
Abstract
Small non-coding RNAs (sncRNAs) derived from tRNAs are known as tRNA-derived small RNAs (tsRNAs). These tsRNAs are further categorized into tRNA-derived fragments (tRFs) and tRNA halves (tiRNAs), which play significant roles in the various molecular mechanisms underlying certain human diseases. However, the generation of tsRNAs and their potential roles during Dengue virus (DENV) infection is not yet known. Here, we performed small RNA sequencing to identify the generation and alterations in tsRNAs expression profiles of DENV-infected Huh7 cells. Upon DENV infection, tRNA fragmentation was found to be increased. We identified a significant number of differentially expressed tsRNAs during DENV infection. Interestingly, the 3′tRF population showed upregulation, while the i-tRF population exhibited downregulation. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed to analyze the impact of differentially expressed tsRNAs on DENV pathogenesis. Our results suggest that differentially expressed tsRNAs are involved in transcriptional regulation via RNA polymerase II promoter and metabolic pathways. Overall, our study contributes significantly to our understanding of the roles played by tsRNAs in the complex dynamics of DENV infection.
Similar content being viewed by others

Introduction
Dengue is considered as the leading cause of arthropod-borne viral disease in humans. It is endemic in more than 100 nations across tropical and subtropical parts of the world1,2. There are four dengue virus serotypes: DENV-1, DENV-2, DENV-3, and DENV-4. Each of these can causes an acute illness called as Dengue fever, characterized by high fever and joint pain2. Dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) are the most severe and fatal form of DENV infection. According to WHO, half of the population of the world is at risk of dengue with approximately 100–400 million cases occurring each year. This has a significant negative impact on the health and economy of the affected countries, as no vaccines or antiviral drugs have been developed to combat dengue infection3. There is a necessity to understand the molecular mechanism underlying DENV pathogenesis4.
Noncoding RNAs (ncRNAs) are the functional RNA molecules that are not translated into proteins. They are expressed at lower levels compared to mRNAs but play crucial roles in regulatory functions. Based on their respective roles in biology, ncRNAs are categorized into constitutively expressed infrastructural ncRNAs and regulatory ncRNAs. Constitutively expressed infrastructural ncRNAs consist of ribosomal, transfer, small nuclear, and small nucleolar RNAs, which are involved in chromatin modification, mRNA splicing, transcriptional and translational modulation, and mechanisms involving physiological and biochemical functions5. Regulatory ncRNAs include microRNAs (miRNAs), Piwi-interacting RNAs (piRNAs), small interfering RNAs (siRNAs), long non-coding RNAs (lncRNAs) and tRNA-derived small RNAs (tsRNAs), which is an emerging class of sncRNAs. These tRNA fragments play an important role in regulating transcription and translation machinery6,7,8,9.
tRNA-derived small RNAs (tsRNAs) typically ranges from 15 to 50 nucleotide (nt) in length. They are generated from premature and mature tRNAs by the enzymatic action of ribonucleases such as ELAC2, DICER, RNaseZ, and Angiogenin, etc., under stressed conditions10. Based on their length, tsRNAs are further categorized into tRNA halves (tiRNAs) and tRNA-derived fragments (tRFs). tiRNAs, usually 31–40 nt long, are generated by the action of ribonuclease Angiogenin (ANG), which cleaves the anticodon loop of tRNA to produce either the 5′half or the 3′half. They are typically produced in response to stress stimuli such as hypoxia, oxidative stress, heat shock and cold shock11,12,13,14. Additionally, tiRNAs can be generated in response to sex hormone stimulation, referred to as Sex Hormone dependent tRNA-derived RNAs (SHOT RNAs). These can further be categorised into 5′SHOT RNA and 3′SHOT RNA. 5′SHOT RNA contains phosphate group at the 5′ end and a cyclic phosphate (cP) group at the 3′end while 3′SHOT RNA contains a hydroxyl group at the 5′ end and the amino acid at the 3′end15. tRFs, ranging from 14 to 30 nt in length, are primarily formed by the action of DICER and RNase Z on the D loop (dihydrouridine) and T loop (TѱC) of tRNAs16. Depending on the cleavage sites, tRFs are classified into 3′tRF, 5′tRF, and i-tRF. 5′tRFs are generated through DICER cleavage at the D-loop or at the stem positions between the D-loop and the anticodon loop. They are further sub-categorized into tRF-5a (13–15 nt), tRF-5b (21–23 nt), and tRF-5c (27–30 nt) based on nucleotide length and the site of incision. On the other hand, 3′tRFs are generated by the action of DICER and Angiogenin cleavage at the T-loop or between T-loop and the anticodon loop which are further classified into tRF-3a (19–23 nt) and tRF-3b (20–28 nt). i-tRFs (internal tRFs) are produced from internal regions of mature tRNA. These consists of anticodon loop and the segments of D-loop or T-loop17. Additionally, two more classes of tRFs exist: tRF-1 and tRF-2. tRF-1s are generated by ribonuclease Z (ELAC2) from 3′end of pre-tRNA containing poly-U tail. tRF-2s are generated from anticodon loop and stem sequences (except loop regions). These tRNA-derived small RNAs play crucial roles in regulating cancer, cell-proliferation, apoptosis, infective biology, metabolic disorders, transcription and translation machinery, and viral infection11,12,13,18,19,20,21,22.
Here, we have studied the expression and functional characterization of tsRNAs during DENV infection. Our study reveals that tRNA fragments are indeed differentially expressed during DENV infection. Previous studies has shown the modulation of various RNases during DENV infection23,24, leading us to hypothesize that tRNA fragmentation may be influenced by this modulation, potentially impacting DENV pathogenesis. In this study, we conducted a comprehensive RNA sequencing analysis to determine the differential expression pattern of tsRNAs in DENV-infected Huh7 cell lines relative to mock-infected controls. This research aims to provide insight into the differential expression of tRNA fragments during DENV infection, potentially advancing our understanding of DENV pathogenesis.
Results
DENV infection leads to differential tsRNA expression
Huh7 cells were infected with DENV and harvested at 0 h (2 h post adsorption) and 48 h post infection. RNA sequencing was performed to determine the expression levels of tsRNAs generated upon DENV infection, comparing 0 h and 48 h DENV-infected samples with the mock-infected samples. Triplicate samples were prepared for each condition. Briefly, RNA was isolated from Huh7 cell line using miRNeasy kit (Qiagen). RNA samples were quantified using a Qubit fluorimeter, and integrity was analysed by running them on an Agilent Tapestation 4200 RNA HS Screentape to determine the RNA Integrity Number (RIN). A RIN value from 7 to 10 denotes intact RNA with no degradation, while a RIN value < 5 represents complete degradation with poor quality RNA. All 9 samples displayed RIN value > 7. Total RNA was then pretreated with the rtStar™ tRF and tiRNA Pretreatment Kit to remove the RNA modifications, including the removal of 2′,3′-cyclic phosphate (3′cP) for 3′ adaptor ligation, removal of 5′-OH (hydroxyl group) for 5′ adaptor ligation, and demethylation for efficient reverse transcription. Following pre-treatment, adaptors were ligated, followed by cDNA synthesis. PCR amplified fragments were then size selected on an 8% TBE-PAGE gel to enrich 15–50 bp insert. The size selected reads were then sequenced on the Illumina NGS platform (Novaseq) (Fig. 1a). Raw reads were transformed into sequenced reads by base calling, which were recorded in the FASTQ file. Following quality control (QC), reads were aligned using MINTmap database. MINTmap v 2.0 is the repository of 640 mitochondrial as well as nuclear tRNAs and provides results in the form of exclusive and ambiguous tRNA fragments25. Total raw reads were subjected to MINTmap from which exclusive tRNA fragments were taken into consideration. Exclusive tRNA fragments are those whose sequence is unique to 640 tRNAs and cannot be found in any other genomic locations. In contrast, ambiguous tRNA fragments may be found in other genomic locations as well. Exclusive tsRNAs were considered for further analysis. Reads with < 10 read count were discarded, and differential expression analysis was performed through DEseq2 (Fig. 1b).

RNA sequencing. (a) Library preparation: Huh7 cells were mock-infected, 0 h DENV-infected, and 48 h DENV-infected. The samples were prepared in triplicates. Total RNA was isolated from the samples and analyzed with Bioanalyzer for integrity. Samples with RIN > 7 were used. After pre-treatment, adapter ligation, cDNA synthesis, and PCR amplification, fragments were size-selected on an 8% TBE-PAGE gel. These fragments were then used for RNA sequencing. (b) Sequencing Analysis: Illumina platform was used for paired-end (PE) sequencing. The raw reads were quality-controlled, identified, and quantified using MINTmap database, focusing on exclusive tRNA fragments to avoid ambiguity. Reads with a count less than 10 (< 10 read count) were discarded, and differential expression analysis was performed through DEseq2.
The RNA sequencing analysis suggests a significant increase in tRNA fragmentation in response to DENV infection. In Fig. 2a, the bar graph shows the number of tsRNAs generated across the different samples, while the pie chart represents the proportions of tsRNA generated within each sample. Upon DENV infection, tRNA fragmentation increases, as observed after DENV adsorption (0 h), and it further increases in parallel with DENV replication, as observed at 48 h of DENV infection. A total of 26,737 tsRNAs were significantly identified across all three samples. In Fig. 2b, Venn diagram shows that 595, 1044, and 3880 tsRNAs were specifically expressed in mock-infected, 0 h DENV-infected, and 48 h DENV-infected samples, respectively. Additionally, 92 tsRNAs were common in the 0 h vs mock group, 440 tsRNAs were common in the 48 h vs mock group, and 4338 tsRNAs were common in the 0 h vs 48 h group (Fig. 2b).

tsRNAs generated during DENV infection. (a) Bar graph (upper panel) and pie chart (lower panel) shows the number of tsRNAs generated and their percentage, respectively during the DENV infection in three different samples. (b) Venn diagram represents the unique and common tsRNAs expressed in different samples.
3′tRFs and i-tRFs are predominately differentially expressed during DENV infection
A total of 3826 tsRNAs were identified, consistently expressed across all three samples. These tsRNAs were further subjected to differential expression analysis using the DESeq2 package. The results were sorted using an adjusted P value of ≤ 0.05 and absolute log2 fold change ≥ 2 (absolute fold change ≥ 4). Differential tsRNAs expression was analysed in the two groups: 0 h vs mock and 48 h vs mock. 1041 and 959 tsRNAs were identified as significant tsRNAs that were differentially expressed in 0 h vs mock and 48 h vs mock groups, respectively (Fig. 3a). Out of these, 733 tRFs were found to be common in both groups. Among the commonly shared 733 tsRNAs, the highly abundant tRFs were 3′tRFs, which accounted for 47%, and i-tRFs contributing 38%, followed by 3′half, 5′half, and 5′tRF at 10%, 4%, and 1% respectively (Fig. 3b). It was observed that more than 80% of the 3′tRFs were upregulated, and more than 60% of i-tRFs were downregulated (Fig. 3c). From the bar graph in Fig. 3c, it was also interesting to note that the number of 3′halves and 5′halves (tiRNAs) generated were not equally distributed as a result of tRNA anticodon cleavage. This indicates that following tRNA cleavage in the anticodon loop, either one of the tiRNAs is undergoing degradation, or it is being further cleaved to produce other tRFs. The volcano plots for the 0 h vs mock and 48 h vs mock groups are depicted in Fig. 4a,b. The heatmap illustrates the expression pattern and hierarchical clustering of the top 40 up- and down-regulated tsRNAs in the 0 h vs mock and 48 h vs mock groups (Fig. 4c). Hierarchical clustering results revealed distinguishable differences in the expression profile of differentially expressed tsRNAs. The differentially expressed tsRNAs in 0 h vs mock group and 48 h vs mock group have been provided as the supplementary material (Supplementary Tables S1 and S2).

Differential expression analysis of tsRNAs. (a) Venn diagram represents shared and unique tsRNAs in the 0 h vs mock and in 48 h vs mock group. 733 tsRNAs were found to be commonly shared between both groups. (b) Pie diagram represents the percent modulation of tsRNAs subtypes. 3′tRFs (47%) and i-tRFs (38%) were found to be majorly modulated subtypes. (c) Bar plot shows the distribution of different types of tsRNAs. Most of the upregulated tRFs are 3′tRFs, and most of the downregulated tRFs are i-tRFs.

Expression profile of differentially expressed tsRNAs. (a) Volcano plot of 0 h vs mock and (b) 48 h vs mock group. Each dot represents tsRNAs. The green-colored dots represents down-regulated tsRNAs, the red-colored dot represents up-regulated tsRNAs and the black colored dots represents non-significant tsRNAs. Top 5 up- and down-regulated tsRNAs have been labelled in the plot. (c) Hierarchical clustering heatmap analysis for tsRNAs. Each row represents one tsRNA, and each column represents one sample.
Functional analysis of differentially expressed tsRNAs
In-silico functional analysis of the differentially expressed tsRNAs was performed. miRanda and TargetScan were used to predict the target gene interactions. The interacting genes were selected based on binding energy. Gene Ontology (GO) enrichment analysis, which effectively categorized outcomes into three vital domains: biological process (BP), molecular function (MF), and cellular component (CC), as depicted in Fig. 5.

Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) analysis. The GO and KEGG analysis revealed most enriched terms for Biological Processes (BP) are regulation of transcription from RNA polymerase II promoter and signal transduction. For Molecular Functions (MF), protein binding was identified as the most enriched term, while for Cellular Component (CC), nucleus and cytosol were identified. KEGG pathway analysis showed metabolic pathways and pathways in cancer to be the most enriched terms.
In the context of biological process (BP) analysis, our finding revealed the involvement of genes interacting with tsRNAs in key processes, primarily involved in the regulation of transcription from RNA polymerase II promoter, signal transduction, and apoptotic pathways. In molecular function (MF) analysis, we observed the involvement of genes in processes such as protein binding and metal ion binding. Meanwhile, the cellular component (CC) analysis highlighted their notable presence within cellular compartments, primarily in the cytoplasm and the nucleus. The co-localization observed within both the cytoplasm and nucleus signifies the importance of tsRNAs across diverse biological environments. Furthermore, based on the number of interacting genes, the KEGG pathway analysis revealed the most enriched pathways with an FDR < 0.05, prominently featuring metabolic pathways, pathways linked to cancer, pathways associated with Herpes Simplex Virus-1 infection, pathways of neurodegeneration, neuroactive ligand receptor interaction, and the PI3K-Akt signalling pathway. Most of these pathways converge to the PI3K-Akt signalling pathway, as this pathway acts as a central hub that responds to various cellular stimuli, exerting regulatory control over essential cellular functions such as transcription, translation, proliferation, growth, and survival. The networking of this pathway with the others strongly implies the active involvement of a substantial proportion of tsRNAs in signalling pathways in response to DENV infection.
Discussion
Dengue is an arthropod borne viral disease, which is a leading cause of severe illness and high mortality rates in Latin American and Asian countries according to WHO report December 2023. Owing to the severity of dengue infection, a comprehensive understanding of the pathophysiology and underlying mechanisms triggered by DENV infection is important for developing effective diagnostic and treatment strategies. The role of various novel classes of non-coding RNAs (ncRNAs), including miRNAs, PIWI-interacting RNAs, lncRNAs, and sfRNAs, has widely been studied in the context of DENV infection in various cell lines models and also in patient samples26,27,28,29,30,31,32,33,34. These ncRNAs not only regulate gene expression under physiological circumstances but also contribute during DENV infection and pathogenesis.
Transfer RNA-derived small RNAs (tsRNAs) like tRFs and tiRNAs are a novel class of small RNAs which have been reported to have diverse regulatory roles in various diseases35,36,37,38,39. Diverse studies have unveiled significant characteristics of tsRNAs that enhance their significance as a potent candidates for their roles as biomarkers or therapeutic targets: (1) Conservation across vertebrates, encompassing fish, amphibians, reptiles, birds, mice, non-human primates, and humans, coupled with temporal and tissue-specific expression; (2) Abundant in-vivo presence, extending not solely to tissue cells but also to bodily fluids like serum/plasma, bile, urine, seminal plasma, and amniotic fluid, facilitating their detection and extraction; (3) Limited but pivotal tRNA modifications, such as 5-methylcytidine (m5C) and N2-methylguanosine (m2G), significantly improves their stability14,40,41. Recent advancements in sequencing, software, and database technologies have substantially deepened our understanding of the roles of these fragments across a spectrum of human diseases and physiological and pathological processes38,42,43,44.
The role of tRNA-derived RNA small RNAs (tsRNAs) during Dengue pathogenesis remains unexplored. The levels of various RNases get modulated during DENV infection. Thus, we hypothesized that tRNA fragmentation and their differential expression might play a crucial role during DENV pathogenesis. In this study, we have carried out RNA sequencing for differential expression analysis of tsRNAs in DENV-infected Huh7 cell line.
The results indicate a substantial increase in tRNA fragmentation upon DENV infection, which correlates with the duration of DENV infection (Fig. 2a). Interestingly, a significant number of tsRNAs were also detected in the mock-infected samples, suggesting that tsRNAs are not exclusively generated during stressful conditions but also have roles in normal physiological contexts. Under viral stress conditions, the production of tsRNAs was found to be enhanced, with the highest levels observed at the 48 h time point in DENV-infected Huh7 cells. As depicted in Fig. 2b, some tsRNAs were found to be shared among the three samples while a subset of tsRNAs was specifically present in each sample. Specifically, 3826 tsRNAs were commonly shared across all three conditions. The differential expression was analysed using the selection criteria of an adjusted P-value ≤ 0.05 and an absolute log2 fold change of ≥ 2, which showed 733 tsRNA to be commonly shared in 0 h vs mock and 48 h vs mock groups, while 308 and 226 tsRNAs were found to be uniquely expressed in both groups, respectively (Fig. 3a). Among these 733 tsRNAs, their composition is primarily distributed as follows: 47% are 3′tRFs, 38% are i-tRFs, 10% are 3′tiRNAs, 4% are 5′tiRNAs, and 1% are 5′tRFs (as depicted in Fig. 3b). Notably, more than 80% of 3′tRFs exhibited upregulation, while over 50% of i-tRFs displayed downregulation, as illustrated in Fig. 3c. The differential generation of various categories of tsRNAs strongly suggests the involvement of various RNase activity during DENV infection. Previous reports have indicated that DICER levels decrease upon DENV infection, which inhibits the RNAi machinery45. Interestingly, DICER is also known to be involved in the generation of tsRNAs46,47. Consequently, the downregulation of i-tRFs may be linked to DICER-dependent cleavage, a hypothesis that warrants experimental validation. Furthermore, we have also observed an increase in the levels of Angiogenin during DENV infection23. Angiogenin is known to cleave tRNA within the anticodon loop to generate tiRNAs, also referred to as tRNA halves47. However, the abundance of 3′tiRNAs generated does not equal to that of 5′tiRNAs. Instead, it was observed that 10% and 4% of 3′tiRNAs and 5′tiRNAs, respectively, were generated. This observation suggests two possible scenarios: either 5′tiRNA undergoes further fragmentation to give rise to other tRF subtypes, or it is subjected to degradation. Further investigation is required to elucidate this phenomenon. Also, DICER and Angiogenin are recognized for their roles in cleaving the T-loop of transfer RNA (tRNA), leading to the formation of 3′ tRNA-derived fragments (3′tRFs)10. However, during DENV infection, DICER level decreases and Angiogenin increases, raising the possibility that Angiogenin assumes a more prominent role in generating 3′tRFs in response to DENV infection. This hypothesis gains support from the observation that 3′tRFs are also found to be upregulated during DENV infection.
We also performed in-silico functional analysis of differentially expressed tiRNAs/tRFs using miRanda and TargetScan to predict target gene interactions. Gene Ontology (GO) enrichment analysis categorized outcomes into biological process (BP), molecular function (MF), and cellular component (CC). Biological process (BP) analysis highlighted their involvement in transcription regulation, signal transduction, and apoptosis. Molecular function (MF) analysis showed roles in protein and metal ion binding. Cellular component (CC) analysis revealed presence in vital cellular compartments. Notably, they co-localized in the cytoplasm and nucleus, indicating diverse biological implications.
The KEGG pathway analysis revealed enriched pathways, primarily driven by the number of interacting genes. Notably, these pathways encompassed crucial biological processes, including metabolism, cancer, and Herpes Simplex Virus-1 (HSV-1) infection. A substantial proportion of these genes were found to be involved in nucleotide and amino acid metabolism. This observation aligns with the fact that viral machinery relies on host machinery for translation and replication, underscoring the relevance of these metabolic pathways. Moreover, among the top 10 KEGG pathways, the PI3K-Akt signaling pathway prominently featured. This pathway serves as a critical hub that responds to diverse cellular stimuli, exerting regulatory control over fundamental cellular functions such as transcription, translation, proliferation, growth, and survival. Activation of this pathway occurs when growth factors bind to their receptor tyrosine kinase (RTK) or G protein-coupled receptors (GPCR), leading to the activation of class Ia and Ib PI3K isoforms. These PI3K enzymes generate phosphatidylinositol-3,4,5-triphosphate (PIP3) at the cell membrane, initiating the activation of Akt. Akt, once activated, orchestrates key cellular processes by phosphorylating specific substrates involved in apoptosis, protein synthesis, metabolism, and the cell cycle (Fig. 6). Further analysis unveiled that the PI3K-Akt signaling pathway plays pivotal roles in other essential pathways, including those related to cancer, Herpes Simplex Virus (HSV) infection, and Human Papillomavirus (HPV) infection. The networking of this pathway with other pathways highlights its significance in various cellular contexts and diseases. These findings strongly suggest that a significant proportion of tsRNAs are actively involved in signaling pathways in response to DENV infection.

PI3K-Akt signalling pathway map for differentially expressed tsRNAs target genes, generated using DAVID software. The highlighted elements represent target genes (this image is obtained from KEGG; Kanehisa Laboratories, with permission).
Materials and methods
Small RNA sequencing sample preparation
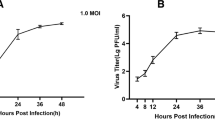
Huh-7 cells (Hepatocellular carcinoma cells) were procured from NCCS, Pune. The cells were cultured and maintained in complete DMEM media (Hyclone, Cytiva) supplemented with 10% FBS (Gibco), and 1% penicillin/streptomycin antibiotic (Gibco) at 37 °C with 5% CO2. The cells were either mock-infected or infected with DENV serotype-2 strain P23085 INDI-60 at a multiplicity of infection (MOI) of 1. Samples were harvested in TRIzol (Invitrogen) at 0 h (2 h post adsorption) and 48 h post infection (hpi). The samples were prepared in triplicates.
RNA extraction and quantification
RNA was isolated from TRIzol extracts. Briefly, 300 µl of chloroform was added to the TRIzol extract. The tubes were then inverted to mix well, followed by 10 min of incubation at room temperature. Subsequently, the tubes were centrifuged at 13,000 RPM at 4 °C for about 10 min. The aqueous supernatant was collected and 600 µl of absolute ethanol was added. Mixed it well and passed through RNA spin columns and centrifuged at 8000 RPM for 30 s. The column was then washed with AW1 and AW2 buffers (Qiagen). After 1 min of empty spin, RNA was finally eluted in nuclease-free water. RNA samples were quantified by NanoDrop (ND-1000). The RNA integrity was determined by the RIN value running on Agilent Tapestation 4200 RNA HS screen tape. Samples with RIN value > 7 were selected for library preparation48.
Library preparation and sequencing
Total RNA was given pre-treatment using the rtStar™ tRF and tiRNA Pretreatment Kit (catalog no. AS-FS-005, Arraystar, MD, USA) to remove internal methylations and terminal modifications for efficient adapter ligation. The small RNA sequencing library was prepared using NEBNext Small RNA Library Prep set for Illumina (catalog no. E7330), followed by adapter ligation using Illumina’s proprietary RT primers and amplification primers. Quality check of the constructed library was performed using Agilent 2100 bioanalyzer. For enrichment of 15–50 nt fragments, the library of 135–160 bp was selected on an 8% TBE-PAGE gel, and the Illumina NovaSeq system was used for sequencing according to the manufacturer’s protocol.
Data analysis and differential expression
Pair-end sequencing was performed using the Illumina NovaSeq system. The original raw data was obtained in FASTQ file format. The quality of the reads was assessed using Fast QC v 0.11.9. Cutadapt v 4.0 was used to remove adapter from the reads. Following trimming and filtering according to the read length of minimum–maximum 15–50 nt, MINTmap database (https://cm.jefferson.edu/mintmap/) was then used to identify and quantify tsRNAs. The MINTmap repository contains 640 mature tRNA sequences (508 true and 102 pseudo human tRNAs from v 1.0 of stRNAdb, the 22 known human mitochondrial tRNAs from NCBI, and 8 exact human tRNA lookalikes that are present in the human nuclear genome)25. Exclusive tRNA fragments were assessed for further analysis. Differential expression profiling of number of observed fragments was done using DEseq2 package. The results were further sorted according to adjusted P-value < 0.05 and absolute log2 fold change ≥ 2 (absolute fold change ≥ 4).
Functional analysis of differentially expressed tsRNAs
The functional role of tsRNAs was determined using TargetScan (www.targetscan.org), and miRanda (www.mirdb.org) to predict the target genes of differentially expressed tsRNAs. Subsequently, the target genes were used for Gene ontology (GO; (http://www.geneontology.org)) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses (http://www.kegg.jp/)49,50,51. Database for Annotation, Visualization, and Integrated Discovery (DAVID version 6.8; https://david.ncifcrf.gov/) was used to generate PI3K-Akt signalling pathway map.
Data availability
All small RNA-seq data generated in this study have been submitted to GEO under the accession number GSE245081.
References
Bonizzoni, M. et al. Complex modulation of the Aedes aegypti transcriptome in response to dengue virus infection. PLoS ONE https://doi.org/10.1371/journal.pone.0050512 (2012).
Butler, M. et al. Cyclin-dependent kinases 8 and 19 regulate host cell metabolism during dengue virus serotype 2 infection. Viruses https://doi.org/10.3390/v12060654 (2020).
Sessions, O. M. et al. Host cell transcriptome profile during wild-type and attenuated dengue virus infection. PLoS Negl. Trop. Dis. https://doi.org/10.1371/journal.pntd.0002107 (2013).
Sinha, S. et al. Dengue virus pathogenesis and host molecular machineries. J. Biomed. Sci. 31 43 (2024).
Eddy, S. R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. https://doi.org/10.1038/35103511 (2001).
Smith, J. L., Jeng, S., McWeeney, S. K. & Hirsch, A. J. A MicroRNA screen identifies the Wnt signaling pathway as a regulator of the interferon response during flavivirus infection. J. Virol. https://doi.org/10.1128/jvi.02388-16 (2017).
Lam, J. K. W., Chow, M. Y. T., Zhang, Y. & Leung, S. W. S. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids https://doi.org/10.1038/mtna.2015.23 (2015).
Wen, W. et al. Cellular microRNA-miR-548g-3p modulates the replication of dengue virus. J. Infect. 70, 631–640 (2015).
Damas, N. D., Fossat, N. & Scheel, T. K. H. Functional interplay between RNA viruses and non-coding RNA in mammals. Non-coding RNA 5, 7 (2019).
Yu, X. et al. tRNA-derived fragments: Mechanisms underlying their regulation of gene expression and potential applications as therapeutic targets in cancers and virus infections. Theranostics https://doi.org/10.7150/thno.51963 (2020).
Tao, E. W. et al. A specific tRNA half, 5’tiRNA-His-GTG, responds to hypoxia via the HIF1α/ANG axis and promotes colorectal cancer progression by regulating LATS2. J. Exp. Clin. Cancer Res. https://doi.org/10.1186/s13046-021-01836-7 (2021).
Elkordy, A. et al. tiRNAs as a novel biomarker for cell damage assessment in in vitro ischemia-reperfusion model in rat neuronal PC12 cells. Brain Res. https://doi.org/10.1016/j.brainres.2019.02.019 (2019).
Kumar, P., Kuscu, C. & Dutta, A. Biogenesis and function of transfer RNA-related fragments (tRFs). Trends Biochem. Sci. https://doi.org/10.1016/j.tibs.2016.05.004 (2016).
Pandey, K. K. et al. Regulatory roles of tRNA-derived RNA fragments in human pathophysiology. Mol. Ther. Nucleic Acids 26, 161–173 (2021).
Honda, S. et al. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. U. S. A. https://doi.org/10.1073/pnas.1510077112 (2015).
Su, Z., Wilson, B., Kumar, P. & Dutta, A. Noncanonical roles of tRNAs: TRNA fragments and beyond. Annu. Rev. Genet. https://doi.org/10.1146/annurev-genet-022620-101840 (2020).
Kumar, P., Anaya, J., Mudunuri, S. B. & Dutta, A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Med. 12, 1–14 (2014).
Zhu, L. et al. Using tRNA halves as novel biomarkers for the diagnosis of gastric cancer. Cancer Biomarkers https://doi.org/10.3233/CBM-182184 (2019).
Soares, A. R. & Santos, M. Discovery and function of transfer RNA-derived fragments and their role in disease. Wiley Interdiscip. Rev. RNA https://doi.org/10.1002/wrna.1423 (2017).
Elkordy, A. et al. Stress-induced tRNA cleavage and tiRNA generation in rat neuronal PC12 cells. J. Neurochem. https://doi.org/10.1111/jnc.14321 (2018).
Yamasaki, S., Ivanov, P., Hu, G. F. & Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. https://doi.org/10.1083/jcb.200811106 (2009).
Tao, E. W., Cheng, W. Y., Li, W. L., Yu, J. & Gao, Q. Y. tiRNAs: A novel class of small noncoding RNAs that helps cells respond to stressors and plays roles in cancer progression. J. Cell. Physiol. https://doi.org/10.1002/jcp.29057 (2020).
Madhry, D. et al. Synergistic correlation between host angiogenin and dengue virus replication. RNA Biol. 20, 805–816 (2023).
Kakumani, P. K. et al. Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J. Virol. https://doi.org/10.1128/jvi.02774-12 (2013).
Loher, P., Telonis, A. G. & Rigoutsos, I. MINTmap: Fast and exhaustive profiling of nuclear and mitochondrial tRNA fragments from short RNA-seq data. Sci. Rep. 7, 1–20 (2017).
Miesen, P., Ivens, A., Buck, A. H. & van Rij, R. P. Small rna profiling in dengue virus 2-infected aedes mosquito cells reveals Viral piRNAs and novel host miRNAs. PLoS Negl. Trop. Dis. 10, 1–22 (2016).
Hess, A. M. et al. Small RNA profiling of Dengue virus-mosquito interactions implicates the PIWI RNA pathway in anti-viral defense. BMC Microbiol. https://doi.org/10.1186/1471-2180-11-45 (2011).
Madhry, D. et al. Role of non-coding RNAs in dengue virus-host interaction. Front. Biosci. Scholar 13, 44–55 (2021).
Wang, Y. & Zhang, P. Recent advances in the identification of the host factors involved in dengue virus replication. Virol. Sin. 32, 23–31 (2017).
Su, Y. et al. microRNAs, the link between dengue virus and the host genome. Front. Microbiol. 12, 1 (2021).
Mishra, R., Kumar, A., Ingle, H. & Kumar, H. The interplay between viral-derived miRNAs and host immunity during infection. Front. Immunol. 10, 1 (2020).
Göertz, G. P. et al. Subgenomic flavivirus RNA binds the mosquito DEAD/H-box helicase ME31B and determines Zika virus transmission by Aedes aegypti. Proc. Natl. Acad. Sci. U. S. A. 116, 19136–19144 (2019).
Bidet, K., Dadlani, D. & Garcia-Blanco, M. A. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog. 10, 1004242 (2014).
Schnettler, E. et al. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and mammalian cells. J. Virol. 86, 13486–13500 (2012).
Kim, H. K. et al. A transfer-RNA-derived small RNA regulates ribosome biogenesis. Nature 552, 57–62 (2017).
Shen, Y. et al. Transfer RNA-derived fragments and tRNA halves: Biogenesis, biological functions and their roles in diseases. J. Mol. Med. 96, 1167–1176 (2018).
Goodarzi, H. et al. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell 161, 790–802 (2015).
Cristodero, M. & Polacek, N. The multifaceted regulatory potential of tRNA-derived fragments. Non-coding RNA Investig. 1, 1–5 (2017).
Deng, J. et al. Respiratory syncytial virus utilizes a tRNA fragment to suppress antiviral responses through a novel targeting mechanism. Mol. Ther. 23, 1622–1629 (2015).
Kiani, J. et al. RNA-mediated epigenetic heredity requires the cytosine methyltransferase Dnmt2. PLoS Genet. 9, 1 (2013).
Godoy, P. M. et al. Large differences in small RNA composition between human biofluids. Cell Rep. https://doi.org/10.1016/j.celrep.2018.10.014 (2018).
Zhu, P., Yu, J. & Zhou, P. Role of tRNA-derived fragments in cancer: novel diagnostic and therapeutic targets tRFs in cancer. Am. J. Cancer Res. 10, 393–402 (2020).
Raina, M. & Ibba, M. TRNAs as regulators of biological processes. Front. Genet. 6, 1–14 (2014).
Kumar, P., Mudunuri, S. B., Anaya, J. & Dutta, A. tRFdb: A database for transfer RNA fragments. Nucleic Acids Res. 43, D141–D145 (2015).
Kakumani, P. K. et al. Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J. Virol. 87, 8870–8883 (2013).
Anderson, P. & Ivanov, P. TRNA fragments in human health and disease. FEBS Lett. 588, 4297–4304 (2014).
Lee, Y. S., Shibata, Y., Malhotra, A. & Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 23, 2639–2649 (2009).
Madhry, D. et al. Various transcriptomic approaches and their applications to study small noncoding RNAs in dengue and other viruses. Integr. Omi. Approaches Infect. Dis. 195–220. https://doi.org/10.1007/978-981-16-0691-5_12 (2021).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkac963 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/28.1.27 (2000).
Acknowledgements
The work is supported by DST-SERB (EEQ/2018/000838, EEQ/2022/000362) to B.V. The research in the Laboratory of Molecular Biology is supported by funding to B.V. through AIIMS Intramural support (AC-31), and DBT (BT/PR39859/MED/29/1519/2020). D.M. acknowledges the fellowship from Council of Scientific and Industrial Research (CSIR), India.
Author information
Authors and Affiliations
Contributions
D.M. and B.V.: conceptualization and methodology; D.M., K.K., V.M., and B.V.: formal analysis; D.M., V.M., and R.R.: investigation; D.M., K.K., V.M., and B.V.: writing–original draft; B.V.: supervision; B.V.: funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Madhry, D., Kumari, K., Meena, V. et al. Unravelling tRNA fragments in DENV pathogenesis: Insights from RNA sequencing. Sci Rep 14, 18357 (2024). https://doi.org/10.1038/s41598-024-69391-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69391-7
- Springer Nature Limited