
Abstract
Denervation, disuse, fasting, and various diseases could induce skeletal muscle atrophy, which results in the decline of life quality and increase of the mortality risk for patients. Noncoding RNAs (ncRNAs) are implicated important in regulating gene expression. Thus, ncRNAs, especially microRNAs and long noncoding RNAs (lncRNAs), have gained widespread attention as crucial players in numerous physiological and pathological processes, including skeletal muscle atrophy. In this review, we comprehensively described the potential of circulating microRNAs as biomarkers, summarized the profiling of microRNAs and lncRNAs in atrophying muscles, as well as discussed the effects and underlying mechanisms of microRNA machinery proteins, microRNAs, and lncRNAs in skeletal muscle atrophy. Considering the large quantity and variety of ncRNAs, the understanding of ncRNAs in muscle atrophy is still very limited. Future studies are needed to elucidate the possibility of ncRNAs as diagnosis biomarkers and therapeutic targets in muscle atrophy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Background
Muscle atrophy is characterized as the decrease in myofiber size, strength, protein content, and total muscle mass [1]. Muscle atrophy can be divided into primary muscular disease and secondary muscular disorders. Primary muscle atrophy is caused by direct diseases of the muscle such as Duchenne muscular dystrophy (DMD) [2] and myotonic dystrophy type 1 (DM1) diseases [3]. Secondary muscular disorders are usually the complications of other diseases, which include chronic kidney diseases (CKD) [4], sepsis [5], diabetes [6], cancers [7], renal and cardiac failure [8], burn injury [9, 10], and HIV/AIDS and neurodegenerative disorders [11]. Additionally, secondary muscular disorders can also occur in healthy individuals under the conditions such as spaceflight, starvation, aging, hindlimb unloading, bed rest, and immobilization [12]. It is well-known that muscle atrophy reduces the quality of life and increases the mortality risk for patients [13]. However, effective treatment methods for muscle atrophy are currently lacking. Thus, there is an urgent need to understand the molecular mechanisms that mediate muscle atrophy, which could greatly contribute to design therapies for alleviating muscle atrophy.
Accumulating evidence shows that noncoding RNAs (ncRNAs) play an important role in regulating distinct steps of muscle atrophy. ncRNAs comprise a large and heterogeneous family including microRNAs (miRs, miRNAs), long noncoding RNAs (lncRNAs), circular RNAs (circRNAs), PIWI-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), and tRNA derivatives. Among them, miRNAs and lncRNAs are the best-studied classes in different physiologic and pathological conditions, including muscle development and muscle diseases. miRNAs, short ncRNAs (∼22 nucleotides), are endogenous and evolutionarily conserved, which mainly repress gene expression posttranscriptionally. One single miRNA has multiple target mRNAs, while individual mRNA can be modulated by numerous miRNAs [14, 15]. miRNAs collectively regulate the expression of 30% of human genes [16]. lncRNAs are a diverse class of noncoding RNAs which are more than 200 nucleotides in length. lncRNAs have been shown vital in regulating gene expression both transcriptionally and posttranscriptionally via various mechanisms. Given that aberrant gene expression underlies muscle atrophy, it is critically important to understand how gene expression is regulated by ncRNAs in response to diverse stresses or diseases which lead to muscle atrophy.
In this review, we will focus upon the ncRNAs (miRNAs and lncRNAs) involved in regulating muscle atrophy and the underlying molecular mechanisms.
2 MicroRNA Machinery Proteins in Muscle Atrophy
It is now evident that miRNAs play important roles in multiple physiological and pathological processes including muscle development, muscle regeneration, and muscle atrophy. After transcription by RNA polymerase II or III, miRNA precursors are catalyzed by DROSHA/DGCR8 complex and exported from the nucleus to cytoplasm by Exportin-5 [17]. Then the enzyme Dicer processes the miRNAs into ~22 nt RNA duplex in cytoplasm, which are loaded onto RNA-induced silencing complex (RISC) and mediate translational repression/mRNA degradation [18, 19].
These proteins involved in miRNA biogenesis and production have been shown important in regulating muscle development and muscle atrophy. Loss of Dicer activity specifically in the myogenic compartment during embryogenesis reduced muscle-specific miRNAs, caused perinatal lethality, and resulted in decreased skeletal muscle mass and abnormal myofiber morphology [20]. Additionally, specific ablation of Dicer1 in postmitotic spinal motor neurons in mice from postnatal day 7 exhibited signs of denervation-related muscle atrophy, including myofiber type grouping, loss of muscle fibers with a large cross-sectional area, and the decreased total fiber diameter [21]. Another miRNA machinery protein Argonaute2 (Ago2) has also been shown important for regulating skeletal muscle atrophy [22]. Crystallin-B (CryAB), a small heat shock protein, interacts with the N and C termini of Ago2 [22]. When the endonuclease activity of Ago2 was significantly repressed through loss of CryAB in mice, the body weight and myofiber cross-sectional area were significantly reduced, while the fibrosis was increased in the skeletal muscle [22]. These results indicated that inhibition of Ago2 caused skeletal muscle atrophy.
In addition, some RNA-binding proteins were also found to negatively regulate miRNA biogenesis. For example, the nuclear factor 90 (NF90; also referred to as ILF3, NFAR1, or DRBP76)-nuclear factor 45 (NF45) complex suppresses miRNA processing through inhibition of pri-miRNA processing [23]. Adult NF90-NF45 double-transgenic mice exhibited skeletal muscle atrophy and centronuclear muscle fibers [24]. Compared with controls, microarray analysis demonstrated that NF90-NF45 overexpression reduced the expression of 23 miRNAs in skeletal muscles, including miR-133a, miR-133b, miR-1, and miR-378 which are reported to promote muscle development [24]. Among them, the processing of pri-miR-133a was found to be suppressed by NF90-NF45 complex [24]. And concomitantly, dynamin 2, a target of miR-133a, is elevated in the muscle of NF90-NF45 double-transgenic mice [24]. Therefore, the upstream regulation of miRNAs plays vital roles in muscle atrophy.
3 MicroRNAs Served as Potential Biomarkers in Muscle Atrophy
The reliable and sensitive blood biomarkers are useful, easily accessible, and convenient for the diagnosis, monitoring, and potential future therapy of diseases. miRNAs are found to be present in blood circulation and have been increasingly suggested as biomarkers for several diseases and clinical conditions [25]. As a consequence of fiber damage during atrophy, muscle-expressed miRNAs have been found to be released into the blood, and their levels are usually correlated with the severity of muscle diseases. Thus, many scientific reports emphasize the possibility of muscle-specific miRNAs as circulating biomarkers for muscle atrophy induced by various stimuli.
Muscle atrophy and weakness are the primary characteristics of Duchenne muscular dystrophy and myotonic dystrophy type 1 patients. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis demonstrated that several muscle-specific miRNAs (miR-1, miR-133a, and miR-206) are increased in the serum of mouse and dog models of DMD [2]. Additional studies indicate that miR-1, miR-133a, and miR-206 are enriched in serum of DMD patients, and their levels were correlated with the severity of DMD disease, indicating that miR-1, miR-133a, and miR-206 are new biomarkers for the diagnosis of DMD and for evaluating the outcomes of therapeutic interventions in humans [26]. By multiplex qRT-PCR analysis of 381 miRNAs in 36 consecutive DM1 patients and 36 healthy controls, a signature of 9 deregulated miRNAs in plasma samples of DM1 patients was identified [3]. miR-133a, miR-193b, miR-191, miR-454, miR-574, miR-885-5p, and miR-886-3p were increased, while miR-27b was decreased in DM1 patients [3]. Among them, miR-133a was suggested to be used as candidate diagnostic biomarker for DM1 [3]. Another study demonstrated that miR-1, miR-133a, miR-133b, and miR-206 were increased in the serum from DM1 patients with progressive muscle atrophy compared to disease-stable DM1 patients [27]. And the levels of miR-1, miR-133a, miR-133b, and miR-206 were correlated with the progression of muscle atrophy in the DM1 patients, supporting their potential as useful and reliable biomarkers for DM1 patients [27].
Muscle atrophy is a common systemic complication of chronic obstructive pulmonary disease (COPD). The expression of muscle-specific miRNAs was determined in serum from 31 COPD patients with muscle atrophy and 14 healthy age-matched controls by qRT-PCR [28]. The expression of miR-1 was reduced in COPD patients compared with controls, but there was no significant difference in the expression of miR-499, miR-208, miR-181, miR-145, miR-206, and miR-133 [28].
Additionally, the serum levels of muscle-specific miRNAs (miR-1, miR-23a, miR-133, miR-206, miR-208b, and miR-499) were all significantly elevated after hindlimb unloading for 7 days in mice, which could induce severe muscle atrophy [29]. Moreover, the serum levels of miR-23a, miR-206, and miR-499 were increased, while miR-1, miR-206, and miR-208b were not changed in 15 healthy human participants after 45 days of head-down bed rest [29]. And the levels of miR-23a, miR-206, and miR-499 were positively correlated with the ratio of soleus volume loss induced by head-down bed rest [29], indicating that circulating miR-23a, miR-206, and miR-499 could be used as candidate biomarkers for the diagnosis of muscle atrophy induced by disuse.
One study selectively characterized the expression of miR-9, miR-206, and miR-132 in serum from spinal muscular atrophy (SMA) mice and patients [30]. Both miR-9 and miR-132 were elevated in the serum from SMA mice and patients [30]. Serum miR-206 was increased in SMA mice compared with controls, but its level in SMA patients has no significant difference [30]. These results indicated the potential of miR-9 and miR-132 as candidate serum biomarkers for SMA.
Collectively, some miRNAs have been identified as possible circulating biomarkers for the diagnosis of DMD, SMA, and DM1 diseases, as well as muscle atrophy induced by hindlimb unloading, head-down bed rest, and COPD disease. However, the specific, sensitive, and reliable biomarkers are still lacking for muscle atrophy.
4 MicroRNAs in Muscle Atrophy
To understand the involvement of miRNAs in muscle atrophy, a large number of miRNA profiling have been performed in atrophying muscles under different conditions such as fasting, denervation, diabetes, disuse, and cancer cachexia. The miRNA signature of muscle atrophy has been found peculiar under each condition [31].
In primary muscle atrophy caused by direct diseases of the muscle, miRNA microarrays in muscle tissues identified 39 miRNAs such as miR-29a, miR-30c, miR-30b, miR-92, miR-29c, miR-423, miR-361, miR-299-3p, and miR-181d which were upregulated in Duchenne muscular dystrophy patients [32]. Sixty-two miRNAs such as miR-16, miR-279, miR-99a, miR-93, miR-455, miR-20b, miR-18a, miR-17-5p, miR-152, miR-106a, and miR-106b were upregulated in facioscapulohumeral muscular dystrophy patients [32]. The levels of miR-1 and miR-133a/b were significantly decreased, while miR-206 was significantly increased in muscles of 12 myotonic dystrophy type 1 patients as compared to 6 healthy controls [33].
Lipopolysaccharide, cancer cachexia, and chronic alcohol exposure are the pathological stimuli for muscle atrophy. Small RNA deep sequencing in pig skeletal muscles analyzed the miRNA expression profiles during lipopolysaccharide-induced wasting [34]. Four miRNAs (miR-146a-5p, miR-221-5p, miR-9860-5p, and miR-148b-3p) were significantly upregulated, while three miRNAs (miR-192, miR-215, and miR-429) were downregulated in the lipopolysaccharide-challenged samples [34]. Cancer cachexia-induced muscle atrophy is a direct cause in the functional decline of cancer patients [35]. By injecting Lewis lung carcinoma cells into C57BL/6 J mice to induce muscle atrophy, miRNA sequencing identified nine dysregulated miRNAs including miR-147-3p, miR-299a-3p, miR-1933-3p, miR-511-3p, miR-3473d, miR-233-3p, miR-431-5p, miR-665-3p, and miR-205-3p in the tibialis anterior muscles injected by Lewis lung carcinoma cells [36]. Utilizing a zebrafish model of muscle atrophy induced by chronic alcohol exposure, miRNA microarray identified that 14 miRNAs were upregulated, while 47 miRNAs were downregulated more than twofold in skeletal muscles [37]. Among them, miR-140-3p was downregulated, whereas miR-146a was upregulated. Interestingly, the potential targets of both miR-140-3p and miR-146a include several members of the Notch signaling pathway [37].
Recently, RNA sequencing was performed to assess the whole transcriptome in mouse models of denervation-induced muscle atrophy [38]. There were 671 differentially expressed miRNAs in gastrocnemius muscles at different time points (1 week, 2 weeks, 4 weeks, and 8 weeks) after nerve injury compared with controls [38]. At an early denervation stage, another miRNA microarray analysis in rats showed that miR-206, miR-195, miR-23a, and miR-30e were differentially expressed in the slow muscles, while other miRNA molecules (miR-214, miR-221, miR-222, miR-152, miR-320, and let-7e) were differentially expressed in the fast muscles compared to controls [39]. These studies indicated that miRNAs were dynamically altered in the progression of muscle atrophy and miRNAs in different types of skeletal muscles respond to the same stimuli in distinct ways.
Amyotrophic lateral sclerosis (ALS) is characterized by the signs of denervation-induced muscle atrophy. In human studies of ALS, miR-206 was elevated in muscles of four early-stage ALS patients [40] and characterized as a potential biomarker for ALS patients [41]. Using small RNA-seq, the expressions of small RNAs in muscle tissues of ALS patients and healthy age-matched controls were compared [42]. Nineteen miRNAs such as miR-100, miR-10a, miR-125a, miR-125b, miR-1260a, miR-128, miR-1291, miR-132, miR-133a, and miR-151a were upregulated, while 10 miRNAs such as miR-126, miR-1285, miR-1303, miR-150, miR-191, and miR-28 were downregulated in the ALS groups [42]. Interestingly, this study did not find changes in the expression of miR-206 in ALS patients [42], which might be due to the differences in study populations.
Spinal cord injury can induce severe skeletal muscle atrophy and the transformation toward fast-twitch, type II fibers. In human, miR-208b and miR-499-5p expressions were progressively declined in skeletal muscle during the first year after spinal cord injury [43]. Moreover, miR-208b and miR-499-5p were inversely correlated with the expression of myostatin, an inhibitor of muscle growth, in human skeletal muscle after spinal cord injury [43]. miR-208b reduced myostatin expression in intact mouse skeletal muscle after spinal cord injury, whereas miR-499-5p had no obvious effect [43].
Addition of dexamethasone (Dex) leads to a distinct atrophic phenotype in differentiated C2C12 myotubes, which is the in vitro model of Dex-induced muscle atrophy [44]. miR-1, miR-322, miR-351, and miR-503-3p were found to be upregulated in Dex-treated C2C12 cells compared to controls, while miR-708 and miR-147 were downregulated [44]. miR-18a expression is declined during C2C12 myoblast differentiation [45]. And in vitro overexpression of miR-18a induces myotube atrophy via the PI3K/AKT pathway through Igf1 [45]. miR-182 expression is dramatically decreased in C2C12 myotubes treated with Dex [46]. miR-182 was enriched in exosomes isolated from the media of C2C12 myotubes, and Dex treatment could increase its abundance in exosomes [46].
In addition to the miRNA profiling studies, functional studies using cellular and animal models have disclosed multiple important miRNAs in muscle atrophy. Spinal and bulbar muscular atrophy (SBMA) is an inherited neurodegenerative disorder caused by the expansion of a polyglutamine repeat in the androgen receptor (AR-polyQ) [47, 48]. SBMA is characterized by proximal muscular atrophy, weakness, contraction fasciculation, and bulbar involvement [49]. miRNA microarray analysis identified that miR-196a, miR-196b, miR-496, miR-323-3p, and miR-29b-3p were upregulated more than twofold in the spinal cords of male SBMA mice expressing full-length human AR with 97 glutamine residues (AR-97Q) compared to the male mice expressing wild-type human AR [50]. Among them, miR-196a was found to enhance the decay of the AR mRNA by silencing CUGBP, Elav-like family member 2 (CELF2) [50]. Further studies demonstrated that adeno-associated virus (AAV) vector-mediated delivery of miR-196a exhibited the strong and continuous inhibition of CELF2 expression and ameliorated the SBMA phenotypes in a mouse model [50]. Importantly, miR-196a was upregulated and the CELF2 mRNA was downregulated in the thoracic spinal cord of patients with SBMA, and miR-196a treatment could downregulate both the AR and CELF2 mRNAs and proteins in the fibroblasts obtained from patients with SBMA [50]. Thus, overexpression of miR-196a can be considered as the potential strategy for treating SBMA. Another report found that miR-298 could ameliorate the phenotype of SBMA in mice [51]. In vitro studies demonstrated that miR-298 directly bound to the 3′-untranslated region (UTR) of the human AR transcripts and reduced AR mRNA and protein levels [51].
miR-1 is specifically expressed in muscles and plays important roles in myogenesis, muscle regeneration, as well as muscle atrophy. High doses of Dex or myostatin (Mstn) induce severe skeletal muscle atrophy [52]. miR-1 was found to be elevated in both C2C12 myotubes and mouse models of Dex-induced atrophy [52]. Both Dex and Mstn could induce miR-1 expression through glucocorticoid receptor (GR) [52]. And miR-1 elevation promotes skeletal muscle atrophy through targeting HSP70 and reducing its levels, which led to decreased phosphorylation of AKT, enhanced activation of FOXO3, and upregulation of MuRF1 and Atrogin-1 [52]. In addition, miR-1 was found to be unchanged in soleus muscle of rats with muscle atrophy induced by hindlimb suspension [53]. Similar to miR-1, miR-133 also has important roles in the myogenesis and muscle development [54, 55]. However, the functional study of miR-133 in muscle atrophy is much more less.
Denervation is a common cause of muscle atrophy, and miR-351, miR-21, and miR-206 have been identified as important regulators of denervation-induced muscle atrophy. Following sciatic nerve transection, miR-351 was gradually reduced with time, and overexpression of miR-351 significantly repressed the decrease of the wet weight ratio and cross-sectional area of the tibialis anterior muscle in rats [56]. Mechanically, miR-351 is able to downregulate TRAF6 expression by directly targeting its 3’-UTR [56] and negatively regulate the two downstream signaling molecules of TRAF6, MuRF1 and MAFBx, in tibialis anterior muscles after sciatic nerve transection [56]. By miRNA profiling in mouse denervated muscles, miR-21 and miR-206 were found to be strongly induced after denervation [31]. Induction of miR-206 and miR-21 in adult mouse muscle contributes to muscle atrophy induced by denervation, whereas repression of miR-206 and miR-21 partially protects against denervation-induced atrophy in vivo [31]. More importantly, luciferase assays confirmed that YY1 was the target gene of miR-21, and eIF4E3 and Pdcd10 were the target genes of both miR-21 and miR-206 in denervated muscles [31]. However, in rats, miR-206 was found to increase the number of differentiating (MyoD1+/Pax7+) satellite cells and counteract denervation-induced atrophy through TGF-β1/Smad3 signaling pathway [57]. Moreover, miR-206 is dramatically increased in a mouse model of amyotrophic lateral sclerosis (ALS), which exhibited denervation and atrophy of targeted muscles [58]. miR-206-deficient mice form normal neuromuscular synapses during development, but loss of miR-206 accelerated ALS progression in mouse model and induced severe skeletal muscle atrophy through targeting histone deacetylase 4 (HDAC4) [58].
A loss of muscle mass during muscle atrophy results from an imbalance of protein synthesis and degradation with a reduction in synthesis. miR-424-5p expression was increased in patients with conditions associated with muscle wasting (COPD patients, patients undergoing aortic surgery, and patients with ICU-acquired weakness) [59]. In mice, overexpression of miR-322 (rodent miR-424 orthologue) promoted muscle atrophy and reduced ribosome RNA levels [59]. Ago2 pull-down assays showed that miR-424-5p bound to mRNAs encoding proteins required for ribosomal RNA transcription and protein synthesis, PolR1A and upstream binding transcription factors [59].
A common clinical feature in patients with severe burns is skeletal muscle atrophy. miR-628 was increased in tibialis anterior muscle after burn injury in rats [9, 10]. Overexpression of miR-628 in rat muscle activates the IRS1/Akt/FoxO3a signaling pathway and promotes cell apoptosis [9]. IRS1 was identified as direct target of miR-628 [9].
Most of miRNAs mentioned above have been shown vital for only one model of muscle atrophy. A systematic study using different models of muscle atrophy identified that miR-29b was elevated in multiple in vivo atrophy models (denervation, Dex, fasting, cancer cachexia, and aging), as well as the in vitro atrophy models (primary myoblasts treated with Dex and myotubes differentiated from C2C12 treated with Dex, TNF-α, or H2O2) [60]. miR-29b overexpression induces muscle atrophy, and its inhibition attenuates muscle atrophy induced by multiple stimuli both in vitro and in vivo [60]. IGF-1 and PI3K(p85α) were identified as the direct targets of miR-29b [60].
miR-23a has also been found to be important in multiple models of muscle atrophy. In patients with chronic kidney disease (CKD), a decline in muscle mass is associated with increased morbidity and mortality [4]. Exercise can ameliorate the phenotype of muscle atrophy induced by CKD [4]. miR-23a was decreased, while miR-27a was unchanged in CKD mice muscle, and resistance exercise elevated miR-23a and miR-27a expression in CKD mouse muscle [61]. Overexpression of miR-23a/miR-27a in CKD mice attenuated muscle loss, improved grip strength, reduced caspase activity, and increased markers of muscle regeneration [61]. In primary satellite cells, PTEN and caspase-7 were identified as targets of miR-23a and FoxO1 was identified as a target of miR-27a [61]. Ectopic expression of miR-23a was sufficient to prevent Dex-induced muscle atrophy both in vitro and in vivo [62]. Furthermore, miR-23a transgenic mice showed resistance against Dex-induced skeletal muscle atrophy [62]. miR-23a repressed the translation of both MAFbx/atrogin-1 and MuRF1 in a 3’ UTR-dependent manner, which were involved in promoting atrophy-associated protein degradation [62]. miR-23a was also reduced both in the atrophying muscles of rats with acute streptozotocin-induced diabetes and the C2C12 myotubes treated with Dex [63]. In-depth study demonstrated that the decrease of miR-23a was due to the attenuation of calcineurin signaling and the promotion of exosome-mediated export of miR-23a caused by atrophy-inducing conditions [63].
Collectively, in vivo studies demonstrated that miR-196a, miR-298, miR-351, miR-23a, and miR-27a suppressed, while miR-1, miR-21, miR-424-5p, miR-628, and miR-29b promoted the progression of muscle atrophy (Fig. 11.1). Particularly, miR-206 suppressed ALS-induced muscle atrophy in mice and denervation-induced muscle atrophy in rats and promoted the denervation-induced muscle atrophy in mice (Fig. 11.1). Future studies based on these results will provide the potential therapeutic targets for muscle atrophy.

MicroRNAs in muscle atrophy. Dex dexamethasone, CKD chronic kidney diseases, ALS amyotrophic lateral sclerosis
5 lncRNAs in Muscle Atrophy
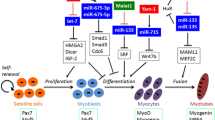
lncRNAs are characterized as noncoding RNA sequences >200 nucleotides [64]. lncRNAs have been regarded as critical epigenetic regulators of gene expression in multiple physiological and pathological conditions [65]. The number of lncRNAs in the human genome is estimated to be no less than protein-coding genes [66]. A substantial number, but not all of the lncRNAs, are transcribed by RNA polymerase II, 5′-capped, spliced, and polyadenylated at the 3′ end, undergoing similar posttranscriptional processing as mRNAs [67]. Compared with miRNAs, little is known about the biological roles of lncRNAs, and even less about their mechanism of action. In mammalian cells, the wide variety of subcellular localizations, expression levels, and stabilities of lncRNAs have been observed and a broad array of diverse mechanisms has been suggested. Based on the examples of well-studied lncRNAs, lncRNAs can either repress or activate gene expression through regulating gene transcription, mRNA stability, pre-mRNA splicing, protein translation, and protein stability [64]. Additionally, lncRNAs can serve as “sponge” RNAs for miRNAs through pairing to miRNAs and titrating them away from their mRNA targets [68]. Similarly, lncRNAs have been reported as a decoy that titrate the protein away from its potential targets, such as lncRNA Gas5 and glucocorticoid receptor [69] and sno-lncRNAs and Fox2 [70]. To date, many studies mainly focused on the physiological function of lncRNAs in muscles, and the number of lncRNAs identified as regulators of muscle atrophy so far is still exiguous. Therefore, our understanding of lncRNAs in muscle atrophy, especially in stress-induced muscle atrophy, is much more limited.
Myogenesis is a complex process required for regeneration and growth of myofibers in adults and begins with the activation and differentiation of muscle stem cells. Multiple lncRNAs were reported to be associated with myogenesis and muscle regeneration. lncRNA SRA [71, 72], H19 [73], MUNC [74], lncMyoD [75], lnc-MD1 [76], lnc-mg [77], MAR1 [78], lnc-YY1 [79], Myolinc [80], and Dum [81] are confirmed as important positive regulators of myogenesis. In contrast, recent studies have shown that certain lncRNAs negatively regulate myogenesis, including SINE-containing lncRNAs [82], Yam-1 [83], Lnc-31 [84], Malat1 [85], and Sirt1 AS lncRNAs [86]. During muscle atrophy, impaired myogenesis is a common underlying mechanism [87]. Thus, the aberrant expression of these myogenesis-related lncRNAs might contribute to muscle atrophy. So far, among the lncRNAs mentioned above, only the roles of lncRNA MAR1 and lnc-mg have been investigated in cellular and animal models of muscle atrophy.
lncRNA MAR1 (muscle anabolic regulator 1) was significantly downregulated in the mouse gastrocnemius muscle during aging and unloading condition [78]. In C2C12 cells, MAR1 was found to promote the myogenic differentiation through serving as the sponges for miR-487b to regulate Wnt5a expression, which is an important factor during myogenesis [78]. Moreover, therapeutic enforced MAR1 expression in skeletal muscle of mice could counteract either age-related muscle atrophy or hindlimb suspension-induced muscle atrophy mice [78].
A myogenesis-associated lncRNA named as lnc-mg is specifically enriched in skeletal muscle and was shown to be induced in muscle stem cell differentiation [77]. According to the in vitro analysis of primary skeletal muscle cells and in vivo analysis of conditional knockout mice, lnc-mg promotes myogenesis by serving as a sponge for miR-125b to elevate the protein abundance of insulin-like growth factor 2 [77]. Conditional knockout of lnc-mg in mouse skeletal muscle results in muscle atrophy and the loss of muscular endurance during exercise [77]. However, muscle loss is not significantly improved after denervation in transgenic mice of lnc-mg [77]. Thus, the rescue effect of lnc-mg on stress-induced skeletal muscle atrophy needs to be carefully elucidated.
Spinal muscular atrophy is an inherited neuromuscular disorder, caused by recessive mutations of the survival motor neuron 1 (SMN1) gene and retention of variable copy numbers of the highly homologous SMN2 gene [88, 89]. lncRNA SMN-AS1 arises from the antisense strand of SMN and is highly enriched in neurons [90]. SMN-AS1 recruited PRC2 to the SMN promoter and transcriptionally repressed SMN expression [90]. Delivery of SMN-AS1 antisense oligonucleotides (ASOs) elevated the SMN expression in patient-derived fibroblast cells, cultured neurons, and a mouse model of severe SMA [90]. Combining SMN-AS1 ASOs with SMN2 splice-switching oligonucleotides additively increased SMN expression and ameliorated SMA in mouse model [90]. Similarly, another independent group also reported that selective disruption of SMN-AS1-mediated PRC2 recruitment could activate SMN and ameliorate SMA phenotypes in mice [91].
In addition to the myogenesis-related lncRNAs as potential candidates, lncRNA profiling has been performed to identify more important lncRNAs in the animal models of muscle atrophy. Severe thermal trauma covering more than 30% of the total body surface area triggers severe muscle atrophy. Microarray was used to determine the lncRNA expression levels in skeletal muscle tissues of three pairs of burned rats at the early flow phase, compared with sham rats [92]. An average of 117 lncRNAs were significantly differentially expressed (1.5-fold) [92]. Recently, the expression patterns of lncRNAs were also detected using RNA sequencing in the mouse gastrocnemius muscle after nerve injury at different time points and compared to that obtained in the control group [38]. There were 664 differentially expressed lncRNAs (75 upregulated and 87 downregulated at 1 week, 78 upregulated and 80 downregulated at 2 weeks, 89 upregulated and 77 downregulated at 4 weeks, and 76 upregulated and 102 downregulated at 8 weeks) in denervated muscle atrophy compared to control groups [38]. Two selected lncRNAs were validated using qRT-PCR and their changes were consistent with the RNA-seq data [38]. Another microarray analysis compares the differentially expressed lncRNAs in gastrocnemius muscle between adult (6-month-old) and aged mice (24-month-old) [78]. And 894 lncRNAs were identified to be downregulated, while 1051 lncRNAs were upregulated more than twofold in aged muscle tissues compared with controls [78].
Collectively, very few lncRNAs including lnc-mg, MAR1, and SMN-AS1 are uncovered to regulate muscle atrophy (Fig. 11.2). And the studies of myogenesis-related lncRNAs and profiling of lncRNAs in muscle atrophy have shown the deserving hints for further investigation of lncRNAs in muscle atrophy.

lncRNAs in muscle atrophy
6 Conclusions and Perspectives
Skeletal muscle atrophy undergoes remarkable adaptations in response to numerous conditions, which significantly diminished quality of life. As we reviewed here, studies published in the past couple years emphasized identifying the potential miRNAs as biomarkers, profiling the changes of miRNAs and lncRNAs, and uncovering the roles and mechanisms of miRNAs and lncRNAs in diverse muscle atrophy.
To date, numerous miRNAs have been found to be altered in the serum of patients with muscle atrophy compared with healthy controls. And several of them have been shown correlated with the different stages and severity of the diseases. However, the possible inconsistencies in the results and the specificity of this kind of biomarker remain the major critical challenges. One of the major reasons is the human subject variability, and therefore recruiting large cohorts of patients could greatly improve the future biomarker studies.
The quantity and variety of miRNAs and lncRNAs are very large, and many of them have been shown changed in atrophying muscles. However, at present, only a few miRNAs and exiguous lncRNAs were investigated in depth. Our current understanding about the mechanisms of miRNAs and especially the lncRNAs are still very limited. Besides, other ncRNAs such as circular RNAs are emerging as the vital regulators of various diseases. One recent RNA sequencing has identified 236 circular RNAs which were differentially expressed in the mouse gastrocnemius muscle after nerve injury at different time points [38] . Although this sequencing data provides a theoretical basis for studying circular RNAs in denervated muscle atrophy, the roles of circular RNAs in muscle atrophy are still unknown [38]. In the immediate future of ncRNA study, deciphering more important ncRNAs in muscle atrophy and uncovering their intrinsic mechanisms are highly needed, which will enhance our ability to gain a better understanding of muscle atrophy and provide novel diagnosis markers and therapeutic targets.
References
Ruegg MA, Glass DJ (2011) Molecular mechanisms and treatment options for muscle wasting diseases. Annu Rev Pharmacol Toxicol 51:373–395. https://doi.org/10.1146/annurev-pharmtox-010510-100537
Mizuno H, Nakamura A, Aoki Y, Ito N, Kishi S, Yamamoto K, Sekiguchi M, Takeda S, Hashido K (2011) Identification of muscle-specific microRNAs in serum of muscular dystrophy animal models: promising novel blood-based markers for muscular dystrophy. PLoS One 6(3):e18388. https://doi.org/10.1371/journal.pone.0018388
Perfetti A, Greco S, Bugiardini E, Cardani R, Gaia P, Gaetano C, Meola G, Martelli F (2014) Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul Disord 24(6):509–515. https://doi.org/10.1016/j.nmd.2014.02.005
Wang XH, Du J, Klein JD, Bailey JL, Mitch WE (2009) Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int 76(7):751–759. https://doi.org/10.1038/ki.2009.260
Gordon BS, Kelleher AR, Kimball SR (2013) Regulation of muscle protein synthesis and the effects of catabolic states. Int J Biochem Cell Biol 45(10):2147–2157. https://doi.org/10.1016/j.biocel.2013.05.039
Bonaldo P, Sandri M (2013) Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6(1):25–39. https://doi.org/10.1242/dmm.010389
Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, Ross JA, Guttridge DC, Lundell L, Fearon KC, Timmons JA (2010) Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med 2(1):1. https://doi.org/10.1186/gm122
von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD (2017) Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol 14(6):323–341. https://doi.org/10.1038/nrcardio.2017.51
Yu Y, Li X, Liu L, Chai J, Haijun Z, Chu W, Yin H, Ma L, Duan H, Xiao M (2016) miR-628 promotes burn-induced skeletal muscle atrophy via targeting IRS1. Int J Biol Sci 12(10):1213–1224. https://doi.org/10.7150/ijbs.15496
Haijun Z, Yonghui Y, Jiake C, Hongjie D (2015) Detection of the MicroRNA expression profile in skeletal muscles of burn trauma at the early stage in rats. Ulus Travma Acil Cerrahi Derg 21(4):241–247. https://doi.org/10.5505/tjtes.2015.80707
Verdijk LB, Dirks ML, Snijders T, Prompers JJ, Beelen M, Jonkers RA, Thijssen DH, Hopman MT, Van Loon LJ (2012) Reduced satellite cell numbers with spinal cord injury and aging in humans. Med Sci Sports Exerc 44(12):2322–2330. https://doi.org/10.1249/MSS.0b013e3182667c2e
Gao Y, Arfat Y, Wang H, Goswami N (2018) Muscle atrophy induced by mechanical unloading: mechanisms and potential countermeasures. Front Physiol 9:235. https://doi.org/10.3389/fphys.2018.00235
Cohen S, Nathan JA, Goldberg AL (2015) Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14(1):58–74. https://doi.org/10.1038/nrd4467
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233. https://doi.org/10.1016/j.cell.2009.01.002
Shukla GC, Singh J, Barik S (2011) MicroRNAs: processing, maturation, target recognition and regulatory functions. Mol Cell Pharmacol 3(3):83–92
Kim VN (2005) MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 6(5):376–385. https://doi.org/10.1038/nrm1644
Yates LA, Norbury CJ, Gilbert RJ (2013) The long and short of microRNA. Cell 153(3):516–519. https://doi.org/10.1016/j.cell.2013.04.003
Chekulaeva M, Filipowicz W (2009) Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol 21(3):452–460. https://doi.org/10.1016/j.ceb.2009.04.009
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136(4):642–655. https://doi.org/10.1016/j.cell.2009.01.035
O’Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, Swanson MS, Harfe BD (2007) Essential role for Dicer during skeletal muscle development. Dev Biol 311(2):359–368. https://doi.org/10.1016/j.ydbio.2007.08.032
Haramati S, Chapnik E, Sztainberg Y, Eilam R, Zwang R, Gershoni N, McGlinn E, Heiser PW, Wills AM, Wirguin I, Rubin LL, Misawa H, Tabin CJ, Brown R Jr, Chen A, Hornstein E (2010) miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci U S A 107(29):13111–13116. https://doi.org/10.1073/pnas.1006151107
Neppl RL, Kataoka M, Wang DZ (2014) Crystallin-alphaB regulates skeletal muscle homeostasis via modulation of argonaute2 activity. J Biol Chem 289(24):17240–17248. https://doi.org/10.1074/jbc.M114.549584
Sakamoto S, Aoki K, Higuchi T, Todaka H, Morisawa K, Tamaki N, Hatano E, Fukushima A, Taniguchi T, Agata Y (2009) The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol 29(13):3754–3769. https://doi.org/10.1128/MCB.01836-08
Todaka H, Higuchi T, Yagyu K, Sugiyama Y, Yamaguchi F, Morisawa K, Ono M, Fukushima A, Tsuda M, Taniguchi T, Sakamoto S (2015) Overexpression of NF90-NF45 represses myogenic MicroRNA biogenesis, resulting in development of skeletal muscle atrophy and centronuclear muscle fibers. Mol Cell Biol 35(13):2295–2308. https://doi.org/10.1128/MCB.01297-14
Wang GK, Zhu JQ, Zhang JT, Li Q, Li Y, He J, Qin YW, Jing Q (2010) Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J 31(6):659–666. https://doi.org/10.1093/eurheartj/ehq013
Cacchiarelli D, Legnini I, Martone J, Cazzella V, D’Amico A, Bertini E, Bozzoni I (2011) miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol Med 3(5):258–265. https://doi.org/10.1002/emmm.201100133
Koutsoulidou A, Kyriakides TC, Papadimas GK, Christou Y, Kararizou E, Papanicolaou EZ, Phylactou LA (2015) Elevated muscle-specific miRNAs in serum of myotonic dystrophy patients relate to muscle disease progress. PLoS One 10(4):e0125341. https://doi.org/10.1371/journal.pone.0125341
Lewis A, Riddoch-Contreras J, Natanek SA, Donaldson A, Man WD, Moxham J, Hopkinson NS, Polkey MI, Kemp PR (2012) Downregulation of the serum response factor/miR-1 axis in the quadriceps of patients with COPD. Thorax 67(1):26–34. https://doi.org/10.1136/thoraxjnl-2011-200309
Wang F, Wang J, He J, Li W, Li J, Chen S, Zhang P, Liu H, Chen X (2017) Serum miRNAs miR-23a, 206, and 499 as potential biomarkers for skeletal muscle atrophy. Biomed Res Int 2017:8361237. https://doi.org/10.1155/2017/8361237
Catapano F, Zaharieva I, Scoto M, Marrosu E, Morgan J, Muntoni F, Zhou H (2016) Altered levels of MicroRNA-9, −206, and −132 in spinal muscular atrophy and their response to antisense oligonucleotide therapy. Mol Ther Nucleic Acids 5(7):e331. https://doi.org/10.1038/mtna.2016.47
Soares RJ, Cagnin S, Chemello F, Silvestrin M, Musaro A, De Pitta C, Lanfranchi G, Sandri M (2014) Involvement of microRNAs in the regulation of muscle wasting during catabolic conditions. J Biol Chem 289(32):21909–21925. https://doi.org/10.1074/jbc.M114.561845
Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S, Flanigan KM, Neely LA, Whitney D, Beggs AH, Kohane IS, Kunkel LM (2007) Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci U S A 104(43):17016–17021. https://doi.org/10.1073/pnas.0708115104
Fritegotto C, Ferrati C, Pegoraro V, Angelini C (2017) Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol Sci 38(4):619–625. https://doi.org/10.1007/s10072-017-2811-2
Zhang J, Fu SL, Liu Y, Liu YL, Wang WJ (2015) Analysis of MicroRNA expression profiles in weaned pig skeletal muscle after lipopolysaccharide challenge. Int J Mol Sci 16(9):22438–22455. https://doi.org/10.3390/ijms160922438
Hauser CA, Stockler MR, Tattersall MH (2006) Prognostic factors in patients with recently diagnosed incurable cancer: a systematic review. Support Care Cancer 14(10):999–1011. https://doi.org/10.1007/s00520-006-0079-9
Lee DE, Brown JL, Rosa-Caldwell ME, Blackwell TA, Perry RA Jr, Brown LA, Khatri B, Seo D, Bottje WG, Washington TA, Wiggs MP, Kong BW, Greene NP (2017) Cancer cachexia-induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol Genomics 49(5):253–260. https://doi.org/10.1152/physiolgenomics.00006.2017
Khayrullin A, Smith L, Mistry D, Dukes A, Pan YA, Hamrick MW (2016) Chronic alcohol exposure induces muscle atrophy (myopathy) in zebrafish and alters the expression of microRNAs targeting the Notch pathway in skeletal muscle. Biochem Biophys Res Commun 479(3):590–595. https://doi.org/10.1016/j.bbrc.2016.09.117
Weng J, Zhang P, Yin X, Jiang B (2018) The whole transcriptome involved in denervated muscle atrophy following peripheral nerve injury. Front Mol Neurosci 11:69. https://doi.org/10.3389/fnmol.2018.00069
Li G, Li QS, Li WB, Wei J, Chang WK, Chen Z, Qiao HY, Jia YW, Tian JH, Liang BS (2016) miRNA targeted signaling pathway in the early stage of denervated fast and slow muscle atrophy. Neural Regen Res 11(8):1293–1303. https://doi.org/10.4103/1673-5374.189195
Di Pietro L, Baranzini M, Berardinelli MG, Lattanzi W, Monforte M, Tasca G, Conte A, Logroscino G, Michetti F, Ricci E, Sabatelli M, Bernardini C (2017) Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci Rep 7(1):9538. https://doi.org/10.1038/s41598-017-10161-z
Waller R, Goodall EF, Milo M, Cooper-Knock J, Da Costa M, Hobson E, Kazoka M, Wollff H, Heath PR, Shaw PJ, Kirby J (2017) Serum miRNAs miR-206, 143-3p and 374b-5p as potential biomarkers for amyotrophic lateral sclerosis (ALS). Neurobiol Aging 55:123–131. https://doi.org/10.1016/j.neurobiolaging.2017.03.027
Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc-Groselj L, Ristic Kovacic S, Curk T, Rogelj B (2018) Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci Rep 8(1):5609. https://doi.org/10.1038/s41598-018-23139-2
Boon H, Sjogren RJ, Massart J, Egan B, Kostovski E, Iversen PO, Hjeltnes N, Chibalin AV, Widegren U, Zierath JR (2015) MicroRNA-208b progressively declines after spinal cord injury in humans and is inversely related to myostatin expression. Physiol Rep 3(11). https://doi.org/10.14814/phy2.12622
Shen H, Liu T, Fu L, Zhao S, Fan B, Cao J, Li X (2013) Identification of microRNAs involved in dexamethasone-induced muscle atrophy. Mol Cell Biochem 381(1–2):105–113. https://doi.org/10.1007/s11010-013-1692-9
Liu C, Wang M, Chen M, Zhang K, Gu L, Li Q, Yu Z, Li N, Meng Q (2017) miR-18a induces myotubes atrophy by down-regulating IgfI. Int J Biochem Cell Biol 90:145–154. https://doi.org/10.1016/j.biocel.2017.07.020
Hudson MB, Rahnert JA, Zheng B, Woodworth-Hobbs ME, Franch HA, Price SR (2014) miR-182 attenuates atrophy-related gene expression by targeting FoxO3 in skeletal muscle. Am J Physiol Cell Physiol 307(4):C314–C319. https://doi.org/10.1152/ajpcell.00395.2013
Kennedy WR, Alter M, Sung JH (1998) Progressive proximal spinal and bulbar muscular atrophy of late onset: a sex-linked recessive trait. Neurology 50(3): 583 and 510 pages following
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352(6330):77–79. https://doi.org/10.1038/352077a0
Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A (1989) X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain 112(Pt 1):209–232
Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, Huang Z, Doi H, Matsumoto S, Kondo N, Iida M, Tohnai G, Tanaka F, Muramatsu S, Sobue G (2012) Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat Med 18(7):1136–1141. https://doi.org/10.1038/nm.2791
Pourshafie N, Lee PR, Chen KL, Harmison GG, Bott LC, Katsuno M, Sobue G, Burnett BG, Fischbeck KH, Rinaldi C (2016) MiR-298 counteracts mutant androgen receptor toxicity in spinal and bulbar muscular atrophy. Mol Ther 24(5):937–945. https://doi.org/10.1038/mt.2016.13
Kukreti H, Amuthavalli K, Harikumar A, Sathiyamoorthy S, Feng PZ, Anantharaj R, Tan SL, Lokireddy S, Bonala S, Sriram S, McFarlane C, Kambadur R, Sharma M (2013) Muscle-specific microRNA1 (miR1) targets heat shock protein 70 (HSP70) during dexamethasone-mediated atrophy. J Biol Chem 288(9):6663–6678. https://doi.org/10.1074/jbc.M112.390369
McCarthy JJ, Esser KA, Peterson CA, Dupont-Versteegden EE (2009) Evidence of MyomiR network regulation of beta-myosin heavy chain gene expression during skeletal muscle atrophy. Physiol Genomics 39(3):219–226. https://doi.org/10.1152/physiolgenomics.00042.2009
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38(2):228–233. https://doi.org/10.1038/ng1725
Huang MB, Xu H, Xie SJ, Zhou H, Qu LH (2011) Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS One 6(12):e29173. https://doi.org/10.1371/journal.pone.0029173
He Q, Qiu J, Dai M, Fang Q, Sun X, Gong Y, Ding F, Sun H (2016) MicroRNA-351 inhibits denervation-induced muscle atrophy by targeting TRAF6. Exp Ther Med 12(6):4029–4034. https://doi.org/10.3892/etm.2016.3856
Huang QK, Qiao HY, Fu MH, Li G, Li WB, Chen Z, Wei J, Liang BS (2016) MiR-206 attenuates denervation-induced skeletal muscle atrophy in rats through regulation of satellite cell differentiation via TGF-beta1, Smad3, and HDAC4 signaling. Med Sci Monit 22:1161–1170
Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR, Olson EN (2009) MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326(5959):1549–1554. https://doi.org/10.1126/science.1181046
Connolly M, Paul R, Farre-Garros R, Natanek SA, Bloch S, Lee J, Lorenzo JP, Patel H, Cooper C, Sayer AA, Wort SJ, Griffiths M, Polkey MI, Kemp PR (2018) miR-424-5p reduces ribosomal RNA and protein synthesis in muscle wasting. J Cachexia Sarcopenia Muscle 9(2):400–416. https://doi.org/10.1002/jcsm.12266
Li J, Chan MC, Yu Y, Bei Y, Chen P, Zhou Q, Cheng L, Chen L, Ziegler O, Rowe GC, Das S, Xiao J (2017) miR-29b contributes to multiple types of muscle atrophy. Nat Commun 8:15201. https://doi.org/10.1038/ncomms15201
Wang B, Zhang C, Zhang A, Cai H, Price SR, Wang XH (2017) MicroRNA-23a and MicroRNA-27a mimic exercise by ameliorating CKD-induced muscle atrophy. J Am Soc Nephrol 28(9):2631–2640. https://doi.org/10.1681/ASN.2016111213
Wada S, Kato Y, Okutsu M, Miyaki S, Suzuki K, Yan Z, Schiaffino S, Asahara H, Ushida T, Akimoto T (2011) Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J Biol Chem 286(44):38456–38465. https://doi.org/10.1074/jbc.M111.271270
Hudson MB, Woodworth-Hobbs ME, Zheng B, Rahnert JA, Blount MA, Gooch JL, Searles CD, Price SR (2014) miR-23a is decreased during muscle atrophy by a mechanism that includes calcineurin signaling and exosome-mediated export. Am J Physiol Cell Physiol 306(6):C551–C558. https://doi.org/10.1152/ajpcell.00266.2013
Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, Dorn GW 2nd, Thum T, Heymans S (2015) Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol 12(7):415–425. https://doi.org/10.1038/nrcardio.2015.55
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, Yang X, Amit I, Meissner A, Regev A, Rinn JL, Root DE, Lander ES (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477(7364):295–300. https://doi.org/10.1038/nature10398
Kapusta A, Feschotte C (2014) Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet 30(10):439–452. https://doi.org/10.1016/j.tig.2014.08.004
Ulitsky I, Bartel DP (2013) lincRNAs: genomics, evolution, and mechanisms. Cell 154(1):26–46. https://doi.org/10.1016/j.cell.2013.06.020
Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4(9):721–726. https://doi.org/10.1038/nmeth1079
Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP (2010) Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal 3(107):ra8. https://doi.org/10.1126/scisignal.2000568
Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL (2012) Long noncoding RNAs with snoRNA ends. Mol Cell 48(2):219–230. https://doi.org/10.1016/j.molcel.2012.07.033
Hube F, Velasco G, Rollin J, Furling D, Francastel C (2011) Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res 39(2):513–525. https://doi.org/10.1093/nar/gkq833
Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, Fuller-Pace FV, Hoffman EP, Tapscott SJ, Sartorelli V (2006) The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell 11(4):547–560. https://doi.org/10.1016/j.devcel.2006.08.003
Dey BK, Pfeifer K, Dutta A (2014) The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28(5):491–501. https://doi.org/10.1101/gad.234419.113
Mueller AC, Cichewicz MA, Dey BK, Layer R, Reon BJ, Gagan JR, Dutta A (2015) MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol Cell Biol 35(3):498–513. https://doi.org/10.1128/MCB.01079-14
Gong C, Li Z, Ramanujan K, Clay I, Zhang Y, Lemire-Brachat S, Glass DJ (2015) A long non-coding RNA, LncMyoD, regulates skeletal muscle differentiation by blocking IMP2-mediated mRNA translation. Dev Cell 34(2):181–191. https://doi.org/10.1016/j.devcel.2015.05.009
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147(2):358–369. https://doi.org/10.1016/j.cell.2011.09.028
Zhu M, Liu J, Xiao J, Yang L, Cai M, Shen H, Chen X, Ma Y, Hu S, Wang Z, Hong A, Li Y, Sun Y, Wang X (2017) Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat Commun 8:14718. https://doi.org/10.1038/ncomms14718
Zhang ZK, Li J, Guan D, Liang C, Zhuo Z, Liu J, Lu A, Zhang G, Zhang BT (2018) A newly identified lncRNA MAR1 acts as a miR-487b sponge to promote skeletal muscle differentiation and regeneration. J Cachexia Sarcopenia Muscle 9:613. https://doi.org/10.1002/jcsm.12281
Zhou L, Sun K, Zhao Y, Zhang S, Wang X, Li Y, Lu L, Chen X, Chen F, Bao X, Zhu X, Wang L, Tang LY, Esteban MA, Wang CC, Jauch R, Sun H, Wang H (2015) Linc-YY1 promotes myogenic differentiation and muscle regeneration through an interaction with the transcription factor YY1. Nat Commun 6:10026. https://doi.org/10.1038/ncomms10026
Militello G, Hosen MR, Ponomareva Y, Gellert P, Weirick T, John D, Hindi SM, Mamchaoui K, Mouly V, Doring C, Zhang L, Nakamura M, Kumar A, Fukada SI, Dimmeler S, Uchida S (2018) A novel long non-coding RNA Myolinc regulates myogenesis through TDP-43 and Filip1. J Mol Cell Biol 10:102. https://doi.org/10.1093/jmcb/mjy025
Wang L, Zhao Y, Bao X, Zhu X, Kwok YK, Sun K, Chen X, Huang Y, Jauch R, Esteban MA, Sun H, Wang H (2015) LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res 25(3):335–350. https://doi.org/10.1038/cr.2015.21
Wang J, Gong C, Maquat LE (2013) Control of myogenesis by rodent SINE-containing lncRNAs. Genes Dev 27(7):793–804. https://doi.org/10.1101/gad.212639.112
Lu L, Sun K, Chen X, Zhao Y, Wang L, Zhou L, Sun H, Wang H (2013) Genome-wide survey by ChIP-seq reveals YY1 regulation of lincRNAs in skeletal myogenesis. EMBO J 32(19):2575–2588. https://doi.org/10.1038/emboj.2013.182
Ballarino M, Cazzella V, D’Andrea D, Grassi L, Bisceglie L, Cipriano A, Santini T, Pinnaro C, Morlando M, Tramontano A, Bozzoni I (2015) Novel long noncoding RNAs (lncRNAs) in myogenesis: a miR-31 overlapping lncRNA transcript controls myoblast differentiation. Mol Cell Biol 35(4):728–736. https://doi.org/10.1128/MCB.01394-14
Han X, Yang F, Cao H, Liang Z (2015) Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J 29(7):3054–3064. https://doi.org/10.1096/fj.14-259952
Wang GQ, Wang Y, Xiong Y, Chen XC, Ma ML, Cai R, Gao Y, Sun YM, Yang GS, Pang WJ (2016) Sirt1 AS lncRNA interacts with its mRNA to inhibit muscle formation by attenuating function of miR-34a. Sci Rep 6:21865. https://doi.org/10.1038/srep21865
Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P (2010) Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One 5(10):e13604. https://doi.org/10.1371/journal.pone.0013604
Lorson CL, Hahnen E, Androphy EJ, Wirth B (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 96(11):6307–6311
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165
d’Ydewalle C, Ramos DM, Pyles NJ, Ng SY, Gorz M, Pilato CM, Ling K, Kong L, Ward AJ, Rubin LL, Rigo F, Bennett CF, Sumner CJ (2017) The antisense transcript SMN-AS1 regulates SMN expression and is a novel therapeutic target for spinal muscular atrophy. Neuron 93(1):66–79. https://doi.org/10.1016/j.neuron.2016.11.033
Woo CJ, Maier VK, Davey R, Brennan J, Li G, Brothers J 2nd, Schwartz B, Gordo S, Kasper A, Okamoto TR, Johansson HE, Mandefro B, Sareen D, Bialek P, Chau BN, Bhat B, Bullough D, Barsoum J (2017) Gene activation of SMN by selective disruption of lncRNA-mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proc Natl Acad Sci U S A 114(8):E1509–E1518. https://doi.org/10.1073/pnas.1616521114
Haijun Z, Yonghui Y, Jiake C (2016) Expression signatures of lncRNAs in skeletal muscles at the early flow phase revealed by microarray in burned rats. Ulus Travma Acil Cerrahi Derg 22(3):224–232. https://doi.org/10.5505/tjtes.2015.04831
Acknowledgments
This work was supported by the grants from National Natural Science Foundation of China (81722008, 91639101 and 81570362 to JJ Xiao), Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to JJ Xiao), the grant from Science and Technology Commission of Shanghai Municipality (17010500100 to JJ Xiao), and the development fund for Shanghai talents (to JJ Xiao).
Competing Financial Interests
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Li, Y., Meng, X., Li, G., Zhou, Q., Xiao, J. (2018). Noncoding RNAs in Muscle Atrophy. In: Xiao, J. (eds) Muscle Atrophy. Advances in Experimental Medicine and Biology, vol 1088. Springer, Singapore. https://doi.org/10.1007/978-981-13-1435-3_11
Download citation
DOI: https://doi.org/10.1007/978-981-13-1435-3_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1434-6
Online ISBN: 978-981-13-1435-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)