
Abstract
Liver cancer is the third leading cause of cancer-related death worldwide. And Hepatocellular carcinoma (HCC) is the most common form of liver cancer. The extreme variability of the clinical outcome caused a major challenge of HCC, which makes it difficult to properly stage the disease and thereby estimate the prognosis. That’s because the rapidly growing tumor displays heterogeneity of genetic and histopathologic characteristics. The risk of HCC may be affected by several known environmental factors such as hepatitis viruses, alcohol, cigarette smoking, and others. The aetiological factors associated with HCC have been well characterized; however, their effects on the accumulation of genomes changes and the influence of ethnic variation in risk factors still remain unclear. Advances in sequencing technologies have enabled the examination of liver cancer genomes at high resolution; somatic mutations, structural alterations, HBV integration, RNA editing and retrotransposon changes have been comprehensively identified. In addition, integrated analyses of trans-omics data have identified diverse critical genes and signaling pathways implicated in hepatocarcinogenesis. These analyses have revealed potential therapeutic targets, and prepared the way for new molecular classifications for clinical application. Therefore, the international collaborations of cancer genome sequencing projects are expected to contribute to an improved understanding of risk assessment, diagnosis and therapy for HCC. This review discusses the contribution of heterogeneity such as aetiological factors, tumor microenvironment, genetic variations, epigenetic changes and signaling pathways in HCC progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
16.1 Introduction
HCC is a leading malignancy worldwide (Torre et al. 2015). Chronic liver damage which may result from chronic hepatitis, liver cirrhosis and fatty liver disease, is closely associated with HCC. Hepatitis virus infection, alcohol intake, aflatoxin B exposure, and some metabolic diseases such as obesity, haemochromatosis and diabetes mellitus are well-known risk factors for HCC (El-Serag 2012; Forner et al. 2012; Yu et al. 2013). The incidence of HCC is high in East Asian and African countries (Torre et al. 2015; El-Serag 2012; Forner et al. 2012; Shaib and El-Serag 2004). Africa and Asian countries (except Japan) have the highest rate of HBV infection in the world (El-Serag 2012). However, the number of patients infected with HCV has been rapidly increasing in Japan and Western countries, especially in the USA where viral hepatitis infection is partly mediated through drug abuse (El-Serag 2012; Forner et al. 2012). With the exception of environmental risk factors, individual genetic predisposition may be linked to the risk of HCC as suggested by the fact that in a relevant percentage of HCC cases, i.e., about 20 % of cases diagnosed in the United States, without known predisposing risk factors, including alcohol use or viral hepatitis, can be identified (El-Serag and Mason 2000). The role of genetic factors in the risk of HCC is supported by strong evidence from animal models, which have enabled the identification of the number and chromosomal location of loci affecting genetic susceptibility to chemically induced hepatocarcinogenesis in both mice and rats (Dragani et al. 1996; Feo et al. 2006). In this Review, we mainly focus on HCC, as HCC showed distinctive genomic alterations at present, which includes estimated risk of HCC according to particular genetic factors.
16.2 Aetiological Factors for HCC
The risk of HCC may be affected by several known environmental factors such as hepatitis viruses, alcohol, cigarette smoking and so on (IARC 2004; Bosch et al. 2004; Kuper et al. 2000; Llovet et al. 2003), among which the prevalence of chronic hepatitis B (HBV) or C (HCV) virus infections plays an identified role in the incidence of HCC. HCC is more prevalent in Southeast Asia and sub-Saharan Africa, where HBV infection is endemic, but HBV-related liver cancer cases also occur in western countries (Bosch et al. 2004; Llovet et al. 2003). Chronic carriers of HBV have up to a 30-fold increased risk of HCC (IARC 1994; Evans et al. 2002; Franceschi et al. 2006). In western countries, HCV infection plays a major role in the pathogenesis of HCC, and it has become more prevalent over the past decades, accompanied by a higher incidence and mortality from HCC (El-Serag and Mason 2000; IARC 1994). The fact that alcohol consumption causes liver cirrhosis and is an independent risk factor for primary liver cancer has been disclosed by a large number of cohort and case–control studies (Kuper et al. 2000; Baan et al. 2007; Ogimoto et al. 2004). And epidemiological studies showed that increasing HCC risks associated with exposure to aflatoxins after adjustment for HBV exposure (IARC 2002). What’s more, cigarette smoking has been causally associated with the risk of HCC (IARC 2004; Kuper et al. 2000), and heavy smoking and heavy drinking was reported to have a multiplicative effect in HCC development (Kuper et al. 2000).
In addition to environmental risk factors, individual genetic predisposition may also play a role in the risk of HCC with the current evidence from epidemiological/genetic studies in human populations, which argues for the important role of monogenic and polygenic factors in determining the risk of HCC development. Rare monogenic syndromes such as alpha1-antitrypsin deficiency, hemochromatosis, acute intermittent, cutanea tarda porphyria, and glycogen storage disease type I as well as hereditary tyrosinemia type I are associated with a high risk of HCC (Andant et al. 2000; Elmberg et al. 2003; Elzouki and Eriksson 1996; Fracanzani et al. 2001; Haddow et al. 2003; Janecke et al. 2001; Ostrowski et al. 1983; Scott 2006; Weinberg et al. 1976). Several common conditions or diseases inherited as polygenic traits e.g. autoimmune hepatitis, type 2 diabetes, non-alcoholic steatohepatitis, hypothyroidism, and a family history of HCC also show an increased risk of HCC compared to the normal population (El-Serag et al. 2006; Hashimoto et al. 2009; Hassan et al. 2009; Werner et al. 2009; Hemminki and Li 2003). Therefore, the increased risk of HCC may not be directly linked to genetic disorders, but instead single germ-line mutations or conditions regulated by complex genetics may cause chronic damage such as liver cirrhosis of the target organ, in turn causing the oncogenic mutations and/or promoting preexisting endogenous or virus- or chemical-induced mutations which lead to HCC. Indeed, similar to those occurring in human liver cirrhosis, conditions of hepatic necrosis and regeneration may promote carcinogen-induced hepatocarcinogenesis, as suggested by the experiments with rodent models (Dragani et al. 1986). Thus, cirrhosis from any cause appears to be the common signaling pathway by which some risk factors exert their hepatocarcinogenesis (Fig. 16.1). Overall, the genetic susceptibility to HCC is characterized by a genetic heterogeneity; With the fact that, a high individual risk of HCC may thus be caused by several unlinked single gene defects, whose carriers are rare in the general population, or by more common conditions inherited by complex genetics.

Aetiological factors for HCC. The risk of HCC may be mainly affected by several known environmental factors, including hepatitis viruses, alcohol, cigarette smoking, and others. In addition to environmental risk factors, individual genetic predisposition may play a role in the risk of HCC as suggested by the fact that in a relevant percentage of HCC cases, i.e.
16.3 Heterogeneity of Tumor Microenvironment in HCC
As a highly heterogeneous disease, HCC displays differences in angiogenesis, extracellular matrix proteins and the immune microenvironment, which contribute to HCC progression. Therefore, a better understanding of its heterogeneity will greatly contribute to the development of strategies for the HCC treatment.
16.3.1 Angiogenic Heterogeneity
HCC has wide variations in vascularity that are dependent upon tumor size (T stage) and histological grade, and angiogenic switch depends on the balance between pro- and antiangiogenic factors at different stages of tumor progression (Baeriswyl and Christofori 2009). Pro-angiogenic factors include VEGF, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), angiopoietin-1 and angiopoietin-2. And anti-angiogenic factors include thrombospondin-1 (TSP1), endostatin, interferon-α, interferon-β and angiostatin. VEGF expression is up-regulated by hypoxia-induced factor-1α (HIF-1α) to switch angiogenic phenotype (Fang et al. 2001). Therefore, HCC is a hypervascularized tumor because of increased angiogenic phenotype (Muto et al. 2015), which is not only required for tumor growth supplied with oxygen and essential nutrients but also facilitates metastasis. A higher level of VEGF mRNA in tumor tissue correlates with increased post-resection recurrences, suggesting that an altered balance between angiogenic stimulators and inhibitors contributes to cancer progression. Therefore, angiogenic heterogeneity is associated with angiogenic molecules such as VEGF, PEDF and HIF-1α (Fig. 16.2a) that could be different among various tumor sizes and time intervals during hepatocarcinogenesis, which needs to be taken into the consideration when we decide to carry out an anti-angiogenic therapy to prevent recurrence in HCC patients (Wu et al. 2007).

Heterogeneity of hepatocellular carcinoma. (a) Angiogenesis (b) immune microenvironment
16.3.2 Heterogeneity of Extracellular Matrix
Extracellular matrix (ECM) components mainly consist of collagen, laminin, fibronectin, glycosaminoglycan and proteoglycan. Because of continuous repatterning of the ECM, HCC tumor cells can invade via direct or indirect interactions among ECM, stroma cells and HCC (Carloni et al. 2014). The major tumor ECM concerned in this process are collagen type IV, lysyl oxidase (LOX) and matricellular proteins (MCPs), whereas MCPs is prime metastatic niches in HCC (Chew et al. 2012; Fang et al. 2013; Wong and Rustgi 2013). Overall, a dynamic ECM contributes to hepatocarcinogenesis. Matrix metalloproteinases (MMPs) were associated with tumor invasion and migration, particularly MMP2, MMP9 and MT1-MMP, which play a pivotal role in the degradation of ECM to facilitate HCC metastasis (Ogasawara et al. 2005). Furthermore, connective tissue growth factor (CTGF) was overexpressed in HCC patients whereas downregulating the expression of CTGF could inhibit HCC growth which could be a potential therapeutic strategy for HCC treatment (Jia et al. 2011). As we all known, epithelial mesenchymal transition (EMT) is an very important step in hepatocarcinogenesis, which involves the interactions between HCC cells and ECM mediated by transforming growth factor-β1(TGF-β1) and/or PDGFR signaling pathway (Dorn et al. 2010).
The heterogeneity of ECM makes it a challenging topic to inhibit ECM proteins due to the various ECM proteins and complex mechanisms. However, it still needs to be considered for the target therapy in which the proteins required to maintain or degrade ECM-related proteins could be used.
16.3.3 Heterogeneity of the Immune Microenvironment
The immune microenvironment in HCC is also found to be heterogeneous. Cell types within or around tumors include cytotoxic T cells (CD8+), regulatory T cells (Treg), natural killer (NK), natural killer T cells (NKT), myeloid-derived suppressor cells (MDSCs) and so on (Fig. 16.2b). These cells can play an important role in promoting or inhibiting HCC progression (Junttila and de Sauvage 2013) (Fig. 16.3).

Somatic alterations in the HCC genome. (a) Some representative somatic mutations in the whole exon domain (exome), which is determined by massively parallel sequencing (b) Some representative somatic mutations in the whole-genome domain, which is performed by whole-genome sequencing (c) representative somatic change of retrotransposons in HCC
CD8+ T cells are found infiltrating among HCC tumor cells, whereas CD4+ T cells are found mainly around the tumor or liver interface (Kasper et al. 2009). Treg cells promote immune suppression by secreting IL-10 and TGF-β and direct contact with tumor cells (Wang et al. 2012). On the other hand, Tregs could inhibit CD8+ T cells responses and would enhance immune responses when the Treg number is low (Huang et al. 2012a). Cytotoxic T cells (CTLs) have the cytotoxicity to kill tumor cells which lead to less immune response against HCC (Gao et al. 2007). Therefore, low number of Tregs and increased number of activated CTLs are associated with a favorable prognosis. A higher frequency of Th17 cells which secret IL-22 are found in advanced HCC patients with poor survival (Zhang et al. 2009; Liao et al. 2013). And a higher expression of IL-22 can activate Stat-3 signaling and promote tumor growth (Jiang et al. 2011).
Some studies have reported that the frequency and cytotoxic function of NK cells to be reduced both in the liver and peripheral blood of HCC patients (Cai et al. 2008; Gao et al. 2009; Hoechst et al. 2009). The reduced NK cell function was associated with lower expression of NK cell receptor, NKG2D (Sha et al. 2014). Invariant natural killer T (iNKT) cells was also shown to be increased in patients produced Interferon-gamma (IFN) to inhibit tumor growth (Crowe et al. 2005). In addition, CD4+ NKT cells produced Th2-cytokine which could also inhibit CD8 T cell expansion and function (Bricard et al. 2009).
Myeloid-derived suppressor cells (MDSCs) are heterogeneous in HCC patients that includes macrophages, dendritic cells, immature granulocytes and early myeloid progenitors. MDSCs could inhibit T cell responses as well as natural killer cell function via the NKp30 receptor (Hoechst et al. 2009). Overall, the development of immunotherapy requires an understanding of the heterogeneous microenvironment, regulation of cytokines at different stages of HCC, and the functional activity of T cells, CTLs, NK cells and MDSCs etc.
16.4 Heterogeneity of HCC Genomes
Progress in sequencing technologies have made it possible to examine liver cancer genomes at high resolution. Somatic mutations, structural alterations, HBV integration, RNA editing, retrotransposon changes and so on have been comprehensively identified. In addition, integrated analyses of genome, transcriptome and methylome data have identified various critical genes and pathways involved in hepatocarcinogenesis, and paved the way for new molecular classifications for clinical application. Furthermore, the international collaborations of cancer genome sequencing projects are expected to contribute to an improved understanding of risk evaluation, diagnosis and therapy strategy for this cancer.
16.4.1 Somatic Alterations in the HCC Genome
Whole-genome and whole-exome sequencing have provided a comprehensive and high-resolution view of somatic genomic alterations in HCC. The liver cancer genome contains multiple types of somatic alterations, including mutations such as single nucleotide substitutions, small insertions and deletions, changes of gene copy numbers, intra-chromosomal rearrangements and inter-chromosomal rearrangements. For the past few years, an increasing appreciation and identification of somatic mutations that drive human tumors have enable us within reach of personalized cancer medicine.
16.4.1.1 Genome-Wide Copy Number Analysis
Somatic DNA copy number changes in human cancers genomes have been detected mainly by array-based comparative genome hybridization methods (CGH). That’s because array-based CGH can enable high-throughput and high-resolution screening of genome-wide DNA copy number changes (Pollack et al. 1999). In addition to well-known oncogenes e.g. MYC and CCND1, and tumour suppressor genes, such as TP53 and RB, liver cancers harbour multiple chromosomal amplifications and deletions, and Shibata et al. have summarized these recurrent copy number alterations on the Table 16.1 (Shibata and Aburatani 2014).
In recent years, several studies reported chromosomal alterations in HCC using array CGH (Chochi et al. 2009; Kakar et al. 2009; Patil et al. 2005; Schlaeger et al. 2008). Guo X et al. discovered significant gains in 5p15.33 and 9q34.2–34.3 and losses in 6q, 9p and 14q in addition to the regions that were previously identified by conventional CGH analyses by a meta-analysis of 159 HCC array CGHs (Chochi et al. 2009; Kakar et al. 2009; Patil et al. 2005; Schlaeger et al. 2008). In a study by Patil et al. (2005), the correlation between DNA copy numbers and gene expression pattern at the 8q region was demonstrated, which was frequently amplified in 49 HCC samples. A study of Roessler et al. (2012) identified ten driver genes that were associated with poor survival by integrating high-resolution array CGH data and gene expression profiles of 256 HCC cases to gain the genes which have the significant correlation between somatic copy number alterations and the whole genome expression patterns. In order to identify potential cancer driver genes, Woo et al. (2009) integrated whole genome copy number profiles of 15 HCC cases with gene expression profiles of 139 HCC cases. They analyzed genes that have a correlation between expression levels and copy number changes, finally discovered 50 potential driver genes that are linked to HCC prognosis.
16.4.1.2 Whole-Exome Sequencing
Advance in sequencing technologies have enabled researchers to explore the liver cancer genome more deeply. Whole exome sequencing (WES) can efficiently identify mutations in protein-coding exons, which are much more easily identifiable than the mutations or variants in non-coding regions. This approach concerns target-enrichment of whole protein-coding exons across the human genome (30–40 Mb, approximately 1 % of the whole human genome) adopting in-solution RNA or oligonucleotide DNA probe hybridization technologies (Gnirke et al. 2009; Hodges et al. 2007) which enable the comprehensive detection of somatic alterations in the protein-coding exons, and have discovered many novel genes involved in liver cancer. In the research of Li M et al., the recurrent inactivating mutations of the ARID2 gene in 18.2 % of HCV-associated HCCs were identified by exomic sequencing of 10 HCV-positive HCCs and analysis of an additional tumour cohort of various aetiological backgrounds (Li et al. 2011). Huang et al. (2012b) sequenced nine pairs of HCCs and their intrahepatic metastases across whole exome to come out with the result that although about 94.2 % substitutions were common in both primary and metastatic tumours, a fraction of mutations were detected in 1.1 % primary or 4.7 % metastatic tumours. Among these mutations, KDM6A, CUL9, RNF139, AKAP4 and FGD6 were only identified in the metastatic tumors of three individuals. Using whole-exome sequencing of 87 HCC cases, Cleary et al. (2013) found recurrent alterations in the NFE2L2–KEAP1 and MLL pathways, while C16orf62 and RAC2 with lower mutation frequencies. According to copy number analysis of 125 HCC cases and whole exome sequencing of 24 of these cases, Guichard et al. (2012) detected novel recurrent mutations in the ARID1A, RPS6KA3, NFE2L2 and IRF2 genes. Interestingly, inactivation of the IRF2 gene was exclusively observed in HBV-related HCC, which led to disruption of TP53 function. In addition, alterations in chromatin remodelers were found in association with alcohol-related HCC.
16.4.1.3 Whole-Genome Sequencing
Many research groups have sequenced the whole liver cancer genome in further attempts to detect all somatic driver events involved in hepatocarcinogenesis. Whole genome sequencing (WGS) can cover almost all the genome sequences in human and detect variants in non-coding regions, copy number alterations, genomic rearrangements, and virus genome integrations except single nucleotide changes (Nakagawa and Shibata 2013). By sequencing HCV-related HCC cases, >16,000 somatic mutations and 26 intra-chromosomal and interchromosomal rearrangements inducing four fusion transcripts were identified, including the TP53, AXIN1, ADAM22, JAK2, KHDRBS2, NEK8, TRRAP and BCORL1 genes, as well as a large number of somatic mutations in genes encoding phosphoproteins and those with bipartite nuclear signals. Through high-resolution analysis, the authors also identified intratumor heterogeneity of the mutations, including inactivation of the TSC complex in a subpopulation of HCV-related HCCs (Totoki et al. 2011). By performed whole-genome sequencing of 27 HCCs and matched normal genomes, Fujimoto et al. showed that 25 of which were associated with HBV or HCV infection. The average number of somatic point mutations at the whole-genome level was 4.2 per Mb. Moreover, several chromatin regulators mutations, including ARID1A, ARID1B, ARID2, MLL, MLL3, BAZ2B, BRD8, BPTF, BRE and HIST1H4B, were identified in 50 % tumors. These mutations were marginally linked to the stage of liver fibrosis and hepatic invasion (Fujimoto et al. 2012). By a whole-genome sequencing study of 88 matched HCC tumor/normal pairs, 81 of which are Hepatitis B virus (HBV) positive, Kan et al. (2013) seeked to identify genetically altered genes and pathways implicated in HBV-associated HCC cases. They found the most frequently mutated oncogene (15.9 %) and the most frequently mutated tumor suppressor (35.2 %) are beta-catenin and TP53, respectively. The Wnt/beta-catenin and JAK/STAT pathways, mutated in 62.5 % and 45.5 % of cases, respectively, are possible to be two major oncogenic drivers in HCC. This research also identified several prevalent and potentially actionable mutations, such as activating mutations of Janus kinase 1 (JAK1) in 9.1 % of patients, suggesting that these genes or pathways could be new therapeutic targets in HCC (Kan et al. 2013).
16.4.2 Somatic Change of Retrotransposons in HCC
The human genome contains a variety of repetitive genome sequences, including tandem repeats and retrotransposons e.g. short interspersed nuclear elements (SINEs) and long interspersed nuclear elements (LINEs). In the human genome, Alu and LINE-1 are major forms of SINEs and LINEs, respectively (Treangen and Salzberg 2012). LINE-1 retrotransposons are a major source of endogenous mutagenesis in humans (Burns and Boeke 2012; Levin and Moran 2011). Retrotransposon insertions can deeply alter gene structure and expression (Levin and Moran 2011; Cordaux and Batzer 2009; Han et al. 2004; Faulkner et al. 2009) and have been identified in nearly 100 cases of diseases (Faulkner 2011; Hancks and Kazazian 2012). LINE-1 activity is consequently suppressed in most somatic cells by methylation of a CpG island in the internal LINE-1 promoter (Coufal et al. 2009; Swergold 1990). By contrast, LINE-1 is often hypomethylated in tumor cells, removing a key obstacle to retrotransposition (Levin and Moran 2011).
Shukla et al. (2013) used enhanced retrotransposon capture sequencing (RC-seq) to detect 19 HCC tumors and matched adjacent liver tissue that were confirmed positive for HBV or HCV infection and elucidated endogenous LINE-1-mediated retrotransposition in the germline and somatic cells of HCC patients. The authors reported two archetypal mechanisms revealing MCC and ST18 as HCC candidate genes. MCC is a highly plausible liver tumor suppressor. However distinct germline LINE-1 or Alu insertions contribute to MCC suppression in tumor and nontumor liver tissue and then activate the WNT/CTNNB1 pathway. The other event is a tumor-specific LINE-1 insertion which activates a potential oncogene, Suppression of tumorigenicity 18 (ST18), in liver tumors (Shukla et al. 2013).
16.4.3 HBV Genome Integrations in the Host Genome
Chronic HBV infection is a major risk factor for HCC, and more than half of HCC cases in the world are attributed to HBV infection. HBV is a DNA virus whose genome can be integrated into the host genome. The integration of the viral DNA sequences affect host gene expression near the integration site and its effect on the integrity of the host genome is associated with virus-mediated hapatocarcinogenesis (Neuveut et al. 2010). By Southern blot analysis or inverse PCR, previous studies identified the integration of HBV DNA sequences into host genomes both in HCC samples and non-tumorous tissues from patients with chronic HBV hepatitis (Brechot et al. 1980). Advanced current genome sequencing technology have enabled researchers to detect virus integration events more comprehensively and at higher resolution than previously.
HBV integration was reportedly observed within or upstream of the TERT gene in tumor tissues in HCC cases with HBV infection (Fujimoto et al. 2012). Furthermore, Sung et al. (2012) reported integration events at the MLL4, CCNE1, SENP5, FN1 and ROCK1 genes. They conducted whole-genome sequencing of 81 HBV-positive and seven HBV-negative HCC samples and found that most HBV breakpoints in HCC were close to active coding genes, which potentially enabled HBV to integrate into the open chromatin region more effectively (Sung et al. 2012). Jiang et al. (2012a) also made comprehensive analyses of HBV-related HCC and their corresponding normal tissues. They found clonal and high-abundance viral integrations in tumor tissue, while many viral integration sites randomly scattered throughout the genome in nontumor liver tissues (Jiang et al. 2012b). These research indicated that a heterogeneous and widespread viral integration landscape in HCC as well as nontumor liver tissue and integration events may cause aberrant expression of genes near the integration sites, alterations of DNA copy number and emergence of fusion genes (Sung et al. 2012; Jiang et al. 2012b). Moreover, recurrent integration of HBV was also detected in the FAR2, ITPR1, MAPK1, IRAK2 and MLL genes (Sung et al. 2012; Gozuacik et al. 2001; Paterlini-Brechot et al. 2003; Murakami et al. 2005; Saigo et al. 2008).
16.4.4 DNA Methylation in HCC
(update) DNA methylation and demethylation is an important mechanism of regulating gene expression and chromatin structure in normal cells. DNA methylase contribute to the methylation of cytosine at CpG islands at the gene promoter region. Aberrant DNA methylation at the gene promoter region is an important mechanism in inactivation of tumor suppressor gene (Nagae et al. 2011; Hendrich and Bird 1998).
Altered DNA methylation is an early event in HCC development. Global hypomethylation has a critical role in increasing chromosomal instability and mainly affected intergenic regions of the genome (Eden et al. 2003). DNA hypermethylation is the state where the methylation of “normally” undermethylated DNA domains, which predominantly consist of CpG islands (Rollins et al. 2006), increases. Abnormal gains of DNA methylation (hypermethylation) of typically unmethylated CpG island-containing promoters can lead to transcriptional repression and loss of gene function. In addition, non-CpG island-containing promoter coding region hypermethylation contribute to genes inactivation (Pogribny and James 2002; Tomasi et al. 2012).
The study of Udali et al. (2015) used array-based DNA methylation and gene expression data of all annotated genes from eight HCC patients undergoing curative surgery to analyze by comparing HCC tissue and homologous cancer-free liver tissue. They identified 159 hypermethylated-repressed, 56 hypomethylated-repressed, 49 hypermethylated-induced, and 30 hypomethylated-induced genes. Notably, promoter DNA methylation proved to be a novel regulatory mechanism for the transcriptional repression of genes e.g. involving the retinol metabolism (ADH1A, ADH1B, ADH6, CYP3A43, CYP4A22, RDH16), one-carbon metabolism (SHMT1), iron homeostasis (HAMP), and potential tumor suppressors (FAM107A, IGFALS, MT1G, MT1H, RNF180).
Nishida et al. (2014) applied Infinium Human Methylation 450 Bead Chip array which can analyze >485,000 CpG sites distributed throughout the genome to analyze comprehensive methylation from 117 liver tissues consisting of 59 HCC and 58 noncancerous livers. They identified 38,330 CpG sites with significant differences in methylation levels between HCCs and nontumors livers (DMCpGs). Among the DMCpGs, 92 % were hypomethylated and only 3051 CpGs (8 %) were hypermethylated in HCC. The DMCpGs were more common within intergenic regions with isolated CpGs. However, DMCpGs that were hypermethylated in HCC were predominantly located within promoter regions and CpG islands.
Shen et al. (2012) analyzed tumor and adjacent nontumor tissues from 62 Taiwanese HCC cases using Illumina methylation arrays which can screen 26,486 autosomal CpG sites. They found that a total of 2324 CpG sites significantly differed in methylation level. Among these CpG sites, 684 CpG sites significantly hypermethylated and 1640 hypomethylated in tumor compared to nontumor tissues. The 684 hypermethylation markers could be utilized for plasma DNA diagnostics. In addition, They identified the top 500 significant CpG sites using a 450 K array from 66 HCC cases. These differential methylations were able to distinguish HCC from adjacent nontumor tissues (Shen et al. 2013).
Previous study (Nishida et al. 2007) reported that extensive methylation is involved in CTNNB1 mutations, while TP53 mutation in HCC is often characterized by chromosomal instability. CpG islands promoter of the tumour suppressor genes CDKN2A and CDKN2B are frequently hypermethylated, leading to inactivation of the RB pathway (Zang et al. 2011). Methylation of the CDKN2A gene promoter occurs in 73 % of HCC tissues (Wong et al. 1999), 56 % of HBV-related HCC, and 84 % of HCV-related HCC (Narimatsu et al. 2004). Moreover, RASSF1A is methylated in up to 85 % of HCCs (Zhang et al. 2002), GSTP1 in 50–90 % (Yang et al. 2003; Zhong et al. 2002), and MGMT in 40 % (Zhang et al. 2003).
16.5 Heterogeneity of Signaling Pathways Affects the Progression of HCC
16.5.1 p53 Gene Pathway
As a tumor suppressor, p53 can initiate cell-cycle arrest, apoptosis, and senescence in response to cellular stress to maintain the integrity of the genome. About 50 % human tumors carry mutant p53, and many p53 mutants facilitate oncogenic functions such as increased proliferation, viability, and invasion or dominant-negative regulate the remaining wild-type p53 (Muller and Vousden 2013).
p53 plays important and unique roles in HCC. A study indicated that ablation of the p53-mediated senescence program in hepatic stellate cells under chronic liver damage promotes liver fibrosis and cirrhosis, which are associated with reduced survival; in addition, loss of p53 enhances the transformation of adjacent epithelial cells into HCC (Lujambio et al. 2013). p53 is mainly regulated by the E3 ubiquitin ligase MDM2. MDM2 binds p53 blocking p53-mediated transcriptional regulation, while simultaneously promoting its degradation (Brown et al. 2011). In addition, the MDM2–p53 binding can be disrupted by a small inhibitor Nutlin-3, which thereby activates p53 dependent apoptosis in different HCC cell lines (Zheng et al. 2010a). Therefore, Inhibition of MDM2–p53 binding could reactivate p53 in cancer cells with wild-type p53 and may offer an effective therapeutic approach for millions of cancer patients (Brown et al. 2011).
16.5.2 Hedgehog Pathway
Hedgehog signaling contributes to many aspects of cell differentiation, organ formation, cancergenesis and cancer metastasis. It is widely accepted that Hedgehog activity plays an important role in the progression of HCC. Many studies report that aberrant activation of Hedgehog signaling promote proliferation, viability, migration and invasion of HCC cells with complex underlying mechanisms (Zheng et al. 2010b, 2012; Lu et al. 2012).
Gli, Smo and PTCH were found to be overexpressed in HCC patients (Che et al. 2012; Jeng et al. 2013; Zhang et al. 2013). Lu et al. (2012) reported that Shh treatment can stimulate Hedgehog signaling to promote HCC cell invasion and migration by increasing GLI1 expression. Sicklick et al. (2006) found overexpression of Smo and an increase in the stoichiometric ratio of Smo to PTCH mRNA levels in HCC, this effect is associated with tumor size and Smo and PTCH may be prognostic marker of HCC. Downstream transcription factors, Gli, affect the proliferation, migration, invasion, angiogenesis, aberrant autophagy and stem cell regeneration in HCC (Zheng et al. 2013). Previous studies have found that GLI1 contributes to the EMT phenotype and intrahepatic metastasis and portal venous invasion of human HCCs (Zheng et al. 2010b). Other studies reported that GLI1 expression in HCC tissues is associated with disease-free survival, overall survival and rapid recurrence (Zheng et al. 2012). In vitro experiments indicated that GLI1 promotes proliferation, viability, colony formation, migration and invasion of Huh7 cells. In addition, inhibition of Hedgeho signaling by GANT61, which is a small-molecule inhibitor of GLI1, led to autophagy. The result demonstrate that Hedgehog signaling is involved in aberrant autophagy of HCC cells (Wang et al. 2013). Furthermore, Several Gli target genes have been identified such as cMyc, Cyclin D1 and FOXM1 (cell proliferation) and Bcl-2 (survival) (Lin et al. 2010). For example, the down-regulation Hedgehog signaling pathways could induce cell arrest at G1 and cause apoptosis by downregulation of Bcl-2 (Chen et al. 2008; Cheng et al. 2009; Kim et al. 2007; Zhang et al. 2011).
16.5.3 Wnt/β-Catenin Signaling
The Wnt/β-catenin signaling pathway is mainly composed of the Wnt protein, Wnt protein ligand frizzled protein, and related regulator proteins such as GSK-3β and β-catenin. Previous study indicated that aberrant activation of WNT signalling is a driving molecular event in many types of tumors, including liver cancers (Polakis 2012). The aberrant Wnt/β-catenin signaling pathway plays an important role in liver physiology and pathology. Various molecular and genetic factors such as CTNNB1, AXIN1 and AXIN2 participate to the aberrant activation of the Wnt/-catenin pathway. Gain-of-function mutations of CTNNB1 which encode for β-catenin are occurred in about 90 % HCCs (Bruix and Sherman 2011). In contrast, loss-of-function mutations of negative regulators such as AXIN1, AXIN2 and APC genes are also observed in such aberrant pathway (Laurent-Puig and Zucman-Rossi 2006). When upstream stimulation activate the pathway, the Wnt protein binds to its ligand and β-catenin accumulates in cells, where β-catenin is activated and transferred into nucleus. In the nucleus, β-catenin dimerizes with the downstream specific transcription factor LEF/TCF, which regulates the transcription of key genes such as cyclin D (Thompson and Monga 2007; Langeswaran et al. 2013).
16.5.4 PI3K/AKT/mTOR Signaling Pathway
The PI3K/AKT/mTOR signaling pathway is a central regulator of various oncogenic processes including cell growth, proliferation, metabolism, survival regulation, antiapoptosis and angiogenesis. It also plays significant function in HCC and is activated in 30–50 % of HCC. There is growing evidence to suggest that activation of the PI3K/AKT/mTOR pathway is associated with less differentiated tumors, earlier tumor recurrence, and worse survival outcomes (Zhou et al. 2010). In normal tissue, this pathway is negatively regulated by the tumor suppressor phosphatase and tensin homolog (PTEN) on chromosome 10. Abnormal PTEN function and expression may lead to excessively activation of the PI3K/AKT/mTOR pathway in HCC (Zhou et al. 2011). Previous study has found that the loss of PTEN and overexpression of pAkt and p-mTOR were linked to the tumor differentiation, TNM stage, intrahepatic metastasis, vascular invasion Ki-67 labeling index, and MMP-2 and MMP-9 upregulation of human HCCs (Chen et al. 2009; Grabinski et al. 2012). Furthermore, Mcl-1, an anti-apoptotic molecule transcribed via a PI3K/Akt dependent pathway, was associated with HCC poor survival (Personeni et al. 2013).
16.5.5 Ras/Raf/MAPK Signaling Pathway
The MAPK intracellular signaling network is often activated in cancer cells. Recent researches show that HCC cells activation and proliferation is known to involve various different signaling pathways as previously mentioned (Laurent-Puig and Zucman-Rossi 2006). Among them, the Ras/Raf/MAPK signaling pathways is one of the most critical pathways in pathogenesis, development and proliferation of HCC and have been extensively investigated (Llovet and Bruix 2008).
The intracellular part of Ras/Raf/MAPK pathway is downstream of several receptor tyrosine kinases such as the EGFR, PDGFR and VEGFR which transmit growth factor signals from the cell membrane to the nucleus regulating multiple cellular functions including cell growth and survival, and differentiation. However, multiple upstream receptors including other receptor tyrosine kinases, integrins, serpentine receptors, heterotrimeric G-proteins, and cytokine receptors are able to activate Ras (Cantrell 2003).
Mechanisms for the increased activity of the Ras/Raf/MAPK signaling pathway in HCC include aberrant upstream signals, inactivation of Raf kinase inhibitor protein and induction by hepatitis viral proteins (Galuppo et al. 2014).
Several components of this pathway are mutated in HCC. Bos (1989) found that about 30 % of HCC bear Ras mutations. The Raf family consists of three isoforms, A-Raf, B-Raf and C-Raf. Overexpression of wild-type C-Raf-1 proto-oncogene has been reported in liver cirrhosis and HCC (Jenke et al. 1994; Huang and Sinicrope 2010). Sorafenib has activity inhibiting B-Raf (Tannapfel et al. 2003). Huynh et al. (2003) found overexpression of MEK1/2 and ERK1/2, and phosphorylation of ERK1/2 in 100 % (46/46), 91 % (42/46) and 69 % (32/46) HCC, respectively.
16.5.6 Notch Signaling
Notch signalling is an evolutionarily conserved pathway that involves in a variety of fundamental cellular processes such as cell fate and differentiation (Artavanis-Tsakonas et al. 1999; Lai 2004). The effects of Notch signaling seem heterogeneous in HCC progression (Strazzabosco and Fabris 2012). Activation of Notch signaling could lead to reduced cell proliferation and tumor growth in HCC (Viatour et al. 2011). And in addition, it also participates in invasion and migration of HCC cells (Zhou et al. 2013). Several researches indicated that NOTCH is activated in mice and human HCC samples (Tschaharganeh et al. 2013; Villanueva et al. 2012). However, other reports found the activation of NOTCH signalling as a suppressor feedback mechanism during HCC progression (Viatour et al. 2011; Qi et al. 2003). These contradictions suggest that biological activities of NOTCH signaling during hepatocarcinogenesis mainly depend on the cellular environment, which is also reported in other tumor types (Radtke and Raj 2003).
16.5.7 KEAP1-NFE2L2 Pathway
A sequence-specific transcriptional factor, encoded by the NFE2L2 gene, upregulates genes associated with oxidative stress and other metabolic pathways (Taguchi et al. 2011). And the level of the NFE2L2 protein is regulated by the ubiquitin proteasome pathway, and KEAP1 functions as an E3 ubiquitin ligase. A study found that NFE2L2 coding for NRF2 a transcription factor crucial for cellular redox homeostasis, was mutated in 6.4 % of HCC (Shibata et al. 2008). The mutation disrupts direct NFE2L2–KEAP1 interaction, or inactivating mutations of the KEAP1 gene are recurrently reported in HCC (Guichard et al. 2012).
16.6 Conclusion
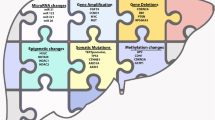
Heterogeneities in aetiological factors, tumor microenvironment, genetic variations and signaling pathways contribute to HCC progression, which makes it difficult to properly stage the disease and thereby estimate the prognosis. Besides the established main role of hepatitis virus infections and of alcohol use in the risk of HCC, multiple genetic factors also play an significant role. Advances in sequencing technologies have guided the examination of HCC genomes into a new view. In addition to copy number changes and mutations, analyses have identified additional genome alterations, including DNA methylation, HBV integration, retrotransposon changes an so on. The integration of data from different levels of global analyses have identified various critical genes and pathways involved in hepatocarcinogenesis. The heterogeneity of HCC makes it difficult to clarify the mechanism of cancer development and to develop effective therapeutics. For future clinical research design, it is essential to take into account how to eliminate the confounding effects from interpatients and intratumor heterogeneity of genome, aetiological factors and tumor microenvironment. Precision medicine based on global genetic analysis will become more and more important to overcome the heterogeneity of HCC. While some genetic profiles or signaling pathways may prove to be potential targets for clinical application. Therefore, targeting these heterogeneity in HCC patients will definitely create a new field for developing personal treatment options.
References
Andant C, Puy H, Bogard C, Faivre J, Soule JC, Nordmann Y, Deybach JC. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol. 2000;32:933–9.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6.
Baan R, Straif K, Grosse Y, Secretan B, El GF, Bouvard V, Altieri A, Cogliano V. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007;8:292–3.
Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19:329–37.
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9.
Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–16.
Brechot C, Pourcel C, Louise A, Rain B, Tiollais P. Presence of integrated hepatitis b virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533–5.
Bricard G, Cesson V, Devevre E, Bouzourene H, Barbey C, Rufer N, Im JS, Alves PM, Martinet O, Halkic N, Cerottini JC, Romero P, Porcelli SA, Macdonald HR, Speiser DE. Enrichment of human CD4+ V(alpha)24/Vbeta11 invariant NKT cells in intrahepatic malignant tumors. J Immunol. 2009;182:5140–51.
Brown CJ, Cheok CF, Verma CS, Lane DP. Reactivation of p53: from peptides to small molecules. Trends Pharmacol Sci. 2011;32:53–62.
Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–2.
Burns KH, Boeke JD. Human transposon tectonics. Cell. 2012;149:740–52.
Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, Shi M, Zhang H, Yang Y, Wu H, Tien P, Wang FS. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol. 2008;129:428–37.
Cantrell DA. GTPases and T cell activation. Immunol Rev. 2003;192:122–30.
Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34:834–43.
Che L, Yuan YH, Jia J, Ren J. Activation of sonic hedgehog signaling pathway is an independent potential prognosis predictor in human hepatocellular carcinoma patients. Chin J Cancer Res. 2012;24:323–31.
Chen XL, Cao LQ, She MR, Wang Q, Huang XH, Fu XH. Gli-1 siRNA induced apoptosis in Huh7 cells. World J Gastroenterol. 2008;14:582–9.
Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, Chen LZ, Tan HX, Li W, Bi J, Zhang LJ. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP-9. Hepatol Res. 2009;39:177–86.
Cheng WT, Xu K, Tian DY, Zhang ZG, Liu LJ, Chen Y. Role of hedgehog signaling pathway in proliferation and invasiveness of hepatocellular carcinoma cells. Int J Oncol. 2009;34:829–36.
Chew V, Tow C, Huang C, Bard-Chapeau E, Copeland NG, Jenkins NA, Weber A, Lim KH, Toh HC, Heikenwalder M, Ng IO, Nardin A, Abastado JP. Toll-like receptor 3 expressing tumor parenchyma and infiltrating natural killer cells in hepatocellular carcinoma patients. J Natl Cancer Inst. 2012;104:1796–807.
Chochi Y, Kawauchi S, Nakao M, Furuya T, Hashimoto K, Oga A, Oka M, Sasaki K. A copy number gain of the 6p arm is linked with advanced hepatocellular carcinoma: an array-based comparative genomic hybridization study. J Pathol. 2009;217:677–84.
Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, Parker JS, Li J, Powers S, Kim H, Fischer S, Guindi M, Ghanekar A, Chiang DY. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–702.
Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703.
Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, Gage FH. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–31.
Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, Hayakawa Y, Godfrey DI, Smyth MJ. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med. 2005;202:1279–88.
Dorn C, Riener MO, Kirovski G, Saugspier M, Steib K, Weiss TS, Gabele E, Kristiansen G, Hartmann A, Hellerbrand C. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int J Clin Exp Pathol. 2010;3:505–14.
Dragani TA, Manenti G, Della PG. Enhancing effects of carbon tetrachloride in mouse hepatocarcinogenesis. Cancer Lett. 1986;31:171–9.
Dragani TA, Canzian F, Manenti G, Pierotti MA. Hepatocarcinogenesis: a polygenic model of inherited predisposition to cancer. Tumori. 1996;82:1–5.
Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455.
Elmberg M, Hultcrantz R, Ekbom A, Brandt L, Olsson S, Olsson R, Lindgren S, Loof L, Stal P, Wallerstedt S, Almer S, Sandberg-Gertzen H, Askling J. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125:1733–41.
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–73.
El-Serag HB, Mason AC. Risk factors for the rising rates of primary liver cancer in the United States. Arch Intern Med. 2000;160:3227–30.
El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol. 2006;4:369–80.
Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (Pizz): is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? Eur J Gastroenterol Hepatol. 1996;8:989–94.
Evans AA, Chen G, Ross EA, Shen FM, Lin WY, London WT. Eight-year follow-up of the 90,000-person Haimen City cohort: I. Hepatocellular carcinoma mortality, risk factors, and gender differences. Cancer Epidemiol Biomarkers Prev. 2002;11:369–76.
Fang J, Yan L, Shing Y, Moses MA. Hif-1alpha-mediated up-regulation of vascular endothelial growth factor, independent of basic fibroblast growth factor, is important in the switch to the angiogenic phenotype during early tumorigenesis. Cancer Res. 2001;61:5731–5.
Fang M, Yuan JP, Peng CW, Pang DW, Li Y. Quantum dots-based in situ molecular imaging of dynamic changes of collagen IV during cancer invasion. Biomaterials. 2013;34:8708–17.
Faulkner GJ. Retrotransposons: mobile and mutagenic from conception to death. Febs Lett. 2011;585:1589–94.
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, Waki K, Hornig N, Arakawa T, Takahashi H, Kawai J, Forrest AR, Suzuki H, Hayashizaki Y, Hume DA, Orlando V, Grimmond SM, Carninci P. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–71.
Feo F, De Miglio MR, Simile MM, Muroni MR, Calvisi DF, Frau M, Pascale RM. Hepatocellular carcinoma as a complex polygenic disease. Interpretive analysis of recent developments on genetic predisposition. Biochim Biophys Acta. 2006;1765:126–47.
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–55.
Fracanzani AL, Taioli E, Sampietro M, Fatta E, Bertelli C, Fiorelli G, Fargion S. Liver cancer risk is increased in patients with porphyria cutanea tarda in comparison to matched control patients with chronic liver disease. J Hepatol. 2001;35:498–503.
Franceschi S, Montella M, Polesel J, La Vecchia C, Crispo A, Dal Maso L, Casarin P, Izzo F, Tommasi LG, Chemin I, Trepo C, Crovatto M, Talamini R. Hepatitis viruses, alcohol, and tobacco in the etiology of hepatocellular carcinoma in Italy. Cancer Epidemiol Biomarkers Prev. 2006;15:683–9.
Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi S, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4.
Galuppo R, Maynard E, Shah M, Daily MF, Chen C, Spear BT, Gedaly R. Synergistic inhibition of HCC and liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and WNT/beta-catenin pathways. Anticancer Res. 2014;34:1709–13.
Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93.
Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–28.
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–9.
Gozuacik D, Murakami Y, Saigo K, Chami M, Mugnier C, Lagorce D, Okanoue T, Urashima T, Brechot C, Paterlini-Brechot P. Identification of human cancer-related genes by naturally occurring hepatitis b virus DNA tagging. Oncogene. 2001;20:6233–40.
Grabinski N, Ewald F, Hofmann BT, Staufer K, Schumacher U, Nashan B, Jucker M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer. 2012;11:85.
Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, Calvo F, Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–8.
Haddow JE, Palomaki GE, McClain M, Craig W. Hereditary haemochromatosis and hepatocellular carcinoma in males: a strategy for estimating the potential for primary prevention. J Med Screen. 2003;10:11–3.
Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–74.
Hancks DC, Kazazian HJ. Active human retrotransposons: variation and disease. Curr Opin Genet Dev. 2012;22:191–203.
Hashimoto E, Yatsuji S, Tobari M, Taniai M, Torii N, Tokushige K, Shiratori K. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroenterol. 2009;44 Suppl 19:89–95.
Hassan MM, Kaseb A, Li D, Patt YZ, Vauthey JN, Thomas MB, Curley SA, Spitz MR, Sherman SI, Abdalla EK, Davila M, Lozano RD, Hassan DM, Chan W, Brown TD, Abbruzzese JL. Association between hypothyroidism and hepatocellular carcinoma: a case-control study in the United States. Hepatology. 2009;49:1563–70.
Hemminki K, Li X. Familial liver and gall bladder cancer: a nationwide epidemiological study from Sweden. Gut. 2003;52:592–6.
Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–47.
Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ, McCombie WR. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–7.
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, Lehner F, Manns MP, Greten TF, Korangy F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807.
Huang S, Sinicrope FA. Sorafenib inhibits STAT3 activation to enhance trail-mediated apoptosis in human pancreatic cancer cells. Mol Cancer Ther. 2010;9:742–50.
Huang Y, Wang FM, Wang T, Wang YJ, Zhu ZY, Gao YT, Du Z. Tumor-infiltrating FoxP3+ Tregs and CD8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion. 2012a;86:329–37.
Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, Fei QL, Chen H, Cai B, Zhou B, Xiao HS, Qin LX, Han ZG. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012b;44:1117–21.
Huynh H, Nguyen TT, Chow KH, Tan PH, Soo KC, Tran E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: its role in tumor progression and apoptosis. BMC Gastroenterol. 2003;3:19.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Hepatitis viruses. IARC Monogr Eval Carcinog Risks Hum. 1994;59:1–255.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr Eval Carcinog Risks Hum. 2002;82:1–556.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco smoke and involuntary smoking. IARC Monogr Eval Carcinog Risks Hum. 2004;83:1–1438.
Janecke AR, Mayatepek E, Utermann G. Molecular genetics of type 1 glycogen storage disease. Mol Genet Metab. 2001;73:117–25.
Jeng KS, Sheen IS, Jeng WJ, Yu MC, Hsiau HI, Chang FY. High expression of sonic hedgehog signaling pathway genes indicates a risk of recurrence of breast carcinoma. Onco Targets Ther. 2013;7:79–86.
Jenke HS, Deml E, Oesterle D. C-raf expression in early rat liver tumorigenesis after promotion with polychlorinated biphenyls or phenobarbital. Xenobiotica. 1994;24:569–80.
Jia XQ, Cheng HQ, Li H, Zhu Y, Li YH, Feng ZQ, Zhang JP. Inhibition of connective tissue growth factor overexpression decreases growth of hepatocellular carcinoma cells in vitro and in vivo. Chin Med J (Engl). 2011;124:3794–9.
Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–9.
Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, Diao J, Yeung S, Jubb A, Ye W, Wu TD, Kapadia SB, de Sauvage FJ, Gentleman RC, Stern HM, Seshagiri S, Pant KP, Modrusan Z, Ballinger DG, Zhang Z. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012a;22:593–601.
Jiang S, Yang Z, Li W, Li X, Wang Y, Zhang J, Xu C, Chen PJ, Hou J, McCrae MA, Chen X, Zhuang H, Lu F. Re-evaluation of the carcinogenic significance of hepatitis B virus integration in hepatocarcinogenesis. PLoS One. 2012b;7:e40363.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54.
Kakar S, Chen X, Ho C, Burgart LJ, Adeyi O, Jain D, Sahai V, Ferrell LD. Chromosomal abnormalities determined by comparative genomic hybridization are helpful in the diagnosis of atypical hepatocellular neoplasms. Histopathology. 2009;55:197–205.
Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, Hauptschein R, Rejto PA, Fernandez J, Wang G, Zhang Q, Wang B, Chen R, Wang J, Lee NP, Zhou W, Lin Z, Peng Z, Yi K, Chen S, Li L, Fan X, Yang J, Ye R, Ju J, Wang K, Estrella H, Deng S, Wei P, Qiu M, Wulur IH, Liu J, Ehsani ME, Zhang C, Loboda A, Sung WK, Aggarwal A, Poon RT, Fan ST, Wang J, Hardwick J, Reinhard C, Dai H, Li Y, Luk JM, Mao M. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–33.
Kasper HU, Drebber U, Stippel DL, Dienes HP, Gillessen A. Liver tumor infiltrating lymphocytes: comparison of hepatocellular and cholangiolar carcinoma. World J Gastroenterol. 2009;15:5053–7.
Kim Y, Yoon JW, Xiao X, Dean NM, Monia BP, Marcusson EG. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res. 2007;67:3583–93.
Kuper H, Tzonou A, Kaklamani E, Hsieh CC, Lagiou P, Adami HO, Trichopoulos D, Stuver SO. Tobacco smoking, alcohol consumption and their interaction in the causation of hepatocellular carcinoma. Int J Cancer. 2000;85:498–502.
Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–73.
Langeswaran K, Gowthamkumar S, Vijayaprakash S, Revathy R, Balasubramanian MP. Influence of limonin on Wnt signalling molecule in HepG2 cell lines. J Nat Sci Biol Med. 2013;4:126–33.
Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–86.
Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 2011;12:615–27.
Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, Velculescu VE, Wang L, Zhou S, Vogelstein B, Hruban RH, Papadopoulos N, Cai J, Torbenson MS, Kinzler KW. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–9.
Liao Y, Wang B, Huang ZL, Shi M, Yu XJ, Zheng L, Li S, Li L. Increased circulating Th17 cells after transarterial chemoembolization correlate with improved survival in stage III hepatocellular carcinoma: a prospective study. PLoS One. 2013;8:e60444.
Lin M, Guo LM, Liu H, Du J, Yang J, Zhang LJ, Zhang B. Nuclear accumulation of glioma-associated oncogene 2 protein and enhanced expression of forkhead-box transcription factor M1 protein in human hepatocellular carcinoma. Histol Histopathol. 2010;25:1269–75.
Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–27.
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–17.
Lu JT, Zhao WD, He W, Wei W. Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta Pharmacol Sin. 2012;33:691–700.
Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–60.
Muller PA, Vousden KH. P53 mutations in cancer. Nat Cell Biol. 2013;15:2–8.
Murakami Y, Saigo K, Takashima H, Minami M, Okanoue T, Brechot C, Paterlini-Brechot P. Large scaled analysis of hepatitis b virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut. 2005;54:1162–8.
Muto J, Shirabe K, Sugimachi K, Maehara Y. Review of angiogenesis in hepatocellular carcinoma. Hepatol Res. 2015;45:1–9.
Nagae G, Isagawa T, Shiraki N, Fujita T, Yamamoto S, Tsutsumi S, Nonaka A, Yoshiba S, Matsusaka K, Midorikawa Y, Ishikawa S, Soejima H, Fukayama M, Suemori H, Nakatsuji N, Kume S, Aburatani H. Tissue-specific demethylation in CpG-poor promoters during cellular differentiation. Hum Mol Genet. 2011;20:2710–21.
Nakagawa H, Shibata T. Comprehensive genome sequencing of the liver cancer genome. Cancer Lett. 2013;340:234–40.
Narimatsu T, Tamori A, Koh N, Kubo S, Hirohashi K, Yano Y, Arakawa T, Otani S, Nishiguchi S. P16 promoter hypermethylation in human hepatocellular carcinoma with or without hepatitis virus infection. Intervirology. 2004;47:26–31.
Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52:594–604.
Nishida N, Nishimura T, Nagasaka T, Ikai I, Goel A, Boland CR. Extensive methylation is associated with beta-catenin mutations in hepatocellular carcinoma: evidence for two distinct pathways of human hepatocarcinogenesis. Cancer Res. 2007;67:4586–94.
Nishida N, Nishimura T, Nakai T, Chishina H, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, Minami Y, Ueshima K, Sakurai T, Kudo M. Genome-wide profiling of DNA methylation and tumor progression in human hepatocellular carcinoma. Dig Dis. 2014;32:658–63.
Ogasawara S, Yano H, Momosaki S, Nishida N, Takemoto Y, Kojiro S, Kojiro M. Expression of matrix metalloproteinases (MMPs) in cultured hepatocellular carcinoma (HCC) cells and surgically resected HCC tissues. Oncol Rep. 2005;13:1043–8.
Ogimoto I, Shibata A, Kurozawa Y, Nose T, Yoshimura T, Suzuki H, Iwai N, Sakata R, Fujita Y, Ichikawa S, Fukuda K, Tamakoshi A. Risk of death due to hepatocellular carcinoma among smokers and ex-smokers, Univariate analysis of JACC study data. Kurume Med J. 2004;51:71–81.
Ostrowski J, Kostrzewska E, Michalak T, Zawirska B, Medrzejewski W, Gregor A. Abnormalities in liver function and morphology and impaired aminopyrine metabolism in hereditary hepatic porphyrias. Gastroenterology. 1983;85:1131–7.
Paterlini-Brechot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Brechot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–6.
Patil MA, Gutgemann I, Zhang J, Ho C, Cheung ST, Ginzinger D, Li R, Dykema KJ, So S, Fan ST, Kakar S, Furge KA, Buttner R, Chen X. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and Jab1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis. 2005;26:2050–7.
Personeni N, Rimassa L, Pressiani T, Destro A, Ligorio C, Tronconi MC, Bozzarelli S, Carnaghi C, Di Tommaso L, Giordano L, Roncalli M, Santoro A. Molecular determinants of outcome in sorafenib-treated patients with hepatocellular carcinoma. J Cancer Res Clin Oncol. 2013;139:1179–87.
Pogribny IP, James SJ. Reduction of p53 gene expression in human primary hepatocellular carcinoma is associated with promoter region methylation without coding region mutation. Cancer Lett. 2002;176:169–74.
Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:a0080052.
Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–6.
Qi R, An H, Yu Y, Zhang M, Liu S, Xu H, Guo Z, Cheng T, Cao X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323–9.
Radtke F, Raj K. The role of notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3:756–67.
Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, Tang ZY, He P, Hunter KW, Thorgeirsson SS, Meltzer PS, Wang XW. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012;142:957–66.
Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–63.
Saigo K, Yoshida K, Ikeda R, Sakamoto Y, Murakami Y, Urashima T, Asano T, Kenmochi T, Inoue I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum Mutat. 2008;29:703–8.
Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, Toedt G, Kleeff J, Ehemann V, Eils R, Lichter P, Schirmacher P, Radlwimmer B. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology. 2008;47:511–20.
Scott CR. The genetic tyrosinemias. Am J Med Genet C: Semin Med Genet. 2006;142C:121–6.
Sha WH, Zeng XH, Min L. The correlation between NK cell and liver function in patients with primary hepatocellular carcinoma. Gut Liver. 2014;8:298–305.
Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115–25.
Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, Yu MW, Chen CJ, Santella RM. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55:1799–808.
Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H, Santella RM. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium Humanmethylation 450 Beadchips. Epigenetics-Us. 2013;8:34–43.
Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11:340–9.
Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–73.
Shukla R, Upton KR, Munoz-Lopez M, Gerhardt DJ, Fisher ME, Nguyen T, Brennan PM, Baillie JK, Collino A, Ghisletti S, Sinha S, Iannelli F, Radaelli E, Dos SA, Rapoud D, Guettier C, Samuel D, Natoli G, Carninci P, Ciccarelli FD, Garcia-Perez JL, Faivre J, Faulkner GJ. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013;153:101–11.
Sicklick JK, Li YX, Jayaraman A, Kannangai R, Qi Y, Vivekanandan P, Ludlow JW, Owzar K, Chen W, Torbenson MS, Diehl AM. Dysregulation of the hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis. 2006;27:748–57.
Strazzabosco M, Fabris L. Notch signaling in hepatocellular carcinoma: guilty in association! Gastroenterology. 2012;143:1430–4.
Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, Poon RT, Fan ST, Chan KL, Gong Z, Hu Y, Lin Z, Wang G, Zhang Q, Barber TD, Chou WC, Aggarwal A, Hao K, Zhou W, Zhang C, Hardwick J, Buser C, Xu J, Kan Z, Dai H, Mao M, Reinhard C, Wang J, Luk JM. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–9.
Swergold GD. Identification, characterization, and cell specificity of a human line-1 promoter. Mol Cell Biol. 1990;10:6718–29.
Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–40.
Tannapfel A, Sommerer F, Benicke M, Katalinic A, Uhlmann D, Witzigmann H, Hauss J, Wittekind C. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 2003;52:706–12.
Thompson MD, Monga SP. Wnt/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298–305.
Tomasi ML, Li TW, Li M, Mato JM, Lu SC. Inhibition of human methionine adenosyltransferase 1A transcription by coding region methylation. J Cell Physiol. 2012;227:1583–91.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara T, Sakamoto H, Wang L, Ojima H, Shimada K, Kosuge T, Okusaka T, Kato K, Kusuda J, Yoshida T, Aburatani H, Shibata T. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–9.
Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46.
Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K, Ladu S, Singer S, Pinna F, Gretz N, Sticht C, Tomasi ML, Delogu S, Evert M, Fan B, Ribback S, Jiang L, Brozzetti S, Bergmann F, Dombrowski F, Schirmacher P, Calvisi DF, Breuhahn K. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology. 2013;144:1530–42.
Udali S, Guarini P, Ruzzenente A, Ferrarini A, Guglielmi A, Lotto V, Tononi P, Pattini P, Moruzzi S, Campagnaro T, Conci S, Olivieri O, Corrocher R, Delledonne M, Choi SW, Friso S. DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clin Epigenetics. 2015;7:43.
Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, Zmoos AF, Mazur PK, Schaffer BE, Ostermeier A, Vogel H, Sylvester KG, Thorgeirsson SS, Grompe M, Sage J. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208:1963–76.
Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, Sole M, Thung S, Stanger BZ, Llovet JM. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660–9.
Wang F, Jing X, Li G, Wang T, Yang B, Zhu Z, Gao Y, Zhang Q, Yang Y, Wang Y, Wang P, Du Z. Foxp3+ regulatory T cells are associated with the natural history of chronic hepatitis B and poor prognosis of hepatocellular carcinoma. Liver Int. 2012;32:644–55.
Wang Y, Han C, Lu L, Magliato S, Wu T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology. 2013;58:995–1010.
Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr. 1976;88:434–8.
Werner M, Almer S, Prytz H, Lindgren S, Wallerstedt S, Bjornsson E, Bergquist A, Sandberg-Gertzen H, Hultcrantz R, Sangfelt P, Weiland O, Danielsson A. Hepatic and extrahepatic malignancies in autoimmune hepatitis. A long-term follow-up in 473 Swedish patients. J Hepatol. 2009;50:388–93.
Wong GS, Rustgi AK. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer. 2013;108:755–61.
Wong IH, Lo YM, Zhang J, Liew CT, Ng MH, Wong N, Lai PB, Lau WY, Hjelm NM, Johnson PJ. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999;59:71–3.
Woo HG, Park ES, Lee JS, Lee YH, Ishikawa T, Kim YJ, Thorgeirsson SS. Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer Res. 2009;69:4059–66.
Wu XZ, Xie GR, Chen D. Hypoxia and hepatocellular carcinoma: the therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22:1178–82.
Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163:1101–7.
Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin Cancer Biol. 2013;23:483–91.
Zang JJ, Xie F, Xu JF, Qin YY, Shen RX, Yang JM, He J. P16 gene hypermethylation and hepatocellular carcinoma: a systematic review and meta-analysis. World J Gastroenterol. 2011;17:3043–8.
Zhang YJ, Ahsan H, Chen Y, Lunn RM, Wang LY, Chen SY, Lee PH, Chen CJ, Santella RM. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol Carcinog. 2002;35:85–92.
Zhang YJ, Chen Y, Ahsan H, Lunn RM, Lee PH, Chen CJ, Santella RM. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation and its relationship to aflatoxin B1-DNA adducts and p53 mutation in hepatocellular carcinoma. Int J Cancer. 2003;103:440–4.
Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–9.
Zhang D, Liu J, Wang Y, Chen J, Chen T. shRNA-mediated silencing of Gli2 gene inhibits proliferation and sensitizes human hepatocellular carcinoma cells towards trail-induced apoptosis. J Cell Biochem. 2011;112:3140–50.
Zhang D, Cao L, Li Y, Lu H, Yang X, Xue P. Expression of glioma-associated oncogene 2 (Gli 2) is correlated with poor prognosis in patients with hepatocellular carcinoma undergoing hepatectomy. World J Surg Oncol. 2013;11:25.
Zheng T, Wang J, Song X, Meng X, Pan S, Jiang H, Liu L. Nutlin-3 cooperates with doxorubicin to induce apoptosis of human hepatocellular carcinoma cells through p53 or p73 signaling pathways. J Cancer Res Clin Oncol. 2010a;136:1597–604.
Zheng X, Yao Y, Xu Q, Tu K, Liu Q. Evaluation of glioma-associated oncogene 1 expression and its correlation with the expression of sonic hedgehog, E-cadherin and S100a4 in human hepatocellular carcinoma. Mol Med Rep. 2010b;3:965–70.
Zheng X, Vittar NB, Gai X, Fernandez-Barrena MG, Moser CD, Hu C, Almada LL, McCleary-Wheeler AL, Elsawa SF, Vrabel AM, Shire AM, Comba A, Thorgeirsson SS, Kim Y, Liu Q, Fernandez-Zapico ME, Roberts LR. The transcription factor GLI1 mediates TGFBETA1 driven EMT in hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS One. 2012;7:e49581.
Zheng X, Zeng W, Gai X, Xu Q, Li C, Liang Z, Tuo H, Liu Q. Role of the hedgehog pathway in hepatocellular carcinoma (review). Oncol Rep. 2013;30:2020–6.
Zhong S, Tang MW, Yeo W, Liu C, Lo YM, Johnson PJ. Silencing of GSTP1 gene by CpG island DNA hypermethylation in HBV-associated hepatocellular carcinomas. Clin Cancer Res. 2002;8:1087–92.
Zhou L, Huang Y, Li J, Wang Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol. 2010;27:255–61.
Zhou Q, Lui VW, Yeo W. Targeting the PI3K/AKT/mTOR pathway in hepatocellular carcinoma. Future Oncol. 2011;7:1149–67.
Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF. The down-regulation of Notch1 inhibits the invasion and migration of hepatocellular carcinoma cells by inactivating the Cyclooxygenase-2/Snail/E-cadherin pathway in vitro. Dig Dis Sci. 2013;58:1016–25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Fang, T., Feng, L., Xia, J. (2016). Heterogeneity of Hepatocellular Carcinoma. In: Wang, X., Baumgartner, C., Shields, D., Deng, HW., Beckmann, J. (eds) Application of Clinical Bioinformatics. Translational Bioinformatics, vol 11. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-7543-4_16
Download citation
DOI: https://doi.org/10.1007/978-94-017-7543-4_16
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-7541-0
Online ISBN: 978-94-017-7543-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)