
Abstract
Cardiovascular diseases (CVDs) as the leading cause of death and morbidity worldwide, have a substantial genetic basis. The personalized medicine (PM) concept tailors therapeutic and preventive strategies according to the genomic, epigenomic, and proteomic profiles of each individual. In the field of cardiology, PM can help cardiologists and health care providers to prevent adverse drug reactions (ADR) and select the best-individualized medication. In addition, a considerable number of cardiomyopathies, arrhythmias, and coronary artery disease (CAD) risk factors have also a genetic basis. In this chapter, we thoroughly reviewed the genetic background in the metabolism of important cardiovascular drugs such as warfarin, clopidogrel, and statins. Moreover, the cardinal role of genetic pathways in the occurrence of cardiomyopathies, arrhythmia, and CAD will be discussed. Finally, the ethical issues and prospective of PM were also reviewed to lighten all aspects of PM in cardiovascular medicine.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara What Will You Learn in This Chapter?In this chapter, we will get familiar with personalized medicine in the field of cardiology, the genetic basis of most common cardiovascular diseases, and the role of genetics in pharmacotherapy. We will also discuss the ethical issues in personalized medicine and the perspective of this field in cardiology.
FormalPara Rationale and ImportancePersonalized medicine is important in early diagnosis; choosing the best treatment options, including the most suitable pharmacotherapy in familial arrhythmias; and preventing adverse drug reactions in the field of cardiology. Recognizing the best treatment and preventive strategy is individualized. It is crucial for healthcare providers to apply the most appropriate approach to patients.
4.1 Introduction
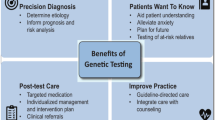
Personalized medicine (PM) is a concept that modifies therapeutic strategies according to each individual’s genomic, epigenomic, and proteomic profiles [1]. The major concept of PM is the treatment and care of patients with a particular condition while considering individual alterations in genetics, exposures, and lifestyle [2]. Cardiovascular diseases (CVD), the most common cause of death all over the world [3], have genetic risk factors, and the pharmacokinetics of cardiology drugs have a broad spectrum of different genotypes [4]. Moreover, genome-wide associated (GWA) studies have revealed several genetic variants that are associated with cardiology conditions such as cardiomyopathies, arrhythmias, and coronary artery diseases. Thus, determining genetic information and applying PM strategies are useful in the effective prevention and treatment of several cardiologic conditions (Fig. 4.1).

Personalized medicine in cardiovascular disease. This figure was created with Biorender.com. All rights and ownership of BioRender content are reserved by BioRender
4.2 Drugs
4.2.1 Warfarin
Warfarin is an anticoagulant that is often prescribed for the treatment and prevention of thromboembolic events in people with prosthetic heart valves, atrial fibrillation, venous thrombosis, and a history of stroke. Warfarin dose requirements, drug response, and risk of bleeding are influenced by environmental factors (such as vitamin K consumption, age, gender, and concurrent medications) and by genetic variations [5]. VKORC1, CYP2C9, and CYP4F2 are considered the main genes that may influence warfarin metabolism and cause genetic variations.
4.2.2 VKORC1
The VKORC1 gene encodes the target enzyme of the warfarin drug, the vitamin K epoxide reductase enzyme, which is responsible for reducing vitamin K epoxide to the active form [6]. A common non-coding variant of VKORC1 that occurs in the promoter region of the gene, c.-1639G>A (rs9923231) polymorphism, affects protein expression and is associated with warfarin sensitivity and lower dose requirements. Patients who are carrying one or two “A” alleles at -1639 require lower warfarin doses than -1639G/G homozygotes [1, 7, 8]. The c.-1639G>A allele frequency shows a discrepancy among different ethnic groups and is more common among Asians, Caucasians, and African Americans [2, 9, 10]. Besides, other less common coding VKORC1 polymorphisms (such as Asp36Tyr) are associated with warfarin resistance and higher dose requirements [4, 11].
4.2.3 CYP2C9
CYP2C9 is a member of the cytochrome P450 superfamily (CYP450) that metabolizes the more potent warfarin S-enantiomer. CYP2C9*1 is the wild-type allele in the “normal metabolizer” phenotype (those with normal enzyme activity and metabolism). Individuals carrying two well-characterized variant alleles, CYP2C9*2 and CYP2C9*3, are known to be more sensitive to warfarin, require lower doses to achieve the therapeutic range, are at a higher risk of bleeding, and take longer to achieve a stable INR compared to normal metabolizers [12, 13]. The maintenance dose requirements of warfarin in patients with *1*1, *2*2, and *3*3 genotypes are reported as 5.28 mg/day, 3.04 mg/day, and 0.5 mg/day, respectively [14]. Other CYP2C9 variants (CYP2C9*5, *6, 8*, and *11), which are more common among African Americans, are also associated with decreased enzyme activity and dose variability [15].
4.2.4 CYP4F2
CYP4F2 is the vitamin K oxidase enzyme and acts as an important counterpart to VKORC1 (vitamin K reductase enzyme), limiting vitamin K accumulation in the liver [16]. A known variant of CYP4F2*3 (c.1297C>T, rs2108622) has been shown to affect enzyme activity and dose requirements of warfarin [17]. Caucasian individuals who carry two “T” alleles require a higher dosage of warfarin (1 mg/day) compared to those with two “C” alleles, which is explained by the reduced function of the enzyme in those with “T” alleles [18]. Thus, including this CYP4F2 variant in warfarin dosing models is helpful in dose prediction in Asians and Europeans, but not in African Americans [19,20,21].
4.2.5 P2Y12 Inhibitors
Clopidogrel is a prodrug, and genetic variants influence the catalytic activity of the CYP P450 isoforms (such as CYP2C19, CYP1A2, CYP2B6, CYP2C9, and CYP3A) and affect the efficiency of active metabolite generation [22]. The most common CYP2C19 loss-of-function alleles are CYP2C19*2 (G681A) and CYP2C19*3 (G636A), and the most common allele that results in increased enzyme activity is CYP2C19*17 [23]. Therefore, based on the CYP2C19 genotypes, patients are categorized as ultrarapid metabolizers (*1/*17, *17/*17), extensive metabolizers (*1/*1), intermediate metabolizers (*1/*2, *1/*3, *2/*17), and poor metabolizers (*2/*2, *2/*3, *3/*3) [24]. The ABCB1 gene polymorphisms are also known to be associated with clinical outcomes in clopidogrel-treated patients [25]; however, the association has been inconsistent across studies, with several studies finding no relationship between ABCB1 variants and the antiplatelet effect of clopidogrel [26]. Prasugrel and ticagrelor are both stronger P2Y12 inhibitors than clopidogrel and lower platelet reactivity more effectively, irrespective of the CYP2C19 genotype [27, 28]. Moreover, polymorphisms of the other isoforms of the CYP450 system appear to not influence prasugrel pharmacokinetics or pharmacodynamics [28].
4.2.6 Statin
Statins, HMG-CoA reductase inhibitors, act by inhibiting cholesterol biosynthesis and increasing low-density lipoprotein cholesterol (LDL-C) uptake by hepatocytes. SLCO1B1 and ABCB1 are proteins that play a role in the transportation of statins. The SLCO1B1 521C (rs4149056) variant is associated with a reduction of the lipid-lowering effect of simvastatin, atorvastatin, lovastatin, and pravastatin. Three ABCB1 gene polymorphisms (1236T, 2677T, and 3435T) have been linked to statin pharmacokinetics and toxicity. HMG-CoA reductase is an important enzyme in cholesterol synthesis and is inhibited by statins within hepatocytes. The H7 haplotype of HMG-CoA reductase is associated with decreased lipid-lowering response to statins [29]. Polymorphisms in the CYP3A gene, such as CYP3A4*22 (rs35599367) and CYP3A5*3 (rs776746), have been shown to reduce CYP3A4 enzyme levels and activity, as well as to affect the pharmacokinetics of simvastatin, atorvastatin, and lovastatin [30, 31].
4.3 Cardiomyopathies
4.3.1 Hypertrophic Cardiomyopathy (HCM)
HCM is one of the common hereditary cardiac diseases, which is associated with two main pathogeneses; the first one is defects in myocardial filaments, associated with sarcomeric genes, and the second one is metabolic and infiltrative disorders [32]. The gene variants that are associated with HCM are the MYH7 gene, which encodes the myosin heavy chain [33], TNNsT2 which encodes cardiac troponin T [34], MYBPC3 which encodes myosin-binding protein C [35], TNNI3 which encodes Cardiac troponin I [36], and FHOD3 which encodes “Formin homology 2 domains containing 3” [37]. Moreover, there are some syndromic genes without isolated left ventricular hypertrophy, including the autosomal recessive GAA gene as Pompe disease [38], and X-linked GLA, which presents as Anderson-Fabry disease [39]. Genotype-positive patients have been shown to present with illness approximately 10 years earlier, to have a greater maximum left-ventricular wall thickness, and to have a higher proportion of family history of HCM and sudden cardiac death than others [40].
4.3.2 Dilated Cardiomyopathy (DCM)
A strong familial component has been reassuringly confirmed in DCM [41]. About 111 genes are associated with DCM. The most associated gene is TTN, which encodes Titin, the largest structural protein of the heart [42]. Another gene variant that is associated with approximately 5% of the causes of DCM is LMNA missense and truncating mutations [43]. LMNA mutations are the main genetic cause of arrhythmogenic DCM.
4.3.3 Restrictive Cardiomyopathy (RCM)
RCM, one of the rarest and poor-prognosis cardiac disorders, is characterized by a normal-sized left ventricle with a hypertrophic atrium. Amyloidosis, as an infiltrative disorder, is the most common cause of RCM. TTR gene variants and APOA1 are the main genetic perturbations in amyloidosis [44]. There is a lack of adequate data about non-infiltrative RCM genes; however, TNNI3, TNNT2, TNNC1, TPM1, TTN, MYH7, MYL2, MYBPC3, MPN, DES, FLNC, LMNA, and BAG3 were labeled as associated genes in RCM [45, 46]. Most of these genes encode sarcomeric proteins. Moreover, CRYAB, which encodes heat-shock proteins (such as crystallin B and BAG3), is also reported in some studies [45, 47].
4.4 Thoracic Aortic Aneurysm/Aortic Dissection (TAAD)
Several causal genes have been identified in syndromic and non-syndromic TAAD. Variants in the smooth muscle contractile (SMC) genes, including ACTA2, MYH11, MYLK, and PRKG1, have been associated with non-syndromic TAAD [48]. Syndromic TAAD is associated with several connective tissue disorders and their corresponding genes, including Marfan syndrome (FBN1), Loeys-Dietz syndrome (TGFBR1, TGFBR2, SMAD3, and TGFB2), Ehlers-Danlos syndrome (COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2), arterial tortuosity syndrome (SLC2A10), and Shprintzen-Goldberg syndrome (SKI) [49]. Marfan syndrome patients with FBN1 mutations have a low risk for acute aortic dissections at diameters less than 5.5 cm and for aneurysms of other arteries [50]. Common genetic variants at 15q21.1, in the FBN1 gene, are associated with an increased risk of TAAD in the general population and are common pathogeneses of aortic disease in Marfan syndrome and sporadic TAAD [51]. Loeys-Dietz syndrome patients with TGFBR1 and TGFBR2 mutations are at higher risk of aortic dissections at aortic diameters less than 5.0 cm, and these patients have aneurysms and dissections of other arteries. Furthermore, studies have shown that TGFBR1 mutation carriers may have a lower risk of aortic dissection with minimal enlargement than TGFBR2 mutation carriers [52]. MYLK encodes the Ca2+/calmodulin-dependent myosin light-chain kinase, which phosphorylates the regulatory light chain in smooth muscle cells to initiate contraction [53]. MYLK missense variants were shown to be associated with earlier-onset aortic events compared to haploinsufficient variants [54].
4.5 Valvopathies
Aortic stenosis (AS) is the narrowing of the aortic valves that leads to obstruction of blood flow from the left ventricle (LV) to the aorta. The incidence of AS is increasing with the aging population. Today, AS is not considered a passive degenerative disease anymore. It is associated with a dynamic, complex, and highly regulated pathobiological process that leads to a multitude of events [55]. The characterization of the whole protein complements of the genome, which is termed “proteome,” is the major goal of proteomics that could improve the patient’s management. Analysis of the cell or tissue is a suitable platform as they are eventually the targets for novel medications and should provide important evidence for treatment discovery.
As a result, lipoproteins and oxidized phospholipids play a significant role in AS that generates inflammation, apoptosis, and calcification of the aortic valve [56]. LPA genetic variants linked to Lp(a) levels are significantly linked to aortic valve calcification and incident AS [57]. Accordingly, to manage the progression of AS, aiming lipoprotein(a) is a potential therapeutic target. The other potential mechanisms are:
-
1.
Calcium deposition: which includes calcium, phosphate, vitamin D, fibroblast growth factor 23 (FGF-23), and PTH; the vitamin D/PTH axis biomarkers are the most verified factors [58]. The N-terminal propeptide of human procollagen type I (PINP), beta carboxy-terminal cross-linking telopeptide of type I collagen (β-CTx), osteocalcin, osteopontin, osteoprotegerin, and fetuin-A are the other suggested factors [59,60,61,62].
-
2.
Inflammation: limited factors are associated with inflammation, which leads to AS. Remarkably, in contrast with CAD, C-reactive protein (CRP) is not associated with the progression of calcified aortic valve disease [63].
-
3.
Cardiac remodeling, B-type natriuretic peptide (BNP), and cardiac troponin are potentially informative about the myocardial consequences of AS. Higher NT proBNP was associated with a higher grade of AS severity and NYHA class [64]. Cardiac troponin was identified as a separate variable associated with mid-wall fibrosis of the myocardium as part of a clinical risk score that predicts cardiovascular events in asymptomatic AS [65]. Biomarkers of extracellular matrix remodeling such as Fibulin-1 are significantly and inversely correlated with AVA index [66].
Personalized medicine contains a multimodal approach that might be especially useful for decision-making in patients with asymptomatic AS rather than patients with AS. Defining which patients could benefit from each therapeutic strategy would be possible with PM, for example, utilizing a transcatheter instead of the surgical aortic valve.
4.5.1 Mitral Valve Replacement
In patients with significant mitral regurgitation (MR) due to floppy mitral valve (FMV)/mitral valve prolapse (MVP), mitral valve replacement is crucial. Due to the significant variability in the size of the mitral annulus, one ring size can’t fit all. The mitral “personalized ring” is a novel device constructed intraoperatively [67]. These “personalized rings” provide excellent support of the mitral annulus, which avoids annular dilatation and paravalvular leak.
4.6 Arrhythmia
4.6.1 Long QT Syndrome (LQTS)
LQTS is defined as QTc ≥480 ms in an asymptomatic patient or a QTc ≥460 ms in the presence of unexplained syncope [68]. Patients with LQTS are at high risk of arrhythmogenic syncope, polymorphous ventricular tachycardia (torsade de pointes), and sudden arrhythmic death [69]. LQT type 1 is caused by loss of function mutation in the KCNQ1 gene which encodes the α-subunit of the slow rectifier current (IKS) [70]. LQT type 2 arises from loss-of-function mutations in KCNH2, which encodes the α-subunit of the rapid rectifier current (IKr) [71]. In contrast, a gain of function in SCN5A will cause LQT type 3, which amplified late sodium current (INa) [72]. Based on LQTS genotyping studies, the best therapeutic option in LQT types 1, 2, and 3 has shown to be β-blockers [73, 74].
4.6.2 Brugada Syndrome (BrS)
Inward sodium current impairment compared with the transient outward potassium current (Ito) in the right ventricular outflow tract is the key pathogenesis of BrS [75]. The most common genetic mutation in BrS, which could be detected in 21% of the patients, is the loss of function in the SCN5A gene. Loss-of-function mutations in SCN5A reduce the overall available sodium current (INa) through either (1) impaired intracellular trafficking of the ion channel to the plasma membrane or (2) through altered gating properties of the channel [76]. CACNA1C, GPD1L, HEY2, PKP2, RANGRF, SCN10A, SCN1B, SCN2B, SCN3B, SLMAP, and TRPM4 are some other rare genes associated with BrS [77]. SCN10A, which encodes α-subunit Nav1.8 sodium channel, is one of the most novel mutations and is responsible for 5 to 16 percent of BrS [78, 79].
4.6.3 Short QT Syndrome (SQTS)
SQTS is defined as QTc ≤330 ms, or QTc interval <360 ms, and at least one of the following conditions: history of cardiac arrest or syncope, family history of sudden cardiac death (SCD) at age 40 or younger, or family history of SQTS [80]. Potassium and calcium channelopathies are the main pathophysiology in SQTS [26, 27]. Gain of function mutations in KCNH2, KCNQ1, and KCNJ2 genes (associated with potassium channels) are responsible for SQT type 1, 2, and 3, respectively [81, 82]. Loss of function in CACNA1C, CACNB2, and CACNA2D1 (associated with calcium channels) leads to SQT 4, 5, and 6, respectively [83, 84].
4.6.4 Idiopathic Ventricular Fibrillation (IVF)
IVF is defined as resuscitated ventricular fibrillation (VF), which had no other causes for VF, that is, metabolic, toxicological, cardiac (including other channelopathies and structural heart disease), respiratory, and infectious causes [68]. IVF is responsible for 6.8% of sudden cardiac death causes [85]. IVF pathophysiology is mainly due to an abnormality affecting the microstructural myocardial or Purkinje system [86]. Several genes have been found in association with IVF, DPP6 was reported in Dutch families [87], CALM1 was reported in a Moroccan family [88], and RYR2 causes a leaky channel at diastolic levels of calcium under non-stress conditions [89].
4.6.5 Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
The main clinical manifestation of CPVT is episodic syncope occurring during exercise or acute emotion in individuals without structural cardiac abnormalities [90]. CPVT1 is caused by a mutation in the RYR2 gene, which encodes the cardiac ryanodine receptor and accounts for 65% of the CVPT cases [91]. RYR2 gene affects intracellular calcium hemostasis and excitation-contraction coupling of the heart [92]. Mutation in the CASQ2 gene, which encodes cardiac calsequestrin (a calcium-buffering protein within the sarcoplasmic reticulum), accounts for 2–5% of the CPVT cases [93]. Some other genes associated with CPVT are TECLR [94], TRDN [95], CALM [96], and CALM2 [97]. ANK2 and KCNJ2 may phenocopy CPVT; hence they are associated with LQT4 and LQT7, respectively [98, 99]. However, no specific gene could be found for almost one-third of CPVT cases [100].
4.6.6 Progressive Cardiac Conduction System Disease (PCCD)
PCCD is defined as impulse conduction progressive delay through the His-Purkinje system with right or left bundle branch block (RBBB or LBBB) [101]. The first reported gene associated with PCCD was SCN5A, which encodes the cardiac sodium channel NaV 1.5 [102]. SCN5A mutations could also be found in BrS type 1; thus, there is a significant overlap between BrS and PCCD. Individuals carrying this mutation may manifest isolated forms of each BrS and PCCD or coexisted forms [103]. Mutations in TRPM4 gene, which encodes a Ca2+-activated but Ca2+-impermeable cation channel [104], are associated with PCCD as well as familial AV block and RBBB [105]. PCCD may be associated with HCM in the presence of mutations in PRKAG2, LAMP2, and GLA; also it may be accompanied by DCM in the occurrence of LMNA, DES, and TNNI3K alterations [106].
4.7 Coronary Artery Disease (CAD)
4.7.1 Genes and Mechanism
In addition to several traditional risk factors (such as smoking, hypertension, diabetes, dyslipidemia, and obesity), a strong genetic basis had been also identified for CAD. According to early GWA studies [107, 108], variants in two loci (LTA and LGALS2) are associated with pathogenesis and increased risk of myocardial infarction (MI). However, later studies failed to show such association between polymorphisms in LTA and LGALS2 and myocardial infarction [109]. In 2007, GWA studies identified SNPs at the 9p21.3 locus, which is located near the CDKN2A and CDKN2B genes and is associated with a 30–40% increased risk of CAD [110, 111].
GWA studies for plasma lipoprotein traits have identified several common single nucleotide polymorphism (SNP) variants that are strongly associated with plasma LDL. Common variants in genes associated with LDL-C levels (PCSK9, LDL-R, APOB, APOE, SORT1, ABCG5-ABCG8, ABO, LPA, and NPC1L1), genes associated with triglyceride levels (LPL, APOA5, ASGR1, ANGPTL4, APOC3, and TRIB1), and the gene encoding cholesteryl ester transfer protein (CETP), which is associated with HDL-C levels, have been linked to CAD [112, 113]. SNPs on chromosome 1P13 have a strong association with LDL and have also been independently linked to CAD and MI [110, 114]. Not all mutations are associated with an increased risk of CAD, in some cases; inactivating mutations may decrease CAD risk in conclusion. PCSK9, NPC1L1, and ASGR1 mutations result in CAD risk reduction by lowering LDL cholesterol levels [11, 12, 115]. Lipoprotein lipase (LPL) hydrolyses lipoprotein-bound triglycerides and reduces triglyceride levels consequently. LPL loss of function is associated with an increased risk of CAD [116]. Apolipoprotein A5 (APOA5) increases LPL activity [116]. In contrast, apolipoprotein C-III (APOC3) and angiopoietin-like 4 (ANGPTL4) reduce LPL activity, and they are associated with CAD [117, 118]. APOA5 mutations increase plasma triglyceride levels [116]; nonetheless, APOC3 loss-of-function has opposite effects, which causes a reduction in plasma triglycerides levels [117].
4.7.2 Premature CAD
GWA studies have identified a considerable number of genetic variants that are associated with premature CAD. Genetic variants in genes such as PCSK9, LDL-R, and NPC1L1 contribute to premature CAD either directly or via traditional cardiovascular risk factors. Variants in locus 9p21.3, which is located on chromosome 9, are also associated with the risk of developing premature CAD [119]. It has been shown that a mutation in LDL receptor (LDLR) may lead to LDL metabolism dysfunction and thus increase the risk of premature CAD [120]. LDLR plays an important role in CAD pathogenesis by increasing LDL cholesterol and triglyceride-rich lipoproteins levels [121].
4.7.3 Vascular Inflammation and Remodeling
The encoding genes of cytokines (CXCL12) [112] and interleukin 6 (IL6) [122] are associated with CAD by vascular inflammation. SH2B3 is one of the novel mutations associated with an increased risk of CAD [122]. SH2B3 mutations trigger an elevation in numerous inflammatory mediators in left ventricle tissues including NRLP12, CCR2, and IFNγ [123]. There are two types of vascular remodeling, constrictive remodeling and expansive remodeling. Constrictive remodeling produces more stable plaque and narrow lumen, in contrast, expansive remodeling causes less stable plaque with no narrowing effect on the lumen [124]. ADAMTS7 is one of the novel genes associated with CAD and plaque formation, but not plaque rupture [125]. MIA3 is another gene associated with CAD, which regulates the levels of large proteins such as collagen VII [126].
4.8 Hypertension
Hypertension is one of the major risk factors for CAD. Based on CHARGE Consortium, ATP2B1, CYP17A1, PLEKHA7, and SH2B3 were associated with systolic blood pressure (SBP); ATP2B1, CACNB2, CSK-ULK3, SH2B3, TBX3-TBX5, and ULK4DBP were associated with diastolic blood pressure (DBP); and ATP2B1 was labeled for hypertension [127]. According to another large-scale study, CYP17A1, CYP1A2, FGF5, SH2B3, MTHFR, c10orf107, ZNF652, and PLCD3 genes caused hypertension [128].
4.9 Recognizing Ethical Issues and How to Deal with Them
Many patients are aware of the benefits of PM although their knowledge of its potential appears to be limited [129]. Patients in oncology request information about PM more frequently than patients with other diseases [130]. Even if patients are aware of the phrase “personalized medicine,” some of them don’t understand the concept of PM [131], which may affect the participation of patients in medical decision-making.
Professionals also describe a lack of knowledge about PM. According to the conducted studies, cardiologists have the lowest information about PM among family physicians, cardiologists, and oncologists [130].
One of the major ethical concerns in this field is data confidentiality not being guaranteed properly [132]. Besides sharing data with the legal system, patients are concerned about data sharing with families in cases where information about a genetic disposition needs to be shared with all at-risk family members.
Test results or the testing process itself can also cause harm for patients. This harm can be caused by professionals’ misinterpretation of the results or making the wrong therapeutic decisions [133]. Besides mentioned issues, the psychological burden from knowing or expecting the assessment results is considerably high. Accordingly, harm to benefits must be evaluated in every patient.
In contrast with clinical practice, in which test results are only beneficial when they provide reliable and actionable evidence that can be used for clinical decisions, there is a lack of evidence in PM results and practice guidelines in this field. The cost-benefit ratio of PM is also questionable whether other treatment interventions would not have a superior benefit. PM costs are supposed as being massive and caused by a much minor proportion of the total patient population [134].
4.10 Digital Twin “Prospective of Precision Medicine in Cardiology”
The idea of digital twins was initially discussed in David Gelernter’s book in the early 1990s [135]. A digital twin is a digital imitation or representation of a physical object, process, or service, but also beyond that. In other words, it is a virtual prototype (data plus algorithms) that will dynamically connect the physical and digital worlds and that will utilize modern technologies, such as smart sensors, data analytics, and artificial intelligence (AI), to monitor system performance, detect and prevent failures, and explore new advancements. A digital twin is intended to make a virtual representation of a physical object, test it, and optimize it in the virtual space, until the virtual representation meets the desired performance, at which point it can be built or enhanced (if already built) in the real world [136]. Collecting real-time data streams from linked clinical, health, and other sensors and combining these mass data with advanced data analytics, cloud computing, and artificial intelligence (including machine learning) will generate highly potent networked computational resources, which could be used in real-world decision-making. Precision cardiology could be established by the utilization of cardiac digital twins (CDT) [137]. This cardiovascular model will maximize the interaction between anatomical and physiologic understanding of the cardiovascular system. Treatment and prevention of cardiovascular disease will be based on precise predictions of both the underlying causes of disease and the pathways; hence, these predictions will be promising with CDT utilization.
References
Vogenberg FR, Isaacson Barash C, Pursel M. Personalized medicine: part 1: evolution and development into theranostics. P T. 2010;35(10):560–76.
Leopold JA, Loscalzo J. Emerging role of precision medicine in cardiovascular disease. Circ Res. 2018;122(9):1302–15. https://doi.org/10.1161/circresaha.117.310782.
GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459–544. https://doi.org/10.1016/s0140-6736(16)31012-1.
Goetz LH, Schork NJ. Personalized medicine: motivation, challenges, and progress. Fertil Steril. 2018;109(6):952–63. https://doi.org/10.1016/j.fertnstert.2018.05.006.
Wadelius M, Pirmohamed M. Pharmacogenetics of warfarin: current status and future challenges. Pharmacogenomics J. 2007;7(2):99–111. https://doi.org/10.1038/sj.tpj.6500417.
Mozaffarian D, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–322. https://doi.org/10.1161/cir.0000000000000152.
Currie G, Delles C. Precision medicine and personalized medicine in cardiovascular disease. Adv Exp Med Biol. 2018;1065:589–605. https://doi.org/10.1007/978-3-319-77932-4_36.
Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363(4):301–4. https://doi.org/10.1056/NEJMp1006304.
Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol. 2003;32(1):1–22. https://doi.org/10.1093/ije/dyg070.
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–6. https://doi.org/10.1001/jama.2017.17219.
Cohen JC, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72. https://doi.org/10.1056/NEJMoa054013.
Stitziel NO, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371(22):2072–82. https://doi.org/10.1056/NEJMoa1405386.
Peloso GM, et al. Rare protein-truncating variants in APOB, lower low-density lipoprotein cholesterol, and protection against coronary heart disease. Circ Genom Precis Med. 2019;12(5):e002376. https://doi.org/10.1161/circgen.118.002376.
Ansell J, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133(6 Suppl):160s–98s. https://doi.org/10.1378/chest.08-0670.
Kasner SE, et al. Warfarin dosing algorithms and the need for human intervention. Am J Med. 2016;129(4):431–7. https://doi.org/10.1016/j.amjmed.2015.11.012.
Ma Z, et al. Clinical model for predicting warfarin sensitivity. Sci Rep. 2019;9(1):12856. https://doi.org/10.1038/s41598-019-49329-0.
Danese E, et al. Impact of the CYP4F2 p.V433M polymorphism on coumarin dose requirement: systematic review and meta-analysis. Clin Pharmacol Ther. 2012;92(6):746–56. https://doi.org/10.1038/clpt.2012.184.
Caldwell MD, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111(8):4106–12. https://doi.org/10.1182/blood-2007-11-122010.
Aithal GP, et al. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353(9154):717–9. https://doi.org/10.1016/s0140-6736(98)04474-2.
Mega JL, et al. Genetics and the clinical response to warfarin and edoxaban: findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet. 2015;385(9984):2280–7. https://doi.org/10.1016/s0140-6736(14)61994-2.
Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics. 2002;12(3):251–63. https://doi.org/10.1097/00008571-200204000-00010.
Jiang X-L, et al. Clinical pharmacokinetics and pharmacodynamics of clopidogrel. Clin Pharmacokinet. 2015;54(2):147–66. https://doi.org/10.1007/s40262-014-0230-6.
Paré G, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med. 2010;363(18):1704–14. https://doi.org/10.1056/NEJMoa1008410.
Scott SA, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94(3):317–23. https://doi.org/10.1038/clpt.2013.105.
Biswas M, et al. Effects of the ABCB1 C3435T single nucleotide polymorphism on major adverse cardiovascular events in acute coronary syndrome or coronary artery disease patients undergoing percutaneous coronary intervention and treated with clopidogrel: a systematic review and meta-analysis. Expert Opin Drug Saf. 2020;19(12):1605–16. https://doi.org/10.1080/14740338.2020.1836152.
Price MJ, Tantry US, Gurbel PA. The influence of CYP2C19 polymorphisms on the pharmacokinetics, pharmacodynamics, and clinical effectiveness of P2Y12 inhibitors. Rev Cardiovasc Med. 2011;12(1):1–12. https://doi.org/10.3909/ricm0590.
Tantry US, et al. First analysis of the relation between CYP2C19 genotype and pharmacodynamics in patients treated with ticagrelor versus clopidogrel. Circ Cardiovasc Genet. 2010;3(6):556–66. https://doi.org/10.1161/CIRCGENETICS.110.958561.
Varenhorst C, et al. Genetic variation of CYP2C19 affects both pharmacokinetic and pharmacodynamic responses to clopidogrel but not prasugrel in aspirin-treated patients with coronary artery disease. Eur Heart J. 2009;30(14):1744–52. https://doi.org/10.1093/eurheartj/ehp157.
Zhang JE, et al. Effects of CYP4F2 genetic polymorphisms and haplotypes on clinical outcomes in patients initiated on warfarin therapy. Pharmacogenet Genomics. 2009;19(10):781–9. https://doi.org/10.1097/FPC.0b013e3283311347.
Perera MA, et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet. 2013;382(9894):790–6. https://doi.org/10.1016/s0140-6736(13)60681-9.
Kitzmiller JP, et al. CYP3A4*22 and CYP3A5*3 are associated with increased levels of plasma simvastatin concentrations in the cholesterol and pharmacogenetics study cohort. Pharmacogenet Genomics. 2014;24(10):486–91. https://doi.org/10.1097/fpc.0000000000000079.
Mazzarotto F, et al. Contemporary insights into the Genetics of hypertrophic cardiomyopathy: toward a new era in clinical testing? J Am Heart Assoc. 2020;9(8):e015473. https://doi.org/10.1161/JAHA.119.015473.
Geisterfer-Lowrance AA, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62(5):999–1006. https://doi.org/10.1016/0092-8674(90)90274-i.
Watkins H, et al. A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat Genet. 1993;3(4):333–7. https://doi.org/10.1038/ng0493-333.
Carrier L, et al. Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat Genet. 1993;4(3):311–3. https://doi.org/10.1038/ng0793-311.
Kimura A, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16(4):379–82. https://doi.org/10.1038/ng0897-379.
Ochoa JP, et al. Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018;72(20):2457–67. https://doi.org/10.1016/j.jacc.2018.10.001.
Martiniuk F, et al. Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower “affinity” for glycogen (GAA 2) and transient gene expression in deficient cells. Am J Hum Genet. 1990;47(3):440–5.
Davies JP, Winchester BG, Malcolm S. Mutation analysis in patients with the typical form of Anderson-Fabry disease. Hum Mol Genet. 1993;2(7):1051–3. https://doi.org/10.1093/hmg/2.7.1051.
Ho CY, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric human cardiomyopathy registry (SHaRe). Circulation. 2018;138(14):1387–98. https://doi.org/10.1161/circulationaha.117.033200.
Pinto YM, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850–8. https://doi.org/10.1093/eurheartj/ehv727.
Herman DS, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28. https://doi.org/10.1056/NEJMoa1110186.
Parks SB, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156(1):161–9. https://doi.org/10.1016/j.ahj.2008.01.026.
Muchtar E, Blauwet LA, Gertz MA. Restrictive cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):819–37. https://doi.org/10.1161/circresaha.117.310982.
Cimiotti D, et al. Genetic restrictive cardiomyopathy: causes and consequences—an integrative approach. Int J Mol Sci. 2021;22(2):558.
Kostareva A, et al. Genetic Spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS One. 2016;11(9):e0163362. https://doi.org/10.1371/journal.pone.0163362.
Brodehl A, et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum Mutat. 2017;38(8):947–52. https://doi.org/10.1002/humu.23248.
Renard M, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2018;72(6):605–15. https://doi.org/10.1016/j.jacc.2018.04.089.
Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473(7347):308–16. https://doi.org/10.1038/nature10145.
Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005;111(11):e150–7. https://doi.org/10.1161/01.cir.0000155243.70456.f4.
LeMaire SA, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43(10):996–1000. https://doi.org/10.1038/ng.934.
Tran-Fadulu V, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46(9):607–13. https://doi.org/10.1136/jmg.2008.062844.
Wang L, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87(5):701–7. https://doi.org/10.1016/j.ajhg.2010.10.006.
Wallace SE, et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet Med. 2019;21(1):144–51. https://doi.org/10.1038/s41436-018-0038-0.
Messika-Zeitoun D, et al. Aortic valve calcification: determinants and progression in the population. Arterioscler Thromb Vasc Biol. 2007;27(3):642–8. https://doi.org/10.1161/01.ATV.0000255952.47980.c2.
Yu B, et al. Pathological significance of lipoprotein(a) in aortic valve stenosis. Atherosclerosis. 2018;272:168–74. https://doi.org/10.1016/j.atherosclerosis.2018.03.025.
Thanassoulis G, et al. Genetic associations with Valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–12. https://doi.org/10.1056/NEJMoa1109034.
Yang ZK, et al. Mineral metabolism disturbances are associated with the presence and severity of calcific aortic valve disease. J Zhejiang Univ Sci B. 2015;16(5):362–9. https://doi.org/10.1631/jzus.B1400292.
Cho HJ, Cho HJ, Kim HS. Osteopontin: a multifunctional protein at the crossroads of inflammation, atherosclerosis, and vascular calcification. Curr Atheroscler Rep. 2009;11(3):206–13. https://doi.org/10.1007/s11883-009-0032-8.
Kiechl S, et al. Osteoprotegerin is a risk factor for progressive atherosclerosis and cardiovascular disease. Circulation. 2004;109(18):2175–80. https://doi.org/10.1161/01.cir.0000127957.43874.bb.
Civitelli R, Armamento-Villareal R, Napoli N. Bone turnover markers: understanding their value in clinical trials and clinical practice. Osteoporos Int. 2009;20(6):843–51. https://doi.org/10.1007/s00198-009-0838-9.
Schafer C, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112(3):357–66. https://doi.org/10.1172/jci17202.
Novaro GM, et al. Clinical factors, but not C-reactive protein, predict progression of calcific aortic-valve disease: the cardiovascular health study. J Am Coll Cardiol. 2007;50(20):1992–8. https://doi.org/10.1016/j.jacc.2007.07.064.
Cimadevilla C, et al. Prognostic value of B-type natriuretic peptide in elderly patients with aortic valve stenosis: the COFRASA-GENERAC study. Heart. 2013;99(7):461–7. https://doi.org/10.1136/heartjnl-2012-303284.
Chin CW, et al. A clinical risk score of myocardial fibrosis predicts adverse outcomes in aortic stenosis. Eur Heart J. 2016;37(8):713–23. https://doi.org/10.1093/eurheartj/ehv525.
Kruger R, et al. Extracellular matrix biomarker, fibulin-1, is closely related to NT-proBNP and soluble urokinase plasminogen activator receptor in patients with aortic valve stenosis (the SEAS study). PLoS One. 2014;9(7):e101522. https://doi.org/10.1371/journal.pone.0101522.
Pitsis A, et al. Mitral valve repair: moving towards a personalized ring. J Cardiothorac Surg. 2019;14(1):108. https://doi.org/10.1186/s13019-019-0926-7.
Priori SG, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the Management of Patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–867. https://doi.org/10.1093/eurheartj/ehv316.
Goldenberg I, Zareba W, Moss AJ. Long QT syndrome. Curr Probl Cardiol. 2008;33(11):629–94. https://doi.org/10.1016/j.cpcardiol.2008.07.002.
Heijman J, et al. Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ Res. 2012;110(2):211–9. https://doi.org/10.1161/circresaha.111.249482.
Sanguinetti MC, et al. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81(2):299–307. https://doi.org/10.1016/0092-8674(95)90340-2.
Kambouris NG, et al. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation. 1998;97(7):640–4. https://doi.org/10.1161/01.cir.97.7.640.
Abu-Zeitone A, et al. Efficacy of different beta-blockers in the treatment of long QT syndrome. J Am Coll Cardiol. 2014;64(13):1352–8. https://doi.org/10.1016/j.jacc.2014.05.068.
Wilde AA, et al. Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation. 2016;134(12):872–82. https://doi.org/10.1161/circulationaha.116.021823.
Antzelevitch C, et al. J-wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. J Arrhythmia. 2016;32(5):315–39. https://doi.org/10.1016/j.joa.2016.07.002.
Kapplinger JD, et al. An international compendium of mutations in the <em>SCN5A</em>−encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. https://doi.org/10.1016/j.hrthm.2009.09.069.
Brugada J, et al. Present status of Brugada syndrome: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72(9):1046–59. https://doi.org/10.1016/j.jacc.2018.06.037.
Hu D, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64(1):66–79. https://doi.org/10.1016/j.jacc.2014.04.032.
Behr ER, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res. 2015;106(3):520–9. https://doi.org/10.1093/cvr/cvv042.
Priori SG, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10(12):e85–108. https://doi.org/10.1016/j.hrthm.2013.07.021.
Bellocq C, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109(20):2394–7. https://doi.org/10.1161/01.cir.0000130409.72142.fe.
Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10(10):571–83. https://doi.org/10.1038/nrcardio.2013.108.
Templin C, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J. 2011;32(9):1077–88. https://doi.org/10.1093/eurheartj/ehr076.
Antzelevitch C, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–9. https://doi.org/10.1161/circulationaha.106.668392.
Waldmann V, et al. Characteristics and clinical assessment of unexplained sudden cardiac arrest in the real-world setting: focus on idiopathic ventricular fibrillation. Eur Heart J. 2018;39(21):1981–7. https://doi.org/10.1093/eurheartj/ehy098.
Haïssaguerre M, et al. Idiopathic ventricular fibrillation: role of Purkinje system and microstructural myocardial abnormalities. JACC Clin Electrophysiol. 2020;6(6):591–608. https://doi.org/10.1016/j.jacep.2020.03.010.
Alders M, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84(4):468–76. https://doi.org/10.1016/j.ajhg.2009.02.009.
Marsman RF, et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2014;63(3):259–66. https://doi.org/10.1016/j.jacc.2013.07.091.
Beach LY, et al. Idiopathic ventricular fibrillation in a 29-year-old man. Circulation. 2017;136(1):112–4. https://doi.org/10.1161/CIRCULATIONAHA.117.029120.
Pflaumer A, Davis AM. Guidelines for the diagnosis and management of Catecholaminergic polymorphic ventricular tachycardia. Heart Lung Circ. 2012;21(2):96–100. https://doi.org/10.1016/j.hlc.2011.10.008.
Ackerman MJ, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European heart rhythm association (EHRA). Heart Rhythm. 2011;8(8):1308–39. https://doi.org/10.1016/j.hrthm.2011.05.020.
Laitinen PJ, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103(4):485–90. https://doi.org/10.1161/01.cir.103.4.485.
Lahat H, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69(6):1378–84. https://doi.org/10.1086/324565.
Bhuiyan ZA, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18(10):1060–6. https://doi.org/10.1111/j.1540-8167.2007.00913.x.
Roux-Buisson N, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21(12):2759–67. https://doi.org/10.1093/hmg/dds104.
Nyegaard M, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91(4):703–12. https://doi.org/10.1016/j.ajhg.2012.08.015.
Makita N, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014;7(4):466–74. https://doi.org/10.1161/circgenetics.113.000459.
Mohler PJ, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421(6923):634–9. https://doi.org/10.1038/nature01335.
Plaster NM, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105(4):511–9. https://doi.org/10.1016/s0092-8674(01)00342-7.
Priori SG, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–63. https://doi.org/10.1016/j.hrthm.2013.05.014.
Baruteau AE, Probst V, Abriel H. Inherited progressive cardiac conduction disorders. Curr Opin Cardiol. 2015;30(1):33–9. https://doi.org/10.1097/hco.0000000000000134.
Schott JJ, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23(1):20–1. https://doi.org/10.1038/12618.
Asatryan B, Medeiros-Domingo A. Emerging implications of genetic testing in inherited primary arrhythmia syndromes. Cardiol Rev. 2019;27(1):23–33. https://doi.org/10.1097/crd.0000000000000203.
Nilius B, et al. Voltage dependence of the Ca<sup>2</sup><sup>+</sup>-activated Cation Channel TRPM4 *. J Biol Chem. 2003;278(33):30813–20. https://doi.org/10.1074/jbc.M305127200.
Stallmeyer B, et al. Mutational spectrum in the Ca(2+)--activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum Mutat. 2012;33(1):109–17. https://doi.org/10.1002/humu.21599.
Asatryan B, Medeiros-Domingo A. Molecular and genetic insights into progressive cardiac conduction disease. EP Europace. 2019;21(8):1145–58. https://doi.org/10.1093/europace/euz109.
Ozaki K, et al. Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-alpha secretion in vitro. Nature. 2004;429(6987):72–5. https://doi.org/10.1038/nature02502.
Ozaki K, et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet. 2002;32(4):650–4. https://doi.org/10.1038/ng1047.
Kimura A, et al. Lack of association between LTA and LGALS2 polymorphisms and myocardial infarction in Japanese and Korean populations. Tissue Antigens. 2007;69:265–9. https://doi.org/10.1111/j.1399-0039.2006.00798.x.
Samani NJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–53. https://doi.org/10.1056/NEJMoa072366.
McPherson R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91. https://doi.org/10.1126/science.1142447.
Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8. https://doi.org/10.1038/ng.784.
Cheng CY, et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nat Commun. 2015;6:6063. https://doi.org/10.1038/ncomms7063.
Myocardial Infarction Genetics, C, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41(3):334–41. https://doi.org/10.1038/ng.327.
Nioi P, et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N Engl J Med. 2016;374(22):2131–41. https://doi.org/10.1056/NEJMoa1508419.
Khera AV, et al. Association of Rare and Common Variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317(9):937–46. https://doi.org/10.1001/jama.2017.0972.
Crosby J, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31. https://doi.org/10.1056/NEJMoa1307095.
Lim GB. Polymorphisms in ANGPTL4 link triglycerides with CAD. Nat Rev Cardiol. 2016;13(5):245. https://doi.org/10.1038/nrcardio.2016.46.
Guo J, et al. Association between 9p21.3 genomic markers and coronary artery disease in East Asians: a meta-analysis involving 9,813 cases and 10,710 controls. Mol Biol Rep. 2013;40(1):337–43. https://doi.org/10.1007/s11033-012-2066-1.
Mabuchi H. Half a century Tales of familial hypercholesterolemia (FH) in Japan. J Atheroscler Thromb. 2017;24(3):189–207. https://doi.org/10.5551/jat.RV16008.
Do R, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–6. https://doi.org/10.1038/nature13917.
Deloukas P, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. https://doi.org/10.1038/ng.2480.
Flister MJ, et al. SH2B3 is a genetic determinant of cardiac inflammation and fibrosis. Circ Cardiovasc Genet. 2015;8(2):294–304. https://doi.org/10.1161/circgenetics.114.000527.
Smits PC, et al. Coronary artery disease: arterial remodelling and clinical presentation. Heart. 1999;82(4):461–4. https://doi.org/10.1136/hrt.82.4.461.
Reilly MP, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377(9763):383–92. https://doi.org/10.1016/s0140-6736(10)61996-4.
Roberts R. A genetic basis for coronary artery disease. Trends Cardiovasc Med. 2015;25(3):171–8. https://doi.org/10.1016/j.tcm.2014.10.008.
Levy D, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87. https://doi.org/10.1038/ng.384.
Newton-Cheh C, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41(6):666–76. https://doi.org/10.1038/ng.361.
Bombard Y, et al. The value of personalizing medicine: medical oncologists’ views on gene expression profiling in breast cancer treatment. Oncologist. 2015;20(4):351–6. https://doi.org/10.1634/theoncologist.2014-0268.
Bonter K, et al. Personalised medicine in Canada: a survey of adoption and practice in oncology, cardiology and family medicine. BMJ Open. 2011;1(1):e000110. https://doi.org/10.1136/bmjopen-2011-000110.
Gray SW, et al. Attitudes of patients with cancer about personalized medicine and somatic genetic testing. J Oncol Pract. 2012;8(6):329–35. https://doi.org/10.1200/jop.2012.000626. 2 p following 335
Hammack CM, Brelsford KM, Beskow LM. Thought leader perspectives on participant protections in precision medicine research. J Law Med Ethics. 2019;47(1):134–48. https://doi.org/10.1177/1073110519840493.
Pearce C, et al. Delivering genomic medicine in the United Kingdom National Health Service: a systematic review and narrative synthesis. Genet Med. 2019;21(12):2667–75. https://doi.org/10.1038/s41436-019-0579-x.
Di Paolo A, et al. Personalized medicine in Europe: not yet personal enough? BMC Health Serv Res. 2017;17(1):289. https://doi.org/10.1186/s12913-017-2205-4.
Gelernter D. Mirror Worlds: or the Day Software Puts the Universe in a Shoebox...How It Will Happen and What It Will Mean.
Bruynseels K, Santoni de Sio F, van den Hoven J. Digital twins in health care: ethical implications of an emerging engineering paradigm. Front Genet. 2018;9:31. https://doi.org/10.3389/fgene.2018.00031.
Corral-Acero J, et al. The ‘Digital Twin’ to enable the vision of precision cardiology. Eur Heart J. 2020;41(48):4556–64. https://doi.org/10.1093/eurheartj/ehaa159.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sheikhy, A., Fallahzadeh, A., Aghaei Meybodi, H.R., Hosseini, K. (2022). Precision Medicine in Cardiovascular Disease Practice. In: Hasanzad, M. (eds) Precision Medicine in Clinical Practice. Springer, Singapore. https://doi.org/10.1007/978-981-19-5082-7_4
Download citation
DOI: https://doi.org/10.1007/978-981-19-5082-7_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-5081-0
Online ISBN: 978-981-19-5082-7
eBook Packages: MedicineMedicine (R0)