
Abstract
Monomer recovery from waste plastic is an essential part of any waste treatment concept. In particular, the depolymerisation of poly(ethylene terephthalate) (PET) by glycolysis and hydrolysis is well established. In addition to the classic products of PET solvolysis, bis(2-hydroxyethyl)terephthalate and terephthalic acid, terephthalic acid amides and rather unconventional terephthalic acid alcohol esters are also obtained. Reactions can take place in ionic liquids or in a microwave oven. Products of depolymerisation can be used as raw materials for virgin polyesters, polyurethanes or bitumen additives. Another group of polyesters that is attracting increasing attention is obtained from biomass. Polymers such as poly(lactic acid), poly(butylene succinate) or polyhydroxyalkanoates are biodegradable. However, the entire effort of production is lost if these materials are returned to the environment untreated. Therefore, recycling of these polymers is an act of resource conservation. Poly(lactic acid) is depolymerised during methanolysis to produce methyl lactate, which can be converted into lactide as a starting material for new polymers. Poly(butylene succinate) and poly(3-hydroxybutyric acid) can be hydrolysed at higher temperatures in the presence of lipases, yielding monomers such as succinic acid, 3-hydroxybutyric acid, and crotonic acid. There are a variety of options for the depolymerisation of polyesters that are examined in this chapter. The increasing number of new polyester monomers requires special solutions for each individual polymer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Poly(ethylene terephthalate)
- Polylactide/Poly(lactic acid)
- Poly(butylene succinate)
- Polyhydroxyalkanoates
- Glycolysis
- Hydrolysis
- Microwave
- Ionic liquids
- Enzymes
- Chemical recycling
1 Introduction
Polyesters comprise a group of polymers obtained by the reaction of dicarboxylic acids and diols. Sometimes block building units are derived from hydroxycarboxylic acids or lactones. The most commonly used polyester is poly(ethylene terephthalate) (PET ), known as the material for soft drink bottles. PET is also used for other food contact applications and for fibres. All aromatic polyesters, including poly(butylene terephthalate) (PBT ) and poly(ethylene naphthalate) (PEN ), have in common that they are made from fossil fuels, although bio-based routes are available [1]. As society strives to decarbonise, carbon sources may become scarce and the need for resource recovery increases.
Bio-based aliphatic polyesters offer an alternative to these fossil fuel-based polymers, such as poly(lactic acid) or polyhydroxyalkanoates. These materials are biodegradable and can be disposed of in composting facilities. This concept is advantageous when plastic is littered; however, all the effort (harvest, work, energy) required to produce these materials is lost when they are biodegraded [2]. Therefore, recycling is also desirable for these polyesters.
The most beneficial way of recycling is to reuse the material as it is (mechanical recycling). However, the material properties are often modified by dyes and other additives. These can affect the optical properties and the intrinsic viscosity, which makes chemical recycling more attractive [3]. In addition, polyesters are generally sensitive to hydrolysis, and the reaction with other protic compounds offers the possibility of recovering monomers or oligomers.
This process is briefly described by the term depolymerisation, which is defined in this chapter as the recovery of monomers or other valuable chemicals rather than the conversion of polymers into fuel. The aim is to obtain defined compounds with a sufficient high purity through highly selective processes. The product is to be used for the production of polymers or other useful materials after minimal processing.
For the depolymerisation of polyesters, solvolytic processes are the most promising options. Glycolysis and hydrolysis are well established for the depolymerisation of PET, while methanolysis can rather be considered a historical process, as the synthesis of PET from dimethyl terephthalate is obsolete. The strategies successfully used for the depolymerisation of PET are also considered for novel polyesters. However, many researchers favour enzymatic pathways, which in turn can provide alternative routes for the degradation of aromatic polyesters.
2 Technologies Used in the Depolymerisation of Polyesters
The depolymerisation of polyesters with sub- or supercritical water has a long tradition [4]. Such methods require high temperatures and pressures, which makes the processes energy intensive and expensive. In addition, sensitive monomers such as ethylene glycol or hydroxyalkanoates could be destroyed in the process [5, 6].
Therefore, strategies have been developed over the years to reduce the heat and pressure requirements. Catalysts have been developed to lower the reaction temperature, as described in the following sections. This trend culminates in the use of lactases and other enzymes [7, 8], pushing reaction temperatures close to ambient temperatures. Although the temperature requirements are drastically reduced, the reaction time can extend to days.
Another strategy is to change the reaction medium. Today, glycolysis is the most studied process for the depolymerisation of PET. One of the main reasons is that the reaction can be carried out below the boiling point of ethylene glycol (196 °C) and therefore pressure reactors are not required. Steam can be used as a reaction medium for the hydrolysis of polyesters [5]. This reduces the pressure requirements but requires temperatures above the melting point of the polymer, which can lead to the destruction of some products.
Most reactions are carried out in autoclaves or in pressureless vessels. With conventional heating, heat is only transferred at the interface between the reactor and reaction medium. From there, the heat is distributed by the thermal conductivity of the medium and by free or forced convection. Reaching thermal equilibrium requires time. Commonly, the reaction time starts when the reaction temperature is reached. By this time, a considerable part of the polymer may already have been degraded [9].
Microwave technology offers an alternative way of heating [10]. Microwave radiation is directly absorbed by various types of materials. Depending on the absorber and the microwave power , rapid heat transfer can be achieved. Water and ethylene glycol are good microwave absorbers because their polar structure is forced by microwave fields into a rotational motion called dipolar polarization. This motion is converted into heat and only material near the absorber is heated. Polymers do not usually act as absorbers. Even if they have a polar structure, they lack the necessary flexibility to follow the electromagnetic field of microwaves. Catalysts with a polar structure can also absorb microwaves and heat up the reaction medium in their vicinity. Ionic catalysts can additionally absorb microwaves by ionic conduction and convert translational energy into heat. Both effects can significantly accelerate the reaction.
3 Depolymerisation of Aromatic Polyesters
The most important aromatic polyester is PET. Therefore, most of this section will deal with this polymer. Besides PET, PBT, and PEN are of interest. The best way of reusing these polymers is mechanical recycling, which can be combined with solid state polycondensation (SSP) to obtain a material appropriate for most applications [11]. However, highly contaminated or degraded materials might become inappropriate for this pathway. Then depolymerisation should be considered.
Recent research focuses on glycolysis, hydrolysis, and aminolysis. The products of all these methods can be used for the production of new polymers. Since the production of PET is a two-step process in which terephthalic acid reacts first with ethylene glycol to form bis(2-hydroxyethyl)terephthalate (BHET), which undergoes in a second step further condensation to high molecular weight PET, terephthalic acid from hydrolysis and BHET from glycolysis can directly be reused in this process.
Many works make use of three values for their description of the depolymerisation process [10]. It is important to understand that these values are often used in a different manner than in common chemical technology. The conversion is not identical with the number of depolymerisation reactions as one would expect. It describes rather the dissolution of polymer, which is commonly seen as the difference between the weight of the polymer before the depolymerisation process starts and the residual polymer afterwards. As PET is insoluble in any common solvent, residue can be recovered by filtration. The molecular weight might be reduced without changing the solubility significantly. Only short chain oligomers are sufficient soluble in the reaction medium used for the depolymerisation. Therefore, the selectivity defines the fraction of desired product in relation to the dissolved fraction of PET. The classical definition assumes that selectivity is reduced when desired product is lost by the formation of undesired by-product. However, low selectivity during the depolymerisation of PET is most likely related to the incomplete reaction of oligomers, which can still be converted into product by changing reaction conditions. That is that the definitions of both conversion and selectivity differ from those commonly used. Only the definition of the yield as the ratio of practical and theoretical amount of product is conventionally used.
3.1 Glycolysis of Poly(Ethylene Terephthalate)
Most attention in the field of PET depolymerisation in recent years has been given to processes associated with glycolysis. Compared to other solvolytic processes, the high boiling point of ethylene glycol at 197 °C allows operation at considerably lower pressure. Below the boiling point, no additional pressure is required. The compatibility of polarity between PET and ethylene glycol reduces the temperature requirement and depolymerisation can be carried out below the melting point of PET. However, this comes at the price of long reaction times, which require the presence of suitable catalysts and advanced reaction systems. Therefore, current research focuses on developing new catalysts and performing glycolysis reactions in ionic liquids or microwave ovens.
The glycolysis reaction itself is the reverse second step of the PET polycondensation described earlier and leads to the formation of BHET as the desired main product (Fig. 9.1). The reaction must be catalysed to proceed at a reasonable rate. Efficient catalysts are ionic materials, such as salts [12], ionic liquids [10, 13, 14], protic ionic salts [15], and deep eutectic solvents (DES) [16, 17]. The product BHET can be used directly for the production of PET [3].

Polycondensation of bis(2-hydroxyethyl) terephthalate (BHET) and glycolysis of poly(ethylene terephthalate) (PET)
Both cation and the anion of the catalyst have a part in the reaction. The cation is coordinated to the ester group and increases its electrophilicity. Liu, et al. [16] reported that the activity of metal ions in deep eutectic solvents decreased in the series Zn2+ > Mn2+ > Co2+ > Ni2+ > Cu2+ > Fe3+ when 1,3-dimethylurea was used, which is consistent with the activity of acetate catalysts providing the series Zn2+ > Mn2+ > Co2+ > Cu2+ > Na+ [18], with Pb2+ being less active than Zn2+ and Mn2+ [19]. The activity series differed slightly in the presence of urea [17]. The anion should have significant Brønstedt basicity, allowing the coordination of the hydroxyl hydrogen of ethylene glycol (Fig. 9.2). The effect can be enhanced by ligands with the ability to form different types of hydrogen bonds with the ethylene glycol . This substantially increases the nucleophilicity of the attacking alcohol. To this end, the catalysts should not have bulky groups that could prevent attack on the polymer chain [10, 13, 16]. Since the reaction is catalysed in both directions, the glycolysis reaction is finished when equilibrium between ethylene glycol , BHET, and oligomers is reached [20].

Proposed poly(ethylene terephthalate)-catalyst complex (Cat: cation, An: anion)
Glycolysis at temperatures below the melting point of PET takes place at the interface between PET and solvent [21]. The initial reaction causes chain scission at the surface without the formation of soluble products, which can be observed as an induction time during which no conversion of PET or formation of BHET is observed. As the number of chain ends increases, the probability of BHET formation increases and a loss in weight of the solid phase is observed. Although the particle size decreases with time, the surface area increases due to the formation of cracks as amorphous regions of the semi-crystalline polymer are affected first [22]. Finally, the remaining oligomers are dissolved by the solvent and the reaction proceeds homogeneously [23].
The equilibrium is strongly affected by the molar ratio between PET and ethylene glycol [15]. It is often observed that a molar ethylene glycol /PET repeat unit ratio of more than 15 is required to raise the BHET yield to its maximum of about 80%. Several papers reported a decreasing BHET yield after exceeding the optimal reaction time, which is explained by the formation of dimers from BHET after complete glycolysis of PET. This behaviour requires explanation, as the equilibrium holds over the entire reaction time (Fig. 9.3). However, it can be observed that BHET is formed at the expense of PET long chains, while the amount of dimers remains constant [24]. This leads to high BHET yields at high ethylene glycol concentrations as long as long PET chains are still present in the reactor. Glycolysis of these last chains at the end of the reaction then reduces the ethylene glycol concentration, causing a shift in the equilibrium from BHET to the dimer. The formation of diethylene glycol and triethylene glycol as by-products from PET long chains can also reduce the ethylene glycol concentration [25]. In addition, the reaction of BHET with ethylene glycol di- and trimers, as well as with water formed during the condensation of ethylene glycol , would reduce the BHET yield. This behaviour is poorly understood and the temporal changes in the molecular weight distribution of PET during glycolysis have not yet been studied. The reaction is endothermic with an enthalpy of reaction of 12 kJ mol−1 at 196 °C [23] and a free energy between −11 and −62 kJ mol−1 at temperatures between 300 and 450 °C [25]. The equilibrium shifts to higher BHET yields at higher temperatures.

Glycolysis of poly(ethylene terephthalate) (PET): Random chain scission takes place on the surface of the PET particle, steadily reducing its size. Soluble oligomers are formed, which become dimers and bis(2-hydroxyethyl)terephthalate (BHET). Initially, the BHET/EG (ethylene glycol ) ratio in the solution is low. As the conversion progresses, the BHET proportion increases and the EG fraction decreases. At the end, an appreciable part of the EG is consumed. Glycolysis of the remaining oligomers consumes even more EG, resulting in the formation of additional dimers from BHET
A kinetic model was developed by Viana, et al. [21] for glycolysis in the presence of zinc acetate, based on a model that considers the reaction of glycol and PET at the interface. The authors found an activation energy of 42 kJ mol−1 at temperatures above 180 °C and 100 kJ mol−1 below this temperature, suggesting that diffusion plays an important role in the reaction at low temperatures. Goje and Mishra [26] found an activation energy of 46 kJ mol−1 and a pre-exponential factor of 1.0 × 105 for a boundary controlled reaction.
The rate of depolymerisation was found to be proportional to the reaction time, while the reaction rate decreased with particle size . The molecular weight of the remaining PET changed only slightly, leading to the assumption that the reaction occurred favourable at the PET-ethylene glycol interface [26]. Reducing of the PET particle size by 40% could reduce the activation energy by 40–50% [18]. Therefore, the shrinking-core model was often adopted for kinetic studies. Choline acetate used as catalyst showed an apparent activation energy of 131 kJ mol−1 and a pre-exponential factor of 1.21 × 1013, which was still higher than that of metal-based catalysts [13]. The activation energy (149 kJ mol−1) of the deep eutectic solvent system zinc acetate/1,3-dimethylurea was even higher [16]. Other researchers assumed first-order kinetics. The catalyst [bmin][OAc], based on 1-butyl-3-methylimidazole, proceeded with an activation energy of 59 kJ mol−1 [20]. The reaction in the presence of Na2CO3 showed first-order kinetics with an activation energy of 185 kJ mol−1 and a pre-exponential factor of 9.4 × 1021 [23].
The parameters that are usually adjusted to optimise BHET yield are temperature, PET/ethylene glycol molar ratio and catalyst concentration. It was found that the most important parameter for the zinc acetate catalysed reaction is the PET/ethylene glycol molar ratio and the other factors have a little influence [12].
Glycolysis without any catalyst requires high temperatures. The reaction gave BHET yields of about 95% after 30–35 min at 450 °C and a pressure of 15.3 MPa under supercritical conditions for different types of PET. Longer reaction times caused a decrease in BHET yield, while the concentrations of the dimer and oligomers rose. The concentrations of diethylene glycol and triethylene glycol increased simultaneously. The reaction under subcritical conditions between 300 and 350 °C required 120 and 70 min, respectively [25].
One of the earliest catalysts used was zinc acetate. A BHET yield of almost 70% was achieved after 1 h at 196 °C [3]. Raising the temperature above the boiling point of ethylene glycol accelerates the reaction. Optimum conditions were found to obtain 85–89% BHET at an ethylene glycol /PET weight ratio of 6:1 after 150 min at 208 °C [12, 27]. A phase transfer reaction in which BHET accumulated in the xylene phase while PET and ethylene glycol formed immiscible droplets, gave 80% BHET and 20% dimer at 220 °C. It was suggested that the separation of BHET from the ethylene glycol phase could shift the equilibrium towards the BHET product [28].
Zinc chloride and didymium chloride (PrNdCl6) required between 7 and 8 h to reach equilibrium conditions and BHET yields of more than 70% were obtained at 197 °C [29]. In ethylene glycol , soluble Na2CO3 gave BHET yields of about 60% after 1 h reaction time at 196 °C [23]. Sodium sulphate, which has a low solubility in ethylene glycol , achieved such BHET yields only after 7 h reaction time [30]. Similar results were obtained for acetic acid, LiOH, and K2SO4. These values are lower than those for zinc salts; however, sodium salts are less toxic than heavy metal ions and therefore provide an environmental viable alternative [3, 30]. For both zinc and sodium salts, it can be assumed that the catalytic activity is limited to the cation.
Heterogeneous catalysts require temperatures above the melting temperature of PET at about 240 °C to support glycolysis, as only the molten PET in such a system can make the required contact with the catalyst. However, the higher temperature should shorten the reaction time and shift the equilibrium towards BHET, thus increasing the yield. The use of silica nanoparticle carrying Mn3O4 provided a BHET yield of more than 90% after 80 min at a temperature of 300 °C and a pressure of 1.1 MPa [31]. A BHET yield of 92% was obtained in the presence of the spinel ZnMn2O4 at a temperature of 260 °C and a pressure of 0.5 MPa after 60 min. The use of CoMn2O4 required 80 min under the same conditions, which was attributed to the smaller surface area and the smaller number of strong acid sites on the surface of this catalyst [24].
The catalytic activity of metal salts acting as Lewis acids can be enhanced by the presence of hydrogen bond acceptors forming deep eutectic solvents. Before the use of deep eutectic solvents was considered, it was already found that the presence of cyclohexyl amine and NaOH in addition to zinc acetate as a catalyst accelerated the reaction rate of glycolysis [26]. The combination of zinc acetate and urea in a ratio of 1–4 gave a BHET yield of 81% after 30 min at 170 °C [17]. It was suggested that the formation of hydrogen bonds between ethylene glycol and amine increased the nucleophilicity of the hydroxyl group and thus facilitated the attack on the PET ester group (Fig. 9.2). A similar result was obtained after 20 min at 190 °C when 1,3-dimethylurea was used instead [16]. The electron-withdrawing effect of the methyl groups in 1,3-dimethylurea increases the basicity of the amine, while more bulky groups sterically prevent any catalytic activity. It was assumed that the ratio of 1–4 was related to the complexation of the metal ion by the amine.
Heterogeneous solid catalysts offer the advantage that they can be easily separated from the reaction solution by filtration or centrifugation. Zinc modified layered double hydroxides gave a maximum BHET yield of 76% after 3 h at a reaction temperature of 196 °C [32].
Sodium titanate nanotubes are comparable to zinc acetate in their catalytic activity. Both catalysts gave comparable results with a BHET yield of about 80% over a time of 2–4 h. Titanate nanotubes gave higher yields for virgin PET, zinc acetate for PET waste. The recovered BHET showed no evidence of the presence of oligomers [33].The reaction most likely proceeded by coordination of the PET carbonyl oxygen at the titanate Lewis acid site . The adjacent oxygen could act as a Lewis base and increase the nucleophilicity of the ethylene glycol [34].
In recent years, magnetic catalysts have been used that could be recovered after glycolysis. The first, nano-sized γ-Fe2O3 (maghemite) provided a BHET yield of 90% after 70 min at 300 °C [35]. The reaction conditions required a pressure of 1.1 MPa. At 255 °C, a yield of more than 80% was still obtained after 80 min. The catalyst acted as a mild acid and had no basic component that could have improved the performance. The nanoscale demobilisation of γ-Fe2O3 on nitrogen-doped graphene combines the Lewis acidity of Fe2O3 with the Brønstedt basicity of the nitrogen bound to the graphene [36]. In addition, terephthalate units are adsorbed on the graphene through strong π-π interactions, which provides a reduced distance between catalyst and PET. Magnetite (Fe3O4) attached to multi-walled carbon nanotubes (MWCNTs) also acted as an excellent catalyst [37]. Both catalysts yielded 100% BHET at a reaction temperature of 195 °C. The Fe3O4-enhanced MWCNTs required only 2 h, while the maghemite-based catalyst took 3 h to complete the reaction. The reaction in the presence of the ionic liquid coated nanoparticles Fe3O4@SiO2@(mim)[FeCl4] resulted in complete conversion after 24 h at 180 °C [38]. These catalysts are recovered after the reaction due to their magnetic properties.
In recent years, research has focussed on ionic liquids, many of which contain zinc or cobalt ions. These can cause concerns about environmental problems arising from the removal of metal ions from the final product [13]. Imidazolium-based ionic liquids also have moderate toxicity. Choline-based ionic liquids are considered less toxic and biodegradable [13].
The Lewis neutral ionic liquids [bmin][Br] and [bmin][Cl] dissolved 1.8 and 2.7 wt% of PET at 180 °C. Solubility increased in the presence of water, which was associated with decreasing pH. Even higher PET solubilities were obtained with [bmin][OAc] and [bmin][AlCl4] [27]. Although PET was partially soluble in [bmin][Br] and [bmin][Cl], little catalytic activity was observed in the glycolysis of PET [14, 20]. The catalytic activity can be increased by the addition of ZnCl2. The resulting catalyst [bmin]ZnCl3 gave a BHET yield of 83% after 2 h of reaction at 190 °C. Similar results are obtained with high concentrations of [bmin]MnCl3 as catalyst. However, the Mn-based catalyst was less effective at low concentrations due to the lower Lewis-acidity of the \( \mathrm{Mn}{\mathrm{Cl}}_3^{-} \)-anion [14]. A BHET yield of 58% was obtained after 3 h at 190 °C when [bmin][OAc] was used as the catalyst [20]. A decreasing pH of the reaction medium could indicate the interaction of ethylene glycol with the ionic liquid, leading to the release of acetic acid. Other imidazolium-based catalysts gave yields of about 80% after 24 h reaction time. The catalyst (dimim)[FeCl4] showed higher catalytic activity than (dimim)2[Fe2Cl6(μ-O)], caused by a higher Brønstedt basicity of the anion and a lower steric hindrance of the cation. Reducing the particle size of the PET dramatically increased the yield by providing a larger surface area for catalyst and ethylene glycol to attack [10]. Choline formate and acetate gave BHET yields of more than 80% in 3 h at 180 °C [13].
The guanidine-based catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) degraded PET at 190 °C in ethylene glycol in a period of less than 10 min. However, the catalyst was rapidly deactivated by atmospheric CO2 and other acidic impurities that may be present in PET waste [39]. This drawback was prevented by forming an organic salt with methanesulfonic acid, resulting in BHET yields of about 90%. No inert gas atmosphere is required and glycolysis can be carried out in an air atmosphere [15]. Similar activity to TBD was observed for the reaction with betaine. [40].
The reaction can be accelerated if microwave radiation is used to heat the reaction medium. The reaction time was reduced from 24 h to 2 h for PET waste using the imidazolium-based catalysts (dimim)[FeCl4] and (dimim)2[Fe2Cl6(μ-O)]. The second catalyst proved to be the more active one, as the polar structure of the iron(III) complex acted as an additional microwave absorption mechanism besides ionic conduction [10].
Since the glycolysis of PET waste is a recycling process, it is important to avoid the formation of production waste from spent reaction medium and catalyst during processing. Therefore, in addition to economic factors , many researchers also investigated the reusability of the reaction system. A pilot plant with a capacity of 200 tons per year could provide BHET at a price of 4.23 €/kg using zinc acetate as the catalyst. Ethylene glycol consumption was identified as the highest cost factor. Up-scaling to 8000 tons per year and reducing the ethylene glycol /PET ratio could reduce the cost to 1.99 €/kg [12]. The glycolysis process with zinc acetate can also tolerate complex waste fractions containing polyolefins from closures and labels, which are solid balls after the process [12]. However, coloured waste fractions led to discoloration of the BHET and require additional purification steps [3].
Cholin-based catalysts are cheaper than imidazolium-based ionic liquids, with a price of about US$1100 per tonne [13]. Due to the lower price and lower toxicity, choline acetate is considered the better catalyst, even though formate has the slightly higher activity.
The recyclability of the catalyst must also be considered for the economic evaluation [10]. In many homogeneous catalytic systems, the catalyst remains in the reaction medium, which is reused after BHET separation. As a result, contaminations can accumulate in the reaction medium and reduce the quality of the product. The ionic liquid [bmin][OAc] was recovered by vacuum distillation after filtration of solid BHET and used six times in succession without loss of activity [20]. The deep-eutectic solvent system zinc acetate/1,3-dimethylurea can be recycled at least 5 time. However, in the last cycle only a quarter of the initial catalyst concentration was still present in the reaction medium. Tighter control of the catalyst loss is required [16]. When using the protic salt formed from methanesulfonic acid and triazabicyclo[4.4.0]dec-5-ene, no loss of activity was observed [15].
Heterogeneous catalysts, such as layered double hydroxides [32], can be removed from the reaction medium by filtration or centrifugation. This allows the replacement of the ethylene glycol and the removal of impurities. Catalysts based on Fe3O4 [37] or γ-Fe2O3 [35, 36], were completely separated from the solvent by their magnetic properties. The recovery of the ionic liquid coated nanoparticles Fe3O4@SiO2@(mim)[FeCl4] required 3 min [38]. These catalysts were reused at least 5 times consecutively without loss of activity.
3.2 Hydrolysis of Poly(Ethylene Terephthalate)
Besides glycolysis, hydrolysis is the most studied process for the depolymerisation of PET. Hydrolysis occurs when PET reacts with water and as a result terephthalic acid and ethylene glycol are formed, both of which can be reused in the production of new PET. This reaction requires high temperatures and pressures when carried out in aqueous solutions under sub- or supercritical conditions. Therefore, catalysts are commonly used to reduce these drastic conditions. Most catalytic approaches use acidic [41,42,43] or basic catalysts [12, 44, 45], but some neutral catalysts are also known [46, 47]. Both acidic and basic catalysts have serious disadvantages. Basic catalysts form terephthalic acid salt solutions that must be neutralized with strong mineral acids to obtain terephthalic acid. Acidic catalysts are commonly strong mineral acids that must be diluted with water to recover terephthalic acid; the mineral acid is disposed of as waste. In addition, like terephthalic acid, these catalysts have the disadvantage of causing corrosion to the equipment. Therefore, attempts have been made to reduce the harsh conditions and instead carry out the reaction in a steam atmosphere [5, 48,49,50]. More recent approaches try to avoid water as a solvent and use less volatile solvents [46, 51, 52] or use enzymes [53,54,55,56] to accelerate the reaction under more ambient conditions.
When hydrolysed in water above the melting point of PET and without a catalyst, an apparent activation energy of 56 kJ mol−1 was observed [4]. The activation energy did not change between 250 and 265 °C when zinc acetate was used as a catalyst. A decreasing activation energy was only observed at a lower temperature, which was explained by a change in the emulsion state [47]. The uncatalysed reaction proceeded in a 75 vol% steam atmosphere with an apparent activation energy of 140 kJ mol−1 and a pre-exponential factor of 1.6 × 109 according to kinetics that most closely follows the models of a contracting cylinder or sphere [49]. Hydrolysis of PET fibres from PVC-coated woven fabrics followed the same models with an activation energy of 56 kJ mol−1 and a pre-exponential factor of 1.3 × 106 [44]. Alkaline hydrolysis of PET with NaOH proceeded with an apparent activation energy of 99 kJ mol−1 [45]. The reaction was diffusion controlled in ethanol/water [57]. In the presence of KOH, an apparent activation energy of 69 kJ mol−1 and a pre-exponential factor of 419 L min−1 cm−2 were observed. The reaction was assumed to be first-order in terms of both PET surface area and KOH concentration [58]. A much higher apparent activation energy of 173 kJ mol−1 was found for the alkaline hydrolysis carried out in ethylene glycol [52].
A value of 89 kJ mol−1, was achieved by developing a modified shrinking core model for the hydrolysis of PET in sulphuric acid, taking into account the partial dissolution of the PET in the acid [42]. Another modified shrinking core model was applied for the reaction in nitric acid. Taking into account the precipitation of terephthalic acid on the PET surface, a value of 101 kJ mol−1 was obtained [43]. The reaction in supercritical CO2 was more likely to proceed by a first-order reaction with an apparent activation energy of about 12.5 kJ mol−1 at 160 °C when sulphated TiO2 was used as a solid acid catalyst [41].
Activation energies ranged from 12.5 to 173 kJ mol−1 depending on the conditions used. Phase boundaries were present in any case; PET was either in the form of solid or molten polymer; the mobile phase was either an aqueous solution, an organic solution, or a gas phase . This implies that in any case mass transfer resistance was present, which had a strong influence on the observed results. It is to be expected that higher activation energies indicate systems with a high degree of chemical control, while lower values indicate an obstruction by phase boundaries.
The hydrolysis of PET in the absence of a catalyst requires high temperatures and pressures. The yield of terephthalic acid was more than 90% at 350 °C after 60 min reaction time [59]. At higher temperatures, the yield decreased because the terephthalic acid was degraded to benzoic acid [60]. The yield of ethylene glycol was lower than that of terephthalic acid . The increasing acid concentration caused the degradation of ethylene glycol . Acetaldehyde, di- and triethylene glycol were found as by-products [59]. Reducing the temperature requires longer reaction times. The reaction was completed after 5 h at 265 °C, with terephthalic acid and ethylene glycol as the main products. A degradation of ethylene glycol was not observed. An equilibrium constant of 0.664 was found, which means that complete hydrolysis of PET cannot be achieved [4].
The addition of zinc or sodium acetate as a catalyst increased the reaction rate by only about 20% compared to the uncatalysed reaction [47]. It was assumed that these catalysts did not directly influence the hydrolysis reaction, but stabilized the emulsion and reduced the droplet size. This was supported by the fact that the apparent activation energy between 250 and 265 °C was equal to the uncatalysed one. When the reaction was catalysed with zinc acetate in xylene, using a minimum amount of water and a detergent to form an emulsion, the average molecular weight of the resulting product at 160 °C was greatly reduced. This process lowered the reaction temperature and pressure and facilitated product recovery, as the recovered ethylene glycol contained little water and PET oligomers were obtained as a fine white powder [46].
The alkaline hydrolysis of PET at 150 °C was completed after 4 h reaction time at a PET/NaOH ratio of 1:2.4. The yield of terephthalic acid was 90% with a purity of more than 95% when PET waste was used [12]. A comparable reaction gave 98% terephthalic acid after 1 h at 200 °C [45]. The reaction temperature was reduced to 80 °C in ethanol/water with a volume ratio of 60–40 with comparable yields [57].
When KOH was used at 160 °C, terephthalic acid with a yield of 91% and ethylene glycol were the only products obtained. The remaining PET hardly changed and retained its high molecular weight even at the end of the reaction, indicating that depolymerisation took place only on the PET surface [58].
Alkaline hydrolysis can also be used to separate PET fibres from poly(vinyl chloride) (PVC) in woven fabrics. The yield of terephthalic acid reached 99% after 2 h at 180 °C in 1 M NaOH solution. Dechlorination of PVC occurred simultaneously and reached 22% at the end of the reaction. Both the hydrolysis of PET and the dechlorination of PVC proceeded independently without the formation of shared by-products [44].
Alkaline hydrolysis in ethylene glycol resulted in complete PET conversion after 1 min at 185 °C. The usual mass transfer resistance caused by the formation of sodium terephthalate at the sample surface was overcome by vigorous stirring. In addition, the molecular weight reduction caused by hydrolytic degradation during processing and aging was also found to shorten the processing time [52]. The reaction with 9 M KOH in Cellusolve (CH3OCH2CH2OH) gave a terephthalic acid yield of 82% after 2.5 h at 120 °C. It was assumed that a higher yield was prevented by product formation on the PET surface, as potassium terephthalate was not soluble in the reaction medium [45].
Concentrated sulphuric acid (18 M) is able to dissolve PET at room temperature. During the slow hydrolysis of the dissolved polymer, more and more end-groups, carboxylic acids and hydroxyl-groups, are formed. The investigation of precipitated product by XRD showed that amorphous regions are dissolved first before the dissolution process also covers PET crystallites [61]. The solubility of PET decreases with the sulphuric acid concentration, which also leads to a delay in the hydrolysis reaction. Complete hydrolysis at 150 °C was achieved at a concentration of 9 mol L−1 after 2 h, whereas with sulphuric acid at a concentration of 3 mol L−1, only 90% of the PET was degraded after 12 h [42, 62]. The reaction of PET in 13 M HNO3 resulted in a weight loss of about 80 wt% after 12 h at 100 °C. Removal of terephthalic acid from the surface was able to accelerate the reaction. Ethylene glycol was oxidized to oxalic acid, which was obtained as the second major product [43].
Hydrolysis carried out in the ionic liquid [Bmin][Cl] in the presence of [HSO3-pmim][HSO4] (where pmim stands for 1-methyl-3-(3-sulfopropyl)-imidazol) as an acid catalyst gave a yield of 89% of terephthalic acid after 4.5 h at 170 °C. Even without any catalyst, the ionic liquid gave a terephthalic acid yield of 76% [51].
The reaction can be strongly accelerated by the use of microwave radiation . The presence of sodium methoxide as catalyst reduces the reaction time in dimethyl sulphoxide and methanol as solvent to 5 min at 70 °C. The reaction yields a mixture of dimethyl and monomethyl terephthalate and terephthalic acid . The terephthalic acid is recovered from the solution after addition of water. The yield was reported to be about 74% for terephthalic acid [63].
An alternative possibility for hydrolysis is the reaction in supercritical CO2. The gas acts as a plasticizer under supercritical conditions, promoting swelling and migration within the polymer. However, it also induces crystallization of amorphous PET, which can slow down the hydrolysis process. The reaction in supercritical CO2 using the solid superacid catalyst \( {\mathrm{SO}}_4^{2-} \)/TiO2 was complete after 5 h at 200 °C at any pressure. Hydronium ions were able to migrate together with dissolved water into the polymer matrix. The reaction took place both on the surface and in the bulk material [41].
High pressure can be avoided if hydrolysis is carried out in a steam atmosphere. The reaction in a fluidised bed requires temperatures above 400 °C to ensure volatilization of the terephthalic acid . At 450 °C, a terephthalic acid yield of about 70% was achieved, while ethylene glycol was almost completely degraded when quartz sand was used as bed material. Up to 24% of the terephthalic acid remained bound in oligomers. Decarboxylation also occurred with the formation of benzoic acid and acetylbenzoic acid [5]. Experiments with 18O-labelled steam showed that at temperatures between 340 and 440 °C, both pyrolysis and hydrolysis occurred in the degradation of PET and other aromatic polyesters. The selectivity of hydrolysis shifted from 46% to 11% in this temperature range when a 75 vol% steam atmosphere was present [49].
Terephthalic acid from the steam hydrolysis of PET can be converted to benzene using CaO as a catalyst. This reaction required 9 h at 450 °C, which could be reduced to 90 min at 550 °C. The benzene yield and purity were between 52 and 65% and between 82 and 89%, respectively. While the benzene yield reached its maximum at 500 °C, the highest purity was obtained at 450 °C. An even higher benzene yield of 74% and a purity of 97% were achieved when a heating rate of 2 K min−1 was applied from 300 to 500 °C [48]. At a heating rate of 5 K min−1, more than 50% of the carbon was converted to benzene. Benzene was obtained as a liquid with a high purity of 99%. Naphthalenes and biphenyls observed during pyrolysis of PET in the presence of CaO are minor impurities and no oxygen-containing compounds were observed [50]. When bottle-PET was hydrolysed at 450 °C and the volatile terephthalic acid was decarboxylated over CaO at 700 °C, benzene was obtained with a yield of 74% and a purity of 96%. The benzene yield from various composites such as X-ray film and magnetic tape decreased to values between 32 and 35%. The main by-product was biphenyl from the secondary reaction of benzene. Oxygenated compounds were negligible [64].
A new way of hydrolytically decomposing PET waste is the use polyester degrading bacteria and their enzymes [65]. In the past, carboxylic acid hydrolases, lipases, serine esterases and carboxylesterases have been reported to be effective in the hydrolysis of PET [56]. Among these, cutinases, which belong to the group of carboxylic acid hydrolases, are the most efficient. It was found that Humicola insolens cutinase (HiC) is more than 20 times faster than other known PET degrading enzymes. One reason for this could be the small size of cutinases compared to other enzymes, which allows better contact with the rigid structure of PET. Terephthalic acid and mono(2-hydroxyethyl)terephthalate (MHET) were the main products of the degradation.
Cutinases are natural polyester hydrolases that have proven to be very efficient in the hydrolysis of PET at a temperature of around 70 °C. Terephthalic acid could then be recovered by filtration and ethylene glycol after distillation. The biodegradation route could be less energy intensive and thus more cost-effective at lower temperature and pressure. However, the biochemical process is more time consuming. Complete hydrolysis of PET film by Thermobifida fusca cutinase TfCut2, expressed by Bacillus subtilis and isolated from E. coli , takes approximately 120 h at 70 °C. With other packaging materials, 50% of the PET was not degraded. The reason for the differences in degradation behaviour was thought to be the inability of the enzyme to attack crystalline and rigid amorphous fractions, neither of which offer sufficient flexibility for an enzymatic reaction [53].
This obstacle is overcome by studying enzymes that are active at temperatures above 70 °C. Degradation of PET by the Thermobifida cellulosilytica derived cutinase Thc_Cut1 caused increasing crystallinity, which may also indicate preferential hydrolysis of the amorphous region [66]. When the thermophilic bacterium Clostridium thermocellum expresses leaf and branch compost cutinase (LCC) at 60 °C, about 62% of the amorphous PET was converted into terephthalic acid and MHET over a period of 14 days [7]. The stability of LCC against aggregation was improved by glycosylation. The stabilized enzyme showed high activity between 70 and 80 °C. The thickness of an amorphous PET film with a crystallinity of 7% was reduced by 60 μm day−1 [54]. With improved thermal stability, LCC was able to depolymerise PET by 90% in less than 10 h in a pilot-plant scale set-up. The recovered terephthalic acid was purified and used to make new PET. The bottles made from this material showed similar properties to those made from commercial PET [67].
The hydrolysis of PET in blends with polyethylene or polyamide is accelerated when Thc_Cut1 is used as an enzyme [66]. The enzymatic hydrolysis of highly crystalline PET fibres requires a pretreatment step. Only after hydrolysis of PET fibres at a temperature of 250 °C and a pressure of 39 bar was HiC able to convert the remaining oligomers into terephthalic acid . The presence of zinc cations as hydrolysis catalyst inhibited the action of the enzyme [55]. Furthermore, MHET [56, 68] and BHET [66] showed inhibitory effects on cutinases. Countermeasures are the addition of Candida Antarctica lipase B (CALB), which degraded MHET to terephthalic acid [56] or the use of an ultrafiltration membrane reactor to continuously remove MHET [68]. A reduced particle size is favourable for the hydrolysis of PET by enzymes [66].
Terephthalic acid can be produced by alkaline hydrolysis at 150 °C at a cost of 2.71 € kg−1. The main cost factor is the wastewater management in terms of the organic content and the salt load due to the neutralisation of the highly basic solution for the precipitation of the terephthalic acid . The price could be reduced to 1.02 € kg−1, if the wastewater is treated within the production plant [12]. The reaction in ethanol/water at 80 °C could reduce greenhouse gas emissions compared to the energetic conversion of PET waste [57].
The recyclability of production waste from PET hydrolysis is rather poor. Wastewater with an extreme pH is often disposed of. When neutralising the reaction solution from alkaline hydrolysis to recover terephthalic acid , saline solutions are produced from which the salt could be recovered. More promising is the use of ionic liquids as the reaction medium, as the two can be easily separated. Ionic liquid and catalyst were reused eight times in a row without loss of catalytic activity [51].
3.3 Aminolysis of Poly(Ethylene Terephthalate)
Poly(ethylene terephthalate ) reacts with ammonia (ammonolyis) or organic amines (aminolysis) to form aromatic amides. Strongly basic amines react faster than amines with a low basicity. The reactivity is reduced by steric hindrance [69]. Depending on the amine used, a catalyst may be required. A catalyst is more likely to be needed for the reaction of lipophilic amines than for hydrophilic amines [70]. Since the amine-group is more nucleophilic than the hydroxyl-group, amino alcohols give always terephthalamides [70].
The reaction takes place below the melting temperature of PET at the particle surface. It was observed that methylamine preferentially attacks amorphous areas, while ammonia attacks both amorphous and crystalline areas from the beginning. This was attributed to the higher nucleophilicity of methylamine [71]. Increasing temperatures cause a softening of the material and facilitate the migration of the amine beyond the surface, which is even more easily penetrated when the temperature exceeds the melting point of PET. The molecular weight of PET has been found to decrease with increasing PET conversion, indicating a random scission process [72].
The apparent activation energy of the aminolysis of PET in ethanolamine was determined to be 153 kJ mol−1 using a modified shrinking core model [72].
Terephthalamides can be obtained from ammonia or various primary or secondary amines. Often, after optimisation the reaction does not require a catalyst [70]. If necessary, catalysts such as zinc acetate [73], sodium sulphate, acetate or bicarbonate [74] are used. Reaction of PET with ammonia under pressure at ambient temperature resulted in complete conversion to terephthalamide after 45 days in the absence of any catalyst and 15 days in the presence of zinc acetate [73]. Reaction with ethanolamine gave bis(hydroxyethyl)terephthalamide (BHETA) after 2 h at 140 °C [70]. Precipitation of the product was observed at high conversion. Reaction of fibres and bottle wastes under reflux in the presence of sodium acetate yielded 91 and 83% of BHETA after 8 h, respectively. The lower rate of degradation of bottle waste was explained by its higher molecular weight and broader molecular weight distribution [75].
The reaction of ethylenediamine with PET required 17 h at 100 °C in the absence of a catalyst. The yield of bis(2-aminoethyl)terephthalamide (BAET ) reached 75% at an ethylene diamine /PET ratio of 16. Lower ratios yielded less BAET. The by-products were α,ω-aminoligo(ethylene terephthalamide )s (AOETs), oligomers from the condensation of BAET. The dimer was the most abundant fraction of the AOETs, although trimers and tetramers were also present [76]. The equilibrium shifted towards the AOET with temperature, reaching about 70% at 250 °C [77]. The reaction of triethylenetetramine with PET gave the corresponding terephthalamide after refluxing for 2 h at 130–140 °C [78].
The guanidine derivative 1,5,7-triazabicyclo [4.4.0] dec-5-ene (TBD) accelerated the aminolysis of PET with various aliphatic and aromatic amines. Aminolysis in the presence of ethylenediamine was completed after 1 h at 120 °C. Aminolysis in aniline took 24 h at 180 °C due to its lower basicity. Although piperidine is a strong base, the reaction was slow due to steric limitations [69]. Aminolysis in ethylenediamine in the presence of a TBD derivative (Fig. 9.4) as catalyst gave complete conversion of PET with BAET as the main product after 10 min at 120 °C. The reaction in ethanolamine required only 3 min at 190 °C and gave BHETA [79].

TBD (1,5,7-triazabicyclo [4.4.0] dec-5-ene) based aminolysis catalyst
The reaction time can also be shortened by using a microwave reactor. The yield of BAET in the presence of ethylenediamine reached a maximum of 30% after 10 min at 250 °C and a pressure of more than 1 MPa. The main product after the complete conversion of PET was AOET. The reaction rate decreased significantly when the reaction was carried out below the melting point of PET [77]. Aminolysis in the presence of ethanolamine and NaHCO3 as catalyst required 5 min for PET fibre waste and 7 min for PET bottle waste to obtain BHETA yields of about 90% under reflux [74]. The reaction time could be reduced to 2 min in the absence of a catalyst at a microwave power of 150 W when an autoclave was used. The temperature rose above the melting temperature of PET and the pressure exceeded 1 MPa. No reaction was observed below 180 °C [72].
The reaction in the presence of ethanolamine catalysed by dibutyltin oxide gave a complete conversion after 60 days in a sand bath heated by solar radiation in the Egyptian summer [80].
The product of PET depolymerized in ethanolamine, BHETA can be used for the production of polyurethanes [81] and corrosion protection paints [80]. The reaction of BAET with cardanol and paraformaldehyde resulted in bis-benzoxazines derivatives, which were converted to thermosets by ring-opening polymerisation (Fig. 9.5). Cardanol is obtained from cashew nut shells and provides a route of bio-based resin production [77].

Thermosets from bis(2-aminoethyl)terephthalamide (BAET ) and cardanol
Products from the aminolysis of PET can be used as additives in asphalt [70]. The in-situ polymerisation of BHETA with methylene diphenyl diisocyanate in bitumen at 90–130 °C resulted in a binder suitable for the construction of roads with high traffic loads [82].
3.4 Solvolysis Using Other Types of Alcohols
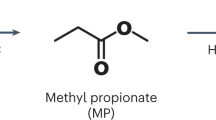
In addition to the reactions described in the sections above, solvolysis can also be carried out in the presence of other alcohols, methanol being the most important. Methanolysis was of some importance in the early days of research, but lost importance when PET production was no longer carried out via the transesterification of dimethyl terephthalate (DMT).
The reaction mechanism is similar to that of the glycolysis process. The same catalytic systems are also applicable. The use of deep eutectic solvents [83] has been reported. A special feature of alcoholysis processes with solvents other than ethylene glycol is that at least two alcohols are present in the system and mixed products can be expected [84, 85]. An ATR-FTIR investigation could show that the Gauche conformation disappeared faster than the Trans conformation during methanolysis. The crystalline fraction also decreased faster than the amorphous fraction [86]. It was found that the transesterification of PET with 2-ethyl-1-hexanol in the presence of choline chloride/zinc acetate as catalyst proceeds by a first-order reaction with an activation energy of 95 kJ mol−1 [83].
The use of methanol requires high pressure due to the low boiling point of methanol (65 °C). The reaction is usually carried out in the supercritical state above the critical point of 239 °C and 8.09 MPa [85]. It is shown that PET cast film was readily penetrated by methanol already at 150 °C, although the low activation energy of 6 kJ mol−1 suggested an impeded mass transfer [86]. However, sufficient solubilisation only occurred under critical conditions. Below the critical pressure, the depolymerisation rate decreased significantly [85]. Below the melting point of PET, degradation occurred mainly at tie molecules reducing the molecular weight to one third. Catalysts can accelerate the formation of oligomers and DMT [87].
Under supercritical conditions, PET is melted and the effect of mass transfer resistance is reduced. It was found that PET dissolution is improved under supercritical conditions [88]. The depolymerisation was completed after 40 min at temperatures between 253 and 273 °C and a pressure of 11 MPa. Most of the by-products are converted into DMT under these conditions and DMT with a purity of 98% could be recovered from waste materials after purification [85]. At a temperature of 300 °C and a pressure of 20 MPa, PET with an average molecular weight of 47 kDa was depolymerised to 3 kDa after 5 min and 1 kDa after 10 min. The main products after 20 min were methyl 2-hydroxyethyl terephthalate, DMT, and terephthalic acid monomethyl ester (TAMME). The yield of TAMME decreased after reaching its maximum after 15 min. The molecular weight distribution was successfully simulated over the reaction time [89]. The optimal conditions for the uncatalysed reaction were found at 298 °C for 112 min, yielding 99.8% DMT [88]. The addition of toluene promoted swelling and increased the DMT yield in the presence of aluminium isopropoxide as catalyst from 67% in pure methanol to 89% in methanol/toluene (80:20) at a temperature of 200 °C [87]. The temperature and time requirements can be reduced by using microwave reactors. Methanolysis in the presence of zinc acetate gave a DMT yield of 87% after 30 min at 180 °C. After reaching a maximum of 1.6 MPa, the pressure dropped to 1.2 MPa during the reaction, caused by the consumption of methanol [84].
The reaction in supercritical ethanol at a temperature of 255 °C and a pressure of 11.65 MPa gave a diethylterephthalate yield of 80% from multilayer packaging PET coated with aluminium and polyethylene [90].
High boiling alcohols do not require high pressure for their reaction with PET. The reaction with 1,4-butanediol and triethylene glycol can be carried out at 220 °C in the presence of zinc acetate as catalyst. The main product after 10 h of the reaction with 1,4-butanediol was bis(4-hydroxybutenyl)terephthalate with the dimer being the main by-product. When triethylene glycol was used, mainly oligomers with molecular weights between 700 and 800 g mol−1 were obtained [91]. Solvolysis in the presence of 2-ethyl-1-hexanol gave a dioctyl terephthalate yield of 84% after 1 h reaction time and with choline chloride/zinc acetate (molar ratio 1:1) as deep eutectic solvent catalyst . The product could replace phthalate plasticizers and reduce the environmental hazard they pose [83].
3.5 Other Aromatic Polyesters
The most common aromatic polyesters besides PET are PBT and PEN. While PBT is used for many applications in the automotive industry and for electrical and electronic equipment, PEN is used as an alternative material to PET for packaging applications due to its better gas barrier properties and higher glass transition temperature. The depolymerisation processes are similar to those for PET.
The hydrolysis of PEN in aqueous ammonia solution at 240 °C was controlled by mass transfer [92]. The activation energy of the hydrolysis of PEN was 95 kJ mol−1 [93], higher than that observed for PET with 56 kJ mol−1 [4]. The activation energies for hydrolysis of PBT and PEN in steam atmosphere were 156 and 161 kJ mol−1, respectively, which were slightly higher than that for PET under the same conditions [49]. For the random scission of PBT in methanol, an activation energy between 84 [9] and 87 kJ mol−1 [94] was determined, which is about 10 kJ mol−1 lower than that for the reaction of PET [9].
Depolymerisation of PEN in hot water is not observed below 227 °C. Yields of 2,6-naphthalene dicarboxylic acid (NDA) and ethylene glycol reached 98 and 86% after 1 h at a temperature of 300 °C [59]. When PEN was heated in water, swelling was first observed, then above 200 °C the state changed to a liquid. This process ended at 270 °C, after which PEN was dissolved and a homogeneous aqueous phase was formed. The hydrolysis of PEN resulted in 83% of NDA and 80% ethylene glycol after 60 min at 260 °C [93].
The reaction of PBT and PEN catalysed by Ca(OH)2 in a steam atmosphere led to the formation of benzene and naphthalene, respectively, as was also observed for PET. The highest product yields were observed at 700 °C for PBT and 600 °C for PEN [95]. Pyrolytic processes were also observed during the hydrolytic degradation of PBT and PEN in a steam atmosphere. Pyrolysis increased drastically from 19 to 73% for PBT hydrolysis between 320 and 440 °C, while the contribution of pyrolysis remained constant at about 75% for PEN [49].
The hydrolysis of PBT in hot water can cause the degradation of terephthalic acid and 1,4-butanediol. Benzoic acid and tetrahydrofuran are observed as products instead [60]. During the hydrolysis of PEN, the resulting NDA can be decarboxylated to form naphthalene, which is oxidised to phthalic anhydride and hydrolysed to phthalic acid (Fig. 9.6) [93].

Degradation of a) poly(butylene terephthalate) (PBT), and b) poly(ethylene naphthalate) (PEN) in hot water
In methanol, PBT was completely depolymerised at 240 °C after 22 min. The highest DMT yield of 95% was observed after 75 min at 290 °C [94]. Shibata, et al. [9] observed complete depolymerisation after 10 min at the same temperature and a pressure of 6 MPa. By the time the reaction temperature was reached, about 50% of the two monomers had already been recovered. Alcoholysis of PBT is faster in methanol than in ethanol or propanol. Complete conversion was achieved in methanol after 20 min at 250 °C, while 25 and 42% of the PBT remained as residue in ethanol and propanol, respectively. The highest yields of dialkyl terephthalates were observed after 75 min at 310 °C with 98.5% of DMT and 76% of diethyl terephthalate [96]. One reason for this behaviour could be the good miscibility of molten PBT with methanol [9].
4 Depolymerisation of Aliphatic Polyesters
In contrast to aromatic polyesters, aliphatic polyesters are biodegradable. These include poly(lactic acid) (PLA , polylactide), poly(butylene succinate) (PBS ), polycaprolactone (PCL), polyhydroxyalkanoates (PHAs) and others. Despite their biodegradability, these polyesters should be recycled to reduce the environmental impact of their production [97]. Most of these polymers can be successfully converted into monomers and other feedstocks for the production of new materials.
4.1 Depolymerisation of Poly(Lactic Acid)
The production of PLA begins with the fermentation of sugary biomass by lacto bacteria. The resulting lactic acid is dimerised to lactide in a conventional chemical process and then further converted to PLA by ring-opening polymerisation. Lactic acid occurs in two enantiomers, both produced by different species of Lactobacillus. Since usually only one of the enantiomers is used for the production of one type of PLA, the aim of depolymerising used PLA is to recover lactide or other lactic acid derivatives without racemisation [98].
The mechanism of catalysed depolymerisation of PLA is often analogous to that of PET glycolysis (Figs. 9.1 and 9.2) with the cation acting as a Lewis acid and the anion increasing the nucleophilicity of the reactant, water or alcohol [99, 100]. The reaction of PLA in the presence of a zinc catalyst is faster than that of PET. It has been suggested that PLA is able to chelate the zinc ion (Fig. 9.7), while the rigid structure of PET prevents chelation [101]. High solubility of PLA is favourable for the reaction. Ionic liquids with a low ability to dissolve PLA showed limited conversion, while the reaction proceeded easily when PLA was sufficiently dissolved. Small polymer chains were more rapidly dissolved and depolymerised by a random scission process than longer ones, resulting in an extraordinary reduction in polydispersity [100].

Chelation of zinc ions by poly(lactic acid) PLA. The R-groups represent PLA chains
Hydrolysis in water and NaOH solution showed comparable activation energies of 82 kJ mol−1 and 72 kJ mol−1, respectively [102]. The activation energy was significantly higher at 134 kJ mol−1 when the reaction was catalysed by [Bmin][OAc] [100]. Methanolysis using a zinc ring-opening catalyst proceeded by random chain scission with an activation energy of (39 and 65) kJ mol−1 [103]. Using an ethylenediamine-zinc(II) complex, an activation energy of about 40 kJ mol−1 was observed [104].
Pyrolysis is the simplest route of PLA depolymerisation, but also the most energy-intensive. In the presence of zinc catalysts at temperatures between 190 and 245 °C, lactide was obtained as the main product with an enantiomeric excess of more than 99% and a yield of up to 92% [105]. However, racemisation occurs in the presence of some fillers. It was observed that in the presence of MgO less than 3% of the lactide was racemised at temperatures below 270 °C. The extent of racemisation increased above this temperature. The presence of CaO resulted in 10–20% racemisation below 250 °C, while above this temperature racemisation decreased to below 2%. It was suggested that racemisation occurred by a backbiting process, while L,L-lactide was obtained by an unzipping reaction [106, 107].
Hydrolysis of PLA in water and NaOH solution gave lactic acid yields between 92% and 98% at 160 °C [102]. Similar results were obtained in the presence of [Bmin][OAc] as catalyst, resulting in a PLA conversion of 94% at a temperature of 130 °C. After precipitation with Ca(CO3), a calcium lactate yield of 76% was obtained [100]. High enantiomeric purity was achieved with Escherichia coli strains [102].
Methanolysis in the presence of KF as catalyst gave methyl lactate yields of more than 99% at 180 °C. The catalyst was able to degrade PLA and poly(bisphenol-A-carbonate) side by side, while PET and nylon-6 interfered with the degradation and reduced the yield of PLA [99]. Reaction in methanol with 4-dimethylaminopyridine (DMAP) as a catalyst resulted in 97% yield of methyl lactate as determined by NMR after heating in a microwave oven at 180 °C for 10 min. The catalysts 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) even achieved yields close to 100% under the same conditions [108]. Microwave methanolysis at 160 °C gave reaction yields of more than 99% after 20 min in the presence of tin(II) 2-ethylhexanoate. High turnover frequencies close to 4 × 104 h−1 were observed [109]. The reaction of PLA with methanol, ethanol and butanol was completed after 2 min at room temperature in the presence of TBD. The reaction of sterically hindered alcohols was strongly delayed due to the space requirements of the catalyst. Acidic alcohols deactivated the basic catalyst [110]. Methanolysis in the presence of zinc acetate gave a methyl lactate yield of 70% after 15 h of heating under reflux [101]. The reaction in tetrahydrofuran using a zinc ring-opening catalyst was completed within 60 min and gave 100% methyl lactate although depolymerisation was already observed at 40 °C [103]. Methyl lactate yields of 100% were obtained from PLA waste after 4 h at 110 °C in the presence of an ethylenediamine-Zn(II) complex as catalyst [104].
The reaction of PLA with diamines gave oligomers when carried out under reflux in xylene. The reaction proceeded with 1,6-diaminohexane without catalyst and after 1 h an oligomer with a molecular weight of 5.5 kg mol−1 was obtained containing both lactic acid and diamine units. Oligomers were also obtained in the presence of tin(II) 2-ethylhexanoate as catalyst when 1,3-propanediol was the degradation agent [111]. Ionic liquids could be recovered from PLA hydrolysis and reused 7 times in a row without loss of activity [100].
Lifecycle assessment showed that chemical recycling and polymerisation of virgin PLA from 1 tonne of waste PLA could reduce the global warming potential by 780 kg CO2-eq. [97]. The energy requirement for the production of lactic acid could be reduced from 55 MJ kg−1 for the production from corn starch to 14 MJ kg−1 for the hydrolysis of PLA [98].
4.2 Depolymerisation of Other Aliphatic Polyesters
Other aliphatic polyesters include PBS, PCL, PHAs, and other mostly biomass derived thermoplastics. Although these materials are often biodegradable, depolymerisation for the purpose of recycling has been an emerging issue in recent years. In this section, only a few examples are presented.
It can be seen that many concepts that were successfully applied to the depolymerisation of PET were also adopted for aliphatic polyesters. The reaction mechanisms found for the reaction catalysed by ionic liquids [112, 113] were analogous to those for PET (Figs. 9.1 and 9.2). The hydrolysis of PBS and poly(butylene succinate/adipate) (PBSA) in hot water proceeded with activation energies of 64 and 58 kJ mol−1 [114]. The low activation energy of 27 kJ mol−1 in the methanolysis of PHB catalysed by [Bmin]FeCl4 [112, 113] indicated mass transport hindrance.
The thermal degradation of PHAs at 190 °C leads to a significant reduction in molecular weight after only 30 min [115]. If PHB was pyrolysed at 310 °C, unsaturated acids are obtained. A yield of about 78% was achieved, including trans-crotonic acid as the main product and 2-pentenoic acid and cis-crotonic acid as by-products. This method can be used to separate PHB from polypropylene, which is degraded at higher temperatures [116].
The hydrolysis of PBS and poly(butylene succinate/adipate) (PBSA) in hot water provided lower acid yields between 180 and 300 °C than comparable experiments from PET. The highest succinic acid yield of 80% was achieved from PBS after about 30 min at temperatures between 270 and 300 °C. In the same temperature range, the succinic acid and adipic acid yields from PBSA reached 65 and 70%, respectively [114].
The most common strategy for the depolymerisation of aliphatic esters is the use of enzymes. Lipases in particular offer the advantage of wide commercial availability [117]. Such lipases were able to reduce the molecular weight of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) from 300 kg mol−1 to below 5 kg mol−1 after 72 h at 37 °C. The reaction of PCL in toluene required 24 h at 50 °C in the presence of Candida antarctica lipase B (CALB) for conversion to di-, tri-, and tetra-oligomers. ε-caprolactone was also observed as a by-product. Depolymerisation proceeded by a random chain scission mechanism. After reaching a minimum after 24 h, the molecular weight increased again significantly [8]. The time required for depolymerisation at moderate temperatures is still too long for technical application. Therefore, higher temperatures are aimed for.
Depolymerisation in the presence of CALB using a twin screw extruder drastically reduced the molecular weight of PBS from 82 kg mol−1 to 2000 g mol−1 after 5 min at a temperature of 120 °C. A maximum succinic acid yield of 44% was achieved, while 1,4-butanediol was not observed among the products. This reaction required catalyst concentrations of up to 10 wt% and the catalyst lost more than 60% of its activity during the process [118]. The depolymerases Est-H and Est-L isolated from the bacterium Roseateles depolymerans TB-87 were able to degrade various aliphatic and aliphatic-aromatic polyesters. Succinic acid and 1,4-butanediol were obtained from PBS. In addition, adipic acid was observed in the degradation of poly(butylene succinate-co-adipate) and terephthalic acid and isophthalic acid in the degradation of poly(butylene succinate/terephthalate/isophthalate)-co-(lactate). The depolymerisation of aliphatic-aromatic polyesters proved to be slower than that of purely aliphatic polyesters [119]. The depolymerisation of poly(hexamethylene succinate-co-hexamethylene-hexylthiosuccinate) with CALB reduced the molecular weight from 78 kg mol−1 to 800 g mol−1. All products were cyclic oligomers. Subsequent ring-opening polymerisation with the same catalyst gave a polymer with a molecular weight of 70 kg mol−1, proving the recyclability of this type of polyester [120].
Methanolysis under reflux in the absence of a catalyst at a temperature of 200 °C and a pressure of 18 bar for a period of 6 h converted PHB to methyl crotonate. The product could be used to obtain propylene and methyl acrylate by metathesis [6]. Methanolysis of PHB in the presence of the ionic liquids [Bmin]FeCl4 and [MIMPS]FeCl4 (1-(3-sulfonic acid)-propyl-3-methylimidazole ferric chloride) as ferromagnetic catalysts gave methyl (3-hydroxybutyrate) yields of 85 and 87% after a reaction time of 3 h at 140 °C, respectively. The purity of the monomer was over 98%. The catalysts were recovered and used six times in succession without loss of activity [112, 113]. The reaction carried out under a microwave heating in the presence of methanol or ethanol gave a mixture of 3-hydroxybutyric acid, crotonic acid, and the 3-alkyl butyric acid ethers [121].
5 Conclusions and Future Outlook
Climate change will force plastics manufacturers to find new sources of carbon in future, when fossil fuels are no longer considered suitable for technical applications. Fossil carbon could be replaced to some extent by biomass. However, it is questionable whether enough biomass can be made available to meet the increasing demand for plastics. Plastic producers could face carbon scarcity in a future world, which would force them to keep the “harvested” carbon in the cycle.
Established polymers such as PET, PBT, and PEN have been developed from monomers that are readily available from fossil fuels. This paradigm could change if biomass becomes the main feedstock for carbon loss replacement. Biomass as a feedstock provides different types of monomers. New polymers, such as PLA, PBS, and PHAs, are the result and others may follow. The structure of the new polymers may depend strongly on the type of biomass used. This could result in an even wider range of polymers than we see today—and all these polymers have their own process requirements for recycling if monomers are to be the target.
We have seen in this chapter that different types of polyesters require different processes for depolymerisation. However, we have also seen that some processes allow the separation of different types of polymers: PET can be separated from PVC [44]; PLA from PET and nylon-6 [99]; PHB from polyolefins [116]. The increasing number of polymers in the waste stream could make the separation of individual polymers more difficult than it is today. Combining chemical recycling methods for monomer recovery may have some advantages, and therefore there is still much work to be done to develop solutions that offer the possibility of fractional depolymerisation and stepwise monomer recovery. To keep track of the increasing number of possible monomers, more of these solutions will be needed in the future.
References
Volanti M, Cespi D, Passarini F, Neri E, Cavani F, Mizsey P, Fozer D. Terephthalic acid from renewable sources: early-stage sustainability analysis of a bio-PET precursor. Green Chem. 2019;21:885–96. https://doi.org/10.1039/C8GC03666G.
Williams ID, Shaw PJ. Reuse in Practice. 4th Symposium on Urban Mining and Circular Economy. Bergamo, Italy; 2018
Lopez-Fonseca R, Duque-Ingunza I, de Rivas B, Arnaiz S, Gutierrez-Ortiz JI. Chemical recycling of post-consumer PET wastes by glycolysis in the presence of metal salts. Polym Degrad Stabil. 2010;95:1022–8. https://doi.org/10.1016/j.polymdegradstab.2010.03.007.
Campanelli JR, Kamal MR, Cooper DG. A kinetic-study of the hydrolytic degradation of polyethylene terephthalate at high-temperatures. J Appl Polym Sci. 1993;48:443–51. https://doi.org/10.1002/app.1993.070480309.
Grause G, Kaminsky W, Fahrbach G. Hydrolysis of poly(ethylene terephthalate) in a fluidised bed reactor. Polym Degrad Stabil. 2004;85:571–5. https://doi.org/10.1016/j.polymdegradstab.2003.10.020.
Spekreijse J, Le Notre J, Sanders JPM, Scott EL. Conversion of polyhydroxybutyrate (PHB) to methyl crotonate for the production of biobased monomers. J Appl Polym Sci. 2015;132:8. https://doi.org/10.1002/app.42462.
Yan F, Wei R, Cui Q, Bornscheuer UT, Liu YJ. Thermophilic whole-cell degradation of polyethylene terephthalate using engineered Clostridium thermocellum. Microb Biotechnol. 2021;14:374–85. https://doi.org/10.1111/1751-7915.13580.
Aris MH, Annuar MSM, Ling TC. Lipase-mediated degradation of poly-epsilon-caprolactone in toluene: Behavior and its action mechanism. Polym Degrad Stabil. 2016;133:182–91. https://doi.org/10.1016/j.polymdegradstab.2016.08.015.
Shibata M, Masuda T, Yosomiya R, Ling-Hui M. Depolymerization of poly(butylene terephthalate) using high-temperature and high-pressure methanol. J Appl Polym Sci. 2000;77:3228–33. https://doi.org/10.1002/1097-4628(20000929)77:14<3228::Aid-app260>3.0.Co;2-g.
Sce F, Cano I, Martin C, Beobide G, Castillo O, de Pedro I. Comparing conventional and microwave-assisted heating in PET degradation mediated by imidazolium-based halometallate complexes. New J Chem. 2019;43:3476–85. https://doi.org/10.1039/c8nj06090h.
Welle F. Twenty years of PET bottle to bottle recycling—An overview. Resour Conserv Recy. 2011;55:865–75. https://doi.org/10.1016/j.resconrec.2011.04.009.
Aguado A, Martinez L, Becerra L, Arieta-araunabena M, Arnaiz S, Asueta A, Robertson I. Chemical depolymerisation of PET complex waste: hydrolysis vs. glycolysis. J Mater Cycles Waste Manage. 2014;16:201–10. https://doi.org/10.1007/s10163-013-0177-y.
Liu YC, Yao XQ, Yao HY, Zhou Q, Xin JY, Lu XM, Zhang SJ. Degradation of poly(ethylene terephthalate) catalyzed by metal-free choline-based ionic liquids. Green Chem. 2020;22:3122–31. https://doi.org/10.1039/d0gc00327a.
Yue QF, Xiao LF, Zhang ML, Bai XF. The glycolysis of poly(ethylene terephthalate) waste: lewis acidic ionic liquids as high efficient catalysts. Polymers. 2013;5:1258–71. https://doi.org/10.3390/polym5041258.
Jehanno C, Flores I, Dove AP, Muller AJ, Ruiperez F, Sardon H. Organocatalysed depolymerisation of PET in a fully sustainable cycle using thermally stable protic ionic salt. Green Chem. 2018;20:1205–12. https://doi.org/10.1039/c7gc03396f.
Liu B, Fu WZ, Lu XM, Zhou Q, Zhang SJ. Lewis acid-base synergistic catalysis for polyethylene terephthalate degradation by 1,3-dimethylurea/Zn(OAc)2 deep eutectic solvent. ACS Sustain Chem Eng. 2019;7:3292–300. https://doi.org/10.1021/acssuschemeng.8b05324.
Wang Q, Yao XQ, Geng YR, Zhou Q, Lu XM, Zhang SJ. Deep eutectic solvents as highly active catalysts for the fast and mild glycolysis of poly(ethylene terephthalate)(PET). Green Chem. 2015;17:2473–9. https://doi.org/10.1039/c4gc02401j.
Kao CY, Cheng WH, Wan BZ. Investigation of catalytic glycolysis of polyethylene terephthalate by differential scanning calorimetry. Thermochim Acta. 1997;292:95–104. https://doi.org/10.1016/s0040-6031(97)00060-9.
Ghaemy M, Mossaddegh K. Depolymerisation of poly(ethylene terephthalate) fibre wastes using ethylene glycol. Polym Degrad Stabil. 2005;90:570–6. https://doi.org/10.1016/j.polymdegradstab.2005.03.011.
Al-Sabagh AM, Yehia FZ, Eissa A, Moustafa ME, Eshaq G, Rabie ARM, ElMetwally AE. Glycolysis of poly(ethylene terephthalate) catalyzed by the lewis base ionic liquid (Bmim)(OAc). Ind Eng Chem Res. 2014;53:18443–51. https://doi.org/10.1021/ie503677w.
Viana ME, Riul A, Carvalho GM, Rubira AF, Muniz EC. Chemical recycling of PET by catalyzed glycolysis: kinetics of the heterogeneous reaction. Chem Eng J. 2011;173:210–9. https://doi.org/10.1016/j.cej.2011.07.031.
Zhou XY, Lu XM, Wang Q, Zhu ML, Li ZX. Effective catalysis of poly(ethylene terephthalate) (PET) degradation by metallic acetate ionic liquids. Pure Appl Chem. 2012;84:789–801. https://doi.org/10.1351/pac-con-11-06-10.
Lopez-Fonseca R, Duque-Ingunza I, de Rivas B, Flores-Giraldo L, Gutierrez-Ortiz JI. Kinetics of catalytic glycolysis of PET wastes with sodium carbonate. Chem Eng J. 2011;168:312–20. https://doi.org/10.1016/j.cej.2011.01.031.
Imran M, Kim DH, Al-Masry WA, Mahmood A, Hassan A, Haider S, Ramay SM. Manganese-, cobalt-, and zinc-based mixed-oxide spinels as novel catalysts for the chemical recycling of poly(ethylene terephthalate) via glycolysis. Polym Degrad Stabil. 2013;98:904–15. https://doi.org/10.1016/j.polymdegradstab.2013.01.007.
Imran M, Kim BK, Han M, Cho BG, Kim DH. Sub- and supercritical glycolysis of polyethylene terephthalate (PET) into the monomer bis(2-hydroxyethyl) terephthalate (BHET). Polym Degrad Stabil. 2010;95:1686–93. https://doi.org/10.1016/j.polymdegradstab.2010.05.026.
Goje AS, Mishra S. Chemical kinetics, simulation, and thermodynamics of glycolytic depolymerization of poly(ethylene terephthalate) waste with catalyst optimization for recycling of value added monomeric products. Macromol Mater Eng. 2003;288:326–36. https://doi.org/10.1002/mame.200390034.
Wang H, Li ZX, Liu YQ, Zhang XP, Zhang SJ. Degradation of poly(ethylene terephthalate) using ionic liquids. Green Chem. 2009;11:1568–75. https://doi.org/10.1039/b906831g.
Guclu G, Kasgoz A, Ozbudak S, Ozgumus S, Orbay M. Glycolysis of poly(ethylene terephthalate) wastes in xylene. J Appl Polym Sci. 1998;69:2311–9. https://doi.org/10.1002/(SICI)1097-4628(19980919)69:12%3C2311::AID-APP2%3E3.0.CO;2-B.
Pingale ND, Palekar VS, Shukla SR. Glycolysis of postconsumer polyethylene terephthalate waste. J Appl Polym Sci. 2010;115:249–54. https://doi.org/10.1002/app.31092.
Shukla SR, Harad AM. Glycolysis of polyethylene terephthalate waste fibers. J Appl Polym Sci. 2005;97:513–7. https://doi.org/10.1002/app.21769.
Imran M, Lee KG, Imtiaz Q, Kim BK, Han M, Cho BG, Kim DH. Metal-oxide-doped silica nanoparticles for the catalytic glycolysis of polyethylene terephthalate. J Nanosci Nanotechnol. 2011;11:824–8. https://doi.org/10.1166/jnn.2011.3201.
Eshaq G, ElMetwally AE. (Mg-Zn)-Al layered double hydroxide as a regenerable catalyst for the catalytic glycolysis of polyethylene terephthalate. J Mol Liq. 2016;214:1–6. https://doi.org/10.1016/j.molliq.2015.11.049.
Lima GR, Monteiro WF, Ligabue R, Santana RMC. Titanate nanotubes as new nanostrutured catalyst for depolymerization of PET by glycolysis reaction. Mater Res-Ibero-Am J Mater. 2017;20:588–95. https://doi.org/10.1590/1980-5373-mr-2017-0645.
Kitano M, Wada E, Nakajima K, Hayashi S, Miyazaki S, Kobayashi H, Hara M. Protonated titanate nanotubes with Lewis and Brønsted acidity: relationship between nanotube structure and catalytic activity. Chem Mat. 2013;25:385–93. https://doi.org/10.1021/cm303324b.
Bartolome L, Imran M, Lee KG, Sangalang A, Ahn JK, Kim DH. Superparamagnetic γ-Fe2O3 nanoparticles as an easily recoverable catalyst for the chemical recycling of PET. Green Chem. 2014;16:279–86. https://doi.org/10.1039/c3gc41834k.
Nabid MR, Bide Y, Fereidouni N, Etemadi B. Maghemite/nitrogen-doped graphene hybrid material as a reusable bifunctional catalyst for glycolysis of polyethylene terephthalate. Polym Degrad Stabil. 2017;144:434–41. https://doi.org/10.1016/j.polymdegradstab.2017.08.033.
Al-Sabagh AM, Yehia FZ, Harding DRK, Eshaq G, ElMetwally AE. Fe3O4-boosted MWCNT as an efficient sustainable catalyst for PET glycolysis. Green Chem. 2016;18:3997–4003. https://doi.org/10.1039/c6gc00534a.
Cano I, Martin C, Fernandes JA, Lodge RW, Dupont J, Casado-Carmona FA, Lucena R, Cardenas S, Sans V, de Pedro I. Paramagnetic ionic liquid-coated SiO2@Fe3O4 nanoparticles—The next generation of magnetically recoverable nanocatalysts applied in the glycolysis of PET. Appl Catal B Environ. 2020;260:118110. https://doi.org/10.1016/j.apcatb.2019.118110.
Fukushima K, Coulembier O, Lecuyer JM, Almegren HA, Alabdulrahman AM, Alsewailem FD, McNeil MA, Dubois P, Waymouth RM, Horn HW, Rice JE, Hedrick JL. Organocatalytic depolymerization of poly(ethylene terephthalate). J Polym Sci Pol Chem. 2011;49:1273–81. https://doi.org/10.1002/pola.24551.
Kim DH, Han DO, In Shim K, Kim JK, Pelton JG, Ryu MH, Joo JC, Han JW, Kim HT, Kim KH. One-pot chemo-bioprocess of PET depolymerization and recycling enabled by a biocompatible catalyst, betaine. ACS Catal. 2021;11:3996–4008. https://doi.org/10.1021/acscatal.0c04014.
Li XK, Lu H, Guo WZ, Cao GP, Liu HL, Shi YH. Reaction kinetics and mechanism of catalyzed hydrolysis of waste PET using solid acid catalyst in supercritical CO2. AICHE J. 2015;61:200–14. https://doi.org/10.1002/aic.14632.
Yoshioka T, Motoki T, Okuwaki A. Kinetics of hydrolysis of poly(ethylene terephthalate) powder in sulfuric acid by a modified shrinking-core model. Ind Eng Chem Res. 2001;40:75–9. https://doi.org/10.1021/ie000592u.
Yoshioka T, Okayama N, Okuwaki A. Kinetics of hydrolysis of PET powder in nitric acid by a modified shrinking-core model. Ind Eng Chem Res. 1998;37:336–40. https://doi.org/10.1021/ie970459a.
Kumagai S, Hirahashi S, Grause G, Kameda T, Toyoda H, Yoshioka T. Alkaline hydrolysis of PVC-coated PET fibers for simultaneous recycling of PET and PVC. J Mater Cycles Waste Manage. 2018;20:439–49. https://doi.org/10.1007/s10163-017-0614-4.
Karayannidis GP, Chatziavgoustis AP, Achilias DS. Poly(ethylene terephthalate) recycling and recovery of pure terephthalic acid by alkaline hydrolysis. Adv Polym Technol. 2002;21:250–9. https://doi.org/10.1002/adv.10029.
Guclu G, Yalcinyuva T, Ozgumu S, Orbay M. Hydrolysis of waste polyethylene terephthalate and characterization of products by differential scanning calorimetry. Thermochim Acta. 2003;404:193–205. https://doi.org/10.1016/s0040-6031(03)00160-6.
Campanelli JR, Cooper G, Kamal MR. Catalyzed-hydrolysis of polyethylene terephthalate melts. J Appl Polym Sci. 1994;53:985–91. https://doi.org/10.1002/app.1994.070530801.
Grause G, Handa T, Kameda T, Mizoguchi T, Yoshioka T. Effect of temperature management on the hydrolytic degradation of PET in a calcium oxide filled tube reactor. Chem Eng J. 2011;166:523–8. https://doi.org/10.1016/j.cej.2010.11.010.
Kumagai S, Morohoshi Y, Grause G, Kameda T, Yoshioka T. Pyrolysis versus hydrolysis behavior during steam decomposition of polyesters using 18O-labeled steam. RSC Adv. 2015;5:61828–37. https://doi.org/10.1039/c5ra08577b.
Du SC, Valla JA, Parnas RS, Bollas GM. Conversion of polyethylene terephthalate based waste carpet to benzene-rich oils through thermal, catalytic, and catalytic steam pyrolysis. ACS Sustain Chem Eng. 2016;4:2852–60. https://doi.org/10.1021/acssuschemeng.6b00450.
Liu FS, Cui X, Yu ST, Li Z, Ge XP. Hydrolysis reaction of poly(ethylene terephthalate) using ionic liquids as solvent and catalyst. J Appl Polym Sci. 2009;114:3561–5. https://doi.org/10.1002/app.30981.
Ruvolo A, Curti PS. Chemical kinetic model and thermodynamic compensation effect of alkaline hydrolysis of waste poly(ethylene terephthalate) in nonaqueous ethylene glycol solution. Ind Eng Chem Res. 2006;45:7985–96. https://doi.org/10.1021/ie060528y.
Wei R, Breite D, Song C, Grasing D, Ploss T, Hille P, Schwerdtfeger R, Matysik J, Schulze A, Zimmermann W. Biocatalytic degradation efficiency of postconsumer polyethylene terephthalate packaging determined by their polymer microstructures. Adv Sci. 2019;6:10. https://doi.org/10.1002/advs.201900491.
Shirke AN, White C, Englaender JA, Zwarycz A, Butterfoss GL, Linhardt RJ, Gross RA. Stabilizing leaf and branch compost cutinase (LCC) with glycosylation: mechanism and effect on PET hydrolysis. Biochemistry. 2018;57:1190–200. https://doi.org/10.1021/acs.biochem.7b01189.
Quartinello F, Vajnhandl S, Valh JV, Farmer TJ, Voncina B, Lobnik A, Acero EH, Pellis A, Guebitz GM. Synergistic chemo-enzymatic hydrolysis of poly(ethylene terephthalate) from textile waste. Microb Biotechnol. 2017;10:1376–83. https://doi.org/10.1111/1751-7915.12734.
de Castro AM, Carniel A, Nicomedes J, Gomes AD, Valoni E. Screening of commercial enzymes for poly(ethylene terephthalate) (PET) hydrolysis and synergy studies on different substrate sources. J Ind Microbiol Biotechnol. 2017;44:835–44. https://doi.org/10.1007/s10295-017-1942-z.
Ügdüler S, Van Geem KM, Denolf R, Roosen M, Mys N, Ragaert K, De Meester S. Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis. Green Chem. 2020;22:5376–94. https://doi.org/10.1039/D0GC00894J.
Wan BZ, Kao CY, Cheng WH. Kinetics of depolymerization of poly(ethylene terephthalate) in a potassium hydroxide solution. Ind Eng Chem Res. 2001;40:509–14. https://doi.org/10.1021/ie0005304.
Sato O, Arai K, Shirai M. Hydrolysis of poly(ethylene terephthalate) and poly(ethylene 2,6-naphthalene dicarboxylate) using water at high temperature: effect of proton on low ethylene glycol yield. Catal Today. 2006;111:297–301. https://doi.org/10.1016/j.cattod.2005.10.040.
Wang JL, Bei K, Hu ZC, Liu YP, Ma YP, Shen Y, Chou IM, Pan ZY. Depolymerization of waste polybutylene terephthalate in hot compressed water in a fused silica capillary reactor and an autoclave reactor: monomer phase behavior, stability, and mechanism. Polym Eng Sci. 2017;57:544–9. https://doi.org/10.1002/pen.24450.
de Carvalho GM, Muniz EC, Rubira AF. Hydrolysis of post-consume poly(ethylene terephthalate) with sulfuric acid and product characterization by WAXD, 13C-NMR and DSC. Polym Degrad Stabil. 2006;91:1326–32. https://doi.org/10.1016/j.polymdegradstab.2005.08.005.
Yoshioka T, Sato T, Okuwaki A. Hydrolysis of waste PET by sulfuric acid at 150°C for a chemical recycling. J Appl Polym Sci. 1994;52:1353–5. https://doi.org/10.1002/app.1994.070520919.
Mohsin MA, Alnaqbi MA, Busheer RM, Haik Y. Sodium methoxide catalyzed depolymerization of waste polyethylene terephthalate under microwave irradiation. Catal Ind. 2018;10:41–8. https://doi.org/10.1134/s2070050418010087.
Kumagai S, Grause G, Kameda T, Yoshioka T. Simultaneous recovery of benzene-rich oil and metals by steam pyrolysis of metal-poly(ethylene terephthalate) composite waste. Environ Sci Technol. 2014;48:3430–7. https://doi.org/10.1021/es405047j.
Kawai F, Kawabata T, Oda M. Current state and perspectives related to the polyethylene terephthalate hydrolases available for biorecycling. ACS Sustain Chem Eng. 2020;8:8894–908. https://doi.org/10.1021/acssuschemeng.0c01638.
Gamerith C, Zartl B, Pellis A, Guillamot F, Marty A, Acero EH, Guebitz GM. Enzymatic recovery of polyester building blocks from polymer blends. Process Biochem. 2017;59:58–64. https://doi.org/10.1016/j.procbio.2017.01.004.
Tournier V, Topham CM, Gilles A, David B, Folgoas C, Moya-Leclair E, Kamionka E, Desrousseaux ML, Texier H, Gavalda S, Cot M, Guémard E, Dalibey M, Nomme J, Cioci G, Barbe S, Chateau M, André I, Duquesne S, Marty A. An engineered PET depolymerase to break down and recycle plastic bottles. Nature. 2020;580:216–9. https://doi.org/10.1038/s41586-020-2149-4.
Barth M, Wei R, Oeser T, Then J, Schmidt J, Wohlgemuth F, Zimmermann W. Enzymatic hydrolysis of polyethylene terephthalate films in an ultrafiltration membrane reactor. J Membr Sci. 2015;494:182–7. https://doi.org/10.1016/j.memsci.2015.07.030.
Fukushima K, Lecuyer JM, Wei DS, Horn HW, Jones GO, Al-Megren HA, Alabdulrahman AM, Alsewailem FD, McNeil MA, Rice JE, Hedrick JL. Advanced chemical recycling of poly(ethylene terephthalate) through organocatalytic aminolysis. Polym Chem. 2013;4:1610–6. https://doi.org/10.1039/c2py20793a.
Merkel DR, Kuang W, Malhotra D, Petrossian G, Zhong L, Simmons KL, Zhang J, Cosimbescu L. Waste PET chemical processing to terephthalic amides and their effect on asphalt performance. ACS Sustain Chem Eng. 2020;8:5615–25. https://doi.org/10.1021/acssuschemeng.0c00036.
Mittal A, Soni RK, Dutt K, Singh S. Scanning electron microscopic study of hazardous waste flakes of polyethylene terephthalate (PET) by aminolysis and ammonolysis. J Hazard Mater. 2010;178:390–6. https://doi.org/10.1016/j.jhazmat.2010.01.092.
Achilias DS, Tsintzou GP, Nikolaidis AK, Bikiaris DN, Karayannidis GP. Aminolytic depolymerization of poly(ethylene terephthalate) waste in a microwave reactor. Polym Int. 2011;60:500–6. https://doi.org/10.1002/pi.2976.
Jain A, Soni RK. Spectroscopic investigation of end products obtained by ammonolysis of poly (ethylene terephthalate) waste in the presence of zinc acetate as a catalyst. J Polym Res. 2007;14:475–81. https://doi.org/10.1007/s10965-007-9131-9.
Pingale ND, Shukla SR. Microwave-assisted aminolytic depolymerization of PET waste. Eur Polym J. 2009;45:2695–700. https://doi.org/10.1016/j.eurpolymj.2009.05.028.
Shukla SR, Harad AM. Aminolysis of polyethylene terephthalate waste. Polym Degrad Stabil. 2006;91:1850–4. https://doi.org/10.1016/j.polymdegradstab.2005.11.005.
Hoang CN, Dang YH. Aminolysis of poly(ethylene terephthalate) waste with ethylenediamine and characterization of a,u-diamine products. Polym Degrad Stabil. 2013;98:697–708. https://doi.org/10.1016/j.polymdegradstab.2012.12.026.
Sharma P, Lochab B, Kumar D, Roy PK. Sustainable Bis-benzoxazines from cardanol and PET-derived terephthalamides. ACS Sustain Chem Eng. 2016;4:1085–93. https://doi.org/10.1021/acssuschemeng.5b01153.
Leng Z, Sreeram A, Padhan RK, Tan Z. Value-added application of waste PET based additives in bituminous mixtures containing high percentage of reclaimed asphalt pavement (RAP). J Clean Prod. 2018;196:615–25. https://doi.org/10.1016/j.jclepro.2018.06.119.
Nica S, Duldner M, Hanganu A, Iancu S, Cursaru B, Sarbu A, Filip P, Bartha E. Functionalized 1,5,7-triazabicyclo [4.4.0] dec-5-ene (TBD) as novel organocatalyst for efficient depolymerization of polyethylene terephthalate (PET) wastes. Rev Chim. 2018;69:2613–6. https://doi.org/10.37358/rc.18.10.6591.
Tawfik ME, Ahmed NM, Eskander SB. Aminolysis of poly(ethylene terephthalate) wastes based on sunlight and utilization of the end product [bis(2-hydroxyethylene) terephthalamide] as an ingredient in the anticorrosive paints for the protection of steel structures. J Appl Polym Sci. 2011;120:2842–55. https://doi.org/10.1002/app.33350.
Shamsi R, Abdouss M, Sadeghi GMM, Taromi FA. Synthesis and characterization of novel polyurethanes based on aminolysis of poly(ethylene terephthalate) wastes, and evaluation of their thermal and mechanical properties. Polym Int. 2009;58:22–30. https://doi.org/10.1002/pi.2488.
Padhan RK, Gupta AA. Preparation and evaluation of waste PET derived polyurethane polymer modified bitumen through in situ polymerization reaction. Constr Build Mater. 2018;158:337–45. https://doi.org/10.1016/j.conbuildmat.2017.09.147.
Zhou L, Lu XM, Ju ZY, Liu B, Yao HY, Xu JL, Zhou Q, Hu YF, Zhang SJ. Alcoholysis of polyethylene terephthalate to produce dioctyl terephthalate using choline chloride-based deep eutectic solvents as efficient catalysts. Green Chem. 2019;21:897–906. https://doi.org/10.1039/c8gc03791d.
Siddiqui MN, Redhwi HH, Achilias DS. Recycling of poly(ethylene terephthalate) waste through methanolic pyrolysis in a microwave reactor. J Anal Appl Pyrolysis. 2012;98:214–20. https://doi.org/10.1016/j.jaap.2012.09.007.
Yang Y, Lu YJ, Xiang HW, Xu YY, Li YW. Study on methanolytic depolymerization of PET with supercritical methanol for chemical recycling. Polym Degrad Stabil. 2002;75:185–91. https://doi.org/10.1016/s0141-3910(01)00217-8.
Andanson JM, Kazarian SG. In situ ATR-FTIR spectroscopy of poly(ethylene terephthalate) subjected to high-temperature methanol. Macromol Symp. 2008;265:195–204. https://doi.org/10.1002/masy.200850521.
Kurokawa H, Ohshima M, Sugiyama K, Miura H. Methanolysis of polyethylene terephthalate (PET) in the presence of aluminium tiisopropoxide catalyst to form dimethyl terephthalate and ethylene glycol. Polym Degrad Stabil. 2003;79:529–33. https://doi.org/10.1016/s0141-3910(02)00370-1.
Liu QL, Li RS, Fang T. Investigating and modeling PET methanolysis under supercritical conditions by response surface methodology approach. Chem Eng J. 2015;270:535–41. https://doi.org/10.1016/j.cej.2015.02.039.
Goto M, Koyamoto H, Kodama A, Hirose T. Degradation kinetics of polyethylene terephthalate in supercritical methanol. AICHE J. 2002;48:136–44. https://doi.org/10.1002/aic.690480114.
Favaro SL, Freitas AR, Ganzerli TA, Pereira AGB, Cardozo AL, Baron O, Muniz EC, Girotto EM, Radovanovic E. PET and aluminum recycling from multilayer food packaging using supercritical ethanol. J Supercrit Fluids. 2013;75:138–43. https://doi.org/10.1016/j.supflu.2012.12.015.
Mansour SH, Ikladious NE. Depolyrnerization of poly(ethylene terephthalate) wastes using 1,4-butanediol and triethylene glycol. Polym Test. 2002;21:497–505. https://doi.org/10.1016/s0142-9418(01)00115-5.
Arai R, Zenda K, Hatakeyama K, Yui K, Funazukuri T. Reaction kinetics of hydrothermal depolymerization of poly(ethylene naphthalate), poly(ethylene terephthalate), and polycarbonate with aqueous ammonia solution. Chem Eng Sci. 2010;65:36–41. https://doi.org/10.1016/j.ces.2009.03.023.
Bei K, Ma PX, Wang JL, Li K, Lyu JH, Hu ZC, Chou IM, Pan ZY. Depolymerization of poly(ethylene naphthalate) in fused silica capillary reactor and autoclave reactor from 240 to 280°C in subcritical water. Polym Eng Sci. 2017;57:1382–8. https://doi.org/10.1002/pen.24523.
Huang J, Yang JH, Chyu MK, Wang QM, Zhu ZB. Continuous-distribution kinetics for degradation of polybutylene terephthalate (PBT) in supercritical methanol. Polym Degrad Stabil. 2009;94:2142–8. https://doi.org/10.1016/j.polymdegradstab.2009.09.011.
Yoshioka T, Grause G, Otani S, Okuwaki A. Selective production of benzene and naphthalene from poly(butylene terephthalate) and poly(ethylene naphthalene-2,6-dicarboxylate) by pyrolysis in the presence of calcium hydroxide. Polym Degrad Stabil. 2006;91:1002–9. https://doi.org/10.1016/j.polymdegradstab.2005.08.010.
Yang JH, Huang J, Chyu MK, Wang QM, Xiong DL, Zhu ZB. Degradation of poly(butylene terephthalate) in different supercritical alcohol solvents. J Appl Polym Sci. 2010;116:2269–74. https://doi.org/10.1002/app.31649.
Maga D, Hiebel M, Thonemann N. Life cycle assessment of recycling options for polylactic acid. Resour Conserv Recycl. 2019;149:86–96. https://doi.org/10.1016/j.resconrec.2019.05.018.
Piemonte V, Sabatini S, Gironi F. Chemical recycling of PLA: a great opportunity towards the sustainable development? J Polym Environ. 2013;21:640–7. https://doi.org/10.1007/s10924-013-0608-9.
Alberti C, Damps N, Meissner RRR, Hofmann M, Rijono D, Enthaler S. Selective degradation of end-of-life poly(lactide) via alkali-metal-halide catalysis. Adv Sustain Syst. 2020;4:9. https://doi.org/10.1002/adsu.201900081.
Song XY, Wang H, Yang XQ, Liu FS, Yu ST, Liu SW. Hydrolysis of poly(lactic acid) into calcium lactate using ionic liquid [Bmin][OAc] for chemical recycling. Polym Degrad Stabil. 2014;110:65–70. https://doi.org/10.1016/j.polymdegradstab.2014.08.020.
Sanchez AC, Collinson SR. The selective recycling of mixed plastic waste of polylactic acid and polyethylene terephthalate by control of process conditions. Eur Polym J. 2011;47:1970–6. https://doi.org/10.1016/j.eurpolymj.2011.07.013.
Chauliac D, Pullammanappallil PC, Ingram LO, Shanmugam KT. A combined thermochemical and microbial process for recycling polylactic acid polymer to optically pure l-lactic acid for reuse. J Polym Environ. 2020;28:1503–12. https://doi.org/10.1007/s10924-020-01710-1.
Roman-Ramirez LA, McKeown P, Jones MD, Wood J. Poly(lactic acid) degradation into methyl lactate catalyzed by a well-defined Zn(II) complex. ACS Catal. 2019;9:409–16. https://doi.org/10.1021/acscatal.8b04863.
Román-Ramírez LA, McKeown P, Shah C, Abraham J, Jones MD, Wood J. Chemical degradation of end-of-life poly(lactic acid) into methyl lactate by a Zn(II) complex. Ind Eng Chem Res. 2020;59:11149–56. https://doi.org/10.1021/acs.iecr.0c01122.
Noda M, Okuyama H. Thermal catalytic depolymerization of poly(L-lactic acid) oligomer into LL-lactide : effects of Al, Ti, Zn and Zr compounds as catalysts. Chem Pharm Bull. 1999;47:467–71. https://doi.org/10.1248/cpb.47.467.
Fan YJ, Nishida H, Mori T, Shirai Y, Endo T. Thermal degradation of poly(L-lactide): effect of alkali earth metal oxides for selective L,L-lactide formation. Polymer. 2004;45:1197–205. https://doi.org/10.1016/j.polymer.2003.12.058.
Fan YJ, Nishida H, Shirai Y, Endo T. Control of racemization for feedstock recycling of PLLA. Green Chem. 2003;5:575–9. https://doi.org/10.1039/b304792j.
Alberti C, Damps N, Meissner RRR, Enthaler S. Depolymerization of end-of-life poly(lactide) via 4-dimethylaminopyridine-catalyzed methanolysis. ChemistrySelect. 2019;4:6845–8. https://doi.org/10.1002/slct.201901316.
Hofmann M, Alberti C, Scheliga F, Meißner RRR, Enthaler S. Tin(ii) 2-ethylhexanoate catalysed methanolysis of end-of-life poly(lactide). Polym Chem. 2020;11:2625–9. https://doi.org/10.1039/D0PY00292E.
Leibfarth FA, Moreno N, Hawker AP, Shand JD. Transforming polylactide into value-added materials. J Polym Sci Pol Chem. 2012;50:4814–22. https://doi.org/10.1002/pola.26303.
Plichta A, Lisowska P, Kundys A, Zychewicz A, Debowski M, Florjanczyk Z. Chemical recycling of poly(lactic acid) via controlled degradation with protic (macro)molecules. Polym Degrad Stabil. 2014;108:288–96. https://doi.org/10.1016/j.polymdegradstab.2014.03.006.
Song XY, Wang H, Wang C, Liu FS, Yu ST, Liu SW, Song ZY. Chemical recycling of bio-based poly(3-hydroxybutyrate) wastes under methanolysis condition catalyzed by Fe-containing magnetic ionic liquid. J Polym Environ. 2019;27:862–70. https://doi.org/10.1007/s10924-018-1347-8.
Song XY, Liu FS, Wang H, Wang C, Yu ST, Liu SW. Methanolysis of microbial polyester poly(3-hydroxybutyrate) catalyzed by Bronsted-Lewis acidic ionic liquids as a new method towards sustainable development. Polym Degrad Stabil. 2018;147:215–21. https://doi.org/10.1016/j.polymdegradstab.2017.12.009.
Tsuji H, Yamamura Y, Ono T, Saeki T, Daimon H, Fujie K. Hydrolytic degradation and monomer recovery of poly(butylene succinate) and poly(butylene succinate/adipate) in the melt. Macromol React Eng. 2008;2:522–8. https://doi.org/10.1002/mren.200800027.
Sin MC, Tan IKP, Annuar MSM, Gan SN. Characterization of oligomeric hydroxyalkanoic acids from thermal decomposition of palm kernel oil-based biopolyester. Int J Polym Anal Charact. 2011;16:337–47. https://doi.org/10.1080/1023666x.2011.588306.
Norrrahim MNF, Ariffin H, Hassan MA, Ibrahim NA, Nishida H. Performance evaluation and chemical recyclability of a polyethylene/poly(3-hydroxybutyrate-co-3-hydroxyvalerate) blend for sustainable packaging. RSC Adv. 2013;3:24378–88. https://doi.org/10.1039/c3ra43632b.
Rodriguez-Contreras A, Calafell-Monfort M, Marques-Calvo MS. Enzymatic degradation of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) by commercial lipases. Polym Degrad Stabil. 2012;97:597–604. https://doi.org/10.1016/j.polymdegradstab.2012.01.007.
Jbilou F, Dole P, Degraeve P, Ladaviere C, Joly C. A green method for polybutylene succinate recycling: depolymerization catalyzed by lipase B from Candida antarctica during reactive extrusion. Eur Polym J. 2015;68:207–15. https://doi.org/10.1016/j.eurpolymj.2015.04.039.
Shah AA, Eguchi T, Mayumi D, Kato S, Shintani N, Kamini NR, Nakajima-Kambe T. Degradation of aliphatic and aliphatic-aromatic co-polyesters by depolymerases from Roseateles depolymerans strain TB-87 and analysis of degradation products by LC-MS. Polym Degrad Stabil. 2013;98:2722–9. https://doi.org/10.1016/j.polymdegradstab.2013.10.003.
Yagihara T, Matsumura S. Enzymatic synthesis and chemical recycling of novel polyester-type thermoplastic elastomers. Polymers. 2012;4:1259–77. https://doi.org/10.3390/polym4021259.
Yang X, Odelius K, Hakkarainen M. Microwave-assisted reaction in green solvents recycles PHB to functional chemicals. ACS Sustain Chem Eng. 2014;2:2198–203. https://doi.org/10.1021/sc500397h.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Grause, G. (2022). Depolymerisation of Fossil Fuel and Biomass-derived Polyesters. In: Fang, Z., Smith Jr., R.L., Xu, L. (eds) Production of Biofuels and Chemicals from Sustainable Recycling of Organic Solid Waste. Biofuels and Biorefineries, vol 11. Springer, Singapore. https://doi.org/10.1007/978-981-16-6162-4_9
Download citation
DOI: https://doi.org/10.1007/978-981-16-6162-4_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-6161-7
Online ISBN: 978-981-16-6162-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)