
Abstract
The fast scanning AFM combination with super resolution optical technique allow co-localized imaging and manipulation with sub-diffraction resolution in a few seconds. The hybrid technique opens up new fields of in-situ dynamic study in living cells, enzymatic reactions, fibril growth and biomedical research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction
Atomic force microscopy (AFM) is a versatile technology for the study of biological samples. A flexible cantilever is used as a force sensor for measuring extremely small forces between the AFM tip and sample surfaces. Dynamic mode AFM using small imaging forces allows nondestructive measurement. That makes AFM more popular in the study of native biomolecules than Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM). However, slow imaging speed hinders the use of AFM, comparing to an optical microscope, SEM and TEM. Apart from the superb lateral and vertical resolution, researchers expect to know sample changes over time during the cellular bioprocess, such as protein folding and unfolding, DNA assembly, DNA-protein interaction and drug-cell interaction, etc. Most of the reactions are too fast to be recorded by an AFM which usually takes more than one minute per frame. The dynamic imaging at single-molecule level in a bio-system is difficult to resolve until the invention of high speed AFM and super-resolution optical microscopy.
Many AFM specialists devote considerable efforts in developing new imaging systems to increase the scanning rate of AFM. To date, the commercially available high speed AFM enables several tens Hertz frame rate [1]. The fast scan techniques allow the AFM tip to trace the dynamic bioprocess with a tiny scan force without sacrificing the image qualities. The high speed AFM is able to investigate the structure and dynamics of single molecules, indicating the molecular movement and diffusion dynamics. High speed AFM enabled many tremendous discoveries recently, such as the nature of myosin V motor mechanism [2], the diffusion dynamics of protein through a membrane [3], and the killing process of an anti-microbial peptide towards Escherichia coli (E. coli) [4]. More and more dynamic process at the nanoscale have been captured and reported as high scanning speed and the wide application in air, vacuum, and liquids [5]. In the life science researches, the optical microscopes are widely used and record the images in sub-second. However, the use of classical optical microscopy for the single-molecule image is limited by the Abbe diffraction of light, which depends on the wavelength of light λ and the numerical aperture of the optical system (NA): Resolution x,y = λ/2NA [6]. To overcome the Abbe limit, several advanced fluorescence methods have been developed to obtain the super-resolution, either by photo-activation, photo-switching, or by selective deactivation of individual fluorophores in fluorescence microscopy. The development of stimulated emission depletion (STED) microscopy, photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM) allow widespread applications in the study of biological samples with sub-Abbe limit, 20–30 nm [6].
Both AFM and optical microscopy provide different information. The AFM detect the topology and mechanical properties of the investigated samples. For the objects having the similar size and mechanical properties, it is difficult to distinguish and specify them simply by AFM images. Moreover, a high-resolution of AFM is not enough to detect the binding of very tiny ligands. On the other hand, an optical microscope can obtain dynamic bioprocess with the specific fluorescence labeling. However, the structure and the mechanical details are missing even at a super-resolution and high signal-to-noise ratios. The coupling of AFM and optical techniques meet the demands for the achievement of the specification, structure, and mechanical information simultaneously. The details of high speed AFM and super-resolution microscopy can be seen in the literature [7]. In this chapter, we mainly focused on the combination of the high speed AFM and super-resolution optical techniques, such as AFM-STED, AFM-STORM and AFM-PALM, for imaging of living systems, and for precise manipulations for demanding experiments and cutting-edge researches fundamentally in situ and in real time.
2 High Speed AFM
High speed AFM is scanning faster than normal AFM, and is getting popular in life sciences. Commercial available high speed AFM also called as ultra speed AFM (JPK) or fast scan AFM (Bruker), all of them comes out with outstanding designs for good quality images in short sample acquisition time. The core techniques for the new designs is to develop a system which can acquire the data with time resolution and spatial resolution at the same time. Generally, fast scan always comes at the expense of an increase of imaging forces, and thus results in image quality unacceptable and may damage the tip or samples easily.
2.1 A High Bandwidth Z–Scanner
A high sampling frequency requires a fast scanning control system, including a high speed scanner and a big bandwidth for the feedback loop [5]. A high resonant frequency of z-scanner is required because Z-movement needs to be faster than the X and Y movement. As Z-response time needs to be less than the pixel time in the X-Y movement. Assuming an image is captured in scan size L × L with scan lines N and pixels N each scan line in scan time t. The scan rate in X-direction ν x = 2LN/t, and a pixel time is t p = L/Nν x = t/2 N 2. Therefore, the Z-scanner needs to move in Z-direction a frequency f z = 2 N 2/t to follow the surface up and down to resolve each pixel in X-direction. If L = 300 nm, N = 100, and t = 20 ms, then one needs a Z-scanner with resonance frequency f > = 1 MHz. The X-Y movement is relatively slow and does not require very high resonance frequency. Generally, the resonance frequency of the Z-scanner is influenced by the size and shape of a scanner. Usually a sample scanner is much bigger and moves slower than a tip scanner. Basically, a smaller scanner has a higher resonance frequency than a larger one. This is why most AFM use separated X-Y and Z-scanner system, while Z-scanner is fixed at the tip side. Some type of AFM fix both the X-Y and a separated Z-scanner on the tip side, also called as tip-scan AFM (JPK NanoWizard®). The minimal requirement for Z-scanner resonance frequency also defines the minimal feedback bandwidth. The shorter acquisition image time, the bigger bandwidth for feedback control system.
2.2 A Small Cantilever
To improve the time resolution, a delicate detecting system is highly demanded for faster information flow from the surface. Obtaining more information per time unit needs more interactions from the sample surfaces. A smaller cantilever should deliver information faster with similar interactions comparing with a bigger cantilever [8]. A small laser spot and a small spring constant are required to reduce the acquisition time. However, this increases the noise level. In dynamic mode AFM, the acquisition time is influenced by the amplitude reading time, the sensor and scanner response time, and the error correction time through the feedback controller. The amplitude detector needs to be fast enough to catch several oscillation cycles at X and Y pixel to detect the phase delay and get an accurate amplitude value. A sensor response time is expressed as Q/πf0 [9]. A cantilever with low Q factor and high resonance frequency delivers shorter response time. A cantilever with small Q factor is less sensitive to surface interactions and needs less energy to restore the dampened oscillations than one with high Q factor. The AFM tip detaching from the surface also delay the sensor response time, called parachuting [10].
High speed AFM is easy for survey scan at single-molecule level. High speed AFM can be applied to detect particulate contaminants to reveal contamination during DNA purification [11]. The single-molecular sensitivity enables High speed AFM to detect DNA molecules without amplification required in PCR, and no chemical labels are required in High speed AFM. Using High speed AFM, trace levels of filamentous contamination were discovered. High speed AFM provides an alternative to characterize and quantify trace contaminants in biomolecular reagents and instruments in the cases when other conventional techniques fail. Importantly, High speed AFM provides highly accurate information of the dsDNA molecule size, which is related to the nature of the contamination sources [11].
For the dynamic study, DNA-protein interactions, such as short-range slide of protein along DNA and hop of protein on DNA, can be visualized by high speed AFM, which were usually inaccessible by other high resolute microscopies. High speed AFM provides nanoscale and sub-second temporal resolution figures of single molecules [12]. The individual protein motion was measured using single-particle tracking mode, and it was observed that the composition of imaging-buffer has a significant effect on the random walk of proteins. The non-Brownian diffusion was investigated in the absence and in the presence of nucleotide cofactors. It was found out that the Brownian motion was hindered and prevented as the double-stranded DNA was immobilized on the surface of proteins. However, RAD54 monomers showed a diffusive behavior, which is not related to the motor activity [12].
High speed AFM imaged and visualized that M5-HMM was advancing progressively with ∼36 nm steps in Fig. 6.1 [2]. Consecutive high-speed AFM images show the progressive movement of M5-HMM in 1 μM ATP solution at 7 fps [13]. Molecular motor mechanism is also investigated by high-speed AFM [14], which shows that the rotorless F1-ATPase still rotates; the three β subunits cyclically display conformational changes in the contraclockwise direction in the separated α3/β3 stator ring, which shows similarity to the rotation of rotary shaft in F1. The stator ring showed unidirectional structure basis. These findings suggest and imply the cooperative interaction between subunits in other hexameric ATPases.

High-speed AFM images of walking M5-HMM. (a), AFM images of M5-HMM movement in 1 mM ATP solution. (b), Image of two-headed M5-HMM bound to actin filaments. (c), Explanation of (d) and (e). (d) & (e), Time-resolved AFM images of hand over hand movement in 1 mM ATP solution and in 2 mM ATP. All images were captured at 146.7 ms per frame. Scale bar: (a) 30 nm; (d) 50 nm; (e) 30 nm. Reprinted with permission from Kodera et al. [2] © Springer Nature (2010)
Figure 6.2 shows the application of high speed AFM for observing the protein movement on DNA in 0.5 sec interval (a) [12] and (b) GltPh elevator domain movements in 1 sec interval [15]. The improved speed enables the observation of many biological phenomena. Meanwhile, high speed AFM is able to capture images at 1 frame per second (fps) for the investigation of cell membrane reconstruction, crystal dynamics of purple membrane and annexin V. With the smaller cantilevers, small biomolecules such as DNA can be imaged by 1 fps, while larger biomolecules like bacteria can be imaged at 10 s for every frame [4]. However, this improved speed is used in the scan sizes are up to ~ 400 nm, and it is still not rapid enough to understand the fast large-scale biological phenomenon. A scan of 500 nm × 500 nm, imaging with 100 × 100 pixel resolution at 10 fps requires a scan rate of 1 mm/s, while the line scan rate reaches 1 kHz. For a scan of 10 μm × 10 μm, imaging at 1 mm/s with 256 × 256 pixel resolution, it takes 5.12 s. This requires the support of a scanner with large scan range. The scan rate of 1 mm/s is the standard rate, and to pursue a frame rate of 10 Hz is challenging to current techniques.

A force-feedback high speed AFM has been developed using a commercial available dimensional micro-actuated silicon probe based on a cantilever-based optical interfacial microscope, which is shown in Fig. 6.3 [9]. This protocol is successful in obtaining large images of E. coli biofilm morphology using zero Newton force as feedback at about 1 fps in air. In this method, the removal of bending error of cantilever by self-actuating imposes a counter force to the sample, which causes sample deformation. Both the AFM tip and the sample may be contaminated or damaged during this process. Moreover, the use of sample scanner limits the fast scan speed. There is plenty room to improve the scan speed to 30 fps.

A schematic diagram of the force-feedback high speed AFM. (a) Conceptual diagrams. (Step 1) The tip contacts the surface of sample. (Step 2) The cantilever bending occurs when tip encounters the sample. (Step 3) The biological structure was compressed and deformed due to the force-feedback. (Step 4) The activation of a feedback loop involving the piezo tube enables the sample to move down. (b) Feedback system. The feedback controller 1 is used for the force-feedback loop involving ZnO stack to obtain force images. The feedback controller 2 is the morphological feedback loop involving the piezo tube. Reprinted with permission from Kim et al. [9] © Elsevier (2016)
3 Combination of High Speed AFM with Super–Resolution Optical Techniques for Imaging
The combined techniques of high speed AFM and high-resolution optical microscopy open the door to image the fast-changing biological phenomena, such as DNA methylation, protein folding and unfolding, bacteriolysis and so on, which provide fundamental experimental data and images for further understanding of the fast-changing biological phenomena and verifying the hypothesis. Although high speed AFM [2] and high speed microscopy [16] have been developed, the correlative real-time imaging of live cells in situ is still challenging.
A setup of AFM combined with a top view microscope has conflict in acquiring high-resolution images of biological samples in liquids. To acquire high-resolution optical images, the objective with a high NA is required to be close to the sample, while the AFM imaging needs the tip to approach the sample. It is hard to get high-resolution optical images with an AFM cantilever between the objective and the sample surfaces. The design of inverted optical microscope coupling with high speed AFM solved this problem. The AFM scan head rests on the top of an inverted microscope providing a platform for any optical access to the sample from below. The setup offers the possibility to integrate any type of fluorescence microscopes, such as epifluorescence, confocal, total internal reflection fluorescence (TIRF) and Fluorescence resonance energy transfer (FRET).
The hybrid high speed AFM/fluorescence microscopy was used to investigate the cell surface structural changes of the living HeLa and 3 T3 fibroblast cell [17]; the cell response to the tip stimulation with soluble agents [18]; the morphological changes of hippocampal neurons at 0.2 fps [19]; the lipid membrane response to charged polymer nanoparticles [20]; the diffusion of lens cell membrane proteins at the cell surface [21] and mechanical properties of elastomeric proteins [22]. Furthermore, the combination of DNA manipulation and high speed AFM was used to image different biochemical reactions, such as enzymatic reaction and DNA structural changes [23].
The combination of high speed AFM and TIRF microscopy was used to simultaneously image the dynamics of biological samples, such as the effect and mechanism of chitinase A on chitin microfibrils at 3 fps (Fig. 6.4) [19]; rhodamine-labeled actin filaments at 8.3 fps [14]; the morphology changes of B. subtilis cell caused by lysozyme [24]; eukaryotic COS-7 and HeLa cell response to insulin [25].

Real-time high speed -AFM/TIRFM imaging of chitinase A labeled with Cy3 moving on a chitin microfibril. Reprinted with permission from Uchihashi et al. [19] © Elsevier (2016)
A single chitinase A molecule advancing to the top-right was captured by high speed AFM images at 3 fps with 150 × 35 pixels. The TIRF images (40 × 15 pixels) confirmed the right movement of chitinase A molecule [19]. Several techniques have been developed over the past decades for super-resolution fluorescence microscopy including STED, PALM, and STOM [26, 27]. All these techniques have shown their strength and are promising for biological research. This review demonstrates the current state of high speed AFM development combined with these super-resolution techniques, as well as their application in biomedical research.
3.1 High Speed AFM with STED
STED microscopy generates images with super-resolution through selective deactivation of the fluorophores. As the illumination area at the focal point was minimized, the resolution of target imaging was improved significantly [28]. A typical STED microscope uses a co-aligned scanning pair of laser pulsed beams at the same focus position and controls the fluorescence through the overlap of the point spread functions (PSF) [29]. STED theory was first proposed in 1994 by Hell [30], while the first STED data was published in 2000 [31]. At first, a laser is used to excite the fluorescent molecules to emit the spontaneous fluorescence, which occurs at nanosecond time scale. In the process, an electron is excited into an excited electronic state from the ground state. Before a photon is released, a second laser pulse (STED beam) is introduced to force the excited electron to relax into a dark state, which depletes the excited state at picosecond time scale [30]. The STED laser has a phase modulation to control the special intensity distribution, resulting in a donut PSF at the focal plane [32]. Overlapping the standard PSF of the first laser and the donut depletion of the second laser results in a suppressed outer PSF (Fig. 6.5). An effective PSF fluorescence is achieved by using a filter to exclude the red-shift fluorescence. Increase the intensity of the STED beam can increase the lateral resolution to less than 50 nm.

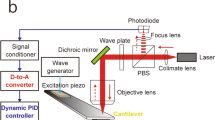
Scheme of an AFM and STED based system showing the major components. The sketch includes the time-shifted pulse of the excitation source and the STED beam, the detector unit, and the AFM placed on an inverted microscope. The layout is adapted from JPK Instruments AG Technical Note: combining AFM with super-resolution STED microscopy system
Integration of AFM with STED enables high-resolution fluorescence, topographical, and nano-mechanical imaging of biological samples in a physiological environment. This protocol provides fundamental understanding of numerous comprehensive studies in medicine, biology and material science. Using this method, microtubes have been investigated within antibody labeled COS7 cells in PBS solutions [33]. In Fig.6.6, it can be observed that the STED image (Fig. 6.6e), 3D AFM height image (Fig. 6.6c) and a Young’s modulus map (Fig. 6.6f) of the same target area on a cell is consistent with each other. Microtubules which are visible in the STED image can be observed in the 3D AFM height image and traced in the Young’s modulus map. The combined technology of the fast speed AFM and a high resolute STED can be applied to different kinds of samples and improve the multifunction and versatility of high speed AFM.

Confocal image (a) and STEM image (b) of Atto 647 N labeled microtubules in COS7 cells in PBS. 3D height image extracted from AFM mapping (c) and calculated elasticity map (f) correlated with the STED (e) and confocal image (d) (both linear deconvolved) collected from the same area. Scale bars in (a) and (b): 2 μm, axes bars in (c)-(f): 3 μm. Reprinted with permission from [33] © Harke et al.; Springer Nature (2012)
3.2 High Speed AFM with dSTORM
STORM is a high-resolution imaging technique, which was proposed in 2006 by Zhuang’s group to investigate DNA and DNA-protein complexes with a resolution of 20-nm [34]. A fluorescence image is re-constructed from high-accuracy localization of photoswitchable fluorophores. There are a series of imaging cycles involved in the STORM imaging process and system. In each cycle, there are only a small fraction of the fluorophores being switched on. Each of the active fluorophores is separated by a distance exceeding the Abbe diffraction limit, so that it is optically distinguishable from the other fluorophores. This enables the determination of these fluorophores’ position with nanometer accuracy. In a repeating imaging cycle, different fluorophores are switched on. The multiple imaging cycles are used to determine the positions of the fluorophores, and are applied to reconstruct the overall image. This method is based on the discovery of the photoactivable molecule (green fluorescent protein) that can be activated and relaxed after emission to a stable dark state that could be re-activated [35].
Integration of AFM with direct STORM holds great potential for conducting correlated nanoscale biomolecular and structural characterization on living samples under physiological conditions [36]. AFM provides exceptional surface resolution, while STORM reveals internal cellular structures at single-molecule level. This combination is particularly useful for actin filaments and microtubules, which are important in intra-cellular transport and cell stability. A hybrid AFM-STORM was used to correlate the density of localizations with the 3D topography along (F-)actin cytoskeletal filament, which is shown in Fig. 6.7 [36]. The combination of high speed AFM and super-resolution optical detection system can provide complementary information, which enables the structural and functional nanoscale characterization of living samples.

Resolution comparison of AFM (a), dSTORM (c), and TIRF (e) images of correlated filaments. (b), (d), and (f) are the cross-sectional profiles for (a), (c) and (e) respectively. (g) Overlap of possible dSTORM map in blue and 3D AFM image in yellow−brown. (h) Suggestion of F-actin arrangements based on AFM analysis. (i) Maximum height profile of the filament according the AFM image showing below. (j) Corresponding AFM/PALM images of E. coli expressing RNA polymerase−mEos2. Reprinted with permission from Odermatt et al. [36] © American Chemical Society (2015)
Figure 6.7 compared AFM, dSTORM, and total internal reflection fluorescence microscope (TIRF) resolution through correlative imaging the same filaments via AFM, dSTORM, and TIRF respectively. It was found that dSTORM provided comparable values to AFM when the width of the actin bundles was measured, indicating the comparability of AFM and dSTORM. However, AFM provides more resolute images than dSTORM when the filament diverged (Fig. 6.7a−c).
3.3 High Speed AFM with PALM
PALM and STORM are based on the same principle, using mathematical models to reconstruct a sub-diffraction limit from many sets of images [37]. Numerous sparse subsets of protein molecules which are photoactivatable fluorescent with caged synthetic fluorophores were activated, localized, and then bleached. A small fraction of particular molecules are photoactivated, while the majority of the population remains in the dark state. The new computational algorithm enables fast imaging of cellular structures with 20 nm resolution to be in less than 10 s [38], which is useful for the dynamic imaging of molecules and cells.
The correlative images of bacteria and mammalian cells in aqueous conditions can be obtained through AFM-PALM [36]. Figure 6.8 shows real-time AFM and PALM images of live CHO-K1 cells in a physiological environment. The paxillin-mEos2 cluster changes were monitored by PALM series, while the cell membrane dynamics were shown in AFM images. The upper corner of the entire cell was chosen for real-time analysis, as shown in Fig. 6.8a. Figure 6.8b showed that filopodia extended gradually followed by the extension of the lamellipodium, as the AFM images were obtained every minute. Figure 6.8c shows the PALM images recorded after AFM imaging. Figure 6.8d−g zoomed in to show paxillin clusters in certain regions. In Fig. 6.8d, there were paxillin clusters on the upper left at 28.2 min, while the paxillin clusters disappeared at 31.6 as shown in Fig. 6.8e, while new ones appeared at 30.7 and 33.3 min in Fig. 6.8f, g. In (d) and (e), it was observed that paxillin clusters disappeared from the top left corner (arrow), and new paxillin clusters were generated within 3 min in (f) and (g) (arrows).

Real-time AFM/PALM images of live mammalian cells. (a) Paxillin-mEos2 expression in CHO-K1 cell under TIRF illumination off-state. Overlap part indicates AFM overview of the upper part of CHO-K1 cell. (b) Time-resolved AFM images indicating the filopodia protrusion and lamellopodia extension of normal cell at frame rate 1 min−1. (c) The paxillin-mEos2 clusters recognized in live-cell by PALM real-time images. The measured area is the same as the white square shown in (a). (d) and (e) Zoom-in image of the highlighted green square at different time in (c). (f) and (g) Zoom-in image of the highlighted red area in (c) at different time. Reprinted with permission from Odermatt et al. [36] © American Chemical Society (2015)
The combination of AFM with dSTORM, PALM and STED was used to give complementary information about the same biological sample from different aspects. Chemically fixed bacterial cells (Fig. 6.7j) were imaged using AFM and dSTORM. E. coli expressing the fusion protein RNP- mEOS2 was firstly measured by AFM and followed by PALM. PALM was chosen over dSTORM in this case, because PALM can be utilized with lower laser power, which is more suitable for living cell imaging. The PALM provided the level of expression, while AFM provided the 3D bacteria morphology. Interestingly, there was no fusion protein for one bacteria in the PALM image (Fig. 6.7j). In the respective AFM image, it is shown that the bacterium without fusion protein is situated on the top of two other bacteria. This bacterium may be out of the illumination volume, so the respective fusion proteins were not found in the PALM image. This example showed the advantage of the AFM/PALM combination technique, which produced a better interpretation of the microscopy result than AFM or PALM alone.
The combination of dSTORM, PALM and STED and AFM techniques to image biologically relevant phenomenon shows the comparison of the two techniques and complement the weaknesses of each technique. In the actin filament imaging and measurement, AFM provided the information of dimension and location, which could be correlated to the labeling density along the actin filament as imaged by the high resolute microscopy.
4 Hyphenated High Speed AFM and Super–Resolution Optical Techniques for Manipulation
4.1 AFM Nanomanipulation
AFM nanomanipulation is utilizing tips to pull, push, scratch and move the object to know the mechanical properties of the targets. Using an AFM tip as a tool, it is able to tailor biological samples at the single-molecule level. Figure 6.9 showed some examples of AFM nanomanipulation. Single-stranded or double-stranded DNA molecules were immobilized on a substrate and then were precisely cut by an AFM tip [39]. The AFM tips are successfully used for DNA pickup from a chromosome [40, 41] or from a substrate by kneading, and then subjected to single-molecule PCR and sequencing [42,43,44,45]. AFM also can be used to move virus or nanoparticles to form a pattern on a surface [46]. Cells can be picked up for the study of cell-cell interactions with the assistance of an optical microscope [47]. Apart from mechanical scratching, pulling, or pushing the surface molecules, structures decoration can be made by a method called dip-pen nanolithography, where the tip is used as a pen to directly deposit collections of ink materials onto the substrate [48, 49].

AFM nanomanipulation at the single-molecule level. (a)–(d), DNA dissection and kneading. Reprinted with permission from Hu et al. [45] © American Chemical Society (2015). (e), Dissection of the chromosome. Reprinted with permission from Fotiadis et al. [41] © Elsevier (2002). (f), Dip-pen Nanolithography for positioning scission of single DNA molecules with a nonspecific endonuclease. Reprinted with permission from Li et al. [49] © American Chemical Society (2015)
4.2 Force Spectroscopy
Modification of both the AFM tip and the sample surfaces made AFM very useful in biological applications [50]. Single-molecule force spectroscopy techniques have been used to study the folding/unfolding of proteins [51], the elasticity of polysaccharide [52], DNA base pair interactions [53], interactions between proteins, between DNA and proteins, and between antibody and antigen. Molecular recognitions can be easily done by tagging a molecule (DNA, RNA, ligand, receptor, peptide, lipids, etc), or virus on the AFM tip for imaging or biosensing (Fig. 6.10).

Schematic of AFM resting on an inverted microscope for fast force mapping and force spectroscopy
4.3 Fast Force Mapping
For a survey study, force mapping, like Force Volume technique (Bruker), at a large scale usually takes more than 1 h and comes with low-resolution topology image, which presents a big challenge to investigating the short-term changes of living cells under physiological conditions. Long time scanning has problems in drift and time-resolution for in situ reactions. Fortunately, new techniques, Peakforce tapping mode AFM (QNM and ScanAsyst, Bruker), QI mode AFM (JPK Instruments AG), and Fast Force Mapping mode AFM (Oxford Instruments), can do fast force mapping in several minutes. The principles are similar to dynamic AFM, which avoids lateral forces by intermittently contacting the sample, and a maximum force is used as feedback. They are able to measure the mechanical properties and to image cells without changing their surface under liquid conditions. Topology, stiffness, adhesion and Youngs’ modulus of samples are all obtained at one time scan when following the procedure after calibration of the cantilever’s spring constant. QI mode AFM is contact mode using contact point to define the height [54]. Small cantilevers are reported to increase the speed of force mapping by 10–100 times to resolve the dynamic process of embryonic fibroblast. With this technique, it is very convenient to study the cell adhesion, dynamics of drug-cell interactions, cell-cell interactions [55].
For the mechanical study, a high-resolution optical microscope helps to localize the AFM tip on the targets. Height image along with mechanical images are collected by fast force mapping over the desired area and then overlaid with the optical image. A modified AFM tip specify the molecules through force mapping to realize double localization. Recently, PeakForce mapping was used to measure the binding step of single viruses over living animal cells [56]. In this case, the confocal laser scanning microscopy was integrated with an AFM to target MDCK cells. The wild-type cell is nonfluorescent, while the TVA-mCherry labeled cells expressed the virus acceptors. The AFM tip was tagged with a single virus and then used to map the cell surfaces. Only cells expressing TVA receptors (mCherry fluorescence channel) were found to interact with the virus, with these binding events appearing as bright pixels in the adhesion image (Fig. 6.11). The combination provides a tool to quantify how molecules interfere with viral entry, and therefore aid the invention of novel drugs against virus infection.

Mapping virus binding to MDCK–TVA cells using correlative confocal microscopy and Peak Force Tapping AFM. Reprinted with permission from Alsteens et al. [56] © Springer Nature (2010)
4.4 AFM Manipulation – STED
With the combination of high resolute microscopies, more detailed properties can be discovered. For example, nano-manipulation can be done through combined STED and high speed AFM technology. AFM images can be overlaid with STED fluorescent image. The STED image can be utilized as a map to target the sample precisely with high-resolution [57]. AFM software was applied to control the tip to the desired bead according to the information provided by STED image. As a result, the AFM tip was manipulated to move a single bead along a designed path and the image was obtained through STED before and after dragging. Thereafter, the STED images are combined together in green and red for analysis (Fig. 6.12b). The red and green colors represent the STED images before and after AFM dragging, respectively. Therefore, the yellow beads indicate the stationary status during the dragging, and the movement path of the dragged bead was observed as the arrows shown in Fig. 6.12e. The fast imaging mode of a STED microscope helps in measuring the nano-manipulation with high-resolution. It can be observed that the AFM manipulation only moved one selected bead while other beads were stationary.

AFM Sub-diffraction Nanomanipulation. STED images show before (c, red channel) and after (d, green channel) AFM manipulation of a bead. The overlay of images (b and e) give a yellowish color for unmoved beads, and the dislocation of one bead from red to green position. This is impossible to be distinguished by confocal images. Reprinted with permission from [57] © Chacko et al.; PLOS (2013)
The integration of high speed AFM with high-resolution as STED provides a protocol to study the dynamic process of interactions between a functionalized particle and a target. A dye-labelled enzyme or drug molecules can be easily localized by STED images. Followed by an AFM manipulation push them to a target DNA, protein, or cell. Fast scanning further gain more information happened in short times.
4.5 AFM Nano–Manipulation – Optical Spectroscopy
Combining nano-manipulation with super-resolution optics is crucial for demanding experiments at single-molecule scale. The methodologies for achieving meaningful results with high signal-to-noise ratios in the study of individual molecules or single nanostructures are challenging. Bringing the AFM tip into a laser focus expands the classical spectroscopy to nanoscale with precise location. The integration enables new discoveries and experimental methods for both characterization and fabrication of nanomaterials, such as tip-enhanced near-field optical microscopy and tip-enhanced Raman spectroscopy (TERS). A simple application is to stretch a protein or DNA molecule which is positioned in the laser focus. Both optical and mechanical properties are probed simultaneously.
TERS was introduced and applied to achieve Raman spectroscopy with extraordinarily high spatial resolution [58]. The protocol uses metal-coated tips or solid metal, or tips attached to nanoparticles such as gold, silver, or platinum. The AFM tip is positioned in a laser beam above the sample surface, and manipulated stationary at a z-position, for localized nanoscale enhancement of the Raman signals (Fig. 6.13). With the help of fast force mapping, the spatial resolution is in the single nanometer regime [59]. The experimental and applications can be seen in review [58].

Schematic of the AFM nanomanipulation for tip-enhanced Raman spectroscopy or nanoantenna
The nanoantenna is usually fabricated by complicated protocols, such as electron beam lithography to have the shape of a small rod or triangle [60]. Plasmonic nanoantennas are able to provide controllable and improved light-matter interactions and strong coupling between localized sources and far-field radiation at the nanoscale [61]. AFM nano-manipulation facilitates the fabrication and the research of nanoantenna. The colloidal nanoparticle can be easily positioned close to another nanoparticle (Fig. 6.13) or coupling to other nanostructures like a waveguide [62]. Bringing the newly formed active nanostructures into the laser focus enables the research of surface plasmon resonance, such as electrical generation of surface plasmon polaritons [63]. The nanoantenna can be used as biosensors for biotechnological applications, such as label-free estimation of DNA hybridization, dynamic quantification of nucleic acid-protein interactions, medical diagnostics, environmental pollution control, insecticide, bacteria, and virus detection [60].
4.6 AFM Manipulation – TIRF Microscopy
As aforementioned, the integration of AFM with high-resolution optical techniques has the advantage of dual localization. The localization of the AFM tip in optical microscope view is important for precise specification and controlling the AFM tip for dragging, pushing or pressing an object. The optical microscopy records the process in video rate. The AFM in return gives more information through imaging or fast force mapping. TIRF microscopy enhances image contrast by producing a thin evanescent field in the total internal reflection of excitation light at the glass-sample interface, which has a Z-resolution of 70–200 nm depends on the refractive index [64]. TIRF microscopy is usually used to study the dynamic event in an aqueous environment that is close to a solid substrate. Selective excitation of the surface-bound fluorophores allows TIRF Microscopy to detect single molecule fluorescence.
A recent application example is that Tan et al. use this hyphenated method to resolve the pinning forces of nanobubbles on a substrate [65]. Surface nanobubbles were first generated through a microfluidic channel using solvent exchange protocols (Fig. 6.14a). Nanobubbles were imaged by AFM and TIRF Microscopy simultaneously to assure the location for bubble pulling. After the AFM tip anchored to the bubble, the bubble-pulling is completed by the stage moving (Fig. 6.14c). Optical images were extracted from the video. The pinning forces can be calculated by a simple equation: F h = σ ∆A, where F is the pinning force, h is the pulling bubble neck, σ is the surface tension, and ∆A is the surface area change due to bubble-pulling. This method opens a window for the study of microbe film, adhesion of cell membrane and biomolecules on the substrates.

AFM manipulation combined with TIRFM for pulling nanobubbles. (a) schematic of setup, bubbles formed inside a microfluidic channel and removed for AFM imaging and manipulation. (b) a nanobubble with the stretched neck. (c) schematic of AFM manipulation. (d) dynamic bubble motion with a hydrophilic tip. (e) dynamic bubble motion with a hydrophobic tip. Reprinted with permission from Tan et al. [65] © American Physical Society (2017)
5 Prospect and Outlook
The high speed AFM and super-resolution optics combination systems are the experimental setups to fulfil the highest demands on mechanical stability, flexibility, and modularity. A tip scan AFM can be easily combined to inverted optical microscopes. The advantage of the integration is superb time and spatial resolution, and including, but not limited to the aforementioned optics, there are some techniques are under developing and will appeal researchers in the biological research field.
FRET is a highly accurate and sensitive method to determine the nanostructures and interactions between molecules, such as lipids, proteins, DNA, sugars, and other polymers. For experiments at the single-molecule level, donor with fluorophore labelling and acceptor molecules can be bound to a molecular chain. The single molecule can be detected and measured, as both substrate and tip are modified with suitable chemicals. The intramolecular forces and the changes in fluorescence of the donor-acceptor system are measured, when the tip was moved away from the surface and the molecule was stretched slowly. This combination provides the fundamental information in terms of distance, interactions, structural changes and optical spectroscopy. It has been applied to investigate the morphology of carbon dots and FRET from carbon dots transferred to riboflavin in the biosensor, respectively [66].
Fluorescence Recovery After Photobleaching (FRAP) is a method to determine the diffusion kinetics of a thin molecular film, which contains cells labeled with fluorescent probes through tissue and cells. It is useful in biological studies in protein binding and cell membrane diffusion, and the characterization of surface structure and free energy. High speed AFM can be combined to examine the phase separating structural changes, such as defects, obstructions, lipid condensation and collapse structures of lipid bilayers [67].
Fluorescence-lifetime imaging microscopy (FLIM) is an imaging technology, which is based on the different fluorescence decay rate of a fluorescent sample. Instead of the fluorophore intensity, the lifetime of the signal was used to generate images, which minimized the effect of photon scattering in the thick layers of samples. High speed AFM is combined to obtain the distribution information of biomolecular conformation and structure changes via FRET [68]. For example, FLIM, Fluorescence correlation spectroscopy (FCS) and AFM were applied to characterize the structure of amyloid β protein (Aβ), which is the possible neurotoxic agent in Alzheimer’s disease (AD).
Another setup combines the high-resolution of AFM with confocal two-photon fluorescence microscope, which has discriminative power and high sensitivity [69]. This scheme provides the capability of obtaining simultaneous morphology and optical images with high-resolution and sensitivity, and was successfully tested using latex beads loaded with dye. The stacking/unstacking of grana membranes in the envelope-free plant chloroplasts was investigated through this scheme. Furthermore, the combination of AFM and two-photon fluorescence microscope enabled a direct comparison between the signaling protein H-Ras distribution in living cells and the cell morphology, which opens up promising applications in the various research area, such as material and life science.
The combined AFM and optical tweezers (AFM-OT, JPK instruments AG) have high potential in biological studies, molecules manipulation, and nanomechanics. The optical tweezers are able to manipulate small dielectric objects and to measure the extremely small forces in the AFM scanning area [70]. This needs to consider the influence of the AFM laser beam which should be minimized and not affect the laser tweezers. AFM-OT techniques allow dual force measurements and provide a platform to integrate other super-resolution optics STED/PALM/STORM/TIRF or confocal fluorescence microscopy. The new technique achieves more data captured simultaneously and opens up new spectrum applications in dynamic studies of single molecules and living cells. DNA unzipping can be studied using the laser beam to trap a bead attached to DNA hairpin at one end while the AFM tip holding the other end of one strand. If a DNA molecule is attached to two beads at both ends, which are trapped by the laser beams, and the AFM tip is tagged by a DNA enzyme, the interactions at DNA-enzyme binding can be sensed by both the AFM and the optical tweezer. The AFM-OT techniques can extend to signaling studies. The optical tweezers position a microbead functionalized with signaling molecules close to cells, and the dynamic process is imaged by the AFM tip. On the other hand, the force can be sensed by the optical tweezers when the cell is stimulated or disturbed by the force or temperature from the AFM tip. Using optical tweezers is convenient to manipulate the sample and triggering cellular response, immune response, and infection process. It is reliable to analyze the resulting changes in cellular structure, dynamics, and mechanical properties using fast-scan AFM.
6 Conclusion
The integration of high speed AFM and super-resolution optics provides super spatial and time resolution simultaneous images for biological studies. The combination is more than the sum of the two parts. Both provide complementary information to each other and time resolution is high enough for recording dynamic processes. There is no need to stain, coat, or freeze samples and experiments can be done in ambient or liquids environments. The hyphenated techniques allow fast acquisition of high-resolution morphology, but also obtaining high-resolution optical spectroscopy at the same interested area. Tip-scanning is a preferred choice for the hyphenated techniques for acquiring stable optical images without stress by shaking the cells. Super-resolution optics provide the specific cellular component with a specific individual fluorophore.
Fast force mapping enables probing mechanical properties of cell surfaces and molecular recognition, such as stiffness, elasticity, and adhesion. AFM nanomanipulation and nanolithography assists the fabrications, patterning, and detections at the single-molecule level. The applications of force spectroscopy range from determining intra-interactions by unfolding or stretching of DNA, protein, polysaccharide, and polymers, molecular recognition, to the interactions between DNA-protein, polymer-substrates, and protein-protein.
With the advantage of the precise positioning, the integration of high performance of AFM nanomanipulation and optical spectroscopy pave the way to advanced optical spectroscopy with high spatial resolution. This is important for the fundamental understanding of the fast-changing biological phenomena. These approaches are promising in great applications in a study on a cellular response, immune signaling, drug selection and so on, which significantly further the knowledge about molecular interactions and dynamic processes in living cells. Precise controlling in positioning, high spatial resolution and super time resolution improve the throughput, flexibility, and reproducibility, and attract research interests across the world in the multidisciplinary field.
References
Huang P, Andersson SB. High speed atomic force microscopy enabled by a sample profile estimator. Appl Phys Lett. 2013;102:213118.
Kodera N, Yamamoto D, Ishikawa R, Ando T. Video imaging of walking myosin V by high-speed atomic force microscopy. Nature. 2010;468:72–6.
Casuso I, Khao J, Chami M, Paul-Gilloteaux P, Husain M, Duneau J-P, et al. Characterization of the motion of membrane proteins using high-speed atomic force microscopy. Nat Nanotechnol. 2012;7:525–9.
Fantner GE, Barbero RJ, Gray DS, Belcher AM. Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat Nanotechnol. 2010;5:280–5.
Yang C, Yan J, Dukic M, Hosseini N, Zhao J, Fantner GE. Design of a high-bandwidth tripod scanner for high speed atomic force microscopy. Scanning. 2016;38:889–900.
Weisenburger S, Sandoghdar V. Light microscopy: an ongoing contemporary revolution. Contemp Phys. 2015;56:123–43.
Fernández-Suárez M, Ting AY. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol. 2008;9:929–43.
Fantner GE, Schitter G, Kindt JH, Ivanov T, Ivanova K, Patel R, et al. Components for high speed atomic force microscopy. Ultramicroscopy. 2006;106:881–7.
Kim BI, Boehm RD. Force-feedback high-speed atomic force microscope for studying large biological systems. Micron. 2012;43:1372–9.
Ando T, Uchihashi T, Kodera N. High-speed AFM and applications to biomolecular systems. Annu Rev Biophys. 2013;42:393–414.
Mikheikin A, Olsen A, Picco L, Payton O, Mishra B, Gimzewski JK, et al. High-speed atomic force microscopy revealing contamination in DNA purification systems. Anal Chem. 2016;88:2527–32.
Sanchez H, Suzuki Y, Yokokawa M, Takeyasu K, Wyman C. Protein–DNA interactions in high speed AFM: single molecule diffusion analysis of human RAD54. Integr Biol. 2011;3:1127–34.
Ando T. High-speed atomic force microscopy coming of age. Nanotechnology. 2012;23:062001.
Uchihashi T, Iino R, Ando T, Noji H. High-speed atomic force microscopy reveals rotary catalysis of Rotorless F1-ATPase. Science. 2011;333:755–8.
Ruan Y, Miyagi A, Wang X, Chami M, Boudker O, Scheuring S. Direct visualization of glutamate transporter elevator mechanism by high-speed AFM. Proc Natl Acad Sci. 2017;114:1584–8.
Shim S-H, Xia C, Zhong G, Babcock HP, Vaughan JC, Huang B, et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci. 2012;109:13978–83.
Suzuki Y, Sakai N, Yoshida A, Uekusa Y, Yagi A, Imaoka Y, et al. High-speed atomic force microscopy combined with inverted optical microscopy for studying cellular events. Sci Rep. 2013;3:2131.
Meister A, Gabi M, Behr P, Studer P, Vörös J, Niedermann P, et al. FluidFM: combining atomic force microscopy and Nanofluidics in a universal liquid delivery system for single cell applications and beyond. Nano Lett. 2009;9:2501–7.
Uchihashi T, Watanabe H, Fukuda S, Shibata M, Ando T. Functional extension of high-speed AFM for wider biological applications. Ultramicroscopy. 2016;160:182–96.
Nievergelt AP, Erickson BW, Hosseini N, Adams JD, Fantner GE. Studying biological membranes with extended range high-speed atomic force microscopy. Sci Rep. 2015;5:11987.
Eghiaian F, Rico F, Colom A, Casuso I, Scheuring S. High-speed atomic force microscopy: imaging and force spectroscopy. FEBS Lett. 2014;588:3631–8.
Zhou H-T, Gao X, Zheng P, Qin M, Cao Y, Wang W. Mechanical properties of elastomeric proteins studied by single molecule force spectroscopy. Acta Phys Sin. 2016;65:188703.
Endo M, Sugiyama H. Single-molecule imaging of dynamic motions of biomolecules in DNA origami nanostructures using high-speed atomic force microscopy. Acc Chem Res. 2014;47:1645–53.
Watanabe H, Uchihashi T, Kobashi T, Shibata M, Nishiyama J, Yasuda R, et al. Wide-area scanner for high-speed atomic force microscopy. Rev Sci Instrum. 2013;84:053702.
Shibata M, Uchihashi T, Ando T, Yasuda R. Long-tip high-speed atomic force microscopy for nanometer-scale imaging in live cells. Sci Rep. 2015;5:8724.
Igarashi K, Uchihashi T, Uchiyama T, Sugimoto H, Wada M, Suzuki K, et al. Two-way traffic of glycoside hydrolase family 18 processive chitinases on crystalline chitin. Nat Commun. 2014;5:3975.
Huang B, Bates M, Zhuang X. Super-Resolution Fluorescence Microscopy. Annu Rev Biochem. 2009;78:993–1016.
Westphal V, Rizzoli SO, Lauterbach MA, Kamin D, Jahn R, Hell SW. Video-rate far-field optical Nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–9.
Nägerl UV, Willig KI, Hein B, Hell SW, Bonhoeffer T. Live-cell imaging of dendritic spines by STED microscopy. Proc Natl Acad Sci. 2008;105:18982–7.
Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–2.
Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci. 2000;97:8206–10.
Tressler C, Stolle M, Fradin C. Fluorescence correlation spectroscopy with a doughnut-shaped excitation profile as a characterization tool in STED microscopy. Opt Express. 2014;22:31154–66.
Harke B, Chacko JV, Haschke H, Canale C, Diaspro A. A novel nanoscopic tool by combining AFM with STED microscopy. Opt Nanosc. 2012;1:3.
Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793–6.
Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 1997;388:355.
Odermatt PD, Shivanandan A, Deschout H, Jankele R, Nievergelt AP, Feletti L, et al. High-resolution correlative microscopy: bridging the gap between single molecule localization microscopy and atomic force microscopy. Nano Lett. 2015;15:4896–904.
Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–5.
Wolter S, Schuttpelz M, Tscherepanow M, van de Linde S, Heilemann M, Sauer M. Real-time computation of subdiffraction-resolution fluorescence images. J Microsc. 2010;237:12–22.
Hansma HG, Vesenka J, Siegerist C, Kelderman G, Morrett H, Sinsheimer RL, et al. Reproducible imaging and dissection of plasmid DNA under liquid with the atomic force microscope. Science. 1992;256:1180–4.
Stark RW, Thalhammer S, Wienberg J, Heckl WM. The AFM as a tool for chromosomal dissection – the influence of physical parameters. Appl Phys A Mater Sci Process. 1998;66:S579–S84.
Fotiadis D, Scheuring S, Müller SA, Engel A, Müller DJ. Imaging and manipulation of biological structures with the AFM. Micron. 2002;33:385–97.
An H, Guo Y, Zhang X, Zhang Y, Hu J. Nanodissection of single- and double-stranded DNA by atomic force microscopy. J Nanosci Nanotechnol. 2005;5:1656–9.
An H, Huang J, Lü M, Li X, Lü J, Li H, et al. Single-base resolution and long-coverage sequencing based on single-molecule nanomanipulation. Nanotechnology. 2007;18:225101.
Lü J-h, Li H-k, An H-j, Wang G-h, Wang Y, Li M-q, et al. Positioning isolation and biochemical analysis of single DNA molecules based on Nanomanipulation and single-molecule PCR. J Am Chem Soc. 2004;126:11136–7.
Hu J, Zhang Y, Gao H, Li M, Hartmann U. Artificial DNA patterns by mechanical Nanomanipulation. Nano Lett. 2002;2:55–7.
Guthold M, Matthews G, Negishi A, Taylor RM, Erie D, Brooks FP, et al. Quantitative manipulation of DNA and viruses with the nanomanipulator scanning force microscope. Surf Interface Anal. 1999;27:437–43.
Herman-Bausier P, Formosa-Dague C, Feuillie C, Valotteau C, Dufrêne YF. Forces guiding staphylococcal adhesion. J Struct Biol. 2017;197:65–9.
Gould P. Lithography: rewriting the rules. Mater Today. 2003;6:34–9.
Li B, Zhang Y, Yan S-H, Lü J-H, Ye M, Li M-q, et al. Positioning scission of single DNA molecules with nonspecific endonuclease based on Nanomanipulation. J Am Chem Soc. 2007;129:6668–9.
Neuman KC, Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat Methods. 2008;5:491–505.
Hughes ML, Dougan L. The physics of pulling polyproteins: a review of single molecule force spectroscopy using the AFM to study protein unfolding. Rep Prog Phys. 2016;79:076601.
Rief M, Oesterhelt F, Heymann B, Gaub HE. Single molecule force spectroscopy on polysaccharides by atomic force microscopy. Science. 1997;275:1295–7.
Rief M, Clausen-Schaumann H, Gaub HE. Sequence-dependent mechanics of single DNA molecules. Nat Struct Biol. 1999;6:346–9.
Formosa-Dague C, Speziale P, Foster TJ, Geoghegan JA, Dufrêne YF. Zinc-dependent mechanical properties of Staphylococcus aureus biofilm-forming surface protein SasG. Proc Natl Acad Sci. 2016;113:410–5.
Eghiaian F, Rigato A, Scheuring S. Structural, mechanical, and dynamical variability of the actin cortex in living cells. Biophys J. 2015;108:1330–40.
Alsteens D, Newton R, Schubert R, Martinez-Martin D, Delguste M, Roska B, et al. Nanomechanical mapping of first binding steps of a virus to animal cells. Nat Nanotechnol. 2017;12:177–83.
Chacko JV, Canale C, Harke B, Diaspro A. Sub-diffraction Nano manipulation using STED AFM. PLoS One. 2013;8:e66608.
Langelüddecke L, Singh P, Deckert V. Exploring the nanoscale: fifteen years of tip-enhanced Raman spectroscopy. Appl Spectrosc. 2015;69:1357–71.
Zhang R, Zhang Y, Dong ZC, Jiang S, Zhang C, Chen LG, et al. Chemical mapping of a single molecule by plasmon-enhanced Raman scattering. Nature. 2013;498:82.
Sadashivappa G, Sharvari N. Nanoantenna-a review. Int J Renew Energy Technol Res. 2015;4:1–9.
Patel SK, Argyropoulos C. Plasmonic nanoantennas: enhancing light-matter interactions at the nanoscale. EPJ Appl Metamater. 2015;2:4.
Cohen M, Shavit R, Zalevsky Z. Observing optical Plasmons on a single nanometer scale. Sci Rep. 2014;4:4096.
Bigourdan F, Hugonin J-P, Marquier F, Sauvan C, Greffet J-J. Nanoantenna for electrical generation of surface Plasmon Polaritons. Phys Rev Lett. 2016;116:106803.
Chan CU, Ohl C-D. Total-internal-reflection-fluorescence microscopy for the study of Nanobubble dynamics. Phys Rev Lett. 2012;109:174501.
Tan BH, An H, Ohl C-D. Resolving the pinning force of Nanobubbles with optical microscopy. Phys Rev Lett. 2017;118:054501.
Dinç S. A simple and green extraction of carbon dots from sugar beet molasses: biosensor applications. Surg Industry. 2016;141:560–4.
Bennun SV, Faller R, Longo ML. Drying and rehydration of DLPC/DSPC symmetric and asymmetric supported lipid bilayers: a combined AFM and fluorescence microscopy study. Langmuir. 2008;24:10371–81.
Koh CJ, Lee M. Structural analysis of amyloid aggregates by multifunctional fluorescence nanoscopy. Curr Appl Phys. 2006;6:e257–e60.
Gradinaru CC, Martinsson P, Aartsma TJ, Schmidt T. Simultaneous atomic-force and two-photon fluorescence imaging of biological specimens in vivo. Ultramicroscopy. 2004;99:235–45.
Pierini F, Zembrzycki K, Nakielski P, Pawłowska S, Kowalewski TA. Atomic force microscopy combined with optical tweezers (AFM/OT). Meas Sci Technol. 2016;27:025904.
Acknowledgements
H. A. acknowledges the support from the National Key R&D Program of China (2016YFD0400800), and the National Natural Science Foundation of China (NSFC-31470090 and NSFC-31571029). H. Y. acknowledges the financial support by the Singapore Ministry of Education Academic Research Fund Tier 1 (R-143-000-A40-114) and project 31371851 supported by NSFC.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Feng, X., Guo, Y., An, H., Yang, H. (2018). The Hyphenated Technique of High Speed Atomic Force Microscopy and Super Resolution Optical Detection System. In: Cai, J. (eds) Atomic Force Microscopy in Molecular and Cell Biology. Springer, Singapore. https://doi.org/10.1007/978-981-13-1510-7_6
Download citation
DOI: https://doi.org/10.1007/978-981-13-1510-7_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1509-1
Online ISBN: 978-981-13-1510-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)