-based bioproducts and biofuels. In particular, microalgal triacylglycerides (TAGs) offer a promising feedstock for biodiesel production (Pienkos and Darzins 2009).

Identification of potential targets for increasing TAG accumulation in microalgae by OMICS approach (Arora et al. 2018)
However, despite the historical and recently renewed interest in algae-based fuels, our understanding of regulatory mechanisms governing algal lipid metabolism, particularly the regulation of fatty acid and TAG accumulation, remains incomplete. Identification of key regulators of genes, proteins, and metabolites triggering algal lipid biosynthesis and accumulation opens the door for genetic and metabolic engineering strategies targeting increased rates and absolute quantity of lipid accumulation. As such, a molecular examination of microalgal lipid accumulation mechanisms has recently intensified. The nitrogen (N)-deprivation response is perhaps the best-characterized inducer of lipid accumulation in microalgae (Yang et al. 2016). In addition to increased total lipid content, N-deprivation can also induce changes in fatty acid chain length and saturation, with resultant TAGs more favorable for biofuel conversion (Guarnieri et al. 2011). Transcriptomic profiling has been widely applied to examine this N-deprivation response, yielding characteristic RNA expression signatures associated with deprivation and concurrent lipid accumulation (Radakovits et al. 2012). Such analyses offer powerful insights into the transcriptional regulation of lipid accumulation, but these analyses do not fully define metabolic regulatory control points. This is especially true in algae and higher plants where post-transcriptional and post-translational regulation plays a critical role in protein expression and metabolic regulation (Gillham et al. 1994). Furthermore, mostly employed endpoint analyses (N-replete vs. N-deplete) unsuccessful to completely elucidate the kinetics of expression, which can both illuminate underlying molecular mechanisms governing phenotypic responses and update strain-engineering strategies necessitating induced gene expression. Time-course proteomic analyses provide a means to observe post-transcriptional responses to N-deprivation and concurrent lipid accumulation, which can both complement and further the mechanistic insights gained from transcriptional analyses. However, to date, the accessibility of such proteomic data is relatively limited, in particular for microalgae, compared to that of transcriptomic analyses (Guarnieri et al. 2011; Le Bihan et al. 2011). Microalgae species that have a high lipid content under nitrogen starvation conditions include P. tricornutum (KaiXian and Borowitzka 1993), and the green algae Chlorella spp. (Illman et al. 2000), Botryococcus braunii (Zhila et al. 2005), and Chlamydomonas reinhardtii (Miller et al. 2010) and Dunaliella salina (Ben-Amotz et al. 1985). The proteome of diatom in nitrogen starvation condition has been compared of nitrogen-replete cells by two-dimensional gel electrophoresis, revealing differences in the responses of central carbon metabolism under nitrogen starvation between diatoms, higher plants, and green algae (Hockin et al. 2012). So far, limited proteomic based studies have been done in microalgae to elucidate the molecular shifts towards lipid accumulation at the proteomic level. Arthrospira platensis is a filamentous cyanobacteria whose high protein content suitable as a nutritive supplement for human and animal diets. Furthermore it is easy to grow and tolerate extreme environmental stresses such as pH, heavy metals, and salinity. In this work, changes in the growth, pigments, and proteome of A. platensis were studied under two major abiotic stresses limiting productivity iron and salinity. The slight concentrations of stresses showed different effects on fresh weight, pigment constituents, growth rate, and soluble protein of this microalgae. A proteomic analysis was performed using SDS gel electrophoresis first to achieve an overview of gross changes, followed by two-dimensional gel electrophoresis as well as mass spectrometry to identify those proteins whose abundance was affected by these environmental stresses. Eighteen protein spots were differentially expressed under the stress conditions. Out of these, six were obtained with increased abundance responding only to Fe2+ stress, and five as a result of NaCl stress alone (Ismaiel et al. 2018). Prototheca zopfii is another algae which is associated with bovine mastitis and human protothecosis. By proteomic study, immunodominant proteins were identified of P. zopfii for development of vaccine against mastitis and protothecosis. Prototheca proteins were separated using 2D-gel electrophoresis. Subsequent immunoblotting with rabbit hyper-immune antiserum revealed 28 immunogenic protein spots. Mass spectrometry analysis revealed 15 immunogenic proteins, including malate dehydrogenase, elongation factor 1-alpha, and heat shock protein 70, which were formerly reported as immunogenic proteins of other eukaryotic pathogens (Irrgang et al. 2015). These proteins could be potential vaccine candidates against the P. zopfii. The current chapter focuses on proteomic analysis of algae and its relevance toward biofuel production and biomedical uses. Without a proteomics study, microalgal omics study is inappropriate. It gives an idea of the particular functional group in both the state (static and dynamic physiological contexts). Targeted proteomics on microalgae has not been reported till now. Proteomics study has used for a better approach for understanding the complexity in the biological world. This can be successfully used for obtaining biodiesel. The motivation behind this proteomic study is to attract attentiveness toward sustainable consumption of microalgae resources for finding indigenous microalgal strains toward exploring their potential role in the production of bioenergy in the country.
3 TAG Synthesis Pathways in Algae
By comparison to plant cells, the production of fatty acids in microalgae is commonly thought to take place in the plastid. Following the pass on to the cytosol, fatty acids enter the TAG assembly pathway catalyzed by enzymes associated with the endoplasmic reticulum. In addition, de novo TAG assembly in the chloroplast has been recently proposed to occur in C. reinhardtii following N-deprivation of a starch-deficient mutant, raising the question of novel TAG assembly pathways in microalgae not observed in seed plants. Until this hypothesis is further corroborated or refuted, one has to consider that parallel TAG assembly pathways in the plastid envelopes and the ER may exist. In fact, an increasing number of putative plastid-targeted acyltransferases in distinct microalgal genomes provide potentially supporting evidence for a chloroplast TAG assembly pathway (Rastogi et al. 2018). Unlike phospholipids found in biological membranes, TAGs do not perform a structural role in cells, but instead serve as a storage form of energy and carbon. After being synthesized, TAGs are deposited in discrete spherical structures, the LDs, located in the cytoplasm or chloroplast. Whether the latter are distinct from plastoglobuli, carotenoid-rich LDs present in plant chloroplasts remain to be seen. Based on predicted orthology of genes identified in algal genomes, at least two major pathways of TAG synthesis have been proposed to function in microalgae: one is the Kennedy or glycerol phosphate pathway, the other is the monoacylglycerol pathway. In both pathways, TAGs are formed by esterification between an acyl-CoA and a glycerol hydroxyl group. In the Kennedy pathway, the first acyl chain is esterified to glycerol-3-phosphate derived from the glycolytic intermediate dihydroxyacetone phosphate (DHAP). The enzyme is glycerol-3-phosphate-acyltransferase (GPAT) which forms lysophosphatidic acid, the substrate for the second acyltransferase, lysophosphatidic acid acyltransferase (LPAAT). This second reaction leads to the formation of phosphatidic acid, which is a central lipid metabolite giving rise to phospholipids and TAGs. Prior to conversion to TAGs, phosphatidic acid is dephosphorylated by a phosphatase to form diacylglycerol (DAG) (Amara et al. 2016). The monoacylglycerol pathway starts with 2-monoacylglycerol, which is converted to DAG by a monoacylglycerol acyltransferase. De novo synthesis of monoacylglycerol in plant cells has been previously described and was shown to be catalyzed by GPAT4 and GPAT6 enzymes, which use dicarboxylic and hydroxy acyl-CoA as acyl-donors for the production of monoacylglycerol (Trentacoste et al. 2013). The monoacylglycerols have been considered initially as precursors for the synthesis of waxy polymers; however, their conversion to DAGs has been reported in Nicotiana bethamina leaves and recently in fruits of Myrica pensylvanica.
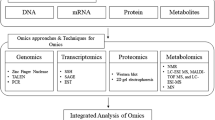
4 Omics Technology
Omic technologies assume a holistic view of the molecules that make up a cell, tissue, or organism. They are targeted mainly identification of genes (genomics), set of all RNA (transcriptomics) and proteins (proteomics) as well as metabolites (metabolomics) in a specific biological sample in a non-targeted and non-biased manner. This can also be referred to as high-dimensional biology; the integration of these techniques is called systems biology.
5 Genomics and Transcriptomics of Microalgae
DNA- and RNA-sequencing studies play a critical role in the quantitative and qualitative improvement of microalgal biomass. The sequence analysis affords useful information about the evolutionary history of the different microalgae groups, thus providing scientific suggestions regarding the role played by particular genes and gene networks. More interestingly, whole-genome sequence information can contribute to our understanding of the molecular mechanisms that microalgae use to adapt to environmental changes, as well as unlocking the potential to develop new and economically important products and technologies (Yao et al. 2017). Genomic and transcriptomic both information can be used to evaluate metabolic pathways and to perform more focused genetic engineering approaches, such as up-regulation or knock out of genes involved in the pathways of interest. Furthermore, a good annotation of the whole genome, alongside to a comparative transcriptomic approach, not only allows to gain insight into the metabolic pathways and their key enzymes, but also to identify regulatory factors and promoters of gene expression. RNA-sequence technology has received a lot of attention nowadays for microalgal worldwide transcriptomic profiling. It is commonly used in transcriptomic analysis of gene expression, predominantly for microalgal strains with potential as biofuel sources. However, inadequate genomic or transcriptomic information for non-model microalgae has limited the understanding of their regulatory mechanisms and hindered genetic improvement to enhance biofuel production (Yao et al. 2017). As such, an optimal microalgal transcriptomic database manufacture is a subject of critical investigation. Dunaliella tertiolecta, a non-model microalgal species, was sequenced by HISEQ 4000 in RNA-Seq studies. The high-quality sequencing data were explored using high-performance computing in a Petascale Data Center. It is subjected to de novo assembly and parallelized mpi BLASTX search with several species. As a result, a transcriptome database was constructed. This enlarged database constructed fueled the RNA-Seq data analysis, which was validated by a nitrogen depletion study that induces lipid production (Yao et al. 2017). Nitrogen depletion (−N) has been used as a technique to stimulate lipid accumulation in many microalgae. Scenedesmus acutus is a promising microalga that can be cultivated in wastewater for biodiesel production (Hawrot-Paw et al. 2020). However, the molecular mechanisms limiting S. acutus lipid accumulation in response to N-deplete remain unidentified. A physiological study determined that N-deplete reduced cell growth and photosynthetic pigments. On the other hand, it stimulated carbohydrate and lipid accumulation (Sirikhachornkit et al. 2018). The transcriptome exploration revealed that glycolysis and starch breakdown were upregulated; on the contrary, gluconeogenesis, photosynthesis, triacylglycerol breakdown, and starch synthesis were downregulated by N-deplete. Under N-deplete, the carbon flux was moved toward lipid synthesis, and the downregulation of lipase genes may contribute to lipid accumulation. A comparative analysis of the N-deplete transcriptomes of microalgae recognized that the downregulation of many lipase genes was a precise mechanism found only in the N-deplete transcriptome of S. acutus. This work unraveled the mechanisms controlling N-deplete induced lipid accumulation in S. acutus and provided new viewpoints for the genetic manipulation of biodiesel-producing microalgae. Biodiesel produced by microalgae may overcome many of the sustainability challenges earlier ascribed to petroleum-based fuels and plant-based biofuels. Here, de novo transcriptomic work for the green microalgae Dunaliella tertiolecta is being studied and screen those genes which were importance for biofuel production. DNA pyro-sequencing next-generation technology used to D. tertiolecta transcripts production. Following size trimming and quality, ~45% of the great quality reads were gathered into isotigs along with 31-fold coverage and singletons. Assembled singletons and sequences were used for BLAST similarity searches and annotated by Gene Ontology (GO) as well as Kyoto Encyclopedia of Genes and Genomes orthology identifiers. These works documented the majority of starch and lipid biosynthesis as well as catabolism pathways in D. tertiolecta (Rismani-Yazdi et al. 2011).
6 Proteomics in Microalgae
The field of proteomics is complementary to genomics as well as transcriptomics in that it delivers additional information on gene expression and regulation. Proteomics comprises the determination of protein–protein interaction and protein expression studies. Furthermore, proteomics studies aim to recognize post-translational modifications of proteins as well as the organization of proteins in multi-protein complexes and their localization in tissues. Technologies are allowing proteomic studies have been in development for numerous decades now (Chandramouli and Qian 2009; Manzoni et al. 2016). A proteomic experiment comprises several steps, which can be taken using altered technological platforms. A critical, primary step is sample preparation and fractionation, which traditionally is being accomplished using two-dimensional gel electrophoresis. Proteins are separated based on their electrical charge in the first dimension (isoelectric focusing) and based on their molecular weight (sodium dodecyl sulfate-polyacrylamide gel electrophoresis,) in the second. The range of 2-DE can be improved by enriching the sample for certain desired proteins. Membrane proteins, for example, can be enriched by a sequential extraction based on the solubilities of proteins in different solutions (Aslam et al. 2017). After sample separation, protein identification is the next step. Proteins of interest can be cut from 2-dimensional gel electrophoresis, trypsinized, and identified by mass spectrometry. Peptide mass data obtained for each sample are queried against a peptide database with known peptide masses of unique proteins (Fig. 11.2). As in transcriptomic methods, proteomics can also be used for the elucidation of certain metabolic pathways. In comparative proteomics, the entire proteome of one sample is compared to that of another sample and kept under altered environmental conditions. Differentially expressed proteins can be identified due to the environmental stimuli and the metabolic pathway they are part of. Compared to the proteomic analysis of specific subcellular compartments or specific protein classes and comparative proteomic studies have so far not been reported. 2-D reference map from C. reinhardtii was represented primarily the soluble sub-proteome (Schmidt et al. 2006). They subsequently used this map to explore high light-induced changes to the proteome, at the same time compared the response of both the wild type and a mutant. Likewise, a comparative quantitative proteomic approach was used to study iron deficiency in C. reinhardtii and found the stress response proteins, such as peroxiredoxin as well as stress-induced light-harvesting protein (Naumann et al. 2007). In studying the effects of cadmium exposure on microalgae C. reinhardtii found a decline in abundance of both large and small subunits of the ribulose-l,5-bisphosphate carboxylase/oxygenase compared to other enzymes involved in photosynthesis (Naumann et al. 2007). Alteration of proteins profile by Cadmium-induced was also investigated in the marine microalga Nannochloropsis oculata (Kim et al. 2005). In Haematococcus pluvialis, a proteomic approach was used to elucidate the microalga’s response to oxidative stress (Gu et al. 2014) and in the halo-tolerant alga Dunaliella salina the molecular basis of salinity tolerance was clarified by proteomics (Kim et al. 2005). Finally, in the perspective of harmful algal bloom prediction, proteome reference maps were built for numerous microalgal species and tested them for their capability for species recognition (Wang et al. 2014). Species specific 2-dimensional gel electrophoresis protein profiles were observed for totally species tested and even distinction between closely related species was possible.

Overview of the proteomic mass spectrometry experimental setup
7 Proteomics: Lipid Production by the Gamma Irradiation Method
Lipid-producing mutant strain of the microalga C. reinhardtii has been developed by gamma irradiation. To induce the mutation, C. reinhardtii was gamma-irradiated at a dose of 400 Gy. After irradiation, the living cells were stained with Nile red. The mutant (Cr-4013) accumulating 20% more lipid than the wild type was selected (Fig. 11.3). Thin-layer chromatography revealed the triglyceride and free fatty acid contents to be markedly increased in Cr-4013. The major fatty acids identified were palmitic acid, oleic acid, linoleic acid, and linolenic acid. Random amplified polymeric DNA analysis showed partial genetic modifications in Cr-4013 (Baek et al. 2016). To ascertain the changes of protein expression in the mutant strain, two-dimensional electrophoresis was conducted. These results showed that gamma radiation could be used for the development of efficient microalgal strains for lipid production.

Time profile of cell growth and total lipid changes in a batch culture of C. reinhardtii and its mutant Cr-4013. Data shown represent the mean values of growth rate based on cell dry weight. Growth rate of wild type (closed circle line), growth rate of mutant Cr-4013 (open circle line), and increased total lipid level inCr-4013 compared with wild type (gray bar)
8 Types of Proteomics
8.1 Expression Proteomics
Expression proteomics is used to study the qualitative and quantitative expression of whole proteins in two different conditions. Like the normal cell and a treated cell can be compared to understand the protein that is accountable for the diseased or stress state or the protein that is expressed due to disease (Chernobrovkin et al. 2014). Typically, expression proteomics studies are used for the exploration of the expression of protein patterns in abnormal cells. For example, tumor tissue samples and the normal tissue comparative study can be analyzed by differential protein expression. 2-D gel electrophoresis and mass spectrometry approaches were used to identify the protein expressional changes, which is present and absent in tumor tissue, when compared with normal tissue. Which are upregulated and downregulated can be identified and characterized protein activities, multi-protein complexes and signaling pathways. Identification of these proteins will give important information about the molecular biology of tumor formation and disease-specific manner for use as diagnostic biomarkers/therapeutic targets.
8.2 Structural Proteomics
Structural proteomics is used to understand the three-dimensional shape and structural complexities of functional proteins. Structural prediction of a protein when its amino acid sequence is determined directly by sequencing or from the gene with a method called homology modelling. Structural proteomics can give detailed information about the structure and function of protein complexes present in a specific cellular organelle (Bai et al. 2016). It is possible to identify all the proteins present in a complex system such as membranes, ribosomes, and cell organelles and to characterize all the protein interactions that can be possible between these proteins and protein complexes (Manjasetty et al. 2012). Different technologies such as NMR spectroscopy and X-ray crystallography were mainly used for structure determination (Meisburger et al. 2017).
8.3 Functional Proteomics
Functional proteomics is used to understand the protein functions as well as molecular mechanisms within the cell, which depend on the identification of the interacting protein partners. The association between unknown protein and partners linking to a specific protein complex involved in a specific mechanism would in fact be powerfully suggestive of its biological function. Furthermore detailed description of the cellular signaling pathways might greatly benefit from the elucidation of protein–protein interactions in-vivo (Cornett et al. 2018).
9 Protein Extraction Methods
Sample preparation is one of the most critical steps in the proteomics study. Good sample preparation can obtain consistent and high-quality results. The efficiency of different protein extraction methods varies depending on the type of samples. Therefore, the protein extraction method needs optimization for different samples, since the amounts and types of non-protein-interfering compounds vary. Algae comprise proteins, carbohydrates, lipids, and nucleic acids in varying proportions and the information is limited on the optimal protein extraction method from algae species. The proteomics investigations of algae are widely used in different fields, mainly including biofuel, biomonitoring, and pollution control. Therefore, the development of an efficient protein extraction method for algae will assist the proteome profiling research in algae (Bleakley and Hayes 2017). Currently, most literature studies focused on the evaluation of protein extraction methods from plant tissues since they contain recalcitrant interferences (polysaccharides, lipids, proteases, oxidative enzymes, and other secondary metabolites) and the presence of cell wall. To date, very few reports investigated protein extraction from algae. Some protein extraction methods can be used for the extraction of proteins from algae (Waghmare et al. 2016).
9.1 Direct Lysis Buffer Method
One milliliter lysis buffer (25 mM tetraethylammonium bromide, 8 M urea, 2% Triton, 0.1% SDS, and complete EDTA-free protease inhibitor tablet (Roche Diagnostics) was added to 100 mg algae sample followed by sonication on ice for 30 min (Feist and Hummon 2015). After centrifugation at 18000 × g for 60 min, the supernatant was stored at −80 °C until use.
9.2 TCA-Acetone Method
In this method, 100 mg of algae sample was resuspended in 1 mL chilled extraction buffer I (10% TCA, 1% polyvinylpyrrolidone, and 2% 2-mercaptoethanol in acetone) and sonicated on ice for 30 min. The mixture was incubated at −20 °C overnight. After centrifugation at 16000 × g for 15 min under 4 °C, the supernatant was discarded and the pellet was washed three times with l mL chilled acetone with 0.07% 2-ME. Between each rinse, the mixture was incubated at −20 °C for 60 min (Niu et al. 2018). The pellet was resuspended in appropriate volume of lysis buffer and incubated at room temperature for 60 min. After centrifugation at 18000 × g for 60 min, the supernatant was stored at −80 °C until use.
9.3 Phenol Method
Phenol extraction was performed according to Wang et al.’s method with some modification. Briefly, 0.5 mL extraction buffer II (30% sucrose, 2% SDS, 5% 2-ME, and 0.1 M Tris, pH 8) was added to 100 mg algae sample. The mixture was sonicated at 4 °C for 30 min. An equal volume of Tris-buffered phenol solution was added and the mixture was well mixed followed by 5 min incubation. The sample was centrifuged at 16000 × g for 5 min under 4 °C and the upper phenolic phase was collected (Awad and Brueck 2020). This procedure was repeated on the residual pellet for two more times and the collection of phenolic phase was combined. Then the solution was precipitated with five volumes of 0.1 M ammonium acetate in 80% methanol at −20 °C overnight. The mixture was centrifuged and the resulting pellet was rinsed with methanol and acetone. The pellet was resuspended in an appropriate volume of lysis buffer and incubated at room temperature for 60 min. After centrifugation at 18000 × g for 60 min, the supernatant was stored at −80 °C until use.
9.4 Phenol/TCA-Acetone Method
Phenol/TCA-acetone method is a combination of TCA-acetone method and the phenol method and it was based on the work of Wang et al. with some modifications. Briefly, 100 mg algae sample was resuspended in 1 mL prechilled extraction buffer I and sonicated in ice for 30 min. After centrifugation at 16000 × g for 5 min under 4 °C, the supernatant was discarded and the pellet was rinsed with 0.1 M ammonium acetate in 80% methanol and acetone with 0.07% 2-ME. Equal volumes of Tris-buffered phenol solution and extraction buffer II were added and the mixture was well mixed followed by 5 min incubation. The sample was centrifuged at 16000 × g for 5 min under 4 °C and the upper phenolic phase was collected (Niu et al. 2018). This procedure was repeated on the residual pellet for two more times and the collection of phenolic phase was combined. Then the solution was precipitated with five volumes of 0.1 M ammonium acetate in 80% methanol at −20 °C overnight. The mixture was centrifuged and the resulting pellet was rinsed with methanol and acetone. The pellet was resuspended in appropriate volume of lysis buffer and incubated at room temperature for 60 min. After centrifugation at 18000 × g for 60 min, the supernatant was stored at −80 °C until use.
10 Technologies of Proteomics
Proteomics mainly used for large-scale experiment. Such type of experiment requires specialized tools, which are developed for the design of the experiment. Three approaches of proteomics technologies have been identified. One is mass spectrophotometer, where the endogenous protein mixtures can be identified along with its analysis (Butterfield and Perluigi 2017). Array-based proteomics, which relates to cDNA microarray and oligonucleotide chips. A third area of proteomic analysis used for localization, metabolism, and physiological parameters of proteins.
10.1 Mass Spectrophotometer
Proteomics through the mass spectrophotometer (MS) has improved the analysis of a number of proteins, which means a number of proteins can be identified in a single experiment (Bruderer et al. 2017). The analysis of protein through MS mainly depends on the breakdown of the protein sample into its constituents with the use of specific enzymes such as sequence protease. The protein as a whole itself is of high molecular weight, which enables the analysis, as the protein cannot be eluted from the gel. For example, C. reinhardtii has been subjected to proteomics studies, in which around 240 proteins have been identified by providing heat shock. Synechocystis 6803 went through the compositional analysis of membrane protein complexes in different growth conditions.
10.2 Array-Based Proteomics
Proteomic array-based analysis used for the identification of a large set of proteins (Betzen et al. 2015). In this method, purified ligands are separated independently. It may be from protein, carbohydrates, or peptide from small molecules such as antibodies or antigens. It can be used for analysis of protein and its expression at the level of protein profiling. Different types of protein microarray formats such as tissue array, reverse-phase array, capture arrays, and lectin arrays have been identified, which are achievement importance in recent years. These tools are used in protein–protein interaction studies, immunological profiling, biomarker discoveries, and vaccine development. Such tools are providing meaningful biological insights into the modern biology of microalgae.
10.3 Next-Generation Proteomic Tools
Two-dimensional gel electrophoresis used for the separation of a large number of protein along with the MALDI-TOF-MS-based; however, they are time consuming and quantitative ability (Altelaar et al. 2013). Nano-liquid mass chromatography techniques have enhanced the quantitative analysis of the protein. Additionally, isobaric tags for relative and absolute quantification method have the capability to identify the serum biomarkers and tissue biomarkers as well as drug resistance markers. Stable isotope labeling by amino acids in cell culture method is one more type of mass spectrometry, which is more proficient toward the cell culture system.
10.4 Quantification Methods
Selected reaction monitoring and are the best tools in the area of biomarker identification. Multiple reaction monitoring based analysis has been used broadly and has grown much significance in the proteomics study, as these methods have substituted some of the expensive approaches of quantification such as antibody-based study like immunoblotting and ELISA. Multiple reaction monitoring achieved on QTOFs as well as Orbit raps are called pseudo-multiple reaction monitoring as well as parallel reaction monitoring (Rauniyar 2015). In comparison, between multiple reaction monitoring and parallel reaction monitoring, the precise fragment ions acquired during acquisition are not possible through parallel reaction monitoring.
11 Post-Translation Modification
Post-translation modifications (PTMs) are used for the cellular processes and the cellular control. Protein activity is restricted by covalent modifiers like phosphate groups or proteolytic cleavage or by ubiquitin moieties. Protein turnover and localization as well as binding interactions can be affected through PTMs. Phosphorylation, methylation, acetylation, lipid modification, glycosylation, and ubiquitination are some of the PTMs which can affect the cellular processes. They directly disturb the protein structure subsequent in the change in the task of the given protein. As such modifications result in the difference in the molecular mass of the amino acid, these differences are significant for being studied in detail. Mass spectrometry, great mass exactness and skill to deal with intricate mixtures are some of the opportunities for describing PTM. A wide explanation of protein stability in the stroma of microalgae, their post-translation modifications and the linking between the two were established. It comprised 2D-gel electrophoresis for the sequestration of the stromal proteins, their arrangement and the description of the PTMs by mass spectrometry joint with bioinformatics action of the data (Mnatsakanyan et al. 2018).
12 Metabolomics Approaches in Microalgae
Metabolites are the end products of cellular regulatory processes. Their levels can be regarded as the ultimate response of biological systems to genetic or environmental changes. Similar to the “genome,” “transcriptome,” and “proteome,” the metabolome denotes to the set of low molecular weight metabolites present in a cell (Deidda et al. 2015). The size of the metabolome differs greatly depending on the organism studied. The metabolic network of S. cerevisiae was formed using genomic, biochemical, and physiological information (Lopes and Rocha 2017). Even within the metabolome of one organism, many different kinds of metabolites exist, with different chemical and physical properties. Moreover, the levels at which various metabolites are present within a cell can cover several magnitudes of concentration. The vastness of the metabolome and its diversity make it technologically impossible to analyze all an organism’s metabolites in a single analysis. Different strategies and methodologies have, therefore, been developed. Metabolite target analysis is restricted to one or a few metabolites related to a specific metabolic reaction and as such does not constitute a high-throughput approach. This strategy is mainly used for screening purposes (Bingol 2018). To elucidate the function of a metabolic pathway, the metabolite profiling approach identifies and quantifies a selected number of predefined metabolites, which belong to a certain class of compounds or to a specific pathway. Metabolic profiling is often used in the context of drug research to study drug candidates (Trifonova et al. 2013). Two approaches have been picked for metabolome analysis, namely metabolic fingerprinting and metabolic profiling. Metabolic fingerprinting is an untargeted analysis of a sample, allowing comparison of metabolite patterns. Metabolomics profiling represents targeted analysis of specific metabolite classes, providing quantitative data for physiological interpretations. Finally, in metabolic fingerprinting, a rapid, global analysis is performed for sample classification according to origin or biological relevance. Quantification and metabolic identification are generally not necessary, allowing for a higher throughput of samples. In algae, most metabolic analyses have so far been focused on the quantification and identification of secondary metabolites with economical value in food science, pharmaceutical industry, and public health, among others. Fatty acids, steroids, carotenoids, polysaccharides, lectins, polyketides, and algal toxins are among the algal products being studied. Environmental metabolomic studies, in contrast—as the application of metabolomics to characterize the metabolic response of an organism to environmental stimuli or stressors—have to date only rarely been carried out in algae. They describe a procedure for cell preparation and metabolite extraction. In extract chromatograms of standardly grown algae, more than 800 metabolites could be detected, with Ala, pyruvate, Glu, glycerolphosphate, and adenosine 5′-monophosphate being among the most prominent peaks. When cells were grown under nutrient deficient conditions (depletion of nitrogen, phosphorus, sulfur, or iron), highly distinct metabolic phenotypes were observed. Multidimensional “omics” technologies provide a platform to enhance our understanding of complex biological systems by describing nearly all biomolecules ranging from DNA to metabolites which is being shown in Table 11.1.
13 Metabolic Engineering in Algae
Genomic manipulation remains restricted to a few select microalgal laboratory models (e.g., C. reinhardtii and Phaeodactylum tricornutum). The increasing curiosity in the area of microalgal biofuels will probably lead to the development of new techniques in other micro-organisms and the establishment of novel model systems. Microalgal trans-genics has been formerly reviewed (Radakovits et al. 2010); nevertheless, the “molecular toolkit” has since expanded due to latest seminal studies. Noteworthy advances contain: (a) the effective expression of transgenes; (b) a new mechanism for gene regulation in microalgae using riboswitches technology; (c) luciferase reporter genes and inducible nuclear promoters (Brogan et al. 2012); and (d) inducible chloroplast gene expression (Doron et al. 2016). Genetic and molecular tool information are mandatory along with metabolic pathways. The sequence analysis will contain the information on metabolomics which opens the part for the metabolic flux examination and the development of metabolic networks. Microalgae having a single cell which are simply cultivable can be concentrated toward metabolic engineering. Moreover, these microalgae perform photosynthesis which aids in understanding the carbon assimilation; this will enhance the research toward bioenergy manufacture. In a recent study, it is deciphered that an microalgae Volvox carteri has been transformed genetically (Umen and Olson 2012). Similarly, trophic conversion of green microalgae C. reinhardtii and P. tricornutum was also described which is a good step in the direction of establishing single gene change. Thus, engineered light-harvesting strains have higher photo-damage resistivity as well as amplified light penetration capacity. The time span from primary transformation to manufacture level is less compared to mammalian-based platforms. It is well recognized that algae is the main source for the manufacture of diverse metabolites and proteins. In numerous studies, C. reinhardtii has been reported as a good source for the manufacture of fatty acids.
14 Future Needs: Integrating “Omics” in Systems Biology
Thanks to the availability of complete genome sequences and the advent of high data content measurement techniques for transcripts, proteins and their interactions and metabolites, a new level of understanding of cells and organisms has become possible. As already mentioned in the introduction, in systems biology, the main goal is to develop a comprehensive and consistent knowledge of a biological system by investigating the behavior of and interaction between its individual components. One of the key steps in this process involves modeling. Once the structure of the system is unraveled, mathematical algorithms allow its dynamics to be modeled. Mathematical models describe the system, but also allow the prediction of the system’s response to perturbations. A framework for systems biology studies was formulated, involving several distinct steps. In a first step, understanding of the structure of the system is required (Lauritano et al. 2019). This involves the identification of the elements of the system, such as gene networks, protein interactions, and metabolic pathways. This knowledge is used to construct an initial model of its behavior (Cooper and Smith 2015). Depending on how much is known about the different components of the system the modeling can be carried out at several levels. Steady-state studies can be done even when knowledge on certain parameters is lacking. Using the available knowledge on the system’s structure theoretical upper and lower limits are calculated, as well as an optimal operation point of the system in steady-state. Other types of analyses allow for an understanding of the dynamic properties of a system, but require knowledge of certain parameters. Secondly, the system is perturbed (genetically or using environmental stimuli) and corresponding responses are measured using high-throughput measurement tools. Data observed at different levels of biological organization are integrated with each other and with the current model of the system. Followingly, the model is adapted in such a way that the experimentally observed phenomena correspond best with the model’s predictions. A new set of perturbations is selected and applied to the model, to distinguish between multiple model hypotheses. These steps are continually repeated, thereby expanding and refining the model until the model’s predictions reflect biological reality. It is obvious that a systems biology analysis requires collective efforts from multiple research areas, such as molecular biology, computer science, and control theory and is therefore not easy to accomplish. Recent and ongoing studies putting the above systems biology framework into practice have focused on bacterial chemotaxis inf. Coli, sugar metabolism in S. cerevisiae, and embryo development in the sea urchin, among others. For algae, no systems biology studies with extensive computational modeling efforts have been reported so far to our knowledge. However, more and more knowledge of the components of algae has become available and the first results on the integration of observations on different levels of biological organization are being published. Metabolite, genomic, and transcriptomic data were used to provide genome-wide insights into the regulation of the metabolic networks utilized by Chlamydomonas reinhardtii under anaerobic conditions associated with H2 production. During acclimation to anoxic conditions the hydroge-nase activity, photosynthesis, cellular respiration, and organic acid accumulation of algal cells were monitored. In conjunction with the formation of fermentation products the levels of transcripts encoding proteins associated with the various fermentation pathways were analyzed using real-time PCR. After establishing that the algae were acclimated to the anoxic conditions microarray analyses were carried out to gain insight in the effects of dark anaerobiosis on transcript abundance in a genome-wide context. Results showed that congruent with elevated H2 production following exposure of the algae to anaerobic conditions was the accumulation of various fermentation products. These findings on metabolome level were augmented with real-time PCR analysis. Results obtained by microarray analysis were in agreement with the real-time PCR data. In addition, genes encoding proteins involved in other metabolic pathways as well as encoding transcription/translation regulatory factors were differentially transcribed (Salama et al. 2019). Although this study cannot be considered a true systems biology approach yet—rather than relating individual elements quantitatively to each other and to the covering system, a qualitative analysis is made—here we see the onset of the integration of data gathered on different levels of biological organization to improve our understanding of (a metabolic network of) a biological system.
15 Applications of Omics Approaches
Omics is a widely used technique in biological fields, mainly applied in Oncology (Tumor biology), Bio-medicine, Agriculture, and Food Microbiology.
15.1 Oncology
Oncology is the study of tumor cell. Tumor metastasis, is the process spread of cancer from one organ to another non-adjacent organ cause death in patients. The main challenge in medicine to describe the molecular and cellular mechanisms of tumor metastasis. Protein expressions analysis and metastatic process help to understand the mechanism of metastasis and facilitate the development of strategies for the therapeutic interventions and clinical management of cancer (Yu and Snyder 2016). Proteomics is a systematic research, the main aim of this research is to characterize the protein expressions, functions of tumor cells and is widely used in biomarker discovery.
15.2 Biomedical Applications
Interactions of microbial pathogens with their hosts is called “infectomics.” It is a very interesting area in proteomics. It deals with the fundamentals of the infections origin and their effect on organs. The key aim of this research is to stop or cure disease at starting level. Advanced diagnostic issues associated to emerging infections, increasing of fastidious bacteria, and generation of patient-tailored phenotypes (Vladareanu et al. 2016).
15.3 Agricultural Applications
The applications of plant proteomics scientific research is still in promising stage. Plant proteomics is also used to know plant–insect interactions that help identify candidate genes involved in the defensive response of plants to herbivore (Van Emon 2016). Population growth and effect of global climate changes imposing severe limits on the sustainability of agricultural crop production.
15.4 Food Microbiology
The use of omics technology in food technology is presented mainly for characterization and standardization of raw materials, process development, and detection of batch-to-batch variations and quality control of the ending product. Further attention is paid to the aspects of food safety, especially regarding biological and microbial safety and the use of genetically modified foods (Walsh et al. 2017).
16 Conclusions
Despite the growing number of completed microalgae genome sequences, only a few examples of genetic engineering of the metabolism for the production of by-products have been reported. Nevertheless, metabolic engineering in model strains of microalgae will likely provide important leads for proof-of-principle studies in the future. Furthermore, the complete genome sequences provide a valuable framework for the huge amounts of marine metagenome projects. Comparative genomics is a powerful tool to unravel previously unknown gene functions. Each algal genome project provides enough genome sequence data to allow comparative analysis of each genome. It is based on small genome repertoires, particularly for unicellular algae; algal genome information should accelerate studies on, for example, the establishment of cellular components, and it will allow us to elucidate cellular and molecular properties that they have in common with land plants or other eukaryotes. Principally omic projects should aim at different microalgae features such as evolution, adaptation, and divergence compared with other species, gene, protein, and metabolite information, and their interaction. This would facilitate an understanding of the biology of microalgae in detail and the application of these concepts in the production of valuable products.
References
Altelaar AM, Munoz J, Heck AJ (2013) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet 14:35
Amara S, Seghezzi N, Otani H, Diaz-Salazar C, Liu J, Eltis LD (2016) Characterization of key triacylglycerol biosynthesis processes in rhodococci. Sci Rep 6:1–13
Arora N, Pienkos PT, Pruthi V, Poluri KM, Guarnieri MT (2018) Leveraging algal omics to reveal potential targets for augmenting TAG accumulation. Biotechnol Adv 36(4):1274–1292
Aslam B, Basit M, Nisar MA, Khurshid M, Rasool MH (2017) Proteomics: technologies and their applications. J Chromatogr Sci 55:182–196
Awad D, Brueck T (2020) Optimization of protein isolation by proteomic qualification from Cutaneotrichosporon oleaginosus. Anal Bioanal Chem 412:449–462
Baek J, Choi J-i, Park H, Lim S, Park SJ (2016) Isolation and proteomic analysis of a Chlamydomonas reinhardtii mutant with enhanced lipid production by the gamma irradiation method. J Microbiol Biotechnol 26:2066–2075
Bai X, Song H, Lavoie M, Zhu K, Su Y, Ye H, Chen S, Fu Z, Qian H (2016) Proteomic study bring new insights into the effect of a dark stress on lipid synthesis in P tricornutum. Sci Rep 6:1–10
Barh D, Zambare V, Azevedo V (2013) Omics: applications in biomedical, agricultural, and environmental sciences. CRC Press, New York
Ben-Amotz A, Tornabene TG, Thomas WH (1985) Chemical profile of selected species of microalgae with emphasis on lipids1. J Phycol 21:72–81
Betzen C, Alhamdani MSS, Lueong S, Schröder C, Stang A, Hoheisel JD (2015) Clinical proteomics: promises, challenges and limitations of affinity arrays. Proteomics–Clin Appl 9:342–347
Bingol K (2018) Recent advances in targeted and untargeted metabolomics by NMR and MS/NMR methods. High-throughput 7:9
Bleakley S, Hayes M (2017) Algal proteins: extraction, application, and challenges concerning production. Foods 6:33
Brogan J, Li F, Li W, He Z, Huang Q, Li C-Y (2012) Imaging molecular pathways: reporter genes. Radiat Res 177:508–513
Bruderer R, Bernhardt OM, Gandhi T, Xuan Y, Sondermann J, Schmidt M, Gomez-Varela D, Reiter L (2017) Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol Cell Proteomics 16(12):2296–2309
Butterfield DA, Perluigi M (2017) Redox proteomics: a key tool for new insights into protein modification with relevance to disease. Antioxid Redox Signal 26(7):277–279
Chandramouli K, Qian P-Y (2009) Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. HGP 2009:239204
Chernobrovkin A, Vicente CM, Visa N, Zubarev RA (2014) Expression proteomics reveals protein targets and highlights mechanisms of action of small molecule drugs. Sct Rep 5:11176
Cooper MB, Smith AG (2015) Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr Opin Plant Biol 26:147–153
Cornett EM, Dickson BM, Krajewski K, Spellmon N, Umstead A, Vaughan RM, Shaw KM, Versluis PP, Cowles MW, Brunzelle J (2018) A functional proteomics platform to reveal the sequence determinants of lysine methyltransferase substrate selectivity. Sci Adv 4:eaav2623
Deidda M, Piras C, Bassareo PP, Dessalvi CC, Mercuro G (2015) Metabolomics, a promising approach to translational research in cardiology. IJC Metabolic Endocrine 9:31–38
Doron L, Segal Na, Shapira M (2016) Transgene expression in microalgae—from tools to applications. Front Plant Sci 7:505
Feist P, Hummon AB (2015) Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int J Mol Sci 16:3537–3563
Gillham NW, Boynton JE, Hauser CR (1994) Translational regulation of gene expression in chloroplasts and mitochondria. Annu Rev Genet 28:71–93
Gu W, Li H, Zhao P, Yu R, Pan G, Gao S, Xie X, Huang A, He L, Wang G (2014) Quantitative proteomic analysis of thylakoid from two microalgae (Haematococcus pluvialis and Dunaliella salina) reveals two different high light-responsive strategies. Sci Rep 4:6661
Guarnieri MT, Pienkos PT (2015) Algal omics: unlocking bioproduct diversity in algae cell factories. Photosynth Res 123:255–263
Guarnieri MT, Nag A, Smolinski SL, Darzins A, Seibert M, Pienkos PT (2011) Examination of triacylglycerol biosynthetic pathways via de novo transcriptomic and proteomic analyses in an unsequenced microalga. PLoS One 6:e25851
Hawrot-Paw M, Koniuszy A, Gałczyńska M, Zając G, Szyszlak-Bargłowicz J (2020) Production of microalgal biomass using aquaculture wastewater as growth medium. Watermark 12:106
Hockin NL, Mock T, Mulholland F, Kopriva S, Malin G (2012) The response of diatom central carbon metabolism to nitrogen starvation is different from that of green algae and higher plants. Plant Physiol 158:299–312
Illman A, Scragg A, Shales S (2000) Increase in chlorella strains calorific values when grown in low nitrogen medium. Enzyme Microb Technol 27:631–635
Irrgang A, Weise C, Murugaiyan J, Roesler U (2015) Identification of immunodominant proteins of the microalgae Prototheca by proteomic analysis. New Microb New Infect 3:37–40
Ismaiel MM, Piercey-Normore MD, Rampitsch C (2018) Proteomic analyses of the cyanobacterium Arthrospira (spirulina) platensis under iron and salinity stress. Environ Exp Bot 147:63–74
Kageyama H, Tanaka Y, Shibata A, Waditee-Sirisattha R, Takabe T (2018) Dimethylsulfoniopropionate biosynthesis in a diatom Thalassiosira pseudonana: identification of a gene encoding MTHB-methyltransferase. Arch Biochem Biophys 645:100–106
KaiXian Q, Borowitzka MA (1993) Light and nitrogen deficiency effects on the growth and composition ofPhaeodactylum tricornutum. Appl Biochem Biotechnol 38:93–103
Kim Y, Yoo W, Lee S, Lee M (2005) Proteomic analysis of cadmium-induced protein profile alterations from marine alga Nannochloropsis oculata. Ecotoxicology 14:589–596
Lauritano C, Ferrante MI, Rogato A (2019) Marine natural products from microalgae: an-omics overview. Mar Drugs 17:269
Le Bihan T, Martin SF, Chirnside ES, van Ooijen G, Barrios-LLerena ME, O'Neill JS, Shliaha PV, Kerr LE, Millar AJ (2011) Shotgun proteomic analysis of the unicellular alga Ostreococcus tauri. J Proteomics 74:2060–2070
Lopes H, Rocha I (2017) Genome-scale modeling of yeast: chronology, applications and critical perspectives. FEMS Yeast Res 17:50
Maghembe R, Damian D, Makaranga A, Nyandoro SS, Lyantagaye SL, Kusari S, Hatti-Kaul R (2020) Omics for bioprospecting and drug discovery from bacteria and microalgae. Antibiotics 9:229
Manjasetty BA, Büssow K, Panjikar S, Turnbull AP (2012) Current methods in structural proteomics and its applications in biological sciences. 3 Biotech 2:89–113
Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, Ferrari R (2016) Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief Bioinform 19:286–302
Meisburger SP, Thomas WC, Watkins MB, Ando N (2017) X-ray scattering studies of protein structural dynamics. Chem Rev 117:7615–7672
Miller R, Wu G, Deshpande RR, Vieler A, Gärtner K, Li X, Moellering ER, Zäuner S, Cornish AJ, Liu B (2010) Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol 154:1737–1752
Mnatsakanyan R, Shema G, Basik M, Batist G, Borchers CH, Sickmann A, Zahedi RP (2018) Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev Proteomics 15:6
Montecinos AE, Couceiro L, Peters AF, Desrut A, Valero M, Guillemin ML (2017) Species delimitation and phylogeographic analyses in the Ectocarpus subgroup siliculosi (Ectocarpales, Phaeophyceae). J Phycol 53:17–31
Naumann B, Busch A, Allmer J, Ostendorf E, Zeller M, Kirchhoff H, Hippler M (2007) Comparative quantitative proteomics to investigate the remodeling of bioenergetic pathways under iron deficiency in Chlamydomonas reinhardtii. Proteomics 7:3964–3979
Niizawa I, Espinaco BY, Leonardi JR, Heinrich JM, Sihufe GA (2018) Enhancement of astaxanthin production from Haematococcus pluvialis under autotrophic growth conditions by a sequential stress strategy. Prep Biochem Biotechnol 48:528–534
Niu L, Zhang H, Wu Z, Wang Y, Liu H, Wu X, Wang W (2018) Modified TCA/acetone precipitation of plant proteins for proteomic analysis. PLoS One 13:e0202238
Pienkos PT, Darzins A (2009) The promise and challenges of microalgal-derived biofuels. Biofuels Bioprod Biorefin 3:431–440
Radakovits R, Jinkerson RE, Darzins A, Posewitz MC (2010) Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell 9:486–501
Radakovits R, Jinkerson RE, Fuerstenberg SI, Tae H, Settlage RE, Boore JL, Posewitz MC (2012) Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropsis gaditana. Nat Commun 3:686
Rai V, Karthikaichamy A, Das D, Noronha S, Wangikar PP, Srivastava S (2016) Multi-omics frontiers in algal research: techniques and progress to explore biofuels in the postgenomics world. Omics: J Integr Biol 20:387–399
Rashidi B, Dechesne A, Rydahl MG, Jørgensen B, Trindade LM (2019) Neochloris oleoabundans cell walls have an altered composition when cultivated under different growing conditions. Algal Res 40:101482
Rastogi RP, Pandey A, Larroche C, Madamwar D (2018) Algal Green Energy–R&D and technological perspectives for biodiesel production. Renew Sustain Energy Rev 82:2946–2969
Rauniyar N (2015) Parallel reaction monitoring: a targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int J Mol Sci 16:28566–28581
Reen CS, Wayne CK, Loke SP, Manickam S, Chuan LT, Yang T (2019) Isolation of protein from Chlorella sorokiniana CY1 using liquid biphasic flotation assisted with sonication through sugaring-out effect. J Oceanol Limnol 37:898–908
Rismani-Yazdi H, Haznedaroglu BZ, Bibby K, Peccia J (2011) Transcriptome sequencing and annotation of the microalgae Dunaliella tertiolecta: pathway description and gene discovery for production of next-generation biofuels. BMC Genomics 12:148
Salama E-S, Govindwar SP, Khandare RV, Roh H-S, Jeon B-H, Li X (2019) Can omics approaches improve microalgal biofuels under abiotic stress? Trends Plant Sci 24:611–624
Sasso S, Stibor H, Mittag M, Grossman AR (2018) The natural history of model organisms: from molecular manipulation of domesticated Chlamydomonas reinhardtii to survival in nature. Elife 7:e39233
Schaum CE (2019) Enhanced biofilm formation aids adaptation to extreme warming and environmental instability in the diatom Thalassiosira pseudonana and its associated bacteria. Limnol Oceanogr 64:441–460
Schmidt M, Geßner G, Luff M, Heiland I, Wagner V, Kaminski M, Geimer S, Eitzinger N, Reißenweber T, Voytsekh O (2006) Proteomic analysis of the eyespot of Chlamydomonas reinhardtii provides novel insights into its components and tactic movements. Plant Cell 18:1908–1930
Sirikhachornkit A, Suttangkakul A, Vuttipongchaikij S, Juntawong P (2018) De novo transcriptome analysis and gene expression profiling of an oleaginous microalga Scenedesmus acutus TISTR8540 during nitrogen deprivation-induced lipid accumulation. Sci Rep 8:3668
Stoffels L, Finlan A, Mannall G, Purton S, Parker BM (2019) Downstream processing of Chlamydomonas reinhardtii TN72 for recombinant protein recovery. Front Bioeng Biotechnol 7:383
Trentacoste EM, Shrestha RP, Smith SR, Glé C, Hartmann AC, Hildebrand M, Gerwick WH (2013) Metabolic engineering of lipid catabolism increases microalgal lipid accumulation without compromising growth. Proc Natl Acad Sci 110:19748–19753
Trifonova O, Lokhov P, Archakov A (2013) Postgenomics diagnostics: metabolomics approaches to human blood profiling. Omics: J Integr Biol 17:550–559
Umen JG, Olson BJ (2012) Genomics of volvocine algae. In: Advances in botanical research. Elsevier, Amsterdam, pp 185–243
Ummalyma SB (2020) Bioremediation and biomass production of microalgae cultivation in river watercontaminated with pharmaceutical effluent. Bioresour Technol 307:123233
Van Emon JM (2016) The omics revolution in agricultural research. J Agric Food Chem 64:36–44
Vladareanu L, Iliescu M, Wang H, Yongfei F, Vladareanu V, Yu H, Smarandache F (2016) CSP and “omics” technology apllied on versatile and intelligent portable platform for modeling complex bio-medical data. In: 2016 international conference on advanced mechatronic systems (ICAMechS), pp 423–428
Waghmare AG, Salve MK, LeBlanc JG, Arya SS (2016) Concentration and characterization of microalgae proteins from Chlorella pyrenoidosa. Bioresour Bioprocess 3:16
Walsh AM, Crispie F, Claesson MJ, Cotter PD (2017) Translating omics to food microbiology. Annu Rev Food Sci Technol 8:113–134
Wang D-Z, Zhang H, Zhang Y, Zhang S-F (2014) Marine dinoflagellate proteomics: current status and future perspectives. J Proteomics 105:121–132
Xu L, Fan J, Wang Q (2019) Omics application of bio-hydrogen production through green alga Chlamydomonas reinhardtii. Front Bioeng Biotechnol 7:201
Yang S, Wang W, Wei H, Van Wychen S, Pienkos PT, Zhang M, Himmel ME (2016) Comparison of nitrogen depletion and repletion on lipid production in yeast and fungal species. Energies 9:685
Yao L, Tan KWM, Tan TW, Lee YK (2017) Exploring the transcriptome of non-model oleaginous microalga Dunaliella tertiolecta through high-throughput sequencing and high performance computing. BMC bioinformatics 18:122
Yu K-H, Snyder M (2016) Omics profiling in precision oncology. Mol Cell Proteomics 15:2525–2536
Zainul Azlan N, Yusof M, Anum Y, Alias E, Makpol S (2019) Chlorella vulgaris improves the regenerative capacity of young and senescent myoblasts and promotes muscle regeneration. Oxid Med Cell Longev 2019:3520789
Zhila NO, Kalacheva GS, Volova TG (2005) Influence of nitrogen deficiency on biochemical composition of the green alga Botryococcus. J Appl Phycol 17:309–315
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sharma, A., Shukla, S., Singh, R.P. (2020). Microalgae: Omics Approaches for Biofuel Production and Biomedical Research. In: Kashyap, B.K., Solanki, M.K., Kamboj, D.V., Pandey, A.K. (eds) Waste to Energy: Prospects and Applications. Springer, Singapore. https://doi.org/10.1007/978-981-33-4347-4_11
Download citation
DOI: https://doi.org/10.1007/978-981-33-4347-4_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-33-4346-7
Online ISBN: 978-981-33-4347-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)