
Abstract
Insulin-like growth factor 1 (IGF-1) is a key anabolic growth factor stimulating phosphatidylinositol 3-kinase (PI3K)/Akt signaling which is well known for regulating muscle hypertrophy. However, the role of IGF-1 in muscle atrophy is less clear. This review provides an overview of the mechanisms via which IGF-1 signaling is implicated in several conditions of muscle atrophy and via which mechanisms protein turnover is altered. IGF-1/PI3K/Akt signaling stimulates the rate of protein synthesis via p70S6Kinase and p90 ribosomal S6 kinase and negatively regulates protein degradation, predominantly by its inhibiting effect on proteasomal and lysosomal protein degradation. Caspase-dependent protein degradation is also attenuated by IGF/PI3K/Akt signaling, whereas evidence for an effect on calpain-dependent protein degradation is inconclusive. IGF-1/PI3K/Akt signaling reduces during denervation-, unloading-, and joint immobilization-induced muscle atrophy, whereas IGF-1/PI3K/Akt signaling seems unaltered during aging-associated muscle atrophy. During denervation and aging, IGF-1 overexpression or injection counteracts denervation- and aging-associated muscle atrophy, despite enhanced anabolic resistance with regard to IGF-1 signaling with aging. It remains unclear whether pharmacological stimulation of IGF-1/PI3K/Akt signaling attenuates immobilization- or unloading-induced muscle atrophy. Exploration of the possibilities to interfere with IGF-1/PI3K/Akt signaling reveals that microRNAs targeting IGF-1 signaling components are promising targets to counterbalance muscle atrophy. Overall, the findings summarized in this review show that in disuse conditions, but not with aging, IGF-1/PI3K/Akt signaling is attenuated and that in some conditions stimulation of this pathway may alleviate skeletal muscle atrophy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Insulin-like growth factor 1 (IGF-1) is a key anabolic growth factor which is involved in tissue development during growth, as well as in adaptation and regeneration of mature tissues and cells. IGF-1 is expressed in multiple isoforms in almost all tissues and cells [1]. It is therefore not surprising that mice deficient in IGF-1 or its receptor show decreased viability, growth deficiency, and malformations in several tissue types [2]. IGF-1 is expressed in the liver, acts locally in an autocrine and paracrine manner on liver cells, but also has a strong endocrine function on other tissues like muscle. In muscle, IGF-1 isoforms that are most abundantly expressed are IGF-1Ea and mechano growth factor (MGF, also referred to as IGF-1Ec in humans or IGF-1Eb in rodents). In skeletal muscle, basal mRNA levels are higher for IGF-1Ea than MGF [3]. Paradoxically, the expression of IGF-1Ea is higher in relative small oxidative myofibers, expressing slow myosin heavy chains (MHCs) than in relative large, low oxidative myofibers expressing fast-type MHCs [4]. Expression of these main IGF-1 isoforms increases substantially in response to mechanical overload by stretching or increased contractile activity [1, 5, 6]. Moreover, IGF-1 expression is also enhanced biochemically by growth hormone (GH), and its half-life time and/or bioactivity is both negatively and positively regulated by several IGF-binding proteins (IGFBPs) as well as by albumin [1, 7, 8]. Since different IGFBPs can compensate for each other [7], single IGFBP measurements provide little evidence regarding the bioavailability of IGF-1.
Both IGF-1 isoforms are derived from the same gene which contains 6 exons. MGF is expressed by alternative splicing of exon 5 and 6 and differs from IGF-1Ea in its E peptide which contains exon 5 and 6 in stead of exon 6 in IGF-1Ea [6]. The IGF-1 domain of IGF-1Ea and MGF, which consists of exon 3 and 4, signals via the IGF-1 receptor (IGF-1R), which is a tyrosine kinase receptor expressed in both myofibers and muscle stem cells (also known as satellite cells). Also the E peptides of IGF-1Ea and MGF E are involved in signaling via the IGF-1R whereby the MGF E peptide is known for its stimulatory effect on satellite cell activation, proliferation, and migration [1, 9, 10]. Moreover, different IGF-1 isoforms exist also due to different promotor start regions upstream of exon 1 or 2 [11]. Transcripts including exon 1 are known as class 1 IGF-1 isoforms, whereas IGF-1 isoforms including exon 2 are referred to as class 2 [11]. Functional differences of the two classes remain however unclear [12]. IGF-1 and insulin share about 50% amino acid homology and can bind each other’s receptors, albeit with lower affinity.
IGF-1 and MGF are well known for their autocrine and paracrine roles during muscle overload and myofiber hypertrophy, however, less is known about how IGF-1 is involved in the induction of muscle atrophy. An important signaling pathway in skeletal muscle atrophy is the IGF-1/phosphatidylinositol 3-kinase (PI3K)/Akt pathway, since this is involved in both protein synthesis and protein degradation [4, 13,14,15,16,17,18,19,20,21,22]. Here we provide an overview of the main signaling pathways via which IGF-1 and MGF modulate the rate of protein synthesis and degradation during muscle atrophy, with particular emphasis on the IGF-1/PI3K/Akt pathway, and how IGF-1 signaling is altered.
2 The Role of IGF-1 in the Regulation of Protein Synthesis and Degradation
Changes in muscle size are the net effect of changes in the rate of protein synthesis and protein degradation. IGF-1 affects both processes, and as such, changes in its signaling have a strong effect on muscle size [4, 13,14,15,16,17,18,19,20,21,22]. In this paragraph, the role of IGF-1 in protein synthesis and different mechanisms of protein breakdown is reviewed.
Binding of IGF-1 to its receptor causes phosphorylation of the intracellular adaptor proteins Shc or insulin receptor substrate 1 (IRS-1), which results in the activation of two main pathways, RAS/RAF/MEK/ERK (also known as mitogen-activated protein kinase (MAPK) signaling) and PI3K/Akt, respectively [21, 23]. IGF-1-induced hypertrophy in rats is prevented by the inhibition of MEK [24], which indicates the requisite for MAPK signaling in hypertrophy in vivo. In myotubes however, inhibition of RAF has been shown to induce hypertrophy [25], suggesting an inhibitory effect of MAPK signaling on hypertrophy in vitro. These observations show that the role of MAPK in protein synthesis and degradation and the underlying mechanisms are not entirely understood. On the other hand, the IGF-1/PI3K/Akt pathway and its anabolic mechanisms underlying myofiber hypertrophy are well established. Translocation of PI3K to phosphorylated IRS-1 results in the phosphorylation of PI3K. Subsequently, this causes the phosphorylation of phosphoinositide-dependent kinase-1 (PDK1) which then phosphorylates the serine/threonine kinase Akt (also known as protein kinase B) [26]. Akt is involved in multiple cellular processes including proliferation, metabolism, and cell size regulation [27]. Because the IGF-1/PI3K/Akt pathway plays a major role in myofiber size, the main focus of this review will be on the role of this pathway during skeletal muscle atrophy.
2.1 Protein Synthesis
Changes in the rate of protein synthesis involve changes in the rate of mRNA transcription and translation, which in muscle are both enhanced by IGF-1 [see for review 13, 28]. IGF-1 increases protein levels of β-catenin (a transcription factor involved in skeletal muscle growth) by phosphorylation of glycogen synthase kinase 3 beta (GSK3β), which prevents atrophy and can even induce hypertrophy in dexamethasone-treated rats [29]. Moreover, IGF-1 has been shown to increase transcription rate of α-skeletal actin during differentiation and myosin heavy chain (MHC) IIB in C2C12 myoblasts and myotubes, respectively [30, 31]. Increased transcription by IGF-1 may be regulated by myogenin and MyoD, which are both transcription factors involved in the expression of actin and myosin, since IGF-1 has been shown to induce myogenin and MyoD expression [32] and both transcription factors increase in the human vastus lateralis after resistance exercise [33]. Note, myogenin has been shown to be stimulated by IGF-1/PI3K/Akt signaling when simultaneously MAPK signaling is inhibited [24, 34]. Indeed, IGF-1 treatment has also been associated with a lack of increase in myogenin and MyoD [31, 35]. These observations show that IGF-1 enhances transcription, but the underlying mechanisms are not entirely clear.
In addition to transcription, the IGF-1/PI3K/Akt pathway stimulates translation by activation of a key anabolic target, the mammalian target of rapamycin (mTOR), which is a kinase that integrates multiple upstream signals, which are not solely derived from IGF-1/PI3K/Akt activation [36]. In addition to IGF-1, another important activator of mTOR is mechanical loading [37], and therefore disuse atrophy is likely to decrease mTOR activity even if IGF-1 signaling would be unaffected. Moreover, mTOR is affected by several other upstream mediators such as energy status or amino acids [36]. Activation of mTOR stimulates the rate of mRNA translation by phosphorylation of 4E-BP (also known as PHAS-1), which prevents its binding (i.e., inactivation) to the eukaryotic initiation factor (eIF) 4E (Fig. 6.1) [17, 38]. Furthermore, activated mTOR also activates p70S6Kinase (p70S6K) which stimulates mRNA translation by phosphorylating ribosomal protein S6 (rpS6) and activation of eukaryotic elongation factor (eEF) 2 [39,40,41,42].

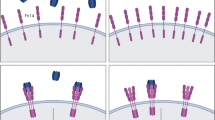
An overview of the key signaling pathways underlying the hypertrophic effects of IGF-1. Stimulation is indicated by arrows, and inhibitory effects are indicated by lines capped by perpendicular lines. Solid lines represent established mechanisms; dashed lines represent mechanisms that have not been consistently proven in myofibers. Colors represent different pathways or downstream targets. Two important signaling pathways induced by IGF-1 are the IGF-1/PI3K/Akt pathway and the IGF-1/Ras/Raf/Mek/Erk pathway. Both pathways result in kinase activation or changes in binding proteins causing enhancement of mRNA translation by regulating ribosomal proteins, eukaryotic initiation factors (eIF), or eukaryotic elongation factors (eEF). Abbreviations: IGF-1 insulin-like growth factor 1, PI3K phosphatidylinositol 3-kinase, PDK1 phosphoinositide-dependent kinase-1, GSK3β glycogen synthase kinase 3 beta, mTOR mammalian target of rapamycin, 4E-BP 4E-binding protein, ERK extracellular signal-regulated kinases
Moreover, PDK1 which is phosphorylated by PI3K and subsequently phosphorylates Akt is also likely to be involved in enhancement of the rate of protein synthesis independent of Akt [26]. The role of PDK1 in skeletal muscle is not fully understood, but evidence from studies on several other cell types, including smooth muscle, suggests that PDK1 can phosphorylate Akt and has also the ability to directly activate p70S6K and p90 ribosomal S6 kinase (p90RSK), both increasing the rate of translation by regulation of rpS6 and eEF2 [26, 40, 41]. In smooth muscle cells, p90RSK is also activated by ERK [26]. In addition, mRNA translation rate is also increased by phosphorylation of Akt which then phosphorylates and inhibits GSK3β which is subsequently no longer able to suppress eIF2B activity [43]. GSK3β has been shown to be required for atrophy in C2C12 myotubes and is involved in both skeletal muscle hypertrophy and atrophy in humans [44, 45]. Moreover, GSK3β may also be inhibited by ERKs as it has been shown in cancer cells that ERKs facilitate the inhibition of GSK3β by [46].
The key regulatory kinases of which the activity is modulated by IGF-1 are p70S6K, p90RSK, and GSK3β, which are all involved in enhancement of the rate of mRNA translation (Fig. 6.1).
2.2 Proteasomal Muscle Protein Degradation
The prime system for muscle protein degradation is the ubiquitin-proteasome system [13, 14, 47, 48]. During protein degradation, contractile proteins are ubiquitinated by the consecutive actions of E1, E2, and E3 enzymes which can then be recognized and subsequently degraded by proteasomes. The gene expression as well as their function in muscle atrophy of two E3 ligases, Muscle Ring Finger 1 (MuRF1) and muscle atrophy F-box (MAFbx, also known as Atrogin-1), has extensively been examined [see for review 48]. Both E3 ligases are particularly involved in the degradation of contractile proteins and eIF3f [49, 50]. During several atrophic conditions, MuRF1 and MAFbx expression levels are increased [48, 51], and these ligases are critical for the enhanced rate of protein degradation as MuRF1- or MAFbx-deficient mice showed a 36% and 56% reduction in denervation-induced muscle atrophy after 14 days, respectively [51]. Expression of MuRF1 and MAFbx is regulated by a group of Forkhead box O (FOXO) transcription factors which stimulate expression of several genes involved in diverse mechanisms of protein degradation, including proteasomal degradation [18, 19]. Transcriptional activation of MuRF-1 and MAFbx expression requires nuclear localization of FOXO transcription factors which is mediated by Akt. Active Akt phosphorylates FOXO transcription factors resulting in their cytoplasmic retention and inactivation of their function as transcription factors in the nucleus [52, 53]. FOXO1, 3, and 4 are the most important FOXO transcription factors involved in muscle atrophy and are all regulated by Akt [18]. Moreover, muscle atrophy induced by IGF-1R and insulin receptor knockout could completely be prevented by the combined knockout of FOXO1, 3, and 4, whereas knockout of single FOXO transcription factors had little or no effect [54], which indicates the importance of all three FOXO factors in muscle atrophy. In short, the IGF-1/PI3K/Akt pathway negatively regulates proteasomal degradation by inactivating FOXO transcription factors and hence the expression of the E3-ligases MAFbx and MuRF-1 (Fig. 6.2).

An overview of the key signaling pathway underlying the anti-atrophic effects of IGF-1. Stimulation is indicated by arrows, and inhibitory effects are indicated by lines capped by perpendicular lines. Solid lines represent established mechanisms; dashed lines represent mechanisms that have not been consistently proven in myofibers. Abbreviations: IGF-1 insulin-like growth factor 1, PI3K phosphatidylinositol 3-kinase, FOXO Forkhead box O transcription factors, MuRF1 Muscle Ring Finger 1, MAFbx muscle atrophy F-box, LC3 microtubule-associated protein 1A/1B-light chain 3, Bnip3 BCL2 interacting protein 3
2.3 Lysosomal Muscle Protein Degradation
Autophagy is another key mechanism for muscle protein degradation [55]. Autophagy concerns the engulfment of cellular particles into autophagosomes which subsequently fuse with lysosomes to be degraded in the acid intralysosomal environment [55]. Several conditions like fasting and denervation result in the upregulation of expression of proteins involved in autophagy [56, 57]. Induced myofiber atrophy by constitutive active FOXO3 was attenuated by knockdown of LC3, a gene involved in autophagy [56]. An accumulation in ubiquitinated proteins was observed in autophagy-deficient mice [58], which suggest that some ubiquitinated proteins are specifically degraded by lysosomal degradation. These observations indicate the involvement of autophagy in muscle atrophy. In addition to its role in muscle atrophy, autophagy is also important for cell maintenance as this mechanism is also responsible for the clearance of misfolded proteins and dysfunctional organelles [55]; therefore both diminished and overactivity of autophagosomes could be harmful to myofibers. The first may affect the quality of myofibers, whereas the second affects the quantity of proteins within myofibers.
IGF-1 has been shown to regulate autophagy since deletion of insulin receptor and IGF-1R in mice increased an autophagic flux [54]. As in proteasomal degradation, deactivation of FOXO transcription factors by Akt is also a key mechanism in autophagy [56, 57]. FOXO3 is involved in the control of autophagosome formation by stimulating expression of two autophagy-related genes, i.e., LC3 and Bnip3 [56, 57]. In addition, the upregulation of several autophagic factors and autophagosome formation induced by fasting or denervation, was abolished by exogenous expression of constitutively active Akt, while inhibition of Akt increased lysosomal proteolysis [56, 57]. The inhibition of total mRNA synthesis while Akt was blocked largely suppressed the increased lysosomal proteolysis caused by Akt inhibition [57], which suggests that FOXO-induced transcription is largely responsible for increased lysosomal proteolysis. Although the effect was relatively small, mTOR inhibition also caused an increase in lysosomal proteolysis, while blocking of mRNA synthesis did not prevent this increase, which indicates that mTOR can also suppress autophagy independent of transcriptional control [57]. These observations are in line with results of a study on acute uremia whereby IGF-1/PI3K/Akt-independent stimulation of mTOR by leucine also suppressed autophagy [59]. These data suggest that an increase in IGF-1/PI3K/Akt signaling inhibits autophagy through activation of predominantly FOXO3 and also mTOR. In addition to fasting and denervation, 4 days of knee joint immobilization in young adult humans caused increased LC3B-II/LC3B-I protein ratios, an indication of increased autophagy, concomitant with decreased pAkt/tAkt levels [60]. This suggests that an increase in autophagy due to reduced IGF-1/PI3K/Akt signaling is associated with unloading. Overall, there is strong evidence that the IGF-1/PI3K/Akt pathway inhibits autophagy during fasting, denervation, and potentially joint immobilization and that this is mediated predominantly through the inactivation of FOXO3 (Fig. 6.2).
However, the role of autophagy in different atrophic conditions is not unambiguous. In contrast to adults, in elderly LC3B-II/LC3B-I protein ratios were unaffected after 4 days of knee joint immobilization [60], which suggests that in aged muscle autophagy may not be induced by joint immobilization. In the long term, after 2 weeks of joint immobilization, no convincing increase in autophagy could be shown in both young adult and elderly. These observations indicate that in elderly joint immobilization does not increase muscle autophagy, whereas in adults autophagy is increased shortly after immobilization and does not occur in the long term [60]. The lack of a long-term effect of unloading is confirmed by a study in which mice were subjected to 91 days of unloading in the International Space Station, which showed no changes in autophagy-related gene expression [61]. In contrast, in adult and old rats undergoing hind limb suspension for 2 weeks, no clear increase in autophagy was observed suggesting that in this unloading model, autophagy may not play a role in the induction of muscle atrophy [62].
Autophagy seems also to be regulated independent of IGF-1/PI3K/Akt signaling, since mice showing aging-associated muscle atrophy, whereby IGF-1 signaling was unchanged, had an increase in autophagic vesicles [63]. This is in line with the effect of lipopolysaccharide (LPS) administration in rat skeletal muscle resulting in acute inflammation, which is associated with proteasomal and lysosomal proteolysis [64, 65]. LPS injection caused a decrease in IGF-1 mRNA expression and Akt phosphorylation [64, 65]. Although blocking of this LPS-induced inflammation restored Akt phosphorylation and autophagy-related protein expression [65], IGF-1 systemic or muscle-specific overexpression could not inhibit the LPS-induced increased autophagy-related gene expression [64]. This also suggests that autophagy is regulated independently of IGF-1 signaling. Indeed, p38 MAPK has been suggested to regulate autophagy [55, 66]. p38 can be stimulated by IGF-1 but also independent of IGF-1 by, for instance, oxidative stress [66].
Overall, there is strong evidence that autophagy is regulated by the IGF-1/PI3K/Akt pathway and is involved in fasting- and denervation-induced atrophy. However, the role of autophagy is not clear in all muscle atrophic conditions and seems to be transient and age dependent.
2.4 Caspase- and Calpain-Dependent Muscle Protein Degradation
2.4.1 Calpain-Dependent Protein Degradation
Calpains are cysteine proteases which are activated by free cytoplasmic calcium and degrade predominantly cytoskeletal proteins [see for review 67]. In skeletal muscle, three different calpain isoforms are mainly expressed, i.e., milli- and micromolar calpains (also referred to as calpain 1 and 2, respectively), which are named after their sensitivity for calcium, and calpain 3, also known as p94 [67]. Although calpains are also able to degrade contractile proteins, they predominantly degrade Z-discs of sarcomeres which makes myofilaments available for degradation by the proteasome [67].
Calpain inhibition prevented immobilization-induced atrophy [68]. Moreover, calpain 3-deficient mice, which exhibit features of limb girdle muscular dystrophy type 2A, showed reduced muscle atrophy when subjected to unloading, suggesting calpain 3 requirement for muscle atrophy [69]. Because of their cooperation with the proteasome, it is conceivable that calpain expression is reduced by IGF-1, similar as E3 ligase expression. Indeed, both in vitro and in vivo studies on myotubes and mature myofibers show that IGF-1 inhibits calpain activity [70, 71]. Moreover, caloric restriction-induced muscle atrophy in neonatal calves was associated with an increase in calpain 1 activity and decrease in IGF-1 protein expression [72]. This observation is in line with that of another study showing that IGF-1 has an inhibitory effect on calpain-dependent proteolysis in dexamethasone-induced L6 myotube atrophy [73], which indicates that IGF-1 attenuates calpain activity. In contrast, another study investigating L6 myotube atrophy using the same calpain blocker in presence or absence of IGF-1 supplementation reported an increase instead of a decrease in myofibrillar protein degradation when calpain activity was blocked [74]. Although there are contrasting results regarding the effect of IGF-1 on calpain-induced proteolysis, the majority of these studies suggest an inhibitory effect of IGF-1 on calpain-dependent protein degradation (Fig. 6.2).
Although the role of IGF-1 in calpain activation is subject to controversy, a few studies have shown some insight in the interaction between calpain activity and Akt. In rat diaphragm muscle ex vivo, it has been shown that activation of calpains reduces Akt activity by lowering the binding of heat shock protein 90 (HSP90) to Akt which preserves Akt activity [67, 75]. Also, a reduction in pAkt in rat soleus muscle was prevented when unloading-induced calpain 1 activation was blocked. [76]. These results indicate that calpain activity reduces Akt phosphorylation. Note that Akt phosphorylation was not affected in calpain 3-deficient mice [69] which suggests calpain isoform specificity for the interaction with Akt activity.
The studies discussed above show that little is known regarding the role of the IGF-1/PI3K/Akt pathway in calpain-dependent protein degradation and to the best of our knowledge, a direct link between IGF-1/PI3K/Akt signaling and calpain activity in skeletal muscle has not been investigated. The data available suggest that calpain 1 but not 3 can inhibit Akt activity and that IGF-1 can inhibit calpain activity, but there is no evidence suggesting that an inhibitory effect of IGF-1 on calpain-dependent muscle protein degradation is mediated by IGF-1/PI3K/Akt signaling.
2.4.2 Caspase-Dependent Protein Degradation
Caspases are proteases, which in particular are involved in apoptosis and inflammation. Caspase-3 is activated in both angiotensin II-induced muscle wasting [77] and chronic kidney disease (associated with muscle wasting) [78]. Moreover, caspase-3 and caspase-9 activities increase during immobilization-induced muscle atrophy [68, 79], and the inhibition of caspase-3 activity prevented immobilization-induced atrophy in the rat soleus [68]. In contrast, no increases in caspase-3, caspase-8, or caspase-9 activities have been observed following limb unloading in both rats and humans [62, 80]. These observations indicate that caspase-mediated protein degradation is involved in several but not all conditions of muscle atrophy. Although support for IGF-1/PI3K/Akt-induced calpain-dependent degradation is scarce, evidence for the IGF-1/PI3K/Akt involvement in the reduction of caspase-dependent protein degradation is more substantial.
Administration of recombinant active caspase-3 to cultured L6 myotubes or rat psoas muscle lysates causes cleavage of myofibrillar proteins resulting in a detectable 14kD actin fragment which is degraded by the proteasome [81]. Serum deprivation also results in enhanced myofibrillar fragmentation which is abolished after inhibition of caspase-3 activity by IGF-1 [81]. Moreover, the inhibitory effect of IGF-1 on caspase-3 activity in L6 myotubes has been shown to be PI3K dependent [81]. These results suggest involvement of caspase-3 in myofibrillar degradation and that this caspase-mediated protein degradation is counterbalanced by IGF-1/PI3K/Akt signaling. In addition to this in vitro evidence, during angiotensin II administration inducing muscle atrophy in mice, IGF-1 signaling reduced, which was indicated by decreased IRS-1 and Akt phosphorylation, while caspase-3-dependent actin degradation increased [77]. Moreover, transgenic mice overexpressing muscle-specific IGF-1 were prevented from caspase-3-mediated actin degradation after angiotensin II treatment [77]. These observations indicate that caspase-3 cleaves myofibrillar proteins resulting in actin fragments which are degraded by the proteasome and that activity of caspase-3 is negatively regulated by IGF-1/PI3K/Akt signaling. The results of these studies are in line with those of other studies suggesting an inhibitory effect of Akt on caspase-3 activation [c.f. 82, 83].
In contrast, rats subjected to hind limb suspension for 2 weeks showed no increases in caspase-3, caspase-8, or caspase-9 activity within their lower leg muscles, while IGF-1 serum levels were slightly decreased [62]. However, since a large fraction of circular IGF-1 is produced by the liver, serum levels do not accurately reflect muscle-specific levels. In addition, as phosphorylated Akt was not decreased during unloading, it cannot be concluded that decreased IGF1/PI3K/Akt signaling is concomitant with a lack in change of caspase activity. This is line with a study showing no changes in both IGF1/PI3K/Akt signaling and caspase-3 mRNA levels following unilateral leg unloading humans [80]. Taken together, IGF-1/PI3K/Akt signaling inhibits caspase-mediated protein degradation (Fig. 6.2). It seems that in atrophic conditions in which IGF-1/PI3K/Akt signaling is unaffected, caspase-dependent protein degradation remains unaffected as well, whereas caspase-mediated protein degradation decreases in atrophic conditions associated with reduced IGF-1/PI3K/Akt signaling.
3 The Role of IGF-1/PI3K/Akt in Skeletal Muscle Atrophy Models
Muscle atrophy is a hallmark of several conditions such as aging, disuse, space flight, and a variety of pathologies. These conditions have in common a reduction in contractile activity of myofibers as well as a reduction in intra- and extracellular mechanical stress and strains to which myofibers are subjected. Despite these similarities, the impact on IGF-1 signaling within muscles varies between different disuse models. Here we discuss the effects of several conditions associated with muscle atrophy on IGF-1 expression and signaling in an attempt to explain the muscle atrophy associated with the corresponding physicochemical conditions.
3.1 Muscle Denervation and IGF-1 Signaling
A widely used model for studying mechanisms underlying muscle atrophy in vivo is muscle denervation which is associated with severe atrophy. Denervation of muscles results in a tremendous loss of muscle activity, retaining little mechanical signaling, however fibrillations occur as side effect [84]. Here we will discuss effects of denervation on IGF-1/PI3K/Akt signaling and how alterations in IGF-1 signaling contribute to denervation-induced atrophy.
Denervation of skeletal muscle has revealed myofiber-type-dependent differences. Three days following denervation in rats, increased IGF-1 mRNA expression levels in fast, glycolytic extensor digitorum longus (EDL) muscle were observed, whereas in slow, oxidative soleus muscle, no changes in IGF-1 mRNA expression were observed [85]. Since calcium-calcineurin signaling regulates IGF-1 mRNA expression [86], the myofiber-type difference in IGF-1 mRNA expression following denervation could well be explained by more fibrillations in fast, glycolytic muscles than in slow, oxidative muscle in the first 3 days following denervation [84]. The increase in IGF-1 mRNA expression in the EDL following denervation was completely blunted at day 7 after denervation [85], suggesting that IGF-1 expression after denervation shows only a transient increase which decays during the first week. A lack of a long-term effect of denervation on IGF-1 mRNA expression has also been shown in rat gastrocnemius muscle 7 weeks after botulin toxin-induced denervation [87]. Moreover, during the first 2 weeks after spinal cord injury in rats, IGF-1 mRNA expression levels in the EDL and soleus muscle were unaltered [85], whereas increased IGF-1 mRNA levels in the plantaris and soleus muscle have been reported after 30 days of spinal cord injury [88]. It seems that IGF-1 mRNA expression is either unaffected or increased after denervation, which depends on muscle type, denervation model, and/or time of measurement.
Regarding the effects of denervation on IGF-1 protein levels, the literature is less ambiguous. IGF-1 protein levels in denervated muscle of rodents or upper leg muscles of humans with spinal cord injury are reduced [89, 90]. In line with these observations, IGF-1R and Akt phosphorylation and protein levels of P13K and IRS-1 have been shown to be decreased after denervation in rodents [88, 89, 91, 92]. Although spinal cord injury is associated with reduced IGF-1 protein levels in human upper leg muscle, Akt phosphorylation was unaltered suggesting a difference between surgical denervation in animal models and human spinal cord injury [90]. Therefore, even though increases in IGF-1 mRNA have been reported following denervation, activity of IGF-1/PI3K/Akt signaling seems to be reduced, with a possible exception after human spinal cord injury.
Besides the observed denervation-related decrease in IGF-1/PI3K/Akt signaling, enhancement of this signaling pathway by either injection of IGF-1 into denervated muscle or transgenic muscle-specific overexpression of IGF-1 in mice has shown to diminish denervation-induced atrophy [19, 93,94,95,96]. Moreover, constitutive expression of activated P13K or Akt also inhibits denervation-induced atrophy in rodents [17, 97]. Similarly, several interventions counterbalancing denervation-induced atrophy are associated with increased Akt phosphorylation [98,99,100,101]. Taken together, IGF-1/PI3K/Akt activity reduces during denervation in adult skeletal muscle, and it is obvious that increasing IGF-1/PI3K/Akt signaling inhibits denervation-induced atrophy.
3.2 Muscle Unloading and IGF-1 Signaling
Unloading of muscles by limb suspension is a disuse model that causes substantial skeletal muscle atrophy. The obvious difference with denervation is the still intact neuronal innervation, but external and internal loads applied to the limbs remain low.
Hind limb suspension (HLS) for 1–2 weeks did not change IGF-1 mRNA levels in rodent soleus, gastrocnemius, or plantaris muscle [102,103,104,105,106,107,108,109]. In contrast to 1–2 weeks after HLS, IGF-1 mRNA expression levels in the soleus and tibialis anterior were decreased after 2 and 3 days of HLS [108, 110]. This suggests that IGF-1 mRNA expression is downregulated during the initial phase of HLS-induced atrophy but is not involved in the longer-term response. At the protein level, IGF-1 expression drops in rat soleus muscle after 2–4 weeks of unloading [111, 112]. In line with reduced IGF-1 protein levels, HLS in rodents for at least 14 days caused decreased phosphorylated Akt levels and/or IRS-1 protein concentrations in soleus and gastrocnemius muscle, indicating that HLS is a strong stimulus for atrophy which is accompanied by reduced IGF-1/PI3K/Akt signaling [17, 110, 111, 113, 114]. In addition to decreased IGF-1 protein levels, an explanation for the reduced Akt phosphorylation and muscle atrophy during unloading may be the increase in ubiquitin ligase Cbl-b expression which results in an elevated ubiquitination of IRS-1 complexes [114]. The contribution of Cbl-b to HLS-induced muscle atrophy is indicated by the observation that Cbl-b-deficient mice are protected from HLS-induced atrophy [114]. To summarize, IGF-1/PI3K/Akt signaling reduces during unloading in different rodent muscles, while IGF-1 mRNA expression is only decreased in the first days of HLS.
Regarding the effectiveness of pharmacological enhancement of IGF-1/PI3K/Akt signaling to counterbalance HLS-induced atrophy, the literature is contradicting. After a period of 1–2 weeks of HLS, muscle-specific overexpression of IGF-1 did not counteract muscle atrophy in mouse soleus, gastrocnemius, and tibialis anterior muscles [107, 115, 116]. These observations are in line with a study showing that systemic injection of both GH and IGF-1 does not attenuate HLS-induced atrophy in rats, however when combined with exercise, muscle atrophy was attenuated [117]. These studies suggest that stimulation of IGF-1 alone is not sufficient to blunt HLS-induced atrophy, which indicates that unloading-induced atrophy is induced by other mechanisms than by reduced IGF-1/PI3K/Akt signaling solely.
In contrast, several studies show that increasing IGF-1/PI3K/Akt signaling can counterbalance HLS-induced atrophy. Overexpression of IGF-1 by DNA electroporation into skeletal muscle or subcutaneous injection of a mixture of IGF-1 and its stabilizing binding protein IGFBP-3 attenuated HLS-induced atrophy in rodents [118, 119]. Also exercise associated with increased IGF-1 and MGF mRNA levels attenuated HLS-induced atrophy in rats [109]. In addition, injections with ghrelin, a growth hormone-releasing peptide, in mice during 2 weeks of HLS enhanced IGF-1/PI3K/Akt signaling in the plantaris but not in soleus muscle, which alleviated atrophy in the plantaris but not in soleus muscle [120].
It seems that HLS-induced muscle atrophy is accompanied by reduced IGF-1/PI3K/Akt signaling as a result of the degradation of IRS-1. Why pharmacological increasing IGF-1/PI3K/Akt signaling alleviates muscle atrophy in some studies but not all remains unsolved. Exercise, however, seems an effective intervention in attenuating unloading-induced muscle atrophy.
3.3 Immobilization and IGF-1 Signaling
Another frequently applied model for disuse and muscle atrophy is joint immobilization, using splints, casts, or surgical staples. The effect of joint immobilization-induced muscle atrophy on IGF-1 expression is however not clear. After ankle and knee immobilization in rodent, rabbit, dog, or human studies, levels of serum IGF-1, muscle protein, or mRNA were not affected [5, 121,122,123,124] or decreased [122, 125,126,127]. Moreover, in human muscle increased levels of IGF-1 mRNA in muscle have been reported upon immobilization [60, 127].
In humans, unilateral knee joint immobilization in 30° knee flexion for 2 weeks in young and old adults was surprisingly related to increased IGF-1 and MGF mRNA levels in m. vastus lateralis, while atrophy was less in old compared to young adults [60, 127]. In contrast, 2 weeks of unilateral knee immobilization in 50° flexion in young adults was associated with a lack of change in serum IGF-1 and mRNA expression levels of IGF-1 as well as MGF in m. vastus lateralis [123]. During immobilization in young adults, serum IGF-1, IGF-1, or MGF mRNA expression increased after administered growth hormone injections, however without attenuating muscle atrophy [123]. When the same protocols were applied to elderly, results were quite similar, except that growth hormone injections and concomitant increases in serum IGF-1, IGF, and MGF mRNA prevented muscle atrophy [124]. These observations indicate that the angle of immobilization affects IGF-1 expression levels and that increased IGF-1 expression levels during immobilization (with or without growth hormone administration) can counterbalance immobilization-induced atrophy in old but not young adults. Since these results only report IGF-1 mRNA expression or serum levels, there is no certainty regarding the activity of the IGF-1/PI3K/Akt pathway. In accordance with the age effect in humans, attenuation of the reduction in Akt phosphorylation as observed during immobilization experiments by losartan supplementation could completely blunt muscle atrophy during 3 weeks of immobilization of the hind limb of old mice [128]. The protective effect of losartan was mainly by maintaining the number of myofibers, which decrease with aging. This might be an explanation for the age-related difference since IGF-1 is antiapoptotic and would therefore be able to inhibit a potential age-related loss of myofibers in immobilization-induced atrophy. Note that losartan treatment does not provide direct evidence for IGF-1/PI3K/Akt signaling since it affects other signaling pathways such as TGF-β signaling as well.
IGF-1R and Akt phosphorylation decreased during immobilization-induced muscle atrophy in young and old mice, which implies blunted IGF-1/PI3K/Akt signaling [122, 128, 129]. Akt phosphorylation also decreased in m. vastus lateralis of young but not adult humans after 2–4 days of knee joint immobilization [60]. In several models of atrophy including immobilization, miR-29b has been shown to be upregulated which downregulates IGF-1/PI3K/Akt signaling [89]. Subsequent in vitro overexpression of IGF-1 or PI3K concomitant with a miR-29b mimic attenuated miR-29b-induced atrophy [89]. Together these studies indicate that loss of IGF-1/PI3K/Akt signaling during joint immobilization contributes to immobilization-induced muscle atrophy although this may not be true for elder humans.
Increased IGF-1 receptor and Akt phosphorylation by angiotensin-(1-7) treatment alleviated immobilization-induced muscle atrophy in mice [129]. In contrast, in vivo overexpression of IGF-1 (viral mediated or induced by growth hormone) improved muscle morphology, indicated by less widened interstitial space, necrotic fibers, and inflammatory cells, but did not reduce myofiber diameter or muscle cross-sectional area during immobilization [125, 130, 131]. Moreover, mice with reduced mTOR activity show muscle atrophy to the same extent as control mice during immobilization [122]. Taken together, some studies on animal models successfully reduced muscle atrophy or morphology by increasing IGF-1 signaling or activation of downstream IGF-1 targets, while other studies did not show any reductions in immobilization-induced muscle atrophy.
From the above it is concluded that IGF-1/PI3K/Akt signaling reduces during joint immobilization. Whether stimulation of IGF-1 signaling plays a role in the maintenance of muscle mass during immobilization-induced muscle atrophy has not been unambiguously established, although in older subjects this may be the case.
3.4 Muscle Aging and IGF-1 Signaling
In addition to primary disuse models, aging is also associated with skeletal muscle atrophy. The loss of skeletal muscle mass and strength during aging, referred to as sarcopenia, is determined by combination of two processes, i.e., loss of myofibers and myofiber atrophy, which have different temporal distributions [132]. As a result of loss of motor units, remaining myofibers are possibly more active as compensation. Whereas under disuse conditions, predominantly type 1 fibers are affected, during aging type 2 myofibers are more susceptible to atrophy and necrosis compared to type 1 myofibers. In aging, the loss of muscle mass is likely due to a reduction in physical activity, oxidative stress, chronic low-grade inflammation, and changes in systemic serum proteins [133]. The chronic state of low-grade inflammation related to aging is associated with increased IL-6 and TNF-α plasma levels [134]. These cytokines can interfere with IGF-1 signaling (see 6.4 Interference with IGF-1 Signaling) and are therefore likely to play a role in aging-associated muscle wasting [135, 136].
IGF-1 serum levels decrease with age, but no differences in IGF-1 serum levels were shown between elderly females with and without sarcopenia [137]. Based on small effects of GH injections on muscle hypertrophy in elderly, while exercise is capable of inducing hypertrophy, several literature-based studies suggest that locally expressed IGF-1 is important in the maintenance of muscle mass, while there is no consistent evidence for a relationship between IGF-1 serum levels and age-related loss of muscle strength [138,139,140]. The role of the IGF-1/PI3K/Akt pathway is discussed below.
Cross-sectional analyses of a large cohort including over 100 human participants and different mouse models, suggest that IGF-1/PI3K/Akt signaling activity is unaffected during aging [141]. Whereas skeletal muscle mRNA levels of IGF-1Ea and MGF reduced with age in mice, this was not evident in skeletal muscle of human subjects. MuRF-1 knockout old mice showed a blunted atrophy but decrease in muscle force, which indicates that proteasomal degradation is essential for maintaining muscle quality during aging. In addition, MuRF-1 and MAFbx mRNA levels did not differ between old sedentary and young human participants [141]. These observations are in line with those of another study showing no change in IGF-1/PI3K/Akt signaling, indicated by unaffected IGF-1R and Akt phosphorylation, in skeletal muscle of klotho mutant mice, a mouse model with an aging-related phenotype showing muscle atrophy [63]. In addition, it was shown that MuRF-1 and MAFbx protein levels in skeletal muscle were not upregulated in klotho mutant mice compared to control mice [63]. Also, no differences in IRS1 phosphorylation did exist between old and young adult rats [142]. Together, these studies indicate that IGF-1/PI3K/Akt signaling is not downregulated with aging and sarcopenia is not the result of increased activation of the ubiquitin-proteasome system.
Note that in old rodent muscles, both similar [62, 63, 143] and lower [142, 144] pAkt/tAkt levels compared to young rodent muscles have been reported. In line with these observations, in biopsies of young and old human subjects, both similar [141] and decreased [145] levels of pAkt/tAkt with age have been reported. The decrease in pAkt/tAkt in aged humans was likely due to increased levels of tAkt, while pAkt levels were not affected, which suggest that in old human muscle, IGF/PI3K/Akt signaling activity is not reduced, but Akt synthesis is upregulated [145]. Although some studies show decreased levels of pAkt/tAkt related to aging, there is not an obvious reduction in IGF-1/PI3K/Akt signaling.
In mice, virus-mediated or transgenic overexpression of IGF-1 can prevent aging-induced muscle atrophy and a decrease in type 2B fiber fraction [146, 147]. Despite elevated IGF-1 expression, sedentary transgenic IGF-1 old mice did not have larger myofiber diameters compared to their aged-matched controls, whereas sedentary transgenic IGF-1 adult mice did show larger myofiber diameters compared to their aged-matched controls [148]. This suggests a decreased anabolic response to IGF-1/PI3K/Akt signaling with age rather than the inability of IGF-1/PI3K/Akt signaling to prevent the aging-associated atrophy. Indeed, overload of hind limb muscles of young, mature, and old rats showed reduced hypertrophy and decreased upregulation of MGF and IGF-1 receptor mRNA with age [149]. This result is in accordance with those of other studies suggesting an impaired anabolic response of the IGF-1/PI3K/Akt pathway in aged rats [142, 150]. From this it can be concluded that the trophic response to IGF-1 decreases with age, but is not completely lost and overexpression of IGF-1 is capable of attenuating aging-related muscle atrophy. Moreover, decreased Akt phosphorylation but no changes in activity of downstream targets of mTOR upon a single bout of resistance exercise were observed in old compared to adult humans, suggesting that the synthesis machinery is not affected by age but rather the IGF-1/PI3K/Akt signaling [151]. A possible explanation is that exercise-induced IGF-1/PI3K/Akt signaling is inhibited by increased levels of IL-6 and TNF-α associated with the chronic low grade of systemic inflammation seen with aging [135, 136].
Regarding the effects of aging, IGF-1/PI3K/Akt signaling does not seem to be reduced during aging-associated muscle atrophy, while IGF-1 overexpression is able to inhibit aging-associated muscle atrophy. However, the anabolic potential of this pathway reduces with age, which might be due to increased interference of pro-inflammatory cytokines.
4 Interference with IGF-1 Signaling
Changes in IGF-1/PI3K/Akt signaling can be the result of decreased IGF-1 expression, bioactivity, receptor availability, or inhibition along its pathway. Insight into the mechanisms affecting IGF-1/PI3K/Akt signaling will reveal possible candidates for counterbalancing reduced IGF-1/PI3K/Akt signaling. Because IGF-1 is involved in many tissues and cell types, clinical interventions should be muscle specific or target a factor which interferes with IGF-1 and has a lesser general effect. Although it is outside the scope of this review to discuss all different interfering factors, a few important ones are pointed out.
AMP-activated kinase (AMPK) interferes with IGF-1 signaling by inhibiting and stimulating the downstream targets mTOR and FOXO3 [152,153,154]. Moreover, AMPK/FOXO3 signaling increased during HLS-induced muscle atrophy in rats [155], which could explain why not all studies report an effect of IGF-1 overexpression on HLS-induced muscle atrophy. Another negative regulator of myofiber size is myostatin, which is a member of the TGF-β family [156]. Myostatin inhibits Akt via Smad3 signaling and has therefore opposite effects compared to IGF-1 [157]. Several types of muscle atrophy are associated with increased myostatin expression (see Chap. 8).
As mentioned before, also pro-inflammatory cytokines like IL-6 and TNF-α can interfere with IGF-1 signaling and likely play a role in muscle atrophy associated with systemic inflammation, such as aging [135, 136]. IL-6 is able to inhibit mTOR, p70S6K, and p90RSK activation in muscle cells, without affecting Akt phosphorylation [158]. TNF-α impairs IGF-1R sensitivity [136] and increases MuRF-1 expression by activating a group of transcriptions factors known as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [13]. Reducing systemic inflammation could counterbalance inflammatory-associated muscle wasting by enhancing the effect of IGF-1. Regular exercise stimulates IGF-1 expression and has an anti-inflammatory effect [159] which attenuates the interference of cytokine signaling with IGF-1 and is therefore a safe and cheap intervention to counterbalance muscle atrophy associated with elevated levels of IL-6 and TNF-α.
Recent studies have shown that microRNAs (miRNAs) are capable of interfering with IGF-1 signaling and thereby play an important role in muscle atrophy [89, 160]. miR-29b negatively regulates IGF-1 and PI3K expression and has been shown to be upregulated in the tibialis anterior, soleus, and EDL muscle in denervation-induced muscle atrophy [89]. Moreover, miR-29b was also upregulated in immobilization, dexamethasone, fasting, cancer cachexia, and aging-induced muscle atrophy [89]. In addition to miR-29b, miR-18a also suppresses IGF-1/PI3K/Akt signaling, and its overexpression induces muscle atrophy [160]. Because of the general role of miR-29b in muscle atrophy (i.e., upregulation in several muscles and atrophic conditions) and the observation that many miRNAs have been shown to be tissue specific [161, 162], miRNAs are promising targets for counterbalancing muscle atrophy. Preclinical and clinical trials in which miRNAs are targeted are currently conducted, although, to the best of our knowledge, not aimed to prevent or restore muscle wasting.
5 Conclusions and Future Perspectives
Here we reviewed the role of IGF-1 signaling in the induction of muscle atrophy and show that in disuse conditions muscle atrophy is in part due to a decline in IGF-1 signaling, whereas with aging-associated muscle atrophy, IGF-1 signaling remains unaffected. Moreover, enhancement of IGF-1/PI3K/Akt in some conditions is an effective strategy to counterbalancing muscle atrophy, however this does not apply to all disuse conditions. Under hypertrophic conditions by mechanical loading, IGF-1/PI3K/Akt signaling increases muscle mass by stimulating protein synthesis and inhibiting protein degradation. Protein synthesis is stimulated by mTOR, which activates p70S6K and p90RSK, which are downstream targets of Akt and PDK1. Akt also stimulates protein synthesis by inhibiting GSK3β activity. During atrophic conditions, protein synthesis is reduced and/or protein degradation is increased. The four main mechanisms in protein degradation are proteasomal-, lysosomal-, and caspase- and calpain-dependent protein degradation. Regarding the role of IGF-1 in protein degradation, it is clear that IGF-1 inhibits proteasomal-mediated muscle protein degradation by lowering the expression of E3-ligases, resulting in attenuated protein ubiquitination. Reductions in expression of E3 ligases are a result of inactivation of FOXO transcription factors by phosphorylated Akt. In addition, FOXO inactivation by phosphorylated Akt also reduces lysosomal degradation. When IGF-1/PI3K/Akt signaling decreases during atrophic conditions, caspase-dependent degradation seems to be reduced as well. Future research is required to obtain more detailed insight in the role of IGF-1/PI3K/Akt signaling on calpain-dependent degradation.
The role of the IGF-1/PI3K/Akt pathway differs between different models of skeletal muscle atrophy. During denervation-induced atrophy, IGF-1/PI3K/Akt signaling activity is reduced, and upregulation of IGF-1/PI3K/Akt signaling counterbalances denervation-induced muscle atrophy. In contrast, during unloading- and joint immobilization-induced atrophy, IGF-1/PI3K/Akt signaling activity is reduced as well, but it remains unclear whether upregulation of the IGF-1/PI3K/Akt pathway is sufficient to attenuate denervation- or joint immobilization-induced muscle atrophy, suggesting that other pathways are involved which cannot be compensated by IGF-1/PI3K/Akt signaling. No obvious downregulation of IGF-1/PI3K/Akt signaling is shown during aging-associated atrophy. Although the anabolic potential of the IGF-1/PI3K/Akt pathway reduces with age, activation of this pathway has the ability to achieve recovery of aging-associated muscle atrophy.
The role of miRNAs in regulation of myofiber size is a novel and promising area for further research. Many miRNAs are tissue specifically expressed and could target IGF-1 signaling components in muscle wasting without affecting its role in many tissues and cell types. Although there is substantial evidence showing that miRNAs can interfere with IGF-1/PI3K/Akt signaling, there remains a lack of knowledge regarding the possibilities to counterbalance muscle atrophy by targeting miRNAs. Because of the general effects of miRNAs in several conditions of muscle atrophy and muscle phenotypes, future studies should aim for more insight in knowledge regarding biological functions of miRNAs and clinical application of altering miRNA activity in prevention and recovery of muscle atrophy. Overall, IGF-1/PI3K/Akt is a key signaling pathway in protein synthesis and degradation, of which its activity is attenuated during several disuse models.
References
Goldspink G (2005) Mechanical signals, IGF-I gene splicing, and muscle adaptation. Physiology 20:232–238. https://doi.org/10.1152/physiol.00004.2005
Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A (1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75(1):59–72
Hameed M, Orrell RW, Cobbold M, Goldspink G, Harridge SD (2003) Expression of IGF-I splice variants in young and old human skeletal muscle after high resistance exercise. J Physiol 547(Pt 1):247–254. https://doi.org/10.1113/jphysiol.2002.032136
van Wessel T, de Haan A, van der Laarse WJ, Jaspers RT (2010) The muscle fiber type-fiber size paradox: hypertrophy or oxidative metabolism? Eur J Appl Physiol 110(4):665–694. https://doi.org/10.1007/s00421-010-1545-0
Yang H, Alnaqeeb M, Simpson H, Goldspink G (1997) Changes in muscle fibre type, muscle mass and IGF-I gene expression in rabbit skeletal muscle subjected to stretch. J Anat 190(Pt 4):613–622
Yang S, Alnaqeeb M, Simpson H, Goldspink G (1996) Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J Muscle Res Cell Motil 17(4):487–495
Duan C, Ren H, Gao S (2010) Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: roles in skeletal muscle growth and differentiation. Gen Comp Endocrinol 167(3):344–351. https://doi.org/10.1016/j.ygcen.2010.04.009
Jaspers RT, van Beek-Harmsen BJ, Blankenstein MA, Goldspink G, Huijing PA, van der Laarse WJ (2008) Hypertrophy of mature Xenopus muscle fibres in culture induced by synergy of albumin and insulin. Pflugers Arch 457(1):161–170. https://doi.org/10.1007/s00424-008-0499-0
Kandalla PK, Goldspink G, Butler-Browne G, Mouly V (2011) Mechano Growth Factor E peptide (MGF-E), derived from an isoform of IGF-1, activates human muscle progenitor cells and induces an increase in their fusion potential at different ages. Mech Ageing Dev 132(4):154–162. https://doi.org/10.1016/j.mad.2011.02.007
Brisson BK, Barton ER (2012) Insulin-like growth factor-I E-peptide activity is dependent on the IGF-I receptor. PLoS One 7(9):e45588. https://doi.org/10.1371/journal.pone.0045588
Adamo ML, Ben-Hur H, LeRoith D, Roberts CT Jr (1991) Transcription initiation in the two leader exons of the rat IGF-I gene occurs from disperse versus localized sites. Biochem Biophys Res Commun 176(2):887–893
Temmerman L, Slonimsky E, Rosenthal N (2010) Class 2 IGF-1 isoforms are dispensable for viability, growth and maintenance of IGF-1 serum levels. Growth Horm IGF Res 20(3):255–263. https://doi.org/10.1016/j.ghir.2010.03.002
Glass DJ (2005) Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37(10):1974–1984. https://doi.org/10.1016/j.biocel.2005.04.018
Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M (2013) Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280(17):4294–4314. https://doi.org/10.1111/febs.12253
Glass DJ (2003) Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol 5(2):87–90. https://doi.org/10.1038/ncb0203-87
Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ (2001) Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 3(11):1009–1013. https://doi.org/10.1038/ncb1101-1009
Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3(11):1014–1019. https://doi.org/10.1038/ncb1101-1014
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3):399–412
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14(3):395–403
Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ (2005) Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 280(4):2737–2744. https://doi.org/10.1074/jbc.M407517200
Florini JR, Ewton DZ, Coolican SA (1996) Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev 17(5):481–517. https://doi.org/10.1210/edrv-17-5-481
Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL (2004) IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab 287(4):E591–E601. https://doi.org/10.1152/ajpendo.00073.2004
Sasaoka T, Ishiki M, Wada T, Hori H, Hirai H, Haruta T, Ishihara H, Kobayashi M (2001) Tyrosine phosphorylation-dependent and -independent role of Shc in the regulation of IGF-1-induced mitogenesis and glycogen synthesis. Endocrinology 142(12):5226–5235. https://doi.org/10.1210/endo.142.12.8543
Haddad F, Adams GR (2004) Inhibition of MAP/ERK kinase prevents IGF-I-induced hypertrophy in rat muscles. J Appl Physiol (1985) 96(1):203–210. https://doi.org/10.1152/japplphysiol.00856.2003
Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ (1999) Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286(5445):1738–1741
Kuemmerle JF (2003) IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase. Am J Physiol Gastrointest Liver Physiol 284(3):G411–G422. https://doi.org/10.1152/ajpgi.00310.2002
Hers I, Vincent EE, Tavare JM (2011) Akt signalling in health and disease. Cell Signal 23(10):1515–1527. https://doi.org/10.1016/j.cellsig.2011.05.004
Huijing PA, Jaspers RT (2005) Adaptation of muscle size and myofascial force transmission: a review and some new experimental results. Scand J Med Sci Sports 15(6):349–380. https://doi.org/10.1111/j.1600-0838.2005.00457.x
Schakman O, Kalista S, Bertrand L, Lause P, Verniers J, Ketelslegers JM, Thissen JP (2008) Role of Akt/GSK-3beta/beta-catenin transduction pathway in the muscle anti-atrophy action of insulin-like growth factor-I in glucocorticoid-treated rats. Endocrinology 149(8):3900–3908. https://doi.org/10.1210/en.2008-0439
Spangenburg EE, Bowles DK, Booth FW (2004) Insulin-like growth factor-induced transcriptional activity of the skeletal alpha-actin gene is regulated by signaling mechanisms linked to voltage-gated calcium channels during myoblast differentiation. Endocrinology 145(4):2054–2063. https://doi.org/10.1210/en.2003-1476
Peters EL, van der Linde SM, Vogel ISP, Haroon M, Offringa C, de Wit GMJ, Koolwijk P, van der Laarse WJ, Jaspers RT (2017) IGF-1 attenuates hypoxia-induced atrophy but inhibits myoglobin expression in C2C12 skeletal muscle myotubes. Int J Mol Sci 18(9). https://doi.org/10.3390/ijms18091889
Feng R, Ma X, Ma J, Jia H, Ma B, Xu L, Liu A (2015) Positive effect of IGF-1 injection on gastrocnemius of rat during distraction osteogenesis. J Orthop Res 33(10):1424–1432. https://doi.org/10.1002/jor.22796
Wilborn CD, Taylor LW, Greenwood M, Kreider RB, Willoughby DS (2009) Effects of different intensities of resistance exercise on regulators of myogenesis. J Strength Cond Res 23(8):2179–2187. https://doi.org/10.1519/JSC.0b013e3181bab493
Tiffin N, Adi S, Stokoe D, Wu NY, Rosenthal SM (2004) Akt phosphorylation is not sufficient for insulin-like growth factor-stimulated myogenin expression but must be accompanied by down-regulation of mitogen-activated protein kinase/extracellular signal-regulated kinase phosphorylation. Endocrinology 145(11):4991–4996. https://doi.org/10.1210/en.2004-0101
Hsu HH, Zdanowicz MM, Agarwal VR, Speiser PW (1997) Expression of myogenic regulatory factors in normal and dystrophic mice: effects of IGF-1 treatment. Biochem Mol Med 60(2):142–148
Laplante M, Sabatini DM (2012) mTOR Signaling. Cold Spring Harb Perspect Biol 4(2). https://doi.org/10.1101/cshperspect.a011593
Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S (2006) The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci U S A 103(12):4741–4746. https://doi.org/10.1073/pnas.0600678103
Bolster DR, Kimball SR, Jefferson LS (2003) Translational control mechanisms modulate skeletal muscle gene expression during hypertrophy. Exerc Sport Sci Rev 31(3):111–116
Nakai N, Kawano F, Oke Y, Nomura S, Ohira T, Fujita R, Ohira Y (2010) Mechanical stretch activates signaling events for protein translation initiation and elongation in C2C12 myoblasts. Mol Cells 30(6):513–518. https://doi.org/10.1007/s10059-010-0147-3
Redpath NT, Foulstone EJ, Proud CG (1996) Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J 15(9):2291–2297
Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20(16):4370–4379. https://doi.org/10.1093/emboj/20.16.4370
Wang L, Proud CG (2002) Regulation of the phosphorylation of elongation factor 2 by MEK-dependent signalling in adult rat cardiomyocytes. FEBS Lett 531(2):285–289
Jefferson LS, Fabian JR, Kimball SR (1999) Glycogen synthase kinase-3 is the predominant insulin-regulated eukaryotic initiation factor 2B kinase in skeletal muscle. Int J Biochem Cell Biol 31(1):191–200
Verhees KJ, Schols AM, Kelders MC, Op den Kamp CM, van der Velden JL, Langen RC (2011) Glycogen synthase kinase-3beta is required for the induction of skeletal muscle atrophy. Am J Physiol Cell Physiol 301(5):C995–c1007. https://doi.org/10.1152/ajpcell.00520.2010
Leger B, Cartoni R, Praz M, Lamon S, Deriaz O, Crettenand A, Gobelet C, Rohmer P, Konzelmann M, Luthi F, Russell AP (2006) Akt signalling through GSK-3beta, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J Physiol 576(Pt 3):923–933. https://doi.org/10.1113/jphysiol.2006.116715
Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, Bargou RC, Qin J, Lai CC, Tsai FJ, Tsai CH, Hung MC (2005) Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell 19(2):159–170. https://doi.org/10.1016/j.molcel.2005.06.009
Kandarian SC, Jackman RW (2006) Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33(2):155–165. https://doi.org/10.1002/mus.20442
Foletta VC, White LJ, Larsen AE, Leger B, Russell AP (2011) The role and regulation of MAFbx/atrogin-1 and MuRF1 in skeletal muscle atrophy. Pflugers Arch 461(3):325–335. https://doi.org/10.1007/s00424-010-0919-9
Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA (2008) The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J 27(8):1266–1276. https://doi.org/10.1038/emboj.2008.52
Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ (2007) The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab 6(5):376–385. https://doi.org/10.1016/j.cmet.2007.09.009
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294(5547):1704–1708. https://doi.org/10.1126/science.1065874
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96(6):857–868
Tran H, Brunet A, Griffith EC, Greenberg ME (2003) The many forks in FOXO’s road. Sci STKE 2003(172):Re5. https://doi.org/10.1126/stke.2003.172.re5
O’Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, Stanford KI, Robinson MM, Cai W, Kleinridders A, Pereira RO, Hirshman MF, Abel ED, Accili D, Goodyear LJ, Nair KS, Kahn CR (2016) Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest 126(9):3433–3446. https://doi.org/10.1172/jci86522
Sandri M (2010) Autophagy in skeletal muscle. FEBS Lett 584(7):1411–1416. https://doi.org/10.1016/j.febslet.2010.01.056
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6(6):458–471. https://doi.org/10.1016/j.cmet.2007.11.001
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6(6):472–483. https://doi.org/10.1016/j.cmet.2007.11.004
Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotz P (2008) Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet 17(24):3897–3908. https://doi.org/10.1093/hmg/ddn292
Garcia de la Serrana D, Fuentes EN, Martin SAM, Johnston IA, Macqueen DJ (2017) Divergent regulation of insulin-like growth factor binding protein genes in cultured Atlantic salmon myotubes under different models of catabolism and anabolism. Gen Comp Endocrinol 247:53–65. https://doi.org/10.1016/j.ygcen.2017.01.017
Suetta C, Frandsen U, Jensen L, Jensen MM, Jespersen JG, Hvid LG, Bayer M, Petersson SJ, Schroder HD, Andersen JL, Heinemeier KM, Aagaard P, Schjerling P, Kjaer M (2012) Aging affects the transcriptional regulation of human skeletal muscle disuse atrophy. PLoS One 7(12):e51238. https://doi.org/10.1371/journal.pone.0051238
Sandona D, Desaphy JF, Camerino GM, Bianchini E, Ciciliot S, Danieli-Betto D, Dobrowolny G, Furlan S, Germinario E, Goto K, Gutsmann M, Kawano F, Nakai N, Ohira T, Ohno Y, Picard A, Salanova M, Schiffl G, Blottner D, Musaro A, Ohira Y, Betto R, Conte D, Schiaffino S (2012) Adaptation of mouse skeletal muscle to long-term microgravity in the MDS mission. PLoS One 7(3):e33232. https://doi.org/10.1371/journal.pone.0033232
White JR, Confides AL, Moore-Reed S, Hoch JM, Dupont-Versteegden EE (2015) Regrowth after skeletal muscle atrophy is impaired in aged rats, despite similar responses in signaling pathways. Exp Gerontol 64:17–32. https://doi.org/10.1016/j.exger.2015.02.007
Iida RH, Kanko S, Suga T, Morito M, Yamane A (2011) Autophagic-lysosomal pathway functions in the masseter and tongue muscles in the klotho mouse, a mouse model for aging. Mol Cell Biochem 348(1-2):89–98. https://doi.org/10.1007/s11010-010-0642-z
Schakman O, Dehoux M, Bouchuari S, Delaere S, Lause P, Decroly N, Shoelson SE, Thissen JP (2012) Role of IGF-I and the TNFalpha/NF-kappaB pathway in the induction of muscle atrogenes by acute inflammation. Am J Physiol Endocrinol Metab 303(6):E729–E739. https://doi.org/10.1152/ajpendo.00060.2012
Gomez-SanMiguel AB, Villanua MA, Martin AI, Lopez-Calderon A (2016) D-TRP(8)-gammaMSH prevents the effects of endotoxin in rat skeletal muscle cells through TNFalpha/NF-KB signalling pathway. PLoS One 11(5):e0155645. https://doi.org/10.1371/journal.pone.0155645
McClung JM, Judge AR, Powers SK, Yan Z (2010) p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol-Cell Physiol 298(3):C542–C549. https://doi.org/10.1152/ajpcell.00192.2009
Huang J, Zhu X (2016) The molecular mechanisms of calpains action on skeletal muscle atrophy. Physiol Res 65(4):547–560
Talbert EE, Smuder AJ, Min K, Kwon OS, Powers SK (2013) Calpain and caspase-3 play required roles in immobilization-induced limb muscle atrophy. J Appl Physiol (1985) 114(10):1482–1489. https://doi.org/10.1152/japplphysiol.00925.2012
Kramerova I, Kudryashova E, Venkatraman G, Spencer MJ (2007) Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet 16(8):1006. https://doi.org/10.1093/hmg/ddm044
McDonagh MB, Fernandez C, Oddy VH (1999) Hind-limb protein metabolism and calpain system activity influence post-mortem change in meat quality in lamb. Meat Sci 52(1):9–18
Wingertzahn MA, Zdanowicz MM, Slonim AE (1998) Insulin-like growth factor-I and high protein diet decrease calpain-mediated proteolysis in murine muscular dystrophy. Proc Soc Exp Biol Med 218(3):244–250
Lu Y, Bradley JS, McCoski SR, Gonzalez JM, Ealy AD, Johnson SE (2017) Reduced skeletal muscle fiber size following caloric restriction is associated with calpain-mediated proteolysis and attenuation of IGF-1 signaling. Am J Physiol Regul Integr Comp Physiol 312(5):R806–r815. https://doi.org/10.1152/ajpregu.00400.2016
Li BG, Hasselgren PO, Fang CH, Warden GD (2004) Insulin-like growth factor-I blocks dexamethasone-induced protein degradation in cultured myotubes by inhibiting multiple proteolytic pathways: 2002 ABA paper. J Burn Care Rehabil 25(1):112–118. https://doi.org/10.1097/01.bcr.0000105100.44745.36
Fernandez C, Sainz RD (1997) Pathways of protein degradation in L6 myotubes. Proc Soc Exp Biol Med 214(3):242–247
Smith IJ, Dodd SL (2007) Calpain activation causes a proteasome-dependent increase in protein degradation and inhibits the Akt signalling pathway in rat diaphragm muscle. Exp Physiol 92(3):561–573. https://doi.org/10.1113/expphysiol.2006.035790
Shenkman BS, Belova SP, Lomonosova YN, Kostrominova TY, Nemirovskaya TL (2015) Calpain-dependent regulation of the skeletal muscle atrophy following unloading. Arch Biochem Biophys 584:36–41. https://doi.org/10.1016/j.abb.2015.07.011
Song YH, Li Y, Du J, Mitch WE, Rosenthal N, Delafontaine P (2005) Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest 115(2):451–458. https://doi.org/10.1172/jci22324
Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE (2006) Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol 17(5):1388–1394. https://doi.org/10.1681/asn.2004100842
Vazeille E, Codran A, Claustre A, Averous J, Listrat A, Bechet D, Taillandier D, Dardevet D, Attaix D, Combaret L (2008) The ubiquitin-proteasome and the mitochondria-associated apoptotic pathways are sequentially downregulated during recovery after immobilization-induced muscle atrophy. Am J Physiol Endocrinol Metab 295(5):E1181–E1190. https://doi.org/10.1152/ajpendo.90532.2008
Gustafsson T, Osterlund T, Flanagan JN, von Walden F, Trappe TA, Linnehan RM, Tesch PA (2010) Effects of 3 days unloading on molecular regulators of muscle size in humans. J Appl Physiol (1985) 109(3):721–727. https://doi.org/10.1152/japplphysiol.00110.2009
Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE (2004) Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest 113(1):115–123. https://doi.org/10.1172/jci18330
Yamaguchi H, Wang HG (2001) The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20(53):7779–7786. https://doi.org/10.1038/sj.onc.1204984
Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE (2004) Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol 15(6):1537–1545
Salafsky B, Bell J, Prewitt MA (1968) Development of fibrillation potentials in denervated fast and slow skeletal muscle. Am J Physiol 215(3):637–643. https://doi.org/10.1152/ajplegacy.1968.215.3.637
Zeman RJ, Zhao J, Zhang Y, Zhao W, Wen X, Wu Y, Pan J, Bauman WA, Cardozo C (2009) Differential skeletal muscle gene expression after upper or lower motor neuron transection. Pflugers Arch 458(3):525–535. https://doi.org/10.1007/s00424-009-0643-5
Alfieri CM, Evans-Anderson HJ, Yutzey KE (2007) Developmental regulation of the mouse IGF-I exon 1 promoter region by calcineurin activation of NFAT in skeletal muscle. Am J Physiol Cell Physiol 292(5):C1887–C1894. https://doi.org/10.1152/ajpcell.00506.2006
Tsai SW, Tung YT, Chen HL, Yang SH, Liu CY, Lu M, Pai HJ, Lin CC, Chen CM (2016) Myostatin propeptide gene delivery by gene gun ameliorates muscle atrophy in a rat model of botulinum toxin-induced nerve denervation. Life Sci 146:15–23. https://doi.org/10.1016/j.lfs.2015.12.056
Kim JA, Roy RR, Kim SJ, Zhong H, Haddad F, Baldwin KM, Edgerton VR (2010) Electromechanical modulation of catabolic and anabolic pathways in chronically inactive, but neurally intact, muscles. Muscle Nerve 42(3):410–421. https://doi.org/10.1002/mus.21720
Li J, Chan MC, Yu Y, Bei Y, Chen P, Zhou Q, Cheng L, Chen L, Ziegler O, Rowe GC, Das S, Xiao J (2017) miR-29b contributes to multiple types of muscle atrophy. Nat Commun 8:15201. https://doi.org/10.1038/ncomms15201
Leger B, Senese R, Al-Khodairy AW, Deriaz O, Gobelet C, Giacobino JP, Russell AP (2009) Atrogin-1, MuRF1, and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients. Muscle Nerve 40(1):69–78. https://doi.org/10.1002/mus.21293
Tando T, Hirayama A, Furukawa M, Sato Y, Kobayashi T, Funayama A, Kanaji A, Hao W, Watanabe R, Morita M, Oike T, Miyamoto K, Soga T, Nomura M, Yoshimura A, Tomita M, Matsumoto M, Nakamura M, Toyama Y, Miyamoto T (2016) Smad2/3 proteins are required for immobilization-induced skeletal muscle atrophy. J Biol Chem 291(23):12184–12194. https://doi.org/10.1074/jbc.M115.680579
Abe T, Kohno S, Yama T, Ochi A, Suto T, Hirasaka K, Ohno A, Teshima-Kondo S, Okumura Y, Oarada M, Choi I, Mukai R, Terao J, Nikawa T (2013) Soy glycinin contains a functional inhibitory sequence against muscle-atrophy-associated ubiquitin ligase Cbl-b. Int J Endocrinol 2013:907565. https://doi.org/10.1155/2013/907565
Shavlakadze T, White JD, Davies M, Hoh JF, Grounds MD (2005) Insulin-like growth factor I slows the rate of denervation induced skeletal muscle atrophy. Neuromuscul Disord 15(2):139–146. https://doi.org/10.1016/j.nmd.2004.10.013
Day CS, Riano F, Tomaino MM, Buranatanitkit B, Somogyi G, Sotereanos D, Huard J (2001) Growth factor may decrease muscle atrophy secondary to denervation. J Reconstr Microsurg 17(1):51–57
Day CS, Buranapanitkit B, Riano FA, Tomaino MM, Somogyi G, Sotereanos DG, Kuroda R, Huard J (2002) Insulin growth factor-1 decreases muscle atrophy following denervation. Microsurgery 22(4):144–151. https://doi.org/10.1002/micr.21742
Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N (2006) Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest 116(11):2945–2954. https://doi.org/10.1172/jci28721
Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S (2002) A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc Natl Acad Sci U S A 99(14):9213–9218. https://doi.org/10.1073/pnas.142166599
Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G, Rizzuto R (2015) The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10(8):1269–1279. https://doi.org/10.1016/j.celrep.2015.01.056
Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, Prodam F, Ronchi G, Fagoonee S, Fornaro M, Chianale F, Baldanzi G, Surico N, Sinigaglia F, Perroteau I, Smith RG, Sun Y, Geuna S, Graziani A (2013) Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 123(2):611–622. https://doi.org/10.1172/jci39920
Kunkel SD, Suneja M, Ebert SM, Bongers KS, Fox DK, Malmberg SE, Alipour F, Shields RK, Adams CM (2011) mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab 13(6):627–638. https://doi.org/10.1016/j.cmet.2011.03.020
Su Z, Hu L, Cheng J, Klein JD, Hassounah F, Cai H, Li M, Wang H, Wang XH (2016) Acupuncture plus low-frequency electrical stimulation (Acu-LFES) attenuates denervation-induced muscle atrophy. J Appl Physiol (1985) 120(4):426–436. https://doi.org/10.1152/japplphysiol.00175.2015
Kachaeva EV, Turtikova OV, Leinsoo TA, Shenkman BS (2010) Insulin-like growth factor 1 and the key markers of proteolysis during the acute period of readaptation of the muscle atrophied as a result of unloading. Biofizika 55(6):1108–1116
Washington TA, White JP, Davis JM, Wilson LB, Lowe LL, Sato S, Carson JA (2011) Skeletal muscle mass recovery from atrophy in IL-6 knockout mice. Acta Physiol (Oxf) 202(4):657–669. https://doi.org/10.1111/j.1748-1716.2011.02281.x
Lomonosova YN, Kalamkarov GR, Bugrova AE, Shevchenko TF, Kartashkina NL, Lysenko EA, Shvets VI, Nemirovskaya TL (2011) Protective effect of L-Arginine administration on proteins of unloaded m. soleus. Biochemistry (Mosc) 76(5):571–580. https://doi.org/10.1134/s0006297911050075
Heinemeier KM, Olesen JL, Haddad F, Schjerling P, Baldwin KM, Kjaer M (2009) Effect of unloading followed by reloading on expression of collagen and related growth factors in rat tendon and muscle. J Appl Physiol (1985) 106(1):178–186. https://doi.org/10.1152/japplphysiol.91092.2008
van der Velden JL, Langen RC, Kelders MC, Willems J, Wouters EF, Janssen-Heininger YM, Schols AM (2007) Myogenic differentiation during regrowth of atrophied skeletal muscle is associated with inactivation of GSK-3beta. Am J Physiol Cell Physiol 292(5):C1636–C1644. https://doi.org/10.1152/ajpcell.00504.2006
Criswell DS, Booth FW, DeMayo F, Schwartz RJ, Gordon SE, Fiorotto ML (1998) Overexpression of IGF-I in skeletal muscle of transgenic mice does not prevent unloading-induced atrophy. Am J Physiol 275(3 Pt 1):E373–E379
Awede B, Thissen J, Gailly P, Lebacq J (1999) Regulation of IGF-I, IGFBP-4 and IGFBP-5 gene expression by loading in mouse skeletal muscle. FEBS Lett 461(3):263–267
Adams GR, Haddad F, Bodell PW, Tran PD, Baldwin KM (2007) Combined isometric, concentric, and eccentric resistance exercise prevents unloading-induced muscle atrophy in rats. J Appl Physiol (1985) 103(5):1644–1654. https://doi.org/10.1152/japplphysiol.00669.2007
Hanson AM, Harrison BC, Young MH, Stodieck LS, Ferguson VL (2013) Longitudinal characterization of functional, morphologic, and biochemical adaptations in mouse skeletal muscle with hindlimb suspension. Muscle Nerve 48(3):393–402. https://doi.org/10.1002/mus.23753
Han B, Zhu MJ, Ma C, Du M (2007) Rat hindlimb unloading down-regulates insulin like growth factor-1 signaling and AMP-activated protein kinase, and leads to severe atrophy of the soleus muscle. Appl Physiol Nutr Metab 32(6):1115–1123. https://doi.org/10.1139/h07-102
Lawler JM, Kwak HB, Kim JH, Lee Y, Hord JM, Martinez DA (2012) Biphasic stress response in the soleus during reloading after hind limb unloading. Med Sci Sports Exerc 44(4):600–609. https://doi.org/10.1249/MSS.0b013e31823ab37a
Mirzoev TM, Tyganov SA, Shenkman BS (2017) Akt-dependent and Akt-independent pathways are involved in protein synthesis activation during reloading of disused soleus muscle. Muscle Nerve 55(3):393–399. https://doi.org/10.1002/mus.25235
Nakao R, Hirasaka K, Goto J, Ishidoh K, Yamada C, Ohno A, Okumura Y, Nonaka I, Yasutomo K, Baldwin KM, Kominami E, Higashibata A, Nagano K, Tanaka K, Yasui N, Mills EM, Takeda S, Nikawa T (2009) Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol Cell Biol 29(17):4798–4811. https://doi.org/10.1128/mcb.01347-08
Park S, Brisson BK, Liu M, Spinazzola JM, Barton ER (2014) Mature IGF-I excels in promoting functional muscle recovery from disuse atrophy compared with pro-IGF-IA. J Appl Physiol (1985) 116(7):797–806. https://doi.org/10.1152/japplphysiol.00955.2013
Pierno S, Camerino GM, Cannone M, Liantonio A, De Bellis M, Digennaro C, Gramegna G, De Luca A, Germinario E, Danieli-Betto D, Betto R, Dobrowolny G, Rizzuto E, Musaro A, Desaphy JF, Camerino DC (2014) Paracrine effects of IGF-1 overexpression on the functional decline due to skeletal muscle disuse: molecular and functional evaluation in hindlimb unloaded MLC/mIgf-1 transgenic mice. PLoS One 8(6):e65167. https://doi.org/10.1371/journal.pone.0065167
Allen DL, Linderman JK, Roy RR, Grindeland RE, Mukku V, Edgerton VR (1997) Growth hormone/IGF-I and/or resistive exercise maintains myonuclear number in hindlimb unweighted muscles. J Appl Physiol (1985) 83(6):1857–1861. https://doi.org/10.1152/jappl.1997.83.6.1857
Alzghoul MB, Gerrard D, Watkins BA, Hannon K (2004) Ectopic expression of IGF-I and Shh by skeletal muscle inhibits disuse-mediated skeletal muscle atrophy and bone osteopenia in vivo. FASEB J 18(1):221–223. https://doi.org/10.1096/fj.03-0293fje
Zdanowicz MM, Teichberg S (2003) Effects of insulin-like growth factor-1/binding protein-3 complex on muscle atrophy in rats. Exp Biol Med (Maywood) 228(8):891–897
Koshinaka K, Toshinai K, Mohammad A, Noma K, Oshikawa M, Ueno H, Yamaguchi H, Nakazato M (2011) Therapeutic potential of ghrelin treatment for unloading-induced muscle atrophy in mice. Biochem Biophys Res Commun 412(2):296–301. https://doi.org/10.1016/j.bbrc.2011.07.086
Kataoka H, Nakano J, Morimoto Y, Honda Y, Sakamoto J, Origuchi T, Okita M, Yoshimura T (2014) Hyperglycemia inhibits recovery from disuse-induced skeletal muscle atrophy in rats. Physiol Res 63(4):465–474
Lang SM, Kazi AA, Hong-Brown L, Lang CH (2012) Delayed recovery of skeletal muscle mass following hindlimb immobilization in mTOR heterozygous mice. PLoS One 7(6):e38910. https://doi.org/10.1371/journal.pone.0038910
Boesen AP, Dideriksen K, Couppe C, Magnusson SP, Schjerling P, Boesen M, Kjaer M, Langberg H (2013) Tendon and skeletal muscle matrix gene expression and functional responses to immobilisation and rehabilitation in young males: effect of growth hormone administration. J Physiol 591(23):6039–6052. https://doi.org/10.1113/jphysiol.2013.261263
Boesen AP, Dideriksen K, Couppe C, Magnusson SP, Schjerling P, Boesen M, Aagaard P, Kjaer M, Langberg H (2014) Effect of growth hormone on aging connective tissue in muscle and tendon: gene expression, morphology, and function following immobilization and rehabilitation. J Appl Physiol (1985) 116(2):192–203. https://doi.org/10.1152/japplphysiol.01077.2013
Lieber RL, Jacks TM, Mohler RL, Schleim K, Haven M, Cuizon D, Gershuni DH, Lopez MA, Hora D Jr, Nargund R, Feeney W, Hickey GJ (1997) Growth hormone secretagogue increases muscle strength during remobilization after canine hindlimb immobilization. J Orthop Res 15(4):519–527. https://doi.org/10.1002/jor.1100150407
Suliman IA, Lindgren JU, Elhassan AM, Diab KM, Adem A (2001) Effects of short- and long-term rat hind limb immobilization on spinal cord insulin-like growth factor-I and its receptor. Brain Res 912(1):17–23
Suetta C, Frandsen U, Mackey AL, Jensen L, Hvid LG, Bayer ML, Petersson SJ, Schroder HD, Andersen JL, Aagaard P, Schjerling P, Kjaer M (2013) Ageing is associated with diminished muscle re-growth and myogenic precursor cell expansion early after immobility-induced atrophy in human skeletal muscle. J Physiol 591(15):3789–3804. https://doi.org/10.1113/jphysiol.2013.257121
Burks TN, Andres-Mateos E, Marx R, Mejias R, Van Erp C, Simmers JL, Walston JD, Ward CW, Cohn RD (2011) Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med 3(82):82ra37. https://doi.org/10.1126/scitranslmed.3002227
Morales MG, Abrigo J, Acuna MJ, Santos RA, Bader M, Brandan E, Simon F, Olguin H, Cabrera D, Cabello-Verrugio C (2016) Angiotensin-(1-7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis Model Mech 9(4):441–449. https://doi.org/10.1242/dmm.023390
Ye F, Mathur S, Liu M, Borst SE, Walter GA, Sweeney HL, Vandenborne K (2013) Overexpression of insulin-like growth factor-1 attenuates skeletal muscle damage and accelerates muscle regeneration and functional recovery after disuse. Exp Physiol 98(5):1038–1052. https://doi.org/10.1113/expphysiol.2012.070722
Stevens-Lapsley JE, Ye F, Liu M, Borst SE, Conover C, Yarasheski KE, Walter GA, Sweeney HL, Vandenborne K (2010) Impact of viral-mediated IGF-I gene transfer on skeletal muscle following cast immobilization. Am J Physiol Endocrinol Metab 299(5):E730–E740. https://doi.org/10.1152/ajpendo.00230.2010
Ballak SB, Degens H, de Haan A, Jaspers RT (2014) Aging related changes in determinants of muscle force generating capacity: a comparison of muscle aging in men and male rodents. Ageing Res Rev 14:43–55. https://doi.org/10.1016/j.arr.2014.01.005
Degens H, Korhonen MT (2012) Factors contributing to the variability in muscle ageing. Maturitas 73(3):197–201. https://doi.org/10.1016/j.maturitas.2012.07.015
Dalle S, Rossmeislova L, Koppo K (2017) The role of inflammation in age-related sarcopenia. Front Physiol 8:1045. https://doi.org/10.3389/fphys.2017.01045
Barbieri M, Ferrucci L, Ragno E, Corsi A, Bandinelli S, Bonafe M, Olivieri F, Giovagnetti S, Franceschi C, Guralnik JM, Paolisso G (2003) Chronic inflammation and the effect of IGF-I on muscle strength and power in older persons. Am J Physiol Endocrinol Metab 284(3):E481–E487. https://doi.org/10.1152/ajpendo.00319.2002
Broussard SR, Zhou JH, Venters HD, Bluthé RM, Freund GG, Johnson RW, Dantzer R, Kelley KW (2001) At the interface of environment-immune interactions: cytokine and growth-factor receptors. J Anim Sci 79(suppl_E):E268–E284. https://doi.org/10.2527/jas2001.79E-SupplE268x
Hofmann M, Halper B, Oesen S, Franzke B, Stuparits P, Tschan H, Bachl N, Strasser EM, Quittan M, Ploder M, Wagner KH, Wessner B (2015) Serum concentrations of insulin-like growth factor-1, members of the TGF-beta superfamily and follistatin do not reflect different stages of dynapenia and sarcopenia in elderly women. Exp Gerontol 64:35–45. https://doi.org/10.1016/j.exger.2015.02.008
Sattler FR (2013) Growth hormone in the aging male. Best Pract Res Clin Endocrinol Metab 27(4):541–555. https://doi.org/10.1016/j.beem.2013.05.003
Giovannini S, Marzetti E, Borst SE, Leeuwenburgh C (2008) Modulation of GH/IGF-1 axis: potential strategies to counteract sarcopenia in older adults. Mech Ageing Dev 129(10):593–601. https://doi.org/10.1016/j.mad.2008.08.001
Hameed M, Harridge SD, Goldspink G (2002) Sarcopenia and hypertrophy: a role for insulin-like growth factor-1 in aged muscle? Exerc Sport Sci Rev 30(1):15–19
Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, Mammucari C, Meskers CG, Pallafacchina G, Paoli A, Pion D, Roceri M, Romanello V, Serrano AL, Toniolo L, Larsson L, Maier AB, Munoz-Canoves P, Musaro A, Pende M, Reggiani C, Rizzuto R, Schiaffino S (2013) Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 14(3):303–323. https://doi.org/10.1007/s10522-013-9432-9
Haddad F, Adams GR (2006) Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J Appl Physiol (1985) 100(4):1188–1203. https://doi.org/10.1152/japplphysiol.01227.2005
Edstrom E, Altun M, Hagglund M, Ulfhake B (2006) Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J Gerontol A Biol Sci Med Sci 61(7):663–674
Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margaritis I, Derijard B (2006) Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech Ageing Dev 127(10):794–801. https://doi.org/10.1016/j.mad.2006.07.005
Leger B, Derave W, De Bock K, Hespel P, Russell AP (2008) Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res 11(1):163–175b. https://doi.org/10.1089/rej.2007.0588
Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL (1998) Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A 95(26):15603–15607
Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N (2001) Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 27(2):195–200. https://doi.org/10.1038/84839
McMahon CD, Chai R, Radley-Crabb HG, Watson T, Matthews KG, Sheard PW, Soffe Z, Grounds MD, Shavlakadze T (2014) Lifelong exercise and locally produced insulin-like growth factor-1 (IGF-1) have a modest influence on reducing age-related muscle wasting in mice. Scand J Med Sci Sports 24(6):e423–e435. https://doi.org/10.1111/sms.12200
Owino V, Yang SY, Goldspink G (2001) Age-related loss of skeletal muscle function and the inability to express the autocrine form of insulin-like growth factor-1 (MGF) in response to mechanical overload. FEBS Lett 505(2):259–263
Funai K, Parkington JD, Carambula S, Fielding RA (2006) Age-associated decrease in contraction-induced activation of downstream targets of Akt/mTor signaling in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 290(4):R1080–R1086. https://doi.org/10.1152/ajpregu.00277.2005
Mayhew DL, Kim JS, Cross JM, Ferrando AA, Bamman MM (2009) Translational signaling responses preceding resistance training-mediated myofiber hypertrophy in young and old humans. J Appl Physiol (1985) 107(5):1655–1662. https://doi.org/10.1152/japplphysiol.91234.2008
Fan J, Yang X, Li J, Shu Z, Dai J, Liu X, Li B, Jia S, Kou X, Yang Y, Chen N (2017) Spermidine coupled with exercise rescues skeletal muscle atrophy from D-gal-induced aging rats through enhanced autophagy and reduced apoptosis via AMPK-FOXO3a signal pathway. Oncotarget 8(11):17475–17490. https://doi.org/10.18632/oncotarget.15728
Nakashima K, Yakabe Y (2007) AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem 71(7):1650–1656
Mounier R, Lantier L, Leclerc J, Sotiropoulos A, Pende M, Daegelen D, Sakamoto K, Foretz M, Viollet B (2009) Important role for AMPKalpha1 in limiting skeletal muscle cell hypertrophy. FASEB J 23(7):2264–2273. https://doi.org/10.1096/fj.08-119057
Zhang SF, Zhang Y, Li B, Chen N (2018) Physical inactivity induces the atrophy of skeletal muscle of rats through activating AMPK/FoxO3 signal pathway. Eur Rev Med Pharmacol Sci 22(1):199–209. https://doi.org/10.26355/eurrev_201801_14118
Lee S-J, McPherron AC (2001) Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci 98(16):9306–9311. https://doi.org/10.1073/pnas.151270098
Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ (2009) Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol-Cell Physiol 296(6):C1258–C1270. https://doi.org/10.1152/ajpcell.00105.2009
Pelosi M, De Rossi M, Barberi L, Musaro A (2014) IL-6 impairs myogenic differentiation by downmodulation of p90RSK/eEF2 and mTOR/p70S6K axes, without affecting AKT activity. Biomed Res Int 2014:206026. https://doi.org/10.1155/2014/206026
Petersen AMW, Pedersen BK (2005) The anti-inflammatory effect of exercise. J Appl Physiol 98(4):1154–1162. https://doi.org/10.1152/japplphysiol.00164.2004
Liu C, Wang M, Chen M, Zhang K, Gu L, Li Q, Yu Z, Li N, Meng Q (2017) miR-18a induces myotubes atrophy by down-regulating IgfI. Int J Biochem Cell Biol 90:145–154. https://doi.org/10.1016/j.biocel.2017.07.020
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12(9):735–739
Liu CG, Calin GA, Meloon B, Gamliel N, Sevignani C, Ferracin M, Dumitru CD, Shimizu M, Zupo S, Dono M, Alder H, Bullrich F, Negrini M, Croce CM (2004) An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci U S A 101(26):9740–9744. https://doi.org/10.1073/pnas.0403293101
Competing Financial Interests
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Timmer, L.T., Hoogaars, W.M.H., Jaspers, R.T. (2018). The Role of IGF-1 Signaling in Skeletal Muscle Atrophy. In: Xiao, J. (eds) Muscle Atrophy. Advances in Experimental Medicine and Biology, vol 1088. Springer, Singapore. https://doi.org/10.1007/978-981-13-1435-3_6
Download citation
DOI: https://doi.org/10.1007/978-981-13-1435-3_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1434-6
Online ISBN: 978-981-13-1435-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)