
Abstract
MicroRNAs (miRNAs), a novel class of post-transcriptional gene regulators, have been demonstrated to be involved in several cellular processes regulating the expression of protein-coding genes. To investigate the mechanisms of miRNA-mediated regulation during the process of muscle atrophy, we performed miRNA microarray hybridization between normal differentiated C2C12 cells and dexamethasone (DEX)-treated C2C12 cells. We observed that 11 miRNAs were significantly up-regulated and six miRNAs were down-regulated in the differentiated C2C12 cells after being treated with DEX. Stem–loop real-time RT-PCR confirmed the differential expression of six selected miRNAs (miR-1, miR-147, miR-322, miR-351, and miR-503*, miR-708). miRNA potential target prediction was accomplished using TargetScan, and many target genes related to muscle growth and atrophy have been reported in previous studies. The results of the current study suggested the potential roles of these differentially expressed miRNAs in skeletal muscle atrophy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Skeletal muscle growth and function are determined by the balance of protein synthesis and breakdown. The increase in skeletal muscle protein catabolism and decrease in protein anabolism will result in skeletal muscle atrophy. Muscle atrophy is characterized by a decrease in muscle mass, protein content, or decreased fiber number, and loss of strength [1]. These phenomena are observed in many physiological and pathological conditions such as microgravity [2], critical illness [3, 4], HIV [5, 6], cancer cachexia [7, 8], and aging [9]. The molecular mechanism of skeletal muscle atrophy is a complex process, which involves many complex signaling pathways. Various animal models have indicated that the ATP-dependent ubiquitin–proteasome pathway (UPP) is primarily responsible for the regulation of skeletal muscle protein degradation in atrophy conditions [10–12]. Other signaling pathways such as IGF1/PI3 K/Akt/Foxo [13], IGF1/PI3 K/Akt/mTOR [14], and PI3 K/AKT/GSK3 [15], as well as TNF-α/NF-κB/MuRF1 [16] are important mechanisms causing muscle atrophy.
MicroRNAs (miRNAs) are a class of highly conserved, endogenous approximately 21-nucleotide-long noncoding RNA molecules that can regulate gene expression at the posttranscriptional level by either degradation or translational repression of a target mRNA [17]. A number of miRNAs have been found in various species and regarded as powerful regulators of gene expression and cellular phenotype. MicroRNAs are initially transcribed as primary miRNAs (pri-miRNAs): the pri-miRNA is cleaved in the nucleus by the RNase III enzyme Dicer to produce an 70-nt long precursor miRNA (pre-miRNA) which has a short stem–loop structure [18]. The pre-miRNA is further cleaved by another RNase III enzyme Dicer to produce the mature form of the miRNA [19]. The mature miRNA then binds to the RNA-induced silencing complex (RISC) which contains multiple proteins, guiding the RISC interaction with target mRNA sequences, most favorably at the 3′ untranslated region (3′UTR), suppressing the process of translation and/or causing the degradation of the targeted mRNA [17]. Growing evidence suggests that miRNAs participate in a series of biological processes such as cell proliferation, apoptosis, immune response, as well as many diseases [20]. In recent studies, several miRNAs have been reported to play important roles in the control of muscle growth and differentiation. Chen et al. [21] found that miR-1 and miR-133 can promote differentiation and proliferation of the cardiac and skeletal muscle cells. Both miR-1 and miR-133 have been identified to participate in cardiac hypertrophy in vitro and in vivo [22, 23].
Dexamethasone (DEX) is the most potent synthetic glucocorticoid, which has purer glucocorticoid activity compared with the naturally occurring cortisol and corticosterone. DEX has been considered as an effective agent to induce the muscle atrophy in vitro or in vivo because of its potential ability to stimulate protein degradation. In this study, we investigated the differential expressions of miRNAs between the normal and DEX-treated C2C12 cells by a mammalian microRNA microarray and discovered the miRNAs which were differentially expressed between the normal and atrophy conditions. The genes functions of which have been demonstrated in skeletal muscle growth and atrophy were predicted to be targets of the differentially expressed miRNAs. These data provide the candidates of miRNA leading us to a better understanding of the molecular pathways regulating skeletal muscle atrophy.
Materials and methods
Cell culture and treatment
Murine C2C12 myoblast cells were maintained in six-well plates at ~80 % confluence in growth media containing Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Gaithersburg, MD) with 1.0 g/l glucose, l-glutamine, penicillin/streptomycin (100 μg/ml) and 10 % FBS (fetal bovine serum). Cells were grown in a humidified 37 °C incubator with 5 % CO2. In order to get a better differentiation potential of C2C12 myoblast, cells were used at low passage (between passages 2 and 4) and induced differentiation by lowering the serum concentration to 2 % HS (horse serum) when reaching about 90 % confluence. C2C12 myotubes, which were differentiated for 72 h, were exposed to DMEM including 50 nM/ml DEX for 48 h. Meanwhile, the cells in the control group were treated with differentiation medium containing 2 % HS. Formation of myotubes was photographed with a Nikon Eclipse TE2000-S inverted microscope (Nikon Instrument, Melville, NY) at 200× magnification. Then, the cells were harvested for RNA isolation. DEX (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 70 % ethanol and diluted to the final treatment concentration using Dulbecco’s Modified Eagle Medium (DMEM). The final concentration of ethanol in medium was <0.07 %. Total RNA was extracted from the C2C12 myotubes using Trizol reagent (Invitrogen, Carlsbad, CA, USA) as described by the manufacturer.
Real-time quantitative PCR for Foxo and MuRF1 gene expression
To identify if we induced the differentiated C2C12 cells into atrophy condition successfully after being treated with the DEX, SYBR Green I based real-time quantitative PCR (RT-qPCR) was employed to evaluate the expression difference of Foxo1, Foxo3a, and MuRF1, three genes which have been identified to be up-regulated during muscle atrophy [11, 13].
The reverse transcription of 2 μg total RNA (three samples for each condition) was performed as described previously [24]. Each RT-qPCR system (20 μl) contained 10 μl 2× SYBR Green master buffer (Toyobo, Osaka, Japan), 0.6 μM of each express primer (Table 1), and 1 μl template cDNA. The PCR reaction was completed on the Roche LightCycler® 480 Real-Time PCR System (Roche, Basel, Switzerland) according to the following program: 95 °C for 2 min; followed by 40 cycles of 94 °C for 20 s, 62 °C for 20 s, and 72 °C for 15 s; then the dissociation curves were generated to insure that a single amplicon was produced for each gene. For each sample, reactions were set up in triplicate to insure the reproducibility of the results, and then the mean Ct value of each triplicate was used for further calculations. The gene expression levels of mouse, Foxo1, Foxo3a, and MuRF1 genes were quantified relative to the expression of 18S by employing the comparative Ct (or ΔCt) value method [25]. The ΔCt values were calculated, and student’s t test was conducted on these averages to identify the expression difference.
Microarray hybridization and data analysis
The total RNA samples described above were sent to a commercial service for hybridization to the Affymetrix GeneChip® miRNA Array, which contains microRNA annotated in Sanger miRNA database version 11 (http://www.mirbase.org/) of 71 organisms and additional human small nuclear RNAs. In brief, one microgram of total RNA from each sample was labeled with the FlashTag Biotin RNA Labeling Kit for Affymetrix GeneChip® miRNA Array (Genisphere, Hatfield, PA, USA). After labeling, the samples were hybridized on Affymetrix GeneChip miRNA arrays according to the manufacturer’s recommendations (Affymetrix, Santa Clara, CA, USA). Hybridization, washing, and scanning of the slides were done according to Affymetrix’s recommendations (Fluidics Protocol FS450_0003). The obtained raw signal intensity files (CEL files) were imported into Partek GS 6.5 software (Partek Inc., St. Louis, MO, USA) and converted to normalized gene signal files by means of the log scale robust multi-array analysis (RMA) [26], which contains four main steps: background correction, quantile normalization, log2 transformation, and median polish summarization. Between-subjects t tests were conducted using the TIGR MultiExperiment Viewer v.4.6 [27], and the raw p values were adjusted by the Benjamini–Hochberg procedure, which controls the false discovery rate (FDR) [28]. The criteria for significance of differentially expressed miRNAs were established as fold change >2 with a Benjamini–Hochberg-corrected p value <0.05. Unsupervised hierarchical clustering analysis for the differentially expressed miRNAs was performed using the software package cluster 3.0. Correlation (uncentered) similarity metric and average linkage algorithm was used in the cluster analysis. The result of cluster was displayed with the program Tree View 1.6.
Design of primers for miRNA RT-PCR
Primer pairs and miRNA sequences used for miRNA RT-PCR are presented in Table 1. The primers for miRNA RT-PCR were designed based on the sequences of miRNA following the method described by Chen et al. [29]. Primers for the RT-PCR included a stem–loop RT primer containing 4–6 nts at the 3′ end complementary to the miRNA molecule, a miRNA-specific forward primer, and a universal reverse primer. Synthetic miRNA oligonucleotides were purchased from Invitrogen (Invitrogen Inc, Shanghai, China).
Real-time quantitative RT-PCR for miRNA expression
A miRNA quantification method, stem–loop reverse transcription followed by SYBR Green Real-time PCR analysis, was used to validate the microarray data. Three independent samples from control and treated myotubes were analyzed. cDNA was synthesized from total RNA using miRNA-specific stem–loop RT primers according to the protocol of our previous studies [30]. Real-time PCR reaction mixture (20 μl) contained 10 μl 2× SYBR Green master buffer (Toyobo, Osaka, Japan), 0.6 μM of each miRNA-specific primer (Table 2) and 1 μl template cDNA. The PCR reaction was completed on the Roche LightCycler® 480 Real-Time PCR System (Roche, Basel, Switzerland) according to the following program: 95 °C for 2 min; Followed by 40 cycles of 94 °C for 20 s, 62 °C for 20 s, and 72 °C for 15 s; then the dissociation curves were generated to insure that a single amplicon was produced for each gene. U6 snRNA (U6 small nuclear RNA) was used as the internal control. The comparative Ct (or ΔCt) value method was used to determine the differences in the expression levels between the DEX-treated group and the control group [25]. The differences in miRNA expression levels between groups were compared using the Student’s t test. A p value <0.05 was considered significant.
Prediction of miRNA target genes
Prediction of putative targets of each miRNA was performed by an available algorithm: TargetScan release version 5.2 (http://www.targetscan.org/), which uses a computational algorithm to identify the predicted mRNA. The predicted genes were sorted based on the context scores. Genes giving absolute values of the context scores of more than 0.20 were pooled as target candidates. Pathway analysis for the predicted target genes was performed using the DAVID (Database for Annotation, Visualization and Integrated Discovery) web-based tool (http://david.abcc.ncifcrf.gov/).
Results
Images obtained from C2C12 skeletal muscle cells cultured for 72 h in horse serum alone, 120 h of horse serum alone, and 72 h of horse serum, followed by an additional 48 h plus 50 nM/ml DEX. Addition of DEX resulted in a distinct atrophic phenotype in differentiated C2C12 myotubes (Fig. 1). To identify if we induced the differentiated C2C12 cells into atrophy condition after being treated with DEX successfully, we evaluated the expression difference of Foxo1, Foxo3a, and MuRF1 gene by means of RT-qPCR analysis. The results showed that these three genes’ mRNA expressions in differentiated C2C12 cells were significantly up-regulated after being treated with DEX (p < 0.05) (Fig. 2). The results also indicated that the differentiated myotubes were induced atrophy successfully and the RNA extracted from these cells could be used for miRNA Array analysis.

Phenotypic changes in C2C12 myotubes following DEX treatment. a C2C12 cells cultured for 72 h of horse serum. b C2C12 cells cultured for 72 h of horse serum followed by an additional 48 h DEX. c C2C12 cells cultured for 120 h of horse serum

Gene expression in differentiated C2C12 cells after being treated with DEX. a Foxo1, b Foxo3a, and c MuRF1 were measured by real-time quantitative PCR to validate the atrophy status. The relative expression level was measured with 2−ΔΔCt value and 18SrRNA was used as a reference gene. Each bar represents the mean of three independent experiments with standard deviation (SD). Asterisk represents significantly difference (p < 0.05)
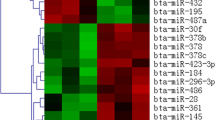
The Affymetirx miRNA chip used for detection of the expression of miRNA contains 6,703 probe sets from 71 species in total, which includes 847 human miRNAs, 609 mouse miRNAs, 351 rat miRNAs and 54 pig miRNAs. There are quadruplicate probes for each miRNA. We compared the expression of these miRNAs between the DEX-treated group and the control group and a total of 17 mmu-miRNAs were differentially expressed with a fold change >2.0 and p value <0.05. In detail, we identified 11 up- and six down-regulated miRNAs in differentiated C2C12 cells after being treated with DEX. The up-regulated miRNAs included miR-133a*, miR-155, miR-192, miR-1, miR-322, miR-351, miR-466f, miR-466j, miR-503*, miR-503, and miR-872. Six down-regulated miRNAs were miR-1192, miR-147, miR-466c-3p, miR-467a*, miR-708, and miR-7a. A clear expression pattern emerged after unsupervised hierarchical clustering analysis of the differentially expressed miRNAs (Fig. 3). A set of six miRNAs with similar expression pattern belonged to the down-regulated group, and another set of 11 miRNAs belonged to the up-regulated group.

Unsupervised hierarchical clustering and tree–view analyses of differentially expressed miRNAs between normal differentiated C2C12 cells and C2C12 cells under atrophic condition. Correlation (uncentered) similarity metric and average linkage algorithm was used in the cluster analysis. Each row represents an individual miRNA, and each column represents a sample. The dendrogram at the top of the image displays similarity of expression among these samples in the same status. Color legend represents the miRNA expression is at the bottom; red indicates high expression levels, whereas green indicates low expression levels. The codes on the legend are log2-transformed values. (Color figure online)
To further validate our microarray results, we examined the expressions of six of the 17 differentially expressed miRNAs (miR-147, miR-708, miR-1, miR-322, miR-351, and miR-503*) by applying the stem–loop RT-PCR based real-time quantitative PCR method (RT-qPCR). The results are shown in Fig. 4, and the results indicated that the expression patterns of the six miRNAs were consistent with the microarray data.

Validation of differentially expressed miRNAs by real-time quantitative PCR analyses. The relative expression level of the six microRNAs (a–f) were measured with 2−ΔΔCt value and U6 snRNA was used as a reference gene. Each bar represents the mean of three independent experiments with standard deviation (SD). Asterisk represents significantly difference (p < 0.05)
Information from miRBase (http://mirbase.org/) showed that several miRNAs are clustered in the genome; we found that mmu-miR-322, mmu-miR-351, and mmu-miR-503, three up-regulated miRNAs, were located in a cluster of miRNA, within a 10-kb sequence on chromosome X (50 401 255-50 407 432). Four other miRNAs, mmu-miR-450a-1, mmu-miR-450a-2, mmu-miR-450b, and mmu-miR-542 are also located in this cluster region. However, no differences in the expression levels of these miRNAs were found in the microRNA array analysis.
Each miRNA can regulate numerous target genes and therefore has the potential to modulate multiple pathways. To explore what targets and pathways may be regulated by the muscle atrophy-associated miRNAs, an available algorithm, TargetScan5.2, was used to predict targets for each miRNA that we identified in this study. By using the target gene prediction tool and the pathway analysis tool, many potential pathways were predicted; the details of the prediction results are shown in Table S1 (Supplementary material). Hundreds of potential targets, including Foxp1, IGF1, SP2, MAP4K3, MAP3K1, Dicer1, PTEN, Foxo3a, and GRK6, were predicted in TargetScan5.2. These potential targets included transcription factors and genes related to DNA repair, cell cycle regulation, microRNA, and signal transduction. Among those signaling pathways, insulin signaling pathway, ubiquitin-mediated proteolysis pathway, MAPK signaling pathway, and mTOR signaling pathway were reported to be involved in the process of muscle growth and atrophy in previous studies.
Discussion
miRNAs constitute a recently identified class of small endogenous noncoding RNAs that act as negative regulators of the protein-coding gene expression and may play roles in both physiological and pathological conditions, including cell division, cell death, differentiation, metabolism, disease, and cancer [31–35]. In the present study, we aimed to analyze the miRNA expression profiles in differentiated C2C12 cells under atrophic condition, and to develop further understanding of the molecular mechanisms involved in the process of muscle atrophy.
By means of the Affymetrix GeneChip® miRNA Array, we observed that the expressions of 17 miRNAs were significantly changed, including 11 up-regulated and six down-regulated ones. Among these miRNAs, six remarkably differentially expressed miRNAs were subsequently identified by stem–loop RT-qPCR, revealing the reliability of our microarray data. The discrepancy of the results between the microarray and RT-qPCR may have been caused by two reasons: first, stem–loop RT-qPCR method is more sensitive than microarray to detect miRNA expression [29, 36]; second, microarray may detect both precursor and mature forms of miRNAs, and stem–loop RT-qPCR method is more specific for mature miRNAs expression level measurement [29, 37]. In this study, consistency between the qPCR and microarray results showed that of the microarray experiment was reliable. This consistency also indicated that the results obtained from the microarray analysis can be used for further study.
In previous studies, many miRNAs have been shown to be associated with muscle growth and development, as well as muscle disease. Eisenberg et al. [38] found that the expression level of miR-455 was significantly increased in several muscular dystrophies models, including the facioscapulohumeral muscular dystrophy (FSHD), limb girdle muscular dystrophy 2A and 2B (LGMD2A-2B), and nemaline myopathy (NM). Myostatin was well known as a negative regulator of muscle growth and control the proliferation of muscle precursor cells. The expression level of Myostatin was significantly increased in almost all muscular atrophy models. Allen et al. [39] identified that the expression of miR-27 was reduced by 50 % in glucocorticoid-induced muscular atrophy, and confirmed that Myostatin was a target gene of miR-27. In this study, an interesting microRNA which related to skeletal muscle development, miR-1, was significantly increased in DEX-induced muscle atrophy. Notably, miR-1 is a skeletal and cardiac muscle-specific microRNA necessary for postmitotic muscle growth and differentiation. Sayed et al. [23] reported that miR-1 is one of the earliest miRNAs down-regulated during muscle hypertrophy, and plausibly through a serum response factor-dependent mechanism. This finding was consistent with our result that miR-1 was significantly increased in muscle atrophy. As known, atrophy and hypertrophy are two opposite conditions that exist in muscle, and the signal pathway involved in these two opposing phenomena are linked, through regulation of the activity of a common set of molecules controlling the cellular pathways; these pathways determine whether the skeletal muscle tissue will respond to defined stimuli with increased protein synthesis and stimulation of cell growth (muscle hypertrophy), or with increased protein breakdown and reduced cell proliferation (muscle atrophy) [40]. In consideration of these results in combination, we speculate that miR-1 also takes part in the process of muscle atrophy, and because of the differential expression level of miR-1 in atrophy and hypertrophy conditions, miR-1 might have great potential to serve as a novel, noninvasive biomarker for detection of muscle atrophy or hypertrophy.
Comparative genomic analyses of miRNAs from different species suggest a variety of mechanisms regulating their expression [17]. Some miRNA genes are clustered in the genome, implying that they are transcribed as a single transcriptional unit [41, 42]. In this study, three up-regulated microRNA, mmu-miR-322, mmu-miR-351, and mmu-miR-503, were reported to locate as a cluster in mouse X chromosome [43], whereas no differential expression was found in other four miRNAs in this cluster. The differences in expression levels of miRNAs in the same cluster were also reported by others in previous studies [44, 45]. Human miR-23b is expressed at much lower levels in all tissues, compared with miR-27b, miR-189, and miR-24, all residing in the same cluster on chromosome 9 [45]. This result suggested that the expression level of each miRNA in the same cluster was controlled by additional mechanisms. In this study, we found that the expression of mmu-miR-322, mmu-miR-351, and mmu-miR-503 were up-regulated. It is indicated that these three miRNAs were involved in the induction of muscle atrophy. Sarkar et al. [46] found that miR-322/424 and 503 were induced during muscle differentiation and promote cell cycle quiescence and differentiation; they also identified that the functions of miR-322/424 and 503 were implemented by down-regulation of Cdc25A, which was a key cell cycle regulator important for the activation of the cell cycle-dependent kinases (Cdks). Therefore, our results indicate that mmu-miR-322, mmu-miR-351, and mmu-miR-503 may also work in the process of muscle atrophy by targeting some unidentified genes.
Furthermore, we predicted the target genes of these differential expressed miRNAs, and several other target genes probably involved in the miRNAs-mediated regulation network were predicted. Among these potential target genes, we noticed that many genes, MAP1A (microtubule-associated protein 1A) (miR-1), IGF1 (insulin-like growth factor 1) (miR-1), IGFBP-5 (insulin-like growth factor binding protein 5) (miR-1192), and MEF2A (myocyte enhancer factor 2A) (miR-155), have been reported to play their respective roles in muscle development and differentiation. Among these genes, IGF1 has been identified as a target gene of miR-1 through experimental verification in previous study [47]. IGF-1 induces hypertrophy by stimulating the phosphatidylinositol 3-kinase (PI3 K)-Akt pathway, resulting in the downstream activation of proteins that are required for protein synthesis [15, 48]. MEF2A is the predominant component of the MEF2 DNA binding complex in differentiating muscle cells, and miR-1 can regulate cardiomyocyte growth responses by negatively regulating the calcium-signaling components calmodulin, MEF2A, and Gata4. A recent study demonstrated that endogenously secreted IGFBP-5 was a differentiation-promoting factor in skeletal muscle cells; it may play a role in modulating the actions of IGF-II on muscle differentiation [49]. Thus, we speculated that these differentially expressed miRNAs might be associated with the growth and atrophy of mouse skeletal muscle by regulating above mentioned target genes and signaling pathways.
In summary, our results revealed an altered miRNA expressional profile between normal differentiated C2C12 cells and C2C12 cells under atrophic conditions, which suggested that these differentially expressed miRNAs might have potential relevance in the growth and atrophy of mouse skeletal muscle induced by DEX.
References
Czerwinski SM, Zak R, Kurowski TT, Falduto MT, Hickson RC (1989) Myosin heavy chain turnover and glucocorticoid deterrence by exercise in muscle. J Appl Physiol 67(6):2311–2315
Adams GR, Caiozzo VJ, Baldwin KM (2003) Skeletal muscle unweighting: spaceflight and ground-based models. J Appl Physiol 95(6):2185–2201
Di Giovanni S, Molon A, Broccolini A, Melcon G, Mirabella M, Hoffman EP (2004) Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann Neurol 55(2):195–206
Price DA, Bassendine MF, Norris SM, Golding C, Toms GL, Schmid ML, Morris CM, Burt AD, Donaldson PT (2006) Apolipoprotein epsilon3 allele is associated with persistent hepatitis C virus infection. Gut 55(5):715–718
Sever JL, Rakusan TA, Campos JM, O’Donnell RM, Price VA (1996) HIV antibody responses in children of HIV-infected mothers. Pediatr AIDS HIV Infect 7(4):246–253
Miró O, Pedrol E, Cebrián M, Masanés F, Casademont J, Mallolas J, Grau JM (1997) Skeletal muscle studies in patients with HIV-related wasting syndrome. J Neurol Sci 150(2):153–159
Baracos VE (2001) Management of muscle wasting in cancer-associated cachexia: understanding gained from experimental studies. Cancer 92(6 Suppl):1669–1677
Mitch WE, Price SR (2001) Transcription factors and muscle cachexia: is there a therapeutic target? Lancet 357(9258):734–735
Brown M, Hasser EM (1996) Complexity of age-related change in skeletal muscle. J Gerontol A Biol Sci Med Sci 51(2):B2117–B2123
Tawa NE Jr, Odessey R, Goldberg AL (1997) Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest 100:197–203
Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18:39–51
Murton AJ, Constantin D, Greenhaf PL (2008) The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim Biophys Acta 1782(12):730–743
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3 K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14(3):395–403
Pallafacehina G, Calabria E, Serrano AL (2002) A protein kinase B-dependent andrapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc Natl Acad Sci USA 14(2):432–438
Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ (2001) Mediation of IGF-1-induced skeletal myotube hypertrophy by PI3 K/Akt/mTOR and PI3 K/Akt/GSK3 pathways. Nat Cell Biol 3(11):1009–1013
Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119(2):285–298
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Kim VN (2004) MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol 14:156–159
Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21(17):4663–4670
Hwang HW, Mendell JT (2006) MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer 94(6):776–780
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38(2):228–233
Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND et al (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13(5):613–618
Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M (2007) MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100(3):416–424
Shen H, Zhao SH, Cao JH, Li XY, Fan B (2011) Porcine MuRF2 and MuRF3: molecular cloning, expression and association analysis with muscle production traits. Mol Biol Rep 38(8):5115–5123
ivak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 25(4):402–408
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31(4):e15
Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J (2006) TM4 microarray software suite. Methods Enzymol 411:134–193
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 57:289–300
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem–loop RT-PCR. Nucleic Acids Res 33(20):e179
Xie SS, Huang TH, Shen Y, Li XY, Zhang XX, Zhu MJ, Qin HY, Zhao SH (2010) Identification and characterization of microRNAs from porcine skeletal muscle. Anim Genet 41(2):179–190
Mendell JT (2005) MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle 4:1179–1184
Miska EA (2005) How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev 15:563–568
Kloosterman WP, Plasterk RH (2006) The diverse functions of microRNAs in animal development and disease. Dev Cell 11:441–450
Shivdasani RA (2006) MicroRNAs: regulators of gene expression and cell differentiation. Blood 108:3646–3653
Wiemer EA (2007) The role of microRNAs in cancer: no small matter. Eur J Cancer 43:1529–1544
Tan PK, Downey TJ, Spitznagel EL Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC (2003) Evaluation of gene expression measurements from commercial microarray platforms. NucleicAcids Res 31(19):5676–5684
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ, Schmittgen TD (2007) Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer 120:1046–1054
Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S et al (2007) Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci USA 104:17016–17021
Allen DL, Loh AS (2011) Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. Am J Physiol Cell Physiol 300(1):C124–C137
Sartorelli V, Fulco M (2004) Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy. Sci STKE 2004(244):re11
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T (2001) Identification of novel genes coding for small expressed RNAs. Science 294(5543):853–858
Lau NC, Lim LP, Weinstein EG, Bartel DP (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294(5543):858–862
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M et al (2007) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129(7):1401–1414
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V (2004) Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5(3):R13
Barad O, Meiri E, Avniel A, Aharonov R, Barzilai A, Bentwich I, Einav U, Gilad S, Hurban P, Karov Y et al (2005) MicroRNA expression detected by oligonucleotide microarrays: system establishment and expression profiling in human tissues. Genome Res 14(12):2486–2494
Sarkar S, Dey BK, Dutta A (2010) MiR-322/424 and -503 are induced during muscle differentiation and promote cell cycle quiescence and differentiation by down-regulation of Cdc25A. Mol Biol Cell 21(13):2138–2149
Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL, Zhu JN, Liu XY, Zhang YY, Li Y, Lin SG et al (2009) Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem Biophys Res Commun 81(4):597–601
Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ et al (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3(11):1014–1019
Ren H, Yin P, Duan C (2008) IGFBP-5 regulates muscle cell differentiation by binding to IGF-II and switching on the IGF-II auto-regulation loop. J Cell Biol 182(5):979–991
Acknowledgments
This study was supported by the National Outstanding Youth Foundation of NSFC (31025026), the NSFC youth foundation (30900792), the creative team project of the Ministry of Education of China (IRT-0831), and the National Key Basic Research Program of China (2012CB124702).
Conflict of interests
The authors have declared that no competing interest exists.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Shen, H., Liu, T., Fu, L. et al. Identification of microRNAs involved in dexamethasone-induced muscle atrophy. Mol Cell Biochem 381, 105–113 (2013). https://doi.org/10.1007/s11010-013-1692-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-013-1692-9