
Abstract
The developmental potential of seven defined types (0–VI) of germ cell tumors (GCT) is determined by the developmental state of the precursor cells from which they originate: the 2C, naïve, or primed state. These basal states are modified by epigenetic changes, particularly genomic imprinting, and by reprogramming, which is mostly due to failure of repression of pluripotency of the precursor cell. Type VI is a new category of neoplasms resembling GCT, resulting from induced pluripotency.
In agreement with the plasticity of developmental states, the same precursor cell may generate different types of GCT, and similar GCT may be derived from different precursors. Developmental plasticity also explains intermediate phenotypes between the defined types of GCT.
The anatomical distribution of extragonadal GCT favors early germ cells, in particular primordial germ cells (PGC), as their precursors.
Most GCT result from dysfunction of the niche of precursor cells; somatic mutation of precursors plays a minor role. Disturbance may be due to internal (and external) systemic factors (genvironment) that will likely affect both gonads, explaining the high incidence of bilaterality of most gonadal GCT, occasionally in combination with extragonadal GCT. Heritable niche-disturbing factors explain familial occurrence of GCT of the same and occasionally different types.
The overwhelming preponderance of gonadal and extragonadal type II GCT (seminomatous and non-seminomatous tumors) in males and 46,XY disorders of sex development are likely due to disturbance of the niche allowing co-expression of OCT4 and TSPY in delayed-matured PGC/gonocytes, giving them survival and proliferative advantage.
This review of recent and classic data on developmental biology of embryonic stem cells, the germline, and GCT provides a biologically plausible and clinically relevant unifying model for all GCT.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Germ cell tumor
- Classification
- Developmental potential
- Epidemiology
- Risk factor
- Anatomical distribution
- Cytogenetics
- Genetics
- Epigenetics
- Molecular biology
- Pathogenesis
- Origin
- Zygote
- Blastomere
- Germline
- Embryonal stem cell
- Induced pluripotency
- 2C state
- Naïve state
- Primed state
- Plasticity of developmental states
- Genomic imprinting
- Reprogramming
3.1 Introduction
Germ cell tumors (GCT) are a seemingly heterogeneous family of neoplasms, whose histological composition likely reflects the developmental potential of the cells from which they are derived.
Recent discoveries on the regulation of developmental states of cells in the early embryo and the germline allow a deeper understanding of the origin and developmental potential of GCT and provide a biologically plausible and clinically relevant basis for their classification.
3.2 Developmental States of Early Embryonic Cells
3.2.1 Restriction Versus Maintaining Developmental Potential
Multicellular organisms develop from a single omnipotent cell, the zygote, through a tightly regulated program of restriction of pluripotency [1], yet for maintaining their kind, they have to preserve totipotency in the germ cell lineage [2]. For full developmental potential, the zygote of placental mammals needs a biparental genomic imprinting (GI) [3–5] and a specifically methylated intact genome with X-inactivation in female cells [6, 7].
Here a brief explanation of GI is appropriate; changes of (global) methylation in the early embryo and the germline will be discussed later in this section. GI is the phenomenon whereby in mammals the expression of some genes depends on maternal or paternal origin, due to parental-specific DNA methylation and histone modification [8]. The GI cycle starts with erasure of the original biparental imprinting pattern of the zygote early in the germ lineage through replacement of methylcytosine by unmethylated cytosine via the base excision repair pathway [9]. Later, during oogenesis and spermatogenesis, respectively, fresh maternal and paternal imprinting patterns are established by de novo methylation of the relevant targets, an estimated 100–200 genes including noncoding RNAs, about 1 % of the genome [10–15] (Fig. 3.1). A variety of human diseases is caused by aberrations of specific imprinted genes through genetic and epigenetic mechanisms (for review [16]).

Cycle of genomic imprinting (GI). Upon fertilization, the zygote acquires a haploid set of paternally imprinted chromosomes from the father and a haploid set of maternally imprinted chromosomes from the mother; the cells of the embryo therefore have a biparental GI pattern. In the germline, GI is erased; during spermatogenesis and oogenesis, respectively, paternal and maternal imprinting is reestablished [746]
Only blastomeres after the first few cleavage divisions, up to the eight-cell stage, may have the full developmental potential (omnipotency) of the zygote [17]. In fact, in the mouse, it is lost beyond the two-cell stage [18]. Later, blastomeres and the embryonal stem cells (ESC) of the inner cell mass (ICM) and epiblast undergo further restriction step by step of their developmental potential, as stem cells are generated with commitment to developing specialized organs and tissues [1], including the germ lineage, which is specified to transfer omnipotency to the next generation [2, 12]. Probably all cells of the ICM and the epiblast are in principle germline competent and thus potentially totipotent/omnipotent [2, 19], although with different efficiency [20].
3.2.2 OCT4: Key Protein in Pluripotency
From studies mainly in the mouse, Oct4 (also known as Pouf1, Oct3, Oct4, and Otf3), a member of the POU-domain family of octamer-binding transcription factors, emerges as the key component in the regulatory network that maintains pluripotency [21–24]. Although not indispensable for the establishment of omnipotency in the zygote, it is required for maintaining pluripotent states in the developing embryo [25]. From the two-cell stage onward [19, 23], after the genome of the zygote is activated, Oct4 is expressed in all cells through the morula stage. Later, in the preimplantation embryo, Oct4 is confined to the ICM and the epiblast, while after implantation, its expression is limited to the primitive ectoderm (Fig. 3.2). Simultaneously with downregulation of Oct4 in the primitive ectoderm during gastrulation, primordial germ cells (PGC), the stem cells of gametogenesis in later life, are formed, with continued expression of Oct4 [26] that is maintained in the developing germ lineage until entry in meiosis. Meiotic oocytes are negative for Oct4; in the mouse, Oct4 is reexpressed in oocytes of the postnatal ovary. In the mouse testis, it is only expressed in type A spermatogonia [27]. In the human embryo, OCT4 expression starts somewhat later than in the mouse, in the eight-cell embryo [28]. OCT4 is normally not expressed in the testis beyond the age of 6 months and thus negative in spermatogonia, i.e., in male germ cells from mitotic arrest onward [29]. In contrast to the mouse, in humans, OCT4 is not expressed in meiotic germ cells both in males and females, and it thus remains also negative after birth in the pre- and postpubertal ovary [29–31]. OCT4 is specific for normal and neoplastic pluripotent cells and not expressed in normal adult human tissues and the large majority of cancers derived from adult tissues [32].

Oct4 expression in the early mouse embryo. The progressive stages of murine preimplantation development, through implantation and gastrulation (embryonic days 0.5–6.5), are schematically represented. Critical genetic and epigenetic events initiated during this period are indicated at the appropriate time points. The expression pattern of Oct4 mRNA and protein in the developing embryos is represented by shading, with the intensity of color reflecting the level of expression. Oct4 is present in the nuclei of all cells through the morula stage. At day 3.5, Oct4 becomes restricted to the inner cell mass (ICM) and, later, at day 4.5, to migrating cells of differentiating primitive endoderm. Following implantation, Oct4 expression is limited to primitive ectodermal cells. Expression in primordial cells is detectable at day 8.5 (not shown) [23]
Notably, in the cells of the ICM and the epiblast of the preimplantation mouse embryo and the germline (collectively, the totipotent ESC) which efficiently contribute to the germline in chimeric embryos [33], Oct4 expression is driven by its distal enhancer. In contrast, in ESC from primitive ectoderm of the postimplantation embryo, which are pluripotent and contribute to the germline with low efficiency, it is driven from its proximal enhancer [26]. In the ESC, both of the pre- and postimplantation embryos and in the germline Oct4 physically partners with Sox2, regardless of the driving enhancer [20, 34]. In humans, OCT4 is coupled with SOX2 in ESC, however, with SOX17 in the germline [35].
OCT4 (6p21–22) [36] is involved in a network of pluripotency factors including among others SOX2 (3q26–27) [37] and NANOG, STELLAR, and GDF3 (12p13) [38] that induce and maintain pluripotency of ESC, repress development of somatic lineages, and regulate cell fate decisions in the early embryo [20, 39].
In various animal models, factors involved specification of early lineages, orchestrated by the pluripotency network, have been identified, such as Ezh2, Sox21, and Cdx2 for trophectoderm [40–42], Sox17 [43] (SOX17 in humans) [44] for primitive endoderm, and TBX3 for mesoderm (in Xenopus) [45]. Oct4 switches partner from Sox2 to Sox17 in the primitive endoderm [34] and in humans also in the germline [35].
3.2.3 Specification and Maintenance of the Germline
In mice timeline at embryonic day 6 (E6), Bmp4 initiates the specification of the germline by inducing Blimp1, Prdm14, and Ap2ɣ in proximal epiblast cells. These three proteins act to repress somatic genes and induce expression of PGC proteins, such as nanos3, re-induce pluripotency genes, and start the epigenetic reprogramming ([35] for review). At E7.25, they form a cluster of 40–50 cells at the base of the allantois due to the homotypic adhesion molecule fragilis. The cells in the center of the cluster with the highest expression of fragilis start to express stella(r) (also known as Dpp3a) and Tnap (the mouse homolog of PLAP) and become recognizable as the first PGC, which on E 8.5, after downregulation of fragilis, start to migrate to the genital ridges, the future gonads [46, 47].
Different from mice, in humans SOX17 is a critical specifier of PGC fate [35], inducing the expression of BLIMP1, which represses endodermal and other somatic genes as in the mouse. SOX17 and BLIMP1 are probably also important in the maintenance of PGC, preventing them from displaying their capacity to totipotency endowed by the expression of OCT4. Both in mice and humans, migrating PGC undergo germline-specific global demethylation, “reprogramming 1,” jointly with upregulation of PRMT5 to protect the vulnerable demethylated genome from damage by transposable elements [7, 35]. In the gonads, these cells, now called gonocytes, undergo “reprogramming 2,” including completion of erasure of parental imprinting [48, 49], a process that is completed within 24 h in the mouse, however, takes several weeks in a locus-specific manner in the human embryo [6]. In the process of global demethylation and erasure of GI of PGC, 5mC is replaced by 5hmC in mouse [50] and man [6].
In the mouse, PGC start migration at E8.5 from the base of the allantois, as mentioned, and reach the genital ridges at E10.5 by passive movement due the folding of the embryo and active migration partly guided by chemotactic factors from the genital ridge [2, 12, 51]. In humans, PGC expressing OCT4 (see above), as well as cKIT (membrane receptor for KIT ligand (KITLG), also known as stem cell factor, crucial for survival and proliferation of PGC), can be recognized in the yolk sac wall from 3 to 4 week postconception (wpc) [52]. PGC are present in the hindgut epithelium, in the mesenchyme of the dorsal mesentery, and in the developing gonadal ridge in wpc 4–6. In wpc 4–5, they leave the gut epithelium by a process resembling epithelial mesenchymal transition (EMT). KITLG activates KIT signaling in the PGC and facilitates their further migration [2], but after establishment of connections between the enteric and sympathetic nervous systems, PGC follow sympathetic nerve fibers toward the gonads. Numerous PGC are still present in the nervous system by wpc14. PGC failing to exit the nerve branches at the gonadal site may continue along the sympathetic trunk along the midline of the body and may end up in other distant localizations including the retroperitoneum (suprarenal region, adrenal glands), abdomen (stomach), anterior mediastinum, heart, lungs, head and neck, and CNS [52, 53]. This is an important observation because these so-called mis-migrated PGC may give rise to GCT in these various extragonadal sites, unless eliminated by apoptosis [54–57] (Fig. 3.3). In the mouse embryo, upon arrival in the genital ridges on E12.5, gonocytes enter a premeiotic stage and upregulate meiotic genes both in female and male embryos. In the male genital ridge, meiosis proceeds no further and the germ cells enter mitotic arrest as G0/G1 prespermatogonia, which resume mitosis only after birth. In contrast, in the female genital ridge, germ cells enter meiotic prophase as oocytes and pass through leptotene, zygotene, and pachytene stages before arresting in diplotene at the time of birth. Germ cells enter meiotic prophase at about the same time not only in the female genital ridge, but also in extragonadal localizations, such as the adrenal gland, in female and male embryos [58, 59].

Horizontal section through abdomen of a human embryo, 8 wpc. Horizontal section of human embryo, CRL= 30 mm, 7 weeks and 6 days pc immunofluorescent labeled against cKit and b-III-tubulin antibody. Survey depicted (a–c), with commencing connectivity of the enteric (ENS) and sympathetic (SNS) nervous system (b). (c) Some sympathetic nerve fibers are found in the adrenal glands (AG), the pancreas (P), and especially in the dorsal mesentery located in the middle of the section. Positive b-III-tubulin reactivity is seen in nerve fibers of ENS, in general in the plexus myentericus (PM), and similar reactivity is observed in the duodenum (d). Furthermore, cKit is also observed in the interstitial cells of Cajal (ICC) of PM. (d) The PGCs in the nerve fibers demonstrate cKit reactivity. (e) Higher magnification of (b) demonstrating b-III-tubulin reactivity of SNS. (f) Higher magnification of boxed area in (c). The larger PGCs, with strong membranous cKit reactivity, are located in close correspondence to the periphery of the individual nerve fibers of the SNS (f, arrows). The small, densely labeled cKit-positive cells outside of the nerve fibers are mast cells (f, arrowheads). Scale bars: (a, b) 500 mm, (c) 200 mm, (d, e) 100 mm, (f) 50 mm [52]
Mouse and human PGC are mortal, nullipotent cells; in vitro exposure to KITLG, LIF, and bFGF reprograms them into totipotent stem cells (EGCs) that can grow indefinitely [60] and can enter the germline efficiently [33, 61].
3.2.4 Plasticity of Pluripotent States
From recent research papers and reviews [17, 19, 20, 62–66], a model emerges of the spectrum of developmental states of the different types of stem cells in the early embryo (mouse and human), how they are regulated at the molecular level in vivo, and how these developmental states can be modeled in vitro depending on culture conditions.
The term “pluripotency,” often used in a more general sense in the quoted papers, as in the legend of Fig. 3.4, is replaced by “developmental potential” throughout this chapter, to avoid confusion with the more specific application of the term “pluripotency” to indicate the developmental potential of cells in the primed state.

Embryonic origin and spectrum of pluripotent stem cell states. The pluripotent cells of a blastocyst between E3.5 and E4.5 can give rise to functionally naive ESC (blue). Between E5.5 and E8.0 postimplantation epiblast can establish EpiSC (orange), which occupy a primed pluripotent state. Additionally, primordial germ cells (PGC), which are the founders of the germline lineage, can give rise to naive EGC (green), which are highly comparable to ESC. Depending on the culture/derivation conditions, these pluripotent stem cells occupy discrete molecular states that can be broadly classed as naive or primed. The most optimized state of naive pluripotency, which closely recapitulates the naive epiblast cells of the blastocyst, is termed ground state. An interchangeable spectrum of pluripotent states may arise that ranges from ground state to primed pluripotency. The state of pluripotency adopted in vitro is primarily dictated by the combination extrinsic signals in the culture environment rather than the developmental source of the pluripotent cells. CH Chiron, PD PD03 [20]
The 2C state represents the full developmental potential (omnipotency) of the zygote, still present in the blastomeres of the two-cell stage of the embryo. These cells have not yet undergone global demethylation, erasure of parental imprinting, and X-inactivation (the latter in female cells) and do not (yet) express Oct4 and Sox2 [19]. In fact, this corresponds to the omnipotent state.
ESC derived from the preimplantation embryo (ICM and epiblast) have the broadest developmental potential, compared to other ESC, with a permissive epigenetic signature, including two active X chromosomes (in female cells), capable of forming embryonal and extraembryonal tissues and efficiently contributing to the germline. They can continuously self-renew; Oct4 expression is driven from the distal enhancer, and Oct4 partners with Sox2. These ESC represent what is called the ground state, naïve state, or totipotent state.
ESC derived from the primitive ectoderm of the postimplantation embryo exhibit reduced/absent expression of many ancillary pluripotency factors, including Klf4, Klf5, Prdm14, Rex1, and Esrrb, due to the attenuated Nanog expression [67]. These cells accumulate epigenetic barriers incompatible with the naïve state, such as female X-inactivation and promoter methylation at pluripotency genes, and thereby resemble the anterior primitive streak [68]. They give rise to somatic lineages and do not readily contribute to extraembryonic tissues and the germline. Their self-renewal capacity is limited, as they progressively differentiate toward stem cells committed to organs and tissues of the embryo proper; Oct4 is driven from the proximal enhancer, and Oct4 partners with Sox2. These ESC represent the so-called primed state or pluripotent state.
The developmental potential of PGC upon reprogramming depends on their epigenetic status. Early PGC, prior to completion of erasure of GI, give rise to EGC with the developmental potential of pluripotent ESC in the primed state. Late PGC with completed erasure of GI give rise to EGC with characteristics of naïve state, totipotent ESC, including a permissive epigenetic signature, the absence of X-inactivation, the activation of Oct4 expression from the distal enhancer, and the combination of Oct4 with Sox2. In human PGC, OCT4 partners with SOX17; upon reprogramming to EGC, OCT4 switches partner with SOX2. In parallel with their changing epigenetic status, PGC will have developmental potentials ranging from the primed to the naïve state.
In vivo, these developmental states are tightly controlled partly by cell autonomous factors (such as retroviral regulatory sequences) [19] but probably more by external cues, like position of ESC in the embryo. Plasticity of the developmental states in vivo is demonstrated by transplantation experiments, for example, cells from the tip of the epiblast become committed to the germline when transplanted in the proximal epiblast [12]. In vitro, naïve state and primed state can alternate depending on the culture conditions [20] (Fig. 3.4). A startling example of plasticity is the phenomenon that probably all ESC from the ICM transiently acquire the omnipotency of two-cell stage embryonic cells, the 2C state [19].
Apart from these physiological pluripotent cells, there are now somatic cells induced to pluripotency (iPSC) by the very factors involved in regulation of pluripotency in the embryo and the germline. This feat was first reported by Takahashi et al., using the same cocktail of pluripotency transcription factors, consisting of Oct4, Sox2, Klf4, and c-Myc, for mouse [69] and human somatic cells [70]. Shortly thereafter, Kim et al. demonstrated that mouse [71] and human [72] neural stem cells (NSC) can be induced to pluripotency by OCT4 alone, probably because these cells endogenously express SOX2, c-MYC, and KLF4. ESC and NSC appear to have many similarities at the transcriptional level [73]. Pluripotent stem cells can also be generated with embryonic stem cell-specific cell cycle regulating miRNAs [74].
iPSC, including those derived from NSC, resemble human ESC as to developmental potential, which means that they produce teratomas in vivo. By proper in vitro conditions, iPSC can be made germline competent [20].
In iPSC, the genomic imprint of the somatic cells from which they are derived is stably retained; however, a low frequency of loss of imprinting can be found, probably acquired in the process of reprogramming [75].
The high degree of plasticity of the developmental potential of stem cells, including iPSC, implies that the actual state of developmental potential of a given cell, rather than the cell type itself, ultimately determines the developmental potential of a stem cell [20]. This being said, it remains that a certain cell type has its characteristic developmental potential, e.g., a blastomere is characterized by omnipotency.
3.3 Developmental Potential of Germ Cell Tumors
Failure of regulation of the developmental potential of stem cells in the early embryo may result in mainly extragonadal tumors early in life reflecting the overall somatic developmental program of the originating cells. Flaws in the control of the developmental potential in the germline may give rise to tumors with a broad spectrum of developmental capacities, mainly in the gonads, and most often beyond childhood. Such gonadal and extragonadal tumors are usually designated with the umbrella-term germ cell tumors (GCT), which shall be used from here on.
Indeed, the predictions above fit with the epidemiology of GCT in infants, adolescents, and adults. Extragonadal GCT occur mainly in neonates and infants, rarely beyond age 6 [76] with an estimated incidence of about 1.5/100,000 for males and females together [77]. Of note, extragonadal GCT are associated with an increased risk for various congenital malformations. In adolescents and adults, GCT are mainly found in the gonads with an incidence of 0.5–12/100,000 for the testis, virtually always malignant, and an incidence of up to 15/100,000 for the ovary, most often benign [78]. Overall GCT are rare, and even in high-incidence countries like Denmark, the lifetime risk for a testicular GCT is only 1 % [79]. It is noteworthy that GCT of the gonads are associated with a risk for impaired fertility.
The rarity of these tumors in any anatomical site in humans, the mouse [80, 81], and other animal species, perhaps with exception of the horse [80, 82, 83], demonstrates how successfully the hazards of dealing with embryonic stem cells and germ cells are coped with, probably because these cells are highly apoptosis prone. To illustrate this point, targeted loss of OCT4 as well as Nanog in PGC in the developing mouse results in apoptosis of these cells [84, 85]. It could well be that these cells can only escape apoptosis if their normally repressed developmental potential unfolds. This mechanism likely plays a role in the origin of many GCT in humans.
GCT can be classified according to their developmental potential [86]. Tumors of a certain developmental type appear to have more features in common, such as age of presentation, anatomical distribution, (cyto)genetic aberrations, and epigenetic characteristics including global methylation and GI status [87] (Table 3.1, Fig. 3.5).

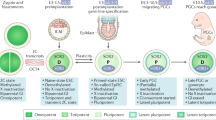
Unifying model of the pathogenesis of GCT based on the hypothesis that the developmental potential of GCT is determined by the developmental state (2C, naïve, primed) of the originating cell. Juxtaposed in the figure are stages of embryogenesis (upper panel), developmental potential of stem cells in subsequent stages of embryonic development and the germline (second panel), critical features of the involved stem cells (third panel), and corresponding GCT types with gender distribution and their histology, reflecting developmental potential (bottom panel, linking Fig. 3.5 to Table 3.1) (abbreviations in order of appearance: DEV.POT. developmental potential, PGC primordial germ cell, iPSC induced pluripotent stem cell, PARAM. parameters, M male, F female, DE-(I) first wave of demethylation, DE-(II) second wave of demethylation, RE remethylation, glob. Meth. global methylation, GI genomic imprinting, X-inact. X-inactivation, D distal enhancer, P proximal enhancer, H human, GCT germ cell tumor, TE teratoma, Im immature, YST yolk sac tumor, SE seminoma, NS non-seminoma, ST spermatocytic tumor, HM hydatidiform mole, DC dermoid cyst)
3.4 Type 0 GCT
3.4.1 Developmental Potential and Incidence
Internal parasitic twins (fetus in fetu) with an incidence of 1/500,000 births [88] and external parasitic twins, such as the epignathus that protrudes from the mouth, are extremely rare. These abnormal growths have the highest, omnipotent, developmental potential of all GCT, essentially not different from a zygote. They may contain well-developed internal organs, limbs [55], and often a vertebral axis [89] and are histologically composed of fully differentiated tissues. The presence of immature tissue or yolk sac tumor (YST) is exceptional [90–93], as is recurrence as YST [92, 94].
3.4.2 Anatomical Distribution
Fetus in fetu is in 80 % of the cases localized in the retroperitoneum and often enclosed in an amniotic sac, sometimes with rudiments of an umbilical cord [95] and extremely rarely placental tissue [92]. Other sites are the skull, hard palate, liver, sacrum, scrotum, and attached to ovary [94] and undescended testis [95]. External parasitic twins are localized at the same sites where conjoined twins are united [96].
3.4.3 Genetics and Pathogenesis
Genetic analyses in some of the more recent cases have with rare exceptions failed to demonstrate differences with the host [97]. These observations are consistent with fetus in fetu and external parasitic twins being monozygotic diamniotic twins [95] lacking a heart and deriving their circulation from the host. Apart from the heart, the brain is also usually missing; in fact, most of the rostral part of the embryo is poorly developed [96].
There are features, such as common anatomical localization and female preponderance, suggesting a continuum and common pathogenesis of conjoined twins, parasitic twins, fetus in fetu, acardiacs, which are considered parasitic twins attached via the placenta, and teratomas [96, 98, 99]. Multiple pregnancies could be the far end of this continuum, as each of the mentioned conditions as well as type I GCT of various anatomical sites [94] and perhaps also dermoid cysts (type IV GCT) [100, 101] is associated with a family history of multiple pregnancies [96] (see below). In fact, over 15 % of cases of fetus in fetu have a family history of twins or double fetus in fetu [94]. The basic defect then would be an increased risk of multiple pregnancies or, more mechanistically phrased, proneness of blastomeres in the 2C state to escape the organizing influence of the developing embryo or rather escape from control of their developmental potential. If the twin fails to develop a functional heart, it will either die or, if it succeeds in getting its circulation from the host, develop as a parasitic twin or a teratoma [96]. The latter may seem far-fetched; however, there is a case report on an oral mature teratoma in a female neonate that contained epididymal tissue. In the tumor, Y-chromosomal DNA was demonstrated by PCR, which was lacking in the peripheral blood of the girl who had a normal female karyotype in peripheral blood lymphocytes (Fig. 3.6). This extraordinary teratoma is probably best regarded as a poorly organized epignathus originating from dizygotic twinning [102], illustrating an exceptional mechanism of origin of teratoma.

Histology and PCR of male epignathus, disguised as teratoma, in a female neonate. Left panel, histology of teratoma with epididymal ducts (arrows); right panel, PCR-amplification of Y-chromosomal DNA. Lane 1: control DNA, female. Lane 2: control DNA, male. Lane 3: 100 bp ladder. Lane 4: DNA extracted from 10 μm thick slides cut from paraffin-embedded teratoma tissue showing ductus epididymis on light microscopy. Lane 5: DNA extracted from paraffin-embedded teratoma tissue showing no ductus epididymis on light microscopy [102]
3.5 Type I GCT
3.5.1 Type I GCT General
3.5.1.1 Developmental Potential
The natural history of type I GCT, emerging from numerous clinical and pathological observations [55, 94, 103–109], is that regardless of anatomical site, they begin during embryonic life as immature teratoma, probably arising from one pluripotent progenitor cell, which may evolve to mature teratoma with trilaminar derivatives. However, an immature teratoma component may contain foci of YST easily overlooked on microscopic examination [106, 110–113], which may eventually overgrow the original teratoma (Fig. 3.7). In fetuses and neonates, and in prenatally resected tumors, YST is virtually always associated with immature teratoma, while pure YST is rare [94, 113]. Thus, these tumors come in three histological variants: pure (immature) teratoma; pure YST, whereby the original teratoma component is probably overgrown by the more aggressive YST component; and combinations of (immature) teratoma and YST. The younger the infant, the more often an immature component is present and the lesser the chance that an overt YST component has developed, irrespective of gender (Table 3.2). YST can take the form of both intraembryonic endodermal derivatives, such as the primitive gut and liver, and extraembryonic structures such as allantois and yolk sac [114], reason for Nogales to advocate the name primitive endodermal tumor instead of YST (Chap. 6).

Testicular type I GCT, in infant of 5 months, composed of mature and immature teratoma with deceptive microscopic foci of YST, difficult to recognize without the aid of immunohistochemistry. (left, H and E ×200; right glypican 3, ×200)
The frequency of the different histological variants differs per anatomical site; however, in population-based registries, teratomas are the most frequent at all sites. In fact, the large majority of type I GCT have a favorable course regardless of degree of immaturity, with the exception of high-grade immature teratomas of the ovary. A YST component, overall present in about 5–10 % of the cases at birth [94, 109], is the only predictor of recurrence at any site [106, 109]. Type I GCT typically lack a seminomatous component, EC, and choriocarcinoma, which are indicative for a type II GCT (see below). Choriocarcinoma may rarely occur in infants, in association with an intracranial type I teratoma [115] or metastatic from placental/gestational choriocarcinoma [116, 117].
Type I GCT may contain OCT4-positive cells, usually in higher-grade immature teratoma components [118, 119], which however are negative for SOX2 and CD30. This is in contrast to EC cells, the stem cells of type II GCT, which typically express these two proteins in addition to OCT4 and other pluripotency proteins, such as NANOG and STELLAR [57, 120]. These OCT4-positive cells may be the stem cells of type I GCT. The lack of expression of CD30 may be explained by the cells being diploid. It was shown that in vitro ESC cells, the normal counterparts of EC cells, only start to express CD30 when they become aneuploid [121]. The rarity of these stem cells in type I GCT suggests that they do not readily self-renew but are rather poised to differentiation, particularly into the various somatic lineages, explaining the usually benign character of these tumors. In fact, the precursor cells seem to be in the primed state.
Type I GCT may contain SOX2-positive cells; however, these are not the OCT4-positive putative stem cells [122], shown in Fig. 3.8.

Type I immature teratoma with OCT4-positive, SOX2- and CD30-negative stem cells. (clockwise, HE, OCT4, SOX2, and CD30; original magnification, ×200)
3.5.1.2 Epidemiology
The age distribution of GCT shows a neonatal peak in the sacrococcygeal area, retroperitoneum, mediastinum, head and neck, brain (apart from the pineal gland), and testis. In the ovary, the early peak is missing; however, GCT do occur from birth through adulthood without interruption. The tumors represented by the early peak are type I GCT, rare tumors, most often occurring in the fetus, neonates, and children under the age of two and seldom beyond age six, with an overall predilection for girls, mainly due to the about 3.5:1 female to male ratio of sacrococcygeal tumors [76]. This skewed sex distribution may be due to global demethylation of PGC taking place earlier in males than in females. As a result, in males, the PGC are more fragile and prone to apoptosis upon mis-migration, whereas in females, the PGC are more robust and therefore have a greater chance to undergo reprogramming, giving them survival advantage outside a proper niche [57]. Such a mechanism would explain the overall slightly increased risk of extragonadal type I and perhaps also type 0 GCT in females [123], not answering the question of why mainly in the sacrococcygeal region.
Exact incidence figures are hard to get because in most cancer registries, only malignant GCT and not teratomas are included. The best approximation is achieved by combining cancer registry data with population-based figures on all GCT obtained in centers specializing in the treatment of these tumors [55, 94, 103, 105, 107, 109, 114]. The Netherlands and Belgian National Cancer Registries report, respectively, 5.2 and 5.4 malignant GCT per million children less than 15 years of age [77]; this figure is 4/million in Germany [107]. Assuming that over half, in fact, up to 70 %, of the type I GCT in children are teratomas, the overall incidence would be 1–1.5/100,000 [77].
Multiplicity of type I GCT is exceedingly rare: one child with a bilateral pure YST of the testis [124]; four cases of bilateral teratoma of the testis [125–127], two of which were brothers with Klinefelter’s syndrome [127]; no bilateral stage I ovarian YST [128, 129]; 1–2 % of ovarian immature teratomas is bilateral [130, 131]; and no published multiple extragonadal cases to the best of our knowledge.
Combinations in one individual of type I GCT with other GCT types do occur: type I GCT may rarely be combined with fetus in fetu (type 0 GCT) among others in the cranial region [132]; ovarian type I GCT may be combined with a type IV GCT in the same ovary and in 11 % in the contralateral ovary [130].
Familial cases of type I GCT have not been described for the testis, apart from the two brothers, both infants, with Klinefelter’s syndrome, mentioned above, with bilateral testicular teratomas [127]. In view of the rarity of bilateral testicular type I teratoma [125, 126] as well as Klinefelter’s syndrome, this is probably not a chance occurrence, suggesting that this syndrome is a risk factor also for type I GCT, in addition to being an established risk factor for type II GCT of the mediastinum and brain.
Ovarian type I GCT may cluster with dermoid cysts of the ovary (type IV GCT). Since the latter may have a familial component, this is probably also true for the type I GCT [130, 133–135].
Finally, there is the family of a mother with an immature teratoma of the ovary coexisting with a newborn baby with an intracranial immature teratoma [132, 136]; Poremba et al. excluded that the tumors were clonally related. Giambartolomei et al. [137] retrieved four families from the literature in which an ovarian type I GCT was combined with one or more type II GCT of the testis (five cases) or ovary (one case).
3.5.1.3 Anatomical Distribution
Type I GCT are most often localized in extragonadal sites along the midline of the body: the sacral region, retroperitoneum (cranially of the kidneys), stomach, anterior mediastinum, heart, head and neck, and brain [94, 138]. This peculiar distribution along the midline, including the brain, is attributed to the migration route of PGC during embryonic development [52, 54, 56, 57]. Others explain it by the relative abundance of ESC (for review [55]) or NSC [139] along the midline of the developing embryo. Type I GCT occur also in the testis, the second most frequent site after the sacral region, and in the ovary. They have never been described in dysgenetic gonads in keeping with the different pathogenesis of type I and type II GCT of the gonads, the latter being derived from transformed, virtually always aneuploid gonocytes in the naïve state, while the former probably originate through direct reprogramming of essentially normal, still methylated, and pre-erased diploid gonocytes to ESC in the primed state.
3.5.1.4 (Cyto)Genetics
Type I (immature) teratoma, either pure or combined with YST, is virtually always diploid, lacking chromosomal rearrangements. However, YST of type I, pure or combined with teratoma, is most often aneuploid, usually (near)diploid, with multiple gains and losses of (parts) of chromosomes. Involved in gains are 1q, 3, 3p, 8q24, 12p13, 20q, and 22; involved in losses are 1p (1p36), 4, 4q, 6q (6q24-qter), 16q, and 20p. Overrepresentation of the whole of 12p or the region 12p11.2–p12.1, typical for type II GCT, is not a feature of type I GCT. As mentioned above, the distal part of 12p and in particular 12p13 may be overrepresented ([111, 140–150], for review [151]). Some of these changes, such as gain of 1q and loss of 1p and 6q, are shared by type II YST and may be related to the phenomenon of progression/differentiation toward YST rather than being specific for type I GCT [152].
Although highly speculative, for some of the chromosomal gains and losses, possibly involved genes have been suggested; gain of 8q24 has been associated with amplification of MYC [142]; gain of 12p13 might involve the pluripotency genes STELLAR, NANOG, and GDF3 [38]; and loss of 1p36 [140] might involve CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastoma [153].
Sporadic case reports describe specific balanced chromosomal translocations in type I GCT. Two infantile sacral teratoma cases had constitutional balanced translocations involving 12q13 probably affecting different genes [146, 154]. In one of the two patients, the genes involved in the translocation t(12;15)(q13;q25) were identified as SUMO-/Sentrin-specific protease 1 gene (SENP1) and the embryonic polarity-related mesoderm development gene (MESDC2) [155]. The resulting fusion protein SEME interferes with the function of MESDC2 as a chaperone for the WNT co-receptors LRP5 and/or LRP6. It is suggested that in both patients, the constitutional translocation was predisposing to the development of the sacral teratoma. In an intrathoracic mature teratoma in a 15-year-old girl, a balanced chromosomal translocation, t(8;22)(p21;q12), was the sole cytogenetic aberration. It resulted in fusion of the genes PPP2R2A and CHEK2, supposedly the initiating event in this teratoma [156].
A genome-wide association study involving type I and II GCT suggests that a variant in BAK1 involved in suppression of apoptosis [157] is associated with gonadal GCT of both types. Type I GCT were not associated with variants in KITLG, SPRY4, and DMRT1 (doublesex and mab-3 related transcription factor 1), which confer an increased risk for type II GCT of the testis [158].
The Wnt/beta-catenin [159] and the TGFbeta/BMP signaling [160] pathways are strongly expressed in type I and II YST, compared to seminoma/dysgerminoma, EC, and choriocarcinoma ([161] for review). Methylation of APC and LOH at 5q21-22 suggests that APC might be involved in the activation of the Wnt pathway [162]. In general, the transcriptome of pediatric YST is enriched for genes associated with a differentiation and proliferation phenotype as compared to seminomatous (type II) GCT [163]. It is likely that the expression patterns of the various GCT types are mostly determined by the cell type(s) present and much less by pathways activated in the process of tumorigenesis. Seminomatous GCT and EC express pluripotency genes, and the various extraembryonic and somatic tissues express genes characteristic for the involved cell lineages [161, 163].
3.5.1.5 Epigenetics, including GI
Type I GCT usually have a biparental GI pattern as in somatic cells. In a small proportion, GI is partially erased, in particular in type I GCT of the testis and ovary [144, 164–167]. These findings on GI support the hypothesis that extragonadal type I GCT may originate from PGC, which are pre-erased or partially erased, corresponding to the methylation status of the genome (see Sect. 3.2.3). Reprogramming of these cells will produce an ESC with the developmental potential of the primed state. It does not exclude their derivation directly from ESC in the primed state, which are also characterized by a biparental GI pattern. Also, a somatic cell with induced pluripotency (iPSC), for example, by reactivation of pluripotency genes, in particular OCT4, as demonstrated for human NSC [72], could theoretically be the precursor of type I GCT. This has been suggested by Scotting and co-workers for GCT of the brain [139, 168, 169], without making the essential distinction between type I and type II GCT [86]. Such iPSC would endow the derived tumors with their own GI pattern [56], as it is stably transmitted to daughter cells [75, 170]. Any degree of loss of imprinting in a type I GCT tumor would suggest that the tumor is derived from a germ cell precursor [166], unless, as reported [169], NSC may also have partial loss of imprinting.
3.5.1.6 Animal Models and Pathogenesis
Apart from humans, pluripotent tumors have been most extensively studied in the mouse. Relevant for human testicular type I GCT are the spontaneous [80, 81] and experimental [171] testicular teratomas in the 129 mouse strain. The spontaneous ovarian teratomas in LT mice [172] are probably a model for human ovarian type I GCT. Teratomas derived from pre- and postimplantation embryos transplanted to various organs, in particular the testis [173] or the kidney [174], might be a model for extragonadal type I GCT in man. Spontaneous extragonadal teratomas in mice [83, 175, 176] are too rare to be practically useful for animal experiments.
The difference between the spontaneous and experimental testicular teratomas as compared to the embryo-derived teratomas is that the gonocytes from which the testicular tumors originate are committed to the germ lineage and not themselves pluripotent [61]. They have to be reprogrammed before being able to form pluripotent tumors [60], a process similar to what happens in the human type I GCT, and to reprogramming of a somatic cell to an iPSC, by converting the nucleus from nullipotent to pluripotent [61].
These different mouse models have a similar developmental potential; when fully developed, they are mainly composed of mature somatic tissues derived from the three germ layers. Immature teratoma and EC cells are less frequent and often minor components; rarer still are extraembryonic lineages. Late takes of embryo transplantation under the kidney capsule consist of parietal YST and occasionally trophoblastic giant cells [177]; these tumors are most often aneuploid [178], just as human type I YST (Fig. 3.9). The observation that in chimeric blastocysts polyploid murine ESC only give rise to extraembryonal lineages (yolk sac and placenta), while the embryo proper is derived from the diploid ESC [179, 180], might explain the restricted developmental potential of aneuploid tumor cells in type I GCT: probably, they are no longer capable to form somatic tissues.

The testicular teratomas in the mouse models originate when a luminal gonocyte or a prespermatogonium in its niche is reprogrammed to an ESC in the primed state, either directly or via an EGC that apparently loses its naïve-state developmental potential [60, 61, 80, 181, 182]. Initially, when proliferating within the seminiferous tubules, the tumor cells stay undifferentiated, as EC cells. When the EC distends and disrupts the tubular wall and invades the testicular interstitium, it starts to differentiate into immature somatic tissues, which gradually develop into mature teratomas [81, 183]. A minority of the tumors will maintain immature teratoma and EC and will be retransplantable in syngeneic hosts. Rare tumors are pure EC from the start, which can readily be transplanted. This same evolution is seen in tumors derived from pre- or postimplantation embryos up to E8. The percentage of tumors with an EC component depends on the strain of the transplanted embryos and is usually higher than in the testicular teratomas [184]. These mouse models, as well as the ovarian teratomas in LT strain mice, resemble human type I GCT. They have the same cells of origin (PGC/gonocytes and ESC), histological evolution, and developmental potential. Whatever the cells of origin, they seem to have or acquire the primed state in view of the developmental potential of the derived tumors.
Probably the most important lesson to be learnt from these models is that disruption of the microenvironment of the pluripotent cell itself suffices to initiate a pluripotent tumor. For gonocytes/prespermatogonia in the developing testis of 129 strain mice, this principle is demonstrated by genital ridge transplantation, as will be discussed in the following.
In 129/Sv mice carrying the loss of function steel mutation (steel or Kitlg is the mouse homolog of KITLG), spontaneous testicular teratomas occur in about 4 % of the animals, twofold the spontaneous rate (2 %) in 129 strain mice lacking this mutation. When from the same animals the genital ridges are transplanted, teratomas develop in over 80 % of the grafted genital ridges, often at multiple sites [171]. This is counterintuitive: loss of PGC with the steel mutation, and even more so by the procedure of the genital ridge transplantation, increases the yield of teratomas. The steel mutation, in the membrane-bound Kitlg [185], and the transplantation procedure are not carcinogenic events acting on the PGC but rather factors that disturb the niche of the PGC, promoting reprogramming of the surviving PGC. Apparently, cell-intrinsic mechanisms for repression of the developmental capacity of gonocytes/prespermatogonia, such as those via Blimp1, Prdm14, and AP2ɣ [186–188] and Dmrt1 [189], are not sufficient when the restraints of the normal tubular environment are disturbed, like Nanos2-/Dmrt-dependent GDNF signaling by Sertoli cells [189]. Remarkably, only male genital ridges produce teratomas; female genital ridges never do [173], probably because in the female genital ridge, the germ cells are blocked in meiosis I and few in numbers. In the male genital ridges, the germ cells are more numerous, premeiotic, and arrested in G0/G1 of mitosis [12]. Contrasting patterns of Dnd1 expression in female and male gonads may also contribute to the different susceptibility to teratoma formation [190].
As for embryo-derived tumors, perfectly normal embryos may turn into teratomas when transplanted into a testis or a kidney [173, 184]. EC cells derived from such tumors, when introduced into the ICM of a blastocyst, can contribute to the normal tissues of the resulting chimeric mouse, demonstrating that the tumor cells when restored to their proper environment may normalize, as well as cause malignant tumors [191].
The importance of genetic factors conditioning the micro-milieu of the niche was demonstrated by crossing susceptibility genes for testicular teratomas into 129 strain mice. As already mentioned, in the original strain, about 2 % of the mice had spontaneous testicular teratomas, introducing the loss of function steel mutation, which reduces the number of PGC and spermatogenesis [192], doubled this percentage [171], and by adding the ter mutation, one third of the mice developed spontaneous testicular teratomas [81]. The gene ter is a recessive gene that causes germ cell deficiency in mice, and in 129/Sv-ter mice, it also enhances the yield of teratomas. Male 129/Sv-ter mice, homozygous for the ter mutation, are sterile and almost always have teratomas, often bilateral [193]. The ter mutation occurs in the Dnd gene, expressed in fetal gonads [190]; in mice, Dnd isoform α is necessary for viability of germ cells including PGC from E8 and for viability of embryos [194]. Specifically, in 129 strain mice homozygous for this mutation, PGC die apoptotically or when they escape apoptosis may be reprogrammed to ESC, which form teratomas. It is even more likely the other way round that some PGC escape apoptosis because they have been reprogrammed [190]. As a corollary, the proneness of 129 mice – and not of other strains [194] – to form teratomas is due to the ease with which PGC of 129 mice PGC are reprogrammed to an ESC in the primed state. This may be due to incompetence of 129 strain mice to adequately suppress reprogramming to pluripotency in germ cells, a process in which among others Dmrt1 expressed in PGC/gonocytes is involved [189].
The variants in KITLG that increase the susceptibility for testicular type II GCT in humans [195, 196] do not seem to affect the incidence of type I GCT [158].
Although derived from PGC, KIT mutations are probably exceptional in type I GCT. In support of this contention, none of the pure immature and mature teratomas of the brain studied by Wang [197], almost certainly type I GCT, had KIT mutations and also very rarely other mutations. This is indeed remarkable since KIT is the crucial survival and proliferation factor for PGC.
In the mouse, germ cells that do not reach the genital ridges die through apoptosis caused by the proapoptotic protein Bax. In Bax-null embryos, large numbers of ectopic (extragonadal) germ cells fail to die [57]. A similar mechanism of impairment of apoptosis of mis-migrated PGC might enhance the development of extragonadal human type I GCT; however, this has not been demonstrated.
The available evidence points to the pathogenesis of type I GCT being foremost “developmental” and not driven by somatic mutations. This implies that the p53-dependent DNA damage response is intact in these tumors, explaining their favorable response to cisplatin-based chemotherapy, just like type II GCT.
3.5.1.7 Summary of the Pathogenesis of Type I GCT
The most likely cells of origin of extragonadal type I GCT are mis-migrated PGC, as these cells have been demonstrated along the midline of the body, indeed in large numbers at the typical sites of these tumors [52]. Most of these PGC die apoptotically; probably only those that are reprogrammed to an ESC manage to survive outside the niches in the gonads, the thymus, and the midline of the brain suitable for PGC. Reprogramming occurs when the mechanisms, with a key role for SOX17, BLIMP1, and OCT4 [35], maintaining the phenotype and suppressing the developmental potential of PGC, break down, probably because of lack of a suitable niche. Since the PGC are pre-erased, reprogramming will result in an ESC in the primed state capable of forming immature somatic tissues that will usually differentiate to fully mature teratoma. YST and very rarely choriocarcinoma are the only other components, which develop from tumor cells that have become aneuploid.
In the testis and ovary, type I GCT originate when diploid, pre-erased gonocytes, and oogonia are reprogrammed to ESC in the primed state due to failure of control of developmental potential by germ cell-intrinsic (DMRT1 in addition to SOX 17, BLIMP1, and OCT4) and niche factors (such as GDNF) [189]. Reprogramming to an ESC can occur directly or via an EGC in which the naïve state is rapidly dismantled [182].
Pathogenesis is mainly developmental; somatic mutations probably play a minor role.
3.5.2 Site-Specific Aspects of Type I GCT
3.5.2.1 Sacral Region
Sacrococcygeal type I GCT, with a frequency of 1/35,000 live births, constitute about 40–50 % of extragonadal type I GCT and are the most frequent neonatal tumor. They are rarely diagnosed beyond the age of 2 years and virtually do not occur after age six [76, 109, 198, 199]. The fact that there are practically no GCT at all in the sacral region past the age of six is in accordance with the absence of type II GCT at this anatomical site. The rare sacrococcygeal type I teratomas in adults probably had their inception before birth and went undetected [55]. There is a strong predilection for girls with a male to female ratio of 1:3.5.
Other congenital disorders occur in up to 25 % of patients with sacrococcygeal type I GCT, including trisomy 21/Down’s syndrome (implying a higher risk for type I GCT in Down’s syndrome), genitourinary malformations, congenital hip dislocation, esophageal atresia and congenital heart disease [114, 198], and duplication of pelvic organs attributable to hindgut twinning [200]. There is a well-documented association with multiple pregnancies, either within the same pregnancy or as a family history of multiple pregnancies [94, 96].
The evolution of these tumors is typical for type I GCT. Starting as immature teratomas prior to birth, they become more mature with time. When completely removed at this stage, which entails removal of the coccyx bone in continuity with the tumor ([114] for review), the child is cured. Incomplete or delayed surgery may allow the tumor to recur as mature or immature teratoma, or by means of tumor progression, to develop a YST component in the primary tumor or in a recurrence. A YST component is found in 5–10 % of the tumors removed before the age of 2 months; thereafter, this figure increases rapidly, and by the age of three, most sacrococcygeal type I GCT are malignant, in principle due to progression to YST [94, 201, 202]. The tendency for malignancy seems somewhat greater in males than in females [201]. Rarely, a somatic-type malignancy may develop such as Wilms’ tumor [203, 204]. Also the type I teratomas of adults may in some 10 % develop a malignant component [205]. Metastases can be local or visceral and are usually composed of YST or less frequently immature teratoma [198, 199]. In contrast to teratoma, YST is aneuploid with the chromosomal aberrations characteristic for YST progression in type I GCT, as discussed.
Sacral teratomas so highly developed that they have a vertebral axis should according to the definition of Willis [206] be classified as parasitic twins. A somewhat less strict definition [96, 98] considers a sacral teratoma with clearly developed limbs as a parasitic twin; in view of the site of attachment, they should be classified as a parasitic pygopagus [96]. Indeed, a personal case, published as sacral teratoma with a classical clinical history, including recurrence as YST (with the characteristic chromosomal aberrations) upon incomplete surgery [207], should probably be reclassified as a parasitic pygopagus, a conjoined twin parasite attached to sacrococcygeal area (Fig. 3.10). This case illustrates the continuum between twinning and the development of a type I GCT and the difficulty pinpointing the cells of origin of these growths. Indeed, some deem it possible that all extragonadal teratomas have originated as twins [96], and at the other end of the spectrum, others consider them as derived from mis-migrated PGC, which have a preference for the rostral and caudal part of the sympathetic nervous system [52]. In between are those who favor the idea that they are derived from an ESC.

Neonate with diploid sacral teratoma/parasitic twin (pygopagus) with a clearly recognizable foot; upon irradical removal recurring as aneuploid YST (right top; H and E, ×100); karyotype with typical loss of 1p and gain of 6q (left bottom); after chemotherapy a small residual mature teratoma was resected (right bottom; H and E, ×40) [207]
Cases like ours [207], and an almost identical one reported by Chen et al. [208], blur the distinction between parasitic twin and teratoma or rather between type 0 and type I GCT. The two types of GCT may be derived from the same or different precursor cells in the 2C, respectively, primed state.
3.5.2.2 Retroperitoneum
In the retroperitoneal region, all GCT under the age of six are probably type I GCT [76]. Perhaps some may be poorly organized included twins (type 0 GCT), as the retroperitoneum is the most common site of fetus in fetu [95].
Five to ten percent of extragonadal type I GCT occur in the retroperitoneum, most of them in the left or right suprarenal region consistent with lateral migration of PGC toward the gonadal ridges [52]. The sex distribution is about equal when several smaller series are combined [104, 209, 210]. Over 10–20 % of tumors are partly or wholly composed of YST, of the remainder, about half have an immature teratoma component and half are completely mature teratomas. The relatively high figure for YST is probably due to the fact that most of the tumors are diagnosed a couple of months after birth.
In postpubertal males, retroperitoneal GCT are virtually always metastatic from unrecognized testicular type II GCT [108, 211–213]. In postpubertal females, retroperitoneal GCT are very rare, usually benign, and probably type I GCT that have remained undetected until after puberty.
3.5.2.3 Stomach
Of the type I GCT, 2–3 % are located in the stomach with a male-to-female ratio of 1:3.7; progression to YST is rare [94]; however, like the type I GCT of the neck, they may metastasize in the form of immature teratoma [94].
3.5.2.4 Mediastinum
The mediastinal type I GCT constitute 2–3 % of the total, most are located in the anterior mediastinum, originating in the thymus [114], and only rarely in the posterior mediastinum. There is a slight preponderance of females [94, 138]. Progression to YST occurs in up to 30 % probably due to surgery several months after birth [214].
3.5.2.5 Heart
Type I GCT of the heart are relatively frequent, 4–7 % of the total, most often located in the pericardial cavity, attached to the great vessels at the base of the heart, and only rarely within the heart itself, very much in accordance with the sites where mis-migrated PGC are found [52]. Males and females are equally affected. Progression to YST occurs in about 5 % [94].
3.5.2.6 Head and Neck
Type I GCT of the head and neck occur in less than 40,000 live births and constitute 10–20 % of all extragonadal type I GCT; the sex distribution is roughly equal. Anatomical localizations are the neck including the thyroid gland (35 %); face (8 %); oro- and nasopharynx and surrounding structures, in particular hard palate and nasopharynx (45 %); and orbit (12 %) [94, 138, 215–217].
They develop during embryonic life and are often diagnosed before birth. The histology is most often mature teratoma, about one third of the cases contain immature teratoma. Immature neural tissue may rarely metastasize to regional lymph nodes and the lungs and on very rare occasions spontaneously mature [218]. About 3 % of the tumors present as pure YST or as teratomas with microscopic foci of YST [94, 216]. In the series of 16 cases described by Lack [215], there were three YST, respectively, in the oropharynx, the nasopharynx, and the floor of the mouth. In two cases, surgery was not carried out immediately after birth but after 6 and 10 months, respectively. Progression to a somatic-type malignancy may occur, in particular neuroblastoma [218]; squamous cell carcinoma has been reported as well [219].
Progression to YST and metastasis did not occur in 51 cases occurring in the neck [217], probably because surgery is done shortly after delivery, preventing the tumors to progress. The low progression rate might raise the suspicion that many of the teratomas are in fact parasitic cephalopagus [96]. Indeed the oral mature teratoma, mentioned before, diagnosed prenatally in a female baby, most likely was a disorganized dizygotic twin [102], which in retrospect should have been classified as epignathus or more formally as parasitic cephalopagus. This is yet another example of a case that blurs the distinction between type 0 and type I GCT.
Oro-nasopharyngeal and cervical teratomas are associated with other congenital disorders in 12 and 6 %, respectively [94].
The highly aggressive sinonasal pluripotent tumors in adults [220–222] are often characterized by chromosomal translocations and will be discussed as type VI GCT.
3.5.2.7 Brain
Intracranial type I GCT constitute about 10–15 % of all type I GCT with an equal sex ratio and 3 % associated with YST [94, 109, 132]. In one third of the cases, the size of tumor obscures the original anatomical localization. When the site can be determined, it is most often cerebral hemisphere (25.5 %), followed by the suprasellar region (23 %), third ventricle (5.6 %), and pineal region (4.4 %) [132]. The tumors may extend into the orbit, neck, face, mouth, or pharynx [94, 132, 138].
3.5.2.8 Testis
Under the age of six, GCT of the testis are practically always of type I [76], amounting to 5–10 % of all type I GCT [94, 109, 223]. Eleven out of the 19 tumors described by De Backer et al. [223] were teratomas, confirming that teratomas are more frequent than YST in unbiased institutional registries [224]; indeed, under the age of 1.5 years, no YST was diagnosed. Four of the 11 teratomas had immature areas; however, none of the tumors was combined with YST or a raised serum alpha-fetoprotein (AFP). Mixed type I GCT, combining teratoma with YST, are rare but do occur also in the testis [224].
In view of the supposed pathogenesis of type I GCT, it is remarkable that mixed tumors are so rare in the testis, at least ten times less frequent than pure YST [224–226]. The presence of immature teratoma may increase the risk of progression toward YST [106, 198]. Probably, when progression occurs early, in a microscopic immature teratoma, the tumor appears as pure YST at clinical presentation; progression in an established teratoma results in a mixed type I GCT combining teratoma with YST. Teratomas may very rarely, also by way of progression, develop PNET as a somatic-type malignancy [227].
Type I GCT of the testis are not associated with germ cell neoplasia in situ (GCNIS) [228] and testicular dysgenesis syndrome (TDS) [229] and do not share the risk factors of testicular type II GCT nor their increasing incidence. Familial susceptibility for prepubertal YST has not been demonstrated [230, 231]. Unlike the type II GCT of the testis, there is no association with single nucleotide polymorphism (SNP) variants of KITLG, SPRY4, and DMRT1, among others. There seems to be an association with a SNP variant of BAK1 [158], suggesting that resistance to apoptosis of primitive germ cells might play a role in the pathogenesis of prepubertal GCT. This is in line with the hypothesis that testicular type I GCT originate through reprogramming of a diploid, methylated, pre-erased, premeiotic PGC to an ESC in the primed state.
3.5.2.9 Ovary
In the ovary, the early neonatal peak in the age distribution of GCT, representing type I GCT, is not apparent [76]; however, it is unlikely that they do not exist. Rather their age distribution is probably broader and overlaps with types II and IV, as shown below.
Among 158 reviewed cases of pure and mixed dysgerminomas of the ovary, by definition type II GCT, the youngest was 4 years old; 6 % were in the age group 0–9 years and 41 % between 10 and 19 [232]. A review of 517 dermoid cysts, type IV GCT of the ovary, showed an almost Gaussian age distribution with no cases under age 10 and 1.5 % under age 15 [233]. From these figures, it can be deduced that the large majority of the 66 pediatric patients through age 15, reported by De Backer et al. [234], had a type I GCT. Six tumors were purely cystic, thus probably type IV GCT, and 12 were type II GCT on histological grounds, leaving 48 type I GCT. Of these, three were pure YST, consistent with the rate of about 5 % YST in other anatomical sites. This makes the ovary the second most frequent site of type I GCT after the sacrococcygeal region, accounting for 15–25 % of all type I GCT.
Apparently, teratomas of the ovary can be of three types: I, II, and IV and taking the type VI teratomas associated with clear cell carcinoma of the ovary (Chap. 6) also into account, four types. The overlapping age distributions and morphological resemblance may pose problems separating them. A morphologically typical dermoid cyst in a patient over 10 years of age is almost certainly a type IV GCT. A solid teratoma or a pure YST, or the combination of the two under age five, is most probably a type I GCT. Any GCT with a dysgerminoma, EC, or choriocarcinoma component, with or without other components, is a type II GCT regardless of age. Teratomas associated with epithelial cancers of the ovary are of type VI. Cases composed of teratoma and/or YST over age 5 could be type I or type II GCT. Separating these malignant GCT is probably not so important clinically. However, for a (partly) solid pure teratoma, in a patient over 5 years, it is crucial to make the distinction, since a type I teratoma is benign, whereas a type II teratoma is malignant. In such cases, the diagnosis needs (cyto)genetic confirmation.
Like for the testicular ones, the assumption is that ovarian type I GCT originate through reprogramming of a diploid, methylated, pre-erased, premeiotic PGC to an ESC in the primed state. Such mitotic germ cells persist in the periphery of the ovary through week 20 gestational age [235, 236]. Oogonia can be present in the cortex of the ovary in the two first years of life before they are finally cleared [237].
3.5.3 Type I GCT Beyond Infancy
In general, type I GCT occur neonatally and in early infancy, in prepubertal individuals. However, GCT with essentially the same developmental potential may become clinically manifest at older ages, also in postpubertal patients. This is obvious for the ovary where the neonatal incidence peak is lacking, and the type I GCT have a broad age range, overlapping with the age distribution of the type II and type IV GCT of the ovary. The existence of prepubertal type I teratomas in the postpubertal testis was recently established [228, 238, 239]. Typically, these teratomas are highly differentiated, lack (cyto)genetic abnormalities in particular gain of the complete short arm of chromosome 12 (12p), and are not associated with GCNIS. Remarkably, they may grossly present as dermoid cysts, sometimes containing hair [238], as the type IV dermoid cysts of the postpubertal ovary almost invariably do. Like type I teratomas, they may, albeit rarely, progress to YST [120]. Zhang et al. [238] have proposed that they have the same pathogenesis as type I, prepubertal teratomas. However, the possibility that they arise later in life from “dormant” germ cells arrested in meiotic prophase, like extragonadal mis-migrated PGC, which can be reprogrammed to the primed state, cannot be excluded [120]. It is likely that type I teratomas may also occur beyond infancy at extragonadal sites, like the mediastinum [55, 240, 241] and brain [242]. Particularly in the mediastinum, postpubertal mature teratomas may have the gross appearance of a dermoid cyst, grossly containing hair and even tooth structures [55, 240, 241] (Fig. 3.11). Microscopically, the cysts are lined by squamous epithelium with pilosebaceous structures and may have glial tissue in the solid parts of the cyst wall. It seems that in these anatomical localizations, ovary, mediastinum, other extragonadal sites, and perhaps also testis, the developmental potential of the teratomas may have intermediate phenotypes between typical type I and type IV GCT. In each site, teratomas occur that are partly dermoid cysts and partly solid teratoma sometimes with immature components. In the ovary, typical type I teratomas may occur side by side with type IV teratomas, both uni- and bilaterally [130]. Remarkably, the incidence of type I GCT beyond infancy in the mediastinum [241] and brain [243] is rather similar in males and females as opposed to type II GCT, which are much more frequent in males than females.

Mediastinal teratoma, late type I GCT, with intermediate phenotype between types I and IV: cyst filled with sebaceous material and hairs
These clinical observations on early and late type I GCT may be explained by the phenomenon that PGC in females and males regardless of anatomical site enter meiotic prophase by default [58, 59]. The only exception are gonocytes in the testis, which within the seminiferous tubules, under the influence of Sertoli cells, undergo mitotic arrest until puberty. The various phenotypes of these tumors, ranging from typical, solid type I GCT to mainly cystic teratomas closely resembling type IV GCT, may be due to epigenetic differences between the originating PGC. It is hypothesized that pre-erased PGC of early infancy, reprogrammed to ESC in the primed state, will give rise to the typical type I GCT phenotype, while PGC that later in life, beyond infancy, have entered meiotic prophase and concomitantly have undergone partial erasure of GI and possibly some degree of maternal imprinting, may form tumors resembling type IV GCT. In fact, these GCT have intermediate phenotypes between type I and type IV GCT. This assumption is supported by the observation that in mice, EGC are totipotent when derived from PGC but that this phenotype is gradually lost in EGC derived from more mature germ cells [61]. Fully fledged type IV GCT seem to occur only in the postpubertal ovary [233].
3.6 Type II GCT
3.6.1 Type II GCT General
3.6.1.1 Developmental Potential
Type II GCT are malignant tumors that come in two variants: first, seminomas (named dysgerminoma in the ovary; germinoma in the brain; seminoma or germinoma in the mediastinum), which are homogeneous neoplasms composed of neoplastic PGC/gonocytes, the default development of type II GCT; second, non-seminomas, which are caricatures of embryonic development, including both somatic and extraembryonic lineages [244]. Non-seminomas arise when a neoplastic PGC/gonocyte is reprogrammed to become an EGC in the naïve state, or, in pathological terms, when a seminomatous cell is reprogrammed to a totipotent EC cell, the stem cell of non-seminomas [245], as originally demonstrated for mouse EC cells by Kleinsmith and Pierce [246]. EC cells may give rise to all lineages of embryogenesis: YST (secreting AFP) and choriocarcinoma (secreting beta-human chorionic gonadotropin (β-HCG)) represent the extra-embryonic tissues; immature and mature teratomas represent somatic tissues of the three germ layers of the embryo in varying degrees of maturation; occasional primitive germ cells represent the germ lineage [247] (Fig. 3.12). These elements are characterized by lineage-specific mRNA [248, 249] and protein expression profiles [161]. In non-seminomas, so-called embryoid bodies can be encountered, which strongly resemble 10-day-old, early presomite human embryos. They show the same expression patterns of both mRNA and proteins as during normal development, like OCT4. Beyond that particular stage, corresponding to the time that in a pregnancy implantation is completed [250], development becomes disorganized, with embryoid bodies turning into patches of EC, YST, trophoblastic giant cells/choriocarcinoma, or teratoma, and disorderly combinations thereof. A possible explanation is that the neoplastic embryo lacks the biparental imprinting pattern of the zygote that is required for proper development of extraembryonic structures and concomitant vascular supply. Mature teratoma may be highly differentiated at the tissue level and even contain organoid structures closely resembling the gut, bronchi, etc., but never fully developed organs as in type 0 GCT or hair and teeth as in type IV GCT. The complete gamut of differentiation lineages, in particular the capacity to develop both embryonic and extraembryonic lineages, the germline competence, and the high capacity of self-renewal of its stem cells (EC cells) characterize type II GCT indeed as totipotent, apparently derived from precursor cells in the naïve state [251].

Germ cell differentiation in non-seminoma: TSPY-positive cells within epithelium of primitive gut-like structure and dispersed in surrounding mesenchymal tissue (TSPY, original magnification 100×) [56]
The mechanism of reprogramming of a seminomatous tumor cell (including the cells of GCNIS) is unknown. It is likely that microenvironmental factors play an important role, suggested by the observation that in the cryptorchidism, the percentage of seminoma depends on the location of the testis: about 90 % in abdominal, about 80 % in inguinal, and about 50 % in scrotal position (both after spontaneous or surgical/hormonal correction of cryptorchidism) [252–254]. The age of clinical presentation of the tumor was the same as in patients with scrotal tumors without a history of cryptorchidism [253]. Recently, it was suggested that interstitial stromal factors like NOGGIN might inhibit bone morphogenetic protein (BMP) in the tumor cell, whereupon reprogramming is initiated via NODAL signaling in two stages [255]. During a maturation phase, a fast-acting NODAL loop stimulates its own activity and temporarily inhibits BMP signaling. During the stabilization phase, a slow-acting NODAL loop, involving WNT signaling [159], reestablishes BMP signaling and the pluripotency circuitry [255]. This is in line with the observations on Cripto, the co-receptor for Nodal [256, 257], which is highly expressed in GCNIS, seminoma, EC, and YST, associated with hypomethylation of the promoter and absent in teratoma where the promoter is hypermethylated [257].
Interestingly, inhibition of BMP is the opposite mechanism from initiation of germline specification in the mouse embryo via expression of Bmp4 ([35] for review). This NODAL-mediated mechanism of reprogramming implies that the tumor cells are exposed to interstitial stromal cells, which is not the case in the intratubular environment, suggesting that within the seminiferous tubule, other factors are involved in reprogramming of GCNIS or intratubular seminoma cells. Moreover, stromal factors are usually not sufficient as primary seminoma is reprogrammed in only about 15 %, giving rise to a mixed non-seminoma with a seminoma component. It seems there is more to be learned about reprogramming of a seminomatous precursor cell to a totipotent EGC.
In all anatomical sites, over half of all primary type II GCT are pure seminomatous tumors. In fact, the younger the patient population, the higher the proportion of seminoma: in dysgenetic gonads and the brain about 80 %, in the ovary 60 %, in the mediastinum 55 %, and in the testis about 50 %. Reprogramming continues even in metastatic seminoma of the testis: 44 % of seminoma metastases eventually develop non-seminoma components [258]. Thus, in the natural history of testicular type II GCT, only 30 % maintain their seminoma phenotype until demise of the patient. These observations suggest that reprogramming is a chance event accumulating over time, whereby in a non-scrotal testis, the chance of reprogramming is deminished, as discussed above.
Seminomatous tumors are by definition pure; the only cells other than neoplastic gonocytes are scattered trophoblastic cells occurring in less than 10 % of the cases [244]. Dysgerminomas in the ovary, mediastinal seminomas, and germinomas of the brain may also contain trophoblastic giant cells in a small percentage [243, 259, 260]. Non-seminomas are often composed of more than one differentiation lineage, in all possible combinations including seminoma, so-called mixed non-seminomas. EC is almost always present and may be the only component, like its derived lineages, thus accounting for pure EC, YST, choriocarcinoma, and teratoma. The frequency of EC attests to the high capacity for self-renewal of these totipotent stem cells of non-seminomas and likely explains the more rapid evolution and earlier clinical manifestation of non-seminomas than seminomas. This is well documented for the testicular type II GCT, where the age distribution for seminomas peaks at 35 years and for the non-seminomas at 25 years. Mixed non-seminomas with a seminoma component, in which reprogramming is delayed because it occurs in already invasive seminoma, peak at the median age of 30 in between non-seminoma and seminoma [261, 262] (Fig. 3.13). Primary type II GCT of the brain, mediastinum, and ovary show the same order in age distribution: for brain, the mean age for germinomas, mixed tumors with a germinoma component, and EC is, respectively, 18, 15, and 12 years [263]; for mediastinum, the mean age for seminomas is about 30 [264] and for non-seminomas 25 years [265]; and for the ovary, the median age of dysgerminoma is 22 years [266], for EC 14 years [267], and for mixed non-dysgerminomas with a dysgerminoma component in between.

Distribution of age of presentation of testicular non-seminoma (dashed line), seminoma (solid line), and non-seminoma with a seminoma component (dotted line) [261]
Somatic tissues of non-seminomas may progress to form somatic-type malignancies that closely resemble their somatic counterparts, as will be discussed per primary site (see also Chap. 12) (for review [244, 268]).
Seminomas and non-seminomas may metastasize to regional lymph nodes and from then on to distant organs, so-called visceral metastasis, in order of frequency: lungs, liver, brain, and bone [258]. Choriocarcinoma has a propensity for blood-borne metastases, which may cause the first clinical manifestation of the tumor [244]. Seminoma cells may at metastatic sites be reprogrammed to non-seminoma in up to 44 %, as mentioned [258]. In non-seminoma, EC cells are the principal metastatic cells; likewise, in somatic cancers, cancer stem cells are the ones that frequently metastasize [269]. This is microscopically apparent as tumor emboli are virtually always composed of EC cells [244]. At the site where these tumor stem cells will eventually lodge, they may resume differentiation, mimicking the histology of the primary tumor. This phenomenon is demonstrated in mouse models of type I GCT, which in this respect are also valid for type II GCT [270] (Fig. 3.14). The level of differentiation at the metastatic site is in general less than in the primary tumor [271], which may be related to the different microenvironment or due to selection of more metastatic EC cells. In support of the latter, the more distant the metastases, the lesser the level of differentiation; in particular, visceral metastases rarely contain teratoma components but often consist only of EC cells that apparently in the metastatic process have progressively been selected toward stemness and loss of differentiation capacity [272].

Inguinal lymph node metastasis of retransplantable embryo-derived teratocarcinoma in the thigh of mouse; left panel, early metastasis, composed of EC cells only (H and E, original magnification ×200); right panel, later metastasis shows somatic differentiation in addition to EC cells (H and E, original magnification ×100) [270]
3.6.1.2 Epidemiology
Over 90 % of type II GCT occur in the testis [76], being the most frequent cancer of males aged 25–45 in Western white Caucasian populations [273]; the remaining develop in dysgenetic gonads/ovary (about 4 %) and in the extragonadal sites: anterior mediastinum/thymus and brain midline/pineal gland (about 3 %) [274]. The youngest age of presentation is in dysgenetic gonads/ovary (from age four with a broad age distribution) [232, 266], followed by brain (children from age 10 to adolescence) [263], mediastinum, and testis (adolescents and adults) [264, 265]. Remarkably, overall the peak age is about 30 for testicular type II GCT: 15 years later than the peak age for those of the brain and 5 years later than for those in the ovary and mediastinum. Type II GCT occur in patients in whom puberty has started or is completed, except for rare cases associated with disorders of sex development (DSD) [275], Down’s syndrome [276], and Klinefelter’s syndrome [277].
In each of the extragonadal sites, males greatly outnumber females with regard to type II GCT [76]. Apparently, type II GCT is very much a disease of adolescent and adult males, probably related to the presence of the TSPY gene on the Y chromosome, as will be explained later on.
The overall global incidence is 1.5/100,000 with a 20-fold difference between areas with the lowest and highest incidence [273]. The global incidence differences and the rising incidence are attributable to the testicular type II GCT [273, 278]. Geographic incidence differences for the other anatomical sites are dwarfed by those of the testis [76].
Testicular and ovarian type II GCT are bilateral, respectively, in 3–5 % [279, 280] and 10–15 % [281, 282]. Rarely, gonadal tumors may be combined with extragonadal type II GCT in the same individual, like in the patient who had a testicular seminoma and a germinoma of the pineal gland [283] and a patient with GCNIS of the testis simultaneous with a mediastinal non-seminoma [212].
Type II GCT have a strong familial component: over 5 % of patients with a testicular type II GCT have a relative with a similar tumor [284]; an estimated 25 % of testicular cases is due to familial susceptibility [285, 286]. Familial clustering is also documented for ovarian tumors, and testicular may cluster with ovarian type II GCT [137]. In one family, a woman with an ovarian dysgerminoma had a brother with a mediastinal EC [287], suggesting a common etiology in these cases. The only difference between sporadic and familial cases is a younger age of clinical manifestation: 2–3 years for testicular tumors [288] and about 7 years for ovarian cases [137].
Gonadal type II and I GCT may cluster in families as discussed under the type I GCT. An intriguing combination was reported by Heimdal et al. [284] of two brothers, one with a non-seminoma (type II) and the other with a spermatocytic tumor (type III). Spermatocytic tumor being so rare, this combination is probably not by chance but due to a pathogenetic commonality.
3.6.1.3 Anatomical Distribution
From the epidemiological data, it appears that type II GCT occur only in the testis, ovary, dysgenetic gonads, anterior mediastinum, most likely arising in the thymus, and midline of the brain with a preference for the pineal gland [76]. Type II GCT localized in the retroperitoneum are not primary tumors as suggested [289] but metastases from unrecognized primary testicular tumors [108, 211–213].
The occurrence of type II GCT in the mediastinum and the midline of the brain is explained by the migration route of PGC, also the explanation proposed for the anatomical distribution of type I GCT [54]. Clearly, the anatomical distribution of type II GCT is much more limited than for type I GCT. Probably initiation and development of the former require specific conditions of the microenvironment, only offered by certain cell types in these sites [56]. The PGC/gonocytes giving rise to type II GCT are hypomethylated, erased, and apoptosis prone and therefore in need of specific supportive cells for their survival: Sertoli cells in the testis, granulosa cells in the dysgenetic gonad/ovary, and perhaps equivalent cells in the thymus and pineal gland. What these supportive cells probably have in common is expression of soluble and membrane-bound KITLG that may activate the KIT receptor expressed on PGC/gonocytes, thereby enhancing their survival and proliferation [290–292]. Moreover, assuming a similar role for AKT in PGC/gonocytes as in EC, KIT signaling may through phosphorylation of OCT4 by AKT be involved in maintenance of the undifferentiated PGC phenotype [293].
Type I GCT occur at all sites of type II GCT and in addition in many more anatomical localizations along the midline of the body and occasionally in organs outside the midline. It seems that the requirements for the development for a type I GCT are less demanding than for a type II GCT, as mentioned. This may be explained by the PGC/gonocytes giving rise to type I GCT still being in an earlier stage and therefore still methylated, pre-erased, or rarely partially erased, and thus less fragile than the more mature, hypomethylated, erased PGC from which the type II GCT originate. Finally, and perhaps most importantly in view of the animal models of type I GCT discussed earlier, the PGC giving rise to type I GCT, because they lack a proper niche, probably do not survive as such but only if reprogrammed to pluripotent, primed state-ESC.
3.6.1.4 (Cyto)Genetics
Testicular type II GCT are virtually always peritriploid [294, 295], whereas in the ovary [296], mediastinum [297], and brain [197, 298], type II GCT may be (near)diploid or (near)tetraploid, reportedly in up to 50 % in the brain [197]. The consistent peritriploidy of the testicular tumors is probably due to the older age of clinical manifestation than at the other sites and the long preceding period of intratubular development with concomitant karyotype evolution.
Regardless of anatomical site, type II GCT are characterized by gain of (parts of) the short arm of chromosome 12, usually in the form of an isochromosome of 12p (i(12p)) [299, 300] (Fig. 3.15). In the testis, it occurs in virtually 100 % [295, 301–304], in the ovary/dysgenetic gonad in about 75 % [296, 305, 306], in the mediastinum in 87 % [307, 308], and in the midline of the brain in 60 % [309, 310]. Also just the more proximal parts of 12p may be involved, as an amplicon, in particular 12p11.2-p12.1, specifically in the invasive components [311–314]. It looks as if the proportion of tumors with 12p gain is inversely related to the age of clinical presentation. The very high proportion of 12p gain in testicular tumors is probably, like their peritriploidy, due to the long period of intratubular karyotype evolution.

Partial karyotype showing four copies of chromosome 12 and one copy of i(12p) (left panel); schematic drawing of chromosome 12 and i(12p) (middle panel); in situ hybridization on cytopreparation of interphase nucleus showing three copies of chromosome 12 and two copies of i(12p) [244]
Isochromosome 12p arises from an erroneous centromeric division during mitotic anaphase preceded by tetraploidization [315] and is of uniparental origin [316]. Among the genes involved in the 12p aberrations are NANOG, STELLAR, GDF3, and EDR1, necessary for maintaining pluripotency; cyclin D2 and KRAS providing proliferative advantage; genes involved in glucose or glycolytic metabolism, including GLUT3, GAPDH, and TPI1 for energy metabolism in a low-oxygen environment [304, 313, 317, 318]; and genes involved in suppression of apoptosis such as EKI1, SOX5, and DAD-R [312, 313]. Expression of these genes maintains the PGC/gonocyte-phenotype of the tumor cells and allows them to survive and proliferate in the proper niches.
With rare exceptions [306], the tumors have over- and underrepresentation of (parts of) chromosomes other than 12p. Gains involve chromosomes X, 7, 8, 12, and 21 and losses the chromosomes Y, 1p, 11, 13, and 18. The overall pattern is consistent with early tetraploidization of the tumor cells, possibly as a result of malfunction of the mitotic-meiotic switch [319], followed by net loss of chromosomes due to nonrandom losses and gains of (parts of) chromosomes [294, 320]. The large stretches involved, often entire chromosome arms, suggest that aberrant meiotic division may have a role in the evolution of the chromosomal aberrations [197].
Type II GCT are chromosomally instable, probably due to their hypomethylated [321] and polyploid genome, and therefore subject to a continuous reallocation of chromosomal material between chromosomes (Fig. 3.16). Upon cell division, this may cause unequal distribution of chromosomal material over the two daughter cells, resulting in different gene dosage [304]. Chromosomal instability likely drives tumor progression of type II GCT, exemplified by the increasing gain of entire chromosome 12, 12p (among others KRAS), 12q (KITLG, located on 12q) [296], and 4q12 (KIT) [322], which renders the neoplastic gonocytes ultimately feeder cell independent and endows them with invasive capacity. Interestingly, in non-seminoma where because of reprogramming KITLG is no longer advantageous to the tumor, 12q13-q22 including KITLG is often deleted [323]. Chromosomal instability may also drive cell fate decisions in the tumor cells, e.g., by influencing the balance of signals promoting germline (BMP) versus embryonal phenotype (NODAL) [255], and thereby the reprogramming of a seminomatous cell to a totipotent EC cell. Finally, in a non-seminoma, it could be involved in lineage determination, e.g., by tipping the balance of normally maternally and paternally expressed genes favoring somatic and extraembryonic/trophoblastic differentiation, respectively.

Spectral karyotype of type II GCT. Normally, each chromosome should have one specific color, e.g., chromosome 12 stains pink. In this case of a type II GCT, chromosome 12 contains fragments of other chromosomes, and fragments of chromosome 12 are present in other chromosomes, such as chromosomes 4 and 10. Asymmetric distribution of chromosomes over daughter cells upon cell division may result in different gene dosage
Mutations and amplifications of oncogenes are rare in type II GCT, with 0.5 mutations per Mb [295] lower than in any other solid cancer of adults [324]. KIT is most frequently mutated and mainly involved in seminomatous GCT: seminoma of the testis, about 30 % [295, 324–329]; dysgerminoma of the ovary, up to 50 % [326, 330–332]; seminoma of the mediastinum, 38 % [333]; and germinomas of the brain, over 50 % [197, 334]. In non-seminomatous tumors, KIT mutations are rare, less than 1.5 %; the same low figure was reported in gonadoblastoma and derived germinomas [332]. Functional studies have shown the KIT mutations to be activating [326, 327], occurring predominantly in the activation loop (exon 17, usually in codons 816, 820, 822, 823, and 825) of the second TK domain [335].
In seminomatous tumors, mutations of KIT appear to be only one of the mechanisms of activation of KIT signaling and its downstream pathways: KRAS/RAF/MEK/ERK and AKT/mTOR. In fact, in these tumors, KIT signaling is always activated either by upregulation of expression or genetically by mutation or amplification [322]. In the TCam-2 seminoma cell line, siRNA-induced reduction of KIT expression reduced the viability of the cells, although only marginally [329].
The quoted studies report that activating KRAS mutations occur in a few percent of type II GCT, more or less at the same rate in seminomatous and non-seminomatous GCT. Earlier studies had found higher figures in testicular seminomas, with subclonal activating KRAS and NRAS mutations in 40 % [325] and activating KRAS mutations in 2/15 cases (15 %) [336] (Fig. 3.17).

KIT/RAS and AKT/mTOR pathway interactions showing frequencies of somatic alterations in key genes. Alteration frequencies are expressed as a percentage of all intracranial GCT patients. Red text, protein positively regulates signaling; blue text, protein negatively regulates signaling; green text, physically interacting protein [197]
KIT and KRAS mutations are mutually exclusive in type II GCT of the brain [334] and perhaps also in those of the testis [324]. Consistent with this observation, “large-scale” gain of 12p (harboring KRAS) seems also to be mutually exclusive with mutations of KIT both in seminomas of the testis [295] and germinomas of the brain [197]. This phenomenon may explain why in non-testicular seminomatous tumors, where gain of 12p is less frequent, KIT mutations are more common.
The fact that KIT mutations are predominantly found in seminomatous tumors and only rarely in non-seminomas suggests that they, like upregulation and amplification of the gene, are in general involved in the progression of seminomatous tumors, rather than in initiation of type II GCT. Further evidences that KIT mutations are most often related to progression of seminomatous tumors include:
-
In gonadoblastoma, the precursor of type II GCT of the dysgenetic gonad KIT mutations occurs in 0.6 %, the same rate as the derived dysgerminomas [332].
-
The copy number of KIT (4q12) is greater in seminoma than in non-seminoma, and high-level amplification of the gene found in a couple of seminomas was not present in adjacent GCNIS [322].
-
In a case of bilateral seminoma, a KIT mutation was found in only one of the two tumors [337].
The report that virtually all bilateral type II GCT, including non-seminomas, had KIT mutations, usually in codon 816 and often with the same mutation on both sides, has strongly incriminated KIT mutations as an initiating event in migrating PGC prior to their reaching the genital ridges [338]. Later studies could not reproduce these findings neither in bilateral tumors of the testis [283, 322] nor of the ovary [330]. Other studies did show more (14/22, 64 %) [339] or less (2/7, 28 %) [340] preference of KIT mutations for bilateral tumors. Whole exome sequencing of 42 testicular type II GCT [295] demonstrated KIT mutations in 3/9 bilateral cases (33 %) and in 3/33 unilateral cases (9 %). It seems, after all, that bilateral cases do have KIT mutations at a somewhat higher rate than unilateral cases, in support of a possible initiating role of this genetic event. Notably, the most frequent mutation in bilateral cases and unilateral cases is probably different, respectively, Y823D and D816V [339]. Also in favor of initiation, the same KIT mutation (A816V) was found in the testicular seminoma and the pineal germinoma of the same patient [283]. This pathogenetically revealing case demonstrates two significant points: initiating KIT mutations may occur in migrating PGC and extragonadal type II GCT of the brain may be derived from mis-migrated PGC.
Indeed, upregulation of KIT signaling [341] and mutant KIT [342] can transform cells; thus, it is very likely that KIT mutations may occasionally be the initiating event in PGC, which during migration depend on KIT signaling for survival and proliferation.
It has been proposed that when a KIT mutation is the initiating event, the transformed PGC will preferentially develop as seminoma [327]. The rarity of KIT mutations in non-seminomas then could be due to the unlikeliness that a seminomatous tumor cell with a KIT mutation is reprogrammed to an EC cell. If this were true, one would expect to find KIT mutations preferentially in GCNIS adjacent to seminomas with a KIT mutation and vice versa; this has not yet been investigated. This hypothesis would also predict the absence of KIT mutations in the seminoma-component of mixed non-seminomas where reprogramming of an invasive seminoma cell has given rise to the non-seminoma component. In the brain 4/8 (50 %) mixed GCT including a germinoma component showed a KIT mutation [197], suggesting that a KIT mutation is compatible with reprogramming in a seminomatous GCT.
Tumor suppressor genes seem to play a modest role, perhaps not surprising in view of the early tetraploidization in the pathogenesis of type II GCT, making loss of heterozygosity less likely to develop. In addition, inactivation of tumor suppressor genes by promoter hypermethylation is rare in type II GCT [343, 344].
The overwhelming male predominance of type II GCT may be explained by the role of co-expression of OCT4 and TSPY [345–347] in their pathogenesis, as it appears from the study of these GCT in DSD patients [290, 291], cryptorchid testis [348], and complete androgen insensitivity (CAIS) [349] (Sect. 3.6.2.1). Notably, TSPY is present in about 35 copies [350] in the GBY region of the male-specific region on Y [351].
Similarly, in the proper niches in the thymus and midline of the brain where sporadic mis-migrated, but not yet demonstrated PGC [52], may survive, co-expression of these two proteins may drive transformation of PGC. In view of the predominance of males, this pathogenetic mechanism is probably the most important but not the only one to explain the origin of type II GCT, as they do occur in phenotypically normal females [352], whereby the possibility of constitutional or chimeric mosaicism for the GBY region of Y has to be kept in mind [353]. In normal females, somatic mutations, in particular KIT mutations, may indeed be the initiating event of type II GCT as illustrated by the study of Hersmus et al. [332], where KIT mutations were found in 53 % of ovarian dysgerminomas of normal women and in only 6 % of dysgerminomas originating in gonadoblastoma in DSD patients.
It seems that type II GCT have at least two fundamentally different pathways of origin: first and foremost, if GBY/TSPY is present, the “developmental pathway,” due to disturbed maturation of PGC leading to co-expression of embryonal proteins in particular OCT4 and early differentiation genes in particular TSPY (followed by overexpression of KITLG in the supportive cells) [290, 291, 348, 349]; second, and much rarer, the “somatic mutation pathway,” in which mutations in oncogenes, in particular in KIT and RAS, are initiating events. In view of the mutation frequency of these two genes in non-seminomatous tumors, the “somatic mutation pathway” seems to occur in less than 2 % of the cases. The frequent mutations in KIT in seminomatous GCT are in general progression related, as discussed. In normal females, obviously lacking GBY/TSPY, initiation of type II GCT by somatic mutations is probably a more common mechanism.
A brief summary of the progression of type II GCT, with emphasis on the various roles of KIT activation, is at this point appropriate. Early progression is sustained by upregulation of KITLG, followed by tetraploidization, whereby probably gain of chromosome 12, with KITLG on 12q and among many others KRAS on 12p, is most significant. In the in situ precursor lesions prior to the development of an invasive tumor, best studied in GCNIS, progression is driven by nonrandom gains and losses of parts of chromosomes harboring genes that promote feeder cell independence, most conspicuously gain of 12p [315]. Activation of KIT (gene on 4q12) signaling via upregulation, mutation, or amplification of the gene is essential in the progression to seminoma. Upon reprogramming of a seminomatous cell into an EC cell, factors favoring seminomatous cells are no longer relevant, as exemplified by the loss of 12q, harboring KITLG in non-seminomas [323].
Mutations common in adult cancers, like in p53, appear in type II GCT when they acquire resistance to cisplatin-based chemotherapy [354] or progress to somatic-type malignancies, most often in (late) recurrences [244, 268, 355]. Of interest is the observation that expression of a specific set of miRNAs, i.e., 372 and 373, might function as an alternative for inactivation of p53 in the pathogenesis of type II GCT [356].
3.6.1.5 Epigenetics: Including GI and miRNAs
Except for teratoma components, type II GCT, including GCNIS and gonadoblastoma, are characterized by global demethylation, erasure of parental imprinting, and the presence of permissive histone modifications [87, 357–364]. Only Alu repeats have been reported to be methylated in non-seminomas [344].
Specifically the developmentally important miRNAs, miR-371-373 as well as miR-302 and miR-367 [356, 365], which have a crucial role in development of embryonic stem cells, are highly expressed in type II GCT, including GCNIS [366].
Global demethylation and this typical expression pattern of miRNAs are part of the phenotype of PGC, underscoring the origin of type II GCT from these germ cells committed to totipotency and confirming the phenotypic similarity of PGC and the cells of the precursors GCNIS [367–370] and gonadoblastoma [290–292], respectively, in the testis and the dysgenetic gonad/ovary.
3.6.1.6 Sensitivity/Resistance/Residual Teratoma/Further Progression
Type II GCT are the solid tumors in adults with the highest sensitivity to DNA-damaging agents, where, e.g., for testicular primary tumors, cure rates >80 % are achieved in disseminated disease [371]. Both seminoma cells and EC cells, the stem cells non-seminoma, are probably highly accessible for DNA-targeting drugs because of their open chromatin structure [344, 360, 372]. EC cells are a factor two to four more sensitive to cisplatin than the clonogenic cells of cell lines derived from common adult cancers [372]. Seminomas are exquisitely sensitive to radiotherapy and cisplatin-based chemotherapy. The typical failure of seminoma cells to repair radiation-induced double-stand breaks, requiring homologous recombination, may be related to premeiotic and embryonic characteristics of the neoplastic gonocyte. A significant change brought about by the reprogramming from seminoma to non-seminoma is that this high radiosensitivity is lost.
Progression from a gonocyte to a type II GCT, particularly in the “developmental pathway,” requires fewer genetic changes than in the development of virtually any other type of cancer. Significantly, the most stringent barrier to immortalization and thereby carcinogenesis is progressive shortening of telomeres due to repression of telomerase activity [373]. This barrier is absent in PGC and ESC, as telomerase is indeed active, and thus immortality is part of their normal [374] and neoplastic phenotype, except for mature teratoma [375–377]. Accordingly, mutations are rare in testicular type II GCT [295, 324]; there is no selective pressure for loss of function of p53 [356], and the DNA-damage response remains intact (for review [167, 354, 378]). In fact, it is hypersensitive, reflecting the physiological situation in germ cells and embryonal cells, characterized by inducibility of wild-type p53 jointly with the absence of p21-induced cell cycle arrest [379], whereby cells with damaged DNA are not repaired but rather eliminated due to a low threshold for apoptosis. This preference of apoptosis over DNA repair is a physiologic mechanism protecting against propagation of repair errors via germ cells into the next generation or via ES cells into the developing embryo.
The open chromatin structure and the hypersensitive germ cell/embryonal phenotype get lost upon somatic differentiation into teratoma [380, 381, 382] of which the adult tissue stem cells are well equipped to survive DNA damage by prolonged G1 and G2 arrest and proficient repair (for review [167, 354, 378]). The phenomenon that residual teratoma after chemotherapy is usually associated with primary tumors with a teratoma component demonstrates that it results from selective survival of somatically differentiated cells rather than induction of somatic differentiation due to chemotherapy [271, 383].
Primary and acquired resistance is relatively rare in type II GCT, probably because of the low mutation rate in these tumors [324, 378]. Mutations involved in resistance have been found in BRAF, correlated with MSI [384], and in the DNA repair gene XRXX2, promoting cisplatin resistance in animal studies [295]. Also mismatch repair deficiency was found correlated with treatment failure [384]. Further molecular mechanisms of (cisplatin)resistance, partly the same as occurring in spontaneous somatic differentiation, are: somatic differentiation accompanied by downregulation of OCT4 (e.g., as a result of hypoxia or treatment with retinoic acid); failure to induce the apoptotic factors Puma and Noxa; changes in the expression levels of miRNAs such as miR-17, miR-106b, and miR-302a or miR-371-373; elevated levels of MDM2 and cytoplasmic translocation of p21 by phosphorylation; activation of the PDGFRβ/PI3K/pAKT pathway [354]. Jointly, these molecular mechanisms explain only the minority of therapy-resistant cases. Development of resistance usually occurs without obvious deviation from typical GCT morphology, mainly as YST but also as EC, choriocarcinoma, and seminoma [385].
A final important mechanism causing resistance of type II GCT is further progression of teratoma and YST due to accumulation of mutations commonly found in adult cancers. Twenty percent of late relapses of testicular type II GCT, defined as recurrences more than 2 years after initial complete response, contain histological elements resulting from further tumor progression with morphologies not typical of type II GCT [386], so-called somatic-type malignancies, which may be derived from teratoma or YST (see also Sect. 3.6.2.2).
In addition to 12p aberrations [387–389], the somatic-type malignancies may have the genetic characteristics of their somatic counterparts, like 2q37 rearrangements in rhabdomyosarcoma, p53 mutations in sarcomas, and t(11;22) translocations in PNET [355]. A somatic-type malignancy confined to a stage I non-seminoma, occurring in about 5 %, does not adversely affect prognosis [390].
Cisplatin-based chemotherapy of non-seminoma is, in fact, a model for stem cell therapy of a solid tumor showing that eradication of the stem cell population does not necessarily cure the patient [268, 391]. The surviving committed stem cells and differentiated cells may possess or acquire clonogenic potential and appear as therapy-resistant recurrence.
3.6.1.7 Animal Models and Cell Lines
Type II GCT are probably unique for humans, as no convincing examples of spontaneous or induced type II GCT have been reported in animals. Neoplasms of primitive germ cells in mice [392] and fish, e.g. experimentally induced in zebrafish [393–395], have been reported; however, reprogramming of a neoplastic primitive germ cell into a totipotent GCT, a key feature of type II GCT, has to the best of our knowledge not been described.
The only experimental model for seminoma is the TCam-2 cell line, derived from a human primary testicular type II GCT with a seminoma component, which can be propagated in vitro and as xenograft in immune-compromised mice [396] due to a BRAF mutation making it more apoptosis resistant than seminoma cells normally are [397, 398]. TCam-2 cells were at the molecular and epigenetic level characterized as seminoma cells [329, 398], including the expression of OCT4 in combination with SOX17 [399]. As expected, TCam-2 cells resemble human PGC, cells committed to totipotency, whose fate is determined by SOX17 and by BLIMP1 that represses differentiation into endodermal and other somatic lineages in PGC by repressing SOX2 [35]. Indeed, by inhibition of BMP, TCam-2 cells could be reprogrammed to an EC phenotype (among others expression of SOX2 instead of SOX17; genome-wide DNA methylation) via NODAL signaling [255, 400, 401], demonstrating in vitro what was first hypothesized from pathological observation [245].
The absence of type II GCT in mice may be explained by the molecular mechanisms of specification and epigenetic modification of the early germline being different in mice and humans. Particularly relevant could be that Oct4 in murine PGC is co-expressed with Sox2, like in mouse ESC, whereas OCT4 in human PGC is co-expressed with SOX17 [35]. Moreover, as mentioned before, epigenetic reprogramming of mouse PGC during embryonic development takes place in 24 h [8], whereas in humans, this process takes several weeks [6, 236]. This situation in humans creates a longer time frame for neoplastic transformation of early germ cells with a totipotent developmental potential, which in addition have the obstacle that they have to switch from SOX17 to SOX2 before being able to revert to an EGC phenotype. The very frequent step of tetraploidization early in the pathogenesis of type II GCT suggests that it is important for maintenance and survival of totipotent tumor cells in the naïve state.
In mice, and possibly also in other animals, the time frame for generating neoplastic totipotent cells is short, probably not long enough for the necessary steps, including polyploidization, to give rise to a tumor of transformed PGC/gonocytes. In mice, PGC/gonocytes can only give rise to neoplasms if they are directly reprogrammed to a diploid, pluripotent ESC in the primed state, probably because it expresses Oct4 in tandem with Sox2, as in ESC. As mentioned before, PGC that are not reprogrammed die apoptotically. The various teratoma models in mice are not representative for type II GCT but rather for type I GCT (Sect. 3.5.1.6). Several features of the models that do help to understand type II GCT are addressed throughout this chapter, where appropriate.
In humans, the PGC/gonocyte phenotype is indeed fairly stable in type II GCT, as at each anatomical site, over half are seminomas, the default state of these tumors. At the same time, it is obvious from the biology and histology of these tumors that reprogramming of a seminomatous cell (GCNIS or seminoma, primary or metastatic) to a totipotent EGC-like cell giving rise to a non-seminoma is a regular, though poorly understood event. To study this phenomenon, more typical seminoma cell lines would be helpful.
Non-seminoma cell lines, derived from in vivo reprogrammed seminomatous precursors, have been less difficult to establish than seminoma cell lines and are readily available. The first clonal cell line, and probably best characterized and intensely studied in vitro and in xenografts, is NT2D1, derived from Tera-2 [402]. It has been widely used, among others non-seminoma cell lines, to study differentiation of EC cells as a model for human embryonic development before human ESC became available [403, 404].
Because of the paucity of models for type II GCT, their pathogenesis has been studied primarily in human tumors in a multidisciplinary approach, combining epidemiology, pathology, (cyto)genetics, cell biology, and molecular approaches, as will be discussed per anatomical site in the following paragraphs.
3.6.2 Specific Aspects of Pathogenesis per Anatomical Site
3.6.2.1 Dysgenetic Gonad
Much of the pathogenesis of type II GCT has been learnt from the study of gonadal dysgenesis in DSD and its typical type II precursor lesion gonadoblastoma, in which the inception of these GCT can be closely followed. It has provided crucial insight into the interactive role of supportive cells and tumor cell-intrinsic factors in the pathogenesis of type II GCT.
Developmental Potential
Gonadoblastoma, originally described by Scully ([405], for review [406]) is composed of two cell types: nonneoplastic immature granulosa cells and gonocytes. The granulosa cells serve as feeder cells, offering a niche for the gonocytes, of which some, actual gonadoblastoma cells have the same atypical morphology as the cells of GCNIS. Gonadoblastoma cells will eventually outgrow the nonneoplastic gonocytes and by way of further progression become feeder-independent invasive dysgerminoma, the counterpart of seminoma of the testis. Most type II GCT of dysgenetic gonads are dysgerminomas (80 %); apparently, reprogramming to non-seminoma occurs in only 20 % of the cases [406].
Epidemiology/Risk Factors
Gonadoblastoma is a rare lesion that develops in patients with certain forms of DSD, bilaterally in 40 % of the cases [405–408]. At high risk are 46,XY patients with mutations in WT1 (including Denys-Drash, Fraser, and WAGR syndromes) [409–413], SRY, SOX9, DHH, ARX, RR5A1, or TSPYL1, resulting in a dysgenetic testis in the presence of Y-chromosomal sequences, although the male gonadal initiation/differentiation pathway is disrupted [275, 408, 414, 415]. Patients with 46,XY/45,XO mosaicism with a high risk for streak gonads [416, 417] may develop gonadoblastoma in up to 50 %. Cases reported in 45,XO patients with a Turner phenotype probably have undetected mosaicism for Y-chromosomal material containing the GBY region with the candidate gene TSPY within the dysgenetic gonad [353, 418, 419].
Anatomical Distribution
Gonadoblastoma and its derived invasive GCT occur at the sites of dysgenetic gonads: intra-abdominal, inguinal, and sometimes scrotal.
(Cyto)Genetics/Epigenetics
The cytogenetic changes in gonadoblastoma are similar to those of GCNIS: early polyploidization followed by the same nonrandom losses and gains of chromosomes. Overrepresentation of 12p material, usually as i(12p), occurs when gonadoblastoma progresses to invasiveness. As opposed to GCNIS, tetraploidization does not always take place [290, 306]. KIT mutations have been identified in dysgerminoma [296, 330], however only once in a dysgerminoma originated from gonadoblastoma [332]. Gonadoblastoma cells have the hypomethylated genome with (partially) erased GI of PGC [360].
Pathogenesis
Typically, gonadoblastoma develops in those forms of DSD, where the presence of the GBY region [351] including the candidate TSPY gene [345–347] is combined with a disturbed gonadal development due to mutations or deletions in genes necessary for the physiological male (46,XY) pathway (initiated by SRY, followed by SOX9/WT1, SF1, and downstream targets) [275, 408, 411, 414], which may be hereditary [420–422]. Disturbed expression of members of this pathway may result in immature supportive stromal cells, resembling granulosa cells [423, 424]. Multiple genes can be affected within a single patient [425]. In this hypovirilized condition, maturation of the gonocytes is delayed, thereby creating a window for co-expression of embryonal genes and early differentiation genes, in particular OCT4 and TSPY [290, 291, 426], which, jointly with enhanced KIT/KITLG signaling [292], promote neoplastic transformation of gonocytes into gonadoblastoma cells in the dysgenetic gonad. A combination of GCNIS and GB has also been reported within a single gonad [427, 428] (Fig. 3.18).

Co-expression of OCT4, TSPY, and KITLG in gonad with gonadoblastoma (left half of the photo’s with granulosa cells and Call-Exner bodies between the neoplastic gonocytes) and GCNIS (right half of the photo’s with neoplastic gonocytes in the spermatogonial niches); clockwise: H and E, OCT4 (brown), KITLG (brown), and TSPY (red), ×200 [427]
Again, the important lesson to be learned from gonadoblastoma is that there are probably two pathogenetic pathways for the origin of dysgerminoma: the first, as in DSD, merely by disturbance of the normal development of the gonad, the “developmental pathway,” related to GBY enabling co-expression of TSPY and OCT4, and the second, much rarer pathway, the somatic mutation pathway involving mutations, particularly in KIT. The somatic mutation pathway is probably more frequent in normal females, as discussed earlier (Sect. 3.6.1.4) [332].
3.6.2.2 Testis
Developmental Potential
Slightly over 50 % of testicular type II GCT are pure seminomas, about 15 % combine seminoma with a non-seminoma component, and the remainder lack seminoma and are composed of one or more embryonal (immature and mature somatic tissues) or extraembryonal (YST and choriocarcinoma) lineages; primitive germ cells are occasionally encountered [247]. Most testicular non-seminomas combine two or more lineages; the most frequent pure non-seminoma is pure EC, followed by pure teratoma; pure choriocarcinoma and pure YST are rare [244]. Somatic-type malignancies occur in primary testicular non-seminomas in 3–6 % [390], in postchemotherapy RPLND in 8 % [429]. In late recurrences, the percentage is over 20 % [386]. Over half of somatic-type malignancies are sarcomas [388], the most frequent types being rhabdomyosarcoma, followed by angiosarcoma, and leiomyosarcoma [430]. Next in frequency are carcinomas of various types and small blue round cell tumors [388, 390, 430–433]. Rarely two somatic-type malignancies develop simultaneously in late recurrences [268]. About 75 % are derived from teratoma ([268] for review); in 25 %, progression has occurred in YST, giving rise to glandular and sarcomatoid YST, mimicking somatic-type malignancy [434, 435].
Epidemiology
The lowest incidence figures of <0.5 for testicular type II GCT are found in Africa and parts of Asia. The incidence is a factor 10–20 higher among most white Caucasian populations in Western societies, like in Europe, North America, Australia, New Zealand, and parts of South America. The highest figures of >12 are recorded for Denmark, Norway, and Switzerland [273, 436–438]. Worldwide, the incidence has increased in the last four decades, in fact more than doubled in most Western and Northern European countries. In earlier low-incidence countries like Spain, Slovakia, and Slovenia, the rates are increasing rapidly and approaching those of Western Europe [273, 278]. In the US, the incidence among Caucasians is 6.6 versus 1.2 in blacks. In both groups, the rates have increased in the past 30 years [76, 439].
The large incidence differences among ethnic groups within the same society, e.g., Caucasians and blacks in the USA [76], demonstrate the importance of genetic factors. On the other hand, the geographic pattern of increasing incidence of testicular type II GCT, the changing incidence among certain immigrant populations [440–442], points to an important causative role of environmental factors, associated with modern, Western lifestyle that probably favor hypovirilization of the developing male embryo [167, 370, 443]. Indeed, the young age of the patients, the bell-shaped age distribution, the decreased risk for testicular type II GCT in Danish men born during World War II [444] and similar birth cohort effects in other populations, and the evidence that type II GCT are derived from gonocytes; all these observations point to their initiation during embryonal development.
Risk Factors
Risk factors for type II GCT of the testis have recently been comprehensively reviewed [445, 244]. Briefly, features of TDS [229] confer a higher risk: cryptorchidism (OR 4.3) [446]), previous inguinal hernia (OR 1.63), as well as hypospadias [447], previous testicular cancer (testicular type II GCT are bilateral in 3–5 %) [448], impaired spermatogenesis [449], and a family history of testicular type II GCT [286] which encompasses urogenital anomalies as in TDS [450]. TDS is considered as a relatively mild disturbance of sex differentiation, due to hypovirilizing factors in utero. This is consistent with the observation that gene mutations that cause DSD in 46,XY individuals, such as AR-mutations, confer a (somewhat) higher risk for type II GCT. Also in individuals with normal sexual development variants in AR, ESR2, HSD17B4, and CYP19A1, involved in steroid signaling or metabolism, confer an increased risk of testicular type II GCT [451, 452].
Factors increasing risk with a lower OR, in the order of 1.3 or less, which also one way or another may relate to disturbed hormonal conditions in utero are maternal bleeding, low birthweight, short gestational age, twin, tall stature, and being first born child. Sibship size is protective, the more siblings the lower risk, OR 0.80, as is late puberty, OR 0.81 [453]. There is sufficient evidence to support a relation between testicular type II GCT and three widely used hormone disruptive organochlorine insecticides (dichlorodiphenyldichloroethylene, cis-nonachlor, and trans-nonachlor), which may have a hypovirilizing influence on a developing male embryo in a specific window of development (masculinization window) [454].
The two identified occupational risks (fire fighting and aircraft maintenance) [455] and cannabis smoking [456] obviously affect males postnatally, in adolescence and adulthood. It might be assumed that these risk factors modify the development of already existing precursor lesions. Cannabis smoking is particularly interesting because it selectively increases the risk of non-seminoma, suggesting that it might promote reprogramming of a seminomatous precursor cell.
Genetic Susceptibility
Familial risk is among the highest in cancers [457, 458]: having a brother with a testicular type II GCT confers a three to eight times and a father a two to four times higher risk. For comparison, having a brother with colon cancer increases ones risk by a factor two. Yet, despite substantial efforts in international collaborations, only few low-penetrance gene mutations were identified by comparing familial and sporadic cases. The first identified risk locus was the gr/gr deletion in azoospermia factor c region of Y [459], and recently a deleterious probably causative germline mutation in PDE11A was discovered in familial and sporadic cases [460]; both mutations explain only a few percent of the familial cases.
Indeed, the small size of affected families, usually a father and a son or two brothers, and the high risk in monozygotic compared to dizygotic twins [457] are consistent with multiple autosomal recessive low-penetrance susceptibility genes [286, 288, 461–463].
The polymorphic gene variants increasing susceptibility to type II GCT shown in recent genome-wide association studies (GWAS) are also consistent with genetic susceptibility being the result of multiple common, relatively low-penetrance gene variants. The first variants (KITLG, BAK1, SPRY4) were demonstrated in 2009 by two independent studies [195, 196]. As of 2015, over 30 variants of genes for proteins which are plausibly involved in the biology of testicular type II GCT have been published, e.g., KIT/KITLG signaling (KITLG, SPRY4, BAK1, PDE11A) in relation to PGC survival and proliferation; DMRT1 variants, involved in sex determination and regulation of meiotic division; genes involved in telomere maintenance, testis differentiation, and sex determination (such as HPGDS), among others [288, 376, 464–467]. Together, these account for about 15 % of the excess familial risk to brothers of testicular type II GCT patients and 22 % of the excess to sons of testicular type II GCT patients [468]. Remarkably, these variants have no association with the established phenotypic risk factors: family history, cryptorchidism, inguinal hernia, age at diagnosis, and bilateral testicular type II GCT [469]. TDS, including cryptorchidism, hypospadias, male infertility, impaired testicular development, and testicular type II GCT [229] may be associated with variants of TGFBR3 and BMP7 [470] and variants in genes involved in steroid signaling but not with the established risk variants for testicular type II GCT.
Importantly, different distribution of variants in KITLG and AR in Caucasian and black populations may partly explain the 20-fold ethnic difference in the incidence of testicular type II GCT [195, 196].
It seems that some of the variants primarily target the PGC/gonocyte, in particular the KIT/KITLG pathway and others primarily the supportive cells (Sertoli and Leydig cells in the testis), and that homozygosity for risk alleles in the two pathways confers the highest risk. For example, men with testicular type II GCT have a 14 times higher chance than controls to be homozygous for the two risk alleles, KITLG (PGC/gonocyte survival and proliferation) and DMRT1 (testicular development) [464].
Pathogenesis: Initiation
There are striking similarities between the pathogenesis of type II GCT in dysgenetic gonads and the testis. Testicular type II GCT are considered part of TDS [229, 471], which as mentioned above also includes cryptorchidism, hypospadias, impaired testicular development, and male infertility, broadly, features of hypovirilization. In fact, the changes in the testis can be considered as a mild form of gonadal dysgenesis [411, 445, 472], supposedly resulting from an interplay between genetic factors, such as mutations in the AR [473], and endogenous and environmental hypovirilizing factors exerting their influence in utero, foremost on the stromal cells of the developing male gonad, so-called genvironmental interactions [474]. Of note, hypovirilization may not only occur during embryonic development, as DMRT1 is required to prevent female reprogramming of the postnatal mammalian testis [475, 476], conceivably with impact on the postnatal development of testicular type II GCT.
In keeping with the above observations, the association between increased risk for testicular type II GCT and impaired fertility might be due to variants within DMRT1 [464], comparable to the role of Dmrt1 in fertility and testicular teratoma formation in 129Sv mice [189]. Similarly, there might be a parallel between the loss of function mutation of steel/Kitlg in this mouse model, impairing fertility and enhancing teratoma formation [185] and variants in KITLG in men. Variants in KITLG resulting in loss of function may disturb the function of Sertoli cells and the germ cell niche, thereby impairing fertility and promoting initiation of type II GCT.
Specifically, the disturbed Sertoli cells/niche might interfere with downregulation of OCT4 in gonocytes relocated from the center of the tubules to the prespermatogonial niche, thereby creating a window for co-expression of OCT4 and TSPY, assumed to respectively protect the germ cells from apoptosis and to stimulate their proliferation [84, 477]. Co-expression of these proteins is in due time accompanied by increased expression of KITLG by the stromal component and/or via an autocrine loop, which supposedly further stimulates the neoplastic transformation. The association of a higher risk for testicular type II GCT with SNP variants in KITLG [195, 196], for example, might be mechanistically explained by interference with KIT signaling in Leydig cells and, as mentioned above, a disturbed interaction between Sertoli cells and gonocytes [348, 377]. Interestingly, one of the likely related variants within KITLG concerns a binding site for p53 [478], whereby the expression level of this allele of KITLG might increase under conditions of stress during early development, such as TDS/DSD [415], via upregulation of p53.
Pathogenesis: Early Development
The early morphological changes in gonocytes undergoing neoplastic transformation have been studied in patients with various degrees of androgen insensitivity, including complete insensitivity [349], and in infants with cryptorchid testes [348]. In both conditions, a continuum from delayed maturation of gonocytes, via pre-GCNIS to GCNIS is observed. In delayed maturation, gonocytes located centrally in the tubules still express OCT4 beyond the normal age limit of 6 months [29]; in pre-GCNIS, gonocytes that have migrated into the spermatogonial niche at the basement membrane fail to switch off OCT4 expression and start to co-express OCT4 and TSPY in a heterogeneous pattern, accompanied by focal expression of KITLG (Fig. 3.19); in GCNIS, gonocytes located in the spermatogonial niche consistently express OCT4, usually with co-expression of TSPY and combined with diffuse expression of KITLG. The GCNIS cells meet the established morphological criteria, including enlarged, angulated nuclei. The change of nuclear morphology in the progression step from pre-GCNIS to GCNIS suggests that polyploidization takes place at this stage. These observations are consistent with the hypothesis that gonocytes moving from the center of the tubules into the prespermatogonial niche do not turn off OCT4, because the malfunctioning Sertoli cells do not give the proper licensing signal toward male gametogenesis [479].

Morphology and expression of OCT3/4, TSPY, and KITLG in delayed-matured gonocytes and pre-GCNIS. Delayed maturation and pre-GCNIS puts gonocytes at risk for malignant transformation without the need of mutations by creating a window for co-expression of OCT3/4, TSPY, and KITLG. (a) Delayed maturation with OCT3-/4-positive germ cells (brown) all in the center of the tubules, gonad of 10-month-old individual. (b) Same area as in A double stained for OCT3/4 (orange) and TSPY (blue) which are not co-expressed within the same cells. (c) Same area as in A and B negative for KITLG. (d) Pre-GCNIS with most of the OCT3-/4-positive germ cells are attached to the basal lamina, gonad of 9-year-old individual. (e) Detail of the same area as in D double stained for OCT3/4 and TSPY, heterogeneity of the germ cells within particular tubules – cells are either positive for both OCT3/4 and TSPY (arrow) or only for TSPY (double arrow). (f) Same area as in E strongly positive for KITLG (red) [349]
Pathogenesis: GCNIS, Progression to Seminoma and Non-seminoma
GCNIS is the common precursor of seminoma and non-seminoma of the testis [480, 481], which is bilateral in 3–5 % of the patients [448, 482, 483]. This high propensity for bilaterality is probably because the germ cell niche is disturbed in both testes as a consequence of TDS. Consistent with this assumption is the fact that in DSD, where the disturbance of the niche is more severe, bilaterality of gonadoblastoma may occur in up to 40 % [405]. GCNIS has a heterogeneous phenotype due to the plasticity of GCNIS cells reflecting different stages of maturation of primitive germ cells [484–487]; a subset of GCNIS cells expresses spermatogonial markers [488]. GCNIS will probably always progress to an invasive type II GCT if left untreated [482, 489], although not proven so far. As yet, no features have been found that predict whether it will progress to (intratubular) seminoma or non-seminoma.
As in dysgenetic gonads, the neoplastic transformation of gonocytes does not require somatic mutations but rather results from a disturbed timing of expression of critical embryonal and differentiation proteins during development, as discussed. Therefore, type II GCT of the testis can be considered developmental tumors [370]. Indeed, somatic mutations are rare and limited to KIT and KRAS [295, 322–326, 490], whereby activating KIT mutations are found in about 25 % of seminomas [327] and rarely in non-seminomas [340]. In seminomas, the KIT pathway is always activated via mutation or amplification of KIT or via overexpression of the protein [322, 491]. KRAS mutations are in a few percent found both in seminomas and non-seminomas. Mutation of KRAS does not seem to occur in combination with high-level amplification of the gene [324] or overexpression of the protein [329]. In testicular type II GCT, KIT and RAS mutations may be mutually exclusive [324].
The much higher frequency of activating KIT mutations in seminoma than non-seminoma suggests that this genetic event does not take place in an early stage of tumor evolution shared by seminoma and non-seminoma. Rather, as discussed before, mutation of KIT is part of progression of seminoma, as is amplification of the gene, and upregulation of its expression [322] resulting in KIT activation, characteristic for seminoma. Since it is related to seminoma progression, it is understandable that non-seminomas rarely harbor KIT mutations, for they offer no advantage after reprogramming of a seminomatous tumor cell into an EC cell. In the rare non-seminomas with a KIT mutation, reprogramming probably took place in a seminomatous tumor cell that already had acquired a KIT mutation. Indeed, one of the first two type II GCT in which an activating KIT mutation was demonstrated was an ovarian mixed dysgerminoma/YST, with the mutation in both components [326].
The observation that KIT mutations are usually found in seminomas that lack large-scale 12p amplifications [295] could mean that for seminomas, KIT mutations and 12p gain are alternative pathways of tumor progression.
The occurrence of the same KIT mutations in bilateral testicular tumors demonstrates that a KIT mutation can be the initiating event taking place in PGC prior to their arrival in the gonadal ridges [283, 339, 340]. Even more convincing, as already referred to, the same KIT mutation was found in the testicular seminoma and the pineal germinoma of the same patient [283].
GCNIS stays more or less dormant until resuming further progression upon hormonal stimulation at puberty [492]. In prepubertal GNCIS, about a quarter of the tumor cells express DMRT1, a key regulator of the mitosis-meiosis switch, whereas in adult cases, this figure drops to a few percent. GCNIS cells that express DMRT1 are not mitotically active and considered dormant [486].
In so-called isolated GCNIS that has not yet given rise to an invasive type II GCT [493] and in GCNIS confined to the spermatogonial niche [304], there is no overrepresentation of 12p (Fig. 3.20). Gain of 12p coincides with the GCNIS cells becoming feeder independent, apparent from their leaving the spermatogonial niche and their capacity to float in the lumen of the seminiferous tubules [304], usually adjacent to an invasive GCT [493] (Fig. 3.21). The next step in the “default development” of a testicular type II GCT is the formation of intratubular seminoma, whereby the GCNIS cells completely fill and distend the seminiferous tubules and oust the Sertoli cells. Intratubular seminoma may contain lymphocytes like invasive seminoma. Though highly proliferative it virtually never shows necrosis, which is practically always present in intratubular non-seminoma. Apparently, intratubular seminoma cells, like normal gonocytes, are well adapted to the intratubular low-oxygen environment [244].

Comparative genomic hybridization on GCNIS cells in the niche (left panel) and on three invasive type II GCT, from left to right EC, teratoma, and YST (right panel). Gain of 12p is absent in GCNIS, while it is present in invasive type II GCT [244]

GCNIS cells in the spermatogonial niche and floating in the seminiferous tubules (direct alkaline phosphatase, ×100)
The trigger for invasive growth of intratubular seminoma is not known. Morphologically, invasion of seminoma has different appearances: one whereby intratubular seminoma extends the seminiferous tubule to the point of breaching the tubular wall [262] and another, microinvasive seminoma in which the tumor cells appear as single cells in the interstitial stroma of the testis [244, 494–496], and combinations of both. Just like intratubular seminoma [262], microinvasive seminoma can be found adjacent to a non-seminoma in about 20 % [494]. Microinvasive growth may be due to expression of matrix metalloproteinase 9 and plasminogen activator, urokinase, by the tumor cells [495].
Upon invasion, seminoma invariably elicits an inflammatory host response, usually composed of lymphocytes, macrophages, plasma cells, and often a granulomatous reaction. Its significance remains controversial. Recently, it was suggested that it is not involved in active immune surveillance [497]. An earlier study demonstrated clonally expanded cytotoxic T cells and evidence of specific and functional T-cell responses operating in seminoma, indicating that the inflammatory infiltrate is indeed involved in the immunological control of the tumor; however, class I MHC molecules could not be demonstrated on the seminoma cells [498], making them invisible to cytotoxic T cells. From histology, the infiltrating lymphocytes seem capable to cause complete regression of seminoma, leaving the scar of a so-called burnt-out tumor. GCNIS and intratubular seminoma may undergo regression as well, whereby the tubules become atrophic and in the end completely fibrosed. This host reaction, and probably the older age of the patients, explains why GCNIS is usually much less extensive in association with seminoma than non-seminoma and even absent in up to 15 % of the cases [262]. The few lymphocytes accompanying GCNIS adjacent to non-seminoma, as opposed to the many adjacent to seminoma, suggest that the host response is indeed elicited by the seminoma, which upon invasion disturbs the mechanisms of immune privilege in the testis [499]. GCNIS, composed of tumor cells that are phenotypically similar to seminoma cells, is probably secondarily involved. It seems less likely that the GCNIS cells themselves, within the intact immunologically privileged testis, trigger a reaction of the host.
The host response is probably clinically relevant in view of the 10 years difference of the median age of presentation of seminoma in patients with AIDS and the general population: respectively, 25 and 35 years. In AIDS patients, seminoma and non-seminoma present at the same age, 25 years, also the age of presentation of testicular non-seminoma in the general population. In addition, a higher risk for disseminated seminoma has been reported in patients with AIDS, suggesting a protective role of an intact immune system [500]. Invasive non-seminoma also contains inflammatory cells; however, the surrounding parenchyma is much less involved than with seminoma, probably explaining why GCNIS is often very extensive and rarely absent [262].
Pathogenesis: Non-seminoma Due to Reprogramming Seminomatous Progenitor Cell
Deviation from the default development of seminoma occurs when a seminomatous cell, either a GCNIS cell or an invasive or metastatic seminoma cell, is reprogrammed to an EC cell, the stem cell of non-seminoma. How often a primary testicular non-seminoma is due to reprogramming in an invasive seminoma (Fig. 3.22) can be estimated from the percentage of mixed non-seminomas with a seminoma component, which is about 15 %. This figure could be an underestimation, since microinvasive seminoma may be overlooked [495, 496]; on the other hand, because a mixed non-seminoma could be a collision tumor of separately developed seminoma and non-seminoma, 15 % may be too high an estimate. The remaining non-seminomas are probably due to reprogramming of an intratubular seminomatous cell. Indeed, the intratubular non-seminoma stage can be demonstrated in the parenchyma adjacent to a non-seminoma in about 15 % of the cases, most often adjacent to small tumors, suggesting that large tumors have overgrown their intratubular precursor [262]. Intratubular reprogramming cannot be explained by downregulation of BMP in GCNIS cells due to interstitial stromal factors [255]. Here, the possibility of unequal distribution of chromosomal material over the two daughter cells [501] might be considered, which would result in a low gene dosage of BMP in one of them, starting off NODAL signaling and thereby reprogramming. In invasive seminoma, the mechanism could be the same or BMP could be downregulated by exposure to interstitial stromal factors like NOGGIN [255].

Foci of EC, teratoma (TE), YST, and choriocarcinoma (CC) in an otherwise typical testicular seminoma demonstrating the phenomenon of reprogramming to non-seminoma in an invasive seminoma. The reprogramming at multiple sites and into different lineages may be due to aneuploidy of the tumor cells, which upon cell division give rise to daughter cells with different chromosomal constitutions (middle panel, H and E ×1; YST, EC, TE, and CC H and E ×200)
With sporadic exceptions, intratubular non-seminoma is composed of pure EC that is partly necrotic and often calcified. The invariable necrosis indicates that EC cells are less adapted to the intratubular low-oxygen environment than GCNIS/seminoma cells. It is conceivable that hypoxia-induced factors like MET trigger invasion of intratubular non-seminoma. Remarkably, only upon invasion, the EC cells start to differentiate and display the totipotent nature of naïve-state EGC due to differentiation-inducing factors in the stroma of the testis and/or loss of differentiation-inhibiting factors in the intratubular microenvironment (Fig. 3.23). Possible stromal factors are TGF-β, FGF, and BMP, which may derepress differentiation-promoting genes by removing polycomb repressive complexes recruited to the promotor sites of these genes by the pluripotency proteins OCT4, SOX2, and NANOG [502]. Noteworthy, spontaneous and experimental teratomas in 129 mice also start as intratubular EC and begin to differentiate when the seminiferous tubules are extended and disrupted [81, 183] (Fig. 3.24).

Differentiation of intratubular non-seminoma upon invasion. Intratubular component consists exclusively of EC (right lower corner); upon invasion development of teratoma elements, YST, and trophoblastic giant cells, in addition to EC. (H and E, ×100)

(a) Intratubular GCT, composed of EC (arrow) in testicular tubule of 19-day fetus of 129 strain mouse. (b) GCT (in 19-day fetus) which has enlarged and ruptured seminiferous tubule in which it arose and is composed of EC and also more differentiated cells forming tubules [81]
Pathogenesis: Summary
Most of the testicular type II GCT have a developmental origin in the context of TDS, a condition of mild undervirilization, whereby inadequate signaling by Sertoli cells interferes with normal maturation of gonocytes, creating a window for co-expression of embryonal and differentiation proteins, in particular OCT4 and TSPY, combined with upregulation of KITLG, resulting in transformation of gonocytes. Polyploidization is an early event, possibly due to dysfunction of the mitotic to meiotic switch [319, 486], providing survival advantage to the gonocytes in the suboptimal niche. In combination with their hypomethylated state [321], it endows the transformed gonocytes with chromosomal instability, which drives tumor progression through nonrandom gains and losses of (parts of) chromosomes, most conspicuously gain of 12p. This chromosomal region harbors a cluster of genes whose products via various mechanisms convey further proliferative and survival advantage to the neoplastic gonocytes, resulting in GCNIS and intratubular seminoma that by default develops into seminoma. Seminomas may harbor mutations predominantly in KIT in up to 25 %, probably most often as a genetic mechanism of tumor progression and only rarely as initiating event. Non-seminoma originates when a neoplastic gonocyte, usually within a seminiferous tubule, is reprogrammed to an EC cell, the totipotent stem cell of non-seminoma, giving rise to intratubular EC that upon invasion of the testicular interstitial tissue may give rise to all extraembryonal and somatic lineages and occasionally the germ lineage.
3.6.2.3 Ovary
Developmental Potential
Ovarian GCT containing dysgerminoma, EC, or choriocarcinoma, either alone or in various combinations with or without YST and/or teratoma, are type II GCT with totipotent developmental potential and can be classified as such on histological grounds. Solid tumors solely composed of (immature) teratoma and/or YST can only be classified with certainty as a type II GCT by demonstrating gain of 12p [503]. However, age and histology do give clues, since in general, pure immature and/or mature solid teratomas are of type I, as they typically lack gain of 12p ([296, 503] for review). Of the pure YST, about 40 % have 12p gains and are therefore type II, while the remaining 60 % are best classified as type I GCT [296]. Tumors combining teratoma and YST can also be of either type; those in infants are likely progressed type I teratomas lacking 12p gain, while the postpubertal ones are likely non-germinomatous type II GCT with 12p gain [296, 305, 503]. As mentioned earlier, in the case of a pure mature solid teratoma, it is important to make this distinction because a type I teratoma is benign and a type II teratoma is malignant.
The ratio between dysgerminoma and non-dysgerminoma, and thus the rate of reprogramming of dysgerminoma, is difficult to establish because usually epidemiologic studies do not specify the histology to the degree that the distinction between type I, II, and IV GCT is possible. The report by Smith et al. [504] is a notable exception: among 1262 malignant ovarian GCT, the 449 pure immature teratomas and at least 110 pure YST (60 % of 183, based on Kraggerud [296]) should be considered as type I GCT, while the 37 teratomas with malignant degeneration should be type IV GCT with a somatic-type malignancy. That leaves 666 type II GCT: 414 dysgerminomas (62 %) and 252 non-dysgerminomas (73 pure YST, 67 mixed GCT, 52 EC, 27 choriocarcinomas, and 33 EC plus teratoma, so-called teratocarcinomas). The percentage of pure dysgerminoma is higher than the slightly over 50 % seminomas within testicular type II GCT. It suggests that reprogramming in ovarian dysgerminoma, which is convincingly documented [505–508] and also apparent from the mixed GCT with a dysgerminoma component [504], occurs less frequently than in GCNIS and seminoma of the testis. The spectrum of histological types in ovarian and testicular non-seminomatous GCT is similar; however, the distribution is different: mixed non-seminoma is the most frequent histology in the testis and pure YST in the ovary; pure choriocarcinoma seems to be more frequent in the ovary than in the testis. In mixed tumors, the percentage of tumors combining dysgerminoma with just immature teratoma or YST seems to be higher than the combination of seminoma with only immature teratoma or YST in the testis (Chap. 6).
Epidemiology
In the ovary, the second most frequent site of type II GCT after the testis, the incidence is about 20-fold lower than in the testis [76, 137, 509, 510], with reportedly a slight decrease over the past 30 years [76, 504]. Data from South East England for about the same period suggest a rate of increase comparable to that of testicular type II GCT [509]. However, the quoted data on incidence trends are not representative for type II GCT, because in neither of the studies, the trends were specified for the different types of GCT of the ovary. The incidence of dysgerminoma in England was reported stable between 1971 and 1984 [511] and in the same period increasing in Los Angeles County [512]. Therefore, the pathogenetically important question whether or not ovarian type II GCT parallel the increasing incidence of those of the testis in recent decades, due to comparable gene-environment interactions, remains open. In the US, dysgerminoma seems to be more frequent in whites and other nonwhites than in blacks [76], suggesting an ethnic influence, just as in testicular type II GCT.
Type II GCT of the ovary may arise before puberty, likely in DSD [275, 513] (see also Sect. 3.6.3). DSD may remain unrecognized, as in the case of two phenotypically normal females having a 46,XX karyotype, who well after puberty were diagnosed, respectively, with gonadoblastoma with a germinoma [514] and gonadoblastoma with a mixed type II GCT [515]. Ninety-five percent of ovarian type II GCT become manifest after puberty in women with a normal 46,XX karyotype [516], a couple of years earlier than in males, in accordance with the earlier onset of puberty in females [232, 266, 517]. Age of presentation is in the typical order: first, the pure non-dysgerminomas, followed by the mixed non-dysgerminomas/dysgerminomas, and then the pure dysgerminomas with a median age of the latter close to 20 years [511]. Dysgerminomas and non-dysgerminomas are bilateral in over 6 % [129, 131, 518–520], a slightly higher figure than for bilateral type II GCT of the testis, probably due to the contribution of gonadoblastoma-related tumors, as 40 % of gonadoblastomas are bilateral [405]. In fact, in a retrospective study, one out of three bilateral dysgerminomas was associated with bilateral gonadoblastoma [521].
Familial cases are rare: among 18 families retrieved from the literature with at least one female with a GCT [137, 522, 523], eight involved only females and ten both females and males (in about 0.2 % of the pedigrees of familial testicular cancer, a female member has a GCT [524]). The information on the histology of the tumors provided in the reviews and the quoted original case reports allows the distinction between GCT of types I and II. In 12 of the families, at least one case concerned a dysgerminoma, combined with various type II GCT of the gonads in close relatives of both sexes; remarkably, one relative had a mediastinal EC. Also noteworthy, in three families, type II GCT clustered with at least one ovarian type I GCT: three pure (immature) teratomas and in two families with pure YST, which may have been of type I. (The remaining family consisted of a woman with an ovarian type I immature teratoma and her baby with a type I immature teratoma of the brain [136].) These families demonstrate two significant points: type II GCT of the ovary may cluster with similar testicular and extragonadal GCT and also with type I GCT of the ovary.
Risk Factors
An established risk for (bilateral) ovarian type II GCT are the various forms of DSD in phenotypic females who have the GBY region, containing TSPY, in their genome, as discussed earlier.
Less certain is that a disturbed hormonal milieu in the mother increases the risk of malignant ovarian GCT in daughters, comparable to the role of hormone disruption in the etiology of TDS and type II GCT of the testis [229, 525]. Reported risks are maternal use of exogenous hormones during pregnancy (OR 3.6), maternal obesity (OR 2.7), early regular menstruation after menarche (OR 1.8), and age at index pregnancy under 20 years (OR 2.8) [526]. Other studies have also reported association of malignant ovarian GCT with reproductive risk factors such as parity, use of contraceptives, ages at first and last births, and time since last birth [527–529]. A more recent study could not link levels of circulating sex hormones with risk of ovarian germ cell cancers [530]. Most studies do not distinguish between type I and II GCT, making the results difficult to interpret.
As discussed above, type II GCT of the ovary lack the strong familial risk of those of the testis. Sporadically, type II and I GCT cluster in families. Both tumor types are so rare that the clustering is probably not by chance, rather these families have susceptibility for both type I and II GCT, pointing to a common cell of origin in different states of developmental potential. The obvious target cell is the PGC/gonocyte, and the familial susceptibility factor might have bearing, for example, on resistance to apoptosis of PGC, such as variants in BAK1, which are associated with a higher risk for type II GCT [195, 196] and perhaps also for type I GCT [158].
In one of the families reported by Huddart [524], a male with a testicular type II GCT clustered with a female with bilateral dermoid cysts (type IV GCT) of the ovary. More recently, a similar family was identified, with a father having a seminoma and his daughter metachronous bilateral dermoid cysts [531]. In both families, it may be a chance occurrence in view of the high frequency of dermoid cysts of the ovary, with bilaterality in 10–15 % of the patients [233, 532] (Chap. 6). It is intriguing though that in both females, the dermoid cysts were bilateral, because in familial cases of dermoid cysts where laterality was stated, 11/28 (39 %) were bilateral, and among the patients who were twins or triplets, 9/12 (75 %) were bilateral [134, 135]. One of identical twins [533] had over the years seven dermoid cysts removed from her left and one from her ovary. These case histories are suggestive of a genetic risk factor for bilaterality of type IV GCT of the ovary, which perhaps could also increase the risk for type II GCT.
(Cyto)Genetics/Epigenetics
Over ninety percent of dysgerminomas are aneuploid, often close to tetraploid [534], and in 77 % have gain of 12p and similar gains and losses of (parts) of other chromosomes as testicular type II GCT (for review [86, 296]). Mixed non-dysgerminomas are also most often aneuploid and have gain of 12p in 68 %. This somewhat lower figure as compared to dysgerminoma is probably due to the fact that part of the mixed GCT combining teratoma and YST is type I GCT. Of the pure YST, 41 % have gain of 12p, indicating that less than half are type II GCT. Pure immature teratomas and mature teratomas have gain of 12p in 5 and 9 %, respectively, and are therefore most often type I GCT (for review [296]). Conversely, one out of ten solid mature teratomas of the ovary is of type II and therefore malignant.
As for the other chromosomes in dysgerminoma, the most common changes are gains from chromosome arms 1p (33 %), 6p (33 %), 12q (75 %), 15q (42 %), 20q (50 %), 21q (67 %), and 22q (58 %); gains of the whole of chromosomes 7 (42 %), 8 (42 %), 17 (42 %), and 19 (50 %); and losses from 13q (58 %) [535]. The strong predominance of gains over losses might be explained by the (near)tetraploidy of most dygerminomas.
Somatic mutations of KIT in exon 17 codon 816 have been found in 27–33 % of dysgerminomas [330–332] and in codon 822 in an additional 20 % [332], adding up to activating mutations in 53 %, always in unilateral and not in bilateral cases. Among 16 DSD patients with GBY in their genome who developed a dysgerminoma, only one case had a KIT mutation, in codon 820; the same mutation was found in the gonadoblastoma from which the dysgerminoma was derived [332]. This may be part of the explanation for the absence of KIT mutations in bilateral dysgerminoma, as bilateral tumors are probably in a substantial proportion derived from gonadoblastoma. KIT mutations are absent in other malignant ovarian GCT, i.e., immature teratoma, YST, and tumors combining these two components. Gain of 12q in dysgerminoma may be related to the localization of KITLG on 12q22, with possible involvement of KITLG in an autocrine loop with KIT. KRAS has hardly been investigated in ovarian type II GCT; in two studied dysgerminomas, it was not mutated [536].
Pathogenesis
Type II GCT of the ovary are derived from hypomethylated, erased, premeiotic PGC/gonocytes in the naïve state (totipotent), present in the early developing gonad, as has been convincingly demonstrated for GCT arising in dysgenetic gonads [275]. It is very likely that the phenotypically identical GCT outside the context of DSD have the same cell of origin. Morphology, immunohistochemistry, and expression studies of dysgerminoma have shown the same profiles as in seminomatous GCT of other anatomical sites and in PGC, with high expression of pluripotency factors, in particular expression of OCT4 in combination with SOX17. The non-germinomas have the same profiles as the type II non-seminomas at other sites, with EC cells expressing OCT4 in combination with SOX2 in addition to other pluripotency factors and the derived lineages showing tissue-specific expression patterns (for review [296]).
Five percent of ovarian type II GCT develop in the context of DSD by the “developmental” pathway, related to the presence of GBY and co-expression of OCT4 and TSPY, consistent with the low rate (0.6 %) of KIT mutation in these tumors [332]. In a molecular analysis of 45 malignant ovarian GCT in patients without signs of DSD, 32 can be classified as type II GCT. In four of these (13 %, two dysgerminomas and two immature teratomas), TSPY was demonstrated in tumor tissue [353]. This would mean that the origin is “developmental,” in association with TSPY, in about 20 % of ovarian type II GCT (5 % DSD and 13 % clinically silent mosaicism for TSPY).
As discussed in the general section on (cyto)genetics of type II GCT (Sect. 3.6.1.4), the KIT mutations found in up to 50 % of dysgerminomas [330, 332] may be initiating, but they are probably most often progression related. In view of the absence of KIT mutations in type II non-germinomatous tumors of the ovary [330], it is likely that the situation is not much different from the testis and that also in the ovary, very few type II GCT are caused by somatic mutations. Thus the initiation pathway in over 80 % of ovarian type II GCT in 46,XX phenotypically normal females remains to be explained.
It is tempting to speculate that in the ovary, the majority of type II GCT develop in the context of mild dysgenesis, comparable to TDS of the male, mainly caused by imbalances of factors regulating gonadal development. It has been shown in a mouse model that downregulation of FOXL2, the key protein for maintenance of the female identity of the gonad, results in reprogramming of the ovary in the male direction, even in adults. Granulosa cells are transformed into Sertoli cells forming tubular structures. Remarkably, if in this situation the expression of FOXL2 is restored, the ovarian identity is repaired [537]. If this would happen during gonadal development, even only transiently, in the stage before oocytes become arrested in the prophase of meiosis I, it would create a hypovirilized testis-like environment favoring disturbance of maturation oogonia/gonocytes and a window for the development of a type II GCT. However, due to the absence of GBY/TSPY, at a much lower rate than in males or in DSD patients carrying GBY in their genome, who have an up to 70-fold risk of developing a type II GCT as compared to normal females [275]. Indeed, insight into the factors involved in the development and maintenance of sexual identity of the gonads (DMRT1, FOX9, and SOXL2) lends credence to the hypothesis that as yet pathogenetically unexplained type II GCT of the ovary have their origin in mild forms of ovarian dysgenesis, possibly even transient, which leave no obvious further phenotypical traces (Fig. 3.25). If this hypothesis were true, it would mean that the large majority ovarian type II GCT have a “developmental” origin.

Role of DMRT1, FOX9, and SOXL2 in development and maintenance of ovarian identity. DMRT1 silences RA-dependent feminization genes to ensure postnatal sex maintenance. During fetal sex determination (1), the bipotential gonad makes a choice between male (blue) and female (pink), largely guided by the presence or absence of Sry. The sexual differentiation machinery downstream of sex determination transforms the undifferentiated gonad into a mature testis or ovary (2), manifested in the formation of Sertoli-cell-containing seminiferous tubules in the male and granulosa-cell-containing ovarian follicles in the female. Postnatal sex maintenance within Sertoli cells (3) is achieved via the silencing of RA signaling-dependent feminization genes (such as Foxl2) by the transcriptional regulator DMRT1. RA is thereby allowed to act in adjacent spermatogonia to promote spermatogenesis within the seminiferous tubule. In Dmrt1 mutant Sertoli cells, however, RA acting through RARa activates feminizing genes and reprograms the Sertoli cell into a granulosa-like cell through the process of transdifferentiation [538]
The much lower incidence of type II GCT in the ovary as compared to the testis has been explained by the lower number of susceptible germ cells in the ovary, and the fact that they are blocked in meiosis I, whereas the gonocytes are more numerous in the testis, are arrested in mitosis [509]. This is probably only part of the explanation in view of the epidemiology of the type II GCT of the mediastinum and brain. One might assume that in these sites, the number of target cells is similar in men and women; moreover, the mis-migrated PGC are blocked in meiosis I in both genders [58, 59]. Yet type II GCT of the mediastinum [241] and brain [539] are much more frequent in males than females. As alluded to earlier, this may be due to men having the GBY region on Y and thus being able to express TSPY in combination with OCT4 in the critical stage of neoplastic transformation of erased PGC. This suggestion is consistent with the high risk of developing a type II GCT in dysgenetic gonads containing GBY/TSPY [275].
Also relevant in this context is the observation that type I GCT have a roughly similar frequency in the ovary and the testis and that the sex distribution, apart from the sacral region, is equal for most extragonadal sites, in particular for the mediastinum and brain [94]. It suggests that indeed the number of susceptible cells, probably mis-migrated, pre-erased PGC for type I GCT, is similar in males and females and that GBY has no role in the pathogenesis of type I GCT.
3.6.2.4 Mediastinum
Developmental Potential
Of the mediastinal GCT in postpubertal male patients, seminomas constitute 32 %, mixed GCT 16 %, EC 4 %, choriocarcinoma 3 %, YST 10 %, and teratoma 35 % [241]. In CGH analysis [165], most mixed GCT in patients older than six had gain of 12p and are therefore type II GCT; in one out of three YST and none of the teratomas in that age group 12p was involved. Thus it can be estimated that in early- and postpubertal patients, seminomas constitute about 55 % and non-seminomas 45 % of mediastinal type II GCT (mixed 28 %, EC 7 %, YST 5 %, choriocarcinoma 5 %). Apart from a higher proportion of pure YST and pure choriocarcinoma, the distribution of histological subtypes is similar to that of the type II GCT of the testis.
Of note, in patients older than six [76, 165], pure (immature) teratoma, pure YST, and mixed GCT combining the two can only with certainty be classified as type I or type II GCT by analysis of 12p status. This is clinically relevant as a type I teratoma is benign and type II teratoma is malignant.
Most intriguingly, 41 patients with Klinefelter’s syndrome who had a type II GCT based on histology and secretion β-HCG showed a very different distribution of histological types from the general adult male population, with seminoma 0 %, mixed non-seminoma 44 %, EC 10 %, choriocarcinoma 15 %, YST 2 %, and (immature) teratoma (29 %) [277]. Not only there were no pure seminomas but also only two of the 12 mixed GCT had a seminoma component. Choriocarcinoma/trophoblastic differentiation, either in pure form or as part of a mixed tumor based on histology or elevated serum β-HCG, was very common: 35/41 (85 %). Considering that over 20 % [540, 541], of all mediastinal GCT, are diagnosed in patients with Klinefelter’s syndrome, this may partly explain the histological differences between testicular and mediastinal type II GCT in adolescent and adult males.
Ten to twenty percent of mediastinal non-seminomas develop a solid somatic-type malignancy [542, 543]. The distribution of histologies is largely similar as in the testis, sarcomas being the most frequent type and among them rhabdomyosarcoma ranking first [430, 543]. In the mediastinal cases, angiosarcomas are more frequent than in the testis [241, 544]. The associated GCT are most often mixed non-seminomas with a (immature) teratoma component and rarely pure YST or seminoma [430]. Patients with Klinefelter’s syndrome may develop somatic-type malignancies other than hematopoietic malignancies.
Hematologic malignancies develop in 2–6 % [543, 545–547], sometimes combined with a sarcoma [388]. These hematological somatic-type malignancies are uniquely associated with mediastinal YST, either pure or as part of a mixed non-seminoma [265, 543, 547–549]; the most frequent types being megakaryoblastic leukemia, followed by malignant and benign histiocytosis and myelomonoblastic leukemia among many other types encompassing virtually all hematopoietic lineages [543]. Except associated with mediastinal non-seminomas, hematologic malignancy has been reported only once, in association with a suprasellar dysgerminoma [550]. Hematologic malignancies are usually diagnosed at a median time of 6 month after primary treatment [549]; about 40 % are synchronous with the primary mediastinal non-seminoma [543]. The rate is comparable in patients with and without Klinefelter’s syndrome, as about 20 % of the hematologic malignancies are diagnosed in patients with this syndrome, indicating that it is indeed the mediastinal localization that predisposes to this somatic-type malignancy. These hematologic malignancies are not treatment related [546, 549] but a peculiar biologic characteristic of mediastinal non-seminomas with a YST component [241, 547].
Added up, the solid and hematologic somatic-type malignancies develop in a quarter of mediastinal non-seminomas, which is about sixfold of the 3–6 % in primary testicular non-seminomas [390]. The higher rate of somatic-type malignancies is possibly due to the larger size at surgery of the non-seminomas in the mediastinum than in the testis [551] and due to the fact that surgery is always preceded by chemotherapy. Indeed, in originally testicular non-seminomas, the percentage of somatic-type malignancies increases to 8 % in postchemotherapy retroperitoneal lymph node dissection specimens [429] and to >20 % in late relapses [386], approaching the rate of somatic-type malignancies in mediastinal non-seminomas.
The poorer prognosis of mediastinal than testicular non-seminomas may be because of the larger size at clinical manifestation and the overall higher rate of somatic-type malignancies, which are largely resistant to the chemotherapy given for non-seminomas [354].
Epidemiology/Risk Factors
With an incidence of about 0.12 in white and 0.05 in black males, which has not increased in the past decades [76], the mediastinal type II GCT constitute 50–70 % of extragonadal type II GCT [552]. Over 95 % occur in men [241, 308], which is true for whites and blacks [76]. The mean age for seminomas is about 30 [264] and for non-seminomas 25 years [265]. The mean age of patients with Klinefelter’s syndrome, always with mediastinal non-seminomas, is 17 years (range 4–31), substantially younger than in non-seminoma patients without Klinefelter’s syndrome. In all Klinefelter cases younger than 12, there was precocious puberty [553], due to β-HCG produced by the tumor [277].
In a large Danish cohort, mediastinal non-seminoma was the only cancer for which Klinefelter’s syndrome conferred a higher risk compared to males without this syndrome, with a relative risk of 67 [554]. Over 20 % of mediastinal type II GCT are associated with Klinefelter’s syndrome [540, 541], meaning that roughly half of the mediastinal type II non-seminomas occur in this context, as pure seminomas do not seem to occur in Klinefelter’s syndrome. The age distribution of Klinefelter patients with mediastinal GCT is bimodal. However, the early peak (between age 4 and 10) is later than in typical type I GCT; moreover, the tumors in the early peak are not the usual type I GCT but more like type II GCT, based on the histological composition and the secretion of β-HCG causing precocious puberty [277]. Remarkably, the two mixed GCT with a seminoma component occurred in two 8-year-old boys. Klinefelter’s syndrome also increases the risk of type II GCT of the brain [555, 556].
Sporadically, mediastinal seminomas have been diagnosed in individuals with Down’s syndrome [557, 558]. Neurofibromatosis 1 [559, 560] and Li-Fraumeni syndrome [561, 562] are not associated with mediastinal type II GCT.
There is one report of a patient with mediastinal type II non-seminoma associated with only GCNIS in one testis, suggesting that the tumors were two independent primary type II GCT [212]. Indeed, contrary to retroperitoneal type II GCT, which are metastatic from unrecognized testicular tumors, mediastinal type II GCT are normally not associated with GCNIS of the testis [211, 563]. König et al. [564] report a mediastinal non-seminoma (“teratocarcinoma”) and a metachronous pituitary stalk germinoma in a patient with Klinefelter’s syndrome, probably both related to the underlying syndrome.
There is one patient, mentioned earlier, with a mediastinal EC who had a sister with an ovarian dysgerminoma [287], demonstrating that in some families, mediastinal type II GCT may cluster with type II GCT at other anatomical sites. To the best of our knowledge, clustering with other GCT types has not been reported [137].
Anatomical Distribution
Primary mediastinal type II GCT are only localized in the anterior or anterosuperior mediastinum in association with the thymus [241]. Occasionally, small tumors are completely localized within the thymus, showing that they had their origin in the thymus itself [241]. Concurring with this conclusion is the observation that in about a quarter of all mediastinal seminomas, remnants of the thymus can be identified in the periphery of the tumor [565]. Thymic cysts have been found in 10 % of seminomas [566] and occasionally in YST of the mediastinum [567], again, indicating that these tumors originate in the thymus. Apparently, in most cases, the thymic origin is obscured by tumor overgrowth.
Mediastinal type II GCT are indeed primary tumors, as testicular type II GCT only rarely metastasize to the anterior mediastinum and only simultaneous with metastases in the visceral mediastinum [568].
(Cyto)Genetics
In a study of 19 malignant mediastinal GCT, 14 were definite type II GCT, based on histology and expression of β-HCG, with the expected distribution of histological types [297]. Four tumors were (near)diploid, six (near)tetraploid, three had a (near)diploid plus a (near)tetraploid stem line, and one tumor had two hypertriploid stem lines. This pattern is completely different from type II GCT of the testis, where on average, the seminomas (and GCNIS) are hypertriploid (DNA index 1.61) and the non-seminomas hypotriploid (DNA index 1.40) [294, 569]. This suggests that the precursor cells do not necessarily undergo tetraploidization and that following that event they undergo less extensive of karyotype evolution, whereby the original (near)diploid tumor cells still may coexist with the derived (near)tetraploid clone.
Results of karyotyping are in agreement with these ploidy data, with chromosome numbers being (near)diploid [307, 570–572], (near)tetraploid [573], (near)diploid plus (near)tetraploid [572], and hypertriploid [574]. Karyotyping, CGH analysis [165], and FISH [308] show i(12p) as the most common structural aberration in type II GCT of the mediastinum; other recurrent changes are gains of chromosomes 21 and X and loss of chromosome 13, similar to type II GCT of other sites. In the study by Schneider [165], two patients had Klinefelter’s syndrome, as did the case karyotyped by Mann [571], partly explaining the extra copies of X. Different from testicular cases is that gain of 12p may be lacking [571, 574] and that fewer chromosomes are involved in gains and losses, again consistent with less extensive karyotype evolution. Mediastinal non-seminomas in patients with Klinefelter’s syndrome may [165] or may not have i(12p) [571].
The demonstration of gain of 12p (in particular i(12p)) in solid and hematologic somatic-type malignancies, often in combination with genetic hallmarks of the somatic cancer, proves their origin from the GCT [575, 576].
Three out of eight mediastinal seminomas (38 %) had activating KIT mutations (exon 17 and codons 818, 820, and 822) [333]; 1/13 seminomas (8 %) had a KRAS mutations (exon 1, codon 13) [336]. Essentially the same pattern of somatic mutations as demonstrated for testicular seminomas [295]). Non-seminomas of the thymus have not been studied for the presence of mutations.
There are no published studies addressing the epigenetics of mediastinal type II GCT.
Pathogenesis
The occasional finding of mediastinal type II GCT entirely within the thymus and the frequent thymus rests in these neoplasms indicates that they have their origin in the thymus. Apparently, this organ offers a niche in which hypomethylated, erased, premeiotic [571] PGC, the precursor cells of type II GCT, may survive [56] as already proposed by Teilum [54]. The fact that mis-migrated PGC have been demonstrated in large numbers in the anterior mediastinum but not in the thymus itself [52] does not rule out this possibility. The tumors probably originate from very few PGC that manage to escape their normal fate of apoptosis by ending up in the thymus. There is circumstantial evidence to suggest that thymic epithelium may have the capacity to support erased, premeiotic PGC. Seminoma cells, the neoplastic counterparts of PGC, tend to home in thymic epithelium [56]. More convincing still is the finding of “seminoma-like” cells enclosed by thymic epithelium in the absence of an accompanying invasive type II GCT [241], resulting in lesions resembling gonadoblastoma of the dysgenetic gonad. Moreover, like Sertoli and granulosa cells, thymic epithelium produces KITLG, the survival and growth factor of PGC [56] (Fig. 3.26).

Seminoma cells homing in thymus epithelium; clockwise: cytokeratin-positive thymus epithelium (brown) enclosing seminoma cells; same area with OCT4-positive nuclei (brown) of seminoma cells; same case expressing KITLG (red) in thymus epithelium (original magnification ×200) [56]
The assumption that mediastinal type II GCT arise from PGC is more credible than the proposed alternatives. Origin from a primordial cell of the thymus (for review [577]) is unlikely in view of the close phenotypic and genetic resemblance of the mediastinal type II GCT with their counterparts in the gonads, of which the origin from PGC/gonocytes is undisputed.
The hypothesis of origin through dissemination of early gonadal lesions, which recapitulate embryonal memory and reverse migrate to thymus [578], was immediately refuted [579]. This idea is conflicting with the absence of testicular type II GCT in patients with Klinefelter’s syndrome combined with their 67-fold risk of mediastinal type II GCT [554]. In addition, the presumed testicular precursor lesions are typically absent in patients with mediastinal type II GCT [211, 563]. Finally, Chaganti et al. [578] stressed the identity of karyotypic changes of testicular and mediastinal type II GCT, which they are not. As discussed, mediastinal type II GCT are characterized by a shorter karyotype evolution than their testicular counterparts. For a mediastinal GCT to be derived from a testicular precursor lesion, one would expect the reverse pattern, with the testicular lesions being in an earlier stage of karyotype evolution than the mediastinal tumors.
Mediastinal type II GCT, like those in the gonads, usually originate via the “developmental” pathway. The almost exclusive male patient population is consistent with a crucial role of TSPY as in the testis and gonadal dysgenesis in 46,XY DSD, in combination with KITLG stimulation. It cannot be excluded that some tumors are initiated by somatic mutations in KIT or in genes of its downstream signaling proteins; however, most KIT mutations are probably engaged in the progression of seminoma. Non-seminomas, also in the mediastinum, result from reprogramming of a seminomatous precursor cell, i.e., a transformed PGC, into an EC cell.
The 67-fold risk of mediastinal type II GCT in Klinefelter’s syndrome [554] supports the notion that these tumors usually have a developmental origin. The syndrome, affecting 1 in 500 males and diagnosed in only a quarter of the cases [277], is characterized by hypergonadotropic hypogonadism, small testes, infertility, gynecomastia, abnormal body habitus, and mild developmental abnormalities due to an abnormal sex chromosomal complement, usually 47,XXY. Relative androgen deficiency at least at the testicular level accelerates degeneration of the testis at the onset of puberty [580]. In adolescents and adults, most tubules become atrophic with very few or no germ cells left. This may explain why testicular type II GCT are rare in these patients with only sporadic cases reported: one seminoma and two non-seminomas [581–583]. It seems that like the normal spermatogenic cells, precursor cells of GCT cannot survive in the defective spermatogonial niche.
At the same time, it is hypothesized that in Klinefelter’s syndrome, the increased levels of gonadotropins, which physiologically stimulate germ cell proliferation, promote malignant transformation of PGC in the thymus [165, 277]. Indeed, the tumors develop at a much younger age than in the testis, before or at the onset of puberty and thus before degeneration of the precursor cells. Hypothetically, the absence of pure seminomas [553] may be due to a similar process of degeneration as in the testis that eventually destroys all precursor cells except those that have undergone reprogramming to EC cells, the more apoptosis-resistant totipotent stem cells of non-seminoma. Supporting this hypothesis, the only mixed GCT that contained a seminoma component were in two 8-year-old boys.
The reportedly higher incidence of type II GCT of the brain in Klinefelter’s syndrome [556] is possibly also caused by the hypergonadotropic stimulation. It remains elusive why as opposed to the mediastinal ones, the brain GCT in Klinefelter’s syndrome have a similar age distribution and histology as in the general population, with more than 80 % seminomas [584].
From a different angle, it has been suggested that the extra copy/copies of chromosome X in Klinefelter’s syndrome play a direct role in the pathogenesis of the type II GCT because the region Xq27 harbors a susceptibility locus for testicular germ cell neoplasms [585]. However, linkage to this locus was not confirmed in a larger set of pedigrees, which included the original 66 families [586], and a candidate gene has not been identified.
Solid somatic-type malignancies are most often due to further tumor progression of somatic components of non-seminomas but may also arise from progression of YST, angiosarcoma, for example [544]. The unique association of mediastinal mixed non-seminomas containing YST, or pure YST, with hematologic malignancies is as yet unexplained.
It has been suggested that the angiosarcomas may arise in myxoid/mesenchymal foci of YST, called magma reticulare by Teilum (quoted in [241]). This tissue has vasoformative capacity, whereby dysplastic spindled and epithelioid cells condense into vessels. In the mouse, the first adult hematopoietic stem cells arise from the endothelium of the major vasculature, in particular the aorta [587, 588]. It is tempting to speculate that the higher incidence of angiosarcoma and hematological malignancies in mediastinal YST (component) are related and that endothelial cells developing in magma reticulare are the source not only of angiosarcoma but also of hematopoietic stem cells [297, 576], which may progress into hematopoietic malignancies (Fig. 3.27). The question remains: why only in mediastinal non-seminomas? Could it be that local factors in the anterior mediastinum, which regulate the specification of adult hematopoietic stem cells in normal embryogenesis, also, in the early stages of tumor development, promote the development of neoplastic hematopoietic stem cells from suitably primed endothelial precursors? Human thymic epithelial cells could be a source of such factors as they reportedly produce granulocyte and macrophage colony-stimulating factors [589].

Mediastinal non-seminoma. Left: primary tumor with florid vascular proliferation (magma reticulare) with groups of blast-like cells suggestive of hematopoietic cells (arrow) (H and E, ×400); right: bone metastasis of same patient with the histology of angiosarcoma 7 months after initial treatment (H and E, ×200)
3.6.2.5 Central Nervous System
Developmental Potential
Type II GCT of the brain have the same totipotent developmental potential as those of other anatomical sites. The proportion of seminomas (called germinomas in the CNS), 80 %, is higher than in other sites, apart perhaps from the dysgenetic (intra-abdominal) gonad. This figure may be a slight overestimation because the histological diagnosis of GCT of the brain is often made on small biopsies not always representing all components of the tumors. It explains the occasional event of recurrences having a different histology from the original tumor, such as a germinoma recurring as YST [590], a YST as growing teratoma [591], and in particular teratoma as germinoma, sometimes after a long interval [592–596].
Most of the non-germinomatous tumors are mixed (54 %), followed by mature teratoma (21 %), immature teratoma (8 %), EC (6 %), YST (3 %), and choriocarcinoma (3 %). Seventy-five percent of the mixed GCT have a germinoma component, most often combined with, usually immature, teratoma, YST and/or EC; a choriocarcinoma component is rare [263]. This observation is consistent with the pathogenetic mechanism, whereby non-germinomas result from reprogramming of a germinomatous precursor cell.
Somatic-type malignancies, sarcoma among others, develop rarely; the reported squamous cell carcinomas may have developed in late type I GCT [263]. There is an exceptional case report, mentioned before, of a black female who presented with a mixed lineage acute myeloid leukemia 4 months after treatment of a suprasellar dysgerminoma, obviously not related to Klinefelter’s syndrome [550].
GCT of the brain (except those of the spinal cord) in patients with Klinefelter’s syndrome have about the same distribution of histological subtypes as in non-Klinefelter cases, with 6/7 being dysgerminoma (86 %), at the median age of 15 years (range 12–35) [584]. This is remarkably different from the mediastinal type II GCT in Klinefelter’s syndrome, which are exclusively non-seminomas, at variance with the general population [553], and may appear before puberty [277]. Their predominant germinoma histology by itself makes it less likely that leukemias develop in association with GCT of the brain in Klinefelter patients. Indeed, this phenomenon has, to our knowledge, not been reported.
In Down’s syndrome, type II GCT of the brain have a lower percentage of germinomas than in the general population (6/11, 55 %), in agreement with the atypical anatomical localization of the tumors, with only one tumor in the pineal gland; of the five non-germinomas, there were four YST and one teratoma [597].
Epidemiology
Historically, for GCT of the brain, a five- to eightfold higher frequency in the Far East than in Western countries has been reported [598–600]. The last WHO classification [243, 601] quotes a twofold difference (0.17 and 0.09 in Japan and the USA, respectively). A recent comprehensive epidemiological study based on four large databases, two from Japan and two from the USA, demonstrates that the incidence is not significantly different in both countries: 0.143 for males and 0.046 for females in Japan and 0.118 for males and 0.030 for females in the USA [539]. In these figures, type I and II GCT are combined; however, >90 % are probably type II, as less than 3 % of the registered cases, often perinatal (immature) teratomas, are below the age of 5 years [602]. Virtually all pineal tumors are type II; indeed, they hardly occur under age six [76]. In Japan, the incidence of CNS GCT has reportedly increased in the 1980s, mostly in males, and has plateaued since [603]. The median age is about 15 years both for males and females [243, 539], whereby patients with germinomas, mixed GCT, and non-germinomas are on average 18, 15, and 12 years, respectively [263].
The overall male-to-female ratio of CNS GCT is about 4:1 [243, 539, 601]. In Japan, all histological types occur predominantly in males [602]. The incidence of non-malignant GCT is similar in males and females, 0.029 and 0.020, respectively. The male-to-female ratio for malignant GCT is 16:1 in the pineal region and 2.1:1 in the rest of the CNS. More than half of all malignant GCT are located in the pineal region [539].
Multifocal tumors in the brain usually involve the pineal gland and the suprasellar region (so-called bifocal tumors) or rarely both basal ganglionic regions, simultaneously or sequentially [243, 604, 605]. Without molecular analysis, it is virtually impossible to prove that the tumors are independent primaries; locoregional metastasis or recurrence is usually the more likely explanation [594].
A series of case reports demonstrates that type II GCT of the brain may be combined with primary, usually type II, GCT at other anatomical sites. The brain tumors were always germinomas located in the pineal gland or immediate vicinity and followed by a seminoma of the testis (three cases) [283, 606, 607], a non-seminoma of the testis [608], a mediastinal non-seminoma [609], or preceded by a mediastinal seminoma [610]. In one case, the brain germinoma was followed by a mediastinal type I YST, in view of the demonstrated 1p36 deletion and the absence of gain of 12p in the latter [611]. In one patient with Klinefelter’s syndrome, the germinoma of the brain was simultaneous with a non-seminoma of the mediastinum, and in another, it was followed by a mediastinal pure choriocarcinoma [612]. In a patient with Down’s syndrome, the germinoma of the brain was simultaneous with an EC of the testis [613].
In view of the relative frequency of testicular and mediastinal type II GCT, the latter are clearly overrepresented, which is plausible: if the conditions are favorable for the development of extragonadal type II GCT, e.g., by hypergonadotropism, they could stimulate their development both in the mediastinum and brain.
In the patient described by Coffey [283], the pineal germinoma had the same KIT mutation as the testicular seminoma diagnosed 6 month later, suggestive for a common cell of origin, as mentioned earlier.
There are occasional reports of familial clustering of type II GCT of the brain: two brothers with a teratoma with germinoma elements in the pineal region [614]; three brothers, one with an EC and two with a dysgerminoma in the pineal region [615]; and a boy with a germinoma of the basal ganglia and his sister with a germinoma of the suprasellar region [616].
Risk Factors
In Klinefelter’s syndrome, type II GCT, which constitute only few percent of brain tumors in the general population, are the most frequent tumors of the brain (median age 16), implicating an increased risk in this syndrome [555]. This is perhaps also true for spinal cord germinomas, although the high age of the patients (median 29 year) suggests that the spinal tumors might be cerebrospinal fluid-borne metastases of subclinical germinomas of the brain [584].
Patients with Down’s syndrome have an increased risk of leukemias and a lower risk of solid cancers [557, 558]; the latter is attributed to overexpression of the DSCR1 and DYRK1a genes on chromosome 21, which suppress the production of VEGF and thereby angiogenesis, which sustains solid tumor growth [617]. Exceptions among the solid tumors are lymphomas and GCT of the brain [557] and possibly the testis [276, 558].
Isolated case reports have associated type II GCT of the brain with neurofibromatosis 1 [618] and multiple congenital melanocytic nevi [619].
Anatomical Distribution
Over 80 % of type II GCT of the brain are located in the pineal gland, suprasellar region (neurohypophysial axis; occasionally within the neurohypophysis), hypothalamus, and the wall of the third ventricle. In these midline structures, the large majority are in male patients, malignant, and germinomas. Germinomas occur also in the basal ganglia, cerebral hemispheres, and in the posterior fossa; however, in these and other atypical, non-midline anatomical sites, the proportion of females, non-germinomas and benign GCT (the latter probably of type I), is somewhat higher [243, 539]. In fact, type I GCT of the brain have an anatomical distribution resembling that of the type II non-germinomas [132]. It seems that germinoma involving the basal ganglia is more frequent in Asian than Western children [604, 605].
The anatomical distribution of type II GCT in Klinefelter’s syndrome, 6/7 in the pineal, suprasellar, and hypothalamic region, is similar as in the general population [584].
In Down’s syndrome, where there is a high percentage of non-germinomas, the tumors lie most often outside the typical midline sites [597].
(Cyto)Genetics
Half of the type II GCT of the brain are (near)diploid and half (near)tetraploid, which is true for germinomas, mixed GCT, and non-germinomas [197]. This finding is consistent with the young age of clinical manifestation of these tumors (median 15 years) and therefore shorter period of karyotype evolution than in their testicular and mediastinal counterparts. The pattern of gains and losses of (parts of) chromosomes is largely similar to that in type II GCT of other anatomical sites: in order of frequency, gain of 12p, 1q, 8, 21, and X, with more defined regions of gain being 12p12, 1q11-q24, and 8q11-q21, and loss of 11q, 18q, and 13 [310]. In addition, Wang et al. [197] found gain of 14q and loss of 10q and 17p, thereby making the picture even more similar to type II GCT of other sites. A comprehensive analysis by SNP microarray further confirms and details this data. The most frequently observed copy number gains were regions on chromosomes 1q (44 %), 2p (37 %), 7q (37 %), 8q (41 %), 12p (59 %), 14 (33 %), 20q (30 %), 21 (63 %), 22 (41 %), and Xq (44 %). Frequently observed copy number losses were regions on chromosomes 1p (26 %), 4q (26 %), 5q (33 %), 9q (30 %), 10q (37 %), 11q (41 %), and 13 (48 %) [298].
Gain of 12p is present in 9/17 (53 %) type II GCT studied by Schneider et al. [310], in 5/15 (33 %) unequivocal type II GCT (germinomas and mixed GCT) in the material of Wang et al. [197], and in 59 % in the study by Terashima et al. [298]. These figures are significantly lower than in other sites, again consistent with less extensive karyotype evolution.
Activating KIT or RAS mutations occur frequently in germinomas (60 %) and less so in non-germinomas (9 %); they are mutually exclusive in both [334]. By next-generation sequencing, Wang etal. [197] confirmed and extended these data. It appears that activating mutations in KIT (47 %) (in order of frequency in exons 17, 11, 18, and 13), KRAS (18 %), and NRAS (6 %) and inactivating mutations in CBL (6 %), a negative regulator of KIT expression, are all mutually exclusive and occur most often in germinomas and mixed GCT that lack gain of 12p. The complementary character of these genetic events and their preferred occurrence in germinomas and mixed GCT indicate that they are probably not initiating but engaged in the progression of germinoma. The AKT/mTOR signaling pathway is activated in about 20 % of cases, mostly by focal amplification of 14q32.33, containing the AKT1 locus, often in tumors lacking gain of 12p. Less frequently, loss of function mutations were identified in BCORL1, MTOR, TP53, SPTA1, KDM2A, and LAMA4 [197]. BCORL1 is a tumor suppressor and a transcriptional corepressor, of which the mutation might interfere with the function of nuclear receptors such as the AR [620].
The study by Terashima et al. [298] highlights frequent gain of PRDM14 on 8q13, which was earlier identified as a susceptibility locus for testicular type II GCT and also frequent aberrations of CCND2 (12p13) and RB1 (13q14) suggesting that the cyclin/CDK-RB-E2F pathway might be involved in the pathogenesis of type II GCT of the brain. Finally, Wang et al. [197] found in their cohort a rare germline variant of JMJD1C in Japanese patients that functions as a chromatin modifier gene interacting with the AR in humans [621] and that in mice is involved in long-term maintenance of male germ cells [622]. This gene variant is reportedly enriched in the Japanese population and fivefold higher in the Japanese patients with GCT of the brain compared to the general population. The authors propose that this variant of JMJD1C might explain the higher incidence of GCT of the brain in Japan [197]. It seems, however, in view of recent incidence figures [539], that there is no significant difference to be explained.
Pathogenesis
The strong phenotypic [352] and genotypic [197, 298, 310, 334, 623] resemblance of type II GCT of the brain and the gonads makes it plausible that they share the same cell of origin: a hypomethylated, erased, premeiotic, totipotent PGC [56], as already proposed by Teilum [54]. Indeed, in 7–14 wpc, human embryos and fetuses mis-migrated PGC that have escaped elimination by apoptosis can be seen in the midline of the CNS. Probably, these PGC have arrived here because they failed to exit the sympathetic trunk at the gonadal site and continued migration in cranial direction along other nerve branches from the sympathetic trunk [52]. Apparently, the midline of the brain, in particular the pineal gland and the suprasellar region, offers a niche with conditions where some of these PGC can survive long enough to give rise to germinoma, which upon reprogramming may go on to develop non-germinoma. The high proportion of pure germinomas (80 %) may be due to the young age of the patients (median age 15 years). Consequently, the time for progression to non-germinoma via reprogramming is short, as in patients with gonadal dysgenesis. This model may also explain why type II GCT in sites away from the midline are rarer and more often non-germinomas. In these non-midline sites, the conditions are supposedly less suitable for neoplastic PGC, favoring precursors that have undergone reprogramming to an ESC-like precursor, with a developmental potential in between the totipotent and the pluripotent state. Indeed, the spectrum of histologies of type II non-germinomas has resemblance to that of type I GCT: relatively high proportions of pure (immature) teratoma, pure YST, and pure choriocarcinoma and rarely pure EC. Yet these tumors occur most often beyond the age of six and have the (cyto)genetic characteristics of type II GCT. In fact, in the brain, particularly away from the midline, there seems to be a gray area with a gradual transition between GCT of type I and II, featuring tumors that are type II by genotype and age but resembling type I by phenotype.
An alternative to the hypothesis that all type II GCT develop from PGC proposes that only germinoma stems from PGC and that the other tumor types develop from corresponding embryonic rests that get incorporated in the developing neural tube through folding errors. In this manner, choriocarcinoma would arise from misplaced trophoblast, YST from patches of yolk sac, and EC and teratoma from fragments of the embryo proper [624]. However, such misplaced elements, contrary to PGC, have never been detected in the developing CNS. Reprogramming of germinoma cells, the neoplastic recapitulation of physiological process in embryonic development, is a more plausible explanation for the origin of the different types of non-germinoma, if only because this mechanism applies also to gonadal type II GCT where misplacement of embryonic rests has no bearing. Nevertheless, the embryonic misplacement hypothesis cannot be totally dismissed. Sporadically, growths have been reported in the head and neck region, including the brain [96], with a morphology in principle compatible with type I or II (immature) teratoma that turned out to be mono- or even dizygotic twins [102]. In such cases, misplacement of a zygote or a blastomere during embryonic development is a plausible pathogenetic mechanism.
More recently, it was proposed by Scotting and colleagues that all GCT of the brain have their origin in NSC that during embryonic development have been induced to pluripotency by activation of OCT4 through demethylation of its promoter region [139, 168]. Indeed, Kim et al. have shown that mouse [71] and human [72] NSC can be induced to pluripotency by activation of OCT4 alone, as mentioned earlier, and when grafted into mice, these stem cells give rise to teratomas. According to Scotting et al., teratomas may develop in the brain via the same mechanism, which subsequently can give rise to all other types: germinoma and the various mixed and pure non-germinomas. To support their hypothesis, they quote a series of case reports to show that each type of GCT can recur as any of the other types of GCT, except germinoma recurring after resection of a YST. None of the cases, however, prove the crucial point of teratoma or rather the stem cells of teratoma, giving rise to germinoma. In two of the quoted cases, the authors of the case reports themselves conclude that the pineal tumor, originally diagnosed as teratoma, was probably mixed and that the germinoma component was missed in the initial intervention. This was histologically likely in the case described by Janzarik et al. [595] and clinically suspected in the case reported by Mao et al. [596]. In the three cases where the original teratoma and the later diagnosed germinoma were anatomically separate, the authors interpret the germinoma as a second primary tumor [592–594], admitting that a late relapse cannot be excluded, as seminoma/germinoma usually has a long protracted clinical course.
The biology of metastasis of type II GCT, of the testis in particular, shows that the seminoma component in a mixed primary non-seminoma disappears over time in the evolution of the tumor [271]. Residual mature teratoma after chemotherapy for non-seminomatous germ cell tumors of the testis occurs significantly less often in the lung than in retroperitoneal lymph node metastases [272]. On the other hand, a pure seminoma may undergo reprogramming to all non-seminomatous components [244], even in metastatic sites, in up to 44 % of the cases [258], as mentioned earlier.
Consistent with the lack of clinical evidence of germinoma originating from teratoma, this phenomenon has never been reported in the decades of research in mouse models of teratoma, starting with Stevens’ seminal work [80]. Also, the many iPSC that have been tested for their developmental potential by grafting into mice have never produced seminomatous tumors [321, 625]. iPSC appear to be in the primed state by default, with a pluripotent development potential, typical for type I GCT. It is therefore very unlikely that induced pluripotent NSC would give rise to type II GCT. They may perhaps be the origin of perinatal/infantile, type I teratoma GCT of the brain, if it is true that NSC, particularly in the midline of the brain (not the typical site of the type I GCT), have partial loss of imprinting [169]. The quoted observations are consistent with specification of the germ lineage being a tightly controlled process that is unlikely to spontaneously occur in a teratoma developed from an iPSC (for review [35]). Reprogramming of PGC to pluri- or totipotency on the other hand is a kind of default pathway for which molecular mechanisms are in place to prevent it from happening ([39], for review [20]). Finally, if all germinomas of the brain were to originate via a teratoma stem cell, one would expect a smaller percentage of the GCT of the brain being pure germinoma/seminoma than in other sites, while in fact in the brain, this figure is the highest. It is suggested that KIT mutations could bias induced pluripotent NSC toward developing germinoma [139]. However, KIT mutations are effect rather than cause of germinoma development, as they are mostly engaged in progression of seminomatous GCT and rarely in initiation of type II GCT, as appears from molecular analysis of GCT of the testis [295, 324] and brain [197].
Like in other sites, probably the majority of type II GCT of the brain have a “developmental” origin with the same arguments in support. The overwhelming preponderance of male patients, like in the thymus, suggests a similar role for co-expression of OCT4, TSPY, and KITLG in maturation-disturbed PGC, as in the hypovirilized conditions in the testis in TDS and in 46,XY gonadal dysgenesis.
The increased risk of type II GCT of the brain in patients with Klinefelter’s and Down’s syndrome is consistent with a developmental origin of these tumors. In both conditions, disturbed development of the gonads causes increased secretion of gonadotropins in the diencephalic centers at the inception of puberty. These hormones are supposed to stimulate the neoplastic transformation of mis-migrated PGC in the midline of the brain [165, 243, 277].
In addition, it has been speculated that overdose of certain, as yet unidentified, genes on chromosomes X and 21 might favor the development of type II GCT. Indeed, both are among the most frequently overrepresented chromosomes in type II GCT in the general population, regardless of anatomical site. Patients with Klinefelter’s and Down’s syndrome seem to have a constitutional “chromosomal advantage” for developing type II GCT. Indeed, the only malignancies for which Klinefelter patients have an increased risk are mediastinal [554] and brain type II GCT [555, 556]. In Down’s syndrome, apart from leukemias and lymphomas, there is only an increased risk for type II GCT of the brain and probably the testis [276] [557], [558].
Probably some tumors are initiated by somatic mutations, as in the earlier quoted patient with the same activating KIT mutation in his testicular seminoma and pineal germinoma [283]. In this case, the mutation has likely occurred in migrating PGC thereby enabling them to not only reach the gonads but also the pineal gland.
Somatic mutations are rare in type II GCT of the brain with 0.50 non-silent mutations per Mb [197], the same figure as in testicular type II GCT [295]. The majority of mutations occur in germinoma and can be explained as involved in the progression of germinoma rather than as initiating events. Initiating mutations should be as frequent in germinoma as in non-germinoma, as the PGC is the precursor for both. The mutation rate in non-germinomas, less than 10 % [197], is therefore a fair indication for the maximum rate of type II GCT of the brain initiated by a somatic mutation. Probably the rare type II GCT in females without mosaicism for Y, where TSPY obviously is not involved in the pathogenesis [352], are more often caused by somatic mutations, particularly in KIT, like in the ovary [332].
Incidental reports on association of NF1 with type II GCT of the brain underscore the pivotal role in the development of these tumors of the KIT/RAS signaling pathway, of which NF1 is a negative regulator (Fig. 3.17).
3.6.3 Type II GCT Before Puberty
Typically, type II GCT develop after puberty. There are three situations in which type II GCT occasionally may occur before puberty: in patients with DSD in the gonads (for review [275], for age distribution of dysgerminomas [232]); in Down’s syndrome, e.g., a seminoma of the testis in a boy of 2 years ([626], for review [276]); and in Klinefelter’s syndrome in the mediastinum, e.g., two 8-year-old boys with a mixed GCT having a seminoma component ([564, 627], for review [277]). What these conditions broadly have in common is the severity of the disturbance of the niches where PGC/gonocytes may home. This is apparent from the gonads in DSD and Klinefelter’s and Down’s syndrome where gonocytes can barely survive and only rarely (in DSD and Down’s syndrome) may differentiate into functional gametes. When gonocytes do survive, in DSD and Down’s syndrome, they have an increased risk for neoplastic transformation; in Klinefelter’s syndrome, transformed PGC probably degenerate before they can produce manifest tumors. What applies to the gonads is probably also true for extragonadal niches: PGC surviving there have a higher risk of neoplastic transformation, particularly when they are reprogrammed to ESC. These precursors may have a developmental potential ranging from primed-state-like, as in early mediastinal GCT in Klinefelter’s syndrome, to fully fledged naïve state, depending on their methylation status at the time of neoplastic transformation.
This generalizing hypothesis does not explain the observation that in Klinefelter’s syndrome mediastinal and not brain type II GCT may occur before puberty and in Down’s syndrome, those of the testis but not those of mediastinum and brain.
3.7 Type III GCT
3.7.1 Developmental Potential
The cells of a spermatocytic tumor, the name proposed in the fourth edition of the WHO classification instead of spermatocytic seminoma [244], resemble postpubertal germ cells with nuclei in three distinct size classes. Those with the smallest nuclei with dense chromatin look like A-dark spermatogonia (considered reserve spermatogonial stem cells); the cells with intermediate and large paler nuclei with finely granular filamentous chromatin resemble A-pale spermatogonia (self-renewing stem cells), B spermatogonia, and leptotene spermatocytes [244, 628–630]. Transcript and protein analyses of spermatocytic tumor cells, reviewed by Waheeb and Hofmann [631] and summarized in Table 3.3, show that they lack markers of embryonic gonocytes and postmeiotic germ cells and express markers of prespermatogonia, spermatocytic stem cells/undifferentiated spermatogonia, and spermatocytes in various combinations, suggesting the phenotype of a postnatal germ cell arrested at any stage of maturation between prespermatogonium and primary spermatocyte. OCT2 expression seems confined to tumor cells resembling A-dark spermatogonia [632]. Rare spermatocytic tumors are composed only of OCT2-expressing tumor cells and thus of neoplastic A-dark spermatogonia with blocked differentiation [632]. In fact, the stem cells of spermatocytic tumors, type III GCT, are committed to spermatogenesis with differentiation capacity limited to premeiotic cells.
So-called anaplastic spermatocytic tumor, a rare variant, has morphological features in common with seminoma [633]. One report describes a metastasizing anaplastic tumor [634]; however, overall this variant behaves as benign as the usual spermatocytic tumor, which only sporadically metastasizes [633]. Exceptionally, with less than 20 published cases, the tumor is associated with a sarcomatous component, usually undifferentiated sarcoma, and rarely rhabdomyo- or chondrosarcoma [635–637]. It is highly malignant and readily metastasizes to regional lymph nodes or, blood-borne, to visceral organs. The sarcoma component is probably the result of progression of the spermatocytic tumor, similar to progression in low-grade leukemias, lymphomas, and sarcomas [636]. The possibility that it has its origin in a germ cell that is reprogrammed to rudimentary somatic differentiation cannot be dismissed.
3.7.2 Epidemiology/Risk Factors
The only population-based epidemiological study finds an incidence of 0.4 per million for spermatocytic tumors, constituting 0.6 % of all testicular cancers in Australia. An increasing incidence over the past 20 years is suggested but has not been found statistically significant; risk factors have not been identified [638]. In about 9 %, the tumor is bilateral, more often metachronous than synchronous [244, 630]. The median age of clinical manifestation is 54 (range 19–92) [638].
3.7.3 Anatomical Distribution
Spermatocytic tumor occurs only in the postpubertal testis [630]. There is one report on a tumor originated in a maldescended testis [639]. Apparently, the tumor develops only if the conditions in the testis are compatible with survival of postnatal germ cells and induction of spermatogenesis.
3.7.4 (Cyto)Genetics
Most spermatocytic tumors are (near)diploid, the second largest group is (near)tetraploid, and a small number is peritriploid [640–642]. The most consistent cytogenetic aberration, present in all studied tumors, is an extra copy of chromosome 9 [643, 644], in which subsequent CGH analysis demonstrated a small amplified region on 9p, containing DMRT1 as the most likely candidate gene involved in tumorigenesis [645]. In passing, DMRT1 has been shown to be an immunohistochemically detectable, useful marker for diagnosing spermatocytic tumor, apparent from Fig. 3.28 [645]. A small number of tumors, usually in the oldest half of the patients, have mutually exclusive, paternal age-related mutations in FGFR3 or HRAS [646]. p53, not expressed in normal postpubertal germ cells, is demonstrated in 80 % of spermatocytic tumors, supposedly related to genomic instability [647].

Exclusively intratubular spermatocytic tumor, the precursor of spermatocytic tumor; no pagetoid involvement of adjacent tubules as in GCNIS (left H and E, ×200; right DMRT1, ×200)
The metastasizing anaplastic spermatocytic tumor published by Mikuz [634] resembled seminoma morphologically but lacked expression of PLAP and OCT4; cytogenetically, it had gain of both chromosome 9 and 12p. This tumor seems a hybrid between seminoma and spermatocytic tumor, whereby the phenotype is partly determined by overexpression of DMRT1, partly by the overdose of the pluripotency genes on 12p, perhaps not adequately repressed by DMRT1 [189]. This is yet another example of the plasticity of developmental states of precursors of GCT, blurring, in this case, the line between type II and type III GCT.
3.7.5 Epigenetics Including GI
An immunohistochemical study found a heterogeneous pattern of DNA methylation and histone modification in spermatocytic tumors, quite different from the regular patterns in normal spermatogenesis, probably because the regulatory signals conveyed by the niche toward the spermatogenetic cells are lacking in the tumors [648].
3.7.6 Pathogenesis and Animal Models
Spermatocytic tumor is not associated with GCNIS [481, 640]; however, it has its own intratubular precursor, at the luminal side of the tight junctions connecting the Sertoli cells, with essentially the same morphology as the adjacent invasive tumor [244, 628–630]. The occasional finding of exclusively intratubular spermatocytic tumor without an invasive component proves that the intratubular part is not due to intratubular extension of the invasive component [244, 630] and indeed the precursor lesion. Unlike GCNIS, it shows no obvious accumulation of precursor cells in the spermatogonial niche, neither as stacking of multiple layers of precursor cells nor as pagetoid extension within the seminiferous tubules to the detriment of spermatogenesis. It is possible that the precursor cells are phenotypically so similar to normal spermatogonia that expansion in the niche is not recognized with light microscopy, including immunohistochemistry. Alternatively, only the spermatogonial tumor stem cells remain in the niche, and upon the earliest differentiation, the tumor cells, like their normal counterparts, move to the lumen of the tubule. Finally, it is possible that the initiated cell lies at the luminal side of the tight junctions. Whatever the initial development, eventually the tumor cells become invasive and apparently independent from the tubular micro-milieu. It is indeed remarkable that fragile, apoptosis-prone cells like spermatogenic cells manage to survive in conditions so alien to them.
Studies in mouse models and human tumors begin to untangle the molecular mechanisms, both in the germ and niche cells, involved in maturation of postnatal male germ cells and controlling the mitosis versus meiosis switch, and how these might bear on the development of spermatocytic tumors.
Glial cell line-derived neurotrophic factor (GDNF), a distant member of the transforming growth factor superfamily, is secreted by Sertoli cells as paracrine factor involved in the regulation of spermatogonial self-renewal and differentiation in mouse and men ([649], for review [631]). Spermatogonial stem cells express the GFRA1/RET receptor complex at the cell surface. Binding of GDNF to this complex upregulates MYCN transcription factor via the PI3K/AKT pathway and FOS transcription factor via the RAS/ERK1/2 pathway (Fig. 3.29), as well as FGFR2 in spermatogonial stem cells. Other niche factors are FGF2, the ligand for FGFR2, produced by Sertoli cells, and CSF-1 secreted by Leydig cells (Fig. 3.30). Downregulation of GDNF in mice causes a Sertoli cell-only phenotype with complete absence of spermatogenic cells. Overexpression causes accumulations of undifferentiated spermatogonia in seminiferous tubules, resembling intratubular spermatocytic tumor, abrogation of spermatogenesis, and tumors in older animals, which are bilateral in over 50 %. By geno- and phenotype, the tumors have intermediate phenotypes between type II seminoma and spermatocytic tumors [650, 651], like the tumor described by Mikuz [634].

Signaling pathways triggered by GDNF in spermatogonial stem cells. GDNF dimerizes and binds to the GFRA1⁄RET receptor complex. (a) Binding of GDNF activates RET, which triggers SRC kinase phosphorylation and the downstream activation of PI3K⁄AKT. Ultimately, the transcription factor MYCN is upregulated. (b) Binding of GDNF also can activate the RAS-mediated signaling pathway, which triggers ERK1⁄2 phosphorylation and upregulation of the transcription factor FOS [631]

A simplified view of the spermatogonial stem cell niche showing the main extrinsic factors driving SSC maintenance and self-renewal. Sertoli cells and spermatogonial stem cells (SSCs) are both attached to the basement membrane (BM). Sertoli cells provide for structural support and produce glial cell line-derived neurotrophic factor (GDNF) and basic fibroblast growth factor (bFGF) which are crucial for SSC self-renewal in vitro and in vivo. Leydig cells (L) and peritubular cells (PE) produce colony-stimulating factor-1 (CSF-1), also essential for self-renewal [631]
DMRT1 is the transcriptional gatekeeper controlling the mitosis versus meiosis decision in male germ cells [652]. It prevents differentiation and meiosis of spermatogonial cells by blocking the transcription of STRA8 and rendering these cells less sensitive to RA-induced meiosis. At the same time, it upregulates SOHLH1, a factor stimulating proliferation of spermatogonial stem cells, and suppresses the pluripotency genes NANOG, SOX2, and OCT4. Decreasing the level of DMRT1 disrupts GDNF signaling, cell cycle control, and pluripotency regulation. In 129Sv mice, but not in other mouse strains, it causes teratoma formation in a dose-dependent manner, probably due to failure to repress pluripotency regulators and reduced GDNF signaling. Postnatally elevated DMRT1 and GDNF signaling blocks differentiation of spermatogonial stem cells, resulting in tumors resembling spermatocytic tumors [189].
From these studies, it appears that both niche factors and factors intrinsic to spermatogonial stem cells may contribute to the formation of spermatocytic tumors. Overexpression of GDNF by Sertoli cells and CSF-1 by Leydig cells, perhaps in response to reduced spermatogenesis in aging men, might explain the old age of clinical manifestation and the relatively high risk of bilateral tumors, like in the mouse model [650, 651]. The niche factors may synergize with cell-intrinsic factors, also acquired with increasing age, such as elevated levels of DMRT1 through gain of chromosome 9 [645], and accumulation of paternal age-related mutations in HRAS and FGFR3 [646].
It is an emerging pattern: like in the other types of GCT, in spermatocytic tumors, initiation is probably primarily due to a developmental deregulation rather than somatic mutations, the latter being mainly progression related.
So-called seminomas have been described as spontaneous tumors in a variety of animals, like the dog [653] and rhinoceros [654], and have been experimentally produced, e.g., in C. elegans [655], zebrafish [393–395], and mice [651]. None of these can be reprogrammed to totipotency like type II seminomas in men and are therefore best regarded as models for spermatocytic tumors, sharing some features with seminomas [629, 653]. In dogs, the often bilateral, DMRT1-positive [656], spermatocytic tumors are frequently combined with nodular hyperplasia or even benign tumors of Leydig and Sertoli cells [653], supporting the idea that disturbance of the hormonal regulation of the spermatogonial niche is a crucial factor in the origin of spermatocytic tumors.
3.8 Type IV GCT
3.8.1 Developmental Potential
Dermoid cysts, type IV GCT, are unique for the ovary as they are derived from meiotic oocytes [657]. Typically, completely mature teratomas, they present as a thin-walled cyst lined with epidermis with attached appendages and filled with sebaceous material and hairs. Usually one solid nodule (Rokitansky’s protuberance) protrudes from the wall into the lumen of the cyst. It is often composed of fat tissue, bone, teeth (with intermediate shapes between deciduous and permanent teeth) [658], and glial tissue and covered with skin with well-developed appendages including hair follicles forming hairs. The nodules mainly contain cranial tissues, suggesting that they represent the rostral part of an attempted embryo; however, virtually any adult tissue can be present. Exceptional tumors are predominantly solid with highly organized structures resembling a fetus, lacking extraembryonic tissues, as is the case in typical dermoid cysts. Benign tumors, such as struma ovarii and carcinoids, may arise within a dermoid cyst. Probably so-called monodermal teratomas similarly have their origin in dermoid cysts, eventually obscured by overgrowth of one tissue type. Epidermoid cysts may be a variant of monodermal teratoma [659], as in the testis. Monodermal teratomas could also originate in type I and II GCT of the ovary, as discussed in the relevant sections. Somatic-type malignancies develop reportedly in 0.2–3 % of dermoid cysts, including, in order of frequency, squamous cell carcinoma (80–90 %), adenocarcinomas (7 %), and sarcomas (7 %). Among the many rare types, PNET is also described (Chap. 6). A peculiar association with dermoid cysts is gliomatosis peritonei [660, 661], which will be discussed in the section on type VI GCT.
Sporadically, dermoid cysts contain immature foci, even more exceptionally combined with YST [133], prompting Yanai-Inbar and Scully to study the relationship between immature teratoma and the dermoid cyst [130]. Among 350 immature teratomas of the ovary submitted to the authors for second opinion, 92 (26 %) contained one or more grossly visible cysts lined by squamous epithelium with pilosebaceous structures. The figure of 26 % is most probably an overestimation because unusual cases are sent for consultation. In 10 % of all 350 cases, there was a dermoid cyst in the contralateral ovary, which is not much different from the percentage of bilaterality for dermoid cysts. In nine cases (aged 17–28, mean 23 years) of immature teratoma, there was a history of prior removal of a dermoid cyst in the same ovary. Four of these nine cases had a dermoid cyst in the other ovary, and three cases had multiple dermoid cysts in the same ovary. In addition, the authors had ten referral cases (aged 15–30, mean 23 years), collected over a period of 23 years, of otherwise typical dermoid cysts with minor immature areas, which did not recur after surgery, as usual for a dermoid cyst.
These cases illustrate a continuum between the typical dermoid cyst, type IV GCT, and type I immature teratomas of infancy. The ten dermoid cysts with small foci of immature teratoma are probably type IV GCT of which not all tissues had fully matured, consistent with the young age of these patients. The nine dermoid cysts recurring as immature teratoma belong probably to the same group as the 92 immature teratomas with macroscopically visible dermoid cysts. These tumors have an intermediate behavior and phenotype between type I and type IV, with an age of presentation in between that of pure immature teratomas and dermoid cysts and bilaterality like in dermoid cysts, whereas bilaterality is rare in pure immature teratomas [130, 131, 662]. These features justify the classification of these GCT as type I tumors beyond infancy, which occur also in the testis and extragonadal sites, in particular in the anteroposterior mediastinum, as discussed earlier.
3.8.2 Epidemiology/Risk Factors
Cancer registries do not provide data on the incidence of dermoid cysts as they are benign tumors. The best approximation is achieved by multiplying the frequency of dermoid cysts relative to cancers of the ovary (from hospital registries) with the incidence of the latter (from cancer registries) in the same region/country, as follows. Among 861 self-referred patients with an ovarian tumor, all of whom were treated in a single hospital (Women’s and Children’s Hospital, Los Angeles, CA), and therefore without obvious referral bias [663], dermoid cysts were the most common tumors with 379 cases (44 %); 211 tumors were malignant (25 %). The incidence of ovarian cancer in Northern America being 8.1 in 2012 [664], the incidence of dermoid cysts of the ovary can be estimated at about 13, making it overall the most common GCT, more frequent even than testicular type II GCT in high-incidence countries.
As for age of presentation, in a review of 517 dermoid cysts from a single institution, the two youngest patients were 10 and 13 years of age, the median age was 30, and the oldest patient 90. There were no prepubertal cases, and the large majority of dermoid cysts occur in the reproductive age between the onset of puberty and menopause [233]. The tumors diagnosed after the menopause probably had their origin during reproductive age [130]. Bilaterality occurred in 10.8 %; in seven cases, the dermoid cyst was associated with a malignant or benign epithelial surface tumor. Upon long-term follow-up, Anteby et al. [133] found 18 bilateral cases among 99 patients; multiple dermoid cysts in a single ovary were found in 9/18 bilateral cases as opposed to 1/81 unilateral cases. The mean age was similar for uni- and bilateral cases: 32.4 and 34.6 years, respectively. However, the age distribution was significantly different: 18/19 cases (95 %) were between 20 and 40 years old in unilateral compared to 61/80 (76 %) in bilateral cases, implying that overall bilateral cases are diagnosed at a younger age. Bilateral cases had a significantly higher risk of developing a recurring dermoid cyst. In familial cases where laterality was stated, 11/28 (39 %) were bilateral, and among the patients who were twins or triplets, 9/12 (75 %) were bilateral [134, 135]. One of the identical twins [533] had over the years seven dermoid cysts removed from her left and one from her right ovary. The age of diagnosis of the first tumor in the familial bilateral cases was known in ten patients; all presented between age 7 and 26, with a median age of 22.5, substantially younger than in nonfamilial bilateral cases [133]. These case histories strongly suggest a genetic predisposition for bilaterality and familial occurrence of type IV GCT of the ovary, which may also confer an increased risk for type II GCT, in view of two families with clustering of bilateral dermoid cysts with testicular seminoma [524, 531].
3.8.3 Anatomical Distribution
Dermoid cysts occur exclusively in the ovaries; on rare occasions, they may become detached from the ovary and reimplanted in either omentum [665], fallopian tube [666], or Douglas’ pouch [667]. In four of the 31 omental and one of the 29 tubal cases, there was also a dermoid cyst in one of the ovaries.
Tumors in the testis and extragonadal sites resembling dermoid cysts have been discussed earlier as type I GCT beyond infancy, with a developmental potential in between that of type I and type IV GCT, probably explained by the arrest of PGC in the prophase of meiosis I at all anatomical sites, except within the seminiferous tubule [58, 59]. As discussed above, GCT with an intermediate phenotype between type I and IV occur also in the ovary.
3.8.4 (Cyto)Genetics
In the study by Surti et al. [668], 93 % of dermoid cysts were diploid, and the remaining 7 % had chromosomal abnormalities including trisomy for chromosomes 7, 8, 12, 15, and X; one case was tetraploid in mosaic form; there were no recurring structural aberrations. Trisomy for chromosomes 8, 12, and X is shared with immature teratomas [668]. Somatic-type malignancies developing in dermoid cysts have the same genetic profiles as the adjacent dermoid cysts, confirming their origin in the teratoma [669]. In general, the somatic-type malignancies have the same genetic changes as their somatic counterparts. For example, malignant struma ovarii has the same BRAF point mutations as papillary carcinomas of the thyroid [670, 671].
Most significantly, the genomic profile and pattern of imprinting of dermoid cysts reflects the stage of meiosis of the oocytes from which they are derived, as will be discussed in the following paragraphs.
3.8.5 Epigenetics Including GI
The process of erasure of biallelic imprinting in the germ lineage and the establishment of the maternal imprint during oogenesis has bearing on the pathogenesis of type IV GCT, the dermoid cysts of the ovary [16, 86]. Pronuclear transplantation experiments [3–5] have demonstrated that gynogenotes, with two haploid sets of maternally imprinted chromosomes, have a relatively good development of the embryo proper but very poor development of trophoblast, particularly the placenta. In contrast, androgenotes with two haploid sets of paternally imprinted chromosomes have a relatively normal development of the placenta but a very poor development of embryonic tissues (Fig. 3.31). The strong preference of the mouse gynogenote for developing somatic tissues is mirrored by the dermoid cyst, which is typically only composed of highly organized mature somatic tissues. Like in the gynogenote, the chromosomes of dermoid cysts lack paternal imprinting but share the maternal imprinting pattern of the oocytes from which they are derived [672]. The further the stage of meiosis of the oocyte, the better the maternal imprinting pattern is established in the derived dermoid cyst, suggesting that GI is a progressive process throughout oogenesis [672].

Compare development of control embryo (a) with that obtained from eggs with two maternal genomes (b) in which a small but well-advanced 25-somite embryo was the maximum development but with poor extraembryonic tissues. The eggs with two paternal nuclei developed maximally to about the 6- to 8-somite stage but with extensive trophoblast development (c). YS yolk sac, TB trophoblast. Scale bar, 1 mm [5]
3.8.6 Pathogenesis and Animal Models
Ever since Linder [673] discovered the parthenogenetic origin of dermoid cysts by looking at allelic loss of isozymes, this mechanism has been reinvestigated with state-of-the-art technology, including study of chromosomal polymorphic markers and various DNA polymorphisms [668, 672, 674–680]. For various reasons, such as inadequate sample preparation, selective growth of host fibroblasts in tissue culture, and a poor noise to signal ratio in the applied assays, contaminating host cells have been interpreted as tumor cells along with genuine neoplastic cells. Therefore, the genomic profile of premeiotic oogonia and host cells being the same, a proportion of dermoid cysts have been misclassified as derived from premeiotic oogonia [657], e.g., in 12 % by Ohama [680] and 25 % by Deka et al. [681]. Kaku et al. [657] have addressed this problem by careful sample preparation and quantitative measurement of the signals from tumor cells and contaminating host cells, enabling them to demonstrate allelic conversions (from hetero- to homozygous and vice versa) even in the presence of somatic cell contamination. They found allelic conversion in all samples taken from Rokitansky’s protuberance; therefore, none of the 64 analyzed dermoid cysts could have been derived from a premeiotic oogonium, 33 stemmed from primary oocytes, 16 from secondary oocytes, and 15 from ova (with endoreduplication) (Fig. 3.32). Apart from the absence of oogonium-derived dermoid cysts, the distribution over the stages of oogenesis was similar as in previous studies [668, 678–680]. The equivalence of numbers of dermoid cysts derived from primary oocytes, secondary oocytes, and ova is unexpected, considering that nearly 300,000 primary oocytes exist through adolescence [682] and only about 20 primary follicles begin to form secondary follicles during the menstrual cycle, in which subsequently secondary oocytes and ova develop. In view of these numbers, one would expect that almost all teratomas would be derived from primary oocytes. This obviously not being the case, Kaku et al. [657] propose that dermoid cysts are not derived from functional oocytes but from primary oocytes that have escaped meiotic arrest and start uncontrolled meiosis. This could explain the lack of premeiotic teratomas and the almost even distribution over the later stages of oogenesis (Fig. 3.33). As meiotic arrest is hormonally regulated [683], this mechanism also clarifies why dermoid cysts occur during reproductive age [233]. Moreover, it is conceivable that the assumed genetic factor underlying proneness for bilaterality and familial occurrence of type IV GCT is also involved in the regulation of meiotic arrest. Thus, it seems probable that, as in other GCT, the origin of type IV GCT is mainly determined by developmental factors, with a minor role for genetic events.

Graphical representation of the origins of mature ovarian cystic teratomas and the relevance of meiotic division. The origins of mature cystic teratomas are conceptually classified into five types. Type I teratomas result from a meiosis I error, in which the segregation of sister chromatids occurs without a preceding mono-oriented separation of bivalent chromosomes, generating biparental diploid cells with homologous DNA recombination. Type II teratomas result from meiosis II errors, in which the nondisjunction of all sister chromatids gives rise to diploid cells with homologous DNA recombination. Type III teratomas occur via endoreduplication of a haploid ovum, which is entirely mono-allelic. Type IV teratomas arise from oogonia. The constitution of chromosomes from type IV teratomas is identical to those from somatic cells. Type V teratomas involve a fusion of two normal haploid ova [657]

The postulated mechanism of human ovarian teratoma formation. The source cells are proposed to be primary oocytes that escaped from meiotic arrest. Subsequent uncontrolled meiotic division could produce ovarian teratomas. PGC primordial germ cell [657]
The rare but well-established phenomenon of ovarian teratomas developing as a fetiform structure (homunculus) [684–686] attests to the close to omnipotent developmental potential (2C state) of some of the precursor cells of ovarian mature teratomas, except for the ability to form trophoblastic tissue. In fact, the principal difference with a type 0 GCT, a parasitic or included twin, is the absence of extraembryonic structures, in agreement with the absence of a paternal imprint. It is conceivable that homunculi develop from precursor cells with the most complete maternal imprinting, closely resembling a zygote. The usual dermoid cysts, mainly composed of tissues from the rostral part of the embryo, with the skin turned inside, might develop from precursor cells with incomplete maternal imprinting. These observations on type IV GCT lead to the speculation that the spatial-temporal organization of embryonic development is somehow related to the progression of maternal imprinting and that a complete maternal imprint is required for developing the entire embryo proper.
At the other end of the spectrum are the poorly developed dermoid cysts, prone to rupture and combined with immature teratoma [130], in the gray zone between type I and type IV GCT, which are probably derived from the least mature primary oocytes next to premeiotic oogonia. The latter cells are the precursors of type I immature teratomas of the ovary as discussed.
3.9 Type V GCT
3.9.1 Developmental Potential
Complete hydatidiform moles, type V GCT, consist of placental tissue only, lacking somatic tissues of the embryo proper. A comprehensive discussion of these abnormal growths of the placenta is beyond the scope of this chapter (for review [687–689]). They are briefly mentioned here to show that complete hydatidiform moles are in the opposite side of the spectrum of developmental potential from dermoid cysts, which are composed of somatic tissues and lack trophoblastic tissue.
Grossly, a complete hydatidiform mole resembles a bunch of grapes, whereby the individual grapes represent enlarged placental villi covered by hyperplastic trophoblast with cyto-nuclear atypia and thus to be considered as dysplastic. The increased size is due to accumulation of fluid, probably caused by defective vasculogenesis and apoptotic degeneration of villous stromal components [690, 691]. Rarely, molar tissue metastasizes to the vagina or lungs. In agreement with its dysplastic nature, it may progress to choriocarcinoma in 2–3 % of cases [692]. Malignant transformation is possibly driven by hypomethylation-associated genomic instability [693], as trophoblast and placenta are hypomethylated compared to somatic tissues (for review) [694]. More specifically, promoter hypermethylation of p16 alone or combined with E-cadherin is associated with progression of hydatidiform mole to choriocarcinoma [695]. In fact, 50 % of all gestational choriocarcinomas originate from complete hydatidiform moles [696].
3.9.2 Epidemiology/Risk Factors
The incidence of gestational trophoblastic disease, mostly complete hydatidiform moles, is 1 in 120 pregnancies in some parts of Asia and South America, more than tenfold higher than in Western societies. Risk factors are pregnancy at young or old age, prior gestational trophoblastic disease, Asian ethnicity, and possibly dietary deficiencies and low socioeconomic status (for review [689]).
A pathogenetically informative genetic risk factor is the presence of maternal mutations of NALP7/NLRP7 on 19q13.4; the protein NALP is a member of the CATERPILLER protein family involved in inflammation and apoptosis. The mutation causes abnormal imprinting with overexpression of the paternal genome, resulting in recurrent familial biparental complete hydatidiform moles and reproductive wastage [697].
3.9.3 Anatomical Distribution
Complete hydatidiform moles develop where pregnancies occur, virtually always in the uterus and occasionally in the fallopian tube as an ectopic pregnancy [698].
3.9.4 (Cyto)Genetics/Epigenetics Including GI
Complete hydatidiform moles are generally diploid with a 46,XX (90 %) or 46,XY (10 %) karyotype [699, 700]; rare cases are tetraploid with four haploid sets of paternal chromosomes [701]. As a consequence of this chromosomal constitution, the genome of these lesions has an exclusively paternal GI, as will be further discussed in the following section.
3.9.5 Pathogenesis and Animal Models
Complete hydatidiform moles are caused by so-called androgenesis with two haploid sets of chromosomes from the father and none from the mother [702]. 46,XX complete hydatidiform moles arise from fertilization of an anuclear empty ovum by one 23,X sperm that replicates its chromosomes; when a 23,Y sperm is involved, this event results in a nonviable zygote. Complete hydatidiform moles with a 46,XY karyotype are the result of fertilization of an empty ovum by two sperm, respectively, with a Y and an X chromosome. Both mechanisms create a zygote with an exclusively paternal imprint, which gives rise to placental tissue only and no somatic tissues of the embryo proper, similar to experimentally produced mouse androgenotes [3, 4].
A partial mole constitutes an intermediate phenotype between a complete mole and a normal pregnancy; it arises when a normal ovum is fertilized by two sperm (69,XXY in 70 %; 69,XXX in 27 %; 69,XYY in 3 %) [703, 704]. Partial moles have enlarged hydropic placental villi in addition to normal villi combined with some development of tissues of the embryo proper [705].
The partial mole and maternal mutations of NALP7/NLRP7 demonstrate the critical importance of the dose of paternally and maternally imprinted genes. Apparently, an overdose of paternally imprinted genes favors placental and severely impairs embryonic development. The apoptosis of stromal cells and the defective vessels (both derived from the embryo proper) observed in very early complete hydatidiform moles [691] and the poorly developed fetal tissues in a partial mole suggest that somatic tissues may be formed but degenerate in an embryo with an exclusively or predominantly paternally imprinted genome.
The precursor cell of the hydatidiform mole has the 2C-state developmental potential, except for the ability to form/maintain somatic tissues of the embryo proper.
3.10 Type VI GCT
3.10.1 Definition
Type VI GCT are defined as neoplasms derived from mature somatic cells or committed stem cells, which resemble GCT as to their developmental potential.
The observation that genetically engineered iPSC may form tumors with the developmental potential of GCT is experimental support for this concept.
3.10.2 Developmental Potential of Genetically Engineered iPSC
The first human iPSC derived from somatic cells [70] and NSC [72] when grafted in mice reportedly produced teratomas with mature somatic derivatives from the three germ layers. In the meantime, it has become clear that iPSC also may give rise to other tumor types such as EC, immature teratoma (often primitive neuroectodermal tissues), YST, and somatic-type malignancies. The composition of the tumors depends on the induced cell type (with varying numbers of somatic mutations) and the genes combined in the transducing vectors (vectors including MYC carry a significant risk of developing malignant GCT) (for review [706, 707, 321, 625]). Each of the consecutive steps in the procedure of induction of pluripotency may contribute to carcinogenesis: integration of gene delivery vectors and transgenes into genomes of the host cells; chromosomal damage during reprogramming; clonal selection for transformed colonies during iPSC expansion; incomplete reprogramming; failure to silence pluripotent networks in differentiated progeny; DNA damage accumulated during cell culture, so-called culture adaptation; and aberrant regulation of the imprinting process [321]. Culture adaptation involves gain of (parts of) chromosomes in particular the chromosomes 12 (12p) [708, 709], 17 (17q) [708–710], 20 (20q11.21) [710–713], and X [708, 714]. In fact, the gains are remarkably similar to those seen in type II GCT [303].
Notably, premature termination of reprogramming in vivo was shown to cause the development of a pediatric cancer (Wilms’ tumor) through altered epigenetic regulation [715]. Also the recipient tissue for the graft is an important factor: human ESC transplanted into mice gave teratomas; however, when transplanted into human fetal tissue grafts in mice, they gave rise to pure EC [716]. The developmental potential of human iPSC with an intact genome is similar to that of human ESC, matching with the primed state of mouse ESC derived from the primitive ectoderm [706] and thus with type I GCT. Genetic aberrations and epigenetic modifications acquired in the derivation of iPSC may result in a higher capacity of self-renewal of the stem cells and thus the development of tumors containing EC or consisting of pure EC [321, 706, 717]. The developmental potential of these iPSC acquires features of the naïve state with totipotent developmental potential corresponding with human erased PGC, mouse ESC derived from the ICM, and the non-seminomatous variants of type II GCT. Seminoma, pure or as part of a mixed tumor, has not been reported. In fact, human iPSC often have a developmental potential somewhere in between that of type I GCT and type II non-seminomas and may be accompanied with somatic-type malignancies, such as can occur in both types of GCT. MYC with its central role in core pluripotency networks, involving NANOG, OCT4, and SOX2, and at the same time being an oncogene [321] is probably crucial for both the change in developmental potential of the iPSC and the causation of somatic malignancies.
3.10.3 Developmental Potential of Spontaneous Type VI GCT
It is well established that in humans there are somatic malignant tumors, apparently not derived from germ cell precursors, in which GCT components develop. Examples are sinonasal teratocarcinosarcomas (for review [220, 718]), cancer of the stomach [719], urothelial cancer [720], and endometrioid adenocarcinomas [721]. GCT arising in association with endometriosis and epithelial cancers of the ovary are comprehensively discussed in Chap. 6. So is gliomatosis peritonei, a rare condition often associated with immature teratoma of the ovary, characterized by the presence of mature glial tissue in the peritoneum, considered implants from the teratoma [661, 722]. It has been suggested that gliomatosis might in rare cases develop directly from peritoneal cells, in particular when associated with endometriosis [661, 723]. Possibly in support of a broad developmental capacity of mesothelial cells, ovarian surface epithelium, scraped from the ovary of postmenopausal women, reportedly expressed early embryonic developmental markers such as stage-specific embryonic antigen-4 (SSEA-4), OCT4, NANOG, and SOX2. When grown in culture, they were claimed to form oocyte-like cells, expressing markers of oocytes, as well as blastocyst-like structures expressing OCT4, SOX2, and NANOG; however when grafted in SCID mice, the ultimate test for pluripotency, no teratomas were formed [724, 725].
There are scattered reports in the literature of highly malignant GCT [726] sometimes in combination with somatic malignancies [727] that for various reasons do not readily fit into the types 0 to V of GCT. They lack the treatment sensitivity of GCT, occur usually at a much higher age than GCT, or at sites that are not compatible with parasitic twinning or mismigration of PGC, such as the foot [728] and the upper arm [729].
The developmental potential of these tumors is not unambiguously that of a type I or II GCT and resembles the potential of tumors produced upon grafting of iPSC. GCT arising in ovarian cancer, for example, may contain EC cells (expressing OCT4 and SOX2 and inconsistently CD30) in addition to polyembryoma, somatic lineages, YST, and choriocarcinoma [730] (Chap. 6). Indeed, strongly suggesting that these GCT components result from induction of pluripotency in somatic (cancer) cells, as will be discussed hereafter.
3.10.4 Epidemiology/Risk Factors
There are no epidemiological studies, as type VI GCT is emerging; the numbers of cases are small, and the patient material is heterogeneous. What the patients have in common is their high age (median 50–60), much higher than usual for GCT, apart from type III, spermatocytic tumor. This is true for the GCT associated with ovarian cancer (Chap. 6), the sinonasal teratocarcinosarcomas [718], and the GCT described by Van Echten et al. [726] and Noguera et al. [727].
3.10.5 Anatomical Distribution
The anatomical distribution is in accordance with the various types of cancer in which development of GCT components occurs, like cancer of the ovary and stomach as mentioned. The sinonasal teratocarcinosarcomas are virtually always located in the nasal cavity and/or ethmoid sinus with occasionally extension into the maxillary sinus or orbit [220]. The location of the tumors described by Van Echten et al. and Noguera et al. was atypical for primary GCT, certainly considering the old age of the patients and included retroperitoneum, posterior mediastinum, and inside the sacrum.
Scotting and colleagues [139, 168] have proposed that GCT of the brain are derived from NSC, induced to pluripotency by activation of OCT4, like the iPSC produced by Kim et al. [72]. However, in terms of developmental potential, epidemiology, anatomical localization, and (cyto)genetics, they fit into the overall pattern of type I and II GCT. Moreover mis-migrated PGC, the most likely precursor cells of these tumors, have been demonstrated in the brain of human embryos [52]. In fact, there are no convincing arguments to assume another cell of origin for GCT of the brain, than the current hypothesis that they are derived from PGC.
3.10.6 (Cyto)Genetics
Little is known on the genetics of GCT originated in somatic cancers. There was no gain of 12p in three sinonasal teratocarcinosarcomas [731]. Thomas et al. [732] demonstrated by ISH an extra copy of 12p13 (a feature of type I GCT) in a subpopulation of cells in a nasal teratocarcinosarcoma.
The three atypical GCT described by Van Echten et al. [726] and Noguera et al. [727] were karyotyped and showed complex balanced translocations, with 6p21 being a common breakpoint in each of them; chromosome 12 was not involved in these cases. The two cases described by Van Echten shared two chromosomal fusions: 6p21::11q13 and 6p22::6q23. Despite considerable efforts, the breakpoints were never fully characterized at the molecular level [733]. The nasal immature teratoma described by Houri et al. [221] was diploid with a balanced translocation t(1;11)(q12;p15).
3.10.7 Epigenetics Including GI
There are no specific data on the epigenetics of type VI GCT; however, in view of the often high age of the patients, it may be assumed that gradual loss of DNA methylation may have resulted in aberrant gene activation [734]. This applies even more to advanced cancers, where epigenetic changes may disrupt the stem cell program [735]. Moreover, mis-regulation of imprinted genes, so-called loss of imprinting, is a frequent and early phenomenon in a large variety of human tumors [736]. In particular, the imprinting of H19 and IGF-II is often lost, e.g., in colorectal cancer [737] and in the normal mucosa of the affected individuals [738] due to hypomethylation. The same genes are also frequently hypomethylated in epithelial cancers of the ovary [739], suggesting that development of a GCT component in epithelial cancers of the ovary might be due to derepression of pluripotency genes, such as OCT4 and SOX2, as has been demonstrated immunohistochemically in these tumors (see Chap. 6) (Fig. 3.34).

Histology of GCT originated in clear cell carcinoma of ovary showing EC and teratoma (a, c) and extraembryonic tissue surrounding an embryoid structure (e); EC cells expressing OCT4 (b, d, and f) (From Nogales and Schuldt Chap. 6)
3.10.8 Pathogenesis and Animal Models
For GCT developing in somatic cancers, various combinations of genetic and epigenetic changes could result in activation of repressed pluripotency genes. This could be a random event; however, it could also specifically target cancer cells with particular mutations or stem cell characteristics. The latter mechanism might apply to nasal teratocarcinosarcoma suggested to originate from transformed stem cells of the sinonasal mucosa [732]. Similarly, the low efficiency of induction of pluripotency in normal somatic cells has been explained as either due to slow stochastic accumulation of events in random cells or due to the targeting of rare (“elite”) cells, probably stem cells [707]. MYC could play a crucial role, since it is a central player in oncogenesis and pluripotency; indeed, more aggressive cancers express both the core pluripotency genes (OCT4, NANOG, SOX2, and KLF4) and MYC-centered networks [740, 741].
The few cytogenetically characterized atypical GCT [221, 726, 727] suggest that breakpoints in certain chromosomal regions might activate the pluripotency program. Most conspicuously, the breakpoint in 6p21–22 in these tumors could involve OCT4 [36]. A breakpoint in 11p15, possibly involving IGF2, was demonstrated in a nasal immature teratoma [221]. Of note, 6p22 was also involved in an atypical teratoid/rhabdoid tumor, a pediatric cancer of the brain. In this tumor, a breakpoint was found in 11p15 as well, likely involving IGF2 implicated in various childhood cancers (Wilms’ tumor, hepatoblastoma, and rhabdomyosarcoma) [742–744]. The overrepresentation of 12p13 in a sinonasal teratocarcinosarcoma [732] might lead to overexpression of the pluripotency cluster NANOG, STELLAR, and GDF3 [38, 745].
The localization of these tumors and the overlap of their chromosomal rearrangements with those of pediatric cancers suggest that committed stem cells in which pluripotency genes are activated due to chromosomal aberrations could be the originating cells.
In terms of morphology and cytogenetics, there is also an overlap with bona fide type I GCT raising the question whether the atypical GCT described here as type VI GCT could be exceptional manifestations of type I GCT. In two sacral type I teratomas where balanced translocations, t(12;15)(q13;q25), were demonstrated, these were constitutional [154, 155]. These are cases relevant for this discussion because they demonstrate that balanced translocations per se can very probably give origin to neoplasms with GCT morphology. The translocation t(8;22)(p21;q12) in an intrathoracic mature teratoma, described by Jin et al. [156], concerned a girl aged 15 and could thus be best considered a type I GCT beyond infancy. It cannot be excluded that balanced translocations are more frequent in these GCT than thus far documented, because they are only detected with dedicated approaches. Further research into this category of tumors will clarify what is now a gray zone between type I GCT beyond infancy and the atypical GCT in the category of type VI GCT, in whose pathogenesis balanced chromosomal translocations seem to play an important role.
Summarizing, type VI GCT are neoplasms that share morphological features with GCT but do not originate from germ cell precursors. They may develop from somatic cells, most often in aggressive cancers, in which by various epigenetic and genetic changes, among others translocations, pluripotency is induced. They come in three variants: GCT as part of somatic neoplasia, de novo by induction of pluripotency in nonneoplastic somatic cells, and most likely in the future as a complication of therapeutic application of human iPSC.
3.11 Integrated View and Summary
3.11.1 GCT, States of Developmental Potential, and Precursor Cells Matched
Type 0 GCT, parasitic and included twins, approach the developmental potential of the zygote and, therefore, must be derived from omnipotent precursor cells in the 2C state, probably blastomeres or ESC similar to mouse ESC from the ICM that happened to be in the 2C state. Familial clustering with twinning supports the hypothesis that these growths are derived from blastomeres that have escaped the organizing influence of the developing embryo or rather the molecular mechanisms that check the omnipotency of these cells.
Type I GCT consist of teratomas with somatic tissues at various levels of maturation representing the three germ layers. YST only develops by way of tumor progression in aneuploid cells that have lost their ability to contribute to somatic lineages of the embryo proper. Tumor stem cells are rarely encountered in these tumors: only in immature components occasional OCT4-positive cells are found, which do not express SOX2. OCT4 is probably driven from the distal enhancer. Germ cell differentiation has not been demonstrated. This developmental potential, the poor self-renewing capacity of the stem cells, which show reduced expression of pluripotency proteins and the germline incompetence of these tumors, is in accordance with a pluripotent precursor cell in the primed state, corresponding to mouse ESC derived from the primitive ectoderm.
In view of the anatomical distribution of the extragonadal type I GCT along the midline of the body, their most likely precursor is a migrating diploid PGC in an early, methylated, pre-erased stage, which has escaped apoptosis because it was reprogrammed to an EGC that acquired the primed state in accordance with its epigenetic status. Neoplastic growth starts during fetal development in keeping with clinical presentation of these tumors at birth or in early infancy, usually before age six. Type I GCT of the gonads, likewise, are derived from methylated, pre- or partially erased, diploid, premeiotic PGC via EGC reprogrammed to ESC in the primed state.
Type II GCT have the broadest developmental potential of human GCT comprising both seminomas, composed of neoplastic, hypomethylated (including both X chromosomes in females), partially to completely erased, premeiotic PGC/gonocytes, as well as non-seminomas, which are caricatures of early embryonic development. The latter develop when the developmental potential of a neoplastic gonocyte is unleashed by reprogramming to an EC cell, the totipotent stem cell of non-seminoma, which may give rise to YST and choriocarcinoma representing the extraembryonic tissues, and also somatic tissues from the three germ layers, from immature to fully mature, and occasionally early germ cell differentiation. EC cells have a high capacity of self-renewal and express many pluripotency markers, such as OCT4, SOX2, NANOG, and LIN28. OCT4 is likely expressed from the proximal enhancer. These characteristics are compatible with the developmental potential of the totipotent or naïve state corresponding to mouse ESC derived from the ICM and preimplantation epiblast.
The precursor cells are more mature PGC (hypomethylated, partially to completely erased, and premeiotic), which can only survive in suitable niches in the gonads, thymus, and midline of the brain. Outside these niches, such cells die apoptotically, explaining the absence of type II GCT at other anatomical sites. When in the gonads the niche functions properly, the gonocytes will differentiate into germ cells. This will not happen in the extragonadal niches, which are incapable of sustaining germ cell development beyond the prophase of meiosis I. A disturbed niche, whether gonadal or extragonadal, may result in delayed maturation of the gonocytes creating, when GBY is present, a window for co-expression of OCT4 and TSPY and accumulation of chromosomal rearrangements, particularly gain of 12p, which maintain the PGC/gonocyte phenotype and totipotent developmental potential of the precursor cells of type II GCT. The crucial role of GBY/TSPY explains the overwhelming male preponderance of type II GCT.
Type III GCT, so-called spermatocytic tumors, which occur only in the testis, have a developmental potential that is limited to postpubertal, premeiotic, spermatogenic cells: A-dark and A-pale spermatogonia, B spermatogonia, and leptotene spermatocytes. The most likely precursor cell is a postpubertal, paternally imprinted spermatogonial cell.
Type IV GCT, dermoid cysts, which occur only in the ovary, are composed of mature somatic tissues mainly from the rostral part of the embryo, often containing teeth; occasional solid variants may resemble a complete fetus; extraembryonic tissues are typically absent. This developmental potential is consistent with a parthenogenetically activated oocyte or ovum with an exclusively maternal genomic imprint as precursor cell that is incapable to support the development of extraembryonic tissues: the “maternal half” of the 2C state.
Type V GCT, hydatidiform moles, are hyperplastic, dysplastic growths composed of placental tissue only. The precursor cell is an empty zygote, fertilized by one sperm, followed by endoreduplication or by two sperm, resulting in a genome that has an exclusively paternal imprint, incapable of sustaining the development of somatic tissues of the embryo proper: the “paternal half” of the 2C state.
Type VI GCT are derived from spontaneous or genetically engineered iPSC that may form somatic tissues with varying degrees of maturation, EC, YST, and occasionally choriocarcinoma. The stem cells of tumors derived from spontaneously induced somatic cells in humans resemble those of type I GCT, with reduced expression of pluripotency genes and limited self-renewal capacity, as in the primed state. Human somatic cells induced to pluripotency in vitro, when assayed in the proper context, may contain large amounts of EC, up to 100 %, in particular if MYC was included in the inducing cocktail. The stem cells of these tumors share characteristics with the naïve-state stem cells of type II GCT; however, PGC or seminoma-like components have never been reported.
3.11.2 Intermediate Phenotypes
Consistent with the plasticity of the developmental states of embryonic stem cells, there are, between the different defined types of GCT, intermediate phenotypes, which will be briefly summarized here.
There is continuum between multiple pregnancies, conjoined twins, parasitic twins, and type I GCT, with intermediate types between type 0 and type I GCT, which could arbitrarily be classified in either type.
Type I GCT and type II GCT have gradual transitions, in particular among prepubertal GCT of the mediastinum in Klinefelter’s and among GCT of the brain in patients with Down’s syndrome, with tumors that are genotypically type II but phenotypically resemble type I, suggesting that the genomic changes typical for type II, particularly gain of 12p, have occurred in a PGC that is still too heavily methylated to allow the full spectrum of the naïve-state developmental potential of a type II GCT.
Type I and type IV GCT may cluster in the same families with an increased risk of bilaterality and multiplicity. The occurrence of tumors composed of immature teratoma with dermoid cysts embedded in the immature teratoma component likely represent a transition form between type I and type IV GCT. It may be hypothesized that such tumors are derived from a precursor cell somewhere in between an oogonium and a type I oocyte, in which maternal GI is not yet completed.
There is at least one published case of a spermatocytic tumor [634] that is intermediate between a type II and a type III GCT, in terms of morphology, chromosomal composition, and behavior: a seminomatous morphology, lacking lymphocytes, gain of 12p and chromosome 9, and metastasis.
The partial mole has an intermediate phenotype between a complete mole (Type V GCT) and a normal pregnancy due to an overdose of paternally imprinted genes in the presence of maternal imprinting.
Type VI tumors have developmental characteristics with features of type I and type II GCT.
Conclusion
GCT should be related to a developmental state rather than a specific cell of origin, as a particular originating cell may assume different developmental states, and different cell types may have the same developmental potential in agreement with the plasticity of developmental states. PGC are a good example: they give rise to type I and type II GCT, respectively from the primed/pluripotent and the naïve/totipotent developmental state, as apparent from the increased risk in Klinefelter’s and Down’s syndrome for both type I and II GCT. As yet, it cannot be excluded that some type I GCT originate from ESC in the primed state. It would not make any difference as to the developmental potential of the tumor. This principle justifies the inclusion of neoplasms, such as those derived from iPSC, in the classification of GCT, even when they are not derived from germ cells, because they share the developmental state of genuine GCT.
The intermediate phenotypes between some of the defined types of GCT attest to the plasticity of the different developmental states from which they had their origin.
References
Slack JM. Origin of stem cells in organogenesis. Science. 2008;322(5907):1498–1501. doi: 322/5907/1498 [pii] 10.1126/science.1162782.
Wylie C. Germ cells. Cell. 1999;96(2):165–174. doi:S0092-8674(00)80557-7 [pii].
Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308(5959):548–50.
McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37(1):179–183. doi:0092-8674(84)90313-1 [pii].
Surani MA, Barton SC, Norris ML. Nuclear transplantation in the mouse: heritable differences between parental genomes after activation of the embryonic genome. Cell. 1986;45(1):127–136. doi:0092-8674(86)90544-1 [pii].
Gkountela S, Li Z, Vincent JJ, Zhang KX, Chen A, Pellegrini M, et al. The ontogeny of cKIT+ human primordial germ cells proves to be a resource for human germ line reprogramming, imprint erasure and in vitro differentiation. Nat Cell Biol. 2013;15(1):113–122. doi:ncb2638 [pii] 10.1038/ncb2638.
Kim S, Gunesdogan U, Zylicz JJ, Hackett JA, Cougot D, Bao S, et al. PRMT5 protects genomic integrity during global DNA demethylation in primordial germ cells and preimplantation embryos. Mol Cell. 2014;56(4):564–579. doi:S1097-2765(14)00787-4 [pii] 10.1016/j.molcel.2014.10.003.
Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117(1–2):15–23. doi:S0925477302001818 [pii].
Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329(5987):78–82. doi:329/5987/78 [pii] 10.1126/science.1187945.
Morison IM, Reeve AE. A catalogue of imprinted genes and parent-of-origin effects in humans and animals. Hum Mol Genet. 1998;7(10):1599–1609. doi:ddb178 [pii].
Surani MA. Reprogramming of genome function through epigenetic inheritance. Nature. 2001;414(6859):122–128. doi:10.1038/35102186 35102186 [pii].
McLaren A. Primordial germ cells in the mouse. Dev Biol. 2003;262(1):1–15. doi:S0012160603002148 [pii].
Durcova-Hills G, Burgoyne P, McLaren A. Analysis of sex differences in EGC imprinting. Dev Biol. 2004;268(1):105–110. doi:10.1016/j.ydbio.2003.12.018 S0012160604000144 [pii].
Morison IM, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet. 2005;21(8):457–465. doi:S0168-9525(05)00166-6 [pii] 10.1016/j.tig.2005.06.008.
Diplas AI, Lambertini L, Lee MJ, Sperling R, Lee YL, Wetmur J, et al. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics. 2009;4(4):235–240. doi:9019 [pii].
Hall JG. Genomic imprinting: review and relevance to human diseases. Am J Hum Genet. 1990;46(5):857–73.
Surani A, Tischler J. Stem cells: a sporadic super state. Nature. 2012;487(7405):43–45. doi:487043a [pii] 10.1038/487043a.
McGrath J, Solter D. Inability of mouse blastomere nuclei transferred to enucleated zygotes to support development in vitro. Science. 1984;226(4680):1317–9.
Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487(7405):57–63. doi:nature11244 [pii] 10.1038/nature11244.
Hackett JA, Surani MA. Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell. 2014;15(4):416–430. doi:S1934-5909(14)00406-8 [pii] 10.1016/j.stem.2014.09.015.
Scholer HR, Balling R, Hatzopoulos AK, Suzuki N, Gruss P. Octamer binding proteins confer transcriptional activity in early mouse embryogenesis. EMBO J. 1989;8(9):2551–7.
Scholer HR, Hatzopoulos AK, Balling R, Suzuki N, Gruss P. A family of octamer-specific proteins present during mouse embryogenesis: evidence for germline-specific expression of an Oct factor. EMBO J. 1989;8(9):2543–50.
Ovitt CE, Scholer HR. The molecular biology of Oct-4 in the early mouse embryo. Mol Hum Reprod. 1998;4(11):1021–31.
Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95(3):379–391. doi:S0092-8674(00)81769-9 [pii].
Wu G, Han D, Gong Y, Sebastiano V, Gentile L, Singhal N, et al. Establishment of totipotency does not depend on Oct4A. Nat Cell Biol. 2013;15(9):1089–1097. doi:ncb2816 [pii] 10.1038/ncb2816.
Yeom YI, Fuhrmann G, Ovitt CE, Brehm A, Ohbo K, Gross M, et al. Germline regulatory element of Oct-4 specific for the totipotent cycle of embryonal cells. Development. 1996;122(3):881–94.
Pesce M, Wang X, Wolgemuth DJ, Scholer H. Differential expression of the Oct-4 transcription factor during mouse germ cell differentiation. Mech Dev. 1998;71(1–2):89–98.
Niakan KK, Eggan K. Analysis of human embryos from zygote to blastocyst reveals distinct gene expression patterns relative to the mouse. Dev Biol. 2013;375(1):54–64. doi:S0012-1606(12)00674-4 [pii] 10.1016/j.ydbio.2012.12.008.
Honecker F, Stoop H, de Krijger RR, Chris Lau YF, Bokemeyer C, Looijenga LH. Pathobiological implications of the expression of markers of testicular carcinoma in situ by fetal germ cells. J Pathol. 2004;203(3):849–57. doi:10.1002/path.1587.
Rajpert-De Meyts E, Hanstein R, Jorgensen N, Graem N, Vogt PH, Skakkebaek NE. Developmental expression of POU5F1 (OCT-3/4) in normal and dysgenetic human gonads. Hum Reprod. 2004;19(6):1338–1344. doi:10.1093/humrep/deh265 deh265 [pii].
Stoop H, Honecker F, Cools M, de Krijger R, Bokemeyer C, Looijenga LH. Differentiation and development of human female germ cells during prenatal gonadogenesis: an immunohistochemical study. Hum Reprod. 2005;20(6):1466–1476. doi:deh800 [pii] 10.1093/humrep/deh800.
Looijenga LH, Stoop H, de Leeuw HP, de Gouveia Brazao CA, Gillis AJ, van Roozendaal KE, et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003;63(9):2244–50.
Stewart CL, Gadi I, Bhatt H. Stem cells from primordial germ cells can reenter the germ line. Dev Biol. 1994;161(2):626–628. doi:10.1006/dbio.1994.1058 S0012-1606(84)71058-X [pii].
Aksoy I, Jauch R, Chen J, Dyla M, Divakar U, Bogu GK, et al. Oct4 switches partnering from Sox2 to Sox17 to reinterpret the enhancer code and specify endoderm. EMBO J. 2013;32(7):938–953. doi:emboj201331 [pii] 10.1038/emboj.2013.31.
Irie N, Weinberger L, Tang WW, Kobayashi T, Viukov S, Manor YS, et al. SOX17 is a critical specifier of human primordial germ cell fate. Cell. 2015;160(1–2):253–268. doi:S0092-8674(14)01583-9 [pii] 10.1016/j.cell.2014.12.013.
Crouau-Roy B, Amadou C, Bouissou C, Clayton J, Vernet C, Ribouchon MT, et al. Localization of the OTF3 gene within the human MHC class I region by physical and meiotic mapping. Genomics. 1994;21(1):241–243. doi:S0888-7543(84)71249-3 [pii] 10.1006/geno.1994.1249.
Schepers GE, Teasdale RD, Koopman P. Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell. 2002;3(2):167–170. doi:S153458070200223X [pii].
Clark AT, Rodriguez RT, Bodnar MS, Abeyta MJ, Cedars MI, Turek PJ, et al. Human STELLAR, NANOG, and GDF3 genes are expressed in pluripotent cells and map to chromosome 12p13, a hotspot for teratocarcinoma. Stem Cells. 2004;22(2):169–79. doi:10.1634/stemcells.22-2-169.
Le Bin GC, Munoz-Descalzo S, Kurowski A, Leitch H, Lou X, Mansfield W, et al. Oct4 is required for lineage priming in the developing inner cell mass of the mouse blastocyst. Development. 2014;141(5):1001–1010. doi:dev.096875 [pii] 10.1242/dev.096875.
Wu FR, Zhang Y, Ding B, Lei XH, Huang JC, Wang CH, et al. H3K27me3 may be associated with Oct4 and Sox2 in mouse preimplantation embryos. Genet Mol Res. 2014;13(4):10121–10129. doi:gmr4191 [pii] 10.4238/2014.December.4.6.
Toyooka Y, Oka S, Fujimori T. Early preimplantation cells expressing Cdx2 exhibit plasticity of specification to TE and ICM lineages through positional changes. Dev Biol. 2016;411(1):50–60. doi:S0012-1606(16)30026-4 [pii] 10.1016/j.ydbio.2016.01.011.
Goolam M, Scialdone A, Graham SJ, Macaulay IC, Jedrusik A, Hupalowska A, et al. Heterogeneity in Oct4 and Sox2 Targets Biases Cell Fate in 4-Cell Mouse Embryos. Cell. 2016;165(1):61–74. doi:S0092-8674(16)30061-7 [pii] 10.1016/j.cell.2016.01.047.
Niakan KK, Ji H, Maehr R, Vokes SA, Rodolfa KT, Sherwood RI, et al. Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self-renewal. Genes Dev. 2010;24(3):312–326. doi:24/3/312 [pii] 10.1101/gad.1833510.
Seguin CA, Draper JS, Nagy A, Rossant J. Establishment of endoderm progenitors by SOX transcription factor expression in human embryonic stem cells. Cell Stem Cell. 2008;3(2):182–195. doi:S1934-5909(08)00327-5 [pii] 10.1016/j.stem.2008.06.018.
Weidgang CE, Russell R, Tata PR, Kuhl SJ, Illing A, Muller M, et al. TBX3 Directs Cell-Fate Decision toward Mesendoderm. Stem Cell Reports. 2013;1(3):248–265. doi:10.1016/j.stemcr.2013.08.002 S2213-6711(13)00068-4 [pii].
Saitou M, Barton SC, Surani MA. A molecular programme for the specification of germ cell fate in mice. Nature. 2002;418(6895):293–300. doi:10.1038/nature00927 nature00927 [pii].
Hogan B. Developmental biology: decisions, decisions! Nature. 2002;418(6895):282–283. doi:10.1038/418282a 418282a [pii].
Matsui Y, Mochizuki K. A current view of the epigenome in mouse primordial germ cells. Mol Reprod Dev. 2014;81(2):160–70. doi:10.1002/mrd.22214.
Ly L, Chan D, Trasler JM. Developmental windows of susceptibility for epigenetic inheritance through the male germline. Semin Cell Dev Biol. 2015;43:96–105. doi:S1084-9521(15)00133-0 [pii] 10.1016/j.semcdb.2015.07.006.
Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339(6118):448–452. doi:science.1229277 [pii] 10.1126/science.1229277.
Freeman B. The active migration of germ cells in the embryos of mice and men is a myth. Reproduction. 2003;125(5):635–43.
Mamsen LS, Brochner CB, Byskov AG, Mollgard K. The migration and loss of human primordial germ stem cells from the hind gut epithelium towards the gonadal ridge. Int J Dev Biol. 2012;56(10–12):771–778. doi:120202lm [pii] 10.1387/ijdb.120202lm.
Mollgard K, Jespersen A, Lutterodt MC, Yding Andersen C, Hoyer PE, Byskov AG. Human primordial germ cells migrate along nerve fibers and Schwann cells from the dorsal hind gut mesentery to the gonadal ridge. Mol Hum Reprod. 2010;16(9):621–631. doi:gaq052 [pii] 10.1093/molehr/gaq052.
Teilum G. Special tumors of ovary and testis and related extragonadal lesions: comparative pathology and histological identification. Copenhagen: Munksgaard; 1976.
Gonzalez-Crussi F. Extragonadal teratomas. atlas of tumor pathology, Second Series. Armed Forces Institute of Pathology: Washington, D.C; 1982.
Oosterhuis JW, Stoop H, Honecker F, Looijenga LH. Why human extragonadal germ cell tumours occur in the midline of the body: old concepts, new perspectives. Int J Androl. 2007;30(4):256–63; discussion 263–4. doi:IJA793 [pii] 10.1111/j.1365-2605.2007.00793.x.
Runyan C, Gu Y, Shoemaker A, Looijenga L, Wylie C. The distribution and behavior of extragonadal primordial germ cells in Bax mutant mice suggest a novel origin for sacrococcygeal germ cell tumors. Int J Dev Biol. 2008;52(4):333–344. doi:072486cr [pii] 10.1387/ijdb.072486cr.
Upadhyay S, Zamboni L. Ectopic germ cells: natural model for the study of germ cell sexual differentiation. Proc Natl Acad Sci U S A. 1982;79(21):6584–8.
McLaren A. Germ cells and germ cell sex. Philos Trans R Soc Lond Ser B Biol Sci. 1995;350(1333):229–33. doi:10.1098/rstb.1995.0156.
Matsui Y, Zsebo K, Hogan BL. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70(5):841–847. doi:0092-8674(92)90317-6 [pii].
Donovan PJ, de Miguel MP. Turning germ cells into stem cells. Curr Opin Genet Dev. 2003;13(5):463–471. doi:S0959437X03001217 [pii].
Hackett JA, Zylicz JJ, Surani MA. Parallel mechanisms of epigenetic reprogramming in the germline. Trends Genet. 2012;28(4):164–174. doi:S0168-9525(12)00014-5 [pii] 10.1016/j.tig.2012.01.005.
Sterneckert J, Hoing S, Scholer HR. Concise review: Oct4 and more: the reprogramming expressway. Stem Cells. 2012;30(1):15–21. doi:10.1002/stem.765.
Irie N, Tang WW, Azim Surani M. Germ cell specification and pluripotency in mammals: a perspective from early embryogenesis. Reprod Med Biol. 2014;13(4):203–215. doi:10.1007/s12522-014-0184-2 184 [pii].
Ferrari F, Apostolou E, Park PJ, Hochedlinger K. Rearranging the chromatin for pluripotency. Cell Cycle. 2014;13(2):167–168. doi:27028 [pii] 10.4161/cc.27028.
Magnusdottir E, Surani MA. How to make a primordial germ cell. Development. 2014;141(2):245–252. doi:141/2/245 [pii] 10.1242/dev.098269.
Festuccia N, Osorno R, Halbritter F, Karwacki-Neisius V, Navarro P, Colby D, et al. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell. 2012;11(4):477–490. doi:S1934-5909(12)00480-8 [pii] 10.1016/j.stem.2012.08.002.
Kojima Y, Kaufman-Francis K, Studdert JB, Steiner KA, Power MD, Loebel DA, et al. The transcriptional and functional properties of mouse epiblast stem cells resemble the anterior primitive streak. Cell Stem Cell. 2014;14(1):107–120. doi:S1934-5909(13)00414-1 [pii] 10.1016/j.stem.2013.09.014.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi:S0092-8674(06)00976-7 [pii] 10.1016/j.cell.2006.07.024.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi:S0092-8674(07)01471-7 [pii] 10.1016/j.cell.2007.11.019.
Kim JB, Sebastiano V, Wu G, Arauzo-Bravo MJ, Sasse P, Gentile L, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136(3):411–419. doi:S0092-8674(09)00071-3 [pii] 10.1016/j.cell.2009.01.023.
Kim JB, Greber B, Arauzo-Bravo MJ, Meyer J, Park KI, Zaehres H, et al. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009;461(7264):649–643. doi:nature08436 [pii] 10.1038/nature08436.
Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298(5593):597–600. doi:10.1126/science.1072530 1072530 [pii].
Miyoshi N, Ishii H, Nagano H, Haraguchi N, Dewi DL, Kano Y, et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell. 2011;8(6):633–8. doi:10.1016/j.stem.2011.05.001.
Hiura H, Toyoda M, Okae H, Sakurai M, Miyauchi N, Sato A, et al. Stability of genomic imprinting in human induced pluripotent stem cells. BMC Genet. 2013;14:32. doi:10.1186/1471-2156-14-32.
Stang A, Trabert B, Wentzensen N, Cook MB, Rusner C, Oosterhuis JW, et al. Gonadal and extragonadal germ cell tumours in the United States, 1973–2007. Int J Androl. 2012;35(4):616–25. doi:10.1111/j.1365-2605.2011.01245.x.
De Backer A. Surgical treatment and outcome of extracranial germ cell tumors in childhood [Doctoral Thesis]. Rotterdam: Erasmus University Medical Center; 2006, ISBN 90-8559-166-X.
Ozgur T, Atik E, Silfeler DB, Toprak S. Mature cystic teratomas in our series with review of the literature and retrospective analysis. Arch Gynecol Obstet. 2012;285(4):1099–101. doi:10.1007/s00404-011-2171-8.
Znaor A, Lortet-Tieulent J, Laversanne M, Jemal A, Bray F. International testicular cancer incidence trends: generational transitions in 38 countries 1900–1990. Cancer Causes Control. 2015;26(1):151–8. doi:10.1007/s10552-014-0486-z.
Stevens LC, Little CC. Spontaneous testicular teratomas in an inbred strain of mice. Proc Natl Acad Sci U S A. 1954;40(11):1080–7.
Stevens LC. A new inbred subline of mice (129-terSv) with a high incidence of spontaneous congenital testicular teratomas. J Natl Cancer Inst. 1973;50(1):235–42.
Slye M. Primary spontaneous tumors of the ovary in mice. Studies on the incidence and heritability of spontaneous tumors in mice. J Cancer Res. 1920;5:205–26.
Phillips RJ, Hulse EV. Two extragonadal teratomas in a mouse, with discussion of possible histogenesis. J Comp Pathol. 1982;92(2):273–284. doi:0021-9975(82)90086-X [pii].
Kehler J, Tolkunova E, Koschorz B, Pesce M, Gentile L, Boiani M, et al. Oct4 is required for primordial germ cell survival. EMBO Rep. 2004;5(11):1078–1083. doi:7400279 [pii] 10.1038/sj.embor.7400279.
Yamaguchi S, Kurimoto K, Yabuta Y, Sasaki H, Nakatsuji N, Saitou M, et al. Conditional knockdown of Nanog induces apoptotic cell death in mouse migrating primordial germ cells. Development. 2009;136(23):4011–4020. doi:136/23/4011 [pii] 10.1242/dev.041160.
Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5(3):210–222. doi:nrc1568 [pii] 10.1038/nrc1568.
Rijlaarsdam MA, Tax DM, Gillis AJ, Dorssers LC, Koestler DC, de Ridder J, et al. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS One. 2015;10(4):e0122146. doi:10.1371/journal.pone.0122146 PONE-D-14-41158 [pii].
Grant P, Pearn JH. Foetus-in-foetu. Med J Aust. 1969;1(20):1016–9.
Willis RA. The borderland of embryology and pathology. Bull N Y Acad Med. 1950;26(7):440–60.
Griscom NT. The Roentgenology of Neonatal Abdominal Masses. Am J Roentgenol Radium Therapy, Nucl Med. 1965;93:447–63.
Du Plessis JP, Winship WS, Kirstein JD. Fetus in fetu and teratoma. A case report and review. S Afr Med J. 1974;48(50):2119–22.
Hopkins KL, Dickson PK, Ball TI, Ricketts RR, O’Shea PA, Abramowsky CR. Fetus-in-fetu with malignant recurrence. J Pediatr Surg. 1997;32(10):1476–1479. doi:S0022-3468(97)90567-4 [pii].
Khadaroo RG, Evans MG, Honore LH, Bhargava R, Phillipos E. Fetus-in-fetu presenting as cystic meconium peritonitis: diagnosis, pathology, and surgical management. J Pediatr Surg. 2000;35(5):721–723. doi:S0022-3468(00)48690-2 [pii] 10.1053/jpsu.2000.6037.
Isaacs H, Jr. Perinatal (fetal and neonatal) germ cell tumors. J Pediatr Surg. 2004;39(7):1003–1013. doi:S002234680400199X [pii].
Hoeffel CC, Nguyen KQ, Phan HT, Truong NH, Nguyen TS, Tran TT, et al. Fetus in fetu: a case report and literature review. Pediatrics. 2000;105(6):1335–44.
Spencer R. Parasitic conjoined twins: external, internal (fetuses in fetu and teratomas), and detached (acardiacs). Clin Anat. 2001;14(6):428–444. doi:10.1002/ca.1079 [pii] 10.1002/ca.1079.
Brand A, Alves MC, Saraiva C, Loio P, Goulao J, Malta J, et al. Fetus in fetu – diagnostic criteria and differential diagnosis – a case report and literature review. J Pediatr Surg. 2004;39(4):616–618. doi:S002234680300959X [pii].
de Lagausie P, de Napoli Cocci S, Stempfle N, Truong QD, Vuillard E, Ferkadji L, et al. Highly differentiated teratoma and fetus-in-fetu: a single pathology? J Pediatr Surg. 1997;32(1):115–116. doi:S0022-3468(97)90112-3 [pii].
Heifetz SA, Alrabeeah A, Brown BS, Lau H. Fetus in fetu: a fetiform teratoma. Pediatr Pathol. 1988;8(2):215–26.
Plattner G, Oxorn H. Familial incidence of ovarian dermoid cysts. Can Med Assoc J. 1973;108(7):892–3.
Feld D, Labes J, Nathanson M. Bilateral ovarian dermoid cysts in triplets. Obstet Gynecol. 1966;27(4):525–8.
Beutel K, Partsch CJ, Janig U, Nikischin W, Suttorp M. Oral mature teratoma containing epididymal tissue in a female neonate. Lancet. 2001;357(9252):283–284. doi:S0140-6736(00)03620-5 [pii] 10.1016/S0140-6736(00)03620-5.
Dehner LP. Gonadal and extragonadal germ cell neoplasia of childhood. Hum Pathol. 1983;14(6):493–511.
Tapper D, Lack EE. Teratomas in infancy and childhood. A 54-year experience at the Children’s Hospital Medical Center. Ann Surg. 1983;198(3):398–410.
Hawkins EP. Pathology of germ cell tumors in children. Crit Rev Oncol Hematol. 1990;10(2):165–179. doi:1040-8428(90)90005-D [pii].
Heifetz SA, Cushing B, Giller R, Shuster JJ, Stolar CJ, Vinocur CD, et al. Immature teratomas in children: pathologic considerations: a report from the combined Pediatric Oncology Group/Children’s Cancer Group. Am J Surg Pathol. 1998;22(9):1115–24.
Schneider DT, Calaminus G, Koch S, Teske C, Schmidt P, Haas RJ, et al. Epidemiologic analysis of 1,442 children and adolescents registered in the German germ cell tumor protocols. Pediatr Blood Cancer. 2004;42(2):169–75. doi:10.1002/pbc.10321.
McKenney JK, Heerema-McKenney A, Rouse RV. Extragonadal germ cell tumors: a review with emphasis on pathologic features, clinical prognostic variables, and differential diagnostic considerations. Adv Anat Pathol. 2007;14(2):69–92. doi:10.1097/PAP.0b013e31803240e6 00125480-200703000-00002 [pii].
De Backer A, Madern GC, Pieters R, Haentjens P, Hakvoort-Cammel FG, Oosterhuis JW, et al. Influence of tumor site and histology on long-term survival in 193 children with extracranial germ cell tumors. Eur J Pediatr Surg. 2008;18(1):1–6. doi:10.1055/s-2007-989399.
Hawkins E, Issacs H, Cushing B, Rogers P. Occult malignancy in neonatal sacrococcygeal teratomas. A report from a Combined Pediatric Oncology Group and Children’s Cancer Group study. Am J Pediatr Hematol Oncol. 1993;15(4):406–9.
Baker BA, Frickey L, Yu IT, Hawkins EP, Cushing B, Perlman EJ. DNA content of ovarian immature teratomas and malignant germ cell tumors. Gynecol Oncol. 1998;71(1):14–18. doi:S0090-8258(98)95102-2 [pii] 10.1006/gyno.1998.5102.
Rescorla FJ, Sawin RS, Coran AG, Dillon PW, Azizkhan RG. Long-term outcome for infants and children with sacrococcygeal teratoma: a report from the Childrens Cancer Group. J Pediatr Surg. 1998;33(2):171–176. doi:S0022-3468(98)90426-2 [pii].
Heerema-McKenney A, Harrison MR, Bratton B, Farrell J, Zaloudek C. Congenital teratoma: a clinicopathologic study of 22 fetal and neonatal tumors. Am J Surg Pathol. 2005;29(1):29–38. doi:00000478-200501000-00004 [pii].
Frazier AL, Weldon C, Amatruda J. Fetal and neonatal germ cell tumors. Semin Fetal Neonatal Med. 2012;17(4):222–230. doi:S1744-165X(12)00059-5 [pii] 10.1016/j.siny.2012.05.004.
Isaacs H, Jr. I. Perinatal brain tumors: a review of 250 cases. Pediatr Neurol. 2002;27(4):249–261. doi:S0887899402004721 [pii].
Blohm ME, Gobel U. Unexplained anaemia and failure to thrive as initial symptoms of infantile choriocarcinoma: a review. Eur J Pediatr. 2004;163(1):1–6. doi:10.1007/s00431-003-1361-1.
McNally OM, Tran M, Fortune D, Quinn MA. Successful treatment of mother and baby with metastatic choriocarcinoma. Int J Gynecol Cancer. 2002;12(4):394–398. doi:ijg01125 [pii].
Ngan KW, Jung SM, Lee LY, Chuang WY, Yeh CJ, Hsieh YY. Immunohistochemical expression of OCT4 in primary central nervous system germ cell tumours. J Clin Neurosci. 2008;15(2):149–152. doi:S0967-5868(07)00017-3 [pii] 10.1016/j.jocn.2006.08.013.
Abiko K, Mandai M, Hamanishi J, Matsumura N, Baba T, Horiuchi A, et al. Oct4 expression in immature teratoma of the ovary: relevance to histologic grade and degree of differentiation. Am J Surg Pathol. 2010;34(12):1842–1848. doi:10.1097/PAS.0b013e3181fcd707 00000478-201012000-00012 [pii].
Oosterhuis JW, Stoop JA, Rijlaarsdam MA, Biermann K, Smit VT, Hersmus R, et al. Pediatric germ cell tumors presenting beyond childhood? Andrology. 2015;3(1):70–7. doi:10.1111/andr.305.
Herszfeld D, Wolvetang E, Langton-Bunker E, Chung TL, Filipczyk AA, Houssami S, et al. CD30 is a survival factor and a biomarker for transformed human pluripotent stem cells. Nat Biotechnol. 2006;24(3):351–357. doi:nbt1197 [pii] 10.1038/nbt1197.
Chang MC, Vargas SO, Hornick JL, Hirsch MS, Crum CP, Nucci MR. Embryonic stem cell transcription factors and D2-40 (podoplanin) as diagnostic immunohistochemical markers in ovarian germ cell tumors. Int J Gynecol Pathol. 2009;28(4):347–55. doi:10.1097/PGP.0b013e318195da86.
Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet. 2008;9(2):129–140. doi: nrg2295 [pii] 10.1038/nrg2295.
Yokomizo S, Tsujimura A, Nonomura N, Kirime S, Takahara S, Okuyama A. Metachronous bilateral testicular tumors in a child. J Urol. 2001;166(6):2341. doi:S0022-5347(05)65584-X [pii].
Abell MR, Holtz F. Testicular neoplasms in infants and children. I. Tumors of germ cell origin. Cancer. 1963;16:965–81.
Carney JA, Kelalis PP, Lynn HB. Bilateral teratoma of testis in an infant. J Pediatr Surg. 1973;8(1):49–54.
Gustavson KH, Gamstorp I, Meurling S. Bilateral teratoma of testis in two brothers with 47, XXY Klinefelter’s syndrome. Clin Genet. 1975;8(1):5–10.
Kurman RJ, Norris HJ. Endodermal sinus tumor of the ovary: a clinical and pathologic analysis of 71 cases. Cancer. 1976;38(6):2404–19.
Gershenson DM. Management of ovarian germ cell tumors. J Clin Oncol. 2007;25(20):2938–2943. doi:25/20/2938 [pii] 10.1200/JCO.2007.10.8738.
Yanai-Inbar I, Scully RE. Relation of ovarian dermoid cysts and immature teratomas: an analysis of 350 cases of immature teratoma and 10 cases of dermoid cyst with microscopic foci of immature tissue. Int J Gynecol Pathol. 1987;6(3):203–12.
Mahdi H, Kumar S, Seward S, Semaan A, Batchu R, Lockhart D, et al. Prognostic impact of laterality in malignant ovarian germ cell tumors. Int J Gynecol Cancer. 2011;21(2):257–262. doi:10.1097/IGC.0b013e31820581e5 00009577-201102000-00011 [pii].
Isaacs Jr H. Fetal intracranial teratoma. A review. Fetal Pediatr Pathol. 2014;33(5–6):289–92. doi:10.3109/15513815.2014.969558.
Anteby EY, Ron M, Revel A, Shimonovitz S, Ariel I, Hurwitz A. Germ cell tumors of the ovary arising after dermoid cyst resection: a long-term follow-up study. Obstet Gynecol. 1994;83(4):605–8.
Kim R, Bohm-Velez M. Familial ovarian dermoids. J Ultrasound Med. 1994;13(3):225–8.
Nezhat C, Kotikela S, Mann A, Hajhosseini B, Veeraswamy A, Lewis M. Familial cystic teratomas: four case reports and review of the literature. J Minim Invasive Gynecol. 2010;17(6):782–786. doi:S1553-4650(10)00333-X [pii] 10.1016/j.jmig.2010.06.006.
Poremba C, Dockhorn-Dworniczak B, Merritt V, Li CY, Heidl G, Tauber PF, et al. Immature teratomas of different origin carried by a pregnant mother and her fetus. Diagn Mol Pathol. 1993;2(2):131–6.
Giambartolomei C, Mueller CM, Greene MH, Korde LA. A mini-review of familial ovarian germ cell tumors: an additional manifestation of the familial testicular germ cell tumor syndrome. Cancer Epidemiol. 2009;33(1):31–36. doi:S1877-7821(09)00038-1 [pii] 10.1016/j.canep.2009.04.015.
Barksdale Jr EM, Obokhare I. Teratomas in infants and children. Curr Opin Pediatr. 2009;21(3):344–9. doi:10.1097/MOP.0b013e32832b41ee.
Tan C, Scotting PJ. Stem cell research points the way to the cell of origin for intracranial germ cell tumours. J Pathol. 2013;229(1):4–11. doi:10.1002/path.4098.
Perlman EJ, Valentine MB, Griffin CA, Look AT. Deletion of 1p36 in childhood endodermal sinus tumors by two-color fluorescence in situ hybridization: a pediatric oncology group study. Genes Chromosomes Cancer. 1996;16(1):15–20. doi:10.1002/(SICI)1098-2264(199605)16:1<15::AID-GCC2>3.0.CO;2-6 [pii] 10.1002/(SICI)1098-2264(199605)16:1<15::AID-GCC2>3.0.CO;2-6.
Bussey KJ, Lawce HJ, Olson SB, Arthur DC, Kalousek DK, Krailo M, et al. Chromosome abnormalities of eighty-one pediatric germ cell tumors: sex-, age-, site-, and histopathology-related differences – a Children’s Cancer Group study. Genes Chromosomes Cancer. 1999;25(2):134–146. doi:10.1002/(SICI)1098-2264(199906)25:2<134::AID-GCC9>3.0.CO;2-Y [pii].
Perlman EJ, Hu J, Ho D, Cushing B, Lauer S, Castleberry RP. Genetic analysis of childhood endodermal sinus tumors by comparative genomic hybridization. J Pediatr Hematol Oncol. 2000;22(2):100–5.
Mostert M, Rosenberg C, Stoop H, Schuyer M, Timmer A, Oosterhuis W, et al. Comparative genomic and in situ hybridization of germ cell tumors of the infantile testis. Lab Investig. 2000;80(7):1055–64.
Schneider DT, Schuster AE, Fritsch MK, Hu J, Olson T, Lauer S, et al. Multipoint imprinting analysis indicates a common precursor cell for gonadal and nongonadal pediatric germ cell tumors. Cancer Res. 2001;61(19):7268–76.
van Echten J, Timmer A, van der Veen AY, Molenaar WM, de Jong B. Infantile and adult testicular germ cell tumors. a different pathogenesis? Cancer Genet Cytogenet. 2002;135(1):57–62. doi:S0165460801006434 [pii].
Veltman IM, Schepens MT, Looijenga LH, Strong LC, van Kessel AG. Germ cell tumours in neonates and infants: a distinct subgroup? APMIS. 2003;111(1):152–160; discussion 60. doi:apm1110119 [pii].
Veltman I, Veltman J, Janssen I, Hulsbergen-van de Kaa C, Oosterhuis W, Schneider D, et al. Identification of recurrent chromosomal aberrations in germ cell tumors of neonates and infants using genomewide array-based comparative genomic hybridization. Genes Chromosomes Cancer. 2005;43(4):367–376. doi:10.1002/gcc.20208.
Zahn S, Sievers S, Alemazkour K, Orb S, Harms D, Schulz WA, et al. Imbalances of chromosome arm 1p in pediatric and adult germ cell tumors are caused by true allelic loss: a combined comparative genomic hybridization and microsatellite analysis. Genes Chromosomes Cancer. 2006;45(11):995–1006. doi:10.1002/gcc.20363.
Harms D, Zahn S, Gobel U, Schneider DT. Pathology and molecular biology of teratomas in childhood and adolescence. Klin Padiatr. 2006;218(6):296–302. doi:10.1055/s-2006-942271.
Palmer RD, Foster NA, Vowler SL, Roberts I, Thornton CM, Hale JP, et al. Malignant germ cell tumours of childhood: new associations of genomic imbalance. Br J Cancer. 2007;96(4):667–676. doi:6603602 [pii] 10.1038/sj.bjc.6603602.
Mosbech CH, Rechnitzer C, Brok JS, Rajpert-De Meyts E, Hoei-Hansen CE. Recent advances in understanding the etiology and pathogenesis of pediatric germ cell tumors. J Pediatr Hematol Oncol. 2014;36(4):263–70. doi:10.1097/MPH.0000000000000125.
Looijenga LH, Rosenberg C, van Gurp RJ, Geelen E, van Echten-Arends J, de Jong B, et al. Comparative genomic hybridization of microdissected samples from different stages in the development of a seminoma and a non-seminoma. J Pathol. 2000;191(2):187–192. doi:10.1002/(SICI)1096-9896(200006)191:2<187::AID-PATH584>3.0.CO;2-T [pii] 10.1002/(SICI)1096-9896(200006)191:2<187::AID-PATH584>3.0.CO;2-T.
Fujita T, Igarashi J, Okawa ER, Gotoh T, Manne J, Kolla V, et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008;100(13):940–949. doi:djn176 [pii] 10.1093/jnci/djn176.
Veltman I, van Asseldonk M, Schepens M, Stoop H, Looijenga L, Wouters C, et al. A novel case of infantile sacral teratoma and a constitutional t(12;15)(q13;q25) pat. Cancer Genet Cytogenet. 2002;136(1):17–22. doi:S0165460801006665 [pii].
Veltman IM, Vreede LA, Cheng J, Looijenga LH, Janssen B, Schoenmakers EF, et al. Fusion of the SUMO/Sentrin-specific protease 1 gene SENP1 and the embryonic polarity-related mesoderm development gene MESDC2 in a patient with an infantile teratoma and a constitutional t(12;15)(q13;q25). Hum Mol Genet. 2005;14(14):1955–1963. doi:ddi200 [pii] 10.1093/hmg/ddi200.
Jin Y, Mertens F, Kullendorff CM, Panagopoulos I. Fusion of the tumor-suppressor gene CHEK2 and the gene for the regulatory subunit B of protein phosphatase 2 PPP2R2A in childhood teratoma. Neoplasia. 2006;8(5):413–8. doi:10.1593/neo.06139.
Kiefer MC, Brauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, et al. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 1995;374(6524):736–9. doi:10.1038/374736a0.
Poynter JN, Hooten AJ, Frazier AL, Ross JA. Associations between variants in KITLG, SPRY4, BAK1, and DMRT1 and pediatric germ cell tumors. Genes Chromosomes Cancer. 2012;51(3):266–71. doi:10.1002/gcc.20951.
Fritsch MK, Schneider DT, Schuster AE, Murdoch FE, Perlman EJ. Activation of Wnt/beta-catenin signaling in distinct histologic subtypes of human germ cell tumors. Pediatr Dev Pathol. 2006;9(2):115–31. doi:10.2350/08-05-0097.1.
Fustino N, Rakheja D, Ateek CS, Neumann JC, Amatruda JF. Bone morphogenetic protein signalling activity distinguishes histological subsets of paediatric germ cell tumours. Int J Androl. 2011;34(4 Pt 2):e218–33. doi:10.1111/j.1365-2605.2011.01186.x.
Alagaratnam S, Lind GE, Kraggerud SM, Lothe RA, Skotheim RI. The testicular germ cell tumour transcriptome. Int J Androl. 2011;34(4 Pt 2):e133–e150; discussion e50–1. doi:10.1111/j.1365-2605.2011.01169.x.
Okpanyi V, Schneider DT, Zahn S, Sievers S, Calaminus G, Nicholson JC, et al. Analysis of the adenomatous polyposis coli (APC) gene in childhood and adolescent germ cell tumors. Pediatr Blood Cancer. 2011;56(3):384–91. doi:10.1002/pbc.22669.
Palmer RD, Murray MJ, Saini HK, van Dongen S, Abreu-Goodger C, Muralidhar B, et al. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 2010;70(7):2911–2923. doi:0008-5472.CAN-09-3301 [pii] 10.1158/0008-5472.CAN-09-3301.
Bussey KJ, Lawce HJ, Himoe E, Shu XO, Heerema NA, Perlman EJ, et al. SNRPN methylation patterns in germ cell tumors as a reflection of primordial germ cell development. Genes Chromosomes Cancer. 2001;32(4):342–352. doi:10.1002/gcc.1199 [pii].
Schneider DT, Schuster AE, Fritsch MK, Calaminus G, Gobel U, Harms D, et al. Genetic analysis of mediastinal nonseminomatous germ cell tumors in children and adolescents. Genes Chromosomes Cancer. 2002;34(1):115–125. doi:10.1002/gcc.10053 [pii].
Sievers S, Alemazkour K, Zahn S, Perlman EJ, Gillis AJ, Looijenga LH, et al. IGF2/H19 imprinting analysis of human germ cell tumors (GCTs) using the methylation-sensitive single-nucleotide primer extension method reflects the origin of GCTs in different stages of primordial germ cell development. Genes Chromosomes Cancer. 2005;44(3):256–64. doi:10.1002/gcc.20237.
Rijlaarsdam MA, Looijenga LH. An oncofetal and developmental perspective on testicular germ cell cancer. Semin Cancer Biol. 2014;29:59–74. doi:S1044-579X(14)00091-1 [pii] 10.1016/j.semcancer.2014.07.003.
Scotting PJ. Are cranial germ cell tumours really tumours of germ cells? Neuropathol Appl Neurobiol. 2006;32(6):569–574. doi:NAN797 [pii] 10.1111/j.1365-2990.2006.00797.x.
Lee SH, Appleby V, Jeyapalan JN, Palmer RD, Nicholson JC, Sottile V, et al. Variable methylation of the imprinted gene, SNRPN, supports a relationship between intracranial germ cell tumours and neural stem cells. J Neuro Oncol. 2011;101(3):419–28. doi:10.1007/s11060-010-0275-9.
Yang J, Cai J, Zhang Y, Wang X, Li W, Xu J, et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader-Willi syndrome. J Biol Chem. 2010;285(51):40303–40311. doi:M110.183392 [pii] 10.1074/jbc.M110.183392.
Stevens LC. Experimental production of testicular teratomas in mice. Proc Natl Acad Sci U S A. 1964;52:654–61.
Stevens LC, Varnum DS. The development of teratomas from parthenogenetically activated ovarian mouse eggs. Dev Biol. 1974;37(2):369–380. doi:0012-1606(74)90155-9 [pii].
Stevens LC. The development of transplantable teratocarcinomas from intratesticular grafts of pre- and postimplantation mouse embryos. Dev Biol. 1970;21(3):364–382. doi:0012-1606(70)90130-2 [pii].
Damjanov I, Solter D. Experimental teratoma. Curr Top Pathol. 1974;59:69–130.
Dunn TB, Andervont HB. Histology of some neoplasms and non-neoplastic lesions found in wild mice maintained under laboratory conditions. J Natl Cancer Inst. 1963;31:873–901.
Artzt K, Damjanov I. Spontaneous extragonadal teratocarcinoma in a mouse. Lab Anim Sci. 1978;28(5):584–6.
van Berlo RJ, Oosterhuis JW, Schrijnemakers E, Schoots CJ, de Jong B, Damjanov I. Yolk-sac carcinoma develops spontaneously as a late occurrence in slow-growing teratoid tumors produced from transplanted 7-day mouse embryos. Int J Cancer. 1990;45(1):153–5.
van Berlo RJ, de Jong B, Oosterhuis JW, Dijkhuizen T, Buist J, Dam A. Cytogenetic analysis of murine embryo-derived tumors. Cancer Res. 1990;50(11):3416–21.
Eakin GS, Behringer RR. Tetraploid development in the mouse. Dev Dyn. 2003;228(4):751–66. doi:10.1002/dvdy.10363.
Eakin GS, Hadjantonakis AK, Papaioannou VE, Behringer RR. Developmental potential and behavior of tetraploid cells in the mouse embryo. Dev Biol. 2005;288(1):150–159. doi:S0012-1606(05)00635-4 [pii] 10.1016/j.ydbio.2005.09.028.
Walt H, Oosterhuis JW, Stevens LC. Experimental testicular germ cell tumorigenesis in mouse strains with and without spontaneous tumours differs from development of germ cell tumours of the adult human testis. Int J Androl. 1993;16(4):267–71.
Gu KL, Zhang Q, Yan Y, Li TT, Duan FF, Hao J, et al. Pluripotency-associated miR-290/302 family of microRNAs promote the dismantling of naive pluripotency. Cell Res. 2016;26(3):350–366. doi:cr20162 [pii] 10.1038/cr.2016.2.
Stevens LC. Testicular teratomas in fetal mice. J Natl Cancer Inst. 1962;28:247–67.
Damjanov I, Solter D. Animal model of human disease: teratoma and teratocarcinoma. Am J Pathol. 1976;83(1):241–4.
Heaney JD, Lam MY, Michelson MV, Nadeau JH. Loss of the transmembrane but not the soluble kit ligand isoform increases testicular germ cell tumor susceptibility in mice. Cancer Res. 2008;68(13):5193–5197. doi:68/13/5193 [pii] 10.1158/0008-5472.CAN-08-0779.
Ohinata Y, Payer B, O’Carroll D, Ancelin K, Ono Y, Sano M, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436(7048):207–213. doi:nature03813 [pii] 10.1038/nature03813.
Magnusdottir E, Dietmann S, Murakami K, Gunesdogan U, Tang F, Bao S, et al. A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat Cell Biol. 2013;15(8):905–915. doi:ncb2798 [pii] 10.1038/ncb2798.
Nakaki F, Hayashi K, Ohta H, Kurimoto K, Yabuta Y, Saitou M. Induction of mouse germ-cell fate by transcription factors in vitro. Nature. 2013;501(7466):222–226. doi:nature12417 [pii] 10.1038/nature12417.
Krentz AD, Murphy MW, Kim S, Cook MS, Capel B, Zhu R, et al. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc Natl Acad Sci U S A. 2009;106(52):22323–22328. doi:0905431106 [pii] 10.1073/pnas.0905431106.
Youngren KK, Coveney D, Peng X, Bhattacharya C, Schmidt LS, Nickerson ML, et al. The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature. 2005;435(7040):360–364. doi:nature03595 [pii] 10.1038/nature03595.
Papaioannou VE, McBurney MW, Gardner RL, Evans MJ. Fate of teratocarcinoma cells injected into early mouse embryos. Nature. 1975;258(5530):70–3.
Yoshinaga K, Nishikawa S, Ogawa M, Hayashi S, Kunisada T, Fujimoto T, et al. Role of c-kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c-kit expression and function. Development. 1991;113(2):689–99.
Noguchi T, Noguchi M. A recessive mutation (ter) causing germ cell deficiency and a high incidence of congenital testicular teratomas in 129/Sv-ter mice. J Natl Cancer Inst. 1985;75(2):385–92.
Bhattacharya C, Aggarwal S, Zhu R, Kumar M, Zhao M, Meistrich ML, et al. The mouse dead-end gene isoform alpha is necessary for germ cell and embryonic viability. Biochem Biophys Res Commun. 2007;355(1):194–199. doi:S0006-291X(07)00186-6 [pii] 10.1016/j.bbrc.2007.01.138.
Rapley EA, Turnbull C, Al Olama AA, Dermitzakis ET, Linger R, Huddart RA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41(7):807–810. doi:ng.394 [pii] 10.1038/ng.394.
Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, Letrero R, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41(7):811–815. doi:ng.393 [pii] 10.1038/ng.393.
Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. 2014;511(7508):241–245. doi:nature13296 [pii] 10.1038/nature13296.
De Backer A, Madern GC, Hakvoort-Cammel FG, Haentjens P, Oosterhuis JW, Hazebroek FW. Study of the factors associated with recurrence in children with sacrococcygeal teratoma. J Pediatr Surg. 2006;41(1):173–181; discussion -81. doi:S0022-3468(05)00763-3 [pii] 10.1016/j.jpedsurg.2005.10.022.
Derikx JP, De Backer A, van de Schoot L, Aronson DC, de Langen ZJ, van den Hoonaard TL, et al. Factors associated with recurrence and metastasis in sacrococcygeal teratoma. Br J Surg. 2006;93(12):1543–8. doi:10.1002/bjs.5379.
Fraumeni Jr JF, Li FP, Dalager N. Teratomas in children: epidemiologic features. J Natl Cancer Inst. 1973;51(5):1425–30.
Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974;9(3):389–98.
Noseworthy J, Lack EE, Kozakewich HP, Vawter GF, Welch KJ. Sacrococcygeal germ cell tumors in childhood: an updated experience with 118 patients. J Pediatr Surg. 1981;16(3):358–364. doi:S0022346881000626 [pii].
Ward SP, Dehner LP. Sacrococcygeal teratoma with nephroblastoma (Wilm’s tumor): a variant of extragonadal teratoma in childhood. A histologic and ultrastructural study. Cancer. 1974;33(5):1355–63.
Biskup W, Calaminus G, Schneider DT, Leuschner I, Gobel U. Teratoma with malignant transformation: experiences of the cooperative GPOH protocols MAKEI 83/86/89/96. Klin Padiatr. 2006;218(6):303–8. doi:10.1055/s-2006-942272.
Miles RM, Stewart Jr GS. Sacrococcygeal teratomas in adult. Ann Surg. 1974;179(5):676–83.
Willis R. The borderland of embryology and pathology. 2 ed. London: Butterworths; 1962.
Oosterhuis JW, Van Berlo RJ, De Jong B, Dam A, Buist J, Tamminga R, et al. Sacral teratoma with late recurrence of yolk sac tumor: human counterpart of embryo or yolk sac derived teratoma? J Urol Pathol. 1993;1:257–67.
Chen YH, Chang CH, Chen KC, Diau GY, Loh IW, Chu CC. Malignant transformation of a well-organized sacrococcygeal fetiform teratoma in a newborn male. J Formos Med Assoc. 2007;106(5):400–402. doi:S0929-6646(09)60326-0 [pii] 10.1016/S0929-6646(09)60326-0.
Billmire DF, Grosfeld JL. Teratomas in childhood: analysis of 142 cases. J Pediatr Surg. 1986;21(6):548–551. doi:S0022346886002014 [pii].
De Backer A, Madern GC, Hazebroek FW. Retroperitoneal germ cell tumors: a clinical study of 12 patients. J Pediatr Surg. 2005;40(9):1475–1481. doi:S0022-3468(05)00424-0 [pii] 10.1016/j.jpedsurg.2005.05.048.
Daugaard G, von der Maase H, Olsen J, Rorth M, Skakkebaek NE. Carcinoma-in-situ testis in patients with assumed extragonadal germ-cell tumours. Lancet. 1987;2(8558):528–530. doi:S0140-6736(87)92922-9 [pii].
Hailemariam S, Engeler DS, Bannwart F, Amin MB. Primary mediastinal germ cell tumor with intratubular germ cell neoplasia of the testis – further support for germ cell origin of these tumors: a case report. Cancer. 1997;79(5):1031–1036. doi:10.1002/(SICI)1097-0142(19970301)79:5<1031::AID-CNCR21>3.0.CO;2-1 [pii].
Fossa SD, Aass N, Heilo A, Daugaard G, Skakkebaek NE, Stenwig AE, et al. Testicular carcinoma in situ in patients with extragonadal germ-cell tumours: the clinical role of pretreatment biopsy. Ann Oncol. 2003;14(9):1412–8.
De Backer A, Madern GC, Hakvoort-Cammel FG, Oosterhuis JW, Hazebroek FW. Mediastinal germ cell tumors: clinical aspects and outcomes in 7 children. Eur J Pediatr Surg. 2006;16(5):318–22. doi:10.1055/s-2006-924647.
Lack EE. Extragonadal germ cell tumors of the head and neck region: review of 16 cases. Hum Pathol. 1985;16(1):56–64.
Dehner LP, Mills A, Talerman A, Billman GF, Krous HF, Platz CE. Germ cell neoplasms of head and neck soft tissues: a pathologic spectrum of teratomatous and endodermal sinus tumors. Hum Pathol. 1990;21(3):309–18.
De Backer A, Madern GC, van de Ven CP, Tibboel D, Hazebroek FW. Strategy for management of newborns with cervical teratoma. J Perinat Med. 2004;32(6):500–8. doi:10.1515/JPM.2004.122.
Azizkhan RG, Haase GM, Applebaum H, Dillon PW, Coran AG, King PA, et al. Diagnosis, management, and outcome of cervicofacial teratomas in neonates: a Childrens Cancer Group study. J Pediatr Surg. 1995;30(2):312–316. doi:0022-3468(95)90580-4 [pii].
Kuhn JJ, Schoem SR, Warnock GR. Squamous cell carcinoma arising in a benign teratoma of the maxilla. Otolaryngol Head Neck Surg. 1996;114(3):447–452. doi:S019459989600109X [pii].
Heffner DK, Hyams VJ. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses A clinicopathologic study of 20 cases. Cancer. 1984;53(10):2140–54.
Houri T, Hashimoto N, Ibayashi N, Mori T, Fujimoto M, Ueda S, et al. Chromosomal translocation, t(1;11)(q12;p15), in an extragonadal immature teratoma. Cancer Genet Cytogenet. 1997;97(1):79–80. doi:S0165460897001969 [pii].
Pai SA, Naresh KN, Masih K, Ramarao C, Borges AM. Teratocarcinosarcoma of the paranasal sinuses: a clinicopathologic and immunohistochemical study. Hum Pathol. 1998;29(7):718–22.
De Backer A, Madern GC, Wolffenbuttel KP, Oosterhuis JW, Hakvoort-Cammel FG, Hazebroek FW. Testicular germ cell tumors in children: management and outcome in a series of 20 patients. J Pediatr Urol. 2006;2(3):197–201. doi:S1477-5131(05)00139-7 [pii] 10.1016/j.jpurol.2005.08.001.
Metcalfe PD, Farivar-Mohseni H, Farhat W, McLorie G, Khoury A, Bagli DJ. Pediatric testicular tumors: contemporary incidence and efficacy of testicular preserving surgery. J Urol. 2003;170(6 Pt 1):2412–2415; discussion 5–6. doi:10.1097/01.ju.0000097383.09743.f9.
Lee SD, Korean Society of Pediatric Urology. Epidemiological and clinical behavior of prepubertal testicular tumors in Korea. J Urol. 2004;172(2):674–678. doi:S0022-5347(05)61714-4 [pii] 10.1097/01.ju.0000129571.13955.6b.
Fan R, Zhang J, Cheng L, Lin J. Testicular and paratesticular pathology in the pediatric population: a 20 year experience at Riley hospital for children. Pathol Res Pract. 2013;209(7):404–408. doi:S0344-0338(13)00086-1 [pii] 10.1016/j.prp.2013.04.002.
Kato K, Ijiri R, Tanaka Y, Toyoda Y, Chiba K, Kitami K. Testicular immature teratoma with primitive neuroectodermal tumor in early childhood. J Urol. 2000;164(6):2068–2069. doi:S0022-5347(05)66968-6 [pii].
Manivel JC, Reinberg Y, Niehans GA, Fraley EE. Intratubular germ cell neoplasia in testicular teratomas and epidermoid cysts. Correlation with prognosis and possible biologic significance. Cancer. 1989;64(3):715–20.
Skakkebaek NE, Rajpert-De Meyts E, Main KM. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum Reprod. 2001;16(5):972–8.
Shu XO, Nesbit ME, Buckley JD, Krailo MD, Robinson LL. An exploratory analysis of risk factors for childhood malignant germ-cell tumors: report from the Childrens Cancer Group (Canada, United States). Cancer Causes Control. 1995;6(3):187–98.
Poynter JN, Radzom AH, Spector LG, Puumala S, Robison LL, Chen Z, et al. Family history of cancer and malignant germ cell tumors in children: a report from the Children’s Oncology Group. Cancer Causes Control. 2010;21(2):181–9. doi:10.1007/s10552-009-9448-2.
Gordon A, Lipton D, Woodruff JD. Dysgerminoma: a review of 158 cases from the Emil Novak Ovarian Tumor Registry. Obstet Gynecol. 1981;58(4):497–504.
Comerci Jr JT, Licciardi F, Bergh PA, Gregori C, Breen JL. Mature cystic teratoma: a clinicopathologic evaluation of 517 cases and review of the literature. Obstet Gynecol. 1994;84(1):22–8.
De Backer A, Madern GC, Oosterhuis JW, Hakvoort-Cammel FG, Hazebroek FW. Ovarian germ cell tumors in children: a clinical study of 66 patients. Pediatr Blood Cancer. 2006;46(4):459–64. doi:10.1002/pbc.20633.
Hartshorne GM, Lyrakou S, Hamoda H, Oloto E, Ghafari F. Oogenesis and cell death in human prenatal ovaries: what are the criteria for oocyte selection? Mol Hum Reprod. 2009;15(12):805–819. doi:gap055 [pii] 10.1093/molehr/gap055.
Anderson RA, Fulton N, Cowan G, Coutts S, Saunders PT. Conserved and divergent patterns of expression of DAZL, VASA and OCT4 in the germ cells of the human fetal ovary and testis. BMC Dev Biol. 2007;7:136. doi:1471-213X-7-136 [pii] 10.1186/1471-213X-7-136.
Byskov AG, Hoyer PE, Yding Andersen C, Kristensen SG, Jespersen A, Mollgard K. No evidence for the presence of oogonia in the human ovary after their final clearance during the first two years of life. Hum Reprod. 2011;26(8):2129–2139. doi:der145 [pii] 10.1093/humrep/der145.
Zhang C, Berney DM, Hirsch MS, Cheng L, Ulbright TM. Evidence supporting the existence of benign teratomas of the postpubertal testis: a clinical, histopathologic, and molecular genetic analysis of 25 cases. Am J Surg Pathol. 2013;37(6):827–35. doi:10.1097/PAS.0b013e31827dcc4c.
Semjen D, Kalman E, Tornoczky T, Szuhai K. Further evidence of the existence of benign teratomas of the postpubertal testis. Am J Surg Pathol. 2014;38(4):580–581. doi:10.1097/PAS.0000000000000186 00000478-201404000-00019 [pii].
Lewis BD, Hurt RD, Payne WS, Farrow GM, Knapp RH, Muhm JR. Benign teratomas of the mediastinum. J Thorac Cardiovasc Surg. 1983;86(5):727–31.
Williamson SR. Germ cell tumors of the mediastinum. In: Marchevsky AM, editor. Pathology of the mediastinum. Cambridge: Cambridge University Press; 2014. p. 146–68.
Stendel R, Pietila TA, Lehmann K, Kurth R, Suess O, Brock M. Ruptured intracranial dermoid cysts. Surg Neurol. 2002;57(6):391–398; discussion 8. doi:S0090301902007231 [pii].
Rosenblum MK. Germ Cell Tumours. In: Louis DN, editor. World Health Organization classification of tumours of the central nervous system. 4th ed. Lyon: IARC Press; 2007. p. 197–204.
Ulbright TM, Amin MB, Balzer B, Berney DM, Epstein JI, Guo C, et al. Germ cell tumours. In: Moch H, Humphrey PA, Ulbright TM, Reuter VE, editors. WHO classification of tumours of the urinary system and male genital organs. 4th ed. Lyon: IARC Press; 2016. p. 189–226.
Oosterhuis JW, Looijenga LH. The biology of human germ cell tumours: retrospective speculations and new prospectives. Eur Urol. 1993;23(1):245–50.
Kleinsmith LJ, Pierce Jr GB. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964;24:1544–51.
Honecker F, Stoop H, Mayer F, Bokemeyer C, Castrillon DH, Lau YF, et al. Germ cell lineage differentiation in non-seminomatous germ cell tumours. J Pathol. 2006;208(3):395–400. doi:10.1002/path.1872.
Looijenga LH, Gillis AJ, Stoop H, Hersmus R, Oosterhuis JW. Relevance of microRNAs in normal and malignant development, including human testicular germ cell tumours. Int J Androl. 2007;30(4):304–314; discussion 14–5. doi:IJA765 [pii] 10.1111/j.1365-2605.2007.00765.x.
Poynter JN, Bestrashniy JR, Silverstein KA, Hooten AJ, Lees C, Ross JA, et al. Cross platform analysis of methylation, miRNA and stem cell gene expression data in germ cell tumors highlights characteristic differences by tumor histology. BMC Cancer. 2015;15:769. doi:10.1186/s12885-015-1796-6 10.1186/s12885-015-1796-6 [pii].
Hoozemans DA, Schats R, Lambalk CB, Homburg R, Hompes PG. Human embryo implantation: current knowledge and clinical implications in assisted reproductive technology. Reprod BioMed Online. 2004;9(6):692–715.
Morgani SM, Canham MA, Nichols J, Sharov AA, Migueles RP, Ko MS, et al. Totipotent embryonic stem cells arise in ground-state culture conditions. Cell Rep. 2013;3(6):1945–1957. doi:S2211-1247(13)00216-7 [pii] 10.1016/j.celrep.2013.04.034.
Batata MA, Whitmore Jr WF, Hilaris BS, Tokita N, Grabstald H. Cancer of the undescended or maldescended testis. AJR Am J Roentgenol. 1976;126(2):302–12. doi:10.2214/ajr.126.2.302.
Batata MA, Chu FC, Hilaris BS, Whitmore WF, Golbey RB. Testicular cancer in cryptorchids. Cancer. 1982;49(5):1023–30.
Halme A, Kellokumpu-Lehtinen P, Lehtonen T, Teppo L. Morphology of testicular germ cell tumours in treated and untreated cryptorchidism. Br J Urol. 1989;64(1):78–83.
Nettersheim D, Jostes S, Sharma R, Schneider S, Hofmann A, Ferreira HJ, et al. BMP inhibition in seminomas initiates acquisition of pluripotency via NODAL signaling resulting in reprogramming to an embryonal carcinoma. PLoS Genet. 2015;11(7):e1005415. doi:10.1371/journal.pgen.1005415 PGENETICS-D-15-00545 [pii].
Spiller CM, Feng CW, Jackson A, Gillis AJ, Rolland AD, Looijenga LH, et al. Endogenous Nodal signaling regulates germ cell potency during mammalian testis development. Development. 2012;139(22):4123–32. doi:10.1242/dev.083006.
Spiller CM, Gillis AJ, Burnet G, Stoop H, Koopman P, Bowles J, et al. Cripto: Expression, epigenetic regulation and potential diagnostic use in testicular germ cell tumors. Mol Oncol. 2016;10(4):526–37. doi:10.1016/j.molonc.2015.11.003.
Bredael JJ, Vugrin D, Whitmore Jr WF. Autopsy findings in 154 patients with germ cell tumors of the testis. Cancer. 1982;50(3):548–51.
Zaloudek CJ, Tavassoli FA, Norris HJ. Dysgerminoma with syncytiotrophoblastic giant cells. A histologically and clinically distinctive subtype of dysgerminoma. Am J Surg Pathol. 1981;5(4):361–7.
Moreira A. Germ cell tumours of the mediastinum. In: Travis W, editor. World Health Organization classification of tumours of the lung, pleura, thymus and heart. 4th ed. Lyon: IARC Press; 2015. p. 244–66.
Pugh R. Testicular tumours – introduction. In: Pugh R, editor. Pathology of the testis. Oxford: Blackwell Scientific; 1976. p. 139–59.
Oosterhuis JW, Kersemaekers AM, Jacobsen GK, Timmer A, Steyerberg EW, Molier M, et al. Morphology of testicular parenchyma adjacent to germ cell tumours. An interim report. APMIS. 2003;111(1):32–40; discussion 1–2.
Matsutani M, Sano K, Takakura K, Fujimaki T, Nakamura O, Funata N, et al. Primary intracranial germ cell tumors: a clinical analysis of 153 histologically verified cases. J Neurosurg. 1997;86(3):446–55. doi:10.3171/jns.1997.86.3.0446.
Moran CA, Suster S. Primary germ cell tumors of the mediastinum: I. Analysis of 322 cases with special emphasis on teratomatous lesions and a proposal for histopathologic classification and clinical staging. Cancer. 1997;80(4):681–90.
Nichols CR, Saxman S, Williams SD, Loehrer PJ, Miller ME, Wright C, et al. Primary mediastinal nonseminomatous germ cell tumors. A modern single institution experience. Cancer. 1990;65(7):1641–6.
Vicus D, Beiner ME, Klachook S, Le LW, Laframboise S, Mackay H. Pure dysgerminoma of the ovary 35 years on: a single institutional experience. Gynecol Oncol. 2010;117(1):23–6. doi:10.1016/j.ygyno.2009.12.024.
Kurman RJ, Norris HJ. Embryonal carcinoma of the ovary: a clinicopathologic entity distinct from endodermal sinus tumor resembling embryonal carcinoma of the adult testis. Cancer. 1976;38(6):2420–33.
Oosterhuis JW, Peeters SH, Smit VT, Stoop H, Looijenga LH, Elzevier HW, et al. Patient with two secondary somatic-type malignancies in a late recurrence of a testicular non-seminoma: illustration of potential and flaw of the cancer stem cell therapy concept. Int J Dev Biol. 2013;57(2–4):153–7. doi:10.1387/ijdb.130141jo.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi:10.1016/j.cell.2011.02.013.
Oosterhuis JW. The metastasis of human teratomas. In: Damjanov I, Solter D, Knowles BB, editors. The human teratomas. Clifton: Humana Press; 1983. p. 137–71.
Oosterhuis JW, Suurmeyer AJ, Sleyfer DT, Koops HS, Oldhoff J, Fleuren G. Effects of multiple-drug chemotherapy (cis-diammine-dichloroplatinum, bleomycin, and vinblastine) on the maturation of retroperitoneal lymph node metastases of nonseminomatous germ cell tumors of the testis. No evidence for De Novo induction of differentiation. Cancer. 1983;51(3):408–16.
De Graaff WE, Oosterhuis JW, Van der Linden S, Homan van der Heide JN, Schraffordt Koops H, Sleijfer DTh. Residual mature teratoma after chemotherapy for non seminomatous germ cell tumors of the testis occurs significantly less often in lung, than in retroperitoneal lymph node metastases. J Urol Pathol. 1991;1:75–81.
Trabert B, Chen J, Devesa SS, Bray F, McGlynn KA. International patterns and trends in testicular cancer incidence, overall and by histologic subtype, 1973–2007. Andrology. 2015;3(1):4–12. doi:10.1111/andr.293.
Arora RS, Alston RD, Eden TO, Geraci M, Birch JM. Comparative incidence patterns and trends of gonadal and extragonadal germ cell tumors in England, 1979 to 2003. Cancer. 2012;118(17):4290–7. doi:10.1002/cncr.27403.
Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006;27(5):468–84. doi:10.1210/er.2006-0005.
Satge D, Sasco AJ, Cure H, Leduc B, Sommelet D, Vekemans MJ. An excess of testicular germ cell tumors in Down’s syndrome: three case reports and a review of the literature. Cancer. 1997;80(5):929–35.
Volkl TM, Langer T, Aigner T, Greess H, Beck JD, Rauch AM, et al. Klinefelter syndrome and mediastinal germ cell tumors. Am J Med Genet A. 2006;140(5):471–81. doi:10.1002/ajmg.a.31103.
Bray F, Richiardi L, Ekbom A, Pukkala E, Cuninkova M, Moller H. Trends in testicular cancer incidence and mortality in 22 European countries: continuing increases in incidence and declines in mortality. Int J Cancer. 2006;118(12):3099–111. doi:10.1002/ijc.21747.
Dieckmann KP, Kulejewski M, Pichlmeier U, Loy V. Diagnosis of contralateral testicular intraepithelial neoplasia (TIN) in patients with testicular germ cell cancer: systematic two-site biopsies are more sensitive than a single random biopsy. Eur Urol. 2007;51(1):175–183; discussion 83–5. doi:10.1016/j.eururo.2006.05.051.
Dieckmann KP, Anheuser P, Sattler F, Von Kugelgen T, Matthies C, Ruf C. Sequential bilateral testicular tumours presenting with intervals of 20 years and more. BMC Urol. 2013;13:71. doi:10.1186/1471-2490-13-71.
Thomas GM, Dembo AJ, Hacker NF, DePetrillo AD. Current therapy for dysgerminoma of the ovary. Obstet Gynecol. 1987;70(2):268–75.
De Palo G, Pilotti S, Kenda R, Ratti E, Musumeci R, Mangioni C, et al. Natural history of dysgerminoma. Am J Obstet Gynecol. 1982;143(7):799–807.
Coffey J, Linger R, Pugh J, Dudakia D, Sokal M, Easton DF, et al. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes Chromosomes Cancer. 2008;47(1):34–42. doi:10.1002/gcc.20503.
Heimdal K, Olsson H, Tretli S, Flodgren P, Borresen AL, Fossa SD. Familial testicular cancer in Norway and southern Sweden. Br J Cancer. 1996;73(7):964–9.
Czene K, Lichtenstein P, Hemminki K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int J Cancer. 2002;99(2):260–6. doi:10.1002/ijc.10332.
Hemminki K, Li X. Familial risk in testicular cancer as a clue to a heritable and environmental aetiology. Br J Cancer. 2004;90(9):1765–70. doi:10.1038/sj.bjc.6601714.
Akyuz C, Koseoglu V, Gogus S, Balci S, Buyukpamukcu M. Germ cell tumours in a brother and sister. Acta Paediatr. 1997;86(6):668–9.
Greene MH, Mai PL, Loud JT, Pathak A, Peters JA, Mirabello L, et al. Familial testicular germ cell tumors (FTGCT) – overview of a multidisciplinary etiologic study. Andrology. 2015;3(1):47–58. doi:10.1111/andr.294.
Hartmann JT, Fossa SD, Nichols CR, Droz JP, Horwich A, Gerl A, et al. Incidence of metachronous testicular cancer in patients with extragonadal germ cell tumors. J Natl Cancer Inst. 2001;93(22):1733–8.
Kersemaekers AM, Honecker F, Stoop H, Cools M, Molier M, Wolffenbuttel K, et al. Identification of germ cells at risk for neoplastic transformation in gonadoblastoma: an immunohistochemical study for OCT3/4 and TSPY. Hum Pathol. 2005;36(5):512–21. doi:10.1016/j.humpath.2005.02.016.
Cools M, van Aerde K, Kersemaekers AM, Boter M, Drop SL, Wolffenbuttel KP, et al. Morphological and immunohistochemical differences between gonadal maturation delay and early germ cell neoplasia in patients with undervirilization syndromes. J Clin Endocrinol Metab. 2005;90(9):5295–303. doi:10.1210/jc.2005-0139.
Stoop H, Honecker F, van de Geijn GJ, Gillis AJ, Cools MC, de Boer M, et al. Stem cell factor as a novel diagnostic marker for early malignant germ cells. J Pathol. 2008;216(1):43–54. doi:10.1002/path.2378.
Lin Y, Yang Y, Li W, Chen Q, Li J, Pan X, et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol Cell. 2012;48(4):627–40. doi:10.1016/j.molcel.2012.08.030.
Oosterhuis JW, Castedo SM, de Jong B, Cornelisse CJ, Dam A, Sleijfer DT, et al. Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab Investig. 1989;60(1):14–21.
Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun. 2015;6:5973. doi:10.1038/ncomms6973.
Kraggerud SM, Hoei-Hansen CE, Alagaratnam S, Skotheim RI, Abeler VM, Rajpert-De Meyts E, et al. Molecular characteristics of malignant ovarian germ cell tumors and comparison with testicular counterparts: implications for pathogenesis. Endocr Rev. 2013;34(3):339–76. doi:10.1210/er.2012-1045.
Oosterhuis JW, Rammeloo RH, Cornelisse CJ, De Jong B, Dam A, Sleijfer DT. Ploidy of malignant mediastinal germ-cell tumors. Hum Pathol. 1990;21(7):729–32.
Terashima K, Yu A, Chow WY, Hsu WC, Chen P, Wong S, et al. Genome-wide analysis of DNA copy number alterations and loss of heterozygosity in intracranial germ cell tumors. Pediatr Blood Cancer. 2014;61(4):593–600. doi:10.1002/pbc.24833.
Atkin NB, Baker MC. Specific chromosome change, i(12p), in testicular tumours? Lancet. 1982;2(8311):1349.
Gibas Z, Prout Jr GR, Sandberg AA. Malignant teratoma of the testis with an isochromosome no. 12, i(12p), as the sole structural cytogenetic abnormality. J Urol. 1984;131(4):762–3.
Castedo SM, de Jong B, Oosterhuis JW, Seruca R, Idenburg VJ, Dam A, et al. Chromosomal changes in human primary testicular nonseminomatous germ cell tumors. Cancer Res. 1989;49(20):5696–701.
Castedo SM, de Jong B, Oosterhuis JW, Seruca R, te Meerman GJ, Dam A, et al. Cytogenetic analysis of ten human seminomas. Cancer Res. 1989;49(2):439–43.
van Echten J, Oosterhuis JW, Looijenga LH, van de Pol M, Wiersema J, te Meerman GJ, et al. No recurrent structural abnormalities apart from i(12p) in primary germ cell tumors of the adult testis. Genes Chromosomes Cancer. 1995;14(2):133–44.
Looijenga LH, Zafarana G, Grygalewicz B, Summersgill B, Debiec-Rychter M, Veltman J, et al. Role of gain of 12p in germ cell tumour development. APMIS. 2003;111(1):161–171; discussion 72–3.
Speleman F, De Potter C, Dal Cin P, Mangelschots K, Ingelaere H, Laureys G, et al. i(12p) in a malignant ovarian tumor. Cancer Genet Cytogenet. 1990;45(1):49–53.
Hamers A, de Jong B, Suijkerbuijk RF, Geurts van Kessel A, Oosterhuis JW, van Echten J, et al. A 46,XY female with mixed gonadal dysgenesis and a 48,XY, +7, +i(12p) chromosome pattern in a primary gonadal tumor. Cancer Genet Cytogenet. 1991;57(2):219–24.
Dal Cin P, Drochmans A, Moerman P, Van den Berghe H. Isochromosome 12p in mediastinal germ cell tumor. Cancer Genet Cytogenet. 1989;42(2):243–51.
Sung MT, Maclennan GT, Lopez-Beltran A, Zhang S, Montironi R, Cheng L. Primary mediastinal seminoma: a comprehensive assessment integrated with histology, immunohistochemistry, and fluorescence in situ hybridization for chromosome 12p abnormalities in 23 cases. Am J Surg Pathol. 2008;32(1):146–55. doi:10.1097/PAS.0b013e3181379edf.
de Bruin TW, Slater RM, Defferrari R, Geurts van Kessel A, Suijkerbuijk RF, Jansen G, et al. Isochromosome 12p-positive pineal germ cell tumor. Cancer Res. 1994;54(6):1542–4.
Schneider DT, Zahn S, Sievers S, Alemazkour K, Reifenberger G, Wiestler OD, et al. Molecular genetic analysis of central nervous system germ cell tumors with comparative genomic hybridization. Mod Pathol Off J U S Can Acad Pathol Inc. 2006;19(6):864–73. doi:10.1038/modpathol.3800607.
Roelofs H, Mostert MC, Pompe K, Zafarana G, van Oorschot M, van Gurp RJ, et al. Restricted 12p amplification and RAS mutation in human germ cell tumors of the adult testis. Am J Pathol. 2000;157(4):1155–66. doi:10.1016/S0002-9440(10)64631-7.
Zafarana G, Gillis AJ, van Gurp RJ, Olsson PG, Elstrodt F, Stoop H, et al. Coamplification of DAD-R, SOX5, and EKI1 in human testicular seminomas, with specific overexpression of DAD-R, correlates with reduced levels of apoptosis and earlier clinical manifestation. Cancer Res. 2002;62(6):1822–31.
Rodriguez S, Jafer O, Goker H, Summersgill BM, Zafarana G, Gillis AJ, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2–p12.1. Oncogene. 2003;22(12):1880–91. doi:10.1038/sj.onc.1206302.
Zafarana G, Grygalewicz B, Gillis AJ, Vissers LE, van de Vliet W, van Gurp RJ, et al. 12p-amplicon structure analysis in testicular germ cell tumors of adolescents and adults by array CGH. Oncogene. 2003;22(48):7695–701. doi:10.1038/sj.onc.1207011.
Geurts van Kessel A, van Drunen E, de Jong B, Oosterhuis JW, Langeveld A, Mulder MP. Chromosome 12q heterozygosity is retained in i(12p)-positive testicular germ cell tumor cells. Cancer Genet Cytogenet. 1989;40(1):129–34.
Sinke RJ, Suijkerbuijk RF, de Jong B, Oosterhuis JW. Geurts van Kessel A. Uniparental origin of i(12p) in human germ cell tumors. Genes Chromosomes Cancer. 1993;6(3):161–5.
Looijenga LH, Gillis AJ, Stoop HJ, Hersmus R, Oosterhuis JW. Chromosomes and expression in human testicular germ-cell tumors: insight into their cell of origin and pathogenesis. Ann N Y Acad Sci. 2007;1120:187–214. doi:10.1196/annals.1411.000.
Korkola JE, Houldsworth J, Bosl GJ, Chaganti RS. Molecular events in germ cell tumours: linking chromosome-12 gain, acquisition of pluripotency and response to cisplatin. BJU International. 2009;104(9 Pt B):1334–8. doi:10.1111/j.1464-410X.2009.08855.x.
Adamah DJ, Gokhale PJ, Eastwood DJ, Rajpert De-Meyts E, Goepel J, Walsh JR, et al. Dysfunction of the mitotic:meiotic switch as a potential cause of neoplastic conversion of primordial germ cells. Int J Androl. 2006;29(1):219–27. doi:10.1111/j.1365-2605.2005.00569.x.
de Jong B, Oosterhuis JW, Castedo SM, Vos A, te Meerman GJ. Pathogenesis of adult testicular germ cell tumors. A cytogenetic model. Cancer Genet Cytogenet. 1990;48(2):143–67.
Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013;19(8):998–1004. doi:10.1038/nm.3267.
McIntyre A, Summersgill B, Grygalewicz B, Gillis AJ, Stoop J, van Gurp RJ, et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 2005;65(18):8085–9. doi:10.1158/0008-5472.CAN-05-0471.
Samaniego F, Rodriguez E, Houldsworth J, Murty VV, Ladanyi M, Lele KP, et al. Cytogenetic and molecular analysis of human male germ cell tumors: chromosome 12 abnormalities and gene amplification. Genes Chromosomes Cancer. 1990;1(4):289–300.
Cutcutache I, Suzuki Y, Tan IB, Ramgopal S, Zhang S, Ramnarayanan K, et al. Exome-wide sequencing shows low mutation rates and identifies novel mutated genes in seminomas. Eur Urol. 2015;68(1):77–83. doi:10.1016/j.eururo.2014.12.040.
Mulder MP, Keijzer W, Verkerk A, Boot AJ, Prins ME, Splinter TA, et al. Activated ras genes in human seminoma: evidence for tumor heterogeneity. Oncogene. 1989;4(11):1345–51.
Tian Q, Frierson Jr HF, Krystal GW, Moskaluk CA. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999;154(6):1643–7. doi:10.1016/S0002-9440(10)65419-3.
Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, et al. KIT mutations are common in testicular seminomas. Am J Pathol. 2004;164(1):305–13. doi:10.1016/S0002-9440(10)63120-3.
Bignell G, Smith R, Hunter C, Stephens P, Davies H, Greenman C, et al. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer. 2006;45(1):42–6. doi:10.1002/gcc.20265.
Goddard NC, McIntyre A, Summersgill B, Gilbert D, Kitazawa S, Shipley J. KIT and RAS signalling pathways in testicular germ cell tumours: new data and a review of the literature. Int J Androl. 2007;30(4):337–348; discussion 49. doi:10.1111/j.1365-2605.2007.00769.x.
Hoei-Hansen CE, Kraggerud SM, Abeler VM, Kaern J, Rajpert-De Meyts E, Lothe RA. Ovarian dysgerminomas are characterised by frequent KIT mutations and abundant expression of pluripotency markers. Mol Cancer. 2007;6:12. doi:10.1186/1476-4598-6-12.
Cheng L, Roth LM, Zhang S, Wang M, Morton MJ, Zheng W, et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011;117(10):2096–103. doi:10.1002/cncr.25794.
Hersmus R, Stoop H, van de Geijn GJ, Eini R, Biermann K, Oosterhuis JW, et al. Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One. 2012;7(8):e43952. doi:10.1371/journal.pone.0043952.
Przygodzki RM, Hubbs AE, Zhao FQ, O’Leary TJ. Primary mediastinal seminomas: evidence of single and multiple KIT mutations. Lab Investig. 2002;82(10):1369–75.
Fukushima S, Otsuka A, Suzuki T, Yanagisawa T, Mishima K, Mukasa A, et al. Mutually exclusive mutations of KIT and RAS are associated with KIT mRNA expression and chromosomal instability in primary intracranial pure germinomas. Acta Neuropathol. 2014;127(6):911–25. doi:10.1007/s00401-014-1247-5.
Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279(30):31655–63. doi:10.1074/jbc.M403319200.
Przygodzki RM, Moran CA, Suster S, Khan MA, Swalsky PA, Bakker A, et al. Primary mediastinal and testicular seminomas: a comparison of K-ras-2 gene sequence and p53 immunoperoxidase analysis of 26 cases. Hum Pathol. 1996;27(9):975–9.
Tate G, Suzuki T, Kishimoto K, Mitsuya T. A c-KIT codon 816 mutation, D816H, in the testicular germ cell tumor: case report of a Japanese patient with bilateral testicular seminomas. Acta Med Okayama. 2005;59(1):33–6.
Looijenga LH, de Leeuw H, van Oorschot M, van Gurp RJ, Stoop H, Gillis AJ, et al. Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res. 2003;63(22):7674–8.
Biermann K, Goke F, Nettersheim D, Eckert D, Zhou H, Kahl P, et al. c-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J Pathol. 2007;213(3):311–8. doi:10.1002/path.2225.
Rapley EA, Hockley S, Warren W, Johnson L, Huddart R, Crockford G, et al. Somatic mutations of KIT in familial testicular germ cell tumours. Br J Cancer. 2004;90(12):2397–2401. doi:10.1038/sj.bjc.6601880 6601880 [pii].
Caruana G, Cambareri AC, Gonda TJ, Ashman LK. Transformation of NIH3T3 fibroblasts by the c-Kit receptor tyrosine kinase: effect of receptor density and ligand-requirement. Oncogene. 1998;16(2):179–90. doi:10.1038/sj.onc.1201494.
Chian R, Young S, Danilkovitch-Miagkova A, Ronnstrand L, Leonard E, Ferrao P, et al. Phosphatidylinositol 3 kinase contributes to the transformation of hematopoietic cells by the D816V c-Kit mutant. Blood. 2001;98(5):1365–73.
Lind GE, Skotheim RI, Lothe RA. The epigenome of testicular germ cell tumors. APMIS. 2007;115(10):1147–1160. doi:APMapm_660.xml [pii] 10.1111/j.1600-0463.2007.apm_660.xml.x.
Okamoto K. Epigenetics: a way to understand the origin and biology of testicular germ cell tumors. Int J Urol. 2012;19(6):504–11. doi:10.1111/j.1442-2042.2012.02986.x.
Tsuchiya K, Reijo R, Page DC, Disteche CM. Gonadoblastoma: molecular definition of the susceptibility region on the Y chromosome. Am J Hum Genet. 1995;57(6):1400–7.
Lau Y, Chou P, Iezzoni J, Alonzo J, Komuves L. Expression of a candidate gene for the gonadoblastoma locus in gonadoblastoma and testicular seminoma. Cytogenet Cell Genet. 2000;91(1–4):160–164. doi:56838 [pii] 56838.
Lau YF, Li Y, Kido T. Gonadoblastoma locus and the TSPY gene on the human Y chromosome. Birth Defects Res C Embryo Today. 2009;87(1):114–22. doi:10.1002/bdrc.20144.
Oosterhuis JW, Stoop H, Dohle G, Boellaard W, van Casteren N, Wolffenbuttel K, et al. A pathologist’s view on the testis biopsy. Int J Androl. 2011;34(4 Pt 2):e14–e19; discussion e20. doi:10.1111/j.1365-2605.2011.01204.x.
Kaprova-Pleskacova J, Stoop H, Bruggenwirth H, Cools M, Wolffenbuttel KP, Drop SL, et al. Complete androgen insensitivity syndrome: factors influencing gonadal histology including germ cell pathology. Mod Pathol Off J U S Can Acad Pathol Inc. 2014;27(5):721–730. doi:modpathol2013193 [pii] 10.1038/modpathol.2013.193.
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423(6942):825–837. doi:10.1038/nature01722 nature01722 [pii].
Page DC. Hypothesis: a Y-chromosomal gene causes gonadoblastoma in dysgenetic gonads. Development. 1987;101(Suppl):151–5.
Hoei-Hansen CE, Sehested A, Juhler M, Lau YF, Skakkebaek NE, Laursen H, et al. New evidence for the origin of intracranial germ cell tumours from primordial germ cells: expression of pluripotency and cell differentiation markers. J Pathol. 2006;209(1):25–33. doi:10.1002/path.1948.
Shahsiah R, Jahanbin B, Rabiei R, Ardalan FA, Sarhadi B, Izadi-Mood N. Malignant ovarian germ cell tumours in gonadal Y chromosome mosaicism. J Clin Pathol. 2011;64(11):973–976. doi:jcp.2011.090738 [pii] 10.1136/jcp.2011.090738.
Jacobsen C, Honecker F. Cisplatin resistance in germ cell tumours: models and mechanisms. Andrology. 2015;3(1):111–21. doi:10.1111/andr.299.
Chaganti RS, Houldsworth J. Genetics and biology of adult human male germ cell tumors. Cancer Res. 2000;60(6):1475–82.
Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124(6):1169–1181. doi:S0092-8674(06)00291-1 [pii] 10.1016/j.cell.2006.02.037.
van Gurp RJ, Oosterhuis JW, Kalscheuer V, Mariman EC, Looijenga LH. Biallelic expression of the H19 and IGF2 genes in human testicular germ cell tumors. J Natl Cancer Inst. 1994;86(14):1070–5.
Netto GJ, Nakai Y, Nakayama M, Jadallah S, Toubaji A, Nonomura N, et al. Global DNA hypomethylation in intratubular germ cell neoplasia and seminoma, but not in nonseminomatous male germ cell tumors. Mod Pathol Off J U S Can Acad Pathol Inc. 2008;21(11):1337–1344. doi:modpathol2008127 [pii] 10.1038/modpathol.2008.127.
Eckert D, Biermann K, Nettersheim D, Gillis AJ, Steger K, Jack HM, et al. Expression of BLIMP1/PRMT5 and concurrent histone H2A/H4 arginine 3 dimethylation in fetal germ cells, CIS/IGCNU and germ cell tumors. BMC Dev Biol. 2008;8:106. doi:1471-213X-8-106 [pii] 10.1186/1471-213X-8-106.
Wermann H, Stoop H, Gillis AJ, Honecker F, van Gurp RJ, Ammerpohl O, et al. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. J Pathol. 2010;221(4):433–42. doi:10.1002/path.2725.
Almstrup K, Nielsen JE, Mlynarska O, Jansen MT, Jorgensen A, Skakkebaek NE, et al. Carcinoma in situ testis displays permissive chromatin modifications similar to immature foetal germ cells. Br J Cancer. 2010;103(8):1269–1276. doi:6605880 [pii] 10.1038/sj.bjc.6605880.
Brait M, Maldonado L, Begum S, Loyo M, Wehle D, Tavora FF, et al. DNA methylation profiles delineate epigenetic heterogeneity in seminoma and non-seminoma. Br J Cancer. 2012;106(2):414–423. doi:bjc2011468 [pii] 10.1038/bjc.2011.468.
Kristensen DG, Nielsen JE, Jorgensen A, Skakkebaek NE, Rajpert-De Meyts E, Almstrup K. Evidence that active demethylation mechanisms maintain the genome of carcinoma in situ cells hypomethylated in the adult testis. Br J Cancer. 2014;110(3):668–678. doi:bjc2013727 [pii] 10.1038/bjc.2013.727.
Killian JK, Dorssers LCJ, Trabert B, Gillis AJM, Cook MB, Wang Y, et al. Malignant testicular germ cell tumor methylomes demonstrate differentiation-dependent somatic reprogramming but persistent erasure of imprinted loci and the pluripotency regulator DPPA3/STELLA. Genome Res. 2016;26: 1490–1504.
Gillis AJ, Stoop HJ, Hersmus R, Oosterhuis JW, Sun Y, Chen C, et al. High-throughput microRNAome analysis in human germ cell tumours. J Pathol. 2007;213(3):319–28. doi:10.1002/path.2230.
Novotny GW, Belling KC, Bramsen JB, Nielsen JE, Bork-Jensen J, Almstrup K, et al. MicroRNA expression profiling of carcinoma in situ cells of the testis. Endocr Relat Cancer. 2012;19(3):365–379. doi:ERC-11-0271 [pii] 10.1530/ERC-11-0271.
Rajpert-De Meyts E, Bartkova J, Samson M, Hoei-Hansen CE, Frydelund-Larsen L, Bartek J, et al. The emerging phenotype of the testicular carcinoma in situ germ cell. APMIS. 2003;111(1):267–278; discussion 78–9. doi:apm1110130 [pii].
Almstrup K, Hoei-Hansen CE, Wirkner U, Blake J, Schwager C, Ansorge W, et al. Embryonic stem cell-like features of testicular carcinoma in situ revealed by genome-wide gene expression profiling. Cancer Res. 2004;64(14):4736–4743. doi:10.1158/0008-5472.CAN-04-0679 64/14/4736 [pii].
Almstrup K, Hoei-Hansen CE, Nielsen JE, Wirkner U, Ansorge W, Skakkebaek NE, et al. Genome-wide gene expression profiling of testicular carcinoma in situ progression into overt tumours. Br J Cancer. 2005;92(10):1934–1941. doi:6602560 [pii] 10.1038/sj.bjc.6602560.
Rajpert-De Meyts E. Developmental model for the pathogenesis of testicular carcinoma in situ: genetic and environmental aspects. Hum Reprod Update. 2006;12(3):303–323. doi:dmk006 [pii] 10.1093/humupd/dmk006.
International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15(2):594–603.
Oosterhuis JW, Andrews PW, Knowles BB, Damjanov I. Effects of cis-platinum on embryonal carcinoma cell lines in vitro. Int J Cancer. 1984;34(1):133–9.
Stampfer MR, Yaswen P. Human epithelial cell immortalization as a step in carcinogenesis. Cancer Lett. 2003;194(2):199–208. doi:S0304383502007073 [pii].
Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18(2):173–179. doi:10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3 [pii] 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3.
Albanell J, Bosl GJ, Reuter VE, Engelhardt M, Franco S, Moore MA, et al. Telomerase activity in germ cell cancers and mature teratomas. J Natl Cancer Inst. 1999;91(15):1321–6.
Turnbull C, Rapley EA, Seal S, Pernet D, Renwick A, Hughes D, et al. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat Genet. 2010;42(7):604–607. doi:ng.607 [pii] 10.1038/ng.607.
Gilbert D, Rapley E, Shipley J. Testicular germ cell tumours: predisposition genes and the male germ cell niche. Nat Rev Cancer. 2011;11(4):278–288. doi:nrc3021 [pii] 10.1038/nrc3021.
Boublikova L, Buchler T, Stary J, Abrahamova J, Trka J. Molecular biology of testicular germ cell tumors: unique features awaiting clinical application. Crit Rev Oncol Hematol. 2014;89(3):366–385. doi:S1040-8428(13)00211-4 [pii] 10.1016/j.critrevonc.2013.10.001.
Koster R, di Pietro A, Timmer-Bosscha H, Gibcus JH, van den Berg A, Suurmeijer AJ, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010;120(10):3594–3605. doi:41939 [pii] 10.1172/JCI41939.
Bauer S, Mühlenberg T, Leahy M, Hoiczyk M, Gauler T, Schuler M, et al. Therapeutic potential of Mdm2 inhibition in malignant germ cell tumours. Eur Urol. 2010;57:679–687.
Koster R, Timmer-Bosscha H, Bischoff R, Gietema JA, De Jong S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell death and disease. 2011;2:e148. doi:10.1038/cddis.2011.33.
Biswal BK, Beyrouthy MJ, Hever-Jardine MP, Amstrong D, Tomlinson CR, Christensen BC, et al. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-Aza Deoxycytidine is associated with global DNA damage-associated p53 activation, anti-pluripotency and DNA demethylation. PLOS ONE. 2012;7:e53003. doi:10.1371/journal.pone.0053003.
Oosterhuis JW, Damjanov I. Treatment of primary embryo-derived teratocarcinomas in mice with cis-diamminedichloroplatinum. Eur J Cancer Clin Oncol. 1983;19(5):695–9.
Honecker F, Wermann H, Mayer F, Gillis AJ, Stoop H, van Gurp RJ, et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol. 2009;27(13):2129–2136. doi:JCO.2008.18.8623 [pii] 10.1200/JCO.2008.18.8623.
Mayer F, Wermann H, Albers P, Stoop H, Gillis AJ, Hartmann JT, et al. Histopathological and molecular features of late relapses in non-seminomas. BJU Int. 2011;107(6):936–43. doi:10.1111/j.1464-410X.2010.09631.x.
Michael H, Lucia J, Foster RS, Ulbright TM. The pathology of late recurrence of testicular germ cell tumors. Am J Surg Pathol. 2000;24(2):257–73.
Gillis AJ, Oosterhuis JW, Schipper ME, Barten EJ, van Berlo R, van Gurp RJ, et al. Origin and biology of a testicular Wilms’ tumor. Genes Chromosomes Cancer. 1994;11(2):126–35.
Motzer RJ, Amsterdam A, Prieto V, Sheinfeld J, Murty VV, Mazumdar M, et al. Teratoma with malignant transformation: diverse malignant histologies arising in men with germ cell tumors. J Urol. 1998;159(1):133–138. doi:S0022-5347(01)64035-7 [pii].
Kum JB, Ulbright TM, Williamson SR, Wang M, Zhang S, Foster RS, et al. Molecular genetic evidence supporting the origin of somatic-type malignancy and teratoma from the same progenitor cell. Am J Surg Pathol. 2012;36(12):1849–1856. doi:10.1097/PAS.0b013e31826df1ab 00000478-201212000-00013 [pii].
Guo CC, Punar M, Contreras AL, Tu SM, Pisters L, Tamboli P, et al. Testicular germ cell tumors with sarcomatous components: an analysis of 33 cases. Am J Surg Pathol. 2009;33(8):1173–8. doi:10.1097/PAS.0b013e3181adb9d7.
Cunningham JJ, Ulbright TM, Pera MF, Looijenga LH. Lessons from human teratomas to guide development of safe stem cell therapies. Nat Biotechnol. 2012;30(9):849–857. doi:nbt.2329 [pii] 10.1038/nbt.2329.
Devouassoux-Shisheboran M, Mauduit C, Tabone E, Droz JP, Benahmed M. Growth regulatory factors and signalling proteins in testicular germ cell tumours. APMIS. 2003;111(1):212–224; discussion 24. doi:apm1110125 [pii].
Neumann JC, Chandler GL, Damoulis VA, Fustino NJ, Lillard K, Looijenga L, et al. Mutation in the type IB bone morphogenetic protein receptor Alk6b impairs germ-cell differentiation and causes germ-cell tumors in zebrafish. Proc Natl Acad Sci U S A. 2011;108(32):13153–13158. doi:1102311108 [pii] 10.1073/pnas.1102311108.
Neumann JC, Lillard K, Damoulis V, Amatruda JF. Zebrafish models of germ cell tumor. Methods Cell Biol. 2011;105:3–24. doi:B978-0-12-381320-6.00001-1 [pii] 10.1016/B978-0-12-381320-6.00001-1.
Basten SG, Davis EE, Gillis AJ, van Rooijen E, Stoop H, Babala N, et al. Mutations in LRRC50 predispose zebrafish and humans to seminomas. PLoS Genet. 2013;9(4):e1003384. doi:10.1371/journal.pgen.1003384 PGENETICS-D-12-02494 [pii].
Mizuno Y, Gotoh A, Kamidono S, Kitazawa S. Establishment and characterization of a new human testicular germ cell tumor cell line (TCam-2). Nihon Hinyokika Gakkai Zasshi. 1993;84(7):1211–8.
Berends JC, Schutte SE, van Dissel-Emiliani FM, de Rooij DG, Looijenga LH, Oosterhuis JW. Significant improvement of the survival of seminoma cells in vitro by use of a rat Sertoli cell feeder layer and serum-free medium. J Natl Cancer Inst. 1991;83(19):1400–3.
de Jong J, Stoop H, Gillis AJ, Hersmus R, van Gurp RJ, van de Geijn GJ, et al. Further characterization of the first seminoma cell line TCam-2. Genes Chromosomes Cancer. 2008;47(3):185–96. doi:10.1002/gcc.20520.
de Jong J, Stoop H, Gillis AJ, van Gurp RJ, van de Geijn GJ, Boer M, et al. Differential expression of SOX17 and SOX2 in germ cells and stem cells has biological and clinical implications. J Pathol. 2008;215(1):21–30. doi:10.1002/path.2332.
Nettersheim D, Gillis AJ, Looijenga LH, Schorle H. TGF-beta1, EGF and FGF4 synergistically induce differentiation of the seminoma cell line TCam-2 into a cell type resembling mixed non-seminoma. Int J Androl. 2011;34(4 Pt 2):e189–203. doi:10.1111/j.1365-2605.2011.01172.x.
Nettersheim D, Westernstroer B, Haas N, Leinhaas A, Brustle O, Schlatt S, et al. Establishment of a versatile seminoma model indicates cellular plasticity of germ cell tumor cells. Genes Chromosomes Cancer. 2012;51(7):717–26. doi:10.1002/gcc.21958.
Andrews PW, Damjanov I, Simon D, Banting GS, Carlin C, Dracopoli NC, et al. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab Investig. 1984;50(2):147–62.
Andrews PW, Fenderson B, Hakomori S. Human embryonal carcinoma cells and their differentiation in culture. Int J Androl. 1987;10(1):95–104.
Andrews PW, Casper J, Damjanov I, Duggan-Keen M, Giwercman A, Hata J, et al. Comparative analysis of cell surface antigens expressed by cell lines derived from human germ cell tumours. Int J Cancer. 1996;66(6):806–816. doi:10.1002/(SICI)1097-0215(19960611)66:6<806::AID-IJC17>3.0.CO;2-0 [pii] 10.1002/(SICI)1097-0215(19960611)66:6<806::AID-IJC17>3.0.CO;2-0.
Scully RE. Gonadoblastoma. A review of 74 cases. Cancer. 1970;25(6):1340–56.
Ulbright TM, Young RH. Gonadoblastoma and selected other aspects of gonadal pathology in young patients with disorders of sex development. Semin Diagn Pathol. 2014;31(5):427–440. doi:S0740-2570(14)00064-1 [pii] 10.1053/j.semdp.2014.07.001.
Houk CP, Hughes IA, Ahmed SF, Lee PA, Writing Committee for the International Intersex Consensus Conference Participants. Summary of consensus statement on intersex disorders and their management. International Intersex Consensus Conference. Pediatrics. 2006;118(2):753–757. doi:118/2/753 [pii] 10.1542/peds.2006-0737.
Cools M, Looijenga LH. Tumor risk and clinical follow-up in patients with disorders of sex development. Pediatr Endocrinol Rev. 2011;9(Suppl 1):519–24.
Hughes IA. Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab. 2008;22(1):119–134. doi:S1521-690X(07)00105-4 [pii] 10.1016/j.beem.2007.11.001.
Subbiah V, Huff V, Wolff JE, Ketonen L, Lang Jr FF, Stewart J, et al. Bilateral gonadoblastoma with dysgerminoma and pilocytic astrocytoma with WT1 GT-IVS9 mutation: A 46 XY phenotypic female with Frasier syndrome. Pediatr Blood Cancer. 2009;53(7):1349–51. doi:10.1002/pbc.22152.
Looijenga LH, Hersmus R, de Leeuw BH, Stoop H, Cools M, Oosterhuis JW, et al. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab. 2010;24(2):291–310. doi:S1521-690X(09)00139-0 [pii] 10.1016/j.beem.2009.10.002.
Tanteles GA, Oakley S, Christian M, O’Neill D, Suri M. Denys-Drash syndrome and gonadoblastoma in a patient with Klinefelter syndrome. Clin Dysmorphol. 2011;20(3):131–5. doi:10.1097/MCD.0b013e328346f6dc.
Looijenga LH. Testicular germ cell tumors. Pediatr Endocrinol Rev. 2014;11(Suppl 2):251–62.
Achermann J. Disorders of sex development. In: Melmed S, editor. Williams textbook of endocrinology. 12th ed. Philadelphia: Elsevier; 2011. p. 868–934.
van der Zwan YG, Biermann K, Wolffenbuttel KP, Cools M, Looijenga LH. Gonadal maldevelopment as risk factor for germ cell cancer: towards a clinical decision model. Eur Urol. 2015;67(4):692–701. doi:S0302-2838(14)00651-4 [pii] 10.1016/j.eururo.2014.07.011.
Davidoff F, Federman DD. Mixed gonadal dysgenesis. Pediatrics. 1973;52(5):725–42.
Sohval AR. “Mixed” gonadal dysgenesis: a variety of hermaphroditism. Am J Hum Genet. 1963;15:155–8.
Ropke A, Pelz AF, Volleth M, Schlosser HW, Morlot S, Wieacker PF. Sex chromosomal mosaicism in the gonads of patients with gonadal dysgenesis, but normal female or male karyotypes in lymphocytes. Am J Obstet Gynecol. 2004;190(4):1059–1062. doi:10.1016/j.ajog.2003.09.053 S0002937803017794 [pii].
Cools M, Boter M, van Gurp R, Stoop H, Poddighe P, Lau YF, et al. Impact of the Y-containing cell line on histological differentiation patterns in dysgenetic gonads. Clin Endocrinol. 2007;67(2):184–192. doi:CEN2859 [pii] 10.1111/j.1365-2265.2007.02859.x.
Maier EM, Leitner C, Lohrs U, Kuhnle U. True hermaphroditism in an XY individual due to a familial point mutation of the SRY gene. J Pediatr Endocrinol Metab. 2003;16(4):575–80.
Isidor B, Capito C, Paris F, Baron S, Corradini N, Cabaret B, et al. Familial frameshift SRY mutation inherited from a mosaic father with testicular dysgenesis syndrome. J Clin Endocrinol Metab. 2009;94(9):3467–3471. doi:jc.2009-0226 [pii] 10.1210/jc.2009-0226.
Shahid M, Dhillon VS, Khalil HS, Haque S, Batra S, Husain SA, et al. A SRY-HMG box frame shift mutation inherited from a mosaic father with a mild form of testicular dysgenesis syndrome in Turner syndrome patient. BMC Med Genet. 2010;11:131. doi:1471-2350-11-131 [pii] 10.1186/1471-2350-11-131.
Hersmus R, Kalfa N, de Leeuw B, Stoop H, Oosterhuis JW, de Krijger R, et al. FOXL2 and SOX9 as parameters of female and male gonadal differentiation in patients with various forms of disorders of sex development (DSD). J Pathol. 2008;215(1):31–8. doi:10.1002/path.2335.
Buell-Gutbrod R, Ivanovic M, Montag A, Lengyel E, Fadare O, Gwin K. FOXL2 and SOX9 distinguish the lineage of the sex cord-stromal cells in gonadoblastomas. Pediatr Dev Pathol. 2011;14(5):391–5. doi:10.2350/10-12-0943-OA.1.
Hersmus R, van der Zwan YG, Stoop H, Bernard P, Sreenivasan R, Oosterhuis JW, et al. A 46,XY female DSD patient with bilateral gonadoblastoma, a novel SRY missense mutation combined with a WT1 KTS splice-site mutation. PLoS One. 2012;7(7):e40858. doi:10.1371/journal.pone.0040858 PONE-D-12-13207 [pii].
Hertel JD, Huettner PC, Dehner LP, Pfeifer JD. The chromosome Y-linked testis-specific protein locus TSPY1 is characteristically present in gonadoblastoma. Hum Pathol. 2010;41(11):1544–1549. doi:S0046-8177(10)00123-1 [pii] 10.1016/j.humpath.2010.04.007.
Hersmus R, Stoop H, White SJ, Drop SL, Oosterhuis JW, Incrocci L, et al. Delayed recognition of Disorders of Sex Development (DSD): a missed opportunity for early diagnosis of malignant germ cell tumors. Int J Endocrinol. 2012;2012:671209. doi:10.1155/2012/671209.
Li Y, Vilain E, Conte F, Rajpert-De Meyts E, Lau YF. Testis-specific protein Y-encoded gene is expressed in early and late stages of gonadoblastoma and testicular carcinoma in situ. Urol Oncol. 2007;25(2):141–146. doi:S1078-1439(06)00166-9 [pii] 10.1016/j.urolonc.2006.08.002.
Carver BS, Shayegan B, Serio A, Motzer RJ, Bosl GJ, Sheinfeld J. Long-term clinical outcome after postchemotherapy retroperitoneal lymph node dissection in men with residual teratoma. J Clin Oncol. 2007;25(9):1033–1037. doi:JCO.2005.05.4791 [pii] 10.1200/JCO.2005.05.4791.
Malagon HD, Valdez AM, Moran CA, Suster S. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol. 2007;31(9):1356–1362. doi:10.1097/PAS.0b013e318033c7c4 00000478-200709000-00008 [pii].
Comiter CV, Kibel AS, Richie JP, Nucci MR, Renshaw AA. Prognostic features of teratomas with malignant transformation: a clinicopathological study of 21 cases. J Urol. 1998;159(3):859–863. doi:S0022-5347(01)63754-6 [pii].
Ganjoo KN, Foster RS, Michael H, Donohue JP, Einhorn LH. Germ cell tumor associated primitive neuroectodermal tumors. J Urol. 2001;165(5):1514–1516. doi:S0022-5347(05)66339-2 [pii].
Michael H, Hull MT, Ulbright TM, Foster RS, Miller KD. Primitive neuroectodermal tumors arising in testicular germ cell neoplasms. Am J Surg Pathol. 1997;21(8):896–904.
Magers MJ, Kao CS, Cole CD, Rice KR, Foster RS, Einhorn LH, et al. “Somatic-type” malignancies arising from testicular germ cell tumors: a clinicopathologic study of 124 cases with emphasis on glandular tumors supporting frequent yolk sac tumor origin. Am J Surg Pathol. 2014;38(10):1396–409. doi:10.1097/PAS.0000000000000262.
Howitt BE, Magers MJ, Rice KR, Cole CD, Ulbright TM. Many postchemotherapy sarcomatous tumors in patients with testicular germ cell tumors are sarcomatoid yolk sac tumors: a study of 33 cases. Am J Surg Pathol. 2015;39(2):251–9. doi:10.1097/PAS.0000000000000322.
Moul JW, Schanne FJ, Thompson IM, Frazier HA, Peretsman SA, Wettlaufer JN, et al. Testicular cancer in blacks. A multicenter experience. Cancer. 1994;73(2):388–93.
Rosen A, Jayram G, Drazer M, Eggener SE. Global trends in testicular cancer incidence and mortality. Eur Urol. 2011;60(2):374–379. doi:S0302-2838(11)00493-3 [pii] 10.1016/j.eururo.2011.05.004.
Shanmugalingam T, Soultati A, Chowdhury S, Rudman S, Van Hemelrijck M. Global incidence and outcome of testicular cancer. Clin Epidemiol. 2013;5:417–427. doi:10.2147/CLEP.S34430 clep-5-417 [pii].
McGlynn KA, Devesa SS, Graubard BI, Castle PE. Increasing incidence of testicular germ cell tumors among black men in the United States. J Clin Oncol. 2005;23(24):5757–5761. doi:23/24/5757 [pii] 10.1200/JCO.2005.08.227.
Ekbom A, Richiardi L, Akre O, Montgomery SM, Sparen P. Age at immigration and duration of stay in relation to risk for testicular cancer among Finnish immigrants in Sweden. J Natl Cancer Inst. 2003;95(16):1238–40.
Montgomery SM, Granath F, Ehlin A, Sparen P, Ekbom A. Germ-cell testicular cancer in offspring of Finnish immigrants to Sweden. Cancer Epidemiol Biomarkers Prev. 2005;14(1):280–282. doi:14/1/280 [pii].
Levine H, Afek A, Shamiss A, Derazne E, Tzur D, Zavdy O, et al. Risk of germ cell testicular cancer according to origin: a migrant cohort study in 1,100,000 Israeli men. Int J Cancer. 2013;132(8):1878–85. doi:10.1002/ijc.27825.
McIver SC, Roman SD, Nixon B, Loveland KL, McLaughlin EA. The rise of testicular germ cell tumours: the search for causes, risk factors and novel therapeutic targets. F1000Res. 2013;2:55. doi:10.12688/f1000research.2-55.v1.
Moller H. Decreased testicular cancer risk in men born in wartime. J Natl Cancer Inst. 1989;81(21):1668–9.
Rajpert-De Meyts E, McGlynn KA, Okamoto K, Jewett MA, Bokemeyer C. Testicular germ cell tumours. Lancet. 2016;387(10029):1762–1774. doi:S0140-6736(15)00991-5 [pii] 10.1016/S0140-6736(15)00991-5.
Cook MB, Akre O, Forman D, Madigan MP, Richiardi L, McGlynn KA. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer – experiences of the mother. Int J Epidemiol. 2009;38(6):1532–1542. doi:dyp287 [pii] 10.1093/ije/dyp287.
Trabert B, Zugna D, Richiardi L, McGlynn KA, Akre O. Congenital malformations and testicular germ cell tumors. Int J Cancer. 2013;133(8):1900–4. doi:10.1002/ijc.28207.
Fossa SD, Chen J, Schonfeld SJ, McGlynn KA, McMaster ML, Gail MH, et al. Risk of contralateral testicular cancer: a population-based study of 29,515 U.S. men. J Natl Cancer Inst. 2005;97(14):1056–1066. doi:97/14/1056 [pii] 10.1093/jnci/dji185.
Moller H, Skakkebaek NE. Risk of testicular cancer in subfertile men: case-control study. BMJ. 1999;318(7183):559–62.
Tollerud DJ, Blattner WA, Fraser MC, Brown LM, Pottern L, Shapiro E, et al. Familial testicular cancer and urogenital developmental anomalies. Cancer. 1985;55(8):1849–54.
Ferlin A, Ganz F, Pengo M, Selice R, Frigo AC, Foresta C. Association of testicular germ cell tumor with polymorphisms in estrogen receptor and steroid metabolism genes. Endocr Relat Cancer. 2010;17(1):17–25. doi:ERC-09-0176 [pii] 10.1677/ERC-09-0176.
Kristiansen W, Andreassen KE, Karlsson R, Aschim EL, Bremnes RM, Dahl O, et al. Gene variations in sex hormone pathways and the risk of testicular germ cell tumour: a case-parent triad study in a Norwegian-Swedish population. Hum Reprod. 2012;27(5):1525–1535. doi:des075 [pii] 10.1093/humrep/des075.
Cook MB, Akre O, Forman D, Madigan MP, Richiardi L, McGlynn KA. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer – experiences of the son. Int J Epidemiol. 2010;39(6):1605–1618. doi:dyq120 [pii] 10.1093/ije/dyq120.
Cook MB, Trabert B, McGlynn KA. Organochlorine compounds and testicular dysgenesis syndrome: human data. Int J Androl. 2011;34(4 Pt 2):e68–e84; discussion e–5. doi:10.1111/j.1365-2605.2011.01171.x.
McGlynn KA, Trabert B. Adolescent and adult risk factors for testicular cancer. Nat Rev Urol. 2012;9(6):339–349. doi:nrurol.2012.61 [pii] 10.1038/nrurol.2012.61.
Daling JR, Doody DR, Sun X, Trabert BL, Weiss NS, Chen C, et al. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer. 2009;115(6):1215–23. doi:10.1002/cncr.24159.
Swerdlow AJ, De Stavola BL, Swanwick MA, Maconochie NE. Risks of breast and testicular cancers in young adult twins in England and Wales: evidence on prenatal and genetic aetiology. Lancet. 1997;350(9093):1723–1728. doi:S0140673697055268 [pii].
Kharazmi E, Hemminki K, Pukkala E, Sundquist K, Tryggvadottir L, Tretli S, et al. Cancer risk in relatives of testicular cancer patients by histology type and age at diagnosis: a joint study from five nordic countries. Eur Urol. 2015;68(2):283–289. doi:S0302-2838(14)01387-6 [pii] 10.1016/j.eururo.2014.12.031.
Nathanson KL, Kanetsky PA, Hawes R, Vaughn DJ, Letrero R, Tucker K, et al. The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am J Hum Genet. 2005;77(6):1034–1043. doi:S0002-9297(07)63387-4 [pii] 10.1086/498455.
Pathak A, Stewart DR, Faucz FR, Xekouki P, Bass S, Vogt A, et al. Rare inactivating PDE11A variants associated with testicular germ cell tumors. Endocr Relat Cancer. 2015;22(6):909–917. doi:ERC-15-0034 [pii] 10.1530/ERC-15-0034.
Heimdal K, Olsson H, Tretli S, Fossa SD, Borresen AL, Bishop DT. A segregation analysis of testicular cancer based on Norwegian and Swedish families. Br J Cancer. 1997;75(7):1084–7.
Hemminki K, Vaittinen P, Dong C, Easton D. Sibling risks in cancer: clues to recessive or X-linked genes? Br J Cancer. 2001;84(3):388–391. doi:10.1054/bjoc.2000.1585 S0007092000915854 [pii].
Valberg M, Grotmol T, Tretli S, Veierod MB, Moger TA, Aalen OO. A hierarchical frailty model for familial testicular germ-cell tumors. Am J Epidemiol. 2014;179(4):499–506. doi:kwt267 [pii] 10.1093/aje/kwt267.
Kanetsky PA, Mitra N, Vardhanabhuti S, Vaughn DJ, Li M, Ciosek SL, et al. A second independent locus within DMRT1 is associated with testicular germ cell tumor susceptibility. Hum Mol Genet. 2011;20(15):3109–3117. doi:ddr207 [pii] 10.1093/hmg/ddr207.
Chung CC, Kanetsky PA, Wang Z, Hildebrandt MA, Koster R, Skotheim RI, et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat Genet. 2013;45(6):680–685. doi:ng.2634 [pii] 10.1038/ng.2634.
Schumacher FR, Wang Z, Skotheim RI, Koster R, Chung CC, Hildebrandt MA, et al. Testicular germ cell tumor susceptibility associated with the UCK2 locus on chromosome 1q23. Hum Mol Genet. 2013;22(13):2748–2753. doi:ddt109 [pii] 10.1093/hmg/ddt109.
Koster R, Mitra N, D’Andrea K, Vardhanabhuti S, Chung CC, Wang Z, et al. Pathway-based analysis of GWAs data identifies association of sex determination genes with susceptibility to testicular germ cell tumors. Hum Mol Genet. 2014;23(22):6061–6068. doi:ddu305 [pii] 10.1093/hmg/ddu305.
Ruark E, Seal S, McDonald H, Zhang F, Elliot A, Lau K, et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet. 2013;45(6):686–689. doi:ng.2635 [pii] 10.1038/ng.2635.
Kratz CP, Han SS, Rosenberg PS, Berndt SI, Burdett L, Yeager M, et al. Variants in or near KITLG, BAK1, DMRT1, and TERT-CLPTM1L predispose to familial testicular germ cell tumour. J Med Genet. 2011;48(7):473–476. doi:jmedgenet-2011-100001 [pii] 10.1136/jmedgenet-2011-100001.
Dalgaard MD, Weinhold N, Edsgard D, Silver JD, Pers TH, Nielsen JE, et al. A genome-wide association study of men with symptoms of testicular dysgenesis syndrome and its network biology interpretation. J Med Genet. 2012;49(1):58–65. doi:jmedgenet-2011-100174 [pii] 10.1136/jmedgenet-2011-100174.
Skakkebaek NE, Holm M, Hoei-Hansen C, Jorgensen N, Rajpert-De Meyts E. Association between testicular dysgenesis syndrome (TDS) and testicular neoplasia: evidence from 20 adult patients with signs of maldevelopment of the testis. APMIS. 2003;111(1):1–9; discussion -11. doi:apm1110103_1 [pii].
Jorgensen A, Lindhardt Johansen M, Juul A, Skakkebaek NE, Main KM, Rajpert-De Meyts E. Pathogenesis of germ cell neoplasia in testicular dysgenesis and disorders of sex development. Semin Cell Dev Biol. 2015;45:124–137. doi:S1084-9521(15)00173-1 [pii] 10.1016/j.semcdb.2015.09.013.
Lottrup G, Jorgensen A, Nielsen JE, Jorgensen N, Duno M, Vinggaard AM, et al. Identification of a novel androgen receptor mutation in a family with multiple components compatible with the testicular dysgenesis syndrome. J Clin Endocrinol Metab. 2013;98(6):2223–2229. doi:jc.2013-1278 [pii] 10.1210/jc.2013-1278.
Looijenga LH, Van Agthoven T, Biermann K. Development of malignant germ cells – the genvironmental hypothesis. Int J Dev Biol. 2013;57(2–4):241–253. doi:130026ll [pii] 10.1387/ijdb.130026ll.
Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature. 2011;476(7358):101–104. doi:nature10239 [pii] 10.1038/nature10239.
Matson CK, Zarkower D. Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat Rev Genet. 2012;13(3):163–174. doi:nrg3161 [pii] 10.1038/nrg3161.
Oram SW, Liu XX, Lee TL, Chan WY, Lau YF. TSPY potentiates cell proliferation and tumorigenesis by promoting cell cycle progression in HeLa and NIH3T3 cells. BMC Cancer. 2006;6:154. doi:1471-2407-6-154 [pii] 10.1186/1471-2407-6-154.
Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, et al. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell. 2013;155(2):410–422. doi:S0092-8674(13)01154-9 [pii] 10.1016/j.cell.2013.09.017.
Gill ME, Hu YC, Lin Y, Page DC. Licensing of gametogenesis, dependent on RNA binding protein DAZL, as a gateway to sexual differentiation of fetal germ cells. Proc Natl Acad Sci U S A. 2011;108(18):7443–7448. doi:1104501108 [pii] 10.1073/pnas.1104501108.
Skakkebaek NE. Possible carcinoma-in-situ of the testis. Lancet. 1972;2(7776):516–517. doi:S0140-6736(72)91909-5 [pii].
Skakkebaek NE, Berthelsen JG, Giwercman A, Muller J. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl. 1987;10(1):19–28.
Berthelsen JG, Skakkebaek NE, von der Maase H, Sorensen BL, Mogensen P. Screening for carcinoma in situ of the contralateral testis in patients with germinal testicular cancer. Br Med J (Clin Res Ed). 1982;285(6356):1683–6.
Dieckmann KP, Loy V. Prevalence of contralateral testicular intraepithelial neoplasia in patients with testicular germ cell neoplasms. J Clin Oncol. 1996;14(12):3126–32.
Rajpert-De Meyts E, Kvist M, Skakkebaek NE. Heterogeneity of expression of immunohistochemical tumour markers in testicular carcinoma in situ: pathogenetic relevance. Virchows Arch. 1996;428(3):133–9.
Sonne SB, Perrett RM, Nielsen JE, Baxter MA, Kristensen DM, Leffers H, et al. Analysis of SOX2 expression in developing human testis and germ cell neoplasia. Int J Dev Biol. 2010;54(4):755–760. doi:082668ss [pii] 10.1387/ijdb.082668ss.
Jorgensen A, Nielsen JE, Almstrup K, Toft BG, Petersen BL, Rajpert-De Meyts E. Dysregulation of the mitosis-meiosis switch in testicular carcinoma in situ. J Pathol. 2013;229(4):588–98. doi:10.1002/path.4154.
Mitchell RT, E Camacho-Moll M, Macdonald J, Anderson RA, Kelnar CJ, O’Donnell M, et al. Intratubular germ cell neoplasia of the human testis: heterogeneous protein expression and relation to invasive potential. Mod Pathol Off J U S Can Acad Pathol Inc. 2014;27(9):1255–1266. doi:modpathol2013246 [pii] 10.1038/modpathol.2013.246.
Gueler B, Sonne SB, Zimmer J, Hilscher B, Hilscher W, Graem N, et al. AZFa protein DDX3Y is differentially expressed in human male germ cells during development and in testicular tumours: new evidence for phenotypic plasticity of germ cells. Hum Reprod. 2012;27(6):1547–1555. doi:des047 [pii] 10.1093/humrep/des047.
Hoei-Hansen CE, Rajpert-De Meyts E, Daugaard G, Skakkebaek NE. Carcinoma in situ testis, the progenitor of testicular germ cell tumours: a clinical review. Ann Oncol. 2005;16(6):863–868. doi:mdi175 [pii] 10.1093/annonc/mdi175.
Sakuma Y, Sakurai S, Oguni S, Hironaka M, Saito K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003;94(6):486–91.
McIntyre A, Gilbert D, Goddard N, Looijenga L, Shipley J. Genes, chromosomes and the development of testicular germ cell tumors of adolescents and adults. Genes Chromosomes Cancer. 2008;47(7):547–57. doi:10.1002/gcc.20562.
Rajpert-De Meyts E. The role of puberty for testicular cancer. In: Bourguignon JP, editor. The Onset of puberty in perspective. Amsterdam: Elsevier Science; 2000. p. 201–13.
Ottesen AM, Skakkebaek NE, Lundsteen C, Leffers H, Larsen J, Rajpert-De Meyts E. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer. 2003;38(2):117–25. doi:10.1002/gcc.10244.
von Eyben FE, Jacobsen GK, Rorth M, Von Der Maase H. Microinvasive germ cell tumour (MGCT) adjacent to testicular germ cell tumours. Histopathology. 2004;44(6):547–554. doi:10.1111/j.1365-2559.2004.01889.x HIS1889 [pii].
von Eyben FE, Jacobsen GK, Skotheim RI. Microinvasive germ cell tumor of the testis. Virchows Arch. 2005;447(3):610–25. doi:10.1007/s00428-005-1257-8.
Browne TJ, Richie JP, Gilligan TD, Rubin MA. Intertubular growth in pure seminomas: associations with poor prognostic parameters. Hum Pathol. 2005;36(6):640–645. doi:S0046817705001620 [pii] 10.1016/j.humpath.2005.03.011.
Hvarness T, Nielsen JE, Almstrup K, Skakkebaek NE, Rajpert-De Meyts E, Claesson MH. Phenotypic characterisation of immune cell infiltrates in testicular germ cell neoplasia. J Reprod Immunol. 2013;100(2):135–145. doi:S0165-0378(13)00126-5 [pii] 10.1016/j.jri.2013.10.005.
Hadrup SR, Braendstrup O, Jacobsen GK, Mortensen S, Pedersen LO, Seremet T, et al. Tumor infiltrating lymphocytes in seminoma lesions comprise clonally expanded cytotoxic T cells. Int J Cancer. 2006;119(4):831–8. doi:10.1002/ijc.21894.
Li N, Wang T, Han D. Structural, cellular and molecular aspects of immune privilege in the testis. Front Immunol. 2012;3:152. doi:10.3389/fimmu.2012.00152.
Goedert JJ, Purdue MP, McNeel TS, McGlynn KA, Engels EA. Risk of germ cell tumors among men with HIV/acquired immunodeficiency syndrome. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1266–1269. doi:16/6/1266 [pii] 10.1158/1055-9965.EPI-07-0042.
Menezes RX, Boetzer M, Sieswerda M, van Ommen GJ, Boer JM. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinformatics. 2009;10:203. doi:1471-2105-10-203 [pii] 10.1186/1471-2105-10-203.
Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6(11):846–856. doi:nrc1991 [pii] 10.1038/nrc1991.
Poulos C, Cheng L, Zhang S, Gersell DJ, Ulbright TM. Analysis of ovarian teratomas for isochromosome 12p: evidence supporting a dual histogenetic pathway for teratomatous elements. Mod Pathol Off J U S Can Acad Pathol Inc. 2006;19(6):766–771. doi:3800596 [pii] 10.1038/modpathol.3800596.
Smith HO, Berwick M, Verschraegen CF, Wiggins C, Lansing L, Muller CY, et al. Incidence and survival rates for female malignant germ cell tumors. Obstet Gynecol. 2006;107(5):1075–1085. doi:107/5/1075 [pii] 10.1097/01.AOG.0000216004.22588.ce.
Lifschitz-Mercer B, Walt H, Kushnir I, Jacob N, Diener PA, Moll R, et al. Differentiation potential of ovarian dysgerminoma: an immunohistochemical study of 15 cases. Hum Pathol. 1995;26(1):62–6.
Parkash V, Carcangiu ML. Transformation of ovarian dysgerminoma to yolk sac tumor: evidence for a histogenetic continuum. Mod Pathol Off J U S Can Acad Pathol Inc. 1995;8(8):881–7.
Aoki Y, Kase H, Fujita K, Tanaka K. Dysgerminoma with a slightly elevated alpha-fetoprotein level diagnosed as a mixed germ cell tumor after recurrence. Gynecol Obstet Invest. 2003;55(1):58–60. doi:68949 68949 [pii].
Cossu-Rocca P, Jones TD, Roth LM, Eble JN, Zheng W, Karim FW, et al. Cytokeratin and CD30 expression in dysgerminoma. Hum Pathol. 2006;37(8):1015–1021. doi:S0046-8177(06)00128-6 [pii] 10.1016/j.humpath.2006.02.018.
Moller H, Evans H. Epidemiology of gonadal germ cell cancer in males and females. APMIS. 2003;111(1):43–46; discussion 6–8. doi:apm1110107 [pii].
Stang A, Trabert B, Wentzensen N, Cook MB, Rusner C, Oosterhuis JW, et al. Burden of extragonadal germ cell tumours in Europe and the United States. Eur J Cancer. 2012;48(7):1116–1117. doi:S0959-8049(12)00215-8 [pii] 10.1016/j.ejca.2012.02.061.
dos Santos SI, Swerdlow AJ. Ovarian germ cell malignancies in England: epidemiological parallels with testicular cancer. Br J Cancer. 1991;63(5):814–8.
Walker AH, Ross RK, Pike MC, Henderson BE. A possible rising incidence of malignant germ cell tumours in young women. Br J Cancer. 1984;49(5):669–72.
Cools M, Stoop H, Kersemaekers AM, Drop SL, Wolffenbuttel KP, Bourguignon JP, et al. Gonadoblastoma arising in undifferentiated gonadal tissue within dysgenetic gonads. J Clin Endocrinol Metab. 2006;91(6):2404–2413. doi:jc.2005-2554 [pii] 10.1210/jc.2005-2554.
Koo YJ, Chun YK, Kwon YS, Lee IH, Kim TJ, Lee KH, et al. Ovarian gonadoblastoma with dysgerminoma in a woman with 46XX karyotype. Pathol Int. 2011;61(3):171–3. doi:10.1111/j.1440-1827.2010.02636.x.
Zhao S, Kato N, Endoh Y, Jin Z, Ajioka Y, Motoyama T. Ovarian gonadoblastoma with mixed germ cell tumor in a woman with 46, XX karyotype and successful pregnancies. Pathol Int. 2000;50(4):332–335. doi:pin1041 [pii].
Talerman A. Germ cell tumors of the ovary. In: Kurman RJ, editor. Blaustein’s pathology of the female genital tract. New York: Springer-Verlag; 1987. p. 659–721.
A L Husaini H, Soudy H, El Din Darwish A, Ahmed M, Eltigani A, A L Mubarak M, et al. Pure dysgerminoma of the ovary: a single institutional experience of 65 patients. Med Oncol. 2012;29(4):2944–8. doi:10.1007/s12032-012-0194-z.
Nogales F. Germ cell tumours. In: Tavassoli FA, editor. World Health Organization classification of tumours: pathology and genetics of tumours of the breast and female genital organs. Lyon: IARC Press; 2003. p. 163–75.
Guillem V, Poveda A. Germ cell tumours of the ovary. Clin Transl Oncol. 2007;9(4):237–243. doi:1027 [pii].
Nogales FF, Dulcey I, Preda O. Germ cell tumors of the ovary: an update. Arch Pathol Lab Med. 2014;138(3):351–62. doi:10.5858/arpa.2012-0547-RA.
Sigismondi C, Scollo P, Ferrandina G, Candiani M, Angioli R, Vigano R, et al. Management of bilateral malignant ovarian germ cell tumors: a MITO-9 retrospective study. Int J Gynecol Cancer. 2015;25(2):203–7. doi:10.1097/IGC.0000000000000358.
Stettner AR, Hartenbach EM, Schink JC, Huddart R, Becker J, Pauli R, et al. Familial ovarian germ cell cancer: report and review. Am J Med Genet. 1999;84(1):43–46. doi:10.1002/(SICI)1096-8628(19990507)84:1<43::AID-AJMG9>3.0.CO;2-2 [pii].
Cyriac S, Rajendranath R, Louis AR, Sagar TG. Familial germ cell tumor. Indian J Hum Genet. 2012;18(1):119–121. doi:10.4103/0971-6866.96679 IJHG-18-119 [pii].
Huddart RA, Thompson C, Houlston R, Huddart RA, Nicholls EJ, Horwich A. Familial predisposition to both male and female germ cell tumours? J Med Genet. 1996;33(1):86.
Sharpe RM, Skakkebaek NE. Testicular dysgenesis syndrome: mechanistic insights and potential new downstream effects. Fertil Steril. 2008;89(2 Suppl):e33–e38. doi:S0015-0282(07)04302-6 [pii] 10.1016/j.fertnstert.2007.12.026.
Walker AH, Ross RK, Haile RW, Henderson BE. Hormonal factors and risk of ovarian germ cell cancer in young women. Br J Cancer. 1988;57(4):418–22.
Horn-Ross PL, Whittemore AS, Harris R, Itnyre J. Characteristics relating to ovarian cancer risk: collaborative analysis of 12 U.S. case-control studies. VI. Nonepithelial cancers among adults. Collaborative Ovarian Cancer Group. Epidemiology. 1992;3(6):490–5.
Adami HO, Hsieh CC, Lambe M, Trichopoulos D, Leon D, Persson I, et al. Parity, age at first childbirth, and risk of ovarian cancer. Lancet. 1994;344(8932):1250–1254. doi:S0140-6736(94)90749-8 [pii].
Albrektsen G, Heuch I, Kvale G. Full-term pregnancies and incidence of ovarian cancer of stromal and germ cell origin: a Norwegian prospective study. Br J Cancer. 1997;75(5):767–70.
Chen T, Surcel HM, Lundin E, Kaasila M, Lakso HA, Schock H, et al. Circulating sex steroids during pregnancy and maternal risk of non-epithelial ovarian cancer. Cancer Epidemiol Biomark Prev. 2011;20(2):324–336. doi:1055-9965.EPI-10-0857 [pii] 10.1158/1055-9965.EPI-10-0857.
Gobbi D, Fascetti Leon F, Aquino A, Melchionda F, Lima M. Metachronous bilateral ovarian teratoma: a germ-line familial disorder and review of surgical management options. J Pediatr Adolesc Gynecol. 2013;26(5):e105–e107. doi:S1083-3188(13)00102-2 [pii] 10.1016/j.jpag.2013.02.006.
Ayhan A, Bukulmez O, Genc C, Karamursel BS, Ayhan A. Mature cystic teratomas of the ovary: case series from one institution over 34 years. Eur J Obstet Gynecol Reprod Biol. 2000;88(2):153–157. doi:S0301211599001414 [pii].
Brown EH, Jr. Identical twins with twisted benign cystic teratoma of the ovary. Am J Obstet Gynecol. 1979;134(8):879–880. doi:0002-9378(79)90860-3 [pii].
Oud PS, Soeters RP, Pahlplatz MM, Hermkens HG, Beck HL, Reubsaet-Veldhuizen J, et al. DNA cytometry of pure dysgerminomas of the ovary. Int J Gynecol Pathol. 1988;7(3):258–67.
Kraggerud SM, Szymanska J, Abeler VM, Kaern J, Eknaes M, Heim S, et al. DNA copy number changes in malignant ovarian germ cell tumors. Cancer Res. 2000;60(11):3025–30.
Ichikawa Y, Yoshida S, Koyama Y, Hirai M, Ishikawa T, Nishida M, et al. Inactivation of p16/CDKN2 and p15/MTS2 genes in different histological types and clinical stages of primary ovarian tumors. Int J Cancer. 1996;69(6):466–470. doi:10.1002/(SICI)1097-0215(19961220)69:6<466::AID-IJC8>3.0.CO;2-2 [pii] 10.1002/(SICI)1097-0215(19961220)69:6<466::AID-IJC8>3.0.CO;2-2.
Lindeman RE, Gearhart MD, Minkina A, Krentz AD, Bardwell VJ, Zarkower D. Sexual cell-fate reprogramming in the ovary by DMRT1. Curr Biol. 2015;25(6):764–771. doi:S0960-9822(15)00066-4 [pii] 10.1016/j.cub.2015.01.034.
DeFalco T. DMRT1 keeps masculinity intact. Dev Cell. 2014;29(5):503–504. doi:S1534-5807(14)00335-9 [pii] 10.1016/j.devcel.2014.05.015.
McCarthy BJ, Shibui S, Kayama T, Miyaoka E, Narita Y, Murakami M, et al. Primary CNS germ cell tumors in Japan and the United States: an analysis of 4 tumor registries. Neuro Oncology. 2012;14(9):1194–1200. doi:nos155 [pii] 10.1093/neuonc/nos155.
Nichols CR, Heerema NA, Palmer C, Loehrer Sr PJ, Williams SD, Einhorn LH. Klinefelter’s syndrome associated with mediastinal germ cell neoplasms. J Clin Oncol. 1987;5(8):1290–4.
Dexeus FH, Logothetis CJ, Chong C, Sella A, Ogden S. Genetic abnormalities in men with germ cell tumors. J Urol. 1988;140(1):80–4.
Kolodziejski L, Duda K, Niezabitowski A, Dyczek S, Staniec B. Occurrence of malignant non-germ cell components in primary mediastinal germ cell tumours. Eur J Surg Oncol J Eur Soc Surg Oncol Br Assoc Surg Oncol. 1999;25(1):54–60. doi:10.1053/ejso.1998.0600.
Neiman RS, Orazi A. Mediastinal non-seminomatous germ cell tumours: their association with non-germ cell malignancies. Pathol Res Pract. 1999;195(8):589–94. doi:10.1016/S0344-0338(99)80010-7.
Contreras AL, Punar M, Tamboli P, Tu SM, Pisters L, Moran C, et al. Mediastinal germ cell tumors with an angiosarcomatous component: a report of 12 cases. Hum Pathol. 2010;41(6):832–7. doi:10.1016/j.humpath.2009.11.008.
Dulmet EM, Macchiarini P, Suc B, Verley JM. Germ cell tumors of the mediastinum. A 30-year experience. Cancer. 1993;72(6):1894–901.
Hartmann JT, Nichols CR, Droz JP, Horwich A, Gerl A, Fossa SD, et al. Hematologic disorders associated with primary mediastinal nonseminomatous germ cell tumors. J Natl Cancer Inst. 2000;92(1):54–61.
Zhao GQ, Dowell JE. Hematologic malignancies associated with germ cell tumors. Expert Rev Hematol. 2012;5(4):427–37. doi:10.1586/ehm.12.24.
Curry WA, McKay CE, Richardson RL, Greco FA. Klinefelter’s syndrome and mediastinal germ cell neoplasms. J Urol. 1981;125(1):127–9.
Nichols CR, Roth BJ, Heerema N, Griep J, Tricot G. Hematologic neoplasia associated with primary mediastinal germ-cell tumors. N Engl J Med. 1990;322(20):1425–9. doi:10.1056/NEJM199005173222004.
Krywicki R, Bowen K, Anderson L, Garland D, Cobb P, Jenkins T, et al. Mixed-lineage acute myeloid leukemia associated with a suprasellar dysgerminoma. Am J Clin Oncol. 1995;18(1):83–6.
Ulbright TM, Loehrer PJ, Roth LM, Einhorn LH, Williams SD, Clark SA. The development of non-germ cell malignancies within germ cell tumors. A clinicopathologic study of 11 cases. Cancer. 1984;54(9):1824–33.
Nichols CR. Mediastinal germ cell tumors. Clinical features and biologic correlates. Chest. 1991;99(2):472–9.
Hasle H, Jacobsen BB, Asschenfeldt P, Andersen K. Mediastinal germ cell tumour associated with Klinefelter syndrome. A report of case and review of the literature. Eur J Pediatr. 1992;151(10):735–9.
Hasle H, Mellemgaard A, Nielsen J, Hansen J. Cancer incidence in men with Klinefelter syndrome. Br J Cancer. 1995;71(2):416–20.
Kaido T, Sasaoka Y, Hashimoto H, Taira K. De novo germinoma in the brain in association with Klinefelter’s syndrome: case report and review of the literature. Surg Neurol. 2003;60(6):553–558; discussion 9.
Queipo G, Aguirre D, Nieto K, Pena YR, Palma I, Olvera J, et al. Intracranial germ cell tumors: association with Klinefelter syndrome and sex chromosome aneuploidies. Cytogenet Genome Res. 2008;121(3–4):211–4. doi:10.1159/000138887.
Satge D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans M. A tumor profile in Down syndrome. Am J Med Genet. 1998;78(3):207–216. doi:10.1002/(SICI)1096-8628(19980707)78:3<207::AID-AJMG1>3.0.CO;2-M [pii].
Hasle H. Pattern of malignant disorders in individuals with Down’s syndrome. Lancet Oncol. 2001;2(7):429–436. doi:S1470-2045(00)00435-6 [pii] 10.1016/S1470-2045(00)00435-6.
Matsui I, Tanimura M, Kobayashi N, Sawada T, Nagahara N, Akatsuka J. Neurofibromatosis type 1 and childhood cancer. Cancer. 1993;72(9):2746–54.
Walker L, Thompson D, Easton D, Ponder B, Ponder M, Frayling I, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. 2006;95(2):233–238. doi:6603227 [pii] 10.1038/sj.bjc.6603227.
Hartley AL, Birch JM, Kelsey AM, Marsden HB, Harris M, Teare MD. Are germ cell tumors part of the Li-Fraumeni cancer family syndrome? Cancer Genet Cytogenet. 1989;42(2):221–226. doi:0165-4608(89)90090-3 [pii].
Hisada M, Garber JE, Fung CY, Fraumeni Jr JF, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90(8):606–11.
Daugaard G, Rorth M, von der Maase H, Skakkebaek NE. Management of extragonadal germ-cell tumors and the significance of bilateral testicular biopsies. Ann Oncol. 1992;3(4):283–9.
Konig R, Schonberger W, Grimm W. [Mediastinal teratocarcinoma and hypophyseal stalk germinoma in a patient with Klinefelter syndrome] Mediastinales Terato-Carzinom und Hypophysenstielgerminom bei einem Patienten mit Klinefelter Syndrom. Klin Padiatr. 1990;202(1):53–6. doi:10.1055/s-2007-1025486.
Moran CA, Suster S, Przygodzki RM, Koss MN. Primary germ cell tumors of the mediastinum: II. Mediastinal seminomas – a clinicopathologic and immunohistochemical study of 120 cases. Cancer. 1997;80(4):691–698. doi:10.1002/(SICI)1097-0142(19970815)80:4<691::AID-CNCR7>3.0.CO;2-Q [pii].
Moran CA, Suster S. Mediastinal seminomas with prominent cystic changes. A clinicopathologic study of 10 cases. Am J Surg Pathol. 1995;19(9):1047–53.
Moran CA, Suster S. Mediastinal yolk sac tumors associated with prominent multilocular cystic changes of thymic epithelium: a clinicopathologic and immunohistochemical study of five cases. Mod Pathol Off J U S Can Acad Pathol Inc. 1997;10(8):800–3.
Kesler KA, Brooks JA, Rieger KM, Fineberg NS, Einhorn LH, Brown JW. Mediastinal metastases from testicular nonseminomatous germ cell tumors: patterns of dissemination and predictors of long-term survival with surgery. J Thorac Cardiovasc Surg. 2003;125(4):913–923. doi:10.1067/mtc.2003.407 S0022522303000990 [pii].
de Graaff WE, Oosterhuis JW, de Jong B, Dam A, van Putten WL, Castedo SM, et al. Ploidy of testicular carcinoma in situ. Lab Investig. 1992;66(2):166–8.
Sandberg AA, Abe S, Kowalczyk JR, Zedgenidze A, Takeuchi J, Kakati S. Chromosomes and causation of human cancer and leukemia. L. Cytogenetics of leukemias complicating other diseases. Cancer Genet Cytogenet. 1982;7(2):95–136. doi:0165-4608(82)90009-7 [pii].
Mann BD, Sparkes RS, Kern DH, Morton DL. Chromosomal abnormalities of a mediastinal embryonal cell carcinoma in a patient with 47,XXY Klinefelter syndrome: evidence for the premeiotic origin of a germ cell tumor. Cancer Genet Cytogenet. 1983;8(3):191–196. doi:0165-4608(83)90134-6 [pii].
Rodriguez E, Mathew S, Reuter V, Ilson DH, Bosl GJ, Chaganti RS. Cytogenetic analysis of 124 prospectively ascertained male germ cell tumors. Cancer Res. 1992;52(8):2285–91.
Kaplan CG, Askin FB, Benirschke K. Cytogenetics of extragonadal tumors. Teratology. 1979;19(2):261–6. doi:10.1002/tera.1420190217.
Oosterhuis JW, van den Berg E, de Jong B, Timens W, Castedo SM, Rammeloo RH, et al. Mediastinal germ cell tumor with secondary nongerm cell malignancy, and extensive hematopoietic activity. Pathology, DNA-ploidy, and karyotyping. Cancer Genet Cytogenet. 1991;54(2):183–195. doi:0165-4608(91)90206-A [pii].
Ladanyi M, Samaniego F, Reuter VE, Motzer RJ, Jhanwar SC, Bosl GJ, et al. Cytogenetic and immunohistochemical evidence for the germ cell origin of a subset of acute leukemias associated with mediastinal germ cell tumors. J Natl Cancer Inst. 1990;82(3):221–7.
Orazi A, Neiman RS, Ulbright TM, Heerema NA, John K, Nichols CR. Hematopoietic precursor cells within the yolk sac tumor component are the source of secondary hematopoietic malignancies in patients with mediastinal germ cell tumors. Cancer. 1993;71(12):3873–81.
Weidner N. Germ-cell tumors of the mediastinum. Semin Diagn Pathol. 1999;16(1):42–50.
Chaganti RS, Rodriguez E, Mathew S. Origin of adult male mediastinal germ-cell tumours. Lancet. 1994;343(8906):1130–1132. doi:S0140-6736(94)90235-6 [pii].
Hasle H, Jacobsen BB. Origin of male mediastinal germ-cell tumours. Lancet. 1995;345(8956):1046.
Wikstrom AM, Hoei-Hansen CE, Dunkel L, Rajpert-De Meyts E. Immunoexpression of androgen receptor and nine markers of maturation in the testes of adolescent boys with Klinefelter syndrome: evidence for degeneration of germ cells at the onset of meiosis. J Clin Endocrinol Metab. 2007;92(2):714–719. doi:jc.2006-1892 [pii] 10.1210/jc.2006-1892.
Beattie LM. Testicular dysgenesis: report of two cases. Can Serv Med J. 1957;8(8):469–79.
Isurugi K, Imao S, Hirose K, Aoki H. Seminoma in Klinefelter’s syndrome with 47, XXY, 15s+ karyotype. Cancer. 1977;39(5):2041–7.
Carroll PR, Morse MJ, Koduru PP, Chaganti RS. Testicular germ cell tumor in patient with Klinefelter syndrome. Urology. 1988;31(1):72–4.
Nakata Y, Yagishita A, Arai N. Two patients with intraspinal germinoma associated with Klinefelter syndrome: case report and review of the literature. AJNR Am J Neuroradiol. 2006;27(6):1204–1210. doi:27/6/1204 [pii].
Rapley EA, Crockford GP, Teare D, Biggs P, Seal S, Barfoot R, et al. Localization to Xq27 of a susceptibility gene for testicular germ-cell tumours. Nat Genet. 2000;24(2):197–200. doi:10.1038/72877.
Crockford GP, Linger R, Hockley S, Dudakia D, Johnson L, Huddart R, et al. Genome-wide linkage screen for testicular germ cell tumour susceptibility loci. Hum Mol Genet. 2006;15(3):443–451. doi:ddi459 [pii] 10.1093/hmg/ddi459.
North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, et al. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002;16(5):661–672. doi:S1074761302002960 [pii].
de Bruijn MF, Ma X, Robin C, Ottersbach K, Sanchez MJ, Dzierzak E. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity. 2002;16(5):673–683. doi:S1074761302003138 [pii].
Le PT, Kurtzberg J, Brandt SJ, Niedel JE, Haynes BF, Singer KH. Human thymic epithelial cells produce granulocyte and macrophage colony-stimulating factors. J Immunol. 1988;141(4):1211–7.
Back MR, Hu B, Rutgers J, French S, Moore TC. Metastasis of an intracranial germinoma through a ventriculoperitoneal shunt: recurrence as a yolk-sac tumor. Pediatr Surg Int. 1997;12(1):24–7. doi:10.1007/BF01194796.
Bi WL, Bannykh SI, Baehring J. The growing teratoma syndrome after subtotal resection of an intracranial nongerminomatous germ cell tumor in an adult: case report. Neurosurgery 2005;56:E191–E194.
Czirjak S, Pasztor E, Slowik F, Szeifert G. Third ventricle germinoma after total removal of intrasellar teratoma. Case report. J Neurosurg. 1992;77(4):643–7. doi:10.3171/jns.1992.77.4.0643.
Ikeda J, Sawamura Y, Kato T, Abe H. Metachronous neurohypophyseal germinoma occurring 8 years after total resection of pineal mature teratoma. Surg Neurol. 1998;49(2):205–208; discussion 8–9. doi:S0090-3019(97)00158-4 [pii].
Sugimoto K, Nakahara I, Nishikawa M. Bilateral metachronous germinoma of the basal ganglia occurring long after total removal of a mature pineal teratoma: case report. Neurosurgery. 2002;50(3):613–616; discussion 6–7.
Janzarik WG, Muller K, Lubbert M, Spreer J, Trippel M, Uhlig I, et al. Occurrence of a germinoma 22 years after resection of a mature cerebral teratoma. J Neuro Oncol. 2008;88(2):217–9. doi:10.1007/s11060-008-9554-0.
Mao Q, Ma L, Pang Z, Liu J. Germinoma occurring 2 years after total resection of an intracranial epidermoid cyst in the pineal region. J Neuro Oncol. 2012;106(2):437–9. doi:10.1007/s11060-011-0683-5.
Maeda Y, Yoshikawa K, Kajiwara K, Ideguchi M, Amano T, Saka M, et al. Intracranial yolk sac tumor in a patient with Down syndrome. J Neurosurg Pediatr. 2011;7(6):604–8. doi:10.3171/2011.3.PEDS10500.
Araki C, Matsumoto S. Statistical reevaluation of pinealoma and related tumors in Japan. J Neurosurg. 1969;30(2):146–9. doi:10.3171/jns.1969.30.2.0146.
Jennings MT, Gelman R, Hochberg F. Intracranial germ-cell tumors: natural history and pathogenesis. J Neurosurg. 1985;63(2):155–67. doi:10.3171/jns.1985.63.2.0155.
Packer RJ, Cohen BH, Cooney K. Intracranial germ cell tumors. Oncologist. 2000;5(4):312–20.
Kuratsu J, Ushio Y. Epidemiological study of primary intracranial tumors: a regional survey in Kumamoto Prefecture in the southern part of Japan. J Neurosurg. 1996;84(6):946–50. doi:10.3171/jns.1996.84.6.0946.
Committee of Brain Tumor Registry of Japan. Report of brain tumor registry of Japan (1969–1996). Neurol Med Chir (Tokyo). 2003;43(Suppl):i–vii.
Kaneko S, Nomura K, Yoshimura T, Yamaguchi N. Trend of brain tumor incidence by histological subtypes in Japan: estimation from the Brain Tumor Registry of Japan, 1973–1993. J Neuro Oncol. 2002;60(1):61–9.
Wong TT, Chen YW, Guo WY, Chang KP, Ho DM, Yen SH. Germinoma involving the basal ganglia in children. Childs Nerv Syst. 2008;24(1):71–8. doi:10.1007/s00381-007-0495-2.
Villani A, Bouffet E, Blaser S, Millar BA, Hawkins C, Bartels U. Inherent diagnostic and treatment challenges in germinoma of the basal ganglia: a case report and review of the literature. J Neuro Oncol. 2008;88(3):309–14. doi:10.1007/s11060-008-9568-7.
Trentini GP, Maiorana A, De Benedittis A. Metachronous seminoma of the pineal region and right testis. Case report. Appl Pathol. 1985;3(3):129–33.
Peat DS, Trowell JE. Testicular seminoma in a patient with pineal germinoma. J Clin Pathol. 1994;47(8):771–2.
Daniel C, Fizazi K, Culine S, Zelek L, Wibault P, Theodore C. Metachronous gonadal and extragonadal primaries, or late relapse of germ cell tumor? Urol Oncol. 2001;6(2):49–52. doi:S1078-1439(00)00091-0 [pii].
Iwata H, Mori Y, Takagi H, Shirahashi K, Shinoda J, Shimokawa K, et al. Mediastinal growing teratoma syndrome after cisplatin-based chemotherapy and radiotherapy for intracranial germinoma. J Thorac Cardiovasc Surg. 2004;127(1):291–293. doi:10.1016/S0022 S0022-5223(03)01300-X [pii].
Bedano PM, Bonnin J, Einhorn LH. Metachronous intracranial germinoma in a patient with a previous primary mediastinal seminoma. J Clin Oncol. 2006;24(15):2386–2387. doi:24/15/2386 [pii] 10.1200/JCO.2005.02.1576.
Benesch M, Schreibmayer N, Ratschek M, Hollwarth M, Lackner H, Urban C. Mediastinal yolk sac tumor ten years after treatment of intracranial germinoma. Med Pediatr Oncol. 2003;40(1):54–6. doi:10.1002/mpo.10031.
Rubinstein LJ. Tumors of the central nervous system. Atlas of tumor pathology, Series 2. Washington D.C.: Armed Forces Institute of Pathology; 1972.
Hashimoto T, Sasagawa I, Ishigooka M, Kubota Y, Nakada T, Fujita T, et al. Down’s syndrome associated with intracranial germinoma and testicular embryonal carcinoma. Urol Int. 1995;55(2):120–2.
Wakai S, Segawa H, Kitahara S, Asano T, Sano K, Ogihara R, et al. Teratoma in the pineal region in two brothers. Case reports. J Neurosurg. 1980;53(2):239–43. doi:10.3171/jns.1980.53.2.0239.
Kido G, Takeuchi T, Tsukiyama T, Nakamura S, Tsubokawa T, Henmi A. Tumor of the pineal region in three brothers. No Shinkei Geka. 1984;12(8):975–80.
Aoyama I, Kondo A, Ogawa H, Ikai Y. Germinoma in siblings: case reports. Surg Neurol. 1994;41(4):313–7.
Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459(7250):1126–1130. doi:nature08062 [pii] 10.1038/nature08062.
Wong TT, Ho DM, Chang TK, Yang DD, Lee LS. Familial neurofibromatosis 1 with germinoma involving the basal ganglion and thalamus. Childs Nerv Syst. 1995;11(8):456–8.
Aker FV, Berkman ZM, Aydingoz I, Hakan T, Toksoy G. Pineal germinoma associated with multiple congenital melanocytic nevi: a unique presentation. Neuropathology. 2005;25(4):336–40.
Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387(6634):733–6. doi:10.1038/42750.
Wolf SS, Patchev VK, Obendorf M. A novel variant of the putative demethylase gene, s-JMJD1C, is a coactivator of the AR. Arch Biochem Biophys. 2007;460(1):56–66. doi:S0003-9861(07)00031-8 [pii] 10.1016/j.abb.2007.01.017.
Kuroki S, Akiyoshi M, Tokura M, Miyachi H, Nakai Y, Kimura H, et al. JMJD1C, a JmjC domain-containing protein, is required for long-term maintenance of male germ cells in mice. Biol Reprod. 2013;89(4):93. doi:biolreprod.113.108597 [pii] 10.1095/biolreprod.113.108597.
Bartkova J, Hoei-Hansen CE, Krizova K, Hamerlik P, Skakkebaek NE, Rajpert-De Meyts E, et al. Patterns of DNA damage response in intracranial germ cell tumors versus glioblastomas reflect cell of origin rather than brain environment: implications for the anti-tumor barrier concept and treatment. Mol Oncol. 2014;8(8):1667–1678. doi:S1574-7891(14)00156-2 [pii] 10.1016/j.molonc.2014.07.001.
Sano K. Pathogenesis of intracranial germ cell tumors reconsidered. J Neurosurg. 1999;90(2):258–64. doi:10.3171/jns.1999.90.2.0258.
Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011;11(4):268–277. doi:nrc3034 [pii] 10.1038/nrc3034.
Matsaniotis N, Karpouzas J, Economou-Mavrou C. Hypothyroidism and seminoma in association with Down’s syndrome. J Pediatr. 1967;70(5):810–2.
Floret D, Renaud H, Monnet P. Sexual precocity and thoracic polyembryoma: Klinefelter syndrome? J Pediatr. 1979;94(1):163.
Masson P. [Not available] Etude sur le seminome. Rev Can Biol. 1946;5(4):361–87.
Scully RE. Spermatocytic seminoma of the testis. A report of 3 cases and review of the literature. Cancer. 1961;14:788–94.
Eble JN. Spermatocytic seminoma. Hum Pathol. 1994;25(10):1035–42.
Waheeb R, Hofmann MC. Human spermatogonial stem cells: a possible origin for spermatocytic seminoma. Int J Androl. 2011;34(4 Pt 2):e296–e305; discussion e. doi:10.1111/j.1365-2605.2011.01199.x.
Lim J, Goriely A, Turner GD, Ewen KA, Jacobsen GK, Graem N, et al. OCT2, SSX and SAGE1 reveal the phenotypic heterogeneity of spermatocytic seminoma reflecting distinct subpopulations of spermatogonia. J Pathol. 2011;224(4):473–83. doi:10.1002/path.2919.
Lombardi M, Valli M, Brisigotti M, Rosai J. Spermatocytic seminoma: review of the literature and description of a new case of the anaplastic variant. Int J Surg Pathol. 2011;19(1):5–10. doi:1066896910388645 [pii] 10.1177/1066896910388645.
Mikuz G, Bohm GW, Behrend M, Schafer G, Colecchia M, Verdorfer I. Therapy-resistant metastasizing anaplastic spermatocytic seminoma: a cytogenetic hybrid: a case report. Anal Quant Cytol Histol. 2014;36(3):177–82.
Floyd C, Ayala AG, Logothetis CJ, Silva EG. Spermatocytic seminoma with associated sarcoma of the testis. Cancer. 1988;61(2):409–14.
True LD, Otis CN, Delprado W, Scully RE, Rosai J. Spermatocytic seminoma of testis with sarcomatous transformation. A report of five cases. Am J Surg Pathol. 1988;12(2):75–82.
True LD, Otis CN, Rosai J, Scully RE, Delprado W. Spermatocytic seminoma of testis with sarcomatous transformation. Am J Surg Pathol. 1988;12(10):806.
Carriere P, Baade P, Fritschi L. Population based incidence and age distribution of spermatocytic seminoma. J Urol. 2007;178(1):125–128. doi:S0022-5347(07)00532-0 [pii] 10.1016/j.juro.2007.03.024.
Stevens MJ, Gildersleve J, Jameson CF, Horwich A. Spermatocytic seminoma in a maldescended testis. Br J Urol. 1993;72(5 Pt 1):657–9.
Muller J, Skakkebaek NE, Parkinson MC. The spermatocytic seminoma: views on pathogenesis. Int J Androl. 1987;10(1):147–56.
Dekker I, Rozeboom T, Delemarre J, Dam A, Oosterhuis JW. Placental-like alkaline phosphatase and DNA flow cytometry in spermatocytic seminoma. Cancer. 1992;69(4):993–6.
Kraggerud SM, Berner A, Bryne M, Pettersen EO, Fossa SD. Spermatocytic seminoma as compared to classical seminoma: an immunohistochemical and DNA flow cytometric study. APMIS. 1999;107(3):297–302.
Rosenberg C, Mostert MC, Schut TB, van de Pol M, van Echten J, de Jong B, et al. Chromosomal constitution of human spermatocytic seminomas: comparative genomic hybridization supported by conventional and interphase cytogenetics. Genes Chromosomes Cancer. 1998;23(4):286–291. doi:10.1002/(SICI)1098-2264(199812)23:4<286::AID-GCC2>3.0.CO;2-6 [pii].
Verdorfer I, Rogatsch H, Tzankov A, Steiner H, Mikuz G. Molecular cytogenetic analysis of human spermatocytic seminomas. J Pathol. 2004;204(3):277–81. doi:10.1002/path.1634.
Looijenga LH, Hersmus R, Gillis AJ, Pfundt R, Stoop HJ, van Gurp RJ, et al. Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res. 2006;66(1):290–302. doi:66/1/290 [pii] 10.1158/0008-5472.CAN-05-2936.
Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, et al. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet. 2009;41(11):1247–1252. doi:ng.470 [pii] 10.1038/ng.470.
Rajpert-De Meyts E, Jacobsen GK, Bartkova J, Aubry F, Samson M, Bartek J, et al. The immunohistochemical expression pattern of Chk2, p53, p19INK4d, MAGE-A4 and other selected antigens provides new evidence for the premeiotic origin of spermatocytic seminoma. Histopathology. 2003;42(3):217–226. doi:1587 [pii].
Kristensen DG, Mlynarska O, Nielsen JE, Jacobsen GK, Rajpert-De Meyts E, Almstrup K. Heterogeneity of chromatin modifications in testicular spermatocytic seminoma point toward an epigenetically unstable phenotype. Cancer Genet. 2012;205(9):425–431. doi:S2210-7762(12)00134-2 [pii] 10.1016/j.cancergen.2012.05.003.
Meng X, Lindahl M, Hyvonen ME, Parvinen M, de Rooij DG, Hess MW, et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287(5457):1489–1493. doi:8297 [pii].
Meng X, de Rooij DG, Westerdahl K, Saarma M, Sariola H. Promotion of seminomatous tumors by targeted overexpression of glial cell line-derived neurotrophic factor in mouse testis. Cancer Res. 2001;61(8):3267–71.
Sariola H, Meng X. GDNF-induced seminomatous tumours in mouse – an experimental model for human seminomas? APMIS. 2003;111(1):192–196; discussion 6. doi:apm1110123 [pii].
Matson CK, Murphy MW, Griswold MD, Yoshida S, Bardwell VJ, Zarkower D. The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev Cell. 2010;19(4):612–624. doi:S1534-5807(10)00428-4 [pii] 10.1016/j.devcel.2010.09.010.
Looijenga LH, Olie RA, van der Gaag I, van Sluijs FJ, Matoska J, Ploem-Zaaijer J, et al. Seminomas of the canine testis. Counterpart of spermatocytic seminoma of men? Lab Investig. 1994;71(4):490–6.
Portas TJ, Hermes R, Bryant BR, Goritz F, Ladds P, Hildebrant TB. Seminoma in a southern white rhinoceros (Ceratotherium simum simum). Vet Rec. 2005;157(18):556–558. doi:157/18/556 [pii].
Subramaniam K, Seydoux G. Dedifferentiation of primary spermatocytes into germ cell tumors in C. elegans lacking the pumilio-like protein PUF-8. Curr Biol. 2003;13(2):134–139. doi:S0960982203000058 [pii].
Bush JM, Gardiner DW, Palmer JS, Rajpert-De Meyts E, Veeramachaneni DN. Testicular germ cell tumours in dogs are predominantly of spermatocytic seminoma type and are frequently associated with somatic cell tumours. Int J Androl. 2011;34(4 Pt 2):e288–e295; discussion e95. doi:10.1111/j.1365-2605.2011.01166.x.
Kaku H, Usui H, Qu J, Shozu M. Mature cystic teratomas arise from meiotic oocytes, but not from pre-meiotic oogonia. Genes Chromosomes Cancer. 2016;55(4):355–64. doi:10.1002/gcc.22339.
Dick HM, Honore LH. Dental structures in benign ovarian cystic teratomas (dermoid cysts). A study of ten cases with a review of the literature. Oral Surg Oral Med Oral Pathol. 1985;60(3):299–307.
Kondi-Pafiti A, Filippidou-Giannopoulou A, Papakonstantinou E, Iavazzo C, Grigoriadis C. Epidermoid or dermoid cysts of the ovary? Clinicopathological characteristics of 28 cases and a new pathologic classification of an old entity. Eur J Gynaecol Oncol. 2012;33(6):617–9.
Roth LM, Talerman A. Recent advances in the pathology and classification of ovarian germ cell tumors. Int J Gynecol Pathol. 2006;25(4):305–20. doi:10.1097/01.pgp.0000225844.59621.9d.
Nogales FF, Preda O, Dulcey I. Gliomatosis peritonei as a natural experiment in tissue differentiation. Int J Dev Biol. 2012;56(10–12):969–74. doi:10.1387/ijdb.120172fn.
Blanchet J, Fouquette B. Case of malignant ovarian cystic teratoma with bilateral immature differentiation. Can Med Assoc J. 1979;120(11):1396–8.
Koonings PP, Campbell K, Mishell Jr DR, Grimes DA. Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol. 1989;74(6):921–6.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi:10.1002/ijc.29210.
Hegde P. Extragonadal omental teratoma: a case report. J Obstet Gynaecol Res. 2014;40(2):618–21. doi:10.1111/jog.12198.
Mazzarella P, Okagaki T, Richart RM. Teratoma of the uterine tube. A case report and review of the literature. Obstet Gynecol. 1972;39(3):381–8.
Ohshima K, Umeda A, Hosoi A, Yamamoto T, Munakata S. Mature cystic teratoma in Douglas’ pouch. Case Rep Pathol. 2015;2015:202853. doi:10.1155/2015/202853.
Surti U, Hoffner L, Chakravarti A, Ferrell RE. Genetics and biology of human ovarian teratomas. I. Cytogenetic analysis and mechanism of origin. Am J Hum Genet. 1990;47(4):635–43.
Devouassoux-Shisheboran M, Vortmeyer AO, Silver SA, Zhuang Z, Tavassoli FA. Teratomatous genotype detected in malignancies of a non-germ cell phenotype. Lab Investig. 2000;80(1):81–6.
Schmidt J, Derr V, Heinrich MC, Crum CP, Fletcher JA, Corless CL, et al. BRAF in papillary thyroid carcinoma of ovary (struma ovarii). Am J Surg Pathol. 2007;31(9):1337–43. doi:10.1097/PAS.0b013e31802f5404.
Wolff EF, Hughes M, Merino MJ, Reynolds JC, Davis JL, Cochran CS, et al. Expression of benign and malignant thyroid tissue in ovarian teratomas and the importance of multimodal management as illustrated by a BRAF-positive follicular variant of papillary thyroid cancer. Thyroid Off J Am Thyroid Assoc. 2010;20(9):981–7. doi:10.1089/thy.2009.0458.
Miura K, Obama M, Yun K, Masuzaki H, Ikeda Y, Yoshimura S, et al. Methylation imprinting of H19 and SNRPN genes in human benign ovarian teratomas. Am J Hum Genet. 1999;65(5):1359–67. doi:10.1086/302615.
Linder D. Gene loss in human teratomas. Proc Natl Acad Sci U S A. 1969;63(3):699–704.
Linder D, Power J. Further evidence for post-meiotic origin of teratomas in the human female. Ann Hum Genet. 1970;34(1):21–30.
Linder D, Hecht F, McCaw BK, Campbell JR. Origin of extragonadal teratomas and endodermal sinus tumours. Nature. 1975;254(5501):597–8.
Linder D, McCaw BK, Hecht F. Parthenogenic origin of benign ovarian teratomas. N Engl J Med. 1975;292(2):63–6. doi:10.1056/NEJM197501092920202.
Carritt B, Parrington JM, Welch HM, Povey S. Diverse origins of multiple ovarian teratomas in a single individual. Proc Natl Acad Sci U S A. 1982;79(23):7400–4.
Parrington JM, West LF, Povey S. The origin of ovarian teratomas. J Med Genet. 1984;21(1):4–12.
Ohama K, Nomura K, Okamoto E, Fukuda Y, Ihara T, Fujiwara A. Origin of immature teratoma of the ovary. Am J Obstet Gynecol. 1985;152(7 Pt 1):896–900.
Ohama K. Androgenesis and parthenogenesis in humans. In:Human genetics. Berlin: Springer-Verlag; 1987. p. 245–9.
Deka R, Chakravarti A, Surti U, Hauselman E, Reefer J, Majumder PP, et al. Genetics and biology of human ovarian teratomas. II. Molecular analysis of origin of nondisjunction and gene-centromere mapping of chromosome I markers. Am J Hum Genet. 1990;47(4):644–55.
Pangas SA. Regulation of the ovarian reserve by members of the transforming growth factor beta family. Mol Reprod Dev. 2012;79(10):666–79. doi:10.1002/mrd.22076.
Zhang M, Su YQ, Sugiura K, Wigglesworth K, Xia G, Eppig JJ. Estradiol promotes and maintains cumulus cell expression of natriuretic peptide receptor 2 (NPR2) and meiotic arrest in mouse oocytes in vitro. Endocrinology. 2011;152(11):4377–4385. doi:en.2011-1118 [pii] 10.1210/en.2011-1118.
Azoury RS, Jubayli NW, Barakat BY. Dermoid cyst of ovary containing fetus-like structure. Obstet Gynecol. 1973;42(6):887–91.
Lee YH, Kim SG, Choi SH, Kim IS, Kim SH. Ovarian mature cystic teratoma containing homunculus: a case report. J Korean Med Sci. 2003;18(6):905–907. doi:200312905 [pii] 10.3346/jkms.2003.18.6.905.
Kuno N, Kadomatsu K, Nakamura M, Miwa-Fukuchi T, Hirabayashi N, Ishizuka T. Mature ovarian cystic teratoma with a highly differentiated homunculus: a case report. Birth Defects Res A Clin Mol Teratol. 2004;70(1):40–6. doi:10.1002/bdra.10133.
Seckl MJ, Sebire NJ, Berkowitz RS. Gestational trophoblastic disease. Lancet. 2010;376(9742):717–729. doi:S0140-6736(10)60280-2 [pii] 10.1016/S0140-6736(10)60280-2.
Lurain JR. Gestational trophoblastic disease II: classification and management of gestational trophoblastic neoplasia. Am J Obstet Gynecol. 2011;204(1):11–18. doi:S0002-9378(10)00852-5 [pii] 10.1016/j.ajog.2010.06.072.
Hui P. Molar pregnancies. In: Kurman RJ, editor. World Health Organization classification of tumours of female reproductive organs. Lyon: IARC Press; 2014. p. 163–6.
Yoshida K, Nagasaka T, Nakashima N, Nishida Y, Saito M, Tomomitsu O. Elucidation of vascular structure of molar villi in complete hydatidiform mole by CD-34 antibody. Int J Gynecol Pathol. 2000;19(3):212–8.
Kim KR, Park BH, Hong YO, Kwon HC, Robboy SJ. The villous stromal constituents of complete hydatidiform mole differ histologically in very early pregnancy from the normally developing placenta. Am J Surg Pathol. 2009;33(2):176–85. doi:10.1097/PAS.0b013e31817fada1.
Ngan HY, Kohorn EI, Cole LA, Kurman RJ, Kim SJ, Lurain JR, et al. Trophoblastic disease. Int J Gynaecol Obstet. 2012;119 Suppl 2:S130–S136. doi:S0020-7292(12)60026-5 [pii] 10.1016/S0020-7292(12)60026-5.
Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300(5618):455. doi:10.1126/science.1083557 300/5618/455 [pii].
Novakovic B, Saffery R. DNA methylation profiling highlights the unique nature of the human placental epigenome. Epigenomics. 2010;2(5):627–38. doi:10.2217/epi.10.45.
Xue WC, Chan KY, Feng HC, Chiu PM, Ngan HY, Tsao SW, et al. Promoter hypermethylation of multiple genes in hydatidiform mole and choriocarcinoma. J Mol Diagn. 2004;6(4):326–334. doi:S1525-1578(10)60528-4 [pii] 10.1016/S1525-1578(10)60528-4.
Hertig AT. Tumors of the female sex organs. Part I hydatidiform mole and choriocarcinoma. Armed Forces Institute of Pathology: Washington, D.C; 1956.
Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38(3):300–302. doi:ng1740 [pii] 10.1038/ng1740.
Sebire NJ, Lindsay I, Fisher RA, Savage P, Seckl MJ. Overdiagnosis of complete and partial hydatidiform mole in tubal ectopic pregnancies. Int J Gynecol Pathol. 2005;24(3):260–264. doi:00004347-200507000-00010 [pii].
Kajii T, Ohama K. Androgenetic origin of hydatidiform mole. Nature. 1977;268(5621):633–4.
Lipata F, Parkash V, Talmor M, Bell S, Chen S, Maric V, et al. Precise DNA genotyping diagnosis of hydatidiform mole. Obstet Gynecol. 2010;115(4):784–794. doi:10.1097/AOG.0b013e3181d489ec 00006250-201004000-00018 [pii].
Fukunaga M, Endo Y, Ushigome S. Clinicopathologic study of tetraploid hydropic villous tissues. Arch Pathol Lab Med. 1996;120(6):569–72.
Wake N, Takagi N, Sasaki M. Androgenesis as a cause of hydatidiform mole. J Natl Cancer Inst. 1978;60(1):51–7.
Jacobs PA, Szulman AE, Funkhouser J, Matsuura JS, Wilson CC. Human triploidy: relationship between parental origin of the additional haploid complement and development of partial hydatidiform mole. Ann Hum Genet. 1982;46(Pt 3):223–31.
Lawler SD, Fisher RA, Pickthall VJ, Povey S, Evans MW. Genetic studies on hydatidiform moles. I. The origin of partial moles. Cancer Genet Cytogenet. 1982;5(4):309–20.
Genest DR. Partial hydatidiform mole: clinicopathological features, differential diagnosis, ploidy and molecular studies, and gold standards for diagnosis. Int J Gynecol Pathol. 2001;20(4):315–22.
Blum B, Benvenisty N. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle. 2009;8(23):3822–3830. doi:10067 [pii] 10.4161/cc.8.23.10067.
Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24(20):2239–2263. doi:24/20/2239 [pii] 10.1101/gad.1963910.
Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25(2):207–215. doi:nbt1285 [pii] 10.1038/nbt1285.
Draper JS, Moore HD, Ruban LN, Gokhale PJ, Andrews PW. Culture and characterization of human embryonic stem cells. Stem Cells Dev. 2004;13(4):325–36. doi:10.1089/scd.2004.13.325.
Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37(10):1099–1103. doi:ng1631 [pii] 10.1038/ng1631.
Werbowetski-Ogilvie TE, Bosse M, Stewart M, Schnerch A, Ramos-Mejia V, Rouleau A, et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat Biotechnol. 2009;27(1):91–97. doi:nbt.1516 [pii] 10.1038/nbt.1516.
Lefort N, Feyeux M, Bas C, Feraud O, Bennaceur-Griscelli A, Tachdjian G, et al. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21 Nat Biotechnol. 2008;26(12):1364–1366. doi:nbt.1509 [pii] 10.1038/nbt.1509.
Spits C, Mateizel I, Geens M, Mertzanidou A, Staessen C, Vandeskelde Y, et al. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat Biotechnol. 2008;26(12):1361–1363. doi:nbt.1510 [pii] 10.1038/nbt.1510.
Inzunza J, Sahlen S, Holmberg K, Stromberg AM, Teerijoki H, Blennow E, et al. Comparative genomic hybridization and karyotyping of human embryonic stem cells reveals the occurrence of an isodicentric X chromosome after long-term cultivation. Mol Hum Reprod. 2004;10(6):461–466. doi:10.1093/molehr/gah051 gah051 [pii].
Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156(4):663–677. doi:S0092-8674(14)00015-4 [pii] 10.1016/j.cell.2014.01.005.
Shih CC, Forman SJ, Chu P, Slovak M. Human embryonic stem cells are prone to generate primitive, undifferentiated tumors in engrafted human fetal tissues in severe combined immunodeficient mice. Stem Cells Dev. 2007;16(6):893–902. doi:10.1089/scd.2007.0070.
Unzu C, Friedli M, Bosman A, Jaconi ME, Wildhaber BE, Rougemont AL. Human Hepatocyte-Derived Induced Pluripotent Stem Cells: MYC Expression, Similarities to Human Germ Cell Tumors, and Safety Issues. Stem Cells Int. 2016;2016:4370142. doi:10.1155/2016/4370142.
Misra P, Husain Q, Svider PF, Sanghvi S, Liu JK, Eloy JA. Management of sinonasal teratocarcinosarcoma: a systematic review. Am J Otolaryngol. 2014;35(1):5–11. doi:S0196-0709(13)00117-8 [pii] 10.1016/j.amjoto.2013.04.010.
Kinjo T, Taniguchi H, Kushima R, Sekine S, Oda I, Saka M, et al. Histologic and immunohistochemical analyses of alpha-fetoprotein – producing cancer of the stomach. Am J Surg Pathol. 2012;36(1):56–65. doi:10.1097/PAS.0b013e31823aafec 00000478-201201000-00008 [pii].
Preda O, Dema A, Iacob M, Goyenaga P, Dulcey I, Aneiros Fernandez J, et al. Urothelial carcinoma of the renal pelvis with simultaneous trophoblastic and malignant clear cell endodermal-type differentiation. Virchows Arch. 2012;460(3):353–6. doi:10.1007/s00428-012-1211-5.
Nogales FF, Bergeron C, Carvia RE, Alvaro T, Fulwood HR. Ovarian endometrioid tumors with yolk sac tumor component, an unusual form of ovarian neoplasm. Analysis of six cases. Am J Surg Pathol. 1996;20(9):1056–66.
Liang L, Zhang Y, Malpica A, Ramalingam P, Euscher ED, Fuller GN, et al. Gliomatosis peritonei: a clinicopathologic and immunohistochemical study of 21 cases. Mod Pathol Off J U S Can Acad Pathol Inc. 2015;28(12):1613–1620. doi:modpathol2015116 [pii] 10.1038/modpathol.2015.116.
Alexander M, Cope N, Renninson J, Hong A, Simpson RH, Hirschowitz L. Relationship between endometriosis, endometrioid adenocarcinoma, gliomatosis peritonei, and carcinoid tumor in a patient with recurrent ovarian teratoma. Int J Gynecol Pathol. 2011;30(2):151–7. doi:10.1097/PGP.0b013e3181f6bcd4.
Virant-Klun I, Zech N, Rozman P, Vogler A, Cvjeticanin B, Klemenc P, et al. Putative stem cells with an embryonic character isolated from the ovarian surface epithelium of women with no naturally present follicles and oocytes. Differentiation. 2008;76(8):843–856. doi:S0301-4681(09)60024-4 [pii] 10.1111/j.1432-0436.2008.00268.x.
Virant-Klun I, Rozman P, Cvjeticanin B, Vrtacnik-Bokal E, Novakovic S, Rulicke T, et al. Parthenogenetic embryo-like structures in the human ovarian surface epithelium cell culture in postmenopausal women with no naturally present follicles and oocytes. Stem Cells Dev. 2009;18(1):137–49. doi:10.1089/scd.2007.0238.
van Echten J, de Jong B, Sinke RJ, Weghuis DO, Sleijfer DT, Oosterhuis JW. Definition of a new entity of malignant extragonadal germ cell tumors. Genes Chromosomes Cancer. 1995;12(1):8–15.
Noguera R, Navarro S, Carda C, Peydro-Olaya A, Llombart-Bosch A. Near-haploidy in a malignant sacrococcygeal teratoma. Cancer Genet Cytogenet. 1999;108(1):70–74. doi:S0165460898001150 [pii].
Chinoy RF, Soman CS, Swaroop D, Badwar RA. Extragonadal malignant teratoma of the foot. Indian J Cancer. 1992;29(2):96–9.
Ait Benali H, Lalya L, Allaoui M, Benmansour A, Elkhanoussi B, Benjelloun S, et al. Extragonadal mixed germ cell tumor of the right arm: description of the first case in the literature. World J Surg Oncol. 2012;10:69. doi:1477-7819-10-69 [pii] 10.1186/1477-7819-10-69.
Garcia-Galvis OF, Cabrera-Ozoria C, Fernandez JA, Stolnicu S, Nogales FF. Malignant Mullerian mixed tumor of the ovary associated with yolk sac tumor, neuroepithelial and trophoblastic differentiation (teratoid carcinosarcoma). Int J Gynecol Pathol. 2008;27(4):515–20. doi:10.1097/PGP.0b013e31817b06c7.
Salem F, Rosenblum MK, Jhanwar SC, Kancherla P, Ghossein RA, Carlson DL. Teratocarcinosarcoma of the nasal cavity and paranasal sinuses: report of 3 cases with assessment for chromosome 12p status. Hum Pathol. 2008;39(4):605–609. doi:S0046-8177(07)00486-8 [pii] 10.1016/j.humpath.2007.09.002.
Thomas J, Adegboyega P, Iloabachie K, Mooring JW, Lian T. Sinonasal teratocarcinosarcoma with yolk sac elements: a neoplasm of somatic or germ cell origin? Ann Diagn Pathol. 2011;15(2):135–139. doi:S1092-9134(10)00009-2 [pii] 10.1016/j.anndiagpath.2010.01.004.
Sinke RJ, Weghuis DO, Suijkerbuijk RF, Tanigami A, Nakamura Y, Larsson C, et al. Molecular characterization of a recurring complex chromosomal translocation in two human extragonadal germ cell tumors. Cancer Genet Cytogenet. 1994;73(1):11–6.
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi:10.1038/nature02625 nature02625 [pii].
Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi:nature05919 [pii] 10.1038/nature05919.
Jelinic P, Shaw P. Loss of imprinting and cancer. J Pathol. 2007;211(3):261–8. doi:10.1002/path.2116.
Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62(22):6442–6.
Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299(5613):1753–1755. doi:10.1126/science.1080902 299/5613/1753 [pii].
Kim HT, Choi BH, Niikawa N, Lee TS, Chang SI. Frequent loss of imprinting of the H19 and IGF-II genes in ovarian tumors. Am J Med Genet. 1998;80(4):391–395. doi:10.1002/(SICI)1096-8628(19981204)80:4<391::AID-AJMG16>3.0.CO;2-H [pii].
Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi:ng.127 [pii] 10.1038/ng.127.
Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143(2):313–324. doi:S0092-8674(10)01058-5 [pii] 10.1016/j.cell.2010.09.010.
Yun K. A new marker for rhabdomyosarcoma. Insulin-like growth factor II. Lab Investig. 1992;67(5):653–64.
Baccarini P, Fiorentino M, D’Errico A, Mancini AM, Grigioni WF. Detection of anti-sense transcripts of the insulin-like growth factor-2 gene in Wilms’ tumor. Am J Pathol. 1993;143(6):1535–42.
Akmal SN, Yun K, MacLay J, Higami Y, Ikeda T. Insulin-like growth factor 2 and insulin-like growth factor binding protein 2 expression in hepatoblastoma. Hum Pathol. 1995;26(8):846–51.
Korkola JE, Houldsworth J, Chadalavada RS, Olshen AB, Dobrzynski D, Reuter VE, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66(2):820–827. doi:66/2/820 [pii] 10.1158/0008-5472.CAN-05-2445.
Hirasawa R, Feil R. Genomic imprinting and human disease. Essays Biochem 2010;48:187–200.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer-Verlag GmbH Germany
About this chapter
Cite this chapter
Oosterhuis, J.W., Looijenga, L.H.J. (2017). Germ Cell Tumors from a Developmental Perspective: Cells of Origin, Pathogenesis, and Molecular Biology (Emerging Patterns). In: Nogales, F., Jimenez, R. (eds) Pathology and Biology of Human Germ Cell Tumors. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-53775-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-662-53775-6_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-53773-2
Online ISBN: 978-3-662-53775-6
eBook Packages: MedicineMedicine (R0)