
Abstract
Lipid droplet accumulation and oxidant stress, once thought to play essential roles in the pathogenesis of nonalcoholic steatohepatitis (NASH), may actually represent parallel epiphenomena. Emerging data now point to nontriglyceride lipotoxicity and complex mechanisms of hepatocyte injury and apoptosis as the major contributors to the disease phenotype currently recognized as NASH. Although specific mediators of hepatic lipotoxicity have not been identified with certainty, abundant evidence from animal studies and recent data in humans indicate that free fatty acids in the liver can serve as substrates for formation of nontriglyceride lipotoxic metabolites that cause liver injury. The accumulation of triglyceride in droplets may actually be protective, and thus therapeutic efforts directed at fat accumulation as a sole endpoint may be misguided. This review examines the new evidence supporting the role of nontriglyceride fatty acid metabolites in causing NASH and how adipose and muscle insulin resistance contribute to hepatic lipotoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipotoxicity is a term that describes the adverse effects of excessive free fatty acids on pancreatic β-cell survival. The term was used without an established definition to describe cellular injury and death from free fatty acids, their metabolites, and even triglyceride [1•, 2••]. Innumerable potential mediators of lipotoxicity can be derived from fatty acids. Sorting through the permutations in fatty acid chain length, number and locations of double bonds, whether they are present in the natural cis or synthetic trans configuration, and how many fatty acids are incorporated into the various metabolites is a major challenge for investigators in the field. Identifying which are direct causes of cellular injury is an area of ongoing study in the field of lipidomics in lipotoxic diseases of the liver, heart, pancreatic islets, and vasculature [3, 4, 5•].
Role of Triglyceride Droplets in Hepatic Lipotoxicity
Through studies in animal models, cell culture systems, and newer data from human studies, it has become clear that triglyceride held within lipid droplets is largely inert. Although the accumulation of lipid droplets was associated with increased markers of endoplasmic reticulum stress, the role of this response in nonalcoholic steatohepatitis (NASH) is uncertain [6]. For example, mice overexpressing diacylglycerol acyltransferase (DGAT)-2, the final enzyme mediating triglyceride formation, had increased fat accumulation and hepatic phosphorylation of protein kinase-like endoplasmic reticulum kinase (PERK), a marker of endoplasmic reticulum stress, yet did not exhibit increases in the proinflammatory pathways of c-Jun N-terminal kinase (JNK) phosphorylation and nuclear factor κB activation, common indicators of insulin resistance and steatohepatitis [6, 7•]. Supporting this observation are studies showing that blocking DGAT2 expression prevented fatty acid–induced triglyceride accumulation but did not prevent lipotoxic injury caused by fatty acids, both in cell culture experiments and in mice [8]. Some difficulties arise in interpreting rodent and cell culture studies that manipulate DGAT2 expression, possibly because of concomitant upregulation of compensatory oxidative disposal pathways [9]. However, a different approach of inhibiting triglyceride incorporation into nascent very-low-density lipoprotein (VLDL) by blocking microsomal triglyceride transfer protein impairs secretion of triglyceride and leads to its accumulation, yet does not appear to be associated with liver injury in animal models.
In humans, it has been particularly difficult to determine if lipid droplet accumulation can cause metabolic abnormalities and cell injury, or if it develops as a parallel process to the events leading to impaired insulin signaling and steatohepatitis. Evidence that fat droplets do not cause hepatic insulin resistance and NASH in humans is provided by several recent studies that identified single nucleotide polymorphisms (SNPs) in the gene for adiponutrin (patatin-like phospholipase domain-containing protein A3, PNPLA3) [10•, 11]. These variations compromise the function of adiponutrin, an enzyme that plays a role in triglyceride turnover, and are associated with the development of hepatic steatosis but not insulin resistance or steatohepatitis.
Given the data showing that formation of triglyceride droplets is not needed for lipotoxic injury, it is not surprising that other studies have now shown that the accumulation of fat is actually a protective response to an increased flux of fatty acids to the liver and the associated lipotoxicity [5•, 12••, 13•]. This response has been demonstrated in cardiomyocytes [14], pancreatic islet cells, and hepatocytes in cell culture, and it has been suggested to be true in humans as well. Nonetheless, temporary storage of triglyceride as fat droplets in tissues such as the liver and muscle is only a temporizing measure, and the triglyceride must still be released at a later time. If a cell is still unable to handle the liberated fatty acids as lipid droplets are turned over, the stored triglyceride could still serve as a source of lipotoxic intermediates [15]
Molecular Mediators of Lipotoxic Liver Injury
If lipid droplets are not directly responsible for cell injury leading to steatohepatitis, then attention must focus on other likely molecular mediators of NASH. Substantial evidence now points to nontriglyceride metabolites of fatty acids as the cause of hepatocellular injury in NASH, similar to the experimental findings in other organs that are targets of lipotoxicity [1•, 5•, 12••].
Direct toxicity from fatty acids has been considered. However, the concentration of intracellular fatty acids is kept fairly constant in the liver [4], even with increased levels in the blood as seen with NASH [16], suggesting that substantial elevations of the fatty acid levels in hepatocytes may not be the direct mediator of lipotoxic injury [2••]. Of course, measurement of tissue fatty acid concentrations at any given time point does not provide information about the net flux through the system. Because they are tightly bound to fatty acid binding proteins (FABP), the levels of fatty acids are mostly a function of FABP concentrations and not reflective of the flow through the system.
Even though free fatty acids are not likely to directly cause steatohepatitis, one potential mechanism of direct fatty acid–induced injury that has been proposed is through the activation of an innate immune response. Saturated fatty acids are ligands for toll-like receptor 4 (TLR-4), leading to a cascade of events precipitating cell death through apoptosis. Inhibition or loss of function of TLR-4 can improve steatohepatitis in mouse models. Another potential direct mechanism may be related to the detergent properties of fatty acids, which by definition are soaps at neutral pH. It could be that through this physico-chemical mechanism, fatty acids destabilize lysosomal membranes, leading to release of cathepsin B and activation of apoptotic pathways [17].
Experimental evidence argues against a direct role for fatty acids in lipotoxicity. For example, studies have demonstrated that formation of acyl coenzyme A (acyl-CoA) is a necessary step in the development of fatty acid–induced lipotoxicity in cell culture. Experiments in a liver cell line and in pancreatic β-cells showed that blocking acyl-CoA synthesis can prevent lipotoxicity caused by palmitic acid, suggesting that either palmitoyl-CoA is toxic or that palmitic acid must be incorporated into another molecule via palmitoyl-CoA to cause lipotoxicity [18••].
Ceramides are one class of fatty acid metabolites that were considered as possible mediators of lipotoxic cellular injury [3, 19]. Ceramides were shown to be important in lipotoxicity in pancreatic islet cells and can increase mitochondrial membrane permeability leading to apoptosis. However, several studies showed that blocking ceramide synthesis in various cell lines does not prevent fatty acid–induced injury in liver cells. Further studies are needed to ascertain whether these results apply to human liver disease, but lipidomic studies have not identified substantially elevated liver ceramide levels in NASH.
Diacylglycerols (DAG) are recognized as major activating ligands of most protein kinase C isoforms, a group of kinases that plays a role in diverse cellular signaling pathways. DAGs are also essential precursors in the synthesis of triglyceride and membrane phospholipids, and thus DAG synthesis is carefully regulated to coordinate these disparate functions [20]. Some studies have suggested that DAG species are major contributors to hepatic lipotoxicity [13•]. However, a lipidomic analysis of liver biopsy specimens from NAFLD patients with simple hepatic steatosis compared with steatohepatitis found no difference between the two groups, although both were higher than levels in normal liver [4]. It may be that the increases reflect increased trafficking of fatty acids through the liver, but do not directly cause the cellular injury of steatohepatitis.
Phosphatidyl choline, or lecithin, constitutes a major component of cell membrane bilayers, lipid droplet envelop monolayers, and is a necessary component of VLDL to facilitate triglyceride secretion from the liver. Lysophosphatidyl choline (LPC) is formed by removal of the middle fatty acid in the glycerol backbone of phosphatidyl choline and has been implicated in lipotoxicity. Although one lipidomic study found only minor increases in hepatic LPC levels in NASH patients [4], another recent study provided compelling evidence that LPC could be an important mediator of hepatic lipotoxicity in NASH [18••]. This study demonstrated increased LPC levels in liver biopsies in a small group of patients with levels increasing in proportion to steatohepatitis severity. The investigators further demonstrated that LPC caused lipotoxic liver injury in liver cell lines, mouse hepatocytes and in vivo in mice. This effect appeared to depend on a G-protein–coupled receptor that ultimately causes apoptosis through mitochondrial membrane depolarization. Lysophosphatidylcholine species can be generated through deacylation of phosphatidyl choline by phospholipase A2 (PLA2) and inhibitors of PLA2 were shown to prevent palmitate-induced lipotoxicity in cell culture. Although this study needs confirmation, it provides multiple lines of evidence for the identity of at least one likely mediator of fatty acid–induced hepatic lipotoxicity. A variety of other similar metabolites of free fatty acids, known and unknown, have also been considered, including lysophosphatidic acid species and phosphatidic acid. Phosphatidic acid, because of its comparatively small surface hydrophilic region, facilitates membrane bending or folding. It also interacts with several intracellular signaling molecules, but the role of these interactions in mediating injury is uncertain.
Metabolic Factors Predisposing to Lipotoxic Liver Injury
The pathways leading to the accumulation of lipid droplets in the liver as well as the pathways leading to lipotoxic injury are upregulated by an increase in free fatty acid availability (Fig. 1). Steatosis, although not pathogenic in itself, is thus a fairly good marker of excessive flow of fatty acids through hepatocytes. The major mechanisms of increased fatty acid exposure and disposal in the liver have been known for more than four decades. Two well-established sources are peripheral lipolysis and de novo lipogenesis (DNL). Adipose tissue triglyceride lipolysis releases free fatty acids into the blood, to be carried to the liver bound to albumin, whereas DNL creates new fatty acids within the liver using primarily excess carbohydrate as a substrate [21]. Adipose tissue is typically the source of most liver fatty acids, supplying 70% of the fatty acids handled by the liver, whereas DNL contributes about 5%. However, Donnelly et al. [22] demonstrated that in patients with NASH, DNL contributes up to a quarter of the fatty acids. This finding may be a consequence of the hyperinsulinemia associated with muscle insulin resistance, which drives a lipogenic program in the liver.

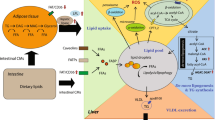
The nontriglyceride lipotoxicity model of nonalcoholic steatohepatitis (NASH). Several free fatty acid metabolites cause hepatic lipotoxicity leading to endoplasmic reticulum (ER) stress, inflammation, apoptosis, necrosis, and liver biopsy findings characteristic of NASH (eg, ballooning and Mallory-Denk bodies). Discerning the role of lipid droplets in the pathogenesis of NASH is challenging because increased delivery of fatty acids or impaired oxidative disposal pathways commonly cause lipid droplet formation and spillover of fatty acids into metabolic pathways, leading to lipotoxic metabolite formation. Triglyceride accumulation is not needed for the development of NASH, and is usually a protective mechanism that prevents lipotoxic injury. Steatosis alone does not progress to NASH. In some individuals, however, the degree of steatosis likely represents a good marker for concomitant lipotoxic injury because both are sequelae of stresses on liporegulation. The major processes predisposing to lipotoxic injury include inappropriate peripheral lipolysis, a common consequence of adipose tissue insulin resistance; excess carbohydrates, typically caused by excessive dietary intake (especially fructose) or impaired disposal of glucose because of muscle insulin resistance; impaired oxidative pathways (eg, downregulation of mitochondrial β-oxidation in the setting of hyperinsulinemia); and impaired formation and/or secretion of triglyceride. Additional fatty acids derive from the release of free fatty acids during lysosomal breakdown of internalized lipoprotein remnants (eg, chylomicron remnants and low-density lipoprotein) and breakdown of lipid-containing autophagosomes, the role of which in NASH is uncertain. Green arrows depict potentially favorable disposal pathways; red arrows depict pathways that may predispose to lipotoxic injury. The role of reactive oxygen species (ROS) in NASH pathogenesis is not established, and increased markers of oxidant stress may be an epiphenomenon reflecting increased flux of fatty acids through oxidative pathways. P450—cytochrome P450 mixed-function oxidases; SER—smooth endoplasmic reticulum; VLDL—very-low-density lipoprotein
Other contributors to the hepatic free fatty acid pool include uptake of short-chain fatty acids delivered after meals from the gut and lysosomal breakdown of lipoprotein remnants. Autophagy, the highly regulated process by which cells catabolize intracellular components using lysosomal enzymes, was recently recognized to be a fate of lipid droplets and source of fatty acids as well, and is discussed further below [23••].What proportion of lipid droplets undergo autophagic catabolism with release of fatty acids into the cytoplasm versus more classical triglyceride lipolysis to release fatty acids is an area of active investigation. Except for low-density lipoprotein (LDL)-receptor mediated uptake of the limited amount of triglyceride in lipoprotein remnants, serum triglycerides are not a major source of fat for the liver. Some of the fatty acids released from chylomicrons and VLDL in the adipose and muscle vasculature for the purpose of their local uptake probably do leak to albumin in the blood and are transported to other sites including the liver, but most are taken up in the tissues where the lipolysis takes place. These points are relevant to treatment. Because most dietary fat does not go directly to the liver, the role of fat restriction in the treatment of NASH is primarily to limit total caloric intake rather than reduce exposure of the liver to fat. Additionally, using drugs to reduce serum triglyceride levels might not have a direct benefit on the liver. In fact, interventions that reduce fasting triglyceride levels by impairing hepatic fat secretion could be counterproductive in terms of liver disease.
When the homeostatic mechanisms regulating fat metabolism are running smoothly (so-called liporegulation [19]), production of lipotoxic metabolites from free fatty acids is minimized. Stresses imposed on this system that alter this homeostasis—whether through increased supply of precursor fatty acids, impaired disposal of fatty acids through oxidative or secretory pathways, or possibly upregulation of pathways that generate lipotoxic intermediates from fatty acids and impaired pathways that dispose of them—could all potentially promote lipotoxicity.
Excessive or Inappropriate Peripheral Lipolysis
Enzymatic hydrolysis of triglyceride stored in adipose tissue is the predominant source of fatty acids delivered to the liver. This highly regulated process is governed by a variety of hormonal, neurologic, and pharmacologic stimuli including insulin, glucagon, catecholamines, natriuretic peptides, tumor necrosis factor α, adenosine, methylxanthines and peroxisome proliferator-activated receptor (PPAR) γ ligands, several neutral lipases, and specific lipid droplet proteins [24, 25]. Insulin resistance in adipose tissue allows inappropriately sustained lipolysis and release of fatty acids, which are sent to the liver at times when the liver is programmed for lipogenesis rather than fat disposal. Although the canonical paradigm has been that fatty acids liberated by adipocyte lipases are destined to be transported to other tissues, newer data suggest that oxidative pathways in adipocytes might also be an important fate, thus playing a role in energy expenditure and thermogenesis [26], although this theory continues to be debated [27]. Because much of the fat broken down in adipose tissue and sent to the liver as free fatty acids returns as triglyceride secreted by the liver, the cycling of fat between adipose tissue, the liver, and back to adipose tissue has been postulated to be important in thermogenesis [28].
It is well recognized, however, that unbridled adipocyte lipolysis can send more fatty acids to the liver than the liver can handle. A common clinical situation in which this occurs is prolonged fasting. Older data demonstrated the adverse impact of fasting on accelerating liver injury. Interestingly, the impact that massive peripheral lipolysis has on the liver can potentially be attenuated by upregulated oxidative metabolism by muscle. Mice with impaired mitochondrial biogenesis are particularly prone to fasting-induced liver steatosis [29] and most mouse strains will develop some degree of fasting-induced steatosis.
Excessive De Novo Lipogenesis
The role of the supply side, either through excessive delivery of fatty acids to the liver or excessive synthesis of fatty acids in the liver (DNL), in the development of NASH has been well described [21]. The synthesis of fatty acids in the liver from the intermediates generated by glycolysis is a major fate of excess carbohydrate presented to the liver from dietary intake and impaired peripheral glucose disposal. Such fatty acids are synthesized as fully saturated molecules, such as palmitate (C16) or stearate (C18), and a portion must undergo desaturation, or the addition of cis-double bonds. This process facilitates the synthesis of triglyceride because the middle position of the glycerol backbone is optimally esterified with an unsaturated fatty acid. Current dietary habits, especially massive amounts of high-fructose corn syrup consumption, may overwhelm the supply side of fatty acid trafficking by forcing excessive synthesis of new saturated fatty acids. Fructose enters the glycolytic pathway downstream from phosphofructokinase, the rate-limiting enzyme for this pathway in the metabolism of glucose, and thus fructose provides an unregulated source of acyl-CoA used as a substrate for DNL [30, 31].
DNL does not occur in isolation, and compensatory mechanisms can be invoked that prevent lipotoxicity from occurring. For example, increased liver triglyceride accumulation in fructose-fed rats can be prevented by stimulating mitochondrial β oxidation of the fatty acids generated [32]. The role of steroyl-CoA desaturase is critically important in facilitating the formation of triglyceride in the absence of dietary unsaturated fatty acids. Recent studies have shown that inhibitors of stearoyl-CoA desaturase-1 (SCD1) diminish triglyceride accumulation but at the expense of increased lipotoxicity [33••]. Previous murine studies have shown that inhibiting SCD-1 is often accompanied by increases in compensatory oxidative pathways, so perhaps as long as these alternative pathways are intact, lipotoxic injury can be averted. Conversely, DNL can be increased by increases in sterol regulatory element binding protein (SREBP)-1c, the transcription factor largely responsible for the expression of enzymes that carry out DNL. Overexpression of SREBP-1c in animal models has been identified as a contributor to lipotoxic liver injury [34, 35].
Lysosomes as a Source of Fatty Acids
Lysosomal lipases are a source of intracellular free fatty acids, releasing them from two major sources of triglyceride. Uptake of lipoprotein remnants such as chylomicron remnants and LDL particles that have been mostly delipidated is mediated by the LDL receptor on hepatocytes and contributes a relatively minor amount of free fatty acid to the intracellular pool. A second lysosomal pathway that may be quite significant as a source of fatty acids is autophagy of lipid droplets followed by the release of fatty acids [23••]. Recently termed lipophagy, this process might even play a larger role in the turnover of hepatocyte lipid droplets than lipolysis mediated by lipases such as adipocyte triglyceride lipase. Understanding the effects of inhibiting intracellular lipases is complicated by the difficulties in knowing if drugs or genetic manipulations affect the liver, adipose tissue, or both. Inhibiting lysosomal lipases with 18 β-glycyrrhetinic acid prevented NAFLD in lard-fed rats and decreased the accumulation of triglyceride in HepG2 cells, suggesting that pharmacologic inhibition of lysosomal fatty acid release could be important in triglyceride turnover [36].
Impaired Oxidative Disposal Pathways of Fatty Acids
Oxidative pathways are collectively an important disposal pathway for free fatty acids. Although these pathways can generate reactive oxygen species, oxidant stress that actually leads to injury is unusual if the endogenous antioxidant mechanisms are appropriately responsive. The smooth endoplasmic reticulum (SER) cytochrome P450 enzymes (CYPs) oxidize a variety of free fatty acids, including unusual branched-chain fatty acids and fatty acids with odd numbers of carbons not readily handled by the mitochondrial β-oxidation enzymes. Often called the microsomal system because during laboratory analysis the liver SER is homogenized into microsomes, this system readily attacks unusual fatty acids and even common fatty acids, initiating their oxidative disposal. The CYPs typically oxidize the aliphatic end of free fatty acids, generating potentially toxic dicarboxylic acids.
Peroxisomes play an important role in the disposal of dicarboxylic acids such as those produced by CYP ω-oxidation of fatty acids, very-long-chain fatty acids, and methyl branched-chain fatty acids. Peroxisomal fatty acid oxidase metabolizes fatty acids through β-oxidation, but unlike mitochondria where oxygen is fully reduced to water, peroxisomal enzymes only partially reduce oxygen to form hydrogen peroxide. Peroxisomes are replete with catalase to deal with this and prevent oxidant-mediated cellular injury. The importance of peroxisomes in preventing lipotoxicity has been shown through manipulation of PPARα, the transcription factor governing the expression of peroxisomal enzymes. PPARα null mice develop severe steatosis and steatohepatitis with prolonged fasting [37–39] and a spontaneous mutation in the Foz mouse strain that prevents disposal of fatty acids through peroxisomal oxidation or triglyceride formation causes fibrosing steatohepatitis on a high fat diet [40]. Conversely, upregulation of peroxisomal β-oxidation with PPARα ligands (eg, fibrates) prevented fat accumulation and NASH in murine models [41]. The experience with these agents in human disease has met with mixed results and is being pursued in clinical trials.
Mitochondrial β-oxidation is the major oxidative fate of free fatty acids. Mitochondrial dysfunction, whether genetic or acquired, is often associated with steatohepatitis [42–44]. Whether this is because of impairment at the level of the liver or a consequence of mitochondrial dysfunction in tissues such as muscle and adipose tissue is unclear. Defects in the mitochondrial electron transport chain can lead to increased reactive oxygen species generation with oxidant-mediated injury if antioxidant mechanisms are overwhelmed. Interestingly, rats selected for poor aerobic capacity have impaired muscle mitochondrial function and hepatic steatosis [45], whereas SNPs that confer increased mitochondrial biogenesis through peroxisome proliferator-activated receptor-γ coactivator-1 are found with increased frequency in high-level athletes. The ability of mitochondria to oxidize fatty acids is strongly regulated by feedback inhibition of carnitine palmitoyl transferase by malonyl CoA, which in turn is generated by acetyl CoA carboxylase (ACC). Mice deficient in this enzyme thus oxidize more fatty acids and are resistant to diet-induced obesity and lipotoxic impairment of insulin signaling [46]. A better understanding of the role of mitochondrial function in the liver, adipose tissue, and muscle appears to be emerging and may lead to new paradigms for the pathogenesis of diabetes, insulin resistance, and exercise tolerance and how these interact in the development of hepatic lipotoxicity.
Impaired Triglyceride Formation and Secretion
Although some fatty acids are needed for maintenance of cellular functions such as membrane synthesis, under normal circumstances most fatty acids that are not metabolized through oxidative pathways are converted to triglyceride for secretion as VLDL or temporary storage in lipid droplets if the secretory pathway is overwhelmed. If the conversion of fatty acids to triglyceride is impaired and compensatory oxidative pathways are not adequate, lipotoxic inhibition of insulin signaling and cellular injury can ensue. This circumstance has been amply demonstrated in animal models [7•, 8, 33••, 47, 48] although equally convincing data in humans is lacking [16].
Once triglyceride is formed, it can either be stored in droplets or packaged in nascent VLDL particles. Both processes are complex and require a repertoire of metabolic machinery that is prone to defects and deficiencies [49]. Understanding the effects of altered triglyceride formation and handling in animal models is complicated by the fact the same processes take place in adipose tissue, and thus a liver phenotype may be caused by multiple effects including liver-specific effects or alterations in the exposure of the liver to fatty acids originating in adipose tissue.
Secretion of fat as triglyceride requires functional apoB100, an ample supply of amino acids to synthesize apoB100, functional microsomal triglyceride transfer protein (MTP) and its partner protein disulfide isomerase, a supply of phosphatidyl choline, and an intact cytoskeletal system to facilitate the secretory process. As such, the process is subject of multiple acquired and genetic defects that can lead to triglyceride accumulation as droplets. Abetalipoproteinemia and hypobetalipoproteinemia, genetic diseases associated with dysfunctional MTP, are associated with steatosis, but not all patients develop liver disease, suggesting the need for additional defects such as impaired mitochondrial β-oxidation to handle fatty acids that might be released through autophagic lipolysis of the excessive lipid droplets.
Conclusions
Emerging from ample animal data and recent human data is a new hypothesis for the pathogenesis of NASH in which the accumulation of excessive triglyceride and the development of lipotoxic cellular injury occur in parallel but independently. Mechanistically, this hypothesis implies that in the setting of excess fatty acids, some people may develop only steatosis whereas others develop steatohepatitis. An important caveat is that steatosis does not “progress” to steatohepatitis, a dogma that has persisted in the field despite longitudinal data to the contrary. Available data indicate that few patients with just steatosis are later found to have steatohepatitis or fibrosis, and one must wonder if tests for steatohepatitis are too insensitive to detect minor but meaningful amounts of lipotoxic liver injury in such individuals. Alternatively, lipotoxic injury could be episodic to the degree that it depends on variations in diet such as consumption of trans fats, a recently recognized cause of steatohepatitis in mice [50•].
The nontriglyceride lipotoxic liver injury hypothesis of the pathogenesis of NASH compels us to focus our therapeutic efforts on reducing the burden of fatty acids that the liver must handle, originating either from peripheral lipolysis or de novo synthesis from excessive carbohydrates. Effective interventions can accomplish this by improving insulin sensitivity at the level of adipose tissue to prevent inappropriate peripheral lipolysis and reducing exposure of the liver to excessive carbohydrates. To this end, reducing carbohydrate consumption through dietary changes and increasing muscle glucose uptake through exercise remain important cornerstones of treatment and prevention of the lipotoxic liver injury we know as NASH.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Anderson N, Borlak J: Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev 2008, 60:311–357. The authors extensively review the mechanisms of hepatic steatosis and nontriglyceride lipotoxicity as well as the intracellular processes leading to lipid droplet accumulation.
•• Malhi H, Gores GJ: Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis 2008, 28:360–369. This article provides an excellent review of hepatic lipotoxicity with a focus on mechanisms of apoptotic cell death in the liver.
Yetukuri L, Katajamaa M, Medina-Gomez G, et al.: Bioinformatics strategies for lipidomics analysis: characterization of obesity related hepatic steatosis. BMC Systems Biology 2007, 1:12.
Puri P, Baillie RA, Wiest MM, et al.: A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46:1081–1090.
• Cusi K: Role of insulin resistance and lipotoxicity in nonalcoholic steatohepatitis. Clin Liver Dis 2009, 13:545–563. This article provides the latest update on lipotoxicity and its adverse effects on the liver and other target organs.
Puri P, Mirshahi F, Cheung O, et al.: Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134:568–576.
• Monetti M, Levin MC, Watt MJ, et al.: Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 2007, 6:69–78. This murine study provided convincing evidence that accumulation of triglyceride droplets in the liver is not responsible for hepatic insulin resistance.
Yamaguchi K, Yang L, McCall S, et al.: Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45:1366–1374.
Choi CS, Savage DB, Kulkarni A, et al.: Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 2007, 282:22678–22688.
• Romeo S, Kozlitina J, Xing C, et al.: Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008, 40:1461–1465. The article describes a genetic analysis of the Dallas Heart Study cohort that identifies a genetic polymorphism replacing an isoleucine with a methionine in adiponutrin and is associated with increased hepatic fat content independent of BMI and diabetes; three people with loss-of-function mutations had the highest liver fat content.
Kotronen A, Johansson LE, Johansson LM, et al.: A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52:1056–1060.
•• Choi SS, Diehl AM: Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol 2008, 19:295–300. This article is an excellent summary of the data linking an excess flux of fatty acids through the liver, and not triglyceride accumulation, in the pathogenesis of NASH.
• Schenk S, Saberi M, Olefsky JM: Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 2008, 118:2992–3002. This article provides a good review focusing on what happens in adipose tissue that predisposes to lipotoxicity in other organs.
Listenberger LL, Han X, Lewis SE, et al.: Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003, 100:3077–3082.
Schaffer JE: Lipotoxicity: when tissues overeat. Curr Opin Lipidol 2003, 14:281–287.
Bradbury MW, Berk PD: Lipid metabolism in hepatic steatosis. Clin Liver Dis 2004, 8:639–671.
Li Z, Berk M, McIntyre TM, et al.: The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47:1495–1503.
•• Han MS, Park SY, Shinzawa K, et al.: Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res 2008, 49:84–97. This multifaceted study provides good evidence for the identity of one possible mediator of lipotoxicity.
Unger RH: Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003, 144:5159–5165.
Shi Y, Cheng D: Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am J Physiol Endocrinol Metab 2009, 297:E10–E18.
Musso G, Gambino R, Cassader M: Recent insights into hepatic lipid metabolism in nonalcoholic fatty liver disease (NAFLD). Prog Lipid Res 2009, 48:1–26.
Donnelly KL, Smith CI, Schwarzenberg SJ, et al.: Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005, 115:1343–1351.
•• Singh R, Kaushik S, Wang Y, et al.: Autophagy regulates lipid metabolism. Nature 2009, 458:1131–1135. The authors describe autophagy (lipophagy) as a mechanism of triglyceride turnover in hepatocytes.
Brasaemle DL: Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 2007, 48:2547–2559.
Granneman JG, Moore HP: Location, location: protein trafficking and lipolysis in adipocytes. Trends Endocrinol Metab 2008, 19:3–9.
Ahmadian M, Duncan RE, Sul HS: The skinny on fat: lipolysis and fatty acid utilization in adipocytes. Trends Endocrinol Metab 2009, 20:424–428.
Lafontan M: Advances in adipose tissue metabolism. Int J Obes (Lond) 2008, 32(Suppl 7):S39–S51.
Prentki M, Madiraju SR: Glycerolipid metabolism and signaling in health and disease. Endocr Rev 2008, 29:647–676.
Leone TC, Lehman JJ, Finck BN, et al.: PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 2005, 3:e101.
Ouyang X, Cirillo P, Sautin Y, et al.: Fructose consumption as a risk factor for nonalcoholic fatty liver disease. J Hepatol 2008, 48:993–999.
Hernandez C, Lin JD: A sweet path to insulin resistance through PGC-1β. Cell Metab 2009, 9:215–216.
Rajasekar P, Anuradha CV: Fructose-induced hepatic gluconeogenesis: effect of L-carnitine. Life Sci 2007, 80:1176–1183.
•• Li ZZ, Berk M, McIntyre TM, Feldstein AE: Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturease. J Biol Chem 2009, 284:5637–5644. The authors report evidence that triglyceride does not cause lipotoxic injury in the liver, although it can cause endoplasmic reticulum stress.
Nakayama H, Otabe S, Ueno T, et al.: Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism 2007, 56:470–475.
Shimano H: SREBPs: physiology and pathophysiology of the SREBP family. Febs J 2009, 276:616–621.
Wu X, Zhang L, Gurley E, et al.: Prevention of free fatty acid-induced hepatic lipotoxicity by 18β-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology 2008, 47:1905–1915.
Leone TC, Weinheimer CJ, Kelly DP: A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A 1999, 96:7473–7478.
Fan CY, Pan J, Usuda N, et al.: Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem 1998, 273:15639–15645.
Reddy JK, Hashimoto T: Peroxisomal β-oxidation and peroxisome proliferator-activated receptor α: an adaptive metabolic system. Annu Rev Nutr 2001, 21:193–230.
Arsov T, Larter CZ, Nolan CJ, et al.: Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem Biophys Res Commun 2006, 342:1152–1159.
Ip E, Farrell GC, Robertson G, et al.: Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38:123–132.
Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al.: Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120:1183–1192.
Caldwell SH, Chang CY, Nakamoto RK, Krugner-Higby L: Mitochondria in nonalcoholic fatty liver disease. Clin Liver Dis 2004, 8:595–617.
Pessayre D: Role of mitochondria in nonalcoholic fatty liver disease. J Gastroenterol Hepatol 2007, 22(Suppl 1):S20–S27.
Wisløff U, Najjar SM, Ellingsen Ø, et al.: Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 2005, 307:418–420.
Choi CS, Savage DB, Abu-Elheiga L, et al.: Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A 2007, 104:16480–16485.
Gutiérrez-Juárez R, Pocai A, Mulas C, et al.: Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J Clin Invest 2006, 116:1686–1695.
Miyazaki M, Flowers MT, Sampath H, et al.: Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab 2007, 6:484–496.
Ducharme NA, Bickel PE: Lipid droplets in lipogenesis and lipolysis. Endocrinology 2008, 149:942–949.
• Tetri LH, Basaranoglu M, Brunt EM, et al.: Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol 2008, 295:G987–G995. This article reports the first evidence for the potential role of dietary trans fats in causing NASH.
Disclosure
The author has served as a consultant to Amylin, Astellas, Centocor, and Gilead.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Neuschwander-Tetri, B.A. Nontriglyceride Hepatic Lipotoxicity: The New Paradigm for the Pathogenesis of NASH. Curr Gastroenterol Rep 12, 49–56 (2010). https://doi.org/10.1007/s11894-009-0083-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-009-0083-6