
Abstract
During the synthesis of DNA and chromosomal segregation, the checkpoints monitor the proper progression through the cell cycle and sense any defects. Via the modulation of CDK activity, these activated checkpoints lead to cell cycle arrest. However, any dysregulation in these kinases will impede this characteristic modulation during the cell cycle. Specific complexes of CDK-cyclins are deregulated frequently by mutations associated with tumors, and either unscheduled cell cycle re-entry or continuous proliferation is witnessed due to this deregulation. Furthermore, these two features are seen in most human tumor cells. The CDK1, along with its interacting partners (A and B cyclins), controls progression at the S-G2 and G2-M phases of the cell cycle. The CDK1 shows high expression in multiple cancers including breast cancer. The dysregulation of CDK1 could lead to many possible events, including enhanced tumor growth, increased cancer cell proliferation, and chromosomal instability. Multiple studies have demonstrated that the CDK1 expression levels and function are dysregulated, thus highlighting its participation in the BC progression. It has been witnessed that blocking or silencing the CDK1 could suppress the BC growth, particularly when combined with other anti-cancer agents. Many pan-CDK inhibitors targeting multiple CDKs including CDK1 have been tested at different phases of clinical trials. This chapter highlights the significant role of CDK1, its dysregulation in breast cancer, and the potential treatment available to combat the same.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
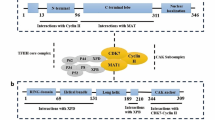
A family of protein kinases responsible for modulating the cell cycle are the CDKs (Poon 2016). CDKs require modulatory subunits to bind them, known as cyclins, to exert their effects. The latter are formed and destroyed at various cell cycle stages in a specific and timely manner, thus regulating the cell cycle properly. The relevance of complex Cdc2 (CDK1) has been discovered in the Schizosaccharomyces pombe (Sofi et al. 2022). The participation of CDK1 in the homologous recombination DNA double-stranded break repair mechanism is also known. In yeast and human cells, the cyclin-dependent kinase activity is required for eliminating DNA double-stranded breaks to form single strands during homologous recombination by recruiting endonucleases Sae2 or CtlP, respectively (Ira et al. 2004; Huertas and Jackson 2009). CDK1 is the most important CDK for maintaining cell cycle control in mammalian cells (Santamaría et al. 2007). During the G1, NHEJ is primarily operational, and during S and G2 phase, HR is in action in yeast and Cdk1 appears to play a crucial part in choosing between the aforementioned methods utilized primarily to repair the ds-breaks (Huertas Sánchez et al. 2008). The tumor suppressor BRCA2 is phosphorylated by CDK1 and CDK2 in humans to regulate its interaction with RAD51. During the S and G2 phases, this connection promotes homologous recombination-dependent repair (Esashi et al. 2005). Deregulation of specific CDK-cyclin complexes is frequently observed as a result of tumor-associated mutations, resulting in either unscheduled cell cycle re-entry or persistent proliferation. Furthermore, most human tumor cells have these two characteristics (Malumbres and Barbacid 2001). The checkpoints monitor the normal course of the cell cycle and detect any problems during DNA synthesis and chromosomal segregation. Furthermore, these active checkpoints cause the cell cycle to be arrested by regulating CDK function. The goal of stopping the cell cycle is to give cells enough time to fix their faults so that they do not proceed to the daughter cells that will be generated. Endogenous genotoxic factors, such as chemicals, free radicals, ionizing radiation, and exogenous products, can cause extensive alterations in the DNA molecule, and DNA damage checkpoints assist to protect cells from such attacks. Furthermore, when these changes occur, they are detected by a signaling pathway, resulting in CDK hindrance and, finally, cell cycle block (Bartek et al. 2004). If the repair process is inefficient due to massive DNA damage caused by checkpoint failure or poor repair machinery caused by genetic flaws in the same pathways, the cells may undergo programmed cell death (apoptosis) or enter senescence (Fig. 9.1).

The CDKs form specific pairs with their respective interacting partners during the various cell cycle phases, thus controlling its progression
On the other side, accumulating DNA mutations can lead to GIN (genomic instability), which can lead to cell transformation and thus cancer (Kastan and Bartek 2004). Unscheduled proliferation, chromosomal instability (CIN), and genomic instability (GIN) are the three most common disorders in the cell cycle. They are caused by either direct or indirect poor control of CDKs (Malumbres and Barbacid 2005). The SAC (spindle assembly checkpoint) is in charge of chromosomal separation after DNA duplication. This signaling system regulates CDK1 activity and protects against chromosomal segregation faults (Kops et al. 2005; Musacchio and Salmon 2007; Pérez de Castro et al. 2007). A defective SAC (spindle assembly checkpoint) can also lead to unequal DNA inheritance. If CIN (numerical chromosomal abbreviations) continue to accumulate and are not addressed, it may lead to tumor growth. A-type cyclins are known to activate CDK1 near the end of interphase to aid mitotic commencement. When the nuclear envelope degrades, the A-type cyclins are destroyed, allowing CDK1-cyclin B complexes to form (Fig. 9.2), which drive the cells into mitosis (Malumbres and Barbacid 2005).

The CDK1 with its interacting partners Cyclin B and Cyclin A is essential in cell cycle regulation and transition at the G2/M phase
Because of their critical function in the progression of the cell cycle, downregulation of CDKs may result in improper homeostasis in specific tissues. On the other hand, by initiating the untimely division in cells (progenitor or stem cells), the hyperactivated CDKs could also aid in the development of tumors (Malumbres and Barbacid 2009). Breast cancer can result from many causes, including mutations in genes that are concerned with the repair of DNA, TSG (p53), and the proto-oncogenes like HER-2, c-myc, as well as cyclin D (Gerger et al. 2007). When BRCA1 and BRCA2 genes are dysregulated, the chance of breast cancer increases. These are known to perform multiple functions, including obstructing the progression of the cell cycle at the S-phase by halting retinoblastoma and possibly CDK2 (Rahman and Stratton 1998; Hashemi et al. 2019). CDK1 is a member of the cell cycle-associated CDK family, which also includes CDK2, CDK4, and CDK6. These CDKs are known to have a direct role in the cell cycle progression and the various phases. Moreover, the cyclins show different concentrations during the different cell cycle phases (Fig. 9.3). The relative levels of CDKs are nearly untouched but their action can be modulated by changes in these cyclin concentrations.

The relative levels of different cyclins during various cell cycle phases. These specific cyclin concentrations are significant for the successful cell cycle progression along with their association with particular CDKs
The uncontrolled cellular growth represents one of the hallmarks of cancer. The cell cycle checkpoint disables and overrides multiple protections implicated in cyclin/CDKs dysregulation in the same way. In several solid tumors, including BC, uncontrolled cell proliferation is witnessed due to cell cycle dysregulation and genetic alterations in the proteins that are involved in the regulation of cell cycle (Hanahan and Weinberg 2011; Brigham et al. 2012). The CDK1, along with its interacting partners (A and B cyclins), controls progression at the S-G2 and G2-M phases of the cell cycle.as shown in Fig. 9.1. Through phosphorylation and dephosphorylation, the CDK1 aids in cell cycle regulation. For inducing apoptosis (programmed cell death), the activated CDK1 has an essential role (Malumbres and Barbacid 2009). Together with A and B-type cyclins, CDK1 kinase modulates the centrosome cycle and the mitotic onset (Fig. 9.4) and represents one of the central modulators of mitosis.

Activation of CDK1 is brought about by B-type cyclin (mainly B1 cyclin), which leads to mitotic entry after successful substrate phosphorylation. The CDK1 inactivation occurs post cyclin B1 destruction, ultimately leading to exit from mitosis
After the successful chromosome condensation and their alignment at the metaphase plate, the CDK1 function is turned down to permit the separation of sister chromatid via separase or separin activation (Musacchio and Salmon 2007). The decondensation of chromosomes, nuclear envelope reformation, and the process of cytokinesis all require this inactivation of CDK1 (Potapova et al. 2006). The CDK1-related cyclins have already been shown to be unstable, and their control is carried out via ubiquitination (Gavet and Pines 2010a, b). Thr14 and Tyr15 phosphorylation of CDK1 inhibits the CDK1/Cyclin B complex during the G2 phase (Gould and Nurse 1989). Tyr15 phosphorylation prevents substrate phosphorylation by blocking CDK1’s ATP-binding site (Li et al. 1995). Phosphorylation of Thr14 prevents ATP binding (Heald et al. 1993).
9.2 Role of CDK1
The CDK1 (also called CDC2 = cell division control protein 2) is a mitotic CDK. After duplicating the DNA, the chromosomal segregation is controlled by SAC (the spindle assembly checkpoint). This process regulates the activity of CDK1 and hinders any defects in the segregation of chromosomes (Kops et al. 2005; Musacchio and Salmon 2007; Pérez de Castro et al. 2007). On similar grounds, impaired SAC (spindle assembly checkpoint) could lead to an equal inheritance of DNA. If not repaired, it could aid in tumor progression due to the accumulation of CIN (numerical chromosomal abbreviations). The A-type cyclins are believed to activate CDK1 toward the interphase end to aid in the start of mitosis. After the degradation of nuclear envelope, the A-type cyclins are dissolved to aid in the CDK1-cyclin B complexes formation, which drives the cells through mitosis (Malumbres and Barbacid 2005). CDK activity has also been recruited for associating the BRCA1 to the MRN [Mre11-Rad50-Nbs1] complex during homologous combination (Chen et al. 2008). The CDK1, 2, 4, and 6 participate in the regulation of cell cycle progression, while CDK7, 8 9 participate in transcription (Izadi et al. 2020). Despite containing the complete complement of interphase CDKs, mice embryos with CDK1 absent do not show division, demonstrating that these CDKs are unable to compensate for the lack of CDK1 (Santamaría et al. 2007). Furthermore, using homologous recombination to replace Cdk1 with Cdk2 ends up causing early embryonic lethality (Satyanarayana et al. 2008), demonstrating that CDK2 cannot replace the role of CDK1, even when the Cdk1 locus is used for its expression. Cyclin A2 knockout causes early embryonic lethality (Murphy et al. 1997), implying that this cyclin’s primary function is to trigger CDK1, the mitotic CDK. A transient delay in interphase was witnessed in human cell lines that lacked CDK1 although there was no hindrance to mitotic entry. Also the mitosis occurring afterward is characterized by several abnormalities (Lau et al. 2021).
Some of the documented functions of CDK1 include participation in Cell division, Checkpoint activation, DNA repair, Apoptotic process, DNA replication, and G2/M transition as shown in Fig. 9.5. In association with cyclin B, the resulting complex CDK1/Cyclin B aids in the progression of the cell cycle at the mitosis phase (Draetta and Beach 1988) and the complex CDK2/Cyclin B modulates the G1 to S transition (Endicott et al. 1999). As per a study’s upshot, the CDK1 removal leads to inaccurate control at G2/M. While as the CDK2 absence does not impact the progression of the cell cycle indicating that other CDKs can compensate for the CDK2 roles (Lau et al. 2021).

Some of the functions of CDK1: Cell division, Checkpoint activation, DNA repair, Apoptotic process, DNA replication, and G2/M transition
9.3 Dysregulation of CDK1 in BC
The uncontrolled cellular proliferation manifests as one of the cancer hallmarks. The same occurs through the cell cycle checkpoint disabling and overriding several safeguards involved with the cyclin/CDKs dysregulation or impairment. It has been seen that the CDK1 shows high expression in multiple cancers, like in the case of BC (Izadi et al. 2020). In the case of MYC-dependent BC patients, CDK1 inhibition is regarded as a potential therapeutic strategy (Izadi et al. 2020). Typically, the CDK1 and cyclin A/B aid the M phase of the cell cycle, but in BC cells, these participate in the programmed cell death of MYC-driven TNBC (Duffy et al. 2015). It has been analyzed through heat map studies that CDK1 and CDK2, 4, 5, and 8 display elevated expression relative to CDK6 and 9 in the case of primary tumors of BC (Sofi et al. 2022).
Roles of CDK1 in mitotic progression have been observed along with the overexpression of Cyclin A2 and B1 (Aaltonen et al. 2009). During the mitotic phase, CDK1/Cyclin B aids in the cell cycle progression (Draetta and Beach 1988). Cyclin B1 has been associated with higher promoter activity of Cyclin B1 as well as the G1/S/G2 cell cycle phases in numerous BC cell lines. Furthermore, enhanced CDK1/Cyclin B1 complex activity has been seen in T-47D and BT-549 cells during the G1 phase (Barrett et al. 2002). docosahexaenoic (DHA) and eicosapentaenoic (EPA) are omega-3 fatty acids that biochemically display anti-cancer effects, and these effects have been examined in several studies. Moreover, both DHA and EPA partially hindered the MDA-MB-231 BC cell proliferation via the CDK1/Cyclin B1 complex obstruction. The duration of G2/M phases is increased after treating the MDA-MB-231 cells with EPA and DHA in the cell cycle. Downregulation of CDK1, Cyclin B1, and Cyclin A was also witnessed as a result of this, and Cyclin B1 phosphorylation was also suppressed and 25C phosphatase reduced, which is known to activate the CDK1 (Barascu et al. 2006). Moreover, as per the study on patients (Chinese Han Women), it was observed that the genetic polymorphisms of genes that code for CDK1 and cyclin B1 could significantly impart the susceptibility to the BC progression and survival in these patients (Li et al. 2013). This also indicated that for BC patients, CDK1 and CDK2 specific activity could be utilized as a prognosis factor. It was observed in a study that CDK1 and CDK2 specific activity could help predict the possible chemotherapy outcome in patients of this study (Kim et al. 2012). Poor five-year relapse-free survival has been observed in patients who exhibit heightened specific activity of CDK1 and CDK2, as shown by Kim and colleagues, and based on the same, the BC patients could be categorized as low and high-risk groups. Thus, a valuable way to predict the outcome of the disease is by monitoring the CDK1 and CDK2 specific activity (Kim et al. 2008). It has been seen that tumor cell growth can be suppressed by employing siRNA molecules or NU2058 and NU6102 (the purine-based inhibitors), both of which lead to CDK1 and CDK2 silencing. It has been observed that apoptosis is induced in both sensitive and resistant cell lines (Johnson et al. 2010).
9.4 The CDK1 and Breast Cancer
Many in vitro and in vivo studies concerned with the function of CDK1 in BC have been carried out; some of the studies have been included in Table 9.1 A heightened expression and activity was seen in G1/S/G2 phases of cell cycle for CDK1/Cyclin B1 in case of certain breast cancer cell lines through an in vitro investigation (Barrett et al. 2002). In another study, it was concluded that evaluating the CDK1 and 2 specific activity could be regarded a significant prognostic value for determining the outcomes in BC subjects, and multiple drugs were used in this in vivo study (Kim et al. 2008). In another in vitro study, Flavopiridol, siRNA were used and the disruption of CDK1 and 2, which in certain BC cell lines resulted in the arrest of cell cycle and apoptosis (Johnson et al. 2010). A study observed that the particular activity of CDK1 and 2 could possibly predict reaction toward the treatment being employed as well as the chances of recurrence and for this in vivo trial, the medicines Paclitaxel, 5-fluorouracil, Epirubicin, and Cyclophosphamide were employed (Kim et al. 2012). As per another study, the in vivo polymorphisms of CDK1 and CCNB1 genes increase the predisposition to BC, disease advancement as well as rate of survival (Li et al. 2013). The growth of TNBC displayed a suppression in SUM149, BT549 MCF-10A cell lines when CDK1 was downregulated by siRNA-laden nanoparticles (Liu et al. 2014). In an in vivo human investigation, increased CDK1 specific activity was linked to early and high recurrence rates (Kim et al. 2014).
In a study using the medications Aminophenazone, Pomalidomide, and Rosoxacin in an in vitro investigation, CDK1 was found to be a diagnostic marker in ductal carcinoma in situ (Ding et al. 2017). The observed phenotype in mouse model with gene-targeted CDK alleles (lacking CDK3) was that in the initial cell divisions, embryonic lethality was witnessed because of deficiency of CDK1 with Cdk1mut/mut type of genotype (Santamaría et al. 2007) and this Cdk1mut allele has been developed by using the insertion of gene trap vector and it represents loss-of-function strain. It has been seen in the study that when the roscovitine, a pan-CDK inhibitor, is administered sequentially preceding doxorubicin treatment is synthetically lethal in the triple-negative breast cancer cells. This inhibitor, when administered, halts the cell cycle in phase G2/M, preparing them for DNA damage. It was observed that this combined treatment approach led to an enhancement in DNA double-stranded breaks and lowered the recruitment of proteins necessary for homologous recombination compared to the solo treatment by doxorubicin. It was also witnessed that by employing this combination therapy, there was a reduction in the tumor volume and an elevated survival was observed in comparison with the solo drug or related treatment in the case of xenograft studies (Jabbour-Leung et al. 2016). It was observed in a study that in the lack of CDK2, the CDK1 acts as G1-S CDK and binds to Cyclin E and when CDK1 is absent, CDK2 binds Cyclin B leading to mitotic entry (Lau et al. 2021) (Table 9.2). It was also seen that although performing all the mitotic roles of CDK1 by CDK2 was not enough in its normal concentrations, the CDK2 overexpression could overcome the mitotic abnormalities that occur due to lack of CDK1 (Lau et al. 2021). The results of CDK1 dysregulation include a robust growth of the tumor, heightened cancer cell proliferation rates, and chromosomal mutability (Barascu et al. 2006), as shown in Fig. 9.6.

The dysregulation of CDK1 could lead to many possible changes/events, including enhanced tumor growth, increased cancer cell proliferation, and chromosomal instability
It was revealed in a study that CDK2 generates less mitotic phosphorylation when compared to CDK1 leading to abnormal late mitotic events and a lack of both CDK1 and 2 results in total abolishment of mitotic entry. It was also elucidated in this study that in the absence of CDK1, the RPE1 human epithelial cell line is unable to undergo mitotic entry unlike cancer cell lines (Lau et al. 2021).
9.5 Therapeutic Implications
The inhibitors directed against CDKs are categorized either as Non-selective or Selective, i.e., either pan-inhibitors or against one single cyclin-dependent kinase, solely based on meticulousness against the CDKs (Ding et al. 2020). Various drugs that are CDK inhibitors in action have entered breast cancer clinical trials and are known to target cell modulators in the cancerous cells, thus furnishing a therapeutic window (Ding et al. 2020). Various pan-CDK inhibitors have been employed in trials to inhibit the activities of CDKS including CDK1. All pan-CDK inhibitors are non-specific in action and produce various undesirable toxicities too. Some of the inhibitors of CDK1 include the following:
-
1.
Flavopiridol (a semi-synthetic flavone) (Kaur et al. 1992; Sedlacek et al. 1996).
-
2.
Roscovitine (a synthetic purine) (Lin et al. 2010).
-
3.
Dinaciclib (Paruch et al. 2010).
These are all pan-CDK inhibitors (Fig. 9.7).

Some of the pan-CDK inhibitors that are also effective against several CDKs including CDK1, and for BC Roscovitine and Dinaciclib trials have been conducted
Roscovitine, Ro-3306, and Dinaciclib are the inhibitors that target CDK1/2 (Lin et al. 2018). Roscovitine inhibits the CDK1 and others (Table 9.3) by directly competing at the ATP-binding sites (Vassilev et al. 2006). Also, Ro-3306 blocks/prevents the G2 to M transition, leading to programmed cell death of tumor cells after CDK1/2 inhibitor exposure for a long time (Xia et al. 2014; D’Andrea 2018). One of the examples is Flavopiridol, a semi-synthetic flavonoid obtained from rohitukine (a chromosome alkaloid). It exerts its anti-cancer effects by inhibiting CDK1, 2, 4, 6, 7, and 9 (Sedlacek et al. 1996; Shapiro 2006) Flavopiridol also known as Alvocidib, a first-generation pan-CDK inhibitor, the primary pan-CDK inhibitor employed in clinical trials. The activities of CDK1, 2, 4, 6, and 7,9 are primarily halted by Flavopiridol (Asghar et al. 2015). In the G1 and G2 phases, Flavopiridol leads to the arrest of the cell cycle and also induces cytotoxicity by blocking CDK7 and CDK9 and c-MYC transcription (Canavese et al. 2012).
The targets of Seliciclib are CDK1, CDK2, CDK5, CDK7, and CDK9 (Whittaker et al. 2004). This inhibitor surfaced as the initial orally available drug (from this class) to become part of the clinical trials due to its relative success in the pre-clinical stage, where its success leads to the onset of apoptosis in tumor cells (Shapiro 2006; Galons et al. 2010; Nanos-Webb et al. 2012). Another example from the pan-CDK inhibitors is provided by Dinaciclib, which is known to inhibit CDK1, 2, 5, and 9 with excellent Rb phosphorylation inhibitory potency (Fig. 9.8), thus showing a better therapeutic index in comparison with the Flavopiridol.

The pan-CDK inhibitors’ working model in general: The drugs like dinaciclib and roscovitine lead to the inhibition of multiple CDKs including CDK1
It must be mentioned that the palbociclib and abemaciclib display very low potency against CDK1, 2, 7, and 9, and with fulvestrant these have been marked for the second-line therapy (Chen et al. 2019). The drug that is orally administered and displays high potency with bioavailability and inhibits CDK4/6 selectivity is Ribociclib. This drug does not display significant activity against CDK2 and CDK1 (Sobhani et al. 2019). The ER+ breast cancer cell proliferation and migration is known to be inhibited by PL (piperlongumine), a novel CDK inhibitor discovered by Jeong et al. The PL is a natural product, and it is obtained from pepper. It hinders the CDK1 and CDK4/6 expression levels and leads to obstruction of the cell cycle at the G2/M phase in order to stop tumorigenesis (Table 9.3) (Asghar et al. 2017).
9.6 Undesirable Effects of Pan-CDK Inhibitors
Several undesirable effects/toxicities have been witnessed due to the use of various pan-CDK inhibitors, including fatigue, myelosuppression, nausea, abnormalities in liver, vomiting, nerve dysfunction, GIT effects, and for these agents lack of predictive biomarkers for the BC patients. Thus, these collapsed before phase second trials. The undesirable effects are shown in Fig. 9.9 (Finn et al. 2016).

Some side effects observed due to consumption of pan-CDK inhibitors in BC patients
9.7 Summary
CDKs require modulatory subunits to bind them, known as cyclins, to exert their effects. The latter are formed and destroyed at various cell cycle stages in a specific and timely manner, thus regulating the cell cycle properly. The uncontrolled cellular proliferation manifests as one of the cancer hallmarks. The same occurs through the cell cycle checkpoint disabling and overriding several safeguards involved with the cyclin/CDKs dysregulation or impairment. In many solid cancers like BC, uncontrolled cell proliferation is witnessed as a result of cell cycle dysregulation and the genetic changes in the proteins involved in cell cycle regulation. CDKs require modulatory subunits to bind them, known as cyclins, to exert their effects. The latter are formed and destroyed at various cell cycle stages in a specific and timely manner, thus regulating the cell cycle properly. Unscheduled proliferation, chromosomal instability (CIN), and genomic instability (GIN) are the three most common disorders in the cell cycle caused by either direct or indirect poor control of CDKs. Because of their critical participation in cell cycle progression, downregulation of CDKs may result in improper homeostasis in specific tissues. On the other hand, by initiating the untimely division in cells (progenitor or stem cells), the hyperactivated CDKs could also aid in the tumor development. CDK1 is a member of the cell cycle-associated CDK family, which also includes CDK2, CDK4, and CDK6. These CDKs play a direct role in the progression of cell cycle. Multiple studies have demonstrated that the CDK1 expression levels and function are dysregulated indicating its potential role in BC progression. It has also been witnessed that by either blocking or silencing the CDK1 could suppress the BC growth, particularly when combined with other anti-cancer agents. The results of CDK1 dysregulation include a robust growth of the tumor, heightened cancer cell proliferation rates, and chromosomal mutability. Furthermore, many studies have been previously carried out to examine the possible roles of CDK1 in the case of BC like the heightened expression of CDK1/Cyclin B1 was witnessed in G1/S/G2phases in breast cancer cell lines. The pan-CDK inhibitors employed for this treatment, however, come with multiple undesirable effects on BC patients. As such combination with other anti-cancer therapeutics for relatively superior outcomes could be a better option for BC patients.
9.8 Further Readings
The readers can have a look upon the following articles for the better understanding of the given topic:
- (i)
- (ii)
The readers can also take a look upon the following visual presentations:
For more insights about the topic, we would suggest detailed findings from the books of Mir MA (2022) https://doi.org/10.1016/C2021-0-02565-7, https://doi.org/10.1016/C2022-0-00074-X Mir MA (2021) https://doi.org/10.52305/WXJL6770, from cancer.net website, https://www.cancer.net/cancer-types/breast-cancer/types-treatment.
References
Aaltonen K et al (2009) High cyclin B1 expression is associated with poor survival in breast cancer. Br J Cancer 100(7):1055–1060
Asghar U et al (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 14(2):130–146
Asghar US et al (2017) Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast CancerCDK4/6 inhibition in triple-negative breast cancer. Clin Cancer Res 23(18):5561–5572
Barascu A et al (2006) CDK1-cyclin B1 mediates the inhibition of proliferation induced by omega-3 fatty acids in MDA-MB-231 breast cancer cells. Int J Biochem Cell Biol 38(2):196–208
Barrett KL et al (2002) Cyclin b1 promoter activity and functional cdk1 complex formation in G1 phase of human breast cancer cells. Cell Biol Int 26(1):19–28
Bartek J et al (2004) Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 5(10):792–804
Brigham et al (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Canavese M et al (2012) Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther 13(7):451–457
Chen L et al (2008) Cell cycle-dependent complex formation of BRCA1· CtIP· MRN is important for DNA double-strand break repair. J Biol Chem 283(12):7713–7720
Chen Y-J et al (2009) A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem 284(44):30695–30707
Chen X et al (2019) Latest overview of the cyclin-dependent kinases 4/6 inhibitors in breast cancer: the past, the present and the future. J Cancer 10(26):6608
D’Andrea ADJDR (2018) Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 71:172–176
Ding Z-H et al (2017) Docking of CDK1 with antibiotic drugs revealed novel therapeutic value in breast ductal cancer in situ. Oncotarget 8(37):61998
Ding L et al (2020) The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci 21(6):1960
Draetta G, Beach DJC (1988) Activation of cdc2 protein kinase during mitosis in human cells: cell cycle-dependent phosphorylation and subunit rearrangement. Cell 54(1):17–26
Duffy MJ et al (2015) Biomarkers in breast cancer: where are we and where are we going? Adv Clin Chem 71:1–23
Endicott JA et al (1999) Cyclin-dependent kinases: inhibition and substrate recognition. Curr Opin Struct Biol 9(6):738–744
Esashi F et al (2005) CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434(7033):598–604
Finn RS et al (2016) Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res 18(1):1–11
Galons H et al (2010) Cyclin-dependent kinase inhibitors: a survey of recent patent literature. Expert Opin Ther Pat 20(3):377–404
Gavet O, Pines JJDC (2010a) Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell 18(4):533–543
Gavet O, Pines JJJOCB (2010b) Activation of cyclin B1–Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol 189(2):247–259
Gerger A et al (2007) A multigenic approach to predict breast cancer risk. Breast Cancer Res Treat 104(2):159–164
Gould KL, Nurse PJN (1989) Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature 342(6245):39–45
Hanahan D, Weinberg RAJC (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hashemi V et al (2019) The role of DEAD-box RNA helicase p68 (DDX5) in the development and treatment of breast cancer. J Cell Physiol 234(5):5478–5487
Heald R et al (1993) Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74(3):463–474
Horiuchi D et al (2012) MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med 209(4):679–696
Huertas P, Jackson SPJJOBC (2009) Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 284(14):9558–9565
Huertas Sánchez P et al (2008) CDK targets Sae2 to control DNA-end resection and homologous recombination
Ira G et al (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431(7011):1011–1017
Izadi S et al (2020) CDK1 in breast cancer: implications for theranostic potential. Anti Cancer Agents Med Chem 20(7):758–767
Jabbour-Leung NA et al (2016) Sequential combination therapy of CDK inhibition and doxorubicin is synthetically lethal in p53-mutant triple-negative breast CancerCDK inhibition sensitizes p53-mutated cells to doxorubicin. Mol Cancer Ther 15(4):593–607
Johnson N et al (2010) Pre-clinical evaluation of cyclin-dependent kinase 2 and 1 inhibition in anti-estrogen-sensitive and resistant breast cancer cells. Br J Cancer 102(2):342–350
Kastan MB, Bartek JJN (2004) Cell-cycle checkpoints and cancer. Nature 432(7015):316–323
Kaur G et al (1992) Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J Natl Cancer Inst 84(22):1736–1740
Kim S et al (2008) Determination of the specific activity of CDK1 and CDK2 as a novel prognostic indicator for early breast cancer. Ann Oncol 19(1):68–72
Kim S et al (2012) Recurrence risk score based on the specific activity of CDK1 and CDK2 predicts response to neoadjuvant paclitaxel followed by 5-fluorouracil, epirubicin and cyclophosphamide in breast cancers. Ann Oncol 23(4):891–897
Kim SJ et al (2014) The cell cycle profiling-risk score based on CDK1 and 2 predicts early recurrence in node-negative, hormone receptor-positive breast cancer treated with endocrine therapy. Cancer Lett 355(2):217–223
Kops GJ et al (2005) On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 5(10):773–785
Lau HW et al (2021) Quantitative differences between cyclin-dependent kinases underlie the unique functions of CDK1 in human cells. Cell Rep 37(2):109808
Li S et al (1995) The cdc2-related kinase, PISSLRE, is essential for cell growth and acts in G2 phase of the cell cycle. Cancer Res 55(18):3992–3995
Li Y et al (2013) Association study of germline variants in CCNB1 and CDK1 with breast cancer susceptibility, progression, and survival among Chinese Han women. PLoS One 8(12):e84489
Lin TS et al (2010) Flavopiridol, fludarabine, and rituximab in mantle cell lymphoma and indolent B-cell lymphoproliferative disorders. J Clin Oncol 28(3):418
Lin ZP et al (2018) Targeting cyclin-dependent kinases for treatment of gynecologic cancers. Front Oncol 8:303
Liu Y et al (2014) Triple negative breast cancer therapy with CDK1 siRNA delivered by cationic lipid assisted PEG-PLA nanoparticles. J Control Release 192:114–121
Malumbres M, Barbacid MJNRC (2001) To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1(3):222–231
Malumbres M, Barbacid MJNRC (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9(3):153–166
Malumbres M, Barbacid MJTIBS (2005) Mammalian cyclin-dependent kinases. Trends Biochem Sci 30(11):630–641
Marais A et al (2010) Cell cycle-dependent regulation of the forkhead transcription factor FOXK2 by CDK· cyclin complexes. J Biol Chem 285(46):35728–35739
Murphy M et al (1997) Delayed early embryonic lethality following disruption of the murine cyclin A2 gene. Nat Genet 15(1):83–86
Musacchio A, Salmon EDJNRMCB (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8(5):379–393
Nakayama S et al (2009) Prediction of paclitaxel sensitivity by CDK1 and CDK2 activity in human breast cancer cells. Breast Cancer Res 11(1):1–10
Nanos-Webb A et al (2012) Targeting low molecular weight cyclin E (LMW-E) in breast cancer. Breast Cancer Res Treat 132(2):575–588
Paruch K et al (2010) Discovery of dinaciclib (SCH 727965): a potent and selective inhibitor of cyclin-dependent kinases. ACS Med Chem Lett 1(5):204–208
Pérez de Castro I et al (2007) A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis 28(5):899–912
Poon RYJCCO (2016) Cell cycle control: a system of interlinking oscillators. Methods Mol Biol:3–19
Potapova TA et al (2006) The reversibility of mitotic exit in vertebrate cells. Nature 440(7086):954–958
Rahman N, Stratton MRJAROG (1998) The genetics of breast cancer susceptibility. Annu Rev Genet 32:95
Reese JM et al (2017) ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget 8(57):96506
Santamaría D et al (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448(7155):811–815
Satyanarayana A et al (2008) Genetic substitution of Cdk1 by Cdk2 leads to embryonic lethality and loss of meiotic function of Cdk2. Development 135(20):3389–3400
Sedlacek H et al (1996) Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int J Oncol 9(6):1143–1168
Shapiro GIJJOCO (2006) Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 24(11):1770–1783
Sobhani N et al (2019) Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cell 8(4):321
Sofi S et al (2022) Cyclin-dependent kinases in breast cancer: expression pattern and therapeutic implications. Med Oncol 39(6):1–16
Vassilev LT et al (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 103(28):10660–10665
Whittaker SR et al (2004) The Cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res 64(1):262–272
Xia Q et al (2014) The CDK1 inhibitor RO3306 improves the response of BRCA-proficient breast cancer cells to PARP inhibition. Int J Oncol 44(3):735–744
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mir, M.A., Haq, B.U. (2023). CDK1 Dysregulation in Breast Cancer. In: Mir, M. (eds) Therapeutic potential of Cell Cycle Kinases in Breast Cancer. Springer, Singapore. https://doi.org/10.1007/978-981-19-8911-7_9
Download citation
DOI: https://doi.org/10.1007/978-981-19-8911-7_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-8910-0
Online ISBN: 978-981-19-8911-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)