
Abstract
Diverse peptides and proteins undergo concentration-dependent self-assembly into one-dimensional cross-β fibril structures. Cross-β fibrils are characterized by self-assembled β-sheet peptides with an intermolecular hydrogen bond network between the constituent peptides that is parallel to the fibril axis with peptide side chains oriented perpendicular to the fibril axis. Cross-β nanoarchitectures are relevant to amyloid protein misfolding disorders, regulated biological structural elements, and designed functional biomaterials. The biological relevance of the cross-β motif has made this protein fold the subject of intense research interest focused on understanding the mechanisms of self-assembly, the structure of the resulting cross-β fibrils and prefibrillar aggregates, and the emergent biological and material properties of these aggregates. Cross-β fibrils are typically non-crystalline high molecular weight aggregates that are often, but not always, highly insoluble in water. These properties make cross-β fibrils challenging targets for analysis. This chapter provides an introduction to current strategies that are applied to interrogating mechanism of assembly, structure, and emergent properties of cross-β nanoarchitectures.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Peptide and protein self-assembly into β-sheet fibrils is a characteristic of both amyloid misfolding disorders and functional biomaterials [1]. Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease are prominent examples of protein misfolding disorders that are characterized by the aberrant self-assembly of proteins or peptides into amyloid fibrils [2]. These fibrils and/or their metastable oligomeric folding intermediates have been shown to be cytotoxic to cells and tissues and are thought to play an important role in the etiology of these amyloid diseases. This relevance to disease has prompted intense scrutiny of the structure and function of amyloid fibrils, including efforts to understand the mechanisms of self-assembly through thermodynamic and kinetic models, the development of tools to more accurately determine the molecular architecture of these materials, and interrogation of the emergent properties of these assemblies and the related prefibrillar aggregates [3, 4]. These efforts have led to discoveries that amyloid fibrils are also found in functional biological materials in which the assembly of the fibrils is carefully regulated [5, 6]. These discoveries have been coupled with efforts to design and engineer novel biomaterials inspired by self-assembled amyloid fibrils for applications in biomedicine and materials [7].
Amyloid fibrils adopt a common quaternary structure that has been termed the cross-β fold . Cross-β fibrils are β-sheet assemblies of peptides and proteins (Fig. 8.1) [8]. These one-dimensional assemblies are unbranched fibrils that are typically ~10 nanometers in width and up to tens or hundreds of micrometers in length. The constituent peptides or proteins adopt parallel or antiparallel β-sheet configurations in which there is an intermolecular hydrogen bond network between the amide backbone entities of neighboring peptides that stabilizes the assembled system. The amide and carbonyl groups are oriented parallel to the fibril axis, which enables the formation of this hydrogen bond network. The side chain groups of the constituent peptides are perpendicular to the fibril axis (Fig. 8.1b), hence the “cross-β” designation. Side chain groups further stabilize the self-assembled fibrils through hydrophobic, Van der Waals, aromatic π-π, and charge-charge interactions. The fibril-forming proteins associated with the known amyloid disorders have no significant sequence or length homology, which has given rise to the hypothesis that all proteins and peptides are capable of forming amyloid fibrils under appropriate concentration and solvent conditions of pH and ionic strength. A great variety of protein and peptide sequences have been demonstrated to form amyloid fibrils, and these fibrils share the common features of the cross-β structure even while the specific structural elements (parallel versus antiparallel β-sheet, length of β-sheet element, the presence/absence of loop structures, etc.) of a given fibril are unique to that sequence. All amyloid fibrils exhibit an X-ray scattering pattern with scattering intensities of 4.8 Å and ~ 10 Å, corresponding to the backbone peptide-peptide distances between hydrogen bonded β-sheets and the intersheet distance between laminated β-sheets, respectively [9]. In addition, amyloid fibrils commonly bind histological staining agents, including Congo red, and exhibit a characteristic red-green birefringence when viewed under polarized light [10, 11].

Illustration of a representative structure of amyloid fibrils of the amyloid-β 1–42 peptide (Aβ42). The N-terminal fragment comprising residues 1–16 is not shown. (a) Five molecules of Aβ42 (shown in red, green, blue, yellow, and magenta) stacked in an amyloid cross-β fibril with the fibril axis indicated. An extensive intermolecular hydrogen bond network composed of the amide groups, which are oriented parallel to the fibril axis, is formed between neighboring peptides. (b) A view of the Aβ42 fibril segment from panel A that has been rotated 90°. This view illustrates the β-sheet-rich structure of these amyloid fibrils, which feature a parallel alignment with a sheet-loop-sheet structure of the constituent peptides. The alignment of the side chain groups perpendicular to the fibril axis becomes readily apparent in this view. This structural model of a segment of Aβ42 fibrils was reproduced from the Protein Data Bank structure reported in Lührs, T.; Ritter, C.; Adrian, M.; Riek-Lohner, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. Proc. Natl. Acad. Sci. USA 2005, 102, 17,342–17,347 (PDB ID Code 2BEG, DOI: https://doi.org/10.2210/pdb2BEG/pdb)
Researchers have committed significant effort to understanding cross-β amyloid fibrils and related biomaterials [9]. Cross-β fibrils have proven to be challenging subjects of study. This is due, in part, to the physical characteristics of cross-β fibrils, which are often insoluble and can be found as part of complex heterogeneous mixtures of subtly different fibril morphologies as well as prefibrillar aggregates. The inherent cytotoxicity of these aggregates has presented challenges to understanding their functional properties. Engineered cross-β biomaterials must be nontoxic, which has placed increased urgency on the need to understand the interactions of cross-β fibrils and their prefibrillar aggregates with cells. Structural understanding has been complicated by the non-crystalline nature of most cross-β fibrils as well as the large molecular mass of these aggregates, which make high-resolution crystallography and solution-state NMR problematic for structural determination. Thus, the development of methods for the characterization of cross-β assemblies has been critical to provide deeper insight into these materials [12, 13]. The objective of this chapter is to present an introduction to current methods used to characterize cross-β fibrils. These methods are divided into those used for interrogation of the self-assembly mechanisms (kinetics and thermodynamics), molecular structure, and the emergent properties of cross-β fibrils.
2 Mechanisms of Cross-β Self-Assembly
2.1 General Mechanistic Considerations
The self-assembly of amyloidogenic peptides into cross-β fibrils typically proceeds in a three-step mechanism (Fig. 8.2) [14]. The first step, nucleation, is often referred to as the “lag phase.” Nucleation is characterized by an associative collapse of monomeric peptides into oligomeric and prefibrillar aggregates [14]. These oligomers give rise to protofibrils, which have the characteristic β-sheet secondary structure of amyloidogenic peptide assemblies. The elongation phase is the second step of amyloid formation. In the elongation phase , addition of monomeric peptide to protofibril results in maturation of the fibril through a lengthening process. Finally, the third phase is marked by a saturation plateau where the monomers in solution have reached equilibrium with the fibrils. The kinetics of amyloid formation typically display a sigmoidal shaped curve in thioflavin T (ThT) assays (discussed in Sect. 2.2). The nucleation phase can be bypassed by “seeding,” where preformed fibrils are added to monomer solutions acting as a template for immediate elongation without the characteristic lag phase, indicating that nucleation is a kinetically slow step in cross-β self-assembly [15]. The aggregation process occurs over a variable time range that is dependent on both peptide/protein identity and environment. Secondary structural conformation changes can occur on the order of milliseconds, while the formation of fibrillar aggregates that are observable to the naked eye can occur on the order of days [4]. Characterization of the kinetic and thermodynamic parameters of cross-β assembly is critical in building mechanistic models of self-assembly [16,17,18]. Methods that are used to interrogate the kinetics and thermodynamics of cross-β assembly are outlined in the following subsections.

A kinetic depiction of the self-assembly of cross-β fibrils showing the initial aggregation into prefibrillar oligomeric aggregates followed by conversion to one-dimensional amyloid fibrils. The initial nucleation of peptides and proteins into oligomers occurs during the “lag phase” which is followed by the exponential elongation of protofibrils into mature cross-β fibrils. These types of kinetics experiments are often conducted using thioflavin T (ThT) as a fluorescent reporter of fibril formation (see Sect. 2.2)
2.2 Fluorescent Reporters of Cross-β Assembly, Including ThT
The kinetics of cross-β amyloid self-assembly are commonly characterized by a thioflavin T (ThT) fluorescence assay [19]. ThT (Fig. 8.3) is a fluorescent dye that has been used for histological staining. ThT has also been useful in studying amyloid assembly since it exhibits a shift in the fluorescence emission spectrum from 510 nm to 480 nm upon binding to β-sheet rich structures, producing an observable change that can be used to conveniently monitor fibril formation as a function of time (Fig. 8.2) [19]. To monitor fibrillization, either the self-assembling peptide is incubated with ThT and the fluorescence signal is read continuously or aliquots of the fibril forming solution are taken at fixed time points and diluted into a ThT-containing solution, and the fluorescence of the resulting solution is recorded. Assuming the concentration of ThT is constant, the fluorescence signal intensity at 480 nm is directly related to the fibril concentration. ThT does not typically bind to prefibrillar oligomeric aggregates; thus this assay reports directly on the degree of fibril formation. As shown in Fig. 8.2, ThT binding assays typically exhibit a sigmoidal curve shape that represents the initial nucleation step as a lag phase, followed by the exponential elongation phase and reaching a final saturation of ThT fluorescence as peptide/protein monomer and fibril reach equilibrium.

Chemical structure of thioflavin T (ThT)
There are some caveats to the use of ThT fluorescence to monitor amyloid self-assembly. First, ThT fluorescence can be induced by binding to molecules other than peptide/protein β-sheets, including DNA or cyclodextrin. ThT fluorescence is thus most useful for in vitro experiments where all components of the mixture can be carefully controlled and quantified. Additionally, ThT fluorescence varies depending on peptide identity and solution pH, ionic strength, and viscosity. This complicates the use of ThT fluorescence for the comparative quantification of absolute fibril concentrations for peptides and proteins with different sequences. Despite these limitations, ThT fluorescence is accepted as a robust tool for the characterization of cross-β self-assembly. While ThT is the most commonly used reporter for fibril self-assembly, others have been developed that have also found utility for the kinetic analysis of self-assembly processes [11].
2.3 Turbidity
Turbidity , a measurement of the amount of light scattered by a given material, is another common method for probing the kinetics of cross-β fibril formation [15]. In this technique, samples are analyzed by UV-Vis spectroscopy, and the degree to which light is scattered by fibrils and other aggregates in solution is quantified [20]. Typically, samples are analyzed at 350–450 nm, where the fibrils do not absorb, so that any changes in transmittance can be attributed to light scattering effects [16]. The main advantages of turbidity measurements for interrogating peptide self-assembly kinetics include simplicity of application, since sample modification or the addition of external reagents is not required, and the high reproducibility of the measurements. However, the sensitivity of this method is low and scattering from the largest insoluble particles dominates the data, which fails to provide a complete picture of all aggregate species in solution. Despite these drawbacks, turbidity measurements are frequently used to characterize the self-assembly kinetics of amyloid systems [21,22,23].
2.4 Infrared Spectroscopy
Fourier-transform infrared spectroscopy (FTIR) has also been used to follow the kinetics of cross-β self-assembly by monitoring the development of the β-sheet structure that is characteristic of these materials as a function of time [24]. Peptides display a prominent amide I band between 1600 and 1700 cm−1 that results from stretching vibrations of the carbonyl group in the peptide backbone [25]. Peptides and proteins lacking a distinct secondary structure have a broad amide I band around 1650 cm−1, while proteins with significant β-sheet secondary structure, such as cross-β fibrils, display a narrower peak from 1620 to 1625 cm−1. It is important to note that the water bending absorption band overlaps with the amide I band, which requires that experiments interrogating peptide/protein secondary structure be carried out in D2O or at a very high peptide concentration. The kinetics of cross-β assembly can be characterized by monitoring the formation of β-sheet structure in the amide I IR region as a function of time. In one illustrative example, the aggregation of Aβ1–28 upon acidification was studied to model how Aβ acts in the acidic lysosomes of cells [26]. In this study, the kinetics of Aβ1–28 aggregation under these conditions was followed by observing an increase in the intensity of the β-sheet band at ~1620 cm−1 accompanied by a decrease in intensity of the random coil band around 1651 cm−1, indicating a loss of random coil structure and transition to β-sheets upon acidification of the environment .
2.5 Circular Dichroism (CD) Spectroscopy
Circular dichroism (CD) is a spectroscopic method commonly used to characterize the secondary structure of peptides and proteins that has also been used to monitor cross-β self-assembly [19]. CD spectra report the differential absorption of left- and right-handed circularly polarized light by chiral molecules [25]. Peptides that lack a clear secondary structure present a very weak CD signal, while β-sheets, α-helices, and random coils present distinct CD spectra that have been used to monitor the formation of β-sheet-rich cross-β structures over time. β-sheets are characterized by a negative CD band at 220 nm. While ThT fluorescence reports selectively on cross-β fibrils without providing information about many prefibrillar aggregates, CD spectra report on formation of β-sheet structure generally in both prefibrillar and fibrillar aggregates [27]. In one example, CD spectroscopy was used to monitor the formation of Aβ fibrils as a function of time in the presence of Cu2+ in low pH environments, where it has been shown that copper ions inhibit β-sheet formation and higher temperatures increase aggregation rates for fibers incubated in the absence of copper ions [27].
2.6 Dynamic Light Scattering (DLS)
Dynamic light scattering (DLS) is another technique that is used to characterize cross-β self-assembly mechanisms by measuring the size of particle aggregates in solution [16]. DLS spectroscopy measures the correlation function of light scattered by particles in solution or suspension which can subsequently be used to determine the translational diffusion coefficient of molecules undergoing Brownian motion to determine the size of particles present [28]. DLS techniques assume that particles are spherical. DLS can therefore be used to monitor amyloid formation as a function of increasing particle size and is especially useful to characterize the early events of self-assembly which includes the formation of nonfibrillar aggregates. Unfortunately, DLS signals are often dominated by the largest particles in solution, making it a low-resolution technique that is often incapable of monitoring monomers or oligomers directly once fibrils begin to appear. Despite these drawbacks, DLS has been useful to interrogate early events in cross-β amyloid aggregation. In one example, DLS was used to measure the kinetics of aggregation of the amyloid-β 40 peptide (Aβ40) at slightly acidic pH values similar to those that would be found in endosomes during cellular update of this peptide [29]. In solutions of Aβ40 under these conditions, DLS was used to detect several early phases of aggregation: an initial rapid burst phase followed by slower phase 1 and phase 2 association phases. In the burst phase, large aggregates with an average hydrodynamic radius (Rh) of 84 nm to 196 nm were observed by DLS in the first 12 minutes of incubation; these aggregates were shown to be amorphous in morphology by correlating atomic force microscopy. The association phases gave rise to smaller spherical aggregates with Rh values that were ~ 36 nm in diameter. This work clearly demonstrates the utility of DLS methods to give insight into early aggregation events in cross-β, specifically showing an initial hydrophobic collapse of peptides into amorphous aggregates followed by reorganization into ordered oligomeric aggregates prior to fibril formation in the case of Aβ40 under these conditions [29].
2.7 Transmission Electron Microscopy (TEM), Atomic Force Microscopy (AFM), and High-Speed AFM (HS-AFM)
A limitation of many of the spectroscopic methods that have been used to study cross-β self-assembly is that they monitor the aggregates that dominate at any given time in the self-assembly process and often fail to capture the complexity of heterogeneous mixtures [30]. They give information about the species that are present in the highest concentration, while species in lower concentrations are overlooked. Microscopic imaging methods, including transmission electron microscopy (TEM) and atomic force microscopy (AFM), have the potential to reflect more accurately the diversity of aggregate species that exist at any given point during self-assembly [30]. This enables deeper mechanistic insight into the heterogeneous structures found in the course of cross-β assembly. TEM and AFM are most often used to structurally characterize the morphology of the aggregates that are formed by peptide self-assembly. Recent advances in high-speed AFM (HS-AFM) have enabled the use of this technique to follow self-assembly over time. For example, HS-AFM has been used to rapidly acquire consecutive images of the elongation of Aβ42 fibrils, enabling the simultaneous observation of structural morphology and elongation kinetics in compiled video files [31, 32].
2.8 Sedimentation Analysis
The thermodynamics of peptide cross-β self-assembly can be quantified using sedimentation analysis [33]. Sedimentation analysis exploits the selective sedimentation of self-assembled aggregates, both cross-β fibrils and prefibrillar aggregates, when subjected to high-speed ultracentrifugation. Under appropriate centrifugal forces, all aggregate structures can be sedimented, leaving monomeric peptides in solution. Quantification of monomeric peptide by high-performance liquid chromatography (HPLC) correlation to a standard curve can be used to extrapolate comparative free energies (ΔΔG°) of self-assembly for specific peptide systems. The concentration of monomer in solution at equilibrium, the critical concentration (Cr), can be related to the equilibrium association constant (Ka) for the addition of a molecule of monomeric peptide to the end of a fibril, which is dependent on both the identity of the peptide and the assembly environment. The association constant, Ka, is the reciprocal of the critical concentration, 1/Cr. The free energy of assembly for that peptide, ΔG°, can be determined from Ka using the standard expression ΔG° = −RT(lnKa). In an application of this method, Nilsson and coworkers have used sedimentation analysis to characterize the relative thermodynamics of nine variants of the amyloid-β 16–22 fragment (Aβ(16–22)) [34, 35]. In the wild-type peptide, residues 19 and 20 are Phe. Variant peptides were prepared in which Phe 19 and 20 were changed to residues of differing hydrophobic and aromatic character in order to understand the possible role of aromatic effects on the thermodynamics of Aβ(16–22) self-assembly. Sedimentation analysis of each of the variant peptides provided ΔΔG° comparisons with the wild-type peptide, giving insight into the thermodynamic role of the Phe side chain groups in promoting self-assembly.
Sedimentation analysis using a range of centrifugal forces has also facilitated insight into the nature of the prefibrillar oligomer species that precede cross-β fibril self-assembly [36]. Prefibrillar oligomers are of interest from both a mechanistic and functional perspective: these oligomers are believed to be the major toxins in many amyloid disorders [37]. Oligomers tend to be soluble aggregates of varying molecular weight. Oligomer species in solution can be separated based on size using a range of centrifugal conditions. Low-speed centrifugation for short time periods can be used to selectively sediment insoluble fibrils and larger aggregates. High-speed centrifugation for longer periods of time can separate smaller aggregate species. Optical density, fluorescence, or related analytical methods can be used to determine sedimentation coefficients for aggregates of varying sizes that can then be used to obtain accurate molecular weights for these aggregates. There are two main techniques in analytical ultracentrifugation: sedimentation equilibrium and sedimentation velocity [38]. Sedimentation equilibrium experiments use short solution columns and low centrifugal speeds to bring the system to equilibrium. This technique has been used to characterize the subunits that form fibrils; however, it is not useful for characterizing fibrils themselves as they are too large to form equilibrium gradients and simply sediment to the vessel floor. Sedimentation velocity is thus used to study the larger fibrils by using hydrodynamic theory to extrapolate the size and shape of molecules moving through a strong centrifugal field [39]. The insight these techniques provide on the size and range of aggregates that exist during cross-β self-assembly is a critical element to the mechanistic puzzle for these processes .
2.9 Electrospray Ionization-Ion Mobility-Mass Spectrometry (ESI-IMS-MS)
Electrospray ionization-ion mobility-mass spectrometry (ESI-IMS-MS) has also been used for characterizing the heterogeneous aggregates generated during cross-β fibrillization to gain understanding of the nature of prefibrillar oligomer species [40]. This technique separates ions based on their shape and/or charge and is therefore capable of separating multiple conformations and/or aggregates of a peptide that have the same m/z ratio. This is significant since many proteins undergo initial misfolding prior to aggregation and the initial oligomeric aggregates are of varying molecular weight and conformation. Prefibrillar oligomers also rapidly interconvert, complicating identification. ESI-IMS-MS has been successfully used to detect distinct conformers of cross-β-forming proteins as well as early aggregation intermediates. In one illustrative example, ESI-IMS-MS analysis of the islet amyloid polypeptide (IAPP) revealed the early formation of β-hairpin conformations, which were hypothesized to be precursors to oligomer aggregation [41, 42]. These and other studies exploiting this technique have provided critical glimpses of elusive early events in cross-β assembly [40].
2.10 Quartz Crystal Microbalance (QCM) Analysis
Quartz crystal microbalance (QCM) methods have been used to characterize the enthalpic and entropic contributions to the overall free energy barriers of fibril elongation [43]. QCM techniques monitor the elongation of fibril seeds that have been deposited on the sensor surface by detecting the change in frequency of the quartz crystal resonator as the hydrodynamic mass of the aggregates increases. This takes advantage of “seeded” elongation in which the addition of fibrils to solutions of monomeric peptide results in immediate elongation of fibrils by monomer without any nucleation phase. This rate of accelerated seeded addition of monomer to fibril corresponds to diffusional motion over a single free energy barrier without any intermediate species. Thus, the rate of change of the hydrodynamic mass of fibrils in QCM experiments is proportional to the rate of fibril elongation. QCM analysis of ten amyloid systems at variable temperature was performed by Buell et al. to derive enthalpic (ΔH≠) and entropic (ΔS≠) contributions to the free energy transition state barriers for elongation of each of these systems [43]. It was found that the enthalpic contributions to these barriers were unfavorable while the entropic contributions were favorable and that these values correspond to peptide/protein characteristics of size and initial folded structure. QCM analysis has been an insightful tool for examining the specific energetic barriers that contribute to the kinetics of amyloid elongation .
2.11 Surface Plasmon Resonance (SPR)
Surface plasmon resonance (SPR) is another biosensor technique that has been used in the analysis of cross-β self-assembly [44]. In SPR analysis of amyloid self-assembly, fibril seed structures have been immobilized to the metal sensor surface, and changes to this surface as monomer is flowed over can be monitored to interrogate fibril elongation as a function of monomer adsorption to the immobilized fibril seed. This analysis facilitates characterization of the kinetics of seeded fibril elongation. Monomeric amyloid peptide has also been immobilized to the sensor surface for the analysis of nucleation kinetics of unseeded aggregation processes [44]. In one example of the use of SPR to assess the kinetics and thermodynamics of fibril extension and dissociation reactions, Aβ fibrils were immobilized on the sensor [45]. The rate of extension by addition of monomer to the fibril ends was measured by flowing monomeric Aβ over the immobilized fibrils. The rate of monomer dissociation from the fibril ends was measured by flowing buffered water over the immobilized fibrils. The rates of the association and dissociation reactions were both found to be proportional to the concentration of Aβ and the initial density of fibers immobilized on the surface of the sensor, consistent with a first-order kinetic model. These experiments also enabled determinations of critical monomer concentrations at equilibrium, which were used to characterize the monomer-fibril association constant, K. SPR was thus demonstrated to be a useful tool for the interrogation of both kinetic and thermodynamic parameters for important cross-β self-assembly processes .
2.12 Isothermal Titration Calorimetry (ITC) and Differential Scanning Calorimetry (DSC)
Isothermal titration calorimetry (ITC) has been used to interrogate the thermodynamics of cross-β peptide and protein self-assembly [46]. ITC can be used to characterize enthalpic and entropic contributions to molecular processes by precisely measuring the heat released to or absorbed from the environment during exothermic and endothermic processes, respectively. These measurements can be used to extrapolate enthalpic and entropic contributions to chemical processes, including peptide self-assembly. Swanekamp et al. have used ITC to characterize the relative enthalpies of the self-assembly of pleated β-sheet nanofibrils and coassembly of enantiomeric peptides into rippled β-sheet nanofibrils [47]. L-Ac-(FKFE)2-NH2 has been shown to self-assemble into pleated β-sheet nanoribbons, while equimolar mixtures of L- and D-Ac-(FKFE)2-NH2 coassembled into rippled β-sheet fibrils with an alternating arrangement of L/D peptides in the fibrils. ITC was used to compare the enthalpies of these reactions by enforcing coassembly through alteration of the charge of the peptides. Specifically, the L-Ac-(FEFE)2-NH2 peptide and L and D-Ac-(FKFK)2-NH2 peptides were prepared; at neutral pH these peptides do not effectively self-assemble due to charge repulsion, but they readily coassemble with complementary charged partners. The ITC experiments were conducted by titrating solutions of either L or D-Ac-(FKFK)2-NH2 peptides into a solution of the L-Ac-(FEFE)2-NH2 peptide and measuring the heat of reaction. The enthalpy for both the L/L and L/D mixtures was favorable, with the L/D mixture showing an enthalpic advantage over the L/L mixture. ITC provided critical insight into the thermodynamic parameters that govern the respective formation of rippled and pleated β-sheet assembly.
Differential scanning calorimetry (DSC) is another tool that has been used to study the thermodynamics of amyloid fibril formation [48]. In this technique, the peptide sample is loaded into the sample cell, and an equal volume of solvent is loaded into the reference cell. As both cells are heated at the same rate, the additional release or absorbance of heat from the sample cell is quantified and compensated for by a feedback heater, giving information about the heat capacity difference between the two cells. DSC analysis of cross-β amyloid materials has given interesting and variable results that depend on both the peptide/protein that comprises the amyloid fibrils and the conditions. Some fibrils (N47A Spc-SH3 domain and WL fibrils of β2-microglobulin (β2-m)) exhibit cooperative thermal transitions that are characteristic of fibril melting. Other fibrils (β2-m, HEWL, and Aβ) show transformation of the fibrils due to presumed transient inter-fibrillar association during heating. These variable outcomes have made DSC a useful method for interrogation of the complex thermodynamics at play in the formation and stability of self-assembled cross-β fibrils .
2.13 In Silico Simulations
Computer modeling and simulation provide critical tools to complement experimental approaches for understanding the fundamental physicochemical parameters that underlie cross-β self-assembly processes [49]. These efforts include the development of computational algorithms to predict the propensity of peptides and proteins to undergo cross-β assembly based on amino acid sequence [50,51,52,53]. These models are based on both phenomenological observations and fundamental physicochemical properties, including hydrophobicity and β-sheet propensity. Simulation approaches have also been harnessed to understand early conformational changes in cross-β assembly as well as the key mechanistic events that occur during nucleation and elongation. Challenges associated with the application of classical molecular dynamics simulations models to peptide/protein self-assembly include managing the high atom count of cross-β systems and achieving meaningful simulations that encompass useful time scales [54]. The use of coarse-grained models, which retain only the necessary information about shape and average interactions, has helped to bridge this gap. For example, coarse-grained computer simulations have accurately replicated the two-step mechanism that includes initial formation of prefibrillar oligomers which convert into β-sheet-rich aggregates that can seed elongation [55, 56]. A detailed treatment of the use of computational simulations to gain insight into cross-β self-assembly processes is beyond the scope of this review. However, the experimental complexities of amyloid self-assembly processes have made in silico approaches of critical importance to understanding the fundamental principles that govern cross-β formation.
3 Structural Characterization of Cross-β Nanomaterials
3.1 Introduction
Structure and function are indelibly connected in biological systems, making interrogation of the structure of cross-β peptide nanomaterials a subject of intense interest. Understanding of the cross-β fold has progressed from the initial recognition that amyloid aggregates are ordered, as evidenced by their Congo red binding properties, to the elucidation of atom-level packing models of cross-β systems [57,58,59]. Techniques that are used to probe the architecture of self-assembled cross-β fibrils and their prefibrillar predecessors range from bulk scale structure analysis by microscopic imaging methods to spectroscopic methods that can provide increasing levels of structural understanding. Spectroscopic methods have further enabled the identification of structural information ranging from secondary structure motifs found in aggregates to β-sheet packing alignment to all-atom models of the aggregates. Structure determination of cross-β amyloid presents significant challenges. Cross-β fibrils tend to be non-crystalline, which complicates the use of high-resolution X-ray diffraction methods. In addition, the high molecular weight of these aggregates likewise complicates the use of solution-phase NMR methods to determine structure. Finally, metastable prefibrillar aggregates that give rise to cross-β structures tend to exist in complex heterogeneous mixtures, which has made interrogation of oligomer structures exceedingly challenging. Innovation in solid-state NMR and electron microscopy have had a paradigm-shifting impact on structural understanding of cross-β nanomaterials. This section will introduce experimental methods that are commonly used to characterize the hierarchical structure of assembled cross-β aggregates, beginning with low-resolution techniques and proceeding to high-resolution methods.
3.2 Circular Dichroism
Circular dichroism (CD) spectroscopy (see Sect. 2.5) is a chiral UV method that provides information about the secondary structure of peptides and proteins [60]. CD spectroscopy can differentiate between secondary structures in chiral protein and peptide systems based on the variable absorption patterns of left-handed and right-handed circularly polarized light. The three-dimensional shape of an asymmetric peptide or protein will differentially absorb left- or right-handed polarized light; the difference of the two spectra provides a characteristic CD spectrum that can assist in the identification of α-helical, β-sheet, and disordered structures (Fig. 8.4). CD spectra of β-sheet motifs exhibit a minimum between 215 and 222 nm [19]. CD is an excellent technique to rapidly determine the secondary structure of assembled peptides and to monitor structural changes in different environmental conditions. CD spectroscopy is a low-resolution technique; however, cross-β systems will exhibit distinct β-sheet character in CD spectra. While it can confirm that self-assembled fibrils are rich in β-sheet secondary structure, CD techniques alone cannot be used as absolute confirmation of a cross-β arrangement in self-assembled nanomaterials. Additional techniques, including TEM or AFM microscopy, must be used in conjunction with CD spectroscopy to provide confirmation of cross-β architecture. CD spectroscopy also has significant drawbacks specifically for the analysis of cross-β assemblies. Many amyloid materials precipitate upon assembly and are high in molecular weight; both of these characteristics can lead to complicated light scattering effects in CD analyses. In spite of these limitations, CD still has an important place among the spectroscopic tools used to assist in the structural characterization of cross-β amyloid assemblies.

Representative CD spectra for α-helical, β-sheet, collagen triple helix, and disordered peptide structures. Adapted with permission from Springer Nature, Greenfield N (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1 (6):2876–2890. https://doi.org/10.1038/nprot.2006.202 (reference [60]), Copyright 2007, Nature Publishing Group
3.3 Vibrational Spectroscopy
3.3.1 Infrared (IR) Spectroscopy
Fourier transform infrared spectroscopy (FTIR) is a vibrational spectroscopy technique that is used to confirm the secondary structure of cross-β aggregates from characteristic molecular vibrations in the amide backbone [61]. Molecular vibrational modes in IR spectroscopy are excited by irradiation with infrared light. IR vibrational transitions in the amide I region, from ~1600 to 1800 cm−1, are distinct for peptides and proteins that adopt α-helical, β-sheet, and disordered structures. IR spectroscopy can be conveniently applied to both solution-phase and solid-phase samples, a significant advantage over CD spectroscopy for the routine analysis of β-sheet assemblies. Cross-β fibrils typically display an amide I stretch between 1615 and 1630 cm−1, consistent with β-sheet secondary structure (Fig. 8.5a). In addition, empirical observations suggest that the parallel or antiparallel orientation of peptide strands in cross-β fibrils can be determined using IR spectroscopy [62]. Antiparallel strands have been shown to display a weak transition between 1670 and 1680 cm−1. This band transition is often not observed in parallel β-sheet structures. Standard IR spectroscopy is similar to CD spectroscopy in that it is a low-resolution spectroscopic method that is primarily useful for corroborating that fibrillar assemblies are rich in β-sheet secondary structure .

(a) Fourier transform infrared spectra overlays of self-assembled l- and d-Aβ(16–22), coassembled l/d-Aβ(16–22), 1-13C labeled l-Aβ(16–22) (labels at Leu 17 and Phe 20), and coassembled 1-13C labeled l-Aβ(16–22) with unlabeled d-Aβ(16–22). These spectra illustrate typical β-sheet amide I signatures for cross-β fibrils (green, orange, and purple lines). In addition, coupling effects due to selective incorporation of 1-13C carbonyl labels are shown (black and blue lines). (b) A structural model for β-sheet strand alignment in putative pleated β-sheets of self-assembled l-Aβ(16–22) with 1-13C labeled positions highlighted in red. The close proximity of the 1-13C labels across strand leads to coupling as evidenced by the line splitting of the major amide I stretch at ~1620 cm−1. (c) A proposed structural model of β-sheet alignment in coassembled l/d-Aβ(16–22) rippled β-sheets. Reproduced with permission from Urban JM, Ho J, Piester G, Fu R, Nilsson BL (2019) Rippled β-Sheet Formation by an Amyloid-β Fragment Indicates Expanded Scope of Sequence Space for Enantiomeric β-Sheet Peptide Coassembly. Molecules 24:1983. https://doi.org/10.3390/molecules24101983 (reference [63]) Copyright 2019 the authors, some rights reserved; exclusive licensee MDPI. Distributed under a Creative Commons Attribution License 4.0 (CC BY) https://creativecommons.org/licenses/by/4.0/
Isotope-edited FTIR spectroscopy (IE-IR) is a technique that provides additional information about strand alignment within β-sheet assemblies [64, 65]. IE-IR requires the incorporation of stable-isotope 1-13C carbonyl labels at selected amino acids in cross-β self-assembling peptides. Incorporating an isotopically labeled carbonyl results in a redshift in vibrational frequency due to the lower vibrational frequency of the heavier 13C isotope. This will lead to band separation of the amide I stretch due to the presence of the labeled carbonyl atom (see Fig. 8.5a for examples of this line splitting). When two isotopically labeled carbonyls are organized directly cross-strand from each other, these carbonyls undergo vibrational coupling, leading to changes in the line splitting patterns relative to uncoupled carbonyl groups (Fig. 8.5a). Strategic incorporation of 1-13C labels can thus be used to characterize strand registry within the cross-β packing structure, a significant enhancement in resolution of structural information over standard FTIR spectroscopy.
In a recent example, IE-IR spectroscopy was applied to the characterization of cross-β systems derived from enantiomers of the Aβ(16–22) peptide and mixtures thereof to compare self-assembly of single enantiomer solutions into pleated β-sheet fibrils and coassembly of equimolar mixtures of L- and D-peptides into two-component rippled β-sheet fibrils [63]. L-Aβ(16–22) was prepared with 1-13C labels incorporated at Leu17 and Phe21, which are expected to be directly aligned in pleated β-sheet assemblies of this peptide (Fig. 8.5b). The IR spectra of the unlabeled L- and D-Aβ(16–22) peptides exhibited typical antiparallel β-sheet spectra with an amide I stretch at ~1620 cm−1 and a weak stretch at ~1690 cm−1 (Fig. 8.5a). The IE-IR spectra of the Leu17/Phe20 1-13C labeled L-Aβ(16–22) peptide displayed a separated amide I band with absorbances at 1598 cm−1 and 1641 cm−1, consistent with a cross-strand coupling interaction between the labeled carbonyl groups. This labeled peptide was then mixed with an equimolar amount of unlabeled D-Aβ(16–22). Mixtures of enantiomeric β-sheet peptides have been predicted to coassemble into so-called “rippled” β-sheet assemblies with alternating arrangements of the L- and D-peptides [66]. In this case, the mixture of the unlabeled D-Aβ(16–22) peptide with the Leu17/Phe20 1-13C labeled L-Aβ(16–22) peptide should provide a strand orientation in which the alternating L/D pattern interferes with the cross-strand coupling of the labeled carbonyl groups (Fig. 8.5c). Indeed, the splitting pattern was altered in this mixture of peptides, with the IR spectrum showing unique absorbances at 1638 cm−1 and 1608 cm−1, consistent with coassembly into L/D patterned rippled β-sheet assembly.
Two-dimensional infrared (2D-IR) spectroscopy is a further refinement of IR analysis that has been used to characterize cross-β assembly [67]. A beneficial aspect of 2D IR spectroscopy is the nonlinearity of the signal strength. Signals in 1D IR spectroscopy are scaled linearly in relation to the concentration and molar absorptivity of the analyte molecule, whereas 2D IR signals are scaled linearly in relation to concentration and quadratically in relation to molar absorptivity. This difference provides sharper spectra and greater sensitivity to secondary structure, and inaccurate baselines from weak absorbers are eliminated. Additional information provided by 2D-IR spectroscopy in the analysis of protein secondary structure includes anharmonic shifts, 2D line shapes, lifetimes, and vibrational dynamics. In a recent application, Zanni and coworkers have leveraged 2D-IR techniques to structurally identify cataract-causing cross-β aggregates [68]. The identification of cross-β in the eye lens using FTIR has been challenging. However, by analyzing 2D-IR spectral features, including diagonal frequency, anharmonic shift, and shifted cross-peak frequencies, Zanni and coworkers were able to differentiate between native β-sheets in juvenile lenses that do not contain cataracts and amyloid cross-β structures in 65-year-old lenses that do contain cataracts. Thus, 2D-IR has the potential to be leveraged in situ for the identification of amyloid in tissues. As with FTIR, 2D-IR provides a higher level of structural detail in cross-β assemblies, enabling the elucidation of strand alignment of peptides within the cross-β fibrils.
3.3.2 Raman Spectroscopy
Raman spectroscopy is an additional form of vibrational spectroscopy used to interrogate the structure of self-assembled cross-β materials [69]. These techniques include normal Raman (NR), deep UV Raman (DUVRR), surface enhanced Raman, and tip-enhanced Raman spectroscopy (TERS), which rely on the Raman effect to obtain structural information. The Raman effect is based on a vibrational frequency difference between the incident and scattered light when a molecule is irradiated with electromagnetic radiation. The spectrum created by Raman spectroscopy is influenced by vibrational modes from the peptide backbone, and aromatic and non-aromatic amino acid side chains, which provides structural information. These techniques can be used to determine the core structure of fibrils, the local environment around aromatic amino acids, the surface organization of amyloid fibrils, and nanoscale structural organization of fibrils. The amide I band from 1600 to 1700 cm−1, the amide II band at ~1550 cm−1, and the amide III band from 1200 to 1340 cm−1 provide information on secondary structure, hydrogen bonding interactions, and aromatic and sulfur side chain environment in the various forms of Raman spectroscopy.
Recently, TERS was used to analyze the differences in structure of assemblies of wild-type Aβ42, the L34T Aβ42 variant that has been shown to form less toxic fibrils, and the G37C Aβ42 variant that gives rise to highly toxic oligomers [70]. TERS exploits atomic force microscopy methodology to obtain structural information of cross-β materials at the single fibril level. In this study, TERS was used to determine that wild-type Aβ42 and the L34T variant assembled into parallel β-sheets while the assemblies formed by the G37C Aβ42 variant were organized into antiparallel β-sheets. This is an interesting demonstration of Raman capability that shows the enhanced range of analysis of Raman compared to closely related IR techniques. Similar to IR methods, Raman spectroscopy can rapidly provide higher-level structural analysis of cross-β materials but falls short of providing atomic-level structural detail .
4 Solid-State NMR (SSNMR)
NMR spectroscopy is an emerging tool to provide detailed structural constraints of self-assembled cross-β systems that can enable the development of molecular models of higher resolution and accuracy [71]. There are several NMR techniques that have been used to help characterize cross-β peptide assemblies. Solution-state NMR experiments have provided structural insight relevant to early events in self-assembly, including structural transitions and early interactions in the self-assembly process [72]. However, solution-state NMR is of limited use in the structural analysis of high molecular weight peptide and protein cross-β aggregates. The high molecular weight and frequent insolubility of cross-β assemblies result in significant line-broadening effects due to anisotropy, which makes standard solution-state NMR unsuitable for the acquisition of correlative data for the construction of higher-resolution cross-β structural models.
Solid-state NMR (SSNMR) spectroscopy has emerged as a critical higher-resolution tool for the resolution characterization of self-assembled cross-β nanomaterials [72, 73]. Magic angle spinning SSNMR techniques are well-suited for the structural interrogation of non-crystalline solids. SSNMR methods exploit pulse sequences that enable the accurate measurement of site-specific intermolecular distances within cross-β assemblies, often between amino acids that are selectively labeled with stable isotopes (including 13C and 15N). These experimental distance measurements have been effectively used as constraints to build accurate high-resolution structural models of cross-β fibrils [74]. SSNMR spectroscopy studies have been critical in the establishment of the fundamental principles of amyloid structure. Figure 8.6 depicts several cross-β fibril structures that have been solved using SSNMR. Early pioneering work by Tycko and others [75, 76] was critical in the development of SSNMR for cross-β structure analysis. In the last two decades, SSNMR has become more broadly applied in this field.

Structural models of cross-β amyloid fibrils developed using correlative solid-state NMR methods. (a) HET-s(218–289) prion fibrils, (b) Aβ1–40 fibrils in Alzheimer’s disease brain tissue, (c) Aβ1–40 D23N “Iowa” mutant fibrils, (d) Aβ1–40 E22D “Osaka” mutant fibrils; E. Humana-synuclein fibrils. Reprinted from Loquet A, El Mammeri N, Stanek J, Berbon M, Bardiaux B., Pintacuda G., Habenstein B. (2018) 3D structure determination of amyloid fibrils using solid-state NMR spectroscopy. Methods 138–139:26–38. https://doi.org/10.1016/j.ymeth.2018.03.014 (reference [74]), Copyright 2018, with permission from Elsevier
5 Diffraction Techniques
X-ray diffraction (XRD) and related fiber diffraction techniques have been used in the structural analysis of cross-β aggregates for over 50 years [77]. X-ray fiber diffraction of cross-β fibrils reveals meridional reflections at 4.7 Å indicative of the β-strand structural repeat parallel to the fibril axis and equatorial reflections at ~10 Å indicative of β-sheet spacing perpendicular to the fibril axis. This characteristic scattering pattern is a fundamental diagnostic confirmation for cross-β assemblies. Wide-angle X-ray scattering (WAXS) is now complemented by small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS), which have been used to reveal higher-order structural features of self-assembled cross-β fibrils [78]. For example, Lynn and coworkers exploited small-angle scattering in the characterization of unique cross-β assemblies formed by the Aβ(16–22) peptide under acidic conditions in mixed water/acetonitrile solvents [79]. Under these assembly conditions, Aβ(16–22) was found to assemble into cross-β nanotubes with tube diameters of ~82 nm reported by electron microscopy measurements. In contrast, SANS and SAXS measurements revealed a more accurate diameter of 52 nm. The higher accuracy of the small-angle scattering experiments is due to the ability to perform these experiments in situ without the need to process the samples. The wider apparent diameter of the tubes in microscopic images is due to collapse of the tubes after deposition and drying on surfaces. The scattering data in these experiments also revealed that the inner and outer walls of the hollow nanotubes had a thickness of 4 nm. Thus, wide and small angle diffraction experiments have proven useful in the determination of higher-order structural elements of cross-β assemblies.
X-ray crystallography has also been applied as a high-resolution technique to visualize the atomic-level packing architecture of crystallized cross-β assemblies [80]. High-resolution crystallography requires crystalline samples, presenting a significant barrier for the use of this methodology in the examination of assembled cross-β fibrils, which tend to be non-crystalline. Nonetheless, some cross-β systems have been shown to form microcrystals that are closely related to their fibrillar polymorphs [59]. In an early example, Eisenberg and coworkers successfully crystallized a fragment of Sup35, GNNQQNY, and were able to use focused X-rays from a synchrotron source to elucidate the packing structure of this amyloid system [81]. In another recent example, Eisenberg and Bowers utilized X-ray microdiffraction crystallography to visualize the molecular packing structures of two amyloid pentapeptides derived from [Leu5]-enkephalin and determined the factors that caused differences in the mode of cross-β assembly for these short peptides [82].
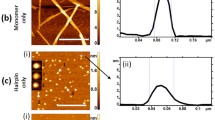
Nowick and coworkers have designed a strategy to template amyloid-forming peptides as macrocyclic β-hairpins, facilitating self-assembly into discrete crystals that have been instructive in understanding the structure of cross-β systems [83]. In an example of this strategy, Nowick and coworkers engineered a derived macrocyclic β-hairpin peptide derived from the Aβ 16–36 fragment to facilitate crystallization (Fig. 8.7a and b) [84]. Crystallization was aided by connection of the N- and C-termini of Aβ(16–36) with a δ-linked ornithine turn unit and incorporation of a cross-strand disulfide bond that replaced Ala21 and Ile31, which stabilized the β-hairpin. In addition, an N-methyl group on Gly33 was introduced to prevent uncontrolled aggregation, further facilitating crystallization. High-resolution X-ray diffraction crystallography revealed the atomic structure of these aggregates, giving insight into possible structures relevant to amyloid prefibrillar oligomers. These methods have been applied to a number of other systems with great success [83]. Thus, insightful engineering of self-assembling peptides along with advances in X-ray diffraction methodology has provided important structural understanding of both cross-β fibrils and their prefibrillar aggregates .

(a) Chemical structure of a putative hairpin structure formed by the Aβ16–36 fragment. (b) Chemical structure of peptide 1, a hairpin of the Aβ16–36 fragment stabilized by peptide macrocyclization by amide bond formation between the δ-amine of Lys 16 and the C-terminus of Val 36. (c) X-ray crystallographic structure of a representative β-hairpin monomer formed by peptide 1 (PDB 6WXM). (d) An overlay of the 11 peptide 1 β-hairpins in the asymmetric unit of the crystallographic structure that illustrates the conformational regularity of each hairpin in the crystal and shows the variability in the loop region. Reprinted with permission from Kreutzer AG, Samdin TD, Guaglianone G, Spencer RK, Nowick JS (2020) X-ray Crystallography Reveals Parallel and Antiparallel β-Sheet Dimers of a β-Hairpin Derived from Aβ16–36 that Assemble to Form Different Tetramers. ACS Chem Neurosci 11 (15):2340–2347. https://doi.org/10.1021/acschemneuro.0cc00290 (reference [84]), Copyright 2020 American Chemical Society
6 Electron Microscopy
Electron microscopy (EM) and atomic force microscopy (AFM) have long been employed in the structural analysis of cross-β amyloid aggregates [85, 32]. Negative stain transmission electron microscopy (TEM) and cryo-EM are fundamental techniques used to observe the physical morphology and geometric parameters of peptide assemblies, including length, width, pitch, etc. Although negative stain TEM provides nanometer-scale images of cross-β materials, it cannot provide information on atomic-scale structure. TEM requires staining with heavy metal salts such as uranyl acetate for increased resolution, and samples need to be deposited onto a surface for imaging. These events can alter the structure of the peptide assembly, sometimes providing an inaccurate representation of the aggregate. AFM often visualizes cross-β materials in a dried state deposited onto a surface, making this technique vulnerable to the same limitations as negative stain TEM. AFM is also possible in solution conditions, which has enabled real-time visualization of nanofiber formation (Sect. 2.7). When imaging low-contrast amyloid fibrils, scanning transmission electron microscopy (STEM) is a powerful method to visualize amyloid structure [86]. STEM is able to directly measure the mass-per-length ratio of individual fibers providing substantial information that can assist in creating a molecular model.
Cryo-EM overcomes many of the disadvantages of other microscopy methods for the analysis of cross-β fibrils [87]. TEM, STEM, and AFM require sample fixation onto flat surfaces and staining, both of which can alter the native structure of an assembly. However, cryo-EM thermally fixates the sample in a thin layer of its original solvent, eliminating problems with sample alteration common to other microscopy methods. In addition, recent advances in cryo-EM data acquisition and processing provide data of sufficient resolution to enable reconstruction of high-resolution packing architectures of cross-β aggregates [88]. Cryo-EM model reconstruction is done from single fibril aggregate analysis at long length scales compared to SSNMR methods, which rely on highly local distance constraints to build models. Cryo-EM has become a transformative tool for structural interrogation of self-assembled cross-β materials, dramatically accelerating the process of generating atomic resolution models of these assemblies.
In a recent example, cryo-EM was used to determine the molecular structure of polymorphic Aβ fibrils obtained from Alzheimer’s brain [89]. Cryo-EM images of these patient-derived fibrils clearly show the polymorphic characteristics of Aβ fibrils (Fig. 8.8a). Electron diffraction data obtained directly from these images revealed symmetry elements and enabled the construction of a high-resolution three-dimensional structure consistent with this data to a resolution of 4.5 Å between the reconstructed map and the model. The constructed map shows two distinct stacks of β-sheets (Fig. 8.8b-e). The model provided atomic-level resolution and refined structural information enabling most of the structural characteristics of brain-derived Aβ fibrils and their polymorphs to be determined. Cryo-EM has the potential to surpass traditional microscopic and spectroscopic techniques with ongoing advances in methodology and as cryo-EM availability increases.

Illustration of the use of cryo-EM techniques to develop models of fibril morphology of Aβ fibrils isolated from Alzheimer’s brain tissue. (a) A cryo-EM image that depicts two distinct fibril types found in Alzheimer’s brain tissue (scale bar is 50 nm). Fibrils of each morphology are labeled with I and II. The asterisk shows a fibril in which a morphology I-like fibril is emerging from a fibril segment that is of morphology II. (b-d) These panels show 3D views of the reconstructed 3D map of fibrils of morphology I at 4.4 Å resolution. Fibrils are composed of two β-sheet stacks, which are shown in gray and blue in these structures. (b) A side view of the reconstructed 3D map. c A cross-sectional view of one molecular layer of the fibril superimposed with the molecular model, illustrating the folded structure of each constituent peptide within the β-sheet assemblies. (d) A side view depiction of a six-layer peptide stack superimposed with the molecular model. This stack is viewed from the perspective indicated by the red arrowhead in panel C. (e) A side view of six molecular layers of the boxed region from panel C, viewed from the perspective indicated by the black arrowhead in panel C. Reprinted with permission from Springer Nature, Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ, Jucker M, Fändrich M (2019) Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue . Nat Commun 10 (1):4760. https://doi.org/10.1038/s41467-019-12683-8 (reference [89]), Copyright 2019, The Authors
7 Emergent Physicochemical Properties of Cross-β Nanomaterials
Cross-β nanomaterials are of great interest due to their emergent physicochemical properties. In biological cross-β amyloid, the physicochemical properties of primary interest are the modes of action that lead to cellular dysfunction. Understanding these modes of action is manifested in efforts to correlate the cytotoxicity of cross-β fibrils and the associated prefibrillar aggregates with the structure of the aggregates. These efforts are as diverse as the diseases that are associated with cross-β amyloid and are specific to the disease states. As such, a detailed discussion of the various methods applied to interrogating the dysfunctional effects of cross-β aggregates is beyond the scope of this chapter. We will instead briefly discuss cross-β structures as biomaterials and provide an introduction to efforts to understand these materials both in terms of their inherent materials properties and their emergent biochemical functions.
Functional cross-β amyloid also possesses interesting physicochemical properties [5, 6]. For example, curli fibrils are components of the complex extracellular matrices produced by Enterobacteriaceae, including Escherichia coli [90]. Curli fibrils are assembled under carefully regulated conditions and mediate adhesion to surfaces, cell aggregation, biofilm formation, and host cell adhesion and invasion. Each of these functional emergent biochemical properties has been the subject of intense study. Again, a detailed discussion of the large body of work that has resulted in elucidation of the emergent physicochemical properties of functional amyloid is beyond the scope of this work. However, this class of amyloid can be used to illustrate the interrogation of cross-β structures using material techniques. As an example, the tensile strength of the interactions between curli fibrils and host fibronectin networks has been probed using AFM cantilever manipulation [91]. Correlation of the fundamental physicochemical properties of cross-β structures with emergent function is now a common mode of interrogation for these types of materials, with the applied methods depending on the function of the material that is being assessed.
Cross-β fibrils that form hydrogel networks are an illustrative example of a system for which the physicochemical properties are of primary significance. Cross-β systems that are engineered to sequester hydrophobic side chain group to the fibril interior while exposing hydrophilic side chain functionality to solvent self-assemble rapidly and at high concentrations entangle to form an emergent hydrogel network [92]. Many of these hydrogel-forming peptides have been designed based on understanding of the structure and mechanism of cross-β self-assembly [93]. The viscoelasticity, or “stiffness” of a hydrogel, is critical to its function. Oscillatory rheology is a critical method used in the interrogation of the emergent viscoelastic properties of these types of hydrogels [94]. These hydrogels have found use in diverse applications that include tissue engineering, regenerative medicine, and drug delivery. Each of these applications has an attendant set of requisite emergent properties that are characterized during the design process. The vast scope of techniques used to assess these properties depends on the property that is being interrogated. We will forego a detailed discussion of these varying techniques.
Finally, cross-β assemblies have been broadly used as scaffolds for the multivalent display of functional chemical groups [95]. These biomaterials have been used for tissue engineering, drug delivery, self-adjuvanting vaccine, antimicrobial, catalytic, and optical applications. The emergent properties that are interrogated for these materials depend on the specific application. For example, a material for tissue engineering may require the multivalent display of a cell-signaling motif on a cross-β scaffold. The relevant biochemical properties that would be assessed would include cellular adhesion and perhaps detection of specific ligand-receptor interactions between the cross-β display and the cells of interest. Detailed discussion of the many properties that are interrogated in these types of cross-β materials is beyond the scope of this chapter. The broad range of applications that cross-β materials have been designed to address illustrates that characterization of these materials must go far beyond typical structural and mechanistic analysis of cross-β assemblies. Cross-β assemblies are an emerging class of next-generation biomaterial for applications across virtually all fields of materials science. As such, it is expected that an ever-increasing range of techniques will be used to assess these materials.
8 Conclusion
Peptide and protein cross-β nanomaterials are the object of increasing fascination and study. The relevance of these supramolecular assemblies to both amyloid disease etiology and evolved biological materials motivates much of this interest. In the last several decades, there has also been an explosion in the development of engineering biomaterials based on peptide and protein cross-β assemblies. Designed cross-β systems have found wide-ranging use as biocompatible materials for regenerative medicine, drug delivery, wound healing, and as self-adjuvanting scaffolds for epitope presentation in vaccines. This broad relevance of cross-β architecture to both medical and materials science has increased the urgency to understand the relationships between peptide and protein sequence and self-assembly properties and to understand the correlations between aggregate structure, mechanism of assembly, and emergent functional properties. There have been transformative advances in understanding the mechanisms of cross-β assembly, in the available methods for the structural characterization of cross-β materials, and in the exploitation of these materials for diverse applications. These advances have dramatically quickened discoveries of the structural and functional properties of cross-β aggregates. The next decade will undoubtedly bring additional innovation, which will lead to transformative acceleration in the study of cross-β structure and the design of next-generation cross-β materials.
References
Ke PC, Zhou R, Serpell LC, Riek R, Knowles TPJ, Lashuel HA, Gazit E, Hamley IW, Davis TP, Fändrich M, Otzen DE, Chapman MR, Dobson CM, Eisenberg DS, Mezzenga R (2020) Half a century of amyloids: past, present and future. Chem Soc Rev 49(15):5473–5509
Harrison RS, Sharpe PC, Singh Y, Fairlie DP (2007) Amyloid peptides and proteins in review. Rev Physiol Biochem Pharmacol 159:1–77
Cawood EE, Karamanos TK, Wilson AJ, Radford SE (2021) Visualizing and trapping transient oligomers in amyloid assembly pathways. Biophys Chem 268:106505
Almeida ZL, Brito RMM (2020) Structure and aggregation mechanisms in amyloids. Molecules 25(5):1195
Levkovich SA, Gazit E, Laor Bar-Yosef D (2021) Two decades of studying functional amyloids in microorganisms. Trends Microbiol 29(3):251–265
Sergeeva AV, Galkin AP (2020) Functional amyloids of eukaryotes: criteria, classification, and biological significance. Curr Genet 66(5):849–866
Abdelrahman S, Alghrably M, Lachowicz JI, Emwas A-H, Hauser CAE, Jaremko M (2020) “What Doesn’t kill you makes you stronger”: future applications of amyloid aggregates in biomedicine. Molecules 25(22):5245
Kajava AV, Squire JM, Parry DAD (2006) β-Structures in fibrous proteins. Adv Protein Chem 73:1–15
Balasco N, Diaferia C, Morelli G, Vitagliano L, Accardo A (2021) Amyloid-like aggregation in diseases and biomaterials: osmosis of structural information. Front Bioeng Biotechnol 9(130)
Westermark GT, Johnson KH, Westermark P (1999) Staining methods for identification of amyloid in tissue. Methods Enzymol 309:3–25
Aliyan A, Cook NP, Martí AA (2019) Interrogating amyloid aggregates using fluorescent probes. Chem Rev 119(23):11819–11856
Castelletto V, Hamley IW (2018) Methods to characterize the nanostructure and molecular Organization of Amphiphilic Peptide Assemblies. Methods Mol Biol 1777:3–21
Vadukul DM, Al-Hilaly YK, Serpell LC (2019) Methods for structural analysis of amyloid fibrils in Misfolding diseases. Methods Mol Biol 1873:109–122
Sahoo BR, Cox SJ, Ramamoorthy A (2020) High-resolution probing of early events in amyloid-β aggregation related to Alzheimer's disease. Chem Commun 56(34):4627–4639
Zhao R, So M, Maat H, Ray NJ, Arisaka F, Goto Y, Carver JA, Hall D (2016) Measurement of amyloid formation by turbidity assay—seeing through the cloud. Biophys Rev 8(4):445–471
Kumar EK, Haque N, Prabhu NP (2017) Kinetics of protein fibril formation: methods and mechanisms. Int J Biol Macromol 100:3–10
Wetzel R (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 39(9):671–679
Wang J, Liu K, Xing R, Yan X (2016) Peptide self-assembly: thermodynamics and kinetics. Chem Soc Rev 45(20):5589–5604
Gade Malmos K, Blancas-Mejia LM, Weber B, Buchner J, Ramirez-Alvarado M, Naiki H, Otzen D (2017) ThT 101: a primer on the use of thioflavin T to investigate amyloid formation. Amyloid 24(1):1–16
Pignataro MF, Herrera MG, Dodero VI (2020) Evaluation of peptide/protein self-assembly and aggregation by spectroscopic methods. Molecules 25(20):4854
Barykin EP, Petrushanko IY, Kozin SA, Telegin GB, Chernov AS, Lopina OD, Radko SP, Mitkevich VA, Makarov AA (2018) Phosphorylation of the amyloid-Beta peptide inhibits zinc-dependent aggregation, prevents Na, K-ATPase inhibition, and reduces cerebral plaque deposition. Front Mol Neurosci 11(302)
Profit AA, Vedad J, Desamero RZB (2017) Peptide conjugates of benzene carboxylic acids as agonists and antagonists of amylin aggregation. Bioconjug Chem 28(2):666–677
Khalili Samani E, Mofid MR, Malakoutikhah M (2020) The effect of terminal groups and halogenation of KLVFF peptide on its activity as an inhibitor of β-amyloid aggregation. J Pept Sci 26(2):e3227
Martial B, Lefèvre T, Auger M (2018) Understanding amyloid fibril formation using protein fragments: structural investigations via vibrational spectroscopy and solid-state NMR. Biophys Rev 10(4):1133–1149
Cristóvão JS, Henriques BJ, Gomes CM (2019) Biophysical and spectroscopic methods for monitoring protein Misfolding and amyloid aggregation. Methods Mol Biol 1873:3–18
Perálvarez-Marín A, Barth A, Gräslund A (2008) Time-resolved infrared spectroscopy of pH-induced aggregation of the Alzheimer Aβ1–28 peptide. J Mol Biol 379(3):589–596
Bin Y, Li X, He Y, Chen S, Xiang J (2013) Amyloid-β peptide (1–42) aggregation induced by copper ions under acidic conditions. Acta Biochim Biophys Sin 45(7):570–577
Radko SP, Khmeleva SA, Suprun EV, Kozin SA, Bodoev NV, Makarov AA, Archakov AI, Shumyantseva VV (2015) Physico-chemical methods for studying amyloid-[beta] aggregation. Biochem Moscow Suppl Ser B 9(3):258–274
Gorman PM, Yip CM, Fraser PE, Chakrabartty A (2003) Alternate aggregation pathways of the Alzheimer β-amyloid peptide: Aβ association kinetics at endosomal pH. J Mol Biol 325(4):743–757
Ruggeri FS, Habchi J, Cerreta A, Dietler G (2016) AFM-based single molecule techniques: unraveling the amyloid pathogenic species. Curr Pharm Des 22(26):3950–3970
Watanabe-Nakayama T, Ono K (2018) High-speed atomic force microscopy of individual Amyloidogenic protein assemblies. Methods Mol Biol 1814:201–212
Watanabe-Nakayama T, Ono K, Itami M, Takahashi R, Teplow DB, Yamada M (2016) High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc Natl Acad Sci 113(21):5835–5840
O'Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, Thiagarajan G, Wetzel R (2006) Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography–based sedimentation assay. Methods Enzymol 413:34–74
Senguen FT, Lee NR, Gu X, Ryan DM, Doran TM, Anderson EA, Nilsson BL (2011) Probing aromatic, hydrophobic, and steric effects on the self-assembly of an amyloid-β fragment peptide. Mol BioSyst 7(2):486–496
Senguen FT, Doran TM, Anderson EA, Nilsson BL (2011) Clarifying the influence of core amino acid hydrophobicity, secondary structure propensity, and molecular volume on amyloid-β 16–22 self-assembly. Mol BioSyst 7(2):497–510
Mok Y-F, Howlett GJ, Griffin MDW (2015) Sedimentation velocity analysis of the size distribution of amyloid oligomers and fibrils. Methods Enzymol 562:241–256
Lee SJC, Nam E, Lee HJ, Savelieff MG, Lim MH (2017) Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chem Soc Rev 46(2):310–323
Pham CLL, Mok Y-F, Howlett GJ (2011) Sedimentation velocity analysis of amyloid fibrils. Methods Mol Biol 752:179–196
Cole JL, Lary JW, Moody T, Laue TM (2008) Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol 84:143–179
Woods LA, Radford SE, Ashcroft AE (2013) Advances in ion mobility spectrometry–mass spectrometry reveal key insights into amyloid assembly. Biochim Biophys Acta 1834(6):1257–1268
Dupuis NF, Wu C, Shea J-E, Bowers MT (2009) Human islet amyloid polypeptide monomers form ordered β-hairpins: a possible direct Amyloidogenic precursor. J Am Chem Soc 131(51):18283–18292
Dupuis NF, Wu C, Shea J-E, Bowers MT (2011) The amyloid formation mechanism in human IAPP: dimers have β-Strand monomer−monomer interfaces. J Am Chem Soc 133(19):7240–7243
Buell AK, Dhulesia A, White DA, Knowles TPJ, Dobson CM, Welland ME (2012) Detailed analysis of the energy barriers for amyloid fibril growth. Angew Chem Int Ed 51(21):5247–5251
White DA, Buell AK, Dobson CM, Welland ME, Knowles TPJ (2009) Biosensor-based label-free assays of amyloid growth. FEBS Lett 583(16):2587–2592
Hasegawa K, Ono K, Yamada M, Naiki H (2002) Kinetic modeling and determination of reaction constants of Alzheimer's β-amyloid fibril extension and dissociation using surface Plasmon resonance. Biochemistry 41(46):13489–13498
Kabiri M, Unsworth LD (2014) Application of isothermal titration calorimetry for characterizing thermodynamic parameters of biomolecular interactions: peptide self-assembly and protein adsorption case studies. Biomacromolecules 15(10):3463–3473
Swanekamp RJ, DiMaio JTM, Bowerman CJ, Nilsson BL (2012) Coassembly of enantiomeric amphipathic peptides into amyloid-inspired rippled β-sheet fibrils. J Am Chem Soc 134(12):5556–5559
Sasahara K, Goto Y (2013) Application and use of differential scanning calorimetry in studies of thermal fluctuation associated with amyloid fibril formation. Biophys Rev 5(3):259–269
Ilie IM, Caflisch A (2019) Simulation studies of Amyloidogenic polypeptides and their aggregates. Chem Rev 119(12):6956–6993
Caflisch A (2006) Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol 10(5):437–444
Ahmed AB, Kajava AV (2013) Breaking the amyloidogenicity code: methods to predict amyloids from amino acid sequence. FEBS Lett 587(8):1089–1095
Huang C, Ghanati E, Schmit JD (2018) Theory of sequence effects in amyloid aggregation. J Phys Chem B 122(21):5567–5578
Wojciechowski JW, Kotulska M (2020) PATH–prediction of Amyloidogenicity by threading and machine learning. Sci Rep 10(1):7721
Michaels TCT, Šarić A, Habchi J, Chia S, Meisl G, Vendruscolo M, Dobson CM, Knowles TPJ (2018) Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation. Annu Rev Phys Chem 69(1):273–298
Šarić A, Chebaro YC, Knowles TPJ, Frenkel D (2014) Crucial role of nonspecific interactions in amyloid nucleation. Proc Natl Acad Sci 111(50):17869–17874
Nguyen P, Derreumaux P (2014) Understanding amyloid fibril nucleation and Aβ oligomer/drug interactions from computer simulations. Acc Chem Res 47(2):603–611
Li D, Liu C (2020) Structural diversity of amyloid fibrils and advances in their structure determination. Biochemistry 59(5):639–646
Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE (2018) A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol 19(12):755–773
Eisenberg DS, Sawaya MR (2017) Structural studies of amyloid proteins at the molecular level. Annu Rev Biochem 86(1):69–95
Greenfield NJ (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1(6):2876–2890
Li H, Lantz R, Du D (2019) Vibrational approach to the dynamics and structure of protein amyloids. Molecules 24(1)
Lomont JP, Ostrander JS, Ho J-J, Petti MK, Zanni MT (2017) Not all β-sheets are the same: amyloid infrared spectra, transition dipole strengths, and couplings investigated by 2D IR spectroscopy. J Phys Chem B 121(38):8935–8945
Urban JM, Ho J, Piester G, Fu R, Nilsson BL (2019) Rippled β-sheet formation by an amyloid-β fragment indicates expanded scope of sequence space for enantiomeric β-sheet peptide Coassembly. Molecules 24(10)
Hiramatsu H, Kitagawa T (2005) FT-IR approaches on amyloid fibril structure. Biochim Biophys Acta 1753(1):100–107
Decatur SM (2006) Elucidation of residue-level structure and dynamics of polypeptides via isotope-edited infrared spectroscopy. Acc Chem Res 39(3):169–175
Pauling L, Corey RB (1953) Two rippled-sheet configurations of polypeptide chains, and a note about the pleated sheets. Proc Natl Acad Sci 39(4):253
Moran SD, Zanni MT (2014) How to get insight into amyloid structure and formation from infrared spectroscopy. J Phys Chem Lett 5(11):1984–1993
Alperstein AM, Ostrander JS, TO Z, Zanni MT (2019) Amyloid found in human cataracts with two-dimensional infrared spectroscopy. Proc Natl Acad Sci 116(14):6602
Kurouski D, Van Duyne RP, Lednev IK (2015) Exploring the structure and formation mechanism of amyloid fibrils by Raman spectroscopy: a review. Analyst 140(15):4967–4980
Bonhommeau S, Talaga D, Hunel J, Cullin C, Lecomte S (2017) Tip-enhanced Raman spectroscopy to distinguish toxic oligomers from Aβ1–42 fibrils at the nanometer scale. Angew Chem Int Ed 56(7):1771–1774
Tycko R (2011) Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem 62(1):279–299
Karamanos TK, Kalverda AP, Thompson GS, Radford SE (2015) Mechanisms of amyloid formation revealed by solution NMR. Prog Nucl Magn Reson Spectrosc 88-89:86–104
Jaroniec CP (2019) Two decades of progress in structural and dynamic studies of amyloids by solid-state NMR. J Magn Reson 306:42–47
Loquet A, El Mammeri N, Stanek J, Berbon M, Bardiaux B, Pintacuda G, Habenstein B (2018) 3D structure determination of amyloid fibrils using solid-state NMR spectroscopy. Methods 138-139:26–38
Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R (2000) Amyloid fibril formation by Aβ16-22, a seven-residue fragment of the Alzheimer’s β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 39(45):13748–13759
Potapov A, Yau W-M, Ghirlando R, Thurber KR, Tycko R (2015) Successive stages of amyloid-β self-assembly characterized by solid-state nuclear magnetic resonance with dynamic nuclear polarization. J Am Chem Soc 137(25):8294–8307
Serpell LC, Fraser PE, Sunde M (1999) X-Ray fiber diffraction of amyloid fibrils. Methods Enzymol 309:526–536
Ricci C, Spinozzi F, Mariani P, Ortore MG (2016) Protein Amyloidogenesis investigated by small angle scattering. Curr Pharm Des 22(26):3937–3949
Lu K, Jacob J, Thiyagarajan P, Conticello VP, Lynn DG (2003) Exploiting amyloid fibril lamination for nanotube self-assembly. J Am Chem Soc 125(21):6391–6393
Toyama BH, Weissman JS (2011) Amyloid structure: conformational diversity and consequences. Annu Rev Biochem 80(1):557–585
Balbirnie M, Grothe R, Eisenberg DS (2001) An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc Natl Acad Sci U S A 98(5):2375–2380
Do TD, LaPointe NE, Sangwan S, Teplow DB, Feinstein SC, Sawaya MR, Eisenberg DS, Bowers MT (2014) Factors that drive peptide assembly from native to amyloid structures: experimental and theoretical analysis of [Leu-5]-Enkephalin mutants. J Phys Chem B 118(26):7247–7256
Kreutzer AG, Nowick JS (2018) Elucidating the structures of amyloid oligomers with macrocyclic β-hairpin peptides: insights into Alzheimer’s disease and other amyloid diseases. Acc Chem Res 51(3):706–718
Kreutzer AG, Samdin TD, Guaglianone G, Spencer RK, Nowick JS (2020) X-ray crystallography reveals parallel and antiparallel β-sheet dimers of a β-hairpin derived from Aβ16–36 that assemble to form different tetramers. ACS Chem Neurosci 11(15):2340–2347
Gras SL, Waddington LJ, Goldie KN (2011) Transmission electron microscopy of amyloid fibrils. Methods Mol Biol 752:197–214
Goldsbury C, Baxa U, Simon MN, Steven AC, Engel A, Wall JS, Aebi U, Müller SA (2011) Amyloid structure and assembly: insights from scanning transmission electron microscopy. J Struct Biol 173(1):1–13
Newcomb CJ, Moyer TJ, Lee SS, Stupp SI (2012) Advances in cryogenic transmission electron microscopy for the characterization of dynamic self-assembling nanostructures. Curr Opin Colloid Interface Sci 17(6):350–359
Fitzpatrick AWP, Saibil HR (2019) Cryo-EM of amyloid fibrils and cellular aggregates. Curr Opin Struct Biol 58:34–42
Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ, Jucker M, Fändrich M (2019) Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat Commun 10(1):4760
Barnhart MM, Chapman MR (2006) Curli biogenesis and function. Annu Rev Microbiol 60(1):131–147
Oh YJ, Hubauer-Brenner M, Gruber HJ, Cui Y, Traxler L, Siligan C, Park S, Hinterdorfer P (2016) Curli mediate bacterial adhesion to fibronectin via tensile multiple bonds. Sci Rep 6(1):33909
Bowerman CJ, Nilsson BL (2012) Review self-assembly of amphipathic β-sheet peptides: insights and applications. Pept Sci 98(3):169–184
Fu K, Wu H, Su Z (2021) Self-assembling peptide-based hydrogels: fabrication, properties, and applications. Biotechnol Adv 49:107752
Yan C, Pochan DJ (2010) Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem Soc Rev 39(9):3528–3540
Distaffen HE, Jones CW, Abraham BL, Nilsson BL (2021) Multivalent display of chemical signals on self-assembled peptide scaffolds. Pept Sci 113(2):e24224
Acknowledgements
The preparation of this chapter was supported by the National Science Foundation (CHE-1904528).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Jones, C.W., Distaffen, H.E., Nilsson, B.L. (2022). Peptide Cross-β Nanoarchitectures: Characterizing Self-Assembly Mechanisms, Structure, and Physicochemical Properties. In: Govindaraju, T., Ariga, K. (eds) Molecular Architectonics and Nanoarchitectonics. Nanostructure Science and Technology. Springer, Singapore. https://doi.org/10.1007/978-981-16-4189-3_8
Download citation
DOI: https://doi.org/10.1007/978-981-16-4189-3_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4188-6
Online ISBN: 978-981-16-4189-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)