
Abstract
Advanced deep sequencing technologies have revolutionized our understanding toward noncoding RNAs (ncRNAs) and have uncovered their various regulatory roles which are performed by the fine-tuning of host gene expression at either epigenetic or transcriptional and posttranscriptional level. Rapid development in various deep sequencing technologies and bioinformatics platforms has targeted ncRNAs for therapeutic purposes. Here we are summarizing various transcriptomic techniques, bioinformatics tools, and databases and their application to understand the modulation of various regulatory ncRNAs in context of dengue and other viral pathophysiologies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Noncoding RNAs
- Dengue virus
- EST
- Next generation sequencing
- Illumina sequencing
- Ion Torrent sequencing
- Pyrosequencing
- Oxford Nanopore
- PacBio sequencing
- CAGE:
-
Cap analysis of gene expression
- EST:
-
Expressed sequence tag
- lncRNA:
-
Long noncoding RNA
- MPSS:
-
Massively parallel signature sequencing
- SAGE:
-
Serial analysis of gene expression
- seq:
-
Sequencing
- sncRNA:
-
Small noncoding RNA
1 Introduction
Viral infections often lead to the modulation of various regulatory RNAs like miRNA, siRNA, tRNA-derived RNA fragments, Piwi RNA, etc. that are called noncoding RNA. These ncRNAs do not code for any protein. Modulation of small ncRNAs (sncRNAs) during viral infection is primarily associated with the regulation of endogenous host gene expression, and expression of some endogenous virus generated noncoding RNA in host cells which may potentially contribute to disease progression. vsRNA and sfRNA are virus-derived small noncoding RNAs which are found to be associated with the evasion of host antiviral response. Table 12.1 depicts various host- and virus-originated noncoding RNAs and their roles during viral infections.
Delayed diagnosis of infectious disease often aggravates the disease condition. Differential expression of sncRNA can prove to be dispensable biomarkers to identify the disease severity during various stages of infection. RNA-based therapeutics represents next-generation sequencing approaches for prospective antiviral and other disease-related therapeutic development. Many RNA therapeutics have already been approved by the FDA such as miravirsen to treat HCV infection and patisiran, givosiran, and MRX34 to treat various tumors. Thus unraveling the transcriptional landscape of various infectious disease is important to build an articulated nexus of regulatory roles of ncRNA during virus-host interaction. Recent advancements in the field of deep sequencing and high-throughput screening have enabled to establish a deeper understanding about RNAome and thus explore the capacities of these sncRNAs. Here we are summarizing various transcriptomic techniques used to study dengue virus and other viruses to study small noncoding RNAs.
2 Tag-Based Approaches
The tag-based approaches have been used for the direct determination of the cDNA sequences. These include expressed sequence tag (EST), serial analysis of gene expression (SAGE), cap analysis of gene expression (CAGE), and massively parallel signature sequencing (MPSS). The expressed sequence tag has proved to be a rapid and efficient means of characterizing the huge sets of gene sequences consisting of 300–1000 bp of DNA, and it is often accumulated in a database as a “single-pass read” that is sufficient in establishing the identity of particular expressed gene. Serial analysis of gene expression (SAGE) is another method used to obtain the qualitative and quantitative level of gene expression profiles in different conditions. The SAGE methodology is considered as an “open architecture” technique, and unlike another array method, it does not need prior information about the genes which needs to be analyzed, and it also reflects the absolute mRNA levels. Further, several SAGE-like methods have also been developed. They can be employed for the genome-wide analysis of DNA copy-number changes, transcription factor targets, and analyzing epigenetic signatures such as methylation patterns and chromatin structure. CAGE is another high-throughput sequencing-based technique that enables us to quantify and identify the expression of 5′ capped RNAs [21]. Likewise, MPSS is an open-ended platform for quantifying in-depth gene expression based on individual mRNA levels in the cell. In MPSS, the identification and characterization of the gene prior to conducting the experiment are not required. It has the routine sensitivity at the level of mRNA molecules, and the datasets are in the digital format that facilitates the management and analysis of data.
3 Chip-Based Approaches
Microarray plays a significant role in transcriptome profiling studies. Microarray is used for DNA mapping, sequencing, and transcript-level analysis [22]. It’s a crucial genomic technology and often used synonymously with DNA microarray and high-throughput gene expression analysis. Gene expression microarray is a nucleic-acid-hybridization-based technique, and a complex mechanism is required to understand the global and parallel analysis of different cellular processes. It’s been more than a decade that microarray has been in use as a gold standard for transcriptome studies in a wide range of settings [23]. Recently, despite the emergence of the next-generation sequencing [24], DNA microarrays still appear an extremely compelling approach due to its quick, precise, and inexpensive detection of the pathogens compared to culture or immunoassay techniques [25, 26]. It is a technique of choice for the detection of altered gene expression upon virus infection and is often used in clinical diagnostic to detect for the presence of existing viruses or new viruses [27, 28].
Tiling arrays are subtypes of microarray which are used to investigate thousands of gene expression in a coordinated fashion, transcriptome mapping, and to identify the transcription factor binding sites [29, 30]. It is a useful tool for the investigation of whole genome or chromosome expression as well as to uncover the various novel RNA expression patterns [31, 32]. Like traditional oligonucleotide microarrays, the probes are designed to the matched parts of the genomic region of interest. The probes get hybridized to the labeled DNA or RNA target molecules which are fixed onto a chip. The experimental technique to identify the site for protein DNA interaction involves the hybridization of immunoprecipitated DNA on a tiling microarray (ChIP-chip experiments) [33]. The genome-tiling microarrays have facilitated the analysis of global expression patterns with or without completely annotated genome in organisms such as prokaryotes, mouse, human, and yeast [34, 35]. In conventional gene-probing microarrays, probes targeting a particular gene give independent measures of the same RNA expression, while when tiling is the strategy applied to the entire genome, the analysis becomes restricted to the annotated genes, and it becomes difficult for the unannotated genes to be analyzed. Therefore, major challenges for tiling array studies are (1) determining transcriptional start and stop sites and (2) predicting whether transcripts are either from long continuous stretches such as genes or from short noncontinuous strands of RNA, i.e., typically ncRNAs [36, 37].
Although microarray-based transcriptional profiling was very much prevalent during the last decade and have made numerous remarkable contributions, with the emergence of new sequencing technologies, its use has become limited. Although there is an advanced microarray application in regard to RNA profiling, however, it has certain limitations in terms of reproducibility, sensitivity, and specificity [38].
4 RNA Sequencing Technologies (Platforms)
4.1 RNA Sequencing
RNA seq is a recently established high-throughput deep sequencing technique which has been used extensively for mapping, quantifying, and identification of novel genes. Small noncoding RNAs has been widely studied these days since these have been found to play a critical role in regulation of gene expression. After microarray, RNA sequencing is slowly procuring its space in the profiling studies and now has become the gold standard for a novel small RNA discovery as well as for small RNA profiling with high throughput, high sensitivity, and reproducibility. Deep RNA sequencing does not need any prerequisite information about the sequence which is a clear advantage over existing approaches like microarrays and qPCR.
4.1.1 Workflow
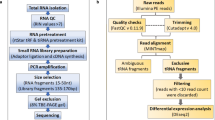
RNA seq workflow typically comprises of small RNA enrichment, RNA assessment and quantitation, small RNA library construction, deep sequencing, and bioinformatics analysis. Figure 12.1 is showing a typical RNA seq workflow.

Workflow of a typical RNA sequencing experiment and analysis
4.1.2 Small RNA Enrichment Methods
Small noncoding RNAs play a significant role in gene expression. In order to analyze them, additional purification steps are required as they are present in very low concentration and thus enrichment methods are employed for their isolation and purification. Two types of enrichment methods can be used, i.e., manual methods and kit-based approaches.
4.1.2.1 Manual Methods for Isolation of sncRNAs
4.1.2.1.1 miRICH Method
miRICH method is variant of TRIzol method used for total RNA isolation. In this method, total RNA is isolated by using TRIzol reagent, and in order to concentrate small RNAs, washing using ethanol is skipped, after the precipitation step and the pellet is overdried. Overdrying the pellet causes aggregation of larger RNA molecules due to high salt concentration; hence larger molecules are barely soluble, while small RNAs can be easily eluted [39].
4.1.2.1.2 Fractionation Approach
Fractionation method makes use of flashPAGE fractionator that works on the principle of gel electrophoresis. sncRNAs are isolated based on their size on a miniature PAGE. A precasted PAGE is inserted into the buffer chamber of the fractionator after addition of lower running buffer; this is followed by addition of upper running buffer. The small RNA fraction will be present in lower running buffer after electrophoresis which further enriched by using flashPAGE cleanup kit [40].
4.1.2.1.3 Multidimensional HPLC for Purification of Small Noncoding RNA
mRNA isolation can be eased by exploiting poly(A) tail, while isolation of sncRNA is difficult because of limited knowledge regarding biochemistry, posttranscriptional processing, ribonucleotide modification, and biological function, hence limiting the ability to obtain RNA in pure form. HPLC can be used to purify all classes of ncRNA from single samples. To achieve complete separation of two ranges, size exclusion chromatography with ion-pair reverse phase chromatography can be used [40].
4.1.2.1.4 p19 Protein-Mediated Isolation of siRNA and Detection of miRNA
Another approach for enrichment of sncRNA is to make use of the property of p19 protein to strongly bind to siRNA derived from plant virus, Carnation Italian ringspot virus (CIRV). The p19 fusion protein designed can be used for isolation of siRNA and detection of miRNA with the help of magnetic beads [41].
4.1.2.1.5 Trans-Kingdom Rapid Affordable Purification of RISC (TRAP) for Isolation of all Classes of Silencing Small Noncoding RNA
Isolation of small noncoding RNAs from total RNA using size separation methods is tedious and may give poor yields. An alternative to total RNA isolation method is the AGO purification technique in which the AGO-associated sncRNA complex is purified in order to isolate small RNA by anion exchange chromatography. The issue of contamination by other RNA species will be overcome by employing this technique, but it requires highly specific antibody against AGO proteins. Anion exchange chromatography makes use of Q Sepharose a positively charged anion exchange matrix to adsorb the RNPs complex, and then the complex is eluted by increasing mild salt concentration. Further analysis is based on the gel electrophoresis technique [42].
4.1.2.2 Kit-Based Methods
Silica-based columns are used for recovery of sncRNAs widely. Different commercial kits are available for isolation of small RNAs like mirVana, miRNeasy mini kit, mirPremier, MasterPure RNA purification kit, and miRCURY as described in Table 12.2. Selection of method depends upon the initial volume of sample, type of sncRNA to be analyzed, ease to use, and price per sample [42].
4.1.3 RNA Quality and Quantity Assessment
For assessment of RNA quality, two methods are used, i.e., denaturing/non-denaturing agarose gel and capillary electrophoresis, which determine intact RNA by identifying 28S and 18S rRNA bands [43]. Quantity of RNA can also be determined by various methods. It includes UV absorbance at wavelength 260 nm, fluorometry which employs fluorescently labeled probe specific to sncRNAs which have to be quantified, and splinted ligation assay where a small RNA-specific bridge oligonucleotide is used to form base pairs with the 5′-end-radiolabeled oligonucleotide and sncRNA [44]. For real-time quantification of small RNA, stem loop PCR is used in order to increase the length of template, and quantification is done using TaqMan probe [45].
Recent development in microfluidic technology has led to development of different instruments which can check RNA integrity as well as quantified RNAs. For example, microfluidic instruments like Agilent 2100 Bioanalyzer, 2200 TapeStation, and Experion can check RNA integrity by calculating 28S /18S ratio, and quantification is done by fluorescent labeling.
4.1.4 Small RNA Library Construction
Small RNA library preparation typically starts with the ligation of DNA adapters. It includes ligation of pre-adenylated adapter oligo at the 3′ end of the sncRNA using a truncated variant of T4 RNA ligase2. It is followed by ligation of adapter oligo at the 5′ end using T4 RNA ligase 1. The sequence is then reverse transcribed into cDNA following ligation. Although direct RNA sequencing is possible, but many instruments are based upon DNA sequencing due to which cDNA library preparation is done and is the crucial step of sequencing. After cDNA preparation, PCR amplification is carried out. For the longer sequences, fragmentation is done using DNase I treatment or sonication before adaptor ligation. But small RNAs can be sequenced directly after adapter ligation since these already exists as a shorter sequence. The adaptor ligation results in loss of strand specificity which is restored by dUTP incorporation in the second strand of cDNA which is then further degraded by uracil-DNA glycosylase (UDG) before the amplification step. Hence, only a single strand is amplified by PCR up to 8–12 cycles. Finally, single-end sequencing (from one end) or paired-end sequencing (from both ends) can be carried out in a high-throughput manner. The read sequences obtained are then aligned with the reference genome present in the database; else de novo assembly is carried out for the reads.
4.1.5 Bioinformatics Analysis
Once the raw sequencing data is generated, it is then preprocessed and normalized which involve trimming of adaptors, alignment, outlier removal, RNA class filtering, and generation of unique reads. Normalization involves comparison of expression levels across libraries. There are two commonly used small RNA database: (1) miRBase comprises information of all miRNA sequences and annotations; (2) Rfam is an open-access database which includes information about tRNA, rRNA, snoRNA, etc.
4.1.6 Challenges
Library Preparation Challenges
RNA seq contains various manipulation stages during cDNA library preparation, which can complicate the profiling procedure of the transcripts. Sometimes, sequencing data is incomparable with the qPCR, Northern blot, and microarray results which predominantly can happen due to the bias that occurs during different library preparation steps. Many identical short reads are amplified from cDNA libraries which give a reflection of abundant RNA species and thus culminate in false results.
Small RNA ligation bias is another attribute for library preparation which is particularly dependent upon T4 RNA ligase during adaptor ligation. It has been suggested that randomization of the adapter sequences near ligation junction might result in bias reduction. Illumina sequencing identifies antisense as well as overlapping transcripts which are unwanted. To avoid this, dUTP is incorporated during the generation of second cDNA synthesis prior to adaption ligation. The strand containing dUTP then finally gets digested leaving only a single strand for amplification.
Bioinformatics challenges include storage, retrieval, and processing of large amount of data which are very much essential for error reduction during result analysis. Background noise also occurs due to incomplete processing of RNAs.
4.2 Second-Generation Sequencing Platforms.
4.2.1 Pyrosequencing
This technique is based on the principle of sequence by synthesis approach which detects released pyrophosphate (PPi) during DNA synthesis. To detect the nucleic acid synthesis, four enzymatic reactions take place in a sequential manner. The primer is first hybridized with the biotin-labeled single-stranded DNA template. The hybridized primer template is then mixed with the enzymes: DNA polymerase, ATP sulfurylase, luciferase, and apyrase, and the substrates adenosine 5′-phosphosulfate (APS) and luciferin. Each dNTP is added separately to the reaction mixture. With the incorporation of a single nucleotide, one PPi is released which is then quantified in an equimolar concentration to the incorporated nucleotide. ATP sulfurylase forms ATP from PPi in the presence of APS. ATP and luciferase catalyze the conversion of luciferin to oxyluciferin which produces visible light which is accountable for the total amount of ATP generated. The produced light is detected by a photon detection device, with a maximum wavelength of 560 nm. ATP and unincorporated dNTPs are continuously degraded by apyrase [46].
Currently, pyrosequencing has been widely used in single nucleotide polymorphism (SNP) genotyping identification of bacteria, fungal, and viral typing. Moreover, this method can be used for the determination of difficult secondary structures and for the identification of mutations. Other possible applications are DNA methylation analysis and whole genome sequencing [47].
4.2.2 Ion Torrent Sequencing
Ion Torrent also requires sequence amplification, but it is the first technique that uses electrochemical detection, not camera scanning and fluorescence detection. Ion Torrent is done with an Ion Personal Genome Machine (PGM). Firstly, the DNA sample is prepared from fragmented RNA and then linked together. The library is then clonally amplified by emulsion PCR onto beads known as Ion Sphere particles. The beads are placed into proton-sensing wells on a semiconductor sequencing chip so that one bead can roughly fit into hundreds of millions of wells. The chip is then submerged into a nucleotide solution and results in the release of protons and a corresponding change in pH. This change in pH is documented by the PGM to determine whether the right nucleotide was used in the process or not, let alone if a nucleotide was added at all. As sequencing occurs, each of the four bases is introduced sequentially. A clear indication of the correct nucleotide being added is the presence of voltage. There will be no voltage found if the wrong nucleotide is added, and there will be double the voltage if two nucleotides are added. The more nucleotides present, the greater the increase in voltage and pH [48].
This technique can be used for targeted DNA and RNA sequencing, exome sequencing, viral typing, bacterial typing, aneuploidy and CNV analysis, and small RNA and miRNA sequencing [49].
4.2.3 Illumina Sequencing
It is often known as Solexa sequencing based on the name of the founder Solexa. It is based upon sequencing by synthesis approach and reversible dye terminators that are fluorescently labeled. These terminators are incorporated to the new DNA strand and stop DNA synthesis. These are then detected by the camera. Each type of dNTP is differently fluorophore-labeled, and therefore each base addition gives off different light which is detected by laser [50].
It is the next-generation sequencing that is used for the small RNA discovery, metagenomics, methylation profiling, transcriptome analysis, genome-wide profiling, and protein-RNA/DNA interaction analysis [51].
4.2.4 ABI SOLiD Sequencing
This principle is based upon the principle of sequencing by ligation. PCR-amplified target sequence is anchored by agarose beads onto a glass surface. Fluorescently labeled oligonucleotides are then added for sequential ligation mediated by enzyme DNA ligase. Once labeled oligonucleotides are annealed; fluorescent gets removed from the fragment due to the formation of phosphonothioate bond between the bases. The removal of fluorophore from 5′ site makes that site vacant for upcoming nucleotide ligation. This allows the formation of different fluorescent peaks corresponding to different nucleotides [52].
It is used for targeted sequencing, identification of small RNAs, epigenome analysis, and chromatin- immunoprecipitation etc. [53].
4.3 Third-Generation Sequencing Platforms
4.3.1 Oxford Nanopore Sequencing
Oxford sequencing is based upon the principle of small variations in electrical conductance that is generated when a DNA strand or a nucleotide passes through a pore of 1.5 nm size. The nanopore is immersed in a conducting fluid along with tiny wells layered with DNA polymerase which catches the hold of the exposed DNA and makes it pass through the nanopore. Each nucleotide passage through the pore creates varying degrees of obstruction that leads to the generation of varying electric current. This small variation in the electric current defines the characteristic of each nucleotide. This change of current is recorded to detect the sequence [54].
This technique confers precision and sensitivity and is cost-effective, and also, RNA can be directly sequenced with this technique. Very long 10–50-kb-sized fragments can also be sequenced. It can be used for metagenomics analysis, de novo assembly, sequencing of long transcripts, etc. [55].
4.3.2 PacBio Sequencing
PacBio is again based upon the principle of sequencing by synthesis approach which makes use of specialized SMRT chips for sequencing. All the four bases are fluorescently labeled with different fluorophores. Upon binding of the polymerase to the template on the SMRT chip, each incorporating base will be irradiated with different fluorescent colors. The peak and wavelength of the fluorescent emission are then recorded for the sequencing [11, 12].
Unlike first- and second-generation sequencing, this technique requires no PCR amplification and can sequence even longer reads. This technique is used for de novo sequencing of genomes and transcriptomes, detecting alternative splicing isoforms, epigenetic modifications, analysis of different mutations, etc. [56] (Table 12.3).
5 Data Processing and Analysis
Next-generation sequencing (NGS) technologies have been emerging with the potential to explore small RNA (sRNA) transcriptomes and their associated roles. It offers a reliable and high-throughput approach for the identification and quantification of various sRNA classes. The steps required for RNA sequence analysis of sncRNAs has been summarized in Fig 12.2. Various tools are available online for the analysis of a particular small RNA from sequencing data, shown in Table 12.4. Various tools are also available for the integrated analysis of more than type of RNA analysis which are depicted in Table 12.5.

Various steps and tools used in RNA sequencing data analysis. (The figure is adopted from [57])
6 Available Databases for Studying Small Noncoding RNAs
-
1.
miRBase: It consists of the miRNA database with information about sequence, genomic location, and predicted targets of miRNA [78].
-
2.
MirZ: It consists of the miRNA database with information about sequence-based miRNA profiles and predicted targets [79].
-
3.
IsomiR database: It mainly has information about miRNAs and isomiRs with respect to the reads; isomiRs assigned to miRNAs belong to human 293 T cells, with miRNA annotation from miRBase [80].
-
4.
siRNAdb: It is a database about siRNA (small interfering RNA) that contains information about experimentally verified and predicted siRNAs, sequence, and the literature links [81].
-
5.
piRNABank: This database is useful for information on piRNA about their sequence, cluster, and homology searching [82].
-
6.
snoRNA-LBME-db: It is a database in which information about snoRNAs and scaRNAs are available with respect to their sequence, expression information, as well as predicted targets including base pairing information [83].
-
7.
Rfam: It consists of the data about snRNAs, snoRNAs, and miRNAs and about the sequence families of other structural RNAs [84].
-
8.
NONCODE: This database contains data about miRNAs, piRNAs, snoRNAs, and scaRNAs. NONCODE is linked with GenBank [85].
-
9.
RNAdb: There is information about miRNAs, piRNAs, snoRNAs, and other ncRNAs that has their sequence with links to literature and other databases[86].
-
10.
deepBase: It is a miRNA, piRNA, endo-siRNA, nasRNA, pasRNA, easRNA, and rasRNA database with the data information about their sequence from different tissues and for computationally predicted snRNAs [87].
-
11.
fRNAdb: It consists of data about various annotated and predicted ncRNAs of various lengths from different sources [88].
7 Applications of Various Transcriptomic Approaches in Context of Dengue and Other Viruses
Expressed sequence tags (ESTs) emerged as the first high-throughput technique to gene expression and annotation of genome. To investigate host gene expression response upon viral infection, EST approach was used as molecular tool. Barón et al. in 2010 compared the differential gene expression profile in the midgut of refractory and susceptible lines of Aedes aegypti mosquitoes by infecting with dengue-2 virus [89]. The annotation of EST-identified differentially expressed genes belongs to immune response and metabolism. Another EST-based study by Guo et al. in 2009 in response dengue-2 virus infection in Aedes albopictus was analyzed. They identified seven ESTs; among them five were overexpressed in susceptibility, and two were overexpressed in refractory lines [90].
Microarray analysis is regularly used for the analysis of altered gene expression upon virus infections. It is often used in clinical diagnosis to detect the presence of known viruses or to find new viruses [27, 28]. In addition, microarray has been used in the detection and identification of seven pathogenic viruses from the Flaviviridae family; these viruses are the yellow fever (YF), West Nile (WN), Japanese encephalitis (JE), and the dengue strain 1–4 viruses [91]. DNA microarray was used for the detection of dual infection with different DENV serotypes [92]. Likewise, high-density resequencing microarrays (RMAs) are another variant of microarray which has been used for rapid identification and molecular analysis of bacteria and viruses. It also enables to predict for the biomarkers during different clinical outcomes of dengue infection [93, 94]. RMA, microarray technique, has also been proven useful for the rapid and accurate identification of pathogens, specifically for the Rhabdoviridae family [95]. However, this technology often has certain limitations in the context of sensitivity, specificity, and reproducibility [38]. Tiling array which is a variant of microarray has a wide range of applications and had been used widely in the detection of multiple foodborne RNA viruses including multiple coxsackievirus serotype (A and B) strains and multiple hepatitis A virus genotype strains. It has also been used in differentiating the virus serotypes [96]. Further, the customized tiling array technique has been utilized into accurate identification of non-polyadenylated RNA in the case of human cytomegalovirus (HCMV) [97].
Transcriptome approach is also used to investigate transcriptional patterns associated with the dengue progression by analyzing the RNA-Seq of peripheral blood mononuclear cells. Patients with varying severity of dengue infection were compared with the patients with other febrile illnesses (OFIs) and the healthy controls. Researchers have collected the sample from individuals; RNA sequencing was performed by constructing a library using Illumina HiSeq 2500. Further, analysis of their data reveals the direct molecular mechanism of bleeding due to decreased platelet count in dengue patients [98]. RNA seq has been used in various studies to explore the potential of noncoding RNA as biomarkers and antiviral targets by analyzing the expression profiles of various noncoding RNAs during different viral infections [99,100,101,102,103,104,105]. An RNA seq study was carried out by Castillo et al. [2], where miRNA profiling in DENV-infected primary macrophages was done which identifies miR-3614-5p as an antiviral target [2]. piRNA profiling was done in DENV-infected Asian tiger mosquito and midgut [101]. On the other hand, differential expression of lncRNA during DENV infection was also carried out in which lncRNA was proven to be the potential biomarkers for disease progression [106].
Next-generation sequencing has been widely used nowadays for profiling and discovery of small noncoding RNAs in various infectious diseases. It has been used for profiling of small noncoding RNAs in dengue virus-infected Aedes mosquito cells to discover specific viral small noncoding RNAs that facilitate viral replication [107].
Illumina has been widely used to determine an active small interfering RNA-based antiviral response in case of West Nile virus-infected mosquitos [108] and to discover microRNA-like small RNA which autoregulates replication of dengue-2 virus in mosquito cells [109]. A study by Samuel et al. in 2018 carried out multiplex sequencing of DENV serotypes using Illumina and nanopore sequencing simultaneously. They found that multiplex sequencing was robust and generated full genome coverage for all DENV isolates in first attempt unlike single plex approach which failed to produce several amplicons [55]. In a meta-analysis study, microarray, Affymetrix, and Illumina datasets were compared which revealed the modulation of extracellular matrix and cell junctions during DENV infection which might prove to be the gene signatures of dengue infection [110]. Illumina has been also used to sequence tRNA-derived fragments during infection of respiratory syncytial viral infection [111]. In HIV-infected cells, novel miRNAs and piRNAs have been identified by using next-generation sequencing platform illumine [112].
With the advancements in omics technologies, third-generation sequencing platforms like nanopore can be used for direct sequencing of RNA. Analysis of viral transcriptome using conventional RNA sequencing methods is more complicated because of complexity in splicing patterns, overlapping reading frames, and high gene density. To overcome these problems, Depledge et al. in 2019 used nanopore sequencing to directly sequence RNA from host and viral RNA to study host-pathogen interaction during herpes simplex virus infection [113].
8 Conclusion and Future Perspectives
Before 2005, noncoding RNA field was very much scarcely studied. But with the emerging next-generation sequencing technologies, the number of studies increased in the last decade. Various tools and techniques have been introduced to make the ncRNA study to be more comprehensive. This chapter has reviewed the current and emerging approaches for decoding the spectrum of ncRNA functions with the primary focus on virus-related tools and techniques. The field of miRNA is the extensively studied field followed by siRNA and piRNA. While various bioinformatics tools and databases are available for miRNA, siRNA, piRNA, snoRNA, etc., for many ncRNAs, there is a lack of tools and databases for their study which primarily includes lncRNA, circRNA, virus-associated RNAs, etc. This could be one reason for impeding the associated research. Moreover, several tools, techniques, and databases have recently been introduced which help to dig deeper into the possible functions of ncRNAs. Due to extensive expansion of various transcriptome analysis tools, it is expected that various roles of noncoding RNAs are to be discovered in the future which will lead to better understanding of various virus pathophysiologies and development of noncoding RNA-based diagnosis and therapeutics.
References
Wu S, He L, Li Y, Wang T, Feng L, Jiang L, Zhang P, Huang X (2013) MiR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J Infect 67(4):329–341. https://doi.org/10.1016/j.jinf.2013.05.003
Castillo JA, Castrillón JC, Diosa-Toro M, Betancur JG, St Laurent G, Smit JM, Urcuqui-Inchima S (2016) Complex interaction between dengue virus replication and expression of miRNA-133a. BMC Infect Dis 16(1):29. https://doi.org/10.1186/s12879-016-1364-y
Firmino N, Martinez VD, Rowbotham DA, Enfield KSS, Bennewith L, Lam WL (2017) HPV status is associated with altered PIWI-interacting RNA expression pattern in head and neck cancer. Oral Oncol 55:43–48. https://doi.org/10.1016/j.oraloncology.2016.01.012.HPV
Léger P, Lara E, Jagla B, Sismeiro O, Mansuroglu Z, Coppée JY, Bonnefoy E, Bouloy M (2013) Dicer-2- and Piwi-mediated RNA interference in Rift Valley fever. J Virol 87(3):1631–1648. https://doi.org/10.1128/JVI.02795-12
Wang Q, Lee I, Ren J, Ajay SS, Lee YS (2013) Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol Ther 21(2):368–379. https://doi.org/10.1038/mt.2012.237
Schorn AJ, Gutbrod MJ, LeBlanc C, Martienssen R (2017) LTR-retrotransposon control by tRNA-derived small RNAs. Cell 170(1):61–71.e11. https://doi.org/10.1016/j.cell.2017.06.013
Watkins NJ, Bohnsack MT (2012) The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. WIREs RNA 3(June):397–414. https://doi.org/10.1002/wrna.117
Falaleeva M, Surface J, Shen M, de la Grange P, Stamm S (2015) SNORD116 and SNORD115 change expression of multiple genes and modify each other’s activity. Gene 572(2):266–273
Yu F, Bracken CP, Pillman KA, Lawrence DM, Goodall GJ, Callen DF, Neilsen PM (2015) p53 represses the oncogenic Sno-MiR-28 derived from a SnoRNA. PLoS One 10(6):1–20. https://doi.org/10.1371/journal.pone.0129190
Ma X, Han N, Shao C, Meng Y (2017) Transcriptome-wide discovery of PASRs (promoter-associated small RNAs) and TASRs (terminus-associated small RNAs) in Arabidopsis thaliana. PLoS One 12(1):1–22. https://doi.org/10.1371/journal.pone.0169212
Seila AC, Calabrese JM, Levine SS, Yeo GW, Peter B, Flynn RA, Young RA, Sharp PA (2009) Divergent transcription from active promoters. Science 322(5909):1849–1851. https://doi.org/10.1126/science.1162253.Divergent
Wang M, Beck CR, English AC, Meng Q, Buhay C, Han Y, Doddapaneni HV, Yu F, Boerwinkle E, Lupski JR, Muzny DM, Gibbs RA (2015a) PacBio-LITS: a large-insert targeted sequencing method for characterization of human disease-associated chromosomal structural variations. BMC Genomics 16(1):1–12. https://doi.org/10.1186/s12864-015-1370-2
Wang Y, Yao JIE, Meng H, Yu Z, Wang Z, Yuan X, Chen H, Wang A (2015b) A novel long non-coding RNA, hypoxia-inducible factor-2 α promoter upstream transcript, functions as an inhibitor of osteosarcoma stem cells in vitro. Mol Med Rep 11:2534–2540. https://doi.org/10.3892/mmr.2014.3024
Fan J, Cheng M, Chi X, Liu X, Yang W (2019) A human Long non-coding RNA LncATV promotes virus replication through restricting RIG-I – mediated innate immunity. Front Immunol 10(July):1–8. https://doi.org/10.3389/fimmu.2019.01711
Zhang L, Xu L, Zhang C, Lu Y, Ji T, Ling L (2017) uc38 induces breast cancer cell apoptosis via PBX1. Am J Cancer Res 7(12):2438–2451
Iiott NE, Heward JA, Roux B, Tsitsiou E, Fenwick PS, Lenzi L, Goodhead I, Hertz-Fowler C, Heger A, Hall N, Donnelly LE, Sims D, Lindsay MA (2014) Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Commun 5:3979. https://doi.org/10.1038/ncomms4979
Greene J, Baird A, Brady L, Lim M, Gray SG, Mcdermott R, Finn SP (2017) Circular RNAs: biogenesis, function and role in human diseases. Front Mol Biosci 4(June):1–11. https://doi.org/10.3389/fmolb.2017.00038
Tagawa T, Gao S, Koparde VN, Gonzalez M, Spouge JL, Serquiña AP, Lurain K, Ramaswami R, Uldrick TS, Yarchoan R, Ziegelbauer JM (2018) Discovery of Kaposi’s sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA. Proc Natl Acad Sci U S A 115(50):12805–12810. https://doi.org/10.1073/pnas.1816183115
Göertz GP, van Bree JWM, Hiralal A, Fernhout BM, Steffens C, Boeren S, Visser TM, Vogels CBF, Abbo SR, Fros JJ, Koenraadt CJM, van Oers MM, Pijlman GP (2019) Subgenomic flavivirus RNA binds the mosquito DEAD/H-box helicase ME31B and determines Zika virus transmission by Aedes aegypti. Proc Natl Acad Sci U S A 116(38):19136–19144. https://doi.org/10.1073/pnas.1905617116
Schuessler A, Funk A, Lazear HM, Cooper DA, Torres S, Daffis S, Jha BK, Kumagai Y, Takeuchi O, Hertzog P, Silverman R, Akira S, Barton DJ, Diamond MS, Khromykh AA (2012) West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J Virol 86(10):5708–5718. https://doi.org/10.1128/jvi.00207-12
Prusty BK, Gulve N, Govind S, Krueger GRF, Feichtinger J, Larcombe L, Aspinall R, Ablashi DV, Toro CT (2018) Active HHV-6 infection of cerebellar Purkinje cells in mood disorders. Front Microbiol 9(August):1–12. https://doi.org/10.3389/fmicb.2018.01955
Takahashi H et al (2014) 5′ end-centered expression profiling using cap-analysis gene expression (CAGE) and next-generation sequencing Hazuki. Nature Protocol 7(3):542–561. https://doi.org/10.1038/nprot.2012.005.5
Schena M, Shalon D, Davis RW, Brownt P (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270(October):467–470
Searfoss GH, Ryan TP, Jolly RA (2005) The role of transcriptome analysis in pre-clinical toxicology. Curr Mol Med 5(February 2015):52–64
Mutz K, Heilkenbrinker A, Lo M, Stahl F (2013) Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol 24:22–30. https://doi.org/10.1016/j.copbio.2012.09.004
Huyghe A, Francois P, Schrenzel J (2020) Characterization of microbial pathogens by DNA microarray. Infect Genet Evol 9(January):987–995. https://doi.org/10.1016/j.meegid.2008.10.016
Parnell GP, Tang BM, Nalos M, Armstrong NJ, Huang SJ, Booth DR, Mclean AS (2013) Identifying key regulatory genes in the whole blood of septic patients to monitor underlying immune dysfunctions. Shock 40(3):166–174. https://doi.org/10.1097/SHK.0b013e31829ee604
Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, Derisi JL (2002) Microarray-based detection and genotyping of viral pathogens. PNAS 99(24):15687–15692
Wang D, Urisman A, Liu Y, Springer M, Ksiazek TG, Erdman DD, Mardis ER, Hickenbotham M, Magrini V, Eldred J, Latreille JP, Wilson RK, Ganem D, Derisi JL (2003) Viral discovery and sequence recovery using DNA microarrays. PLoS Biol 1(2):257–260. https://doi.org/10.1371/journal.pbio.0000002
Bertone P, Gerstein M, Snyder M (2005) Applications of DNA tiling arrays to experimental genome annotation and regulatory pathway discovery. Chromosom Res 13:259–274
Yazaki J et al (2009) Mapping the genome landscape using tiling array technology. Curr Opin Plant Biol 10(5):534–542. https://doi.org/10.1016/j.pbi.2007.07.006.Mapping
Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, Tammana H, Gingeras TR (2004) Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res 14:331–342. https://doi.org/10.1101/gr.2094104.Sanger
Lawrence JG, Hendrix RW, Lawrence JG, Hendrix RW (2001) Where are the pseudogenes in bacterial genomes ? Trends Microbiol 9(11):535–540
Buck MJ, Lieb JD (2004) ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83(3):349–360. https://doi.org/10.1016/j.ygeno.2003.11.004
Zhu H et al (1998) Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Microbiology 95(November):14470–14475
de Saizieu A et al (1998) Bacterial transcript imaging by hybridization of total RNA to oligonucleotide arrays. Nat Biotechnol 16:45–48
Li H (2001) Cluster-Rasch models for microarray gene expression data. Genome Biol 2:1–13
Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, B. EL. (1996) Expression monitoring by hybridization to high- density oligo nucleotide arrays. Nat Biotechnol 14:1675–1680
Murphy D (2020) Gene expression studies using microarrays. Adv Physiol Educ 26(4):256–270
Choi C, Yoon S, Moon H, Bae Y, Kim C, Diskul-na-ayudthaya P, Van Ngu T, Munir J, Han J, Park S, Moon J, Song S, Ryu S (2018) mirRICH, a simple method to enrich the small RNA fraction from over-dried RNA pellets. RNA Biol 6286:763–772. https://doi.org/10.1080/15476286.2018.1451723
Chaudhary S, Chaudhary PS, Vaishnani TA (2016) Small RNA extraction using fractionation approach and library preparation for NGS platform. J Adv Res Biotechnol 1:7
Jin G, Cid M, Poole CB, McReynolds LA (2010) Protein mediated miRNA detection and siRNA enrichment using p19. BioTechniques 48(6):xvii–xxiii. https://doi.org/10.2144/000113364
Grentzinger T, Oberlin S, Schott G, Handler D, Svozil J, Barragan-borrero V, Humbert A, Duharcourt S, Brennecke J, Voinnet O (2020) A universal method for the rapid isolation of all known classes of functional silencing small RNAs. Nucleic Acid Res 48(14):1–15. https://doi.org/10.1093/nar/gkaa472
Becker C, Hammerle-Fickinger A, Riedmaier I, Pfaffl MW (2010) mRNA and microRNA quality control for RT-qPCR analysis. Methods 50(4):237–243. https://doi.org/10.1016/j.ymeth.2010.01.010
Nilsen TW (2013) Splinted ligation method to detect small RNAs. Cold Springer Harbor Laboratory Press, Harbor, NY, pp 54–59. https://doi.org/10.1101/pdb.prot072611
Caifu C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33(20):1–9. https://doi.org/10.1093/nar/gni178
Fakruddin M, Chowdhury A (2012) Pyrosequencing-an alternative to traditional sanger sequencing. Am J Biochem Biotechnol 8(1):14–20. https://doi.org/10.3844/ajbbsp.2012.14.20
Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW (2007) Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res 17(8):1195–1201. https://doi.org/10.1101/gr.6468307
Merriman B, Torrent I, Rothberg JM (2012) Progress in ion Torrent semiconductor chip based sequencing. Electrophoresis 33(23):3397–3417. https://doi.org/10.1002/elps.201200424
Fujimoto M, Moyerbrailean GA, Noman S, Gizicki JP, Ram ML, Green PA, Ram JL (2014) Application of ion torrent sequencing to the assessment of the effect of alkali ballast water treatment on microbial community diversity. PLoS One 9(9):1–9. https://doi.org/10.1371/journal.pone.0107534
Gamez S, Antoshechkin I, Mendez-Sanchez SC, Akbari OS (2020) The developmental transcriptome of aedes albopictus, a major worldwide human disease vector. G3: Genes, Genomes. Genetics 10(3):1051–1062. https://doi.org/10.1534/g3.119.401006
Amar L, Benoit C, Beaumont G, Vacher CM, Crepin D, Taouis M, Baroin-Tourancheau A (2012) MicroRNA expression profiling of hypothalamic arcuate and paraventricular nuclei from single rats using Illumina sequencing technology. J Neurosci Methods 209(1):134–143. https://doi.org/10.1016/j.jneumeth.2012.05.033
Ondov BD, Varadarajan A, Passalacqua KD, Bergman NH (2008) Efficient mapping of applied Biosystems SOLiD sequence data to a reference genome for functional genomic applications. Bioinformatics 24(23):2776–2777. https://doi.org/10.1093/bioinformatics/btn512
Li S, Wang H, Qi Y, Tu J, Bai Y, Tian T, Huang N, Wang Y, Xiong F, Lu Z, Xiao Z (2011) Assessment of nanomaterial cytotoxicity with SOLiD sequencing-based microRNA expression profiling. Biomaterials 32(34):9021–9030. https://doi.org/10.1016/j.biomaterials.2011.08.033
Jain M, Olsen HE, Paten B, Akeson M (2016) The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17(1):1–11. https://doi.org/10.1186/s13059-016-1103-0
Stubbs S, Blacklaws B, Yohan B, Yudhaputri F, Schwem B, Salvaña E, Destura R, Myint K, Sasmono RT, Frost S (2018) A Nanopore-based method for generating complete coding region sequences of dengue virus in resource-limited settings. Virol J. https://doi.org/10.1101/499111
Rhoads A, Au KF (2015) PacBio sequencing and its applications. Genom Proteom Bioinf 13(5):278–289. https://doi.org/10.1016/j.gpb.2015.08.002
Ruano P, Delgado LL, Picco S, Villegas L, Tonelli F, Merlo M, Rigau J, Diaz D, Masuelli M (2016) We are IntechOpen, the world’s leading publisher of Open Access books Built by scientists, for scientists TOP 1%. Intech, tourism, 13. https://www.intechopen.com/books/advanced-biometric-technologies/liveness-detection-in-biometrics
Fasold M, Langenberger D, Binder H, Stadler PF, Hoffmann S (2011) DARIO: a ncRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acid Res 39(May):112–117. https://doi.org/10.1093/nar/gkr357
Axtell MJ (2013) ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19:740–751. https://doi.org/10.1261/rna.035279.112.of
Chen C-j, Servant N, Toedling J, Sarazin A, Marchais A, Duvernois-berthet E, Colot V, Voinnet O, Heard E, Ciaudo C, Barillot E, Strasbourg D, Strasbourg F, Upr C (2012) ncPRO-seq: a tool for annotation and profiling of ncRNAs in sRNA-seq data. Bioinformatics 28(23):3147–3149. https://doi.org/10.1093/bioinformatics/bts587
Aransay AM, Hackenberg M (2011) miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acid Res 39(April):132–138. https://doi.org/10.1093/nar/gkr247
Williamson V, Kim A, Xie B, Mcmichael GO, Gao Y, Vladimirov V (2012) Detecting miRNAs in deep-sequencing data: a software performance comparison and evaluation. Brief Bioinform 14(1):36–45. https://doi.org/10.1093/bib/bbs010
Gao D, Middleton R, Rasko JEJ, Ritchie W (2013) Bioinformatics applications note Sequence analysis miREval 2.0: a web tool for simple microRNA prediction in genome sequences. Bioinformatics 29(24):3225–3226. https://doi.org/10.1093/bioinformatics/btt545
Ronen R, Gan I, Modai S, Sukacheov A, Dror G, Halperin E, Shomron N (2010) miRNAkey: a software for microRNA deep sequencing analysis. Bioinformatics 26(20):2615–2616. https://doi.org/10.1093/bioinformatics/btq493
Sun Z, Evans J, Bhagwate A, Middha S, Bockol M, Yan H, Kocher J (2014) CAP-miRSeq: a comprehensive analysis pipeline for microRNA sequencing data. BMC Genomics 15:1–10
Zheng L, Xu W, Liu S, Sun W, Li J, Wu J, Yang J, Qu L (2016) tRF2Cancer: a web server to detect tRNA-derived small RNA fragments (tRFs) and their expression in multiple cancers. Nucleic Acid Res 44(May):185–193. https://doi.org/10.1093/nar/gkw414
Selitsky SR, Sethupathy P (2015) tDRmapper: challenges and solutions to mapping, naming, and quantifying tRNA-derived RNAs from human small RNA-sequencing data. BMC Bioinf 16:1–13. https://doi.org/10.1186/s12859-015-0800-0
Guo Q, Qu X, Jin W (2015) PhaseTank: genome-wide computational identification of phasiRNAs and their regulatory cascades. BBA – Gene Regul Mech 31(2):284–286. https://doi.org/10.1093/bioinformatics/btu628
Lu Y, Baras AS, Halushka MK (2018) miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinformatics 19:1–12
Wan C, Gao J, Zhang H, Jiang X, Zang Q, Ban R, Zhang Y, Shi Q (2017) Sequence analysis CPSS 2.0: a computational platform update for the analysis of small RNA sequencing data. Bioinformatics 33(February):3289–3291. https://doi.org/10.1093/bioinformatics/btx066
Zhao F, Lang H, Wang Z, Zhang T, Zhang D, Wang R, Lin X, Liu X, Shi P, Pang X (2019) Human novel MicroRNA Seq-915_x4024 in keratinocytes contributes to skin regeneration by suppressing scar formation. Mol Ther Nucleic Acid 14(March):410–423. https://doi.org/10.1016/j.omtn.2018.12.016
Rahman R, Gautam A, Bethune J, Sattar A, Fiosins M, Magruder DS, Capece V, Shomroni O, Bonn S (2018) Oasis 2: improved online analysis of small RNA-seq data. BMC Bioinf 19:1–10
Stocks MB, Mohorianu I, Beckers M, Paicu C, Moxon S, Thody J, Dalmay T, Moulton V (2018) Sequence analysis the UEA sRNA Workbench (version 4.4): a comprehensive suite of tools for analyzing miRNAs and sRNAs. Bioinformatics 34(May):3382–3384. https://doi.org/10.1093/bioinformatics/bty338
Cristina G, Rueda A, Barturen G, Lebr R, Oliver L, Hackenberg M (2015) sRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acid Res 43(May):467–473. https://doi.org/10.1093/nar/gkv555
Valladares O, Kuksa PP, Amlie-wolf A, Wang L, Leung YY (2018) SPAR: small RNA-seq portal for analysis of ˇ Katani c. Nucleic Acid Research 46(May):36–42. https://doi.org/10.1093/nar/gky330
Liu Q, Ding C, Lang X, Guo G, Chen J (2019) Small noncoding RNA discovery and profiling with sRNA tools based on high-throughput sequencing. Brief Bioinform 00(July):1–11. https://doi.org/10.1093/bib/bbz151
Gebert D, Hewel C, Rosenkranz D (2017) Unitas: the universal tool for annotation of small RNAs. BMC Genomics 18:1–14. https://doi.org/10.1186/s12864-017-4031-9
Bonnal RJP, Rossi RL, Carpi D, Ranzani V, Abrignani S, Pagani M (2015) MiRiadne: A web tool for consistent integration of miRNA nomenclature. Nucleic Acids Res 43(W1):W487–W492. https://doi.org/10.1093/nar/gkv381
Hausser J, Berninger P, Rodak C, Jantscher Y, Wirth S, Zavolan M (2009) MirZ: an integrated microRNA expression atlas and target prediction resource. Nucleic Acids Res 37(Suppl. 2):266–272. https://doi.org/10.1093/nar/gkp412
Zhang Y, Zang Q, Xu B, Zheng W, Ban R, Zhang H, Yang Y, Hao Q, Iqbal F, Li A, Shi Q (2016) IsomiR Bank: a research resource for tracking IsomiRs. Bioinformatics 32(13):2069–2071. https://doi.org/10.1093/bioinformatics/btw070
Truss M, Swat M, Kielbasa SM, Schäfer R, Herzel H, Hagemeier C (2005) HuSiDa – the human siRNA database: an open-access database for published functional siRNA sequences and technical details of efficient transfer into recipient cells. Nucleic Acids Res 33(DATABASE ISS):108–111. https://doi.org/10.1093/nar/gki131
Sai Lakshmi S, Agrawal S (2008) piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res 36(Suppl. 1):173–177. https://doi.org/10.1093/nar/gkm696
Lestrade L, Weber MJ (2006) snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34(Database issue):158–162. https://doi.org/10.1093/nar/gkj002
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR (2003) Rfam: an RNA family database. Nucleic Acids Res 31(1):439–441. https://doi.org/10.1093/nar/gkg006
Xiyuan L, Dechao B, Liang S, Yang W, Shuangsang F, Hui L, Haitao L, Chunlong L, Wenzheng F, Runsheng C, Yi Z (2017) Using the NONCODE database resource. Curr Protoc Bioinformatics 2017:12.16.1–12.16.20. https://doi.org/10.1002/cpbi.25
Pang KC, Stephen S, Dinger ME, Engström PG, Lenhard B, Mattick JS (2007) RNAdb 2.0 – an expanded database of mammalian non-coding RNAs. Nucleic Acids Res 35(SUPPL. 1):178–182. https://doi.org/10.1093/nar/gkl926
Yang JH, Shao P, Zhou H, Chen YQ, Qu LH (2009) deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res 38(Suppl.1):123–130. https://doi.org/10.1093/nar/gkp943
Kin T, Yamada K, Terai G, Okida H, Yoshinari Y, Ono Y, Kojima A, Kimura Y, Komori T, Asai K (2007) fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res 35(SUPPL. 1):145–148. https://doi.org/10.1093/nar/gkl837
Barón OL, Ursic-bedoya RJ, Lowenberger CA, Ocampo CB (2010) Differential gene expression from midguts of refractory and susceptible lines of the mosquito, Aedes aegypti, infected with Dengue-2 virus. J Insect Sci 10:1–23
Guo X, Yang F, Zhao T, Dong Y, Lu B (2009) Initial analysis of gene expressions in response to dengue-2 virus infection in Aedes albopictus (Diptera: Culicidae). J Agric Urban Entomol 26(4):157–165. https://doi.org/10.3954/1523-5475-26.4.157
Falk KI, Lindegren G, Mouzavi-jazi M (2005) DNA microarray technique for detection and identification of seven Flaviviruses pathogenic for man. J Gen Virol 540(August):528–540. https://doi.org/10.1002/jmv.20489
Díaz-Badillo A, Muñoz M d L, Perez-Ramirez G, Altuzar V, Burgueño J, Mendoza-Alvarez JG, Martínez-Muñoz JP, Cisneros A, Navarrete-Espinosa J, Sanchez-Sinencio F (2014) A DNA microarray-based assay to detect dual infection with two dengue virus serotypes. Sensors (Switzerland) 14(5):7580–7601. https://doi.org/10.3390/s140507580
Loke P, Hammond SN, Leung JM, Kim CC, Batra S, Balmaseda A, Harris E (2010) Gene expression patterns of dengue virus-infected children from Nicaragua reveal a distinct signature of increased metabolism. PLoS Negl Trop Dis 4(6):1–12. https://doi.org/10.1371/journal.pntd.0000710
Simmons CP, Popper S, Dolocek C, Nguyen T, Chau B, Griffiths M, Thi N, Dung P, Long TH, Hoang DM, Chau NV, Thi L, Thao T, Hien TT, Relman DA, Farrar J (2007) Patterns of host genome – wide gene transcript abundance in the peripheral blood of patients with acute dengue hemorrhagic fever. J Infect Dis 195:1097–1107. https://doi.org/10.1086/512162
Dacheux L, Berthet N, Dissard G, Holmes EC, Delmas O, Larrous F, Guigon G, Dickinson P, Faye O, Sall AA, Old IG, Kong K, Kennedy GC, Manuguerra J, Cole ST, Gessain A (2010) Application of broad-Spectrum resequencing microarray for. J Virol 84(18):9557–9574. https://doi.org/10.1128/JVI.00771-10
Ayodeji M, Kulka M, Jackson SA, Patel I, Mammel M, Cebula TA, Goswami BB (2009) A microarray based approach for the identification of common foodborne viruses. Open Virol J 3:7–20
Huang Y, Warden C, Zhu H (2013) Accurate identification of non-polyadenylated RNA using a custom human cytomegalovirus tiling array. J Virol Methods 187(1):90–93. https://doi.org/10.1016/j.jviromet.2012.09.020
Banerjee A, Shukla S, Pandey AD, Goswami S, Bandyopadhyay B, Ramachandran V, Das S, Malhotra A, Agarwal A, Adhikari S, Rahman M, Chatterjee S, Bhattacharya N, Basu N, Pandey P, Sood V, Vrati S (2017) RNA-Seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with disease progression in dengue patients. Transl Res 186:62–78.e9. https://doi.org/10.1016/j.trsl.2017.06.007
Bao S, Jia L, Zhou X, Zhang ZG, Wu HWL, Yu Z, Ng G, Fan Y, Wong DSM, Huang S, Wang To KK, Yuen KY, Yeung ML, Song YQ (2018) Integrated analysis of mRNA-seq and miRNA-seq for host susceptibilities to influenza A (H7N9) infection in inbred mouse lines. Funct Integr Genomics. https://doi.org/10.1007/s10142-018-0602-3
Barral-Arca R, Gómez-Carballa A, Cebey-López M, Currás-Tuala MJ, Pischedda S, Viz-Lasheras S, Bello X, Martinón-Torres F, Salas A (2020) Rna-seq data-mining allows the discovery of two long non-coding rna biomarkers of viral infection in humans. Int J Mol Sci. https://doi.org/10.3390/ijms21082748
Hess AM, Prasad AN, Ptitsyn A, Ebel GD, Olson KE, Barbacioru C, Monighetti C, Campbell CL (2011) Small RNA profiling of dengue virus-mosquito interactions implicates the PIWI RNA pathway in anti-viral defense. BMC Microbiol. https://doi.org/10.1186/1471-2180-11-45
Lin YT, Kincaid RP, Arasappan D, Dowd SE, Hunicke-Smith SP, Sullivan CS (2010) Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA. https://doi.org/10.1261/rna.1967910
Lu S, Zhu N, Guo W, Wang X, Li K, Yan J, Jiang C, Han S, Xiang H, Wu X, Liu Y, Xiong H, Chen L, Gong Z, Luo F, Hou W (2020) RNA-Seq revealed a circular RNA-microRNA-mRNA regulatory network in Hantaan virus infection. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2020.00097
Shi J, Hu N, Mo L, Zeng Z, Sun J, Hu Y (2018) Deep RNA sequencing reveals a repertoire of human fibroblast circular RNAs associated with cellular responses to herpes simplex virus 1 infection. Cell Physiol Biochem. https://doi.org/10.1159/000491471
Wong AMG, Kong KL, Tsang JWH, Kwong DLW, Guan XY (2012) Profiling of Epstein-Barr virus-encoded microRNAs in nasopharyngeal carcinoma reveals potential biomarkers and oncomirs. Cancer. https://doi.org/10.1002/cncr.26309
Wang XJ, Jiang SC, Wei HX, Deng SQ, He C, Peng HJ (2017) The differential expression and possible function of long noncoding RNAs in liver cells infected by dengue virus. Am J Trop Med Hyg 97(6):1904–1912. https://doi.org/10.4269/ajtmh.17-0307
Miesen P, Ivens A, Buck AH, van Rij RP (2016) Small RNA profiling in dengue virus 2-infected Aedes Mosquito cells reveals viral piRNAs and novel host miRNAs. PLoS Negl Trop Dis 10(2):1–22. https://doi.org/10.1371/journal.pntd.0004452
Göertz GP, Fros JJ, Miesen P, Vogels CBF, van der Bent ML, Geertsema C, Koenraadt CJM, van Rij RP, van Oers MM, Pijlman GP (2016) Noncoding subgenomic Flavivirus RNA is processed by the mosquito RNA interference machinery and determines West Nile virus transmission by Culex pipiens mosquitoes. J Virol 90(22):10145–10159. https://doi.org/10.1128/jvi.00930-16
Hussain M, Asgari S (2014) MicroRNA-like viral small RNA from dengue virus 2 autoregulates its replication in mosquito cells. Proc Natl Acad Sci U S A 111(7):2746–2751. https://doi.org/10.1073/pnas.1320123111
Afroz S, Giddaluru J, Abbas MM, Khan N (2016) Transcriptome meta-analysis reveals a dysregulation in extra cellular matrix and cell junction associated gene signatures during dengue virus infection. Sci Rep 6(September):1–12. https://doi.org/10.1038/srep33752
Zhou J, Liu S, Chen Y, Fu Y, Silver AJ, Hill MS, Lee I, Lee YS, Bao X (2017) Identification of two novel functional tRNA-derived fragments induced in response to respiratory syncytial virus infection. J Gen Virol 98(7):1600–1610. https://doi.org/10.1099/jgv.0.000852
Chang ST, Thomas MJ, Sova P, Green RR, Palermo RE, Katze MG (2013) Next-generation sequencing of small RNAs from HIV-infected cells identifies phased microRNA expression patterns and candidate novel microRNAs differentially expressed upon infection. MBio 4(1):1–10. https://doi.org/10.1128/mBio.00549-12
Depledge DP, Srinivas KP, Sadaoka T, Bready D, Mori Y, Placantonakis DG, Mohr I, Wilson AC (2019) Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat Commun 10(1):754. https://doi.org/10.1038/s41467-019-08734-9
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Madhry, D. et al. (2021). Various Transcriptomic Approaches and Their Applications to Study Small Noncoding RNAs in Dengue and Other Viruses. In: Hameed, S., Fatima, Z. (eds) Integrated Omics Approaches to Infectious Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-16-0691-5_12
Download citation
DOI: https://doi.org/10.1007/978-981-16-0691-5_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0690-8
Online ISBN: 978-981-16-0691-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)