
Abstract
Overnutrition, usually with obesity and genetic predisposition, lead to insulin resistance, which is an invariable accompaniment of nonalcoholic fatty liver disease (NAFLD). The associated metabolic abnormalities, pre- or established diabetes, hypertension and atherogenic dyslipidemia (clustered as metabolic syndrome) tend to be worse for nonalcoholic steatohepatitis (NASH), revealing it as part of a continuum of metabolic pathogenesis. The origins of hepatocellular injury and lobular inflammation which distinguish NASH from simple steatosis have intrigued investigators, but it is now widely accepted that NASH results from liver lipotoxicity. The key issue is not the quantity of liver fat but the type(s) of lipid molecules that accumulate, and how they are “packaged” to avoid subcellular injury. Possible lipotoxic mediators include free (unesterified) cholesterol, saturated free fatty acids, diacylglycerols, lysophosphatidyl-choline, sphingolipids and ceramide. Lipid droplets are intracellular storage organelles for non-structural lipid whose regulation is influenced by genetic polymorphisms, such as PNPLA3. Cells unable to sequester chemically reactive lipid molecules undergo mitochondrial injury, endoplasmic reticulum (ER) stress and autophagy, all processes of interest for NASH pathogenesis. Lipotoxicity kills hepatocytes by apoptosis, a highly regulated, non-inflammatory form of cell death, but also by necrosis, necroptosis and pyroptosis; the latter involve mitochondrial injury, oxidative stress, activation of c-Jun N-terminal kinase (JNK) and release of danger-associated molecular patterns (DAMPs). DAMPs stimulate innate immunity by binding pattern recognition receptors, such as Toll-like receptor 4 (TLR4) and the NOD-like receptor protein 3 (NLRP3) inflammasome, which release a cascade of pro-inflammatory chemokines and cytokines. Thus, lipotoxic hepatocellular injury attracts inflammatory cells, particularly activated macrophages which surround ballooned hepatocytes as crown-like structures. In both experimental and human NASH, livers contain cholesterol crystals which are a second signal for NLRP3 activation; this causes interleukin (IL)-1β and IL18 secretion to attract and activate macrophages and neutrophils. Injured hepatocytes also liberate plasma membrane-derived extracellular vesicles; these have been shown to circulate in NASH and to be pro-inflammatory. The way metabolic dysfunction leads to lipotoxicity, innate immune responses and the resultant pattern of cellular inflammation in the liver are likely also relevant to hepatic fibrogenesis and hepatocarcinogenesis. Pinpointing the key molecules involved pharmacologically should eventually lead to effective pharmacotherapy against NASH.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
Nonalcoholic fatty liver disease (NAFLD) is highly prevalent (15–45%) in North and South America, Europe and the Asia-Pacific region, but only 10–25% of cases develop nonalcoholic steatohepatitis (NASH), which can lead to cirrhosis, liver failure and hepatocellular carcinoma (HCC). The pathology of NASH includes steatosis, hepatocellular degeneration/cell death (ballooning, Mallory hyaline, apoptosis), lobular mixed-cell inflammation (macrophages, neutrophils, lymphocytes) and fibrosis. NASH is more likely than simple steatosis to cause liver fibrosis. We and others have previously reviewed the relationship of steatosis to overnutrition [1,2,3,4,5,6,7], with resultant insulin resistance, hyperinsulinemia, hyperglycemia, metabolic syndrome and hypoadiponectinemia. Less is known about hepatocyte cell death and inflammatory recruitment in NASH, despite their defining importance for perpetuating liver injury, hepatic fibrogenesis and hepatocarcinogenesis [8]. Here we consider the origins of hepatocyte injury and resultant patterns of cell death and inflammatory recruitment in NASH. Lipotoxicity is central to current understanding of these processes. It is a form of tissue injury associated with inflammation and wound healing, also thought to be central to the pathogenesis of type 2 diabetes [1, 5, 9].
In lipotoxic liver injury, hepatocytes die by apoptosis, necrosis and necroptosis, and possibly pyroptosis, a more recently recognised form of pro-inflammatory cell death related to inflammasomes (discussed later). An additional phenomenon is that some hepatocytes exhibit the unusual appearance of “ballooning”, one of the diagnostic criteria for NASH [3]. Ballooning may reflect cytoskeletal injury as a result of caspase 3 activation with inability to complete programmed cell death (“undead cells”) or to enter the cell cycle (senescence) [10]. The interactive roles of cell stressors (oxidative, endoplasmic reticulum [ER], altered membrane fluidity resulting from altered lipid content), intracellular signalling pathways (c-Jun N-terminal kinase [JNK], nuclear factor kappa-B [NF-κB]) [8], responses to stress (autophagy [discussed in Chap. 10], senescence), and subcellular injury to mitochondria, lipid droplets (the intracellular storage site for potentially toxic lipids) and plasma membrane are all potentially relevant to NASH pathogenesis. How varying patterns of hepatocyte injury/cell death incite an inflammatory response, particularly by liganding pattern recognition receptors that activate innate immunity (sterile inflammation), interacting with “inputs” from gut microbiota [discussed in Chaps. 8 and 9], stressed adipose and immune/inflammatory cells activated by other processes could be central to the pathological dichotomy between NASH and simple steatosis.
The focus of the present chapter will be on connections between lipotoxic injury to hepatocytes and liver inflammation. Several excellent reviews on sterile liver inflammation and hepatocyte cell death, both of central interest to NASH, have been published recently; for more detailed molecular discussion, the reader is referred to them [5, 11,12,13].
3.2 What Is Lipotoxicity, and How Does It Fit into a Framework for NASH Pathogenesis?
Unger coined the term “lipotoxicity” in 1994 to describe the pattern of lipid molecule-induced cell injury that occurs in pancreatic β-cells in type 2 diabetes (diabetes) and muscle in metabolic syndrome [9]. Since the first lipidomic data from human NAFLD and NASH livers were published in 2007 and 2009 [14, 15], coupled with the development of animal models where metabolic syndrome determines experimental steatohepatitis [16], the concept that NASH could be a form of liver lipotoxicity has gained ground [1,2,3,4,5,6,7, 17]. In 2012, Cusi proposed the term “liver lipotoxicity” be used instead of NASH [5]. We agree with this important conceptual reorientation of NASH (versus “not-NASH” NAFLD), but before adopting the term “liver lipotoxicity” to replace “NASH”, it would be useful to know which lipid(s) is/are responsible. Effectively lowering their hepatic levels should reverse NASH.
The central issue in NASH pathogenesis is what is different in livers showing NASH, as opposed to the larger proportion of NAFLD patients whose liver pathology, simple steatosis or steatosis with minor non-specific inflammation (often termed “not-NASH” NAFLD), is less commonly associated with fibrosis progression [3, 8]. Two decades ago it was proposed that NASH reflected operation of a second tier of injury and inflammation-inducing pathways, such as oxidative stress and Th1 cytokine responses (e.g. tumor necrosis factor-alpha [TNF-α]) in a liver that was fatty because of overweight and diabetes (the “first hit”). The second hit was presumed to originate from outside the liver, such as from gut-derived endotoxin (demonstrated experimentally by Yang and colleagues from the Diehl group in 1997 [18] or from inflamed adipose tissue in obesity. This 2-hit hypothesis, eloquently articulated by Day and James in 1998 [19], proved useful to focus attention on injury mechanisms in NASH. With hindsight it now seems simplistic because the metabolic milieu leading to steatosis can also lead “directly” to NASH, as we and others discuss elsewhere [2, 5]. The jaded 2-hit concept is still cited [20]. However, writing in the first book on NAFLD (2003), Bass and Merriman articulated the idea that the lipids which accumulate in NASH could themselves (directly) mediate the disease process [1]. Greg Gores’ group then showed that free fatty acid (FFA) lipotoxicity could give rise to TNF-α [21], and by 2007 the concept of “good fat/bad fat” was editorialised in Hepatology [22]. Most working in the field now agree that Bass and Merriman were correct. There are multiple responses to lipotoxicity, and these descend through a web of interactions and reactions, a “multiple hit” concept as conceived by Tilg [23], to result in NASH.
3.3 Which Lipid Molecules Accumulate in NASH?
If analogy is drawn with Koch’s postulates for determining the infectious aetiology of a disease, similar lines of evidence (or “rules”) could apply for implicating endogenous toxic molecules as the cause of a pattern of tissue injury. These are encapsulated in Box 3.1.
Box 3.1: Rules for Pathogenicity of Lipotoxic Molecules in NASH
-
Rule 1 Phenotypic association. By phenotypic association, it is reasoned that the causative lipotoxin will be present in human livers showing NASH but not in simple steatosis.
-
Rule 2 Congruent explanation. The reasons why this lipotoxin accumulates will be consistent with a metabolic pathogenesis and/or be explained by genetic predisposition.
-
Rule 3 Therapy proves causation. Ideally, removal of the lipotoxic lipid should reverse NASH pathology, noting that the outcome is likely to be simple steatosis, not a lean liver.
-
Rule 4 Direct lipotoxicity. The putative lipotoxin should be directly toxic to hepatocytes, the cells most conspicuously injured in NASH.
-
Rule 5 Lipotoxicity causes NASH. The way the putative lipotoxin injures hepatocytes must be pro-inflammatory, that is, the injury has an outcome (or form of cell death) that leads to recruitment of macrophages, lymphocytes, and neutrophils to the liver.
3.4 Which Models Should We “Trust” for Interpretation of NASH Pathogenesis Studies?
Before balancing the evidence for and against candidate lipotoxins in NASH, a few words about animal models are salient (animal models are discussed more fully in relation to obesity-related hepatocarcinogenesis by Lau and colleagues in Chap. 11). While the ideal “model” of a human disease is the affected patient, repeated tissue sampling and the challenges (including potential hazards) of experimental interventions generally limit the study of NASH pathogenesis in man to “static measurements”, that is, blood or liver samples usually obtained at a single time. The study of Vilar-Gomez et al. [24] is an important rare exception [24]. The next best model might be one that replicates, as faithfully as possible, the preconditions (other than species) for development of human NASH. As reviewed earlier [16], these desirable attributes, and why we think they are prerequisites, are tabulated (Table 3.1).
In addition to the “metabolic determinants” listed in Table 3.1, suitable models should exhibit the pathological spectrum of human NAFLD, which under varying circumstances spans simple steatosis, steatohepatitis (NASH), pericellular fibrosis (noting that rodents are unlikely to develop cirrhosis), and hepatocarcinogenesis (in rats and mice, as well as in an apparently substantial proportion of patients with NASH, HCC does not proceed through advanced liver fibrosis).
While opinions vary widely on the relative merits and weaknesses of numerous NAFLD or steatohepatitis models, we are of the opinion that models whose only virtue is “providing NASH pathology” should not be used uncritically to progress concepts of NASH pathogenesis. For example, rodents with methionine and choline deficiency, the MCD model, was developed by the author’s group in 1996 in rats [25] and 2000 in mice [26], as an attempt to provide a model with steatohepatitis, the pathology that is not observed in obese rodents with defects in leptin or the leptin receptor (obesity is associated with simple steatosis) or those fed high fat diet without other nutrient excess. However, Rinella and Green, confirmed by Rizki and Maher, showed that 20–40% weight loss experienced by MCD-fed mice is associated with insulin sensitivity [27, 28], whereas insulin resistance is a sine qua non for pathogenesis of NASH [29, 30]. The similar choline-deficient defined L-amino acid (CDAA) diet of Matsumoto et al. [31], popularised by Miura and Brenner [32], appears to be less “nutritionally challenged”, but animals still fail to gain excessive weight or develop glucose intolerance; by one biochemical measurement, CDAA-fed mice may have impaired insulin signaling, but physiological insulin resistance has not been demonstrated [31]. Use of the term “NASH” to describe results in MCD or CDAA mice (more than 10 articles in 2014) is curious since, in 2005, the Editor of Gastroenterology specifically instructed the authors to use the more appropriate term “nutritional steatohepatitis” [33].
Other models have provided interesting insights into how “mal-processing” of certain molecules, particularly cholesterol, can lead to steatohepatitis [34,35,36,37]. These elegant studies indicate what can happen under certain conditions, but do not prove that the same thing does happen in humans with NASH. Likewise, diets that promote overnutrition (for example, those rich in simple carbohydrates, particularly fructose or sucrose [38,39,40], and those that combine excess carbohydrate, fat and cholesterol (“western” or “atherogenic” diets) are most likely to result in NASH [37, 41,42,43], whereas “uni-dimensional diets”, such as high fat or high sucrose without cholesterol generally cause simple steatosis. On the other hand, for rodents a diet containing 1–2% (wt/wt) cholesterol, which Ginsberg described 10 years ago as the human equivalent of eating >100 big Macs a day [44], is physiologically unrealistic; it is debatable whether such “cholesterol toxicity” is akin to NASH, and the liver histology is not convincing.
Here we draw heavily on studies that have used animal models of overnutrition in which the metabolic determinants of NASH (insulin resistance, hyperglycemia and metabolic syndrome) are present. Such models include rodents and pigs that are deliberately hyper-alimented (e.g. by in-dwelling gastric devices) [45, 46], and rodents with genetically-determined appetite defects, such as melanocortin 4 receptor (Mc4r) and Alms1 mutant mice [42, 47, 48]. The latter is the murine equivalent of Alström syndrome [49], a rare monozygotic form of extreme childhood obesity with diabetes, cardiovascular disease and cirrhosis. Appetite-defective mice are particularly useful as they, like children with monozygotic morbid obesity, “can’t stop eating” and soon become quite inactive [50].
3.5 Triglyceride, the Most Abundant Lipid in Fatty Livers, Does Not Injure Hepatocytes
Steatosis is defined by stainable fat in hepatocytes, most of which is triacylglycerol or triglyceride (TG), or by an increase in hepatic TG content (>5.5%) determined, for example, by proton magnetic resonance spectroscopy (MRS). While correlations between hepatic TG content and development of insulin resistance have been derived in human studies [51], these investigations have been based on MRS evidence of steatosis, not on lipid analyses. There is no experimental evidence that TG impairs insulin receptor signalling or has any noxious effect on hepatocytes.
Within hepatocytes, TG is usually stored efficiently in lipid droplets (discussed later). TG is formed by transacylation of diacylglycerols (DG), catalyzed by diacylglyceride acyltransferases (DGAT); DGAT expression protects hepatocytes from palmitic acid-induced lipotoxicity and steatohepatitis caused by MCD diet. Thus, in DGAT2-deficient animals or with DGAT2 knockdown, liver injury (serum alanine aminotransferase [ALT] level) and resultant fibrogenesis were exaggerated, whereas strategies to enhance TG synthesis were protective [52, 53].
3.6 FFAs Are Lipotoxic in vitro But Seem Unlikely to Cause NASH
Until recently, FFAs have been the favoured lipotoxin in NASH [54,55,56], but the evidence for this is based on in vitro studies of lipotoxicity (as mentioned above) and the MCD model. FFA, including but not confined to saturated FFA (satFFA), accumulate in all human NAFLD livers, irrespective of disease phenotype [14, 15, 56]. There is no difference in hepatic FFA levels between livers showing NASH and simple steatosis in humans or mice and opossums with metabolic syndrome [37, 57] (Itoh 2012 is an exception [58]).
In vitro, satFFA are more lipotoxic than mono-unsaturated or poly-unsaturated long-chain or very long-chain fatty acids. However, neither the published human nor foz/foz mouse analyses indicate important differences in saturation status between NASH and simple steatosis. In mice lacking elongation of long-chain fatty acids family member 6 (Elovl6), which catalyzes formation of stearate (C18:0) from palmitate (C16:0) and oleate (C18:1) from palmitoleate (C16:1), and also promotes expression of sterol-regulatory element-binding protein 1c (SREBP1c), there was no effect on obesity-related liver fat but improved insulin sensitivity attributable to low abundance of DAG [59]; the relevance to human NASH is unclear. An interesting observation has been a relative paucity of polyunsaturated C20-C22 fatty acids in FFA, triacylglycerides and phospholipids in NASH [14, 60]. These reductions include arachidonic (20:4n-6) (the source of eicosanoids), eicosapentanoic (20:5n-3), and docosahexanoic (22:6n-3) acids. This paucity is potentially relevant to liver inflammation because it could change the balance in lipid mediators from antiinflammatory to pro-inflammatory. In mice with forced hepatic overexpression of PNPLA3I148M [61, 62], a polymorphism associated with frequency and severity of human NAFLD [63, 64], relative depletion of polyunsaturated C20-C22 fatty acids was observed among triacylglycerols [65]. Most recently, Chiappini et al. reported the opposite finding, depletion of long-chain fatty acids attributable to decreased activity of the fatty acid desaturase 1 (FADS1) [66]; this particularly altered phospholipid synthesis.
satFFA cytotoxicity to hepatocytes is an experimental paradigm for liver lipotoxicity and resultant inflammation. Some key points are summarised in Box 3.2. satFFA, such as palmitic acid (C16:0) and stearic acid (C18:0), are more toxic than mono-unsaturates, such as palmitoleic (C16:1) and oleic acid (C18:1); the latter protect liver cells against saturated FFA lipotoxicity, possibly by promoting their incorporation into TG [54, 67]. Mono-unsaturates also decrease lysophosphatidylcholine (LPC) content [68], which appears to be an essential mediator of FFA lipotoxicity. Lipotoxicity also involves activation of JNK possibly independent of oxidative stress. In turn, JNK activation injures mitochondria leading to oxidative stress, a self-amplifying step in JNK signal transduction.
Box 3.2: Key Points for FFA Toxicity.
-
1.
Saturates are more toxic than unsaturates.
-
2.
Mono-unsaturates confer protective effects.
-
3.
Toxicity proceeds through lysophosphatidylcholine (LPC) formation.
-
4.
Lipotoxicity involves JNK activation.
-
5.
JNK-dependant mitochondrial injury occurs.
-
6.
Secondary oxidative stress causes further damage to the cell.
In studies from Gores’ lab using primary murine hepatocytes, which validated key findings in well-differentiated human hepatocytes [69], data confirmed operation of the JNK-mitochondrial cell death pathway in satFFA lipotoxicity via PUMA. They also showed that satFFA lipotoxicity proceeds through the formation of LPC, which had been suggested as the lipotoxic mediator in palmitic acid toxicity studies by Han et al. [68].
3.7 Phospholipids: Lysophosphatidylcholine Is a Candidate Lipotoxin in NASH
Like palmitic acid, LPC produces hepatocyte injury in vitro, and it also induces hepatitis in vivo [68]. Han and colleagues found increased LPC content in five NASH cases, higher than in seven with simple steatosis or 12 lean livers; few other lipidomic analyses in NAFLD mention LPC. Chiappini et al. recently found decreased phosphatidylcholine-to-phosphatidylethanolamines rates in human NASH livers [66]. They demonstrated experimentally in HCC cell lines and primary human hepatocytes that lipid mixes from NASH patients can strikingly induce cellular toxicity [66].
3.8 Does Ceramide Play a Role?
Ceramide is the prototypic sphingolipid formed in the ER by condensation of palmitoyl CoA (or other fatty acid moiety) with sphingosine. The rate of ceramide synthesis by this pathway depends on availability of satFFA, but, in addition, de novo synthesis of ceramide can be rapidly generated from sphingomyelin by sphingomyelinase. Such rapid generation of ceramide has been implicated in apoptosis by the death ligands, TNF-α and Fas ligand (FasL) [70]. Ceramide can also play a role in insulin resistance. A prevailing concept is that satFFA lead to formation of ceramide, accumulation of which kills pancreatic β-cells (and neurons) by death-ligand-induced apoptosis. However, Wei et al. showed that ceramide is not essential for satFFA lipotoxicity in liver cells [71], and Han and colleagues showed that ceramide synthesis inhibitors do not modulate palmitic acid-induced lipoapoptosis to hepatocytes [68]. On the other hand, phospholipase A2 (PLA2) inhibitors blocked cell death in these experiments, suggesting a role of PLA2 or its product LPC. Lipidomic analyses of human or murine NAFLD livers have usually failed to identify ceramide accumulation [72], and bariatric surgery improved NASH without lowering circulating ceramide levels [73]. Sphingosine 1-phosphate (S1P), a derivative of ceramide, contributes to macrophage-related inflammation in some contexts. Mauer and colleagues recently showed that the S1P antagonist, FTY720 reduced inflammation and fibrosis in a high fat, high cholesterol dietary model of steatohepatitis in mice [74].
3.9 Diacylglycerols
DAG activates atypical protein kinase C (PKC) isoforms, which have been implicated in the molecular pathogenesis of insulin resistance. By activating NF-κB, PKCs are also pro-inflammatory. Puri et al. found increased hepatic DAG levels in NAFLD versus lean livers, but no difference between NASH and not-NASH fatty livers [14]. In foz/foz mice fed normal rodent chow to cause steatosis or atherogenic diet to cause NASH, hepatic DAG levels increased compared to lean controls, but values were similar in NASH and simple steatosis [75, 76]. Gorden et al.. found increased DAG species, particularly in phospholipids between normal versus NAFLD livers [77, 78], but without specificity to NASH phenotype (see Rule 1, Box 3.1).
3.10 Cholesterol Is a Strong Contender for a Lipotoxin Causing NASH
Three lipidomic analyses found that free cholesterol (FC) is increased in NASH livers but not simple steatosis [60, 76, 77, 79]. Caballero et al used filipin which fluoresces blue upon binding FC but not cholesterol esters [15]. They observed that all 14 NASH livers fluoresced for FC within hepatocytes, versus 4 of 17 with simple steatosis. In foz/foz mice, atherogenic dietary intake caused NASH with major increases of hepatic cholesterol ester and FC; chow-fed foz/foz mice with simple steatosis showed no such increase [76]. Atherogenic diet-fed Wt mice exhibited a transient increase in FC, after which adaptation occurred with return of hepatic FC to normal; this was associated with simple steatosis, not NASH [76].
Epidemiological studies reveal a positive association of dietary cholesterol intake with cirrhosis (irrespective of aetiology) and HCC [80]. Some but not all nutritional studies confirm diets high in saturated fat and cholesterol are associated with NASH [81]. On the other hand, intake of fructose or simple carbohydrates is more consistently identified, and there is a reproducible inverse relationship with micronutrients [82]. Japanese workers showed a strong effect of atherogenic diet (7.5% cocoa butter, 1.25% cholesterol) on NASH in rodents [43], and the co-requirement for cholesterol as well as saturated fat, often with simple carbohydrate, has been found repeatedly [17, 40, 41, 83].
Appetite-defective animals eat more and readily become obese, particularly when fed high fat diet. Mc4r mutant mice fed a 60% fat diet (0.04% cholesterol) develop liver and adipose inflammation, with a pattern of crown-like structures (CLSs) of macrophages around hepatocytes that exhibited large lipid droplets [58]. In our studies, modulation of the cholesterol content of atherogenic diet (0, 0.2–2.0% wt/wt) caused a stepwise increase in hepatic FC content, and this was associated with corresponding increases in serum ALT, hepatocyte apoptosis, activated macrophage and neutrophil accumulation, and NASH severity estimated by NAFLD Activity Score (NAS). In ABCB4 mutant opossums (ABCB4 encodes a canalicular transporter of bile acids and glutathione conjugates), consumption of a “western diet” (0.7% cholesterol) caused hepatic cholesterol accumulation, atherogenic dyslipidemia and NASH [37]. In low density lipoprotein receptor (LDLR) knockout mice, cholesterol content of the diet was a key determinant of whether liver pathology was steatosis or steatohepatitis [36], and this was associated with increased hepatic cholesterol content [34, 35].
3.11 Dysregulation of Hepatic Cholesterol Homeostasis Occurs in NASH
The liver is the central organ for bodily cholesterol homeostasis. Cholesterol synthesis is regulated in response to “need”, as perceived by nuclear receptors such as SREBP2 [84]. SREBP2, the master regulator of cholesterol homeostasis, is upregulated by insulin. Three studies confirm upregulation of hepatic SREBP2 in human NASH [14, 15, 60], and similar changes occur in obese, diabetic foz/foz mice with NASH [75, 76]. In the rodent model, HMG-CoA reductase enzyme activity was correspondingly suppressed, as expected by the regulatory role of SREBP2. Min et al. [60] measured circulating metabolites of cholesterol synthesis (desmosterol:cholesterol ratio) and decreased hepatic HMG-CoA phosphoprotein, and concluded that cholesterol synthesis is increased in NASH but not simple steatosis [60]; this finding is counter-intuitive, given the observed upregulation of SREBP2. The different results between the mouse and human studies could reflect species or methodological differences.
Tracer studies indicate that most liver lipid in human NASH arises from peripheral sources, albeit an increase in de novo synthesis (hepatic lipogenesis) also occurs [85]. Hepatocytes express three cholesterol uptake pathways: scavenger receptor B1 (SRB1), CD36 (uptake pathway for cholesterol and FFAs) and LDLR. LDLR expression is not increased in human NASH [60], but CD36-enriched extracellular vesicles (EVs) circulate in diabetes and metabolic syndrome, and in patients with NASH (Chen B, Farrell GC, Teoh NC – unpublished data). Further, hepatic CD36 expression is upregulated in obese, diabetic mice with NASH [86].
FC is highly reactive. Thus, cells form fatty acyl esters (CEs) catalyzed by acyl-CoA:cholesterol transferase (ACAT) 2, and also sequester FC into lipid droplets. While hepatic expression of ACAT2 increases in NASH (human and mouse), the reverse pathway of esterolysis, catalyzed by cholesterol ester hydrolase (CEH), is also upregulated [60, 76].
A uniquely important pathway for FC disposition in the liver is biotransformation into bile acids. The rate-limiting steps are cytochromes P450 (CYP) 7A1 and 2B1 in ER, and CYP27A in mitochondria. Profound dysregulation of cholesterol homeostasis occurs in NASH: CYP7A1 expression (human liver) and Cyps7a1, 2b1 and 27a (mouse liver) are all near-totally suppressed in NASH, but not in simple steatosis [60, 76].
Hepatocytes rid themselves of cholesterol by passage into blood (via basolateral ATP-binding cassette transporter 1 [ABCA1]) or bile, directly via ABCG5/G8 heterodimers, or as bile acids via ABCB11 (bile salt export pump, BSEP) and ABCB4 (mdr2). ABCA1 is downregulated in human NASH [60], and profound suppression of ABCB11, ABCB4 and ABCG5/8 occurs in foz/foz mice with NASH [76].
In summary, while details may vary between species, it is clear that cholesterol inputs (uptake or synthesis) are increased in NASH versus simple steatosis. Cholesterol ester hydrolysis is also increased, but biotransformation and egress of cholesterol (directly or as bile acids) is profoundly impaired. The net effect is FC trapping in hepatocytes. Van Rooyen et al. showed that insulin concentrations that circulate in insulin-resistant animals are sufficient to upregulate SREBP2 and LDLR, and to downregulate ABCB11 in murine primary hepatocytes [76].
Recently, it has become apparent that hepatic cholesterol trapping in NASH leads to precipitation of cholesterol crystals. To study the conditions that lead to this change in physical state, Ioannou and colleagues [87] fed mice diet high in fat and with increasing amounts of cholesterol for 6 months [87]. Mice fed diets with 0.5% cholesterol or higher developed NASH with fibrosis whereas those fed lower cholesterol high fat diet showed simple steatosis (Fig. 3.1). It was evident that cholesterol crystal-laden lipid droplet remnants from dead or dying hepatocytes were processed by lysosomal enzymes within Kupffer cells. These Kupffer cells (and presumably also recruited macrophages) formed the centre of CLSs, which stained positive for NLRP3 and active caspase 1 (see later section on inflammasome). This phenomenon could be modelled in culture: thus, HepG2 cells exposed to LDL-cholesterol developed crystals in lipid droplet membranes that upregulated TNF, NLRP3 and IL-1β in co-cultured macrophages, with secretion of IL-1β [87].

(A) Mice were fed high fat (15%) diet with increasing cholesterol content. At 0.5% cholesterol, the number of hepatic cholesterol crystals (black line) increased, associated with macrophage crown-like structures (blue line), and Sirius Red positive area for fibrosis (red line). (*) P < 0.01 and (+) P < 0.05 compared with 0% cholesterol group. (B) Representative mouse liver sections after 6 months on each diet. The first column shows blue-coloured birefringent crystalline material within lipid droplets, which are cholesterol crystals stained by filipin (×200 magnification). Second column is hematoxylin and eosin-stained liver sections. Third column shows TNF-α positive macrophages (within crown-like structures), and the last column exhibits Sirius Red positive areas for fibrosis. (Adopted from Ioannou et al. [87], with permission)
Atorvastatin inhibits HMG-CoA reductase but this and other statins have no beneficial effect on NASH pathology [88, 89]; this may be because depletion of hepatic cholesterol upregulates a hepatic cholesterol re-uptake pathway in which Niemann Pick C1-like protein 1 (NPC1L1) transports cholesterol across biliary (and intestinal) epithelium. Conversely, blockade of NPC1L1 by ezetimibe upregulates cholesterol synthesis, and ezetimibe appears ineffective therapy in NASH [90]. We used combination atorvastatin and ezetimibe to return hepatic FC to normal levels in atherogenic diet-fed foz/foz mice [57]. There were commensurate reductions in serum ALT, hepatocyte apoptosis, macrophage activation and severity of NASH pathology, including liver fibrosis. Combination atorvastatin and ezetimibe also removed cholesterol crystals [79] (Fig. 3.2). Thus, cholesterol accumulation (with crystal formation) correlates with NASH pathology, and cholesterol removal (FC and crystals) cures NASH.

Representative liver sections from foz/foz mice fed atherogenic diet (23% fat, 0.2% cholesterol) treated with ezetimibe or atorvastatin, or their combination versus vehicle controls. Liver sections were stained with hematoxylin and eosin, Sirius Red for fibrosis, F4/80 for pro-inflammatory macrophages (inflammation), and unstained frozen sections viewed with polarized light for birefringent cholesterol crystals. Compared to vehicle controls, there were less cholesterol crystals in the atorvastatin- and ezetimibe-treated groups, and cholesterol crystals were nearly abolished in combination-treated mice. As a result, liver histology improved, and inflammation and fibrosis were less in the drug treatment groups. Black and white scale bars 50 μm and 100 μm, respectively. (Adopted from Ioannou et al. [79], with permission)
3.12 Free Cholesterol Is Directly Lipotoxic to Primary Hepatocytes
Caballero and colleagues showed that the mitochondrial cholesterol transporter, steroidogenic acute regulatory protein (StAR) is upregulated in NASH [15], serving as a pathway for mitochondrial cholesterol accumulation. In livers of foz/foz mice with NASH, FC partitioned into mitochondria, and to a lesser extent plasma membrane and ER [91]. Ultrastructural studies confirmed mitochondrial damage in this model (swelling, fragmentation of cristae, formation of crystalline material in the matrix), and similar findings have been documented in human NASH since 1999 [92, 93]. Human studies have also found low hepatic ATP levels in NASH [94], which is consistent with mitochondrial injury.
To establish whether FC is directly lipotoxic, we incubated primary murine hepatocytes with unmodified human LDL and showed dose-dependent FC uptake over 24 h [91] (Fig. 3.3). FC loading caused cell injury, apoptosis and necrosis, redox stress, mitochondrial membrane pore transition with cytochrome c leakage into cytosol, and rapid decline in cellular ATP content. These processes were linked mechanistically to JNK1 activation; thus, Jnk1 −/− hepatocytes were refractory to FC lipotoxicity, and specific JNK inhibitors blocked both apoptosis and necrosis. Mitochondrial protectants (cyclosporine A) and caspase 3/7 inhibitors also rescued hepatocytes from FC lipotoxicity, whereas the ER stress chaperone, 4-phenylbutyric acid, failed to exert any protective effect. The cholesterol loading of HepG2 cells recently reported by Ioannou et al. [87], in which cholesterol crystals were recognised in lipid droplet remnants, also support direct toxicity of cholesterol to hepatocyte-like cells.

Free cholesterol accumulation causes lipotoxicity in hepatocytes. (A) Primary murine hepatocytes incubated with human LDL showed dose-dependent free cholesterol (FC) uptake over 24 h, (B) with proportionate LDL release indicating hepatocellular injury with FC loading. Wildtype (WT) hepatocytes showed increased apoptosis (Höechst 33342 positive nuclei) and necrosis (propidium iodide positive cells) with FC-loading; (C) JNK1 and (D) TLR4 deletion protected cells from both apoptosis and necrosis. (E) Hepatocyte release of the danger-associated molecular pattern, high-mobility group box 1 protein (HMGB1), increased with FC-loading of WT hepatocytes, and (F) anti-HMGB1 protected cells from both apoptosis and necrosis. (*) P < 0.05 compared with 0 μM LDL. (†) P < 0.05 compared to WT. (%) P < 0.05 compared to no addition. (Adopted from Gan et al. [91], with permission)
In summary, among potential lipotoxins causing NASH, FC conforms to the rules of phenotypic association, accumulation by pathways related to metabolic pathogenesis of NASH, is directly toxic to hepatocytes, and its therapeutic removal cures NASH and reverses liver fibrosis (see Box 3.1). Recent data also support a role for selective changes in long-chain fatty acid composition, particularly in selected phospholipid DAGs [66, 95], with alterations in PC:PE ratio that may affect membrane permeability. Whether PNPLA3I148M (discussed later) could underlie such changes requires further study. Meanwhile, it remains possible that fatty acid chain length and accumulation of free cholesterol are both important mechanistically and act synergistically to cause NASH. In order to consider the implications of lipotoxicity for inflammatory recruitment, consideration of the sites of hepatocellular injury in lipotoxic NASH is critical.
3.13 Subcellular Sites of Hepatocyte Injury in NASH
satFFA, LPC and FC lipotoxicity are all associated with mitochondrial membrane pore transition (MPT) that causes disruption of cellular respiration, generation of oxidative stress, and cytochrome c leakage from the matrix into the cytosol where it activates the apoptosome [96]. Provided there is sufficient energy (ATP) required for the final execution steps that involve caspase 3/7-mediated cleavage of the cytoskeleton and cell movement, this causes of apoptosis. When ATP is depleted, programmed cell death is aborted, terminating in necroptosis (caspase 8 is not involved; mixed lineage kinase domain-like pseudokinase [MLKL] is recruited and binds to the receptor-interacting protein 1 [RIP1]/RIP3 complex), or necrosis. Necrosis (sometimes referred to as type 2 necrosis or oncosis) also occurs with more rapid permeabilization of the plasma membrane and dispersion of ion gradients within the cell, a disorganized form of cell death that liberates cell contents. A recent study reported that mitochondrial DNA (mtDNA) was released in mice subjected to a high fat diet, although steatohepatitis was not well-demonstrated in this study model [97]. The authors then showed that mtDNA activated TLR9 (discussed later), and proposed this as a pathway to macrophage recruitment in NASH.
ER is a site of FC deposition in NASH [91], and ER stress is favoured as a pathway to apoptosis (via C/EBP homologous protein [CHOP]-mediated cleavage of Bid) and inflammation (via JNK activation in type 2 diabetes). Triggering of one ER stress response factor but not others, and without CHOP expression, has been described in human NASH [98], and operation of ER stress was reported with MCD steatohepatitis [99]. Legry, Leclercq and colleagues conducted a set of studies in foz/foz and ob/ob mice, measuring elements of its activation, creating ER stress experimentally, and opposing it pharmacologically [100]. The results provide a strong case against ER stress being conspicuous in steatohepatitis related to obesity, diabetes and metabolic syndrome (NASH) [100, 101]. Likewise, in FC lipotoxicity to hepatocytes there was no increased expression of CHOP, and 4-phenylbutyric acid failed to protect hepatocytes [91]. Finally, in three randomized-controlled clinical trials, ursodeoxycholic acid (which opposes ER stress) was ineffective at improving NASH pathology [102,103,104].
Ballooned hepatocytes and apoptotic bodies arise from caspase 3/7-mediated lysis of the cytoskeleton. Membrane-bound vesicles are also shed from activated or injured/dying cell types present in NASH livers. Very small vesicles (30–100 mm) are known as exosomes, larger ones (100–1000 nm) are microparticles (MPs); the term extracellular vesicles (EVs) will be used here to encompass both sizes. EVs circulate in hepatic ischemia-reperfusion injury [105], an acute type of sterile liver inflammation, and they have been detected in humans with NAFLD [106]. Povero and colleagues noted that the circulating titre of EVs increases during the transition of steatosis to steatohepatitis in the CDAA model [107]; levels correlated with hepatocyte cell death, fibrosis and angiogenesis. These EVs contained asialoglycoprotein receptor 1 (ASGPR1), a protein unique to hepatocytes, and miR-122 and 192 which are associated with chronic liver disease. Circulating EVs have also been reported to contain mitochondria or mtDNA in experimental fatty and alcoholic liver injury [97, 108]. We recently found that EVs shed from hepatocytes subjected to FC lipotoxicity contain high-mobility group box 1 protein (HMGB1), and activate Kupffer cells via an HMGB1- and TLR4-dependent process (Fig. 3.3, and Gan L, Farrell GC – unpublished data). This paracrine process links lipotoxicity to inflammatory cell activation in NASH via the intermediary of EVs and soluble HMGB1. The shedding of EVs from liver cells can also activate hedgehog signalling [109], which has implications for inflammatory recruitment and fibrogenesis [107]. Recently, EVs released from lipotoxic cells were shown to be internalised by both hepatocytes and macrophages; within those cells, NF-κB-mediated upregulation of NLRP3, pro-caspase-1 and pro-interleukin-1 was demonstrated, indicating another link between lipotoxicity and pro-inflammatory pathways in NASH [110].
Using in vitro models, it has now been shown that the PNPLA3I148M variant preferentially localises to lipid droplets and is associated with defective remodelling activity, potentially enhancing TG accumulation in lipid droplets [111]. Such lipid droplet “dysfunction” could lead to lipotoxicity indirectly because of “suboptimal storage” of toxic lipids, such as FC [87], and this has now been observed experimentally [66]. Autophagy is another important intracellular pathway, targeting cell constituents to the lysosome for degradation. Mark Czaja and colleagues identified the operation of autophagy for lipid turnover (lipophagy) in fatty liver disease, and the importance of autophagy in opposing apoptosis and influencing insulin sensitivity indicates a possible role in pathogenesis of NASH (elegantly reviewed in Amir and Czaja) [112].
3.14 Hepatocyte Injury and Cell Death in NASH: Relationship to Inflammation
3.14.1 Serum ALT and Ferritin
Circulation of hepatocyte proteins such as ALT and ferritin is evidence of liver injury in NAFLD, but serum ALT level has low sensitivity and specificity for distinguishing NASH from simple steatosis. Maximos et al. found that NAFLD patients with raised ALT had worse adipose insulin resistance, lower plasma adiponectin and higher liver triglyceride (by MRS) than those with normal ALT, but there was no correlation with ballooning, inflammation or fibrosis [51]. Unlike in viral or autoimmune hepatitis, serum ALT values in NASH (and alcoholic hepatitis) rarely exceed tenfold the upper limit of normal (~300 U/L). One reason may be that apoptosis is the predominant form of cell death [113], another is that necrosis tends to be focal rather than zonal or extensive.
3.14.2 Ballooned Hepatocytes
Ballooned hepatocytes are one of three criteria used to calculate the NAS [114], and in global assessment of “NASH vs not NASH” pathology. Further, their number correlates with fibrotic outcome [115]. Ballooned hepatocytes are large, clear cells with “blurred” plasma membrane, possibly reflecting cytoskeletal damage; they lack cytokeratin 8/18 (CK8/18) [116] (suggesting caspase 3 activation) and are ubiquitin positive. They also express hedgehog signalling ligands, which by analogy with Drosophila melanogaster may indicate cells have initiated a cell death program but are unable to complete the process (the term “undead” cells has been used) possibly because of deletion of caspase 9 [117]. Hedgehog ligands are chemotactic and could activate stellate cells directly [118]. It is therefore of interest that hedgehog pathway activation correlates with histologic severity of NAFLD, specifically with ballooning, portal inflammation and fibrosis severity [119, 120].
3.14.3 Apoptosis Versus Necrosis and Necroptosis
As reviewed by Luedde and colleagues recently [12], apoptosis is a physiological form of programmed cell death during development and tissue remodelling. It does not release cell contents other than within larger membrane-bound vesicles known as apoptotic bodies. These are typically engulfed by neighbouring cells without an inflammatory response, but often with a “wound healing” response of tissue regeneration and matrix deposition (fibrosis). Feldstein and colleagues showed that hepatocyte apoptosis is prominent in NASH livers [57, 121]. Furthermore, M30, the caspase 3/7-mediated cleavage product of CK8/18, circulates in NASH to a great extent than in simple steatosis [122]. In mice, the number of M30 positive hepatocytes is high in NASH and negligible in simple steatosis [57]. However, while apoptosis is abundant, it is not the only form of cell death that occurs in lipotoxicity or NASH.
In vitro studies of FC lipotoxicity showed a dose-dependent relationship between hepatocyte FC content and necrosis, as well as apoptosis [91]. In addition to release of ALT and ferritin, other evidence suggests that necrosis occurs in vivo in NASH, as summarized in Box 3.3. Thus, whole length CK8/18 (among other hepatocyte-specific proteins) circulates [122], and the inflammatory infiltrate that surrounds lipid-laden hepatocytes in NASH contains polymorphonuclear leukocytes (neutrophils) as well as macrophages; these may be a response to lipid peroxidation or to release of neutrophil chemokines. Neutrophils release perforins and other proteolytic enzymes that kill cells by necrosis [123]. Finally, experiments in rodent livers showing NASH have found increased expression of RIP3 (a marker of necrosis) and MLKL (a marker of necroptosis) [124].
Box 3.3: Evidence that Hepatocyte Necrosis or Necroptosis Occur in NASH.
-
1.
Serum ALT increases.
-
2.
CK8/18 circulates.
-
3.
Neutrophils are present.
-
4.
RIP3 and MLKL expression increase.
3.14.4 Potential Role of Danger-Associated Molecular Patterns (DAMPs)
Release of HMGB1 in FC lipotoxicity experiments is likely a response to oxidative stress or necrosis [91], as has been documented in ischemia-reperfusion injury [125]. HMGB1 is an archetypical DAMP that can activate cell death on neighbouring hepatocytes by binding to TLR4, and it also binds and activates TLR9 and receptor for advanced glycation endproducts (RAGE) [125, 126]. HMGB1 antiserum reduced cell death in FC-loaded hepatocytes, while hepatocytes from Tlr4 −/− mice, as well as those from Jnk1 −/− animals, were refractory to FC lipotoxicity [91] (Fig. 3.3). HMGB1 also readily binds TLR4 on Kupffer cells and macrophages, a possible pathway that links hepatocyte necrotic cell death to inflammation in NASH [91]. Ganz and colleagues found hepatic levels of HMGB1 increased during intake of a high fat high cholesterol diet, when steatohepatitis was present at 8 weeks [127]. Consistent with the proposed role of this DAMP in NASH pathogenesis, Li and colleagues documented HMGB1 release and TLR4 involvement in feed-forward hepatocyte injury during the early stages of NAFLD caused by intake of a high fat, 2% cholesterol diet [128].
3.14.5 Sterile Inflammation in NASH
While hepatocytes may generate inflammation in NASH by release of DAMPs that signal through pattern recognition receptors [91, 127], inflammation can kill hepatocytes. In alcoholic hepatitis, the origins of liver inflammation appear to be the gut microbiota [129]. Thus, excessive alcohol compromises intestinal permeability, with resultant absorption of bacterial pathogen-activated molecular patterns (PAMPs) such as endotoxin (lipopolysaccharide). These interact with TLR4 and possibly other TLRs, while bacterial CpG DNA ligands TLR9 to activate NF-κB. We have reviewed the evidence that NF-κB is an essential pro-inflammatory “trigger” in NASH [8]. Its activation results in production and release of chemokines and cytokines that promote a cellular inflammatory response. Normal hepatocytes are not killed by TNF-α. However, under certain conditions, including FC loading of mitochondria and glutathione (GSH) depletion caused by oxidative stress, TNF-α can activate hepatocyte apoptosis via caspase 8-mediated death signaling [130]. The evidence that the gut microbiome could play a role in NASH pathogenesis is reviewed in Chaps. 8 and 9.
3.14.6 Role of the NLRP3 Inflammasome
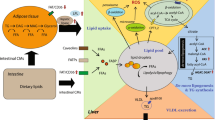
Inflammasomes are intracellular pattern recognition receptors that require a “double trigger” to be assembled (Fig. 3.4). The first signal is MYD88 mediated, and leads to NF-κB-mediated induction of inflammasome components such as NLRP3, pro-caspase 1 and pro-IL-1β. Upon exposure to a second signal, NLRP3 recruits apoptosis-associated speck-like protein containing CARD (ASC) as the scaffold for dimerization of pro-caspase 1, converting it to its active enzyme. Caspase 1 then cleaves pro-IL-1β and pro-IL18 to form the active cytokines. Secreted IL18 attracts and activates macrophages. IL-1β indirectly attracts and then activates neutrophils; it also has direct effects on fibrosis by activating stellate cells.

Inflammasomes are intracellular pattern recognition receptors that require a “double trigger” to be assembled. Abbreviations: ASC apoptosis-associated speck-like protein containing a card, HSC hepatic stellate cell, IL1 interleukin 1, LPS lipopolysaccharide, MYD88 myeloid differentiation primary response gene 88, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, NLRP3 NOD-like receptor protein 3, ROS reactive oxygen species, TLR toll-like receptor, TNF tumour necrosis factor
The MYD88-signaling molecules that could provide a signal 1 in NASH include RAGE, TLR2, TLR4, TLR9, oxidative stress, and, more controversially, saturated FFAs such as palmitic acid [131]. Signal 2 can be provided by foreign particulate matter, uric acid crystals (gout) [132], and cholesterol crystals (as in atheroma) [133]. In the presence of a second signal, increased NLRP3 (expressed in hepatocytes as well as kupffer cells) is activated to generate cellular inflammation with macrophages and neutrophils. NLRP3 inflammasome activation also causes pyroptosis, a form of cell death exhibiting features both of apoptosis (DNA fission into ~200 kDa oligonucleotide “ladders”) and necrosis (plasma membrane pores). Like necrosis and necroptosis, pyroptosis allows leakage of intracellular contents through such pores to promote inflammation.
Mice defective in inflammasome components (NLRP3, CARD, caspase 1) are protected from MCD steatohepatitis and high fat diet-induced NAFLD. The identification of cholesterol crystals in human NASH and two animal models provides a rationale for NLRP3 activation in the relevant metabolic context [79, 87]. In mice fed a high fat and cholesterol added diet, the number of cholesterol crystals increased from 0.5% cholesterol in the diet, while severity of steatohepatitis and resultant liver fibrosis was proportional to the number of crystals [87] (Fig. 3.1). Conversely, in foz/foz mice, the number of cholesterol crystals remaining in livers after treatment with cholesterol-lowering agents correlated with the number of residual F4/80 positive macrophages, neutrophils and extent of liver fibrosis [79]. In both models, active caspase 1 and NLRP3 co-located with cholesterol crystals, and such expression was no longer present when crystals were dissolved as the result of cholesterol-lowering therapy.
If cholesterol crystal-related NLRP3 inflammasome activation is central to the pathogenesis of NASH, NLRP3 inhibitors might provide a new therapeutic opportunity [134], as shown by Szabo and colleagues for the IL-1β receptor inhibitor, anakinra, in alcohol-related fatty liver disease [135]. We used the small molecule NLRP3 inhibitor, MCC950 [134], to test this proposal in atherogenic diet-fed foz/foz mice with NASH. We showed that NLRP3 blockade abolishes liver inflammation during development of NASH, with beneficial effects on liver fibrogenesis [136]. Outstanding challenges are to develop NLRP3 inhibitors that can be administered safely to humans with NASH, and to show that they reverse established NASH and fibrosis, as well as arrest its development.
3.14.7 Other Pattern Recognition Receptors
TLR4 is upregulated in human as well as experimental forms of NAFLD [124, 137]. Its likely importance for NASH is reviewed elsewhere [138, 139]. A pro-inflammatory role has also been suggested for TLR3 [131, 138], but there do not appear to be data indicating its upregulation in human NASH. We have recently reported that TLR9 is upregulated in human NASH compared to livers showing simple steatosis [124]. Unlike TLRs 2 and 4, which are located on the cell surface, TLR9 is present in the endosome of macrophages and, to a lesser extent hepatocytes [124]. In rodents, TLR9 appears to govern M1 activation of macrophages, but its role on human macrophages is less clear. Mice deficient in TLR9 are protected from the milder forms of fatty liver disease generated by a high fat diet [97, 124], and also from CDAA-induced steatohepatitis [32]. Neither is associated with obesity, and it is entirely possible that this apparent protection is because Tlr9 −/− mice are smaller in body weight (less over-nourished). We crossed Tlr9 −/− with foz/foz mice. The resultant obese diabetic mice lacking TLR9 appeared minimally protected against NASH [124]; most critically, resultant liver fibrosis was the same in TLR9-deficient and intact obese animals (Fig. 3.5).

Hepatic Sirius red-positive areas for fibrosis did not change with TLR9 deletion in appetite-defective (hyperphagic) foz/foz mice or wildtype (Wt) littermates fed an atherogenic diet (23% fat, 0.2% cholesterol)
RAGE is another NF-κB-mobilising receptor of potential relevance to signal 1 for NLRP3 activation. RAGE signals in response to ligation by advanced glycosylation end-products (AGE) that circulate in diabetes and have been linked to diabetic complications. AGE is also found in some charcoal grilled food products. Upregulation of RAGE has been demonstrated in a murine dietary model of NAFLD; its deletion protected mice from exacerbation by a high AGE-containing diet, but not from NASH caused by a HF/HC diet in the absence of AGE [140, 141].
3.14.8 Liver Inflammatory Phenotype
In steatohepatitis, the lobular mixed cell inflammatory infiltrate is comprised of activated (M1) macrophages, lymphocytes and neutrophils. In NASH, macrophages aggregate around ballooned or fat-laden hepatocytes in what are referred to as crown-like structures (CLSs). The same macrophage foci are found in inflamed adipose with type 2 diabetes and metabolic obesity [142, 143]. In NASH, their localization around injured hepatocytes infers signals expressed by or released from those cells are important for their recruitment. Within CLSs, cells exhibit markers of pro-inflammatory (M1) macrophages. Tracking studies suggest most of the expansion in numbers of activated macrophages in NASH is from bone marrow-derived monocytes, and a smaller proportion are derived from resident macrophages (Kupffer cells) [144]. Macrophage chemokines from both the CCL2 and CCL5 families are produced by fatty livers [145]. Such chemokines play an important role in macrophage recruitment and activation in NASH. A CCR2/5 antagonist has recently been shown to improve liver fibrosis in human NASH [146], although it failed to impact NASH pathology.
Among candidate sentinel cell populations responsible for sensing DAMPs in fatty liver injury, Kupffer cells and dendritic cells appear most likely involved. In an experimental system of acute sterile inflammation, CD11b positive cells (likely macrophages) were essential for detection of danger signals [11, 147]. Lymphocytes are among the less well-characterized inflammatory cells that accumulate in NASH livers. In a choline-deficient high fat diet murine model which exhibited insulin resistance and NASH, activated intrahepatic CD8 T cells, natural killer T (NKT) cells and inflammatory cytokines were detected [148]. The authors proposed that CD8 T cells and NKT cells but not myeloid cells promote development of NASH by interactions with hepatocytes (inflammation causes liver injury). In a different murine model (high fat high carbohydrate diet), NKT cells and CD8 T cells were also both required for significant liver injury and hepatic infiltration with macrophages [149].
Neutrophils are a neglected feature of NASH inflammation. By release of reactive oxygen species (ROS) and growth factors, they could augment the effect of M1 macrophages in activating stellate cells to promote fibrosis. The mechanisms that promote neutrophil recruitment and retention in NASH are poorly understood [11, 150]. In acute forms of sterile liver inflammation, such as thermal injury, ATP-induced activation of the P2X7 receptor and the NLRP3 inflammasome are involved (discussed in Kubes and Mehal [11]). In the LDLR−/− model, neutrophil-derived myeloperoxidase contributed importantly to the inflammatory phenotype of fatty liver disease [151].
3.15 Does Inflammation Arise from Other Tissues or from the Liver Itself?
With overnutrition, the essential precondition for NASH, adipose depots are the primary site for energy storage in the body. Adipose stores energy in the form of TG, but is unable to store cholesterol efficiently. By taking up and storing excess glucose and FFAs, adipose counters the potentially toxic effects of these circulating nutrients. The tissue response to chronic energy surplus include adipocyte hypertrophy and hyperplasia. However, there is a limit for adipocyte enlargement, adipose expansion and lipid storage (Haczeyni F review in progress). In metabolic obesity associated with NASH, hypertrophic adipocytes exhibit stress signals and start degenerating [152]. The cellular mechanisms of such degeneration and death of stressed adipocytes exhibit similarities to those seen in lipotoxic hepatocytes, as discussed earlier. Further, the consequences of emission of stress signals and cell degeneration/death of lipotoxic adipocytes and hepatocytes are similar, inflammatory recruitment [142]. Thus, both lipid-engorged adipocytes and hepatocytes attract a group of classically-activated, pro-inflammatory macrophages to form CLSs, as discussed earlier for livers with NASH [58, 79] (Fig. 3.6). The properties of macrophage CLSs around degenerating adipocytes or hepatocytes are also similar. The presence of adipose inflammation, and the loss of highly differentiated adipocyte functions, such as secretion of the insulin-sensitizing adipokine, adiponectin, contribute to the development of adipose insulin resistance in NAFLD [153]. A recent human study found that NAFLD patients with raised serum ALT had more severe adipose insulin resistance, lower serum adiponectin and higher hepatic TG levels than those with normal ALT levels, although there was no correlation with the histological indices of NASH [51]. In mice fed HFD 16 weeks, both hepatic and adipose insulin resistance developed with adipose but not liver inflammation [154]. Early (but not late) depletion of Kupffer cells attenuated adiposity and adipose inflammation and insulin resistance. These kind of data indicate links between liver and adipose responses to overnutrition in terms of insulin resistance and inflammation, now termed “metaflammation”, but it seems likely that pro-inflammatory pathways operate in each tissue separately rather than simply “spilling over” from adipose to liver [153, 155]. This proposal is supported by our recent observations of separate effects of obeticholic acid on adipose morphometry and inflammation in relation to hepatic steatosis and inflammation in different models of NAFLD and NASH [156].

F4/80 positive macrophages in liver and adipose tissue of appetite-defective foz/foz mice fed an atherogenic diet (23% fat, 0.2% cholesterol). In both tissues, pro-inflammatory macrophages aggregate around injured hepatocytes and adipocytes in what we termed “crown-like structures”, as a feature of NASH and metabolic obesity
3.16 Therapeutic Relevance and New Directions
Over the last 20 years, the Holy Grail of research into NASH pathogenesis has been to find a critical juncture at which the pathology and clinical course of NASH separate from simple steatosis, the more benign and more common NAFLD phenotype. The rationale is to design mechanism-based and effective drug treatment for NASH because currently there is none. Correction of the metabolic preconditions for NASH, obesity, insulin resistance and metabolic syndrome, does reverse NASH, but the non-pharmacological approaches required for success (lifestyle intervention with 10% reduction in body weight [24]; bariatric surgery [157]) are often not accessed, not achieved or not sustained. To date, empirical approaches or drugs aimed at lipid partitioning are either not effective (metformin) or marginally so (thiazolidinediones) [158]. Similar comments apply to antioxidants (vitamin E), TNF-α release inhibitors (pentoxifylline), FXR agonists (obeticholic acid) and ER stress blockade (ursodeoxycholic acid) [89]. The literature on individual cholesterol-lowering agents has been reviewed [88,89,90], and is no longer encouraging, but use of combination statin plus ezetimibe does not appear to have been studied in NASH. Recent evidence of the synergistic effect for lowering cardiovascular risk [159], the impressive results of animal studies [57], and recent discovery of cholesterol crystals in NASH [79] indicates that this is a logical approach to NASH therapy worthy of clinical trial.
While it remains possible that the inflammation found in NASH originates from outside the liver [23], in inflamed adipose tissue or is provoked by PAMPs generated by the gut microbiota through a “leaky gut” (Chaps. 8 and 9), the authors’ view is that we need look no further than the processes involved with hepatocyte lipotoxicity. If this is the case, preventing accumulation of lipotoxic lipids (like FC) is the logical approach to prevent NASH or to reverse its earliest stages. Unless there is substantial weight reduction, the outcome is likely to be simple steatosis, not a non-fatty liver. We are not convinced that LIGHT from NKT cells [148], or IL-1β [32] are responsible for clinically relevant lipid accumulation in overnourished individuals with NASH (inflammation begets steatosis) [23, 136]. If this is the case, continued use of 2 points improvement of the NAS as the endpoint of NASH drug trials, as recommended by American Association for Liver Disease (AASLD) and mandated by the Federal Drug Authority (FDA) may be inappropriate because 3 of the 7 possible points are allocated to steatosis. Resolution of NASH, preferably with reversal of fibrosis, is more relevant [160].
The sequence of molecular signalling and subcellular injury by which lipotoxicity injures hepatocytes in NASH now presents a more logical “palette” of potential therapeutic opportunities than does the global issue of hepatic lipid partitioning, though it remains possible that newer approaches to improve glycemic control in type 2 diabetes with incretin-based therapies, and agents that improve muscle and adipose insulin sensitivity (PPAR-α/δ [161] or PPAR-δ agonists [162]) could be useful. JNK inhibitors, mitochondrial protectants, antiHMGB1 strategies, TLR4 blockade and NLRP3 inhibitors are all worth exploring. The most appropriate models in which to test such agents are animals with NASH that is attributable to metabolic syndrome.
References
Bass NM, Merriman RB. Fatty acid metabolism and lipotoxicity in the pathogenesis of NAFLD/NASH. In: Farrell GC, George J, Hall P de la M, McCullough AJ, editors. Fatty liver disease: NASH and related disorders. Malden: Blackwell Publishing; 2005. p. 109–22.
Larter CZ, Chitturi S, Heydet D, Farrell GC. A fresh look at NASH pathogenesis. Part 1: the metabolic movers. J Gastroenterol Hepatol. 2010;25:672–90.
Farrell GC, McCullough AJ, Day CP. Non-alcoholic fatty liver disease: A practical guide. Somerset: Wiley; 2013. p. 1–313.
Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–23.
Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–25. e716.
Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55:885–904.
Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1769–77.
Farrell GC, Van Rooyen D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut and Liver. 2012;6:149.
Lee Y, Hirose H, Ohneda M, Johnson J, McGarry JD, Unger RH. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci (USA). 1994;91:10878–82.
Hirsova P, Gores GJ. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. 2015;1:17–27.
Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–72.
Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147:765–83. e764.
Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets. 2011;10:509–36.
Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–90.
Caballero F, Fernández A, De Lacy AM, Fernández-Checa JC, Caballería J, García-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol. 2009;50:789–96.
Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–48.
Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–88.
Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci. 1997;94:2557–62.
Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5.
Henao-Mejia J, Elinav E, Jin C-C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179.
Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology. 2004;40:185–94.
McClain CJ, Barve S, Deaciuc I. Good fat/bad fat. Hepatology. 2007;45:1343–6.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367–8. e365.
Weltman MD, Farrell GC, Liddle C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology. 1996;111:1645–53.
Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067.
Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, et al. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res. 2006;47:2280–90.
Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40:47–51.
Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–5.
Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–9.
Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int J Exp Pathol. 2013;94:93–103.
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology. 2010;139:323–34. e327.
dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-κB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663–74.
Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lütjohann D, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–86.
Wouters K, van Bilsen M, van Gorp PJ, Bieghs V, Lütjohann D, Kerksiek A, et al. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett. 2010;584:1001–5.
Bieghs V, Verheyen F, van Gorp PJ, Hendrikx T, Wouters K, Lütjohann D, et al. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One. 2012;7:e34378.
Chan J, Sharkey FE, Kushwaha RS, VandeBerg JF, VandeBerg JL. Steatohepatitis in laboratory opossums exhibiting a high lipemic response to dietary cholesterol and fat. Am J Physiol Gastrointest Liver Physiol. 2012;303:G12–9.
Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–95.
Pickens MK, Yan JS, Ng RK, Ogata H, Grenert JP, Beysen C, et al. Dietary sucrose is essential to the development of liver injury in the methionine-choline-deficient model of steatohepatitis. J Lipid Res. 2009;50:2072–82.
Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301:G825–34.
Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology. 2013;57:81–92.
Itoh M, Suganami T, Nakagawa N, Tanaka M, Yamamoto Y, Kamei Y, et al. Melanocortin 4 receptor–deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol. 2011;179:2454–63.
Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–403.
Ginsberg HN. Is the slippery slope from steatosis to steatohepatitis paved with triglyceride or cholesterol? Cell Metab. 2006;4:179–81.
Qg D, She H, Cheng JH, French SW, Koop DR, Xiong S, et al. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology. 2005;42:905–14.
Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, et al. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology. 2009;50:56–67.
Arsov T, Larter CZ, Nolan CJ, Petrovsky N, Goodnow CC, Teoh NC, et al. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem Biophys Res Commun. 2006;342:1152–9.
Farrell GC, Mridha AR, Yeh MM, Arsov T, Van Rooyen DM, Brooling J, et al. Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int. 2014;34:1084–93.
Arsov T, Silva DG, O’bryan MK, Sainsbury A, Lee NJ, Kennedy C, et al. Fat aussie—a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol. 2006;20:1610–22.
Haczeyni F, Barn V, Mridha AR, Yeh MM, Estevez E, Febbraio MA, et al. Exercise improves adipose function and inflammation and ameliorates fatty liver disease in obese diabetic mice. Obesity. 2015;23:1845–55.
Maximos M, Bril F, Portillo Sanchez P, Lomonaco R, Orsak B, Biernacki D, et al. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology. 2015;61:153–60.
Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci (USA). 2003;100:3077–82.
Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–74.
Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–51.
Cazanave SC, Gores GJ. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin Lipidol. 2010;5:71–85.
Neuschwander-Tetri BA. Nontriglyceride hepatic lipotoxicity: the new paradigm for the pathogenesis of NASH. Curr Gastroenterol Rep. 2010;12:49–56.
Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, et al. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol. 2013;59:144–52.
Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, et al. Hepatic crown-like structure: a unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS One. 2013;8:e82163.
Bruce CR, Febbraio MA. It’s what you do with the fat that matters! Nat Med. 2007;13:1137–8.
Min H-K, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15:665–74.
Boursier J, Diehl AM. Patatin-like phospholipase domain-containing protein 3 and liver disease: opportunities to unravel mechanisms underlying statistical associations. Hepatology. 2015;61:18–20.
Smagris E, BasuRay S, Li J, Huang Y, Lai KV, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108–18.
Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:645–55.
Zain SM, Mohamed R, Mahadeva S, Cheah PL, Rampal S, Basu RC, et al. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum Genet. 2012;131:1145–52.
Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130.
Chiappini F, Coilly A, Kadar H, Gual P, Tran A, Desterke C, et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci Rep. 2017;7:44658.
Nolan CJ, Larter CZ. Lipotoxicity: why do saturated fatty acids cause and monounsaturates protect against it? J Gastroenterol Hepatol. 2009;24:703–6.
Han MS, Park SY, Shinzawa K, Kim S, Chung KW, Lee J-H, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res. 2008;49:84–97.
Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2012;302:G77–84.
Garcia-Ruiz C, Mari M, Colell A, Morales A, C Fernandez-Checa J. Metabolic therapy: lessons from liver diseases. Curr Pharm Des. 2011;17:3933–44.
Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–81.
Kotronen A, Seppänen-Laakso T, Westerbacka J, Kiviluoto T, Arola J, Ruskeepää A-L, et al. Hepatic stearoyl-CoA desaturase (SCD)-1 activity and diacylglycerol but not ceramide concentrations are increased in the nonalcoholic human fatty liver. Diabetes. 2009;58:203–8.
Anjani K, Lhomme M, Sokolovska N, Poitou C, Aron-Wisnewsky J, Bouillot J-L, et al. Circulating phospholipid profiling identifies portal contribution to NASH signature in obesity. J Hepatol. 2015;62:905–12.
Mauer AS, Hirsova P, Maiers JL, Shah VH, Malhi H. Inhibition of sphingosine 1-phosphate signaling ameliorates murine nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2017;312:G300–13.
Van Rooyen DM, Farrell GC. SREBP-2: a link between insulin resistance, hepatic cholesterol, and inflammation in NASH. J Gastroenterol Hepatol. 2011;26:789–92.
Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology. 2011;141:1393–403. e1395.
Gorden DL, Myers DS, Ivanova PT, Fahy E, Maurya MR, Gupta S, et al. Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. J Lipid Res. 2015;56:722–36.
Gorden DL, Ivanova PT, Myers DS, McIntyre JO, VanSaun MN, Wright JK, et al. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS One. 2011;6:e22775.
Ioannou GN, Van Rooyen DM, Savard C, Haigh WG, Yeh MM, Teoh NC, et al. Cholesterol-lowering drugs cause dissolution of cholesterol crystals and disperse Kupffer cell crown-like structures during resolution of NASH. J Lipid Res. 2015;56:277–85.
Yu L, Morishima C, Ioannou GN. Dietary cholesterol intake is associated with progression of liver disease in patients with chronic hepatitis C: analysis of the hepatitis C antiviral long-term treatment against cirrhosis trial. Clin Gastroenterol Hepatol. 2013;11:1661–6. e1663.
Marchesini G, Petta S, Dalle Grave R. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: pathophysiology, evidence, and practice. Hepatology. 2016;63:2032–43.
Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1961–71.
Serviddio G, Bellanti F, Villani R, Tamborra R, Zerbinati C, Blonda M, et al. Effects of dietary fatty acids and cholesterol excess on liver injury: a lipidomic approach. Redox Biol. 2016;9:296–305.
Horton JD, Goldstein JL, Brown MS. SREBPs activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343.
Larter CZ, Yeh MM, Van Rooyen DM, Teoh NC, Brooling J, Hou JY, et al. Roles of adipose restriction and metabolic factors in progression of steatosis to steatohepatitis in obese, diabetic mice. J Gastroenterol Hepatol. 2009;24:1658–68.
Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res. 2017;58:1067–79.
Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res. 2013;52:175–91.
Wong VW-S, Chitturi S, Wong GL-H, Yu J, Chan HL-Y, Farrell GC. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol Hepatol. 2016;1:56–67.
Musso G. Ezetimibe in the balance: can cholesterol-lowering drugs alone be an effective therapy for NAFLD? Diabetologia. 2014;57:850–5.
Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, Farrell GC. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J Hepatol. 2014;61:1376–84.
Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430–4.
Sanyal AJ, Campbell–Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92.
Cortez-Pinto H, Chatham J, Chacko V, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–64.
Chiappini F, Desterke C, Bertrand-Michel J, Guettier C, Le Naour F. Hepatic and serum lipid signatures specific to nonalcoholic steatohepatitis in murine models. Sci Rep. 2016;6:31587.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58.
Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 2016;126:859.
Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–76.
Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, et al. CCAAT/enhancing binding protein β deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–17.
Legry V, Van Rooyen DM, Lambert B, Sempoux C, Poekes L, Español-Suñer R, et al. Endoplasmic reticulum stress does not contribute to steatohepatitis in obese and insulin-resistant high-fat-diet-fed foz/foz mice. Clin Sci. 2014;127:507–18.
Leclercq IA, Van Rooyen DM, Farrell GC. Hepatic endoplasmic reticulum stress in obesity: deeper insights into processes, but are they relevant to nonalcoholic steatohepatitis? Hepatology. 2011;54:2261–6.
Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23:1464–7.
Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rössle M, Cordes HJ, et al. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–9.
Ratziu V, De Ledinghen V, Oberti F, Mathurin P, Wartelle-Bladou C, Renou C, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54:1011–9.
Teoh NC, Ajamieh H, Wong HJ, Croft K, Mori T, Allison AC, et al. Microparticles mediate hepatic ischemia-reperfusion injury and are the targets of Diannexin (ASP8597). PLoS One. 2014;9:e104376.
Kornek M, Lynch M, Mehta SH, Lai M, Exley M, Afdhal NH, et al. Circulating microparticles as disease-specific biomarkers of severity of inflammation in patients with hepatitis C or nonalcoholic steatohepatitis. Gastroenterology. 2012;143:448–58.
Povero D, Panera N, Eguchi A, Johnson CD, Papouchado BG, de Araujo Horcel L, et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cells via microRNA targeting peroxisome proliferator-activated receptor-γ. Cell Mol Gastroenterol Hepatol. 2015;1:646–63. e644.
He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology. 2017;66:220–34.
Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, et al. Liver cell–derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136:320–30. e322.
Cannito S, Morello E, Bocca C, Foglia B, Benetti E, Novo E, et al. Microvesicles released from fat-laden cells promote activation of hepatocellular NLRP3 inflammasome: a pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS One. 2017;12:e0172575.
Ruhanen H, Perttilä J, Hölttä-Vuori M, Zhou Y, Yki-Järvinen H, Ikonen E, et al. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55:739–46.
Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:159–66.
Eguchi A, Feldstein AE. Adipocyte cell death, fatty liver disease and associated metabolic disorders. Dig Dis. 2014;32:579–85.
Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21.
Gramlich T, Kleiner DE, McCullough AJ, Matteoni CA, Boparai N, Younossi ZM. Pathologic features associated with fibrosis in nonalcoholic fatty liver disease. Hum Pathol. 2004;35:196–9.
Lackner C, Gogg-Kamerer M, Zatloukal K, Stumptner C, Brunt EM, Denk H. Ballooned hepatocytes in steatohepatitis: the value of keratin immunohistochemistry for diagnosis. J Hepatol. 2008;48:821–8.
Kakisaka K, Cazanave SC, Werneburg NW, Razumilava N, Mertens JC, Bronk SF, et al. A hedgehog survival pathway in ‘undead’ lipotoxic hepatocytes. J Hepatol. 2012;57:844–51.
Bijlsma MF, Damhofer H, Roelink H. Hedgehog chemotaxis is mediated by smoothened located outside the primary cilium. Sci Signal. 2012;5:ra60.
Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55:1711–21.
Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, et al. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol. 2011;224:401–10.
Castro RE, Ferreira DM, Afonso MB, Borralho PM, Machado MV, Cortez-Pinto H, et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J Hepatol. 2013;58:119–25.
Joka D, Wahl K, Moeller S, Schlue J, Vaske B, Bahr MJ, et al. Prospective biopsy-controlled evaluation of cell death biomarkers for prediction of liver fibrosis and nonalcoholic steatohepatitis. Hepatology. 2012;55:455–64.
Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. 2007;35:757–66.
Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, et al. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci. 2017;131:2145–59.
Huebener P, Pradere J-P, Hernandez C, Gwak G-Y, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125:539.
Gao B. Innate immunity and steatohepatitis: a critical role of another toll (TLR-9). Gastroenterology. 2010;139:27.
Ganz M, Bukong TN, Csak T, Saha B, Park J-K, Ambade A, et al. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat–cholesterol–sugar diet model in mice. J Transl Med. 2015;13:193.
Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–30.
Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–44.
Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF-and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–98.
Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–89.
Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature. 2010;464:1357.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476.
Mridha AR, Wree A, Robertson AA, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66:1037–46.
Sharifnia T, Antoun J, Verriere TG, Suarez G, Wattacheril J, Wilson KT, et al. Hepatic TLR4 signaling in obese NAFLD. Am J Physiol Gastrointest Liver Physiol. 2015;309:G270–8.
Miura K, Ohnishi H. Role of gut microbiota and toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:7381.
Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. 2013;28:38–42.
Leung C, Herath CB, Jia Z, Andrikopoulos S, Brown BE, Davies MJ, et al. Dietary advanced glycation end-products aggravate non-alcoholic fatty liver disease. World J Gastroenterol. 2016;22:8026.
Goodwin M, Herath C, Jia Z, Leung C, Coughlan MT, Forbes J, et al. Advanced glycation end products augment experimental hepatic fibrosis. J Gastroenterol Hepatol. 2013;28:369–76.
Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55.
Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. 2008;49:1562–8.
Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300.
Camps J, Joven J. Chemokine ligand 2 and paraoxonase-1 in non-alcoholic fatty liver disease: the search for alternative causative factors. World J Gastroenterol. 2015;21:2875.
Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754–67.
Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol. 2010;184:4470–8.
Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–64.
Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P, et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun. 2017;1:299.
Heymann F, Tacke F. Immunology in the liver – from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88.
Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS One. 2012;7:e52411.
Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res. 2013;54:2423–36.
Caputo T, Gilardi F, Desvergne B. From chronic overnutrition to metaflammation and insulin resistance: adipose tissue and liver contributions. FEBS Lett. 2017;591:3061.
Lanthier N, Molendi-Coste O, Cani PD, van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J. 2011;25:4301–11.
Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. 2016;57:2099–114.
Haczeyni F, Poekes L, Wang H, Mridha AR, Barn V, Geoffrey Haigh W, et al. Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity. 2017;25:155–65.
Klebanoff MJ, Corey KE, Chhatwal J, Kaplan LM, Chung RT, Hur C. Bariatric surgery for nonalcoholic steatohepatitis: a clinical and cost-effectiveness analysis. Hepatology. 2017;65:1156–64.
Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta-analysis. JAMA Intern Med. 2017;177:633–40.
Gryn SE, Hegele RA. Ezetimibe plus simvastatin for the treatment of hypercholesterolemia. Expert Opin Pharmacother. 2015;16:1255–62.
Wong VWS, Chan WK, Chitturi S, Chawla Y, Dan YY, Duseja A, et al. The Asia-Pacific working party on nonalcoholic fatty liver disease guidelines 2017 part 1: definition, risk factors and assessment. J Gastroenterol Hepatol. 2017;
Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator− activated receptor− α and− δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–59.
Haczeyni F, Wang H, Barn V, Mridha AR, Yeh MM, Haigh WG, et al. The selective peroxisome proliferator–activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun. 2017;1:663.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Farrell, G.C., Haczeyni, F., Chitturi, S. (2018). Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. In: Yu, J. (eds) Obesity, Fatty Liver and Liver Cancer. Advances in Experimental Medicine and Biology, vol 1061. Springer, Singapore. https://doi.org/10.1007/978-981-10-8684-7_3
Download citation
DOI: https://doi.org/10.1007/978-981-10-8684-7_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8683-0
Online ISBN: 978-981-10-8684-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)