
Abstract
Epigenetic modifications are known to alter the activation pattern of some genes and not the per se DNA sequence. Stress to the plant causes epigenetic alterations in the plant either as hyper- or as hypo-methylation of certain DNA sequences. To overcome or to counter the various abiotic stress conditions, the plant’s defense machinery including cellular signaling pathways gets regulated by several stress-responsive genes, which in turn are regulated by various mechanisms including DNA and chromatin modifications, and also through different small RNA-based mechanisms. There is a sudden spurt in the epigenetic studies aiming to find their role in the imposition of various types of abiotic stress tolerance in different plant species, mainly due to the quick advancements in the high-throughput NGS technologies. Many reports associating the DNA methylation response with that of various abiotic stress adaptations are available in many legume species like soybean, chickpea, pigeon pea, Medicago, lotus, peanut, and common beans using these techniques. These legumes have shown tolerance to several abiotic stresses because of unique epigenetic variations, which are present in the natural populations. Understanding the epigenetic mechanism regulating the tolerance to the abiotic stresses will help plant breeders in the development of more resilient and climate-smart varieties, giving higher yields under varied abiotic stresses. This chapter covers the current status of a novel and promising field of epigenetics in legume crops, especially for the imposition of different abiotic stress tolerance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
Legumes are unique in the sense that they can enhance soil fertility through natural nitrogen fixation ability and thereby help in the overall agricultural sustainability. Several grain legumes are the staple food and the key protein source, especially for the poor residing in developing and underdeveloped countries (Mishra et al. 2021). Ironically, most of the legumes suffer heavily due to the negative impact of many abiotic stresses like heat, cold, drought (water-deficit stress), and salinity (Bhalani et al. 2019; Sarkar et al. 2014, 2016). Thus, to sustain such important crops, there is a need to opt for novel approaches like epigenetics to develop the climate-resilient and abiotic stress-tolerant varieties in the legumes. Next-generation sequencing (NGS) technologies are giving an option for rapid and cost-effective omics technologies in many legumes including chickpea, mung bean, lentils, pigeon pea, peas, soybean, Medicago, etc. for the identification of key genes and regulatory pathways involved with different abiotic stress tolerance (Dasgupta et al. 2021; Mishra et al. 2020; Nawade et al. 2018). Various studies have proved the associations between methylation levels and different abiotic stresses, suggesting the pronounced role of epigenetic mechanisms in plant adaptability (Malabarba et al. 2021; Windels et al. 2021). Thus, understanding their role in the abiotic stress tolerance mechanism is very important to have improved productivity (Ramu et al. 2016).
In general, epigenetics (meaning above genetics) is being referred to as any heritable alteration which is unable to modify the DNA sequence(s) or genetic code yet causes modified gene expression and altered phenotype. However, the concept of epigenetics is constantly changing, and its exact definition is always debated (Deans and Maggert 2015). Epigenetic change results in the modification of the chromatin structure, which in turn affects the transcription pattern of the cells. Epigenetic regulation mechanisms can be largely classified into three groups, viz., DNA methylation, histone modification, and RNA interference (RNAi) (Saraswat et al. 2017).
Plants being sessile in nature are exposed continuously to the environmental vagaries and experience stresses of different kinds such as availability of water and nutrient, temperature and light regimes, and salinity (Patel et al. 2016, 2017; Reddy et al. 2020). Adaptation to these stressors needs constant dynamic changes in the plants at both morphological and molecular levels. To overcome such environmental vagaries, plants have developed several strategies including epigenetic regulation for better survivability (Saraswat et al. 2017; Shanker and Venkateswarlu 2011). Several epigenetic mechanisms including abiotic stress responses were identified mainly from the model plants like Arabidopsis (Pecinka et al. 2020) and rice (La et al. 2011). The knowledge derived from these species is being used to understand the similar phenomenon in legumes too (Chinnusamy and Zhu 2009; Gutzat and Scheid 2012). Also, reports mentioning the changes in the DNA methylation pattern in different plant species are available for different stresses such as water-deficit stress (Kapazoglou et al. 2013), temperature stress (Naydenov et al. 2015), and continuous cropping (Liang et al. 2019).
The most deeply studied model legumes at the genomic levels include soybean, Medicago truncatula, and lotus (Cañas and Beltrán 2018; Mochida et al. 2010; Ramesh et al. 2019). Lately, a few other legumes like Phaseolus vulgaris (common bean), chickpea, and cowpea are also being deeply investigated at the genomic level (Lobaton et al. 2018; Mishra et al. 2021, 2022; Timko et al. 2008; Varshney et al. 2013). Recently, many legume crops like pigeon pea, lentil, mung bean, peas, beans, Medicago, lotus, peanut, chickpea, and soybean have been sequenced, and the amount of genomic sequence data is increasing with each passing year (Ahmad et al. 2020; Bosamia et al. 2020; Garg et al. 2014; Mishra et al. 2020). Further, due to the rapid increase in the relatively cheap genomic technologies (including epigenome analysis), such studies even in the non-model organism have also been possible.
Under abiotic stress, plants respond differently involving multiple mechanisms through massive differential gene expressions and nuclear organizations including epigenetic changes (Budak et al. 2015). Nevertheless, studies delineating the role of epigenetics in the imposition of abiotic stresses in legumes are not very deeply understood, to date (Niederhuth and Schmitz 2014). Epigenetic variations in the DNA have been reported in response to many abiotic stresses (Pandey et al. 2016). Yet, the precise role of various enzymes catalyzing the active DNA methylations or other modifications has not been thoroughly understood.
Legumes generally have large genome sizes, many TEs, repeat regions, and numerous high-copy-number genes, and to understand their functions, legume breeding should include novel -omics technologies including the use of epigenetic approaches while going for the development of new high-yielding and climate-resilient varieties (Bosamia et al. 2015; Mishra et al. 2015; Salgotra and Gupta 2019). With this backdrop, this chapter gives an in-depth overview of various epigenetic studies in different legume species with a detailed focus on the role of epigenetics in abiotic stress responses in legumes.
4.2 Epigenetics and Major DNA Methylation Mechanisms
The “epigenetic landscape” and “epigenetics” terms were coined by Conrad Waddington way back during the early 1940s. Gene expression can be regulated through various epigenetic mechanisms like chromatin modifications (e.g., histone acetylation, methylation, phosphorylation, and ubiquitylation) and DNA modifications (e.g., cytosine methylation) (Gibney and Nolan 2010). These epigenetic modifications are prompted by various developmental and/or environmental reasons, which then modify the chromatin architecture without changing the DNA sequence(s) (Chinnusamy and Zhu 2009). The detailed understanding of the role of epigenetic factors regulating various abiotic stresses is still very limited.
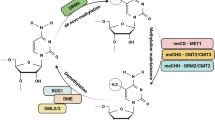
Among various epigenetic mechanisms, DNA methylation and posttranslational histone modifications (PHM) are the deeply studied DNA modification mechanisms. In the case of DNA methylation, a methyl group gets added from S-adenosyl-l-methionine to the fifth C of the cytosine ring, which results in the formation of 5-methylcytosine (5mC). In plant system, DNA methylation is reported to occur in three sequence contexts, viz., (1) symmetric CG, (2) symmetric CHG, and (3) asymmetric CHH, where H can be A, T, or C base or except G any other base (Malabarba et al. 2021). Process-wise DNA methylation can be of three types, viz., (1) de novo methylation, (2) maintenance of methylation, and (3) demethylation, which involves several enzymes (Fig. 4.1).

Outline of DNA methylation and demethylation operating in the plants
4.2.1 De Novo Methylation
The process uses domains rearranged methyltransferase-2 (DRM2), which gets controlled by RNA-directed DNA methylation (RdDM) pathway (Law and Jacobsen 2010; Matzke and Mosher 2014), wherein Pol IV (RNA pol IV) transcribes single-stranded RNAs (ssRNAs) which then form double-stranded RNA intermediates (dsRNAs) by RNA-dependent RNA polymerase 2 (RDR2). Afterward, DCL3 (RNase III-class DICER-LIKE 3) cleaves the dsRNAs to form 24-nt small interfering RNAs (siRNAs), which get incorporated into AGO4 (ARGONAUTE 4) and base-paired with Pol V and produce scaffold RNA and use DDR protein complex. This comprises proteins such as defective in RNA-directed DNA methylation 1 (DRD1), RNA-directed DNA methylation 1 (RDM1), and defective in meristem silencing 3 (DSM1) that stabilize the Pol V and chromatin interaction using MORC protein complex. Pol V then guides the AGO4 to the chromatin (Wierzbicki et al. 2009). These ultimately result in the DRM2 recruitment, which is followed by methylation of specific DNA base(s) (Matzke and Mosher 2014; Huiming Zhang and Zhu 2011). The precise function of the RdDM pathway indirect gene methylation and regulation is still not very clear. Rather, this pathway targets some repetitive sequences and transposable elements (TEs), which then controls the activation and repression of close-by gene(s) (Sigman and Slotkin 2016). The methylation of CHH context (de novo) of mostly heterochromatin regions especially that of TEs was regulated by the CMT2-dependent pathway (Zemach et al. 2013), which acts in a siRNA-independent way and is dependent on decreased in DNA methylation 1 (DDM1) chromatin remodeler.
4.2.2 Maintenance of Methylation
This is very much dependent on the sequence contexts; for example, methylation of CG context is reportedly maintained by methyltransferase 1 (MET1) and decrease in DNA methylation 1 (DDM1), whereas CHG by chromomethylase 2 and 3 (CMT2 and CMT3), and CHH by DRM2 and CMT2 (Chan et al. 2005).
4.2.3 DNA Demethylation
Demethylation can be through either active or passive ways, wherein passive demethylation denotes loss of methylation during DNA replication due to the inactivity of the demethylating enzyme (Zhu 2009). This process is being regulated by four bifunctional 5mC DNA glycosylases, viz., repressor of silencing 1 (ROS1), Demeter (DME), DME-like 2 (DML2), and DML3, which removes the 5mC using base excision repair (BER) pathway (Zhang and Zhu 2012). Due to the antagonistic effect of RdDM and ROS1 activity, some sort of coordination has been reported between DNA methylation and demethylation, which in turn stops the hypermethylation of certain loci (Tang et al. 2016). A 39-nt regulatory element or MEMS (DNA monitoring methylation sequence) is the ROS1 promoter and functions as a putative sensor of MET1 and RdDM pathway. Very high activity of MET1 and RdDM results in the hypermethylation of MEMS, which causes activation of ROS1 demethylase activity and regulates DNA methylation at the whole-genome level (Lei et al. 2015).
4.3 Methylation of Various Regions of the Gene
Gene expression also gets regulated by the methylation in the promotor region via inhibition of transcriptional activators/repressors. This may completely inhibit the tissue-based gene expression (Johnson et al. 2007; Zhang et al. 2006), or this may also regulate specific processes like gene imprinting during seed development or immune-responsive gene regulation (Matzke and Mosher 2014; Zhang et al. 2018a). However, the function of DNA methylation within the gene bodies is still not very clear, and two hypotheses have been proposed about their role, viz., (1) masking of the cryptic transcription sites, which will assist in its isoform splicing (Neri et al. 2017), and (2) reduction in the gene expression variations via exclusion of H2A.Z from the nucleosome (Zilberman et al. 2008). The function of methylation in the TE activity regulations is thoroughly studied, wherein it mainly functions as either TE silencing or a repressor of the transposition by hypermethylation of all the sequence contexts (Sigman and Slotkin 2016).
4.3.1 Histone Modifications
Chromatin accessibility in the gene’s promotor region is observed through histone modifications, especially through methylation or acetylation (Berger 2007; Kouzarides 2007) (Fig. 4.2). A nucleosome consists of eight histone proteins (two copies each of H2A, H2B, H3, and H4 proteins), which are wrapped by 147 bp DNA (Peterson and Laniel 2004) and function through epigenetic modification of various genes by controlling the access and binding of regulatory elements (Berger 2007). Modification of the amino acids present at the N-terminal tails of histone proteins (H3 and H4) is reported, which can either activate the genes via acetylation, phosphorylation, and ubiquitination or repress the genes mainly via methylation (with some exceptions) (Zhao et al. 2019). Even though histones are considered as highly conserved proteins, plants do possess structurally and functionally discrete classes of H2A (H2A.X, H2A.Z) and H3 (H3.3) forms (Deal and Henikoff 2011). Increased H3K9ac (in the heterochromatic chromatin knobs) was found to be associated with an increase in the transcription, while increased H3K9me2 was found to be correlated with a decrease in the transcription of certain stress-responsive genes (Yong Hu et al. 2012). The stress-responsive genes in the plants show transient modifications in the histones under varied stress conditions (Zong et al. 2013).

Schematic representation of epigenetic mechanism and marks operating in plants
4.3.2 Noncoding RNAs and Epigenetic Regulation Under Abiotic Stress
Noncoding RNAs (long ncRNA or small ncRNAs) regulate the opening and closing of the chromatin and are associated with both gene silencing and activation at both transcriptional and posttranscriptional levels. The ncRNAs which are associated with epigenetic regulation (i.e., heterochromatin formation, histone modification, DNA methylation, and gene silencing) are of two major types, viz., the short ncRNAs (<30 nts) and the long ncRNAs (>200 nts). The short ncRNAs are grouped into three major types: (1) microRNAs (miRNAs), (2) short interfering RNAs (siRNAs), and (3) piwi-interacting RNAs (piRNAs) (Salgotra and Gupta 2019). Sequence-specific methylation is known to be caused by ds-RNA and RdDM (Law and Jacobsen 2010), while RNAi was reportedly associated with the RdDM causing cytosine methylation (Meister and Tuschl 2004; Wassenegger et al. 1994). In plants, miRNAs are known to cause RNA silencing and posttranscriptional gene regulation (Lee et al. 1993; Maxwell et al. 2012). Various classes of siRNAs (i.e., antisense siRNAs, heterochromatic siRNAs, trans-acting siRNAs) were known to enable gene silencing through methylation of histone and RdDM (Mosher et al. 2008; Xu et al. 2013).
Several reports have proved the role of ncRNAs in the regulation of gene expression under abiotic stress conditions. Zeller et al. (2009) reported the accumulation of various novel antisense transcripts under abiotic stress situations in the plants, which were the source of siRNAs. Downregulation of certain siRNAs such as Hc-siRNAs (heterochromatic siRNAs), siR441, and siR446 has been reported under abiotic stress situations, which seems essential for the gene regulation, especially under stress conditions (Yan et al. 2011). The miRNAs are known to have a major role under different abiotic stress-like conditions, especially under cold, heat, salinity, etc. (Salgotra and Gupta 2019).
In mung bean, among different sequence contexts, the proportion of mCHH DNA methylation was very high, while in soybean (Schmitz et al. 2013), mCG is the most commonly methylated sequence context. Both mung bean and common bean had maximum cytosine methylation in the CHH sequence context (Do Kim et al. 2015). These two species were known to have diverged nearly 8.0 Ma (million years ago), whereas soybean is considered to have diverged from the mung bean nearly 19.2 Ma (Lavin et al. 2005). Thus, the traces of mCHH-enriched DNA methylation in both mung bean and common bean might have come from their common ancestor (Kang et al. 2017).
The role of the RdDM pathway has been observed regulating the seed parameters (size and weight) in chickpea during the seed development stage, which showed a gradual increase in the methylation of CHH context of TEs along with increased frequency of small RNAs in hypermethylated TEs (Rajkumar et al. 2020). Kurdyukov et al. (2014) have studied a 2HA seed line of Medicago truncatula which was highly embryogenic, and microarray showed downregulation of an ethylene insensitive 3-like gene in 2HA callus. Ethylene is reportedly linked with several developmental processes such as somatic embryogenesis (SE) and various types of stress responses. The Medicago truncatula EIL1 gene (MtEIL1) was found to be epigenetically silenced in the 2HA line, which could be due to the increased level of miRNA targeting its 3′UTR and also due to the methylation of MtEIL1 (Kurdyukov et al. 2014). A plant-specific gene MutS HOMOLOG1 (MSH1) has been used for the RNAi suppression in several plant species including soybean for the production of developmental changes including abiotic stress response along with methylome repatterning. Therefore, RNAi can be a direct means of exploitation of epigenetic variations associated with abiotic stress tolerance in plants (Raju et al. 2018).
4.4 Epigenetics and Abiotic Stress Tolerance in Legumes
4.4.1 Temperature-Stress Tolerance
Climate change induced by various factors including excessive greenhouse gas (GHG) emission has caused many negative impacts on crop plants primarily in the form of heat stress due to global warming. The mean temperature of the earth is reportedly increased to the tune of 0.35 °C from 1979 to 2003 (Venterea 2014; Walther et al. 2002). An increase in the temperature or heat stress is known to cause early flowering (Peñuelas et al. 2009), modified plant architecture (Wahid et al. 2007), poor seed development, decreased dormancy, reduced size, and poor grain yield (Folsom et al. 2014; Long and Ort 2010), and shifting of plant establishment to higher altitudes (Pauli et al. 2012). For the transcriptional machinery, chromatin conformation plays a major role by allowing the access of DNA sequences (Li et al. 2008). Several studies have proved the role of histone acetylation and methylation in the plant’s response to abiotic stresses (Stępiński 2012). In the root meristem of the soybean, changes were recorded for the DNA methylation, histone methylation, and histone acetylation when grown under different temperature regimes.
4.4.1.1 Heat Stress
In response to abiotic stress like heat, the heat-stress factors (HSF) and secondary metabolites have been found to have a great role in managing such stress response (McClung and Davis 2010). In addition, even by the transcriptional reprogramming like by upregulation of kinases and various transcription factors (TF) and downregulation of growth-related genes, such stresses were also managed (Popova et al. 2013). ROS1 was reported to be demethylating all the DNA methylation contexts as in the ros1 mutants all the DNA methylation contexts get hypermethylated (Tang et al. 2016). Heat stress is also known to affect DNA methylation in different plant species as increased global methylation and homologous recombination frequency (Boyko et al. 2010).
Various methylation process proteins [e.g., DRM2, nuclear RNA polymerase D1 (NRPD1)] also show upregulation upon high-temperature stress and increased DNA methylation (Naydenov et al. 2015). The RdDM pathway is also reported to partially regulate the transcriptional response to high-temperature stress (Popova et al. 2013). Interestingly, global DNA methylation induced by heat stress is species and tissue specific. In addition, the result of the impact of heat stress on transgenerational memory was recorded as phenotypic changes over generations by different researchers (Migicovsky et al. 2014; Suter and Widmer 2013). During repeated heat-stress conditions, methylation of histone H3K4 was found to be associated with the persistent expression and hyper-induction of high temperature-responsive genes (Lämke et al. 2016).
In the changing environmental scenario, an increase in the temperature is a major focus of various studies that are involved in unfolding the temperature-dependent genes and pathways (Arya et al. 2017). Temperature is a key factor affecting significantly the flowering in the plant system. Genome annotation has identified four copies of the PIF4 gene in soybean, which were found expressing abundantly under short-day conditions (Wong et al. 2013). Also, eight copies of the SVP gene were identified in soybean, of which high expression of Glyma01G02880.1 and Glyma02G04710.1 has been recorded in shoot apical meristem during the floral transition phase (Wong et al. 2013). In soybean, miRNA 156 and miRNA 172 have shown upregulation during the floral transition stage, when plants were exposed to high temperatures (Li et al. 2015). H3K56ac and H3K4me3 methylation marks are usually recorded in class 3 SDGs, which showed differential expression in shoot apical meristem (SAM) in soybean, during the floral transition (Liew et al. 2013). Besides, several RNAi genes like RNA pol, dsRNA binding (DRB), Dicer-like, AGO, DNA methyltransferase, and DNA glycosylase were upregulated in SAM during the floral transition (Liew et al. 2013).
4.4.1.2 Chilling Stress
More of 5mC and H3K9me2 were recorded through immunostaining in the heterochromatin region of the soybean genome during chilling stress than during the recovery phase. However, indicators of permissive chromatin (i.e., H3K9ac, H4K12ac, and H3K4me) showed weak labeling in the euchromatin region (under stress) over recovered plants (Stępiński 2012). Ivashuta et al. (2002) reported transcriptional activation of specific retrotransposons in Medicago sativa under cold-stress conditions. Hypermethylation was recorded in Cannabis sativa genotypes when exposed to different levels of cold acclimation (Mayer et al. 2015). Epigenetic factors are known to regulate cold-stress tolerance in hemp. Under cold-stress conditions, soluble sugars are found to accumulate and be maintained in higher concentrations in the tolerant hemp genotypes. These genotypes expressed more of COR gene transcripts, which is associated with the de novo DNA methylation. Also, significantly higher methylcytosine levels at COR gene loci were recorded in the tolerant genotypes when deacclimated, thereby confirming the function of locus-specific DNA methylation (Mayer et al. 2015). Relatively more methylation over demethylation was recorded in the cold stress-tolerant genotypes over susceptible chickpea genotype. This could be due to the comparatively higher activation of cold stress-responsive genes in the tolerant genotypes (Rakei et al. 2016).
4.4.2 Drought-Stress Tolerance
Overall increased DNA methylation or hypermethylation has been recorded in both tolerant and sensitive genotypes of faba bean (Abid et al. 2017) and pea (Labra et al. 2002) under drought or water-deficit stress situations. In groundnut, the regulation of the AhDREB1 gene (of AP2/ERF family TF) through acetylation of H3 helped in the positive regulation of water-deficit stress tolerance genes when exposed to the PEG-induced water-deficit stress conditions. Higher AhDREB1 gene expression was recorded when an inhibitor of histone deacetylase (HDAC), i.e., trichostatin (TSA), was used, which then showed water-deficit stress tolerance in the peanut plant (Zhang et al. 2018b). In chickpea, drought stress was able to induce the CaHDZ12 (an HD-Zip TF) activation, and its expression was found to be correlated with that of H3K9ac acetylation in the promoter region (Sen et al. 2017). Cytosine hypomethylation has been reported in the soybean roots under heat-stress conditions (Hossain et al. 2017). Reduced metabolic activity has been recorded in pea plants due to the drought-induced hyper-methylation of some key genes (Labra et al. 2002; Salgotra and Gupta 2019), while higher methylation was recorded in the drought-sensitive horse gram (Macrotyloma uniflorum) genotype than the tolerant genotype (Bhardwaj et al. 2013).
Epigenetic changes such as histone modifications, sRNAs, methylation of DNA, and lncRNAs were known to be associated with gene regulation in faba beans (Meyer 2015). A high degree of association between DNA methylation and gene expression under drought conditions in faba bean suggests the involvement of DNA methylation in the imposition of drought-stress tolerance (Abid et al. 2017). Several water-deficit stress-associated differentially methylated regions (DMRs) were identified by Abid et al. (2017), which became the basis for further understanding the epigenetic regulation of drought stress in faba bean.
In addition, a number of drought-responsive miRNAs are identified in various legumes (Mantri et al. 2013). Under drought and salinity stress, 259 differentially expressed miRNAs have been identified from the root tip tissue of chickpea. Many of these were found to have auxin and other abiotic stress-responsive cis-elements in their promoter region, which in turn imparted abiotic stress tolerance through various phytohormone syntheses (Khandal et al. 2017). In chickpea, under water-deficit stress, miR408 was found to be accumulated (Hajyzadeh et al. 2015), while similar results were also reported for Medicago (Trindade et al. 2010). Interestingly, 24 novel miRNA families have been reported in water-deficit stress-tolerant and -sensitive cowpea genotype (Barrera-Figueroa et al. 2012), and of these, 22 families were reported from soybean too (Kulcheski et al. 2011). Most of the iso-miRNAs showed upregulation during water-deficit stress in sensitive genotypes, while downregulation in tolerant genotypes. miRNAs were known to play a key role in the abiotic stress tolerance in cowpea, and much water-deficit stress-associated microRNAs have been identified (Barrera-Figueroa et al. 2011). Inconsistent miRNA expression was reported in the studied cowpea genotypes and nine recorded predominant or exclusive expression in one of the two studied genotypes, while a few were found regulated under water-deficit stress in only one genotype (Barrera-Figueroa et al. 2011).
The genome sequencing of common bean has generated a lot of information that can provide the much-needed evidence about future PTGS studies (Vlasova et al. 2016). Posttranscriptional regulation in common bean under water-deficit stress is reportedly regulated via a legume-specific miR1514a, which targets the NAC family of TF and generates secondary phasiRNAs (Sosa-Valencia et al. 2017b). The miR1514a showed differential expression levels in the roots of the common bean when exposed to water-deficit stress conditions. In addition, an RNA-seq study has also identified the role of NAC 700 TF in the water-deficit stress, while degradome analysis revealed the two NAC TFs (Phvul.010g121000 and Phvul.010g120700) as the target of miR1514a. In addition, small RNA-seq data indicate the role of only Phvul.010g120700 in the generation and accumulation of phasiRNAs under water-deficit stress conditions (Sosa-Valencia et al. 2017b).
4.4.3 Salinity-Stress Tolerance
In the pigeon pea shoot tissues, a global decline in the methylation levels of DNA has been reported under salinity-stress conditions (Awana et al. 2019). Similarly, imposition of continuous stress for a relatively longer duration has resulted in an increase in the overall DNA demethylation in soybean, mainly in the tolerant genotypes, and this increase corresponded well with that of increased expression of various DNA demethylases (e.g., DML and ROS1). Further, different demethylation studies could identify that the regulatory genes having CG and CHG contexts were more crucial than the CHH for their adaptation to salinity stress (Liang et al. 2019). The salinity stress in Medicago truncatula revealed variations to the tune of 77% in CHH, while CHG and CG showed 9.1% and 13.9% changes, respectively.
Interestingly, no such correlation has been recorded between DNA methylation pattern and level of transcripts for other salinity-stress tolerance-associated key genes, which means that these genes might be regulated by other epigenetics processes (Yaish et al. 2018). On the contrary, four TFs showed induced response under salinity stress in soybean, and of these three showed demethylations in CG and non-CG contexts and also active enrichment of histone marks (H3K4me3 and H3K9ac) along with a reduction in the repressive mark H3K9me2. Thus, the possible interplay was recorded between methylation of DNA and histone modifications when exposed to salinity stress (Song et al. 2012). In the plants of salinity-stressed soybean, cross talk has been reported between histone methylation and acetylation (Liu et al. 2010; Stępiński 2012). The soybean plants under salt-stress condition showed the binding of GmPHD5 (a homeodomain TF) with H3K4me2 marks (salt-induced), which then recruits a complex associated with gene activation having GmISWI (a type of nonhistone proteins and a chromatin remodeling factor) and GmGNAT1 (an acetyltransferase), which selectively acetylates H3K14 for the activated expression of salinity-induced genes (Wu et al. 2011).
The methylation-sensitive amplified polymorphism (MSAP) and enzyme-linked immunosorbent assay (ELISA) showed significantly more methylation in mCG under salinity stress in M. truncatula (Al-Lawati et al. 2016; Yaish et al. 2014). A comparative whole-genome bisulfite sequencing (WGBS) on the DNA isolated from the root tissues of Medicago truncatula under salinity stress and control has revealed higher methylation levels in all sequence contexts, ranging from 3.8% to 10.2%. However, qPCR-based gene expression studies did not find any stable association between mCG methylation levels and transcript abundance of some key genes involved in the imposition of salinity tolerance. Thus, it seems that some other epigenetic controllers are regulating the gene expression under salinity stress (Yaish et al. 2018).
The role of MTases regulating different aspects of plant development including different abiotic stress responses has been unfolded in different legume species using expression analysis. Garg et al. (2014) have identified 16 members of the DNMT2 family in several legume species, and increased expression of DNMT (CaDNMT2) was observed in chickpea shoots under both salt- and drought-stress conditions, suggesting the role of DNMT in abiotic stress response. Overall, under abiotic stress, more transcript was recorded for CMT and DRM genes, signifying the role of stress-induced methylation in chickpea (Garg et al. 2014). In chickpea, salinity stress was found inducing the CaHDZ12, which also showed a correlation with that of H3K9ac acetylation in the promoter region (Sen et al. 2017). Preferential transmission of salinity tolerance and DNA methylation was reported through the female germline. However, paternal dme mutants recorded restoration of paternal memory, indicating that the active DNA methylation in male gametes is essential for the inhibition of paternal inheritance of hyperosmotic priming response (Wibowo et al. 2016). The details of epigenetic response in various legumes during abiotic stress-associated processes are presented in Table 4.1.
4.4.4 Abiotic Stress Tolerance and DNA Demethylation
Several reports are aimed to analyze the changes which occur in the DNA demethylase gene when exposed to various abiotic stresses, and only some mentioning the detailed analysis involving loss-of-function mutations are available (Parrilla-Doblas et al. 2019). Interestingly, some recent studies showed the function of active demethylation of DNA in the inter-generational transmission of “stress memory” helping rapid adaptation to short-term environmental variations called “priming” (Parrilla-Doblas et al. 2019). In addition, the response of a plant to abscisic acid (ABA) has also shown active demethylation of DNA under abiotic stress conditions. Still, various factors regulating such demethylation during abiotic stress response are superficially known to the scientific community. Additionally, miRNAs are now found to have some role in the active demethylation of DNA of certain genes (Parrilla-Doblas et al. 2019). This has been generally observed during gametophyte development (Slotkin et al. 2009). However, active demethylation of DNA considers enzyme-based elimination of methylated cytosine, by a family of DNA glycosylases (such as DME, ROS1, DML2, and DML3), which was followed by the base excision repair (BER)-dependent process (Penterman et al. 2007; Zhu 2009). This does not only alter genome-wide epigenetics, but also regulate locus-specific genes with abiotic stress tolerance (Hsieh et al. 2009).
4.4.5 Abiotic Stress Tolerance and Epigenetics-Based Breeding Strategies in Legumes
Till now, we have compiled several ways that can be used for the enhancement of abiotic stress tolerance in various legumes. The use of epigenetics and epigenomics in improving the adaptation to abiotic stresses needs a combination of technical and biological innovations so that the breeders can go for the targeted gene-specific modifications of the epigenome for the desired trait improvement. Besides positive impact, stress-based memory may also have a negative impact on yield (Chinnusamy and Zhu 2009). Thus, care must be taken while going for an epigenetic-based approach for the abiotic stress improvement of the crops. We can use the impact prediction models for the epigenetic variations on a plant’s phenotype and performance (Colicchio et al. 2015; Yaodong Hu et al. 2015). Identification of epialleles having an impact on the abiotic stress tolerance traits can result in epigenetic-based breeding of crop plants like the use of mutant lines, recurrent epi-selection, epigenomic selection, and editing (Greaves et al. 2014; Hauben et al. 2009; Lämke and Bäurle 2017; Oakey et al. 2016; Yang et al. 2015).
In soybean, Raju et al. (2018) have proposed a breeding strategy using the MSH1 gene system for the improvement of yield and stability by inducing epigenetic variations. The soybean memory lines (wild type and msh1 acquired) were crossed to develop the epi-lines having wide variations for various yield-related traits. The identified epi-types showed low epi-type × environment (e × E) interactions and thus more stability under varied environments expressing different abiotic stresses (Varotto et al. 2020). The novel epigenetic variations induced by the MSH1 suppression were found to be inherited for at least three generations and can be used for enhancement and stabilization of the overall yield of soybean crops. In addition, several metabolic pathway genes regulating improved adaptation and plasticity (across generations) of the plant are also identified (Fujimoto et al. 2012; Raju et al. 2018; Robertson and Wolf 2012).
In association with classical genetic approaches, the novel sequencing technologies have helped in understanding the epigenetic process at the whole-genome level. Epigenome profiling and epigenome editing will help in the creation of novel epiallelic variants through DNA methylation and chromatin modifications (Springer and Schmitz 2017). Breeders are now preferring to use the mapping of epigenetic marks at genome-wide level (epigenomics), and also identification of epigenetic targets to modify the plants’ epigenomic variability to make them more resilient and climate smart (Lane et al. 2014). There is a need to do large-scale cross-species generation and comparison of epigenetic data in legumes, especially in response to abiotic stresses (Lane et al. 2014). Epigenetic modifications can be attempted either globally or at a specific locus using emerging techniques like CRISPR/Cas9 and dCas (Hilton et al. 2015; Moradpour and Abdulah 2020). The knowledge about the activation and repression of specific chromatin regions (using DNA-binding domains like Zn fingers, TALEs, dCas9) under specific abiotic stress can be used for the gene-specific activation or repression as per the need for the imposition of abiotic stress tolerance in crop plants (Bilichak and Kovalchuk 2016). In addition, a sound prediction model about the impact of epigenetic variations on the plant’s overall performance is needed (Colicchio et al. 2015; Yaodong Hu et al. 2015).
4.5 Conclusions and Future Prospects
In legumes, the epigenetic studies are still in infancy and are mainly targeting the identification of key epigenetic factors in the plant’s developmental and stress-related processes. A major reason for this could be the poor annotation of most of the legume genomes, which are incidentally full of numerous high-copy-number genes having overlapping or distinct functions (Windels et al. 2021). However, in times to come, we expect tremendous growth in legume epigenetic studies for various traits including abiotic stress tolerance. Stably inherited natural or induced epigenetic variations can be used to create climate-smart crops (Vriet et al. 2015). Otherwise, most stress-induced epigenetic modifications show reversion once there is no stress. Still, some of the modifications do show stable inheritance as epigenetic-mediated stress memory does result in long-term adaptations (Sudan et al. 2018). However, detailed studies are needed to find the factors regulating the epiallele stability in crop plants for their further use in a breeding program (Hofmeister et al. 2017). There is a need to develop various mathematical models for the identification of heritable epigenetic phenotypes, for the enhanced efficiency of the breeding program (Tal et al. 2010). Besides, epi-genotyping procedures can be developed for the identification of newly formed epialleles and their inheritance pattern (Hofmeister et al. 2017). Also, more precise epi-mutagenesis and targeted epigenome editing are needed for targeted epigenome editing (Johnson et al. 2014; Springer and Schmitz 2017).
The regions associated with the transposable elements are more prone to methylation under abiotic stress situations. Thus, these regions should be targeted to understand the trend of epigenetic changes at the whole-genome level through cytosine methylation studies (Bruce et al. 2007). Differential DNA methylation has been recorded in different tissues of the soybean (Song et al. 2013), but it is unclear whether the differences were spontaneous or developmentally controlled by differentially methylated regions (DMRs) (Salgotra and Gupta 2019). We expect an increase in the functional studies of various key epigenetic factors that can be enhanced by the recent developments in the CRISPER technologies via the generation of several epigenetic mutants at least in major legume crops. Thus, a better understanding generated about the epigenetic mechanism along with the identification of epialleles will potentially boost the plant’s ability to cope with various abiotic stresses.
There is a need to modify the active DNA demethylation through CRISPER/Cas9 technology to the genes involved in the demethylation pathway. In the future, we need very precise control on the DNA methylation and demethylation of specific genes as epigenome engineering, for targeted abiotic stress tolerance breeding in the legumes (Springer and Schmitz 2017; Stricker et al. 2017). In soybean, most of the DNA methyltransferase genes were found to be expressed at low levels in seed and seem to contribute to the silencing of certain mC genes in the seed tissues. There is a need to do deep analysis about the mC pattern in different tissues under various abiotic stresses to gain an insight into the role of gene methylation, resulting in novel epigenetic gene regulation (Garg et al. 2014). The details of abiotic stress management strategies in legumes using epigenetic approaches are presented in Fig. 4.3.

Comprehensive abiotic stress management strategies in legumes using epigenetic approaches
Although several legume crops (viz., mung bean, lentil, peanut, chickpea, cowpea, pea, Medicago, pigeon pea, lotus, soybean, beans, etc.) have been sequenced, epigenetic studies concerning abiotic stress tolerance are limited to a few species like soybean, chickpea, pigeon pea, cowpea, and beans. There is an urgent need to study more legumes for abiotic stress tolerance using epigenetic approaches. For this, a joint research platform may be developed by various national and international organizations like the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT, India), the International Center for Agriculture Research in the Dry Areas (ICARDA, Lebanon), the International Center for Tropical Agriculture which is an international research and development organization (CIAT, Colombia), and Indian Agricultural Research Institute (IARI, India), working for the improvement of various legume crops for targeted improvement using epigenetic approaches. In the initial stage, crops like mung bean, lentil, peanut, and pea can be targeted, and at a later stage depending on the availability of whole-genome information, more legumes can be added.
References
Abid G, Mingeot D, Muhovski Y, Mergeai G, Aouida M, Abdelkarim S, Aroua I, El Ayed M, M’hamdi M, Sassi K (2017) Analysis of DNA methylation patterns associated with drought stress response in faba bean (Vicia faba L.) using methylation-sensitive amplification polymorphism (MSAP). Environ Exp Bot 142:34–44
Ahmad S, Nawade B, Sangh C, Mishra GP, Bosamia TC, Kumar N, Dobaria JR, Gajera HP (2020) Identification of novel QTLs for late leaf spot resistance and validation of a major rust QTL in peanut (Arachis hypogaea L.). 3 Biotech 10(10):1–13
Al-Lawati A, Al-Bahry S, Victor R, Al-Lawati AH, Yaish MW (2016) Salt stress alters DNA methylation levels in alfalfa (Medicago spp.). Genet Mol Res 15(1):15018299
An YC, Goettel W, Han Q, Bartels A, Liu Z, Xiao W (2017) Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci Rep 7(1):1–14
Arya H, Singh MB, Bhalla PL (2017) Molecular genomic investigations on floral induction using soybean as a model system. In: International symposium on flowering, fruit set and alternate bearing, vol 1229, pp 47–56
Awana M, Yadav K, Rani K, Gaikwad K, Praveen S, Kumar S, Singh A (2019) Insights into salt stress-induced biochemical, molecular and epigenetic regulation of spatial responses in Pigeonpea (Cajanus cajan L.). J Plant Growth Regul 38(4):1545–1561
Barrera-Figueroa BE, Gao L, Diop NN, Wu Z, Ehlers JD, Roberts PA, Close TJ, Zhu J-K, Liu R (2011) Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biol 11(1):1–11
Barrera-Figueroa BE, Gao L, Wu Z, Zhou X, Zhu J, Jin H, Liu R, Zhu J-K (2012) High throughput sequencing reveals novel and abiotic stress-regulated microRNAs in the inflorescences of rice. BMC Plant Biol 12(1):1–11
Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447(7143):407–412
Bhalani H, Thankappan R, Mishra GP, Sarkar T, Bosamia TC, Dobaria JR (2019) Regulation of antioxidant mechanisms by AtDREB1A improves soil-moisture deficit stress tolerance in transgenic peanut (Arachis hypogaea L.). PLoS One 14(5):e0216706
Bhardwaj J, Mahajan M, Yadav SK (2013) Comparative analysis of DNA methylation polymorphism in drought sensitive (HPKC2) and tolerant (HPK4) genotypes of horse gram (Macrotyloma uniflorum). Biochem Genet 51(7):493–502
Bilichak A, Kovalchuk I (2016) Transgenerational response to stress in plants and its application for breeding. J Exp Bot 67(7):2081–2092
Bosamia TC, Mishra GP, Thankappan R, Dobaria JR (2015) Novel and stress relevant EST derived SSR markers developed and validated in peanut. PLoS One 10(6):e0129127
Bosamia TC, Dodia SM, Mishra GP, Ahmad S, Joshi B, Thirumalaisamy PP, Kumar N, Rathnakumar AL, Sangh C, Kumar A (2020) Unraveling the mechanisms of resistance to Sclerotium rolfsii in peanut (Arachis hypogaea L.) using comparative RNA-Seq analysis of resistant and susceptible genotypes. PLoS One 15(8):e0236823
Boyko A, Blevins T, Yao Y, Golubov A, Bilichak A, Ilnytskyy Y, Hollander J, Meins F Jr, Kovalchuk I (2010) Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS One 5(3):e9514
Bruce TJA, Matthes MC, Napier JA, Pickett JA (2007) Stressful “memories” of plants: evidence and possible mechanisms. Plant Sci 173(6):603–608
Budak H, Hussain B, Khan Z, Ozturk NZ, Ullah N (2015) From genetics to functional genomics: improvement in drought signaling and tolerance in wheat. Front Plant Sci 6:1012
Cañas LA, Beltrán JP (2018) Model legumes: functional genomics tools in Medicago truncatula. In: Functional genomics in Medicago Truncatula. Humana, New York, pp 11–37
Chan SW-L, Henderson IR, Jacobsen SE (2005) Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat Rev Genet 6(5):351–360
Chan SW-L, Zhang X, Bernatavichute YV, Jacobsen SE (2006) Two-step recruitment of RNA-directed DNA methylation to tandem repeats. PLoS Biol 4(11):e363
Chinnusamy V, Zhu J-K (2009) Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol 12(2):133–139
Colicchio JM, Miura F, Kelly JK, Ito T, Hileman LC (2015) DNA methylation and gene expression in Mimulus guttatus. BMC Genomics 16(1):1–15
Dasgupta U, Mishra GP, Dikshit HK, Mishra DC, Bosamia T, Roy A, Bhati J, Aski M, Kumar RR, Singh AK (2021) Comparative RNA-Seq analysis unfolds a complex regulatory network imparting yellow mosaic disease resistance in mungbean [Vigna radiata (L.) R. Wilczek]. PLoS One 16(1):e0244593
De la Rosa C, Covarrubias AA, Reyes JL (2019) A dicistronic precursor encoding miR398 and the legume-specific miR2119 coregulates CSD1 and ADH1 mRNAs in response to water deficit. Plant Cell Environ 42(1):133–144
Deal RB, Henikoff S (2011) Histone variants and modifications in plant gene regulation. Curr Opin Plant Biol 14(2):116–122
Deans C, Maggert KA (2015) What do you mean, “epigenetic”? Genetics 199(4):887–896
Do Kim K, El Baidouri M, Abernathy B, Iwata-Otsubo A, Chavarro C, Gonzales M, Libault M, Grimwood J, Jackson SA (2015) A comparative epigenomic analysis of polyploidy-derived genes in soybean and common bean. Plant Physiol 168(4):1433–1447
Folsom JJ, Begcy K, Hao X, Wang D, Walia H (2014) Rice fertilization-Independent Endosperm1 regulates seed size under heat stress by controlling early endosperm development. Plant Physiol 165(1):238–248
Fujimoto R, Sasaki T, Ishikawa R, Osabe K, Kawanabe T, Dennis ES (2012) Molecular mechanisms of epigenetic variation in plants. Int J Mol Sci 13(8):9900–9922
Garg R, Kumari R, Tiwari S, Goyal S (2014) Genomic survey, gene expression analysis and structural modeling suggest diverse roles of DNA methyltransferases in legumes. PLoS One 9(2):e88947
Gibney ER, Nolan CM (2010) Epigenetics and gene expression. Heredity 105(1):4–13
Greaves IK, Groszmann M, Wang A, Peacock WJ, Dennis ES (2014) Inheritance of trans chromosomal methylation patterns from Arabidopsis F1 hybrids. Proc Natl Acad Sci 111(5):2017–2022
Gutzat R, Scheid OM (2012) Epigenetic responses to stress: triple defense? Curr Opin Plant Biol 15(5):568–573
Hajyzadeh M, Turktas M, Khawar KM, Unver T (2015) miR408 overexpression causes increased drought tolerance in chickpea. Gene 555(2):186–193. https://doi.org/10.1016/j.gene.2014.11.002
Hauben M, Haesendonckx B, Standaert E, Van Der Kelen K, Azmi A, Akpo H, Van Breusegem F, Guisez Y, Bots M, Lambert B (2009) Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc Natl Acad Sci 106(47):20109–20114
Hilton IB, D’ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA (2015) Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33(5):510–517
Hofmeister BT, Lee K, Rohr NA, Hall DW, Schmitz RJ (2017) Stable inheritance of DNA methylation allows creation of epigenotype maps and the study of epiallele inheritance patterns in the absence of genetic variation. Genome Biol 18(1):1–16
Hossain MS, Kawakatsu T, Do Kim K, Zhang N, Nguyen CT, Khan SM, Batek JM, Joshi T, Schmutz J, Grimwood J (2017) Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. New Phytol 214(2):808–819
Hsieh T-F, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL, Zilberman D (2009) Genome-wide demethylation of Arabidopsis endosperm. Science 324(5933):1451–1454
Hu Y, Zhang LU, He S, Huang MIN, Tan J, Zhao LIN, Yan S, Li HUI, Zhou KUN, Liang Y (2012) Cold stress selectively unsilences tandem repeats in heterochromatin associated with accumulation of H3K9ac. Plant Cell Environ 35(12):2130–2142
Hu Y, Morota G, Rosa GJM, Gianola D (2015) Prediction of plant height in Arabidopsis thaliana using DNA methylation data. Genetics 201(2):779–793
Ivashuta S, Uchiyama K, Gau M, Shimamoto Y (2002) Linear amplification coupled with controlled extension as a means of probe amplification in a cDNA array and gene expression analysis during cold acclimation in alfalfa (Medicago sativa L.). J Exp Bot 53(367):351–359
Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, Jacobsen SE (2007) The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 17(4):379–384
Johnson LM, Du J, Hale CJ, Bischof S, Feng S, Chodavarapu RK, Zhong X, Marson G, Pellegrini M, Segal DJ (2014) SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 507(7490):124–128
Kang YJ, Bae A, Shim S, Lee T, Lee J, Satyawan D, Kim MY, Lee S-H (2017) Genome-wide DNA methylation profile in mungbean. Sci Rep 7(1):1–8
Kapazoglou A, Drosou V, Argiriou A, Tsaftaris AS (2013) The study of a barley epigenetic regulator, HvDME, in seed development and under drought. BMC Plant Biol 13(1):1–16
Khandal H, Parween S, Roy R, Meena MK, Chattopadhyay D (2017) MicroRNA profiling provides insights into post-transcriptional regulation of gene expression in chickpea root apex under salinity and water deficiency. Sci Rep 7(1):1–14
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Kulcheski FR, de Oliveira LFV, Molina LG, Almerão MP, Rodrigues FA, Marcolino J, Barbosa JF, Stolf-Moreira R, Nepomuceno AL, Marcelino-Guimarães FC (2011) Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics 12(1):1–17
Kurdyukov S, Mathesius U, Nolan KE, Sheahan MB, Goffard N, Carroll BJ, Rose RJ (2014) The 2HA line of Medicago truncatula has characteristics of an epigenetic mutant that is weakly ethylene insensitive. BMC Plant Biol 14(1):1–16
La H, Ding B, Mishra GP, Zhou B, Yang H, del Rosario Bellizzi M, Chen S, Meyers BC, Peng Z, Zhu J-K (2011) A 5-methylcytosine DNA glycosylase/lyase demethylates the retrotransposon Tos17 and promotes its transposition in rice. Proc Natl Acad Sci 108(37):15498–15503
Labra M, Ghiani A, Citterio S, Sgorbati S, Sala F, Vannini C, Ruffini-Castiglione M, Bracale M (2002) Analysis of cytosine methylation pattern in response to water deficit in pea root tips. Plant Biol 4(06):694–699
Lämke J, Bäurle I (2017) Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol 18(1):1–11
Lämke J, Brzezinka K, Altmann S, Bäurle I (2016) A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J 35(2):162–175
Lane AK, Niederhuth CE, Ji L, Schmitz RJ (2014) pENCODE: a plant encyclopedia of DNA elements. Annu Rev Genet 48:49–70
Lavin M, Herendeen PS, Wojciechowski MF (2005) Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54(4):575–594
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11(3):204–220
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75(5):843–854
Lei M, Zhang H, Julian R, Tang K, Xie S, Zhu J-K (2015) Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc Natl Acad Sci 112(11):3553–3557
Li W-X, Oono Y, Zhu J, He X-J, Wu J-M, Iida K, Lu X-Y, Cui X, Jin H, Zhu J-K (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20(8):2238–2251
Li W, Wang P, Li Y, Zhang K, Ding F, Nie T, Yang X, Lv Q, Zhao L (2015) Identification of microRNAs in response to different day lengths in soybean using high-throughput sequencing and qRT-PCR. PLoS One 10(7):e0132621
Liang X, Hou X, Li J, Han Y, Zhang Y, Feng N, Du J, Zhang W, Zheng D, Fang S (2019) High-resolution DNA methylome reveals that demethylation enhances adaptability to continuous cropping comprehensive stress in soybean. BMC Plant Biol 19(1):1–17
Liew LC, Singh MB, Bhalla PL (2013) An RNA-seq transcriptome analysis of histone modifiers and RNA silencing genes in soybean during floral initiation process. PLoS One 8(10):e77502
Lin J-Y, Le BH, Chen M, Henry KF, Hur J, Hsieh T-F, Chen P-Y, Pelletier JM, Pellegrini M, Fischer RL (2017) Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc Natl Acad Sci 114(45):E9730–E9739
Liu C, Lu F, Cui X, Cao X (2010) Histone methylation in higher plants. Annu Rev Plant Biol 61:395–420
Lobaton JD, Miller T, Gil J, Ariza D, de la Hoz JF, Soler A, Beebe S, Duitama J, Gepts P, Raatz B (2018) Resequencing of common bean identifies regions of inter–gene pool introgression and provides comprehensive resources for molecular breeding. Plant Genome 11(2):170068
Long SP, Ort DR (2010) More than taking the heat: crops and global change. Curr Opin Plant Biol 13(3):240–247
Malabarba J, Windels D, Xu W, Verdier J (2021) Regulation of DNA (de)methylation positively impacts seed germination during seed development under heat stress. Genes 12(3):457
Mantri N, Basker N, Ford R, Pang E, Pardeshi V (2013) The role of micro-ribonucleic acids in legumes with a focus on abiotic stress response. Plant Genome 6(3):plantgenome2013-05
Matzke MA, Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15(6):394–408
Maxwell EK, Ryan JF, Schnitzler CE, Browne WE, Baxevanis AD (2012) MicroRNAs and essential components of the microRNA processing machinery are not encoded in the genome of the ctenophore Mnemiopsis leidyi. BMC Genomics 13(1):1–11
Mayer BF, Ali-Benali MA, Demone J, Bertrand A, Charron J (2015) Cold acclimation induces distinctive changes in the chromatin state and transcript levels of COR genes in Cannabis sativa varieties with contrasting cold acclimation capacities. Physiol Plant 155(3):281–295
McClung CR, Davis SJ (2010) Ambient thermometers in plants: from physiological outputs towards mechanisms of thermal sensing. Curr Biol 20(24):R1086–R1092
Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431(7006):343–349
Meyer P (2015) Epigenetic variation and environmental change. J Exp Bot 66(12):3541–3548
Migicovsky Z, Yao Y, Kovalchuk I (2014) Transgenerational phenotypic and epigenetic changes in response to heat stress in Arabidopsis thaliana. Plant Signal Behav 9(2):e27971
Mishra GP, Radhakrishnan T, Kumar A, Thirumalaisamy PP, Kumar N, Bosamia TC, Nawade B, Dobaria JR (2015) Advancements in molecular marker development and their applications in the management of biotic stresses in peanuts. Crop Prot 77:74–86
Mishra GP, Dikshit HK, Kumari J, Tripathi K, Devi J, Aski M, Mehra R, Sarker A, Kumar S (2020) Identification and characterization of novel penta-podded genotypes in the cultivated lentil. Crop Sci 60(4):1974–1985
Mishra GP, Dikshit HK, Tontang MT, Stobdan T, Sangwan S, Aski MS, Singh A, Kumar RR, Tripathi K, Kumar S (2021) Diversity in phytochemical composition, antioxidant capacities, and nutrient contents among mungbean and lentil microgreens when grown at plain-altitude region (Delhi) and high-altitude region (Leh-Ladakh), India. Front Plant Sci 12:1485
Mishra GP, Dikshit HK, Kukreja B, Aski M, Yadava DK, Sarker A, Kumar S (2022) Historical overview of biofortification in crop plants and its implications. In: Biofortification of staple crops. Springer, New York, pp 31–61
Mochida K, Yoshida T, Sakurai T, Yamaguchi-Shinozaki K, Shinozaki K, Tran L-SP (2010) LegumeTFDB: an integrative database of Glycine max, Lotus japonicus and Medicago truncatula transcription factors. Bioinformatics 26(2):290–291
Moradpour M, Abdulah SNA (2020) CRISPR/dC as9 platforms in plants: strategies and applications beyond genome editing. Plant Biotechnol J 18(1):32–44
Mosher RA, Schwach F, Studholme D, Baulcombe DC (2008) PolIVb influences RNA-directed DNA methylation independently of its role in siRNA biogenesis. Proc Natl Acad Sci 105(8):3145–3150
Nawade B, Mishra GP, Radhakrishnan T, Dodia SM, Ahmad S, Kumar A, Kumar A, Kundu R (2018) High oleic peanut breeding: achievements, perspectives, and prospects. Trends Food Sci Technol 78:107–119
Naydenov M, Baev V, Apostolova E, Gospodinova N, Sablok G, Gozmanova M, Yahubyan G (2015) High-temperature effect on genes engaged in DNA methylation and affected by DNA methylation in Arabidopsis. Plant Physiol Biochem 87:102–108
Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F, Oliviero S (2017) Intragenic DNA methylation prevents spurious transcription initiation. Nature 543(7643):72–77
Ni Z, Hu Z, Jiang Q, Zhang H (2013) GmNFYA3, a target gene of miR169, is a positive regulator of plant tolerance to drought stress. Plant Mol Biol 82(1):113–129
Niederhuth CE, Schmitz RJ (2014) Covering your bases: inheritance of DNA methylation in plant genomes. Mol Plant 7(3):472–480
Oakey H, Cullis B, Thompson R, Comadran J, Halpin C, Waugh R (2016) Genomic selection in multi-environment crop trials. G3 6(5):1313–1326
Pandey G, Sharma N, Pankaj Sahu P, Prasad M (2016) Chromatin-based epigenetic regulation of plant abiotic stress response. Curr Genomics 17(6):490–498
Parrilla-Doblas JT, Roldán-Arjona T, Ariza RR, Córdoba-Cañero D (2019) Active DNA demethylation in plants. Int J Mol Sci 20(19):4683
Patel KG, Mandaliya VB, Mishra GP, Dobaria JR, Thankappan R (2016) Transgenic peanut overexpressing mtlD gene confers enhanced salinity stress tolerance via mannitol accumulation and differential antioxidative responses. Acta Physiol Plant 38(7):1–14
Patel KG, Thankappan R, Mishra GP, Mandaliya VB, Kumar A, Dobaria JR (2017) Transgenic peanut (Arachis hypogaea L.) overexpressing mtlD gene showed improved photosynthetic, physio-biochemical, and yield-parameters under soil-moisture deficit Stress in lysimeter system. Front Plant Sci 8:1881
Pauli H, Gottfried M, Dullinger S, Abdaladze O, Akhalkatsi M, Alonso JLB, Coldea G, Dick J, Erschbamer B, Calzado RF (2012) Recent plant diversity changes on Europe’s mountain summits. Science 336(6079):353–355
Pecinka A, Chevalier C, Colas I, Kalantidis K, Varotto S, Krugman T, Michailidis C, Vallés M-P, Muñoz A, Pradillo M (2020) Chromatin dynamics during interphase and cell division: similarities and differences between model and crop plants. J Exp Bot 71(17):5205–5222
Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL (2007) DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci 104(16):6752–6757
Peñuelas J, Rutishauser T, Filella I (2009) Phenology feedbacks on climate change. Science 324(5929):887–888
Peterson CL, Laniel M-A (2004) Histones and histone modifications. Curr Biol 14(14):R546–R551
Popova OV, Dinh HQ, Aufsatz W, Jonak C (2013) The RdDM pathway is required for basal heat tolerance in Arabidopsis. Mol Plant 6(2):396–410
Rajkumar MS, Gupta K, Khemka NK, Garg R, Jain M (2020) DNA methylation reprogramming during seed development and its functional relevance in seed size/weight determination in chickpea. Commun Biol 3(1):1–13
Raju SKK, Shao M, Sanchez R, Xu Y, Sandhu A, Graef G, Mackenzie S (2018) An epigenetic breeding system in soybean for increased yield and stability. Plant Biotechnol J 16(11):1836–1847
Rakei A, Maali-Amiri R, Zeinali H, Ranjbar M (2016) DNA methylation and physio-biochemical analysis of chickpea in response to cold stress. Protoplasma 253(1):61–76
Ramesh SV, Govindasamy V, Rajesh MK, Sabana AA, Praveen S (2019) Stress-responsive miRNAome of Glycine max (L.) Merrill: molecular insights and way forward. Planta 249(5):1267–1284
Ramu VS, Paramanantham A, Ramegowda V, Mohan-Raju B, Udayakumar M, Senthil-Kumar M (2016) Transcriptome analysis of sunflower genotypes with contrasting oxidative stress tolerance reveals individual- and combined-biotic and abiotic stress tolerance mechanisms. PLoS One 11(6):e0157522
Reddy VRP, Das S, Dikshit HK, Mishra GP, Aski M, Meena SK, Singh A, Pandey R, Singh MP, Tripathi K (2020) Genome-wide association analysis for phosphorus use efficiency traits in mungbean (Vigna radiata L. Wilczek) using genotyping by sequencing approach. Front Plant Sci 11:1546
Robertson AL, Wolf DE (2012) The role of epigenetics in plant adaptation. Trends Evol Biol 4(1):e4
Salgotra RK, Gupta M (2019) Exploring the role of epigenetics in cereal and leguminous crops exposed to abiotic stress. In: Epigenetics in plants of agronomic importance: fundamentals and applications. Springer, New York, pp 149–170
Saraswat S, Yadav AK, Sirohi P, Singh NK (2017) Role of epigenetics in crop improvement: water and heat stress. J Plant Biol 60(3):231–240
Sarkar T, Thankappan R, Kumar A, Mishra GP, Dobaria JR (2014) Heterologous expression of the AtDREB1A gene in transgenic peanut-conferred tolerance to drought and salinity stresses. PLoS One 9(12):e110507
Sarkar T, Thankappan R, Kumar A, Mishra GP, Dobaria JR (2016) Stress inducible expression of AtDREB1A transcription factor in transgenic peanut (Arachis hypogaea L.) conferred tolerance to soil-moisture deficit stress. Front Plant Sci 7:935
Schmitz RJ, He Y, Valdés-López O, Khan SM, Joshi T, Urich MA, Nery JR, Diers B, Xu D, Stacey G (2013) Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res 23(10):1663–1674
Sen S, Chakraborty J, Ghosh P, Basu D, Das S (2017) Chickpea WRKY70 regulates the expression of a homeodomain-leucine zipper (HD-Zip) I transcription factor CaHDZ12, which confers abiotic stress tolerance in transgenic tobacco and chickpea. Plant Cell Physiol 58(11):1934–1952
Shanker A, Venkateswarlu B (2011) Abiotic stress in plants: mechanisms and adaptations. BoD–Books on Demand
Shui X-R, Chen Z-W, Li J-X (2013) MicroRNA prediction and its function in regulating drought-related genes in cowpea. Plant Sci 210:25–35
Sigman MJ, Slotkin RK (2016) The first rule of plant transposable element silencing: location, location, location. Plant Cell 28(2):304–313
Slotkin RK, Vaughn M, Borges F, Tanurdžić M, Becker JD, Feijó JA, Martienssen RA (2009) Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136(3):461–472
Song Y, Ji D, Li S, Wang P, Li Q, Xiang F (2012) The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS One 7(7):e41274
Song Q-X, Lu X, Li Q-T, Chen H, Hu X-Y, Ma B, Zhang W-K, Chen S-Y, Zhang J-S (2013) Genome-wide analysis of DNA methylation in soybean. Mol Plant 6(6):1961–1974
Sosa-Valencia G, Romero-Pérez PS, Palomar VM, Covarrubias AA, Reyes JL (2017a) Insights into the function of the phasiRNA-triggering miR1514 in response to stress in legumes. Plant Signal Behav 12(3):e1284724
Sosa-Valencia G, Palomar M, Covarrubias AA, Reyes JL (2017b) The legume miR1514a modulates a NAC transcription factor transcript to trigger phasiRNA formation in response to drought. J Exp Bot 68(8):2013–2026
Springer NM, Schmitz RJ (2017) Exploiting induced and natural epigenetic variation for crop improvement. Nat Rev Genet 18(9):563–575
Stępiński D (2012) Levels of DNA methylation and histone methylation and acetylation change in root tip cells of soybean seedlings grown at different temperatures. Plant Physiol Biochem 61:9–17
Stricker SH, Köferle A, Beck S (2017) From profiles to function in epigenomics. Nat Rev Genet 18(1):51–66
Sudan J, Raina M, Singh R (2018) Plant epigenetic mechanisms: role in abiotic stress and their generational heritability. 3 Biotech 8(3):1–12
Suter L, Widmer A (2013) Phenotypic effects of salt and heat stress over three generations in Arabidopsis thaliana. PLoS One 8(11):e80819
Tal O, Kisdi E, Jablonka E (2010) Epigenetic contribution to covariance between relatives. Genetics 184(4):1037–1050
Tang K, Lang Z, Zhang H, Zhu J-K (2016) The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nat Plants 2(11):1–10
Timko MP, Rushton PJ, Laudeman TW, Bokowiec MT, Chipumuro E, Cheung F, Town CD, Chen X (2008) Sequencing and analysis of the gene-rich space of cowpea. BMC Genomics 9(1):1–20
Trindade I, Capitão C, Dalmay T, Fevereiro MP, dos Santos DM (2010) miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 231(3):705–716
Varotto S, Tani E, Abraham E, Krugman T, Kapazoglou A, Melzer R, Radanović A, Miladinović D (2020) Epigenetics: possible applications in climate-smart crop breeding. J Exp Bot 71(17):5223–5236
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar’an B (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31(3):240–246
Venterea R (2014) Climate change 2014: mitigation of climate change. J Environ Qual 38:837
Vlasova A, Capella-Gutiérrez S, Rendón-Anaya M, Hernández-Oñate M, Minoche AE, Erb I, Câmara F, Prieto-Barja P, Corvelo A, Sanseverino W (2016) Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol 17(1):1–18
Vriet C, Hennig L, Laloi C (2015) Stress-induced chromatin changes in plants: of memories, metabolites and crop improvement. Cell Mol Life Sci 72(7):1261–1273
Wahid A, Gelani S, Ashraf M, Foolad MR (2007) Heat tolerance in plants: an overview. Environ Exp Bot 61(3):199–223
Walther G-R, Post E, Convey P, Menzel A, Parmesan C, Beebee TJC, Fromentin J-M, Hoegh-Guldberg O, Bairlein F (2002) Ecological responses to recent climate change. Nature 416(6879):389–395
Wang P, Shi S, Ma J, Song H, Zhang Y, Gao C, Zhao C, Zhao S, Hou L, Lopez-Baltazar J (2018) Global Methylome and gene expression analysis during early Peanut pod development. BMC Plant Biol 18(1):1–13
Wassenegger M, Heimes S, Riedel L, Sänger HL (1994) RNA-directed de novo methylation of genomic sequences in plants. Cell 76(3):567–576
Wibowo A, Becker C, Marconi G, Durr J, Price J, Hagmann J, Papareddy R, Putra H, Kageyama J, Becker J (2016) Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. Elife 5:e13546
Wierzbicki AT, Ream TS, Haag JR, Pikaard CS (2009) RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat Genet 41(5):630–634
Windels D, Dang TT, Chen Z, Verdier J (2021) Snapshot of epigenetic regulation in legumes. Legume Sci 3(3):e60
Wong CE, Singh MB, Bhalla PL (2013) The dynamics of soybean leaf and shoot apical meristem transcriptome undergoing floral initiation process. PLoS One 8(6):e65319
Wu T, Pi E-X, Tsai S-N, Lam H-M, Sun S-M, Kwan YW, Ngai S-M (2011) GmPHD5 acts as an important regulator for crosstalk between histone H3K4 di-methylation and H3K14 acetylation in response to salinity stress in soybean. BMC Plant Biol 11(1):1–13
Xu C, Tian J, Mo B (2013) siRNA-mediated DNA methylation and H3K9 dimethylation in plants. Protein Cell 4(9):656–663
Yaish MW, Peng M, Rothstein SJ (2014) Global DNA methylation analysis using methyl-sensitive amplification polymorphism (MSAP). In: Arabidopsis protocols. Springer, New York, pp 285–298
Yaish MW, Al-Lawati A, Al-Harrasi I, Patankar HV (2018) Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genomics 19(1):1–17
Yan Y, Zhang Y, Yang K, Sun Z, Fu Y, Chen X, Fang R (2011) Small RNAs from MITE-derived stem-loop precursors regulate abscisic acid signaling and abiotic stress responses in rice. Plant J 65(5):820–828
Yang X, Kundariya H, Xu Y-Z, Sandhu A, Yu J, Hutton SF, Zhang M, Mackenzie SA (2015) MutS HOMOLOG1-derived epigenetic breeding potential in tomato. Plant Physiol 168(1):222–232
Zeller G, Henz SR, Widmer CK, Sachsenberg T, Rätsch G, Weigel D, Laubinger S (2009) Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J 58(6):1068–1082
Zemach A, Kim MY, Hsieh P-H, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153(1):193–205
Zhang H, Zhu J-K (2011) RNA-directed DNA methylation. Curr Opin Plant Biol 14(2):142–147
Zhang H, Zhu J-K (2012) Active DNA demethylation in plants and animals. Cold Spring Harb Symp Quant Biol 77:161–173
Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW-L, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126(6):1189–1201
Zhang H, Lang Z, Zhu J-K (2018a) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19(8):489–506
Zhang B, Su L, Hu B, Li L (2018b) Expression of AhDREB1, an AP2/ERF transcription factor gene from peanut, is affected by histone acetylation and increases abscisic acid sensitivity and tolerance to osmotic stress in Arabidopsis. Int J Mol Sci 19(5):1441
Zhao T, Zhan Z, Jiang D (2019) Histone modifications and their regulatory roles in plant development and environmental memory. J Genet Genomics 46(10):467–476
Zhu J-K (2009) Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet 43:143–166
Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S (2008) Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456(7218):125–129
Zong W, Zhong X, You J, Xiong L (2013) Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol Biol 81(1):175–188
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mishra, G.P., Dikshit, H.K., Devi, J., Aski, M.S., Durgesh, K. (2023). Epigenetics of Abiotic Stress Tolerance in Legumes. In: Muthu Arjuna Samy, P., Ramasamy, A., Chinnusamy, V., Sunil Kumar, B. (eds) Legumes: Physiology and Molecular Biology of Abiotic Stress Tolerance. Springer, Singapore. https://doi.org/10.1007/978-981-19-5817-5_4
Download citation
DOI: https://doi.org/10.1007/978-981-19-5817-5_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-5816-8
Online ISBN: 978-981-19-5817-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)