
Abstract
While germ cell tumors (GCTs) comprise only about 1 % of cancers diagnosed each year in American men, they still constitute the most common malignancy to affect male adolescent and young adults (AYA) in the US and other developed countries. Most commonly, these tumors originate in the testis and comprise more than 95 % of all testicular malignancies. However, GCTs can also arise in extragonadal locations such as the mediastinum, pineal gland, and retroperitoneum. Despite having a nearly universal genetic marker, isochromosome 12p, GCTs are not a singular tumor type but rather a fascinating group of malignancies derived from the malignant transformation of developing germ cells. The pluripotential differentiating capacity of developing germ cells explains the intriguing biology and array of histologies found in GCTs. These histologies mimic the cell and tissue types seen during embryologic and extraembryologic fetal development. In this chapter, the authors review GCT epidemiology, highlighting well-known risk factors such as cryptorchidism as well as newly appreciated risk factors such as infertility and genetic polymorphisms, with plausible explanations of how these characteristics may be interrelated. An overview of the biology of GCT development and differentiation is also presented with an emphasis on recent insights and persistent controversies. The authors provide a detailed review of GCT genetics including potential candidate genes and regions within 12p and other frequent chromosomal aberrations. In the latter part of the chapter, the authors summarize the data supporting and refuting proposed explanations for the exquisite sensitivity of most GCTs to cisplatin, which underlies their unique curability even in the setting of widely metastatic disease. Finally, various mechanisms by which GCTs might acquire or have inherent cisplatin resistance are presented. As such, the reader of this chapter will be left with a solid understanding of the epidemiology, biology, and genetics of this fascinating malignancy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Germ cell tumors
- Testicular cancer
- Isochromosome 12p
- Differentiation
- Cisplatin resistance
- Epidemiology
- Embryologic development
Introduction
Germ cell tumors (GCTs) comprise more than 95 % of testicular neoplasms [1, 2] and account for the most common cancer diagnosis each year among adolescent and young adult (AYA) men in developed countries. Despite being associated with a nearly universal favorable outcome, GCTs display extraordinary diversity in their histologic appearance, clinical presentation, and genetic composition, which relates to their unique biology as the malignant counterpart of a spectrum of tissues involved in human development. Derived from malignant transformation of a germ cell, histologically, GCTs are broadly separated into two histologic subtypes, seminomatous GCT (SGCT) and nonseminomatous GCT (NSGCT). More than 90 % of GCTs originate in the testis, although they can also arise at extragonadal sites, typically midline structures such as the mediastinum, retroperitoneum, and pineal gland. This chapter will review the epidemiology, biology, and genetics of GCTs with a special focus on insights gained over the last decade as well as persistent areas of controversy.
Epidemiology
Since most agencies providing cancer statistics (e.g., American Cancer Society, SEER) report tumor incidence by primary site (e.g., breast, lung, testis, etc.) rather than histologic type, annual crude incidence rates for GCTs must be estimated from knowledge of the annual testicular cancer incidence (8,590 cases per year) [3], the proportion of testicular cancers that are GCTs (>95 %) [1, 4], and the proportion of GCTs that arise outside of the testis (6 %) [5]. Based on these values, we estimate that approximately 9,000 cases of GCT will be diagnosed among men in the United States in 2012. As such, GCTs comprise only slightly more than 1 % of tumors diagnosed in American men each year. The number of cases diagnosed in women is even smaller and is beyond the scope of this review.
It is important to note that while GCTs are rare, they still represent the most common tumor to affect men between the ages of 15 and 34. In addition, the incidence has been increasing by more than 1 % per year over the last few decades [6–8]. The median age at the time of diagnosis ranges from 25 to 29 for NSGCT and from 35 to 39 for SGCT [9]. Similar to histology, primary tumor site also varies by age with pineal gland GCTs nearly always diagnosed before the age of 30 [5]. Incidences also vary by location and ethnic/racial background. For example, the incidence is as high as 10 per 100,000 among men in Scandinavian countries such as Norway compared to approximately 5 per 100,000 among men in the United States and approximately 1 per 100,000 among men in parts of Asia and Africa [6]. GCTs are also significantly more common in Caucasians, compared to African Americans, Hispanic Americans, and Asian Americans [9].
Risk factors for development of GCT are listed in Table 26.1. The most well proven risk factor for testicular GCTs is a history of cryptorchidism. Without surgical correction, approximately 5–10 % of cryptorchid patients will develop testicular GCT [10]. While orchiopexy, particularly when performed early in life (and certainly before puberty) [11] can decrease this risk, it does not eliminate it, and these patients also remain at risk for development of contralateral testicular GCTs. Hypospadias, another common male congenital malformation, was also recently demonstrated to increase the risk of testicular GCT development in a Danish cohort study [12].
Infertility and subfertility have been associated with increased risks of developing testicular GCTs. One study found that 0.25 % of men presenting with infertility were subsequently found to have testicular cancer, a rate approximately 20-fold higher than the SEER estimates (0.01 %) of the incidence among men of similar age and race during the same time period [13]. Patients with disorders of sex development, which commonly coexist with male infertility, also appear to be at increased risk of testicular GCT development [14].
A prior history of GCT or its noninvasive precursor lesion, intratubular germ cell neoplasia of unknown significance (ITGCNU) are additional strong risk factors for the development of GCTs. Approximately 2 % of men in the United States with unilateral testicular GCT will eventually develop contralateral GCT during their lifetime [15]. In Scandinavian countries where testicular cancer has a higher incidence, the risk of contralateral GCT may be even greater [16, 17]. If untreated, IGCNU, identified on testicular biopsy, performed for the workup of infertility or in a patient with a diagnosis of GCT in the contralateral testis, portends an extremely high risk of invasive GCT development (≈50 % within 5 years) [18, 19]. A prior history of extragonadal GCT is also associated with an increased risk of a subsequent testicular GCT diagnosis [20].
Family history constitutes another risk factor for GCT, with brothers of patients with testicular GCTs carrying a 5–12-fold increase in risk and sons accruing an approximately two-fold increase in risk [21–24]. Recently, several germ-line DNA single nucleotide polymorphisms (SNPs) were found to increase the likelihood of developing GCTs, possibly explaining a familial predilection. Genome wide association studies (GWAS) performed by two independent groups demonstrated that patients homozygous for a SNP within the KIT ligand (KITLG/SCF) on chromosome 12q22 carried up to a 4.5-fold increased risk of developing a GCT [25, 26]. In addition, SNPs in downstream effectors of KIT such as the SPRY4 gene on chromosome 5q313 were also associated with an increased risk of developing testicular GCTs [25]. Since KIT signaling is known to play an important role in germ cell development and fertility [27, 28], these findings may provide a plausible pathway-based explanation as to why testicular GCTs may be more common in men with infertility or subfertility.
Some environmental exposures have recently been proposed as risk factors for the development of testicular GCTs. Two retrospective case-control studies found frequent and long-term marijuana use to be more prevalent among patients diagnosed with nonseminomatous (but not seminomatous) testicular cancer as compared to age-matched controls [29, 30]. Other environmental exposures, especially those occurring in utero, such as pesticides, have also been proposed to increase the risk of testicular GCT development [31, 32]. Finally, Klinefelter’s syndrome has been demonstrated to significantly increase the risk for mediastinal but not testicular GCTs [33]. No other mediastinal GCT risk factors have thus far been identified.
Pathobiology and Histology of Germ Cell Tumors
Germ Cell Development
GCTs have a fascinating biology which relates to the pluripotent nature of their cell of origin, the developing germ cell. A brief review of male gonadal development, spermatogenesis, and embryogenesis can be helpful to understanding the biology and range of histologies seen in GCTs. Arising from the embryonic ectoderm, the primordial germ cell (PGC) is first recognized during gastrulation based on its expression of alkaline phosphatase [34]. PGCs subsequently migrate to the genital ridge, where they further develop into gender-distinct gonads, a process dependent upon the presence (male) or absence (female) of stromal expression of the SRY gene, located on the Y chromosome. In males, PGCs in combination with Sertoli cells form seminiferous cords which, along with Leydig cells, subsequently organize into the embryonic gonads by about 2 months of gestational age. PGCs differentiate into gonocytes, which then cease proliferation until after birth. Postnatally, gonocytes begin proliferating again, and mature into undifferentiated Type A spermatogonia by about 3 months of age. Prior to and culminating with the initiation of puberty under stimulation from gonadotrophins, Type A spermatogonia mature into Type B spermatogonia. In AYA men, Type A spermatogonia are postulated to comprise the gonadal stem cells, existing as either Type Ad (dark type), a non-dividing germ cell reserve in case of destruction or loss, or Type Ap (pale type), an actively dividing, possibly self-renewing germ cell population. Following a single mitotic division, Type B spermatogonia become primary spermatocytes, which in turn undergo DNA replication and then two meioses, ultimately resulting in 4 haploid gametes. Spermiogenesis ensues, leading to the formation of mature spermatozoa.
Germ Cell Transformation to GCT and Histologic Differentiation
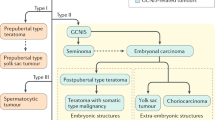
GCTs arise when developing germ cells undergo malignant transformation. The earliest recognizable abnormal histology during this transformation is ITGCNU which is thought to represent the non-invasive precursor to all GCTs [35]. Upon becoming invasive, GCTs are separated into the two histologic categories, seminoma and nonseminoma.
Seminomas are more similar to ITGCNU than nonseminomas, morphologically resembling undifferentiated spermatogonial germ cells and expressing proteins common to germ cells in early development such as placental alkaline phosphatase (PLAP), KIT, and POU5F1 (OCT3/4) [36–38]. Seminomas typically display low mitotic and apoptotic rates and tend to be clinically diagnosed in Stage I (limited to the testis), often curable with orchiectomy alone [1]. Nonseminomas include four distinct histologies (embryonal carcinoma [EC], yolk sac tumor [YST], choriocarcinoma [CC], and teratoma [T]), each of which parallels a different stage of embryonic or extraembryonic development and differentiation. Normal germ cells destined to become gametes are subject to inhibitory signaling that prevents them from undergoing differentiation until fertilization with ova is achieved. Thus, nonseminoma formation can be explained by germ cells undergoing reprogramming during malignant transformation resulting in the acquisition of the capacity for embryonic and extraembryonic differentiation, although in a spatially and temporally aberrant manner. In vitro evidence supports this view as normal PGCs isolated from the mouse and human can be converted into pluripotent cells (embryonic germ cells) following exposure to KIT ligand (stem cell factor), leukemia inhibitory factor (LIF), and basic fibroblast factor (bFGF) [39]. Epigenetic modifications such as DNA methylation and chromatin acetylation may play a role in the reprogramming process [40]. In contrast, SGCTs lack the ability to initiate differentiation.
Differentiation of EC along embryonic (T) or extraembryonic (YST, CC) pathways leads to the decline of pluripotency of the transformed germ cell paralleling the process in embryonic development. For example, expression of POU5F1 in EC is downregulated in T, CC, and YST. [41] EC is considered the malignant counterpart of an early embryo and is pluripotent. ECs display the highest mitotic and apoptotic rates of any GCT histology and have been demonstrated to be genetically similar to embryonic stem cells [42]. The normal zygote at this stage is comprised of an inner cell mass surrounded by trophectoderm. The inner cell mass gives rise to the fetal tissues and the extraembryonic endoderm, whereas the trophectoderm gives rise to the placenta, consisting of an outer syncytiotrophoblast layer and an inner cytotrophoblast layer. CC represents malignant transformation of the placenta, and by definition must contain both the syncytiotrophoblast and cytotrophoblast layers. In contrast, malignant syncytiotrophoblast cells that appear in the absence of cytotrophoblast cells are not considered CC and in fact, can occur in combination with seminoma in the absence of any other nonseminoma component; indeed, they are still considered to be pure seminomas. CCs, like the placenta, produce HCG and are highly vascular in nature.
As normal embryological development continues, the morula undergoes repeated cell divisions and eventually the inner cell mass separates into two layers, an outer epiblast, which gives rise to the three fetal tissue layers (ectoderm, mesoderm, endoderm), and an inner hypoblast, consisting of extraembryonic endoderm, which forms the yolk sac. In the embryo, the yolk sac serves as the initial hematopoietic organ as well as a source of protein synthesis and nutrient transport. Differentiation of malignant germ cells along the yolk sac lineage leads to formation of YST, also known as endodermal sinus tumor. YSTs typically express AFP but not HCG in contrast to CCs, which express HCG but not AFP. EC, as a pluripotent neoplasm, is capable of differentiating into either of these tumor types, and can express both HCG and AFP.
Sperger and colleagues demonstrated the genetic similarity between EC and embryonic stem cells (ESCs) by comparing gene expression signatures of human ESC lines, EC cell lines and primary tumors, yolk sac tumor cell lines and primary tumors, seminomas, somatic cell lines, and normal testis. Upon hierarchical clustering, ESC lines clustered closest with EC tumors as compared to any of the other cell lines or tissues [42]. The original definition of ESCs was based on the expression of specific genes associated with pluripotency regulation including FGF2, POU5F1, THY1, SOX2, EBAF1, ZFP42, and TDGF1. Studies from our lab supported the similarity between ESCs and ECs; we demonstrated all of the aforementioned genes to be expressed by ECs whereas seminomas lacked expression of SOX2, FGF4, EBAF1, and TDGF1 [43, 44]. These data are consistent with the notion that SOX2, FGF4, EBAF1, and TDGF1 play a specific role in pluripotency. Furthermore, the transcription factors known to be important for maintenance of the undifferentiated state, such as POU5F1 (OCT3/4) and NANOG, were upregulated in both ECs and seminomas [43, 44].
Teratomas display somatic differentiation of the three tissue layers of the embryo. Typically, two or three of these layers are represented in a given teratoma. Differentiation can be complete, appearing identical to adult tissue types in the case of mature T, or incomplete, resembling fetal tissue in the case of immature T. Both mature and immature Ts tend to have low rates of mitosis and apoptosis, although this can be more variable in the case of immature Ts. On occasion, Ts can undergo malignant transformation, developing into a secondary somatic malignancy derived from a particular T tissue type. Common secondary somatic malignant histologies include rhabdomyosarcomas, adenocarcinomas, and primitive neuroepithelial tumors (PNET). Tumors that recur after prolonged remissions, known as late relapses, and mediastinal primary nonseminomas tend to have a higher propensity to undergo malignant transformation.
The most frequent nonseminomatous histology is a mixed form, comprised of more than one component (e.g., EC plus T) or a combination of a nonseminoma component with a seminoma [1, 9]. The most common pure nonseminoma histology is EC [1, 9]. Regardless of histologic subtype, a hallmark of all seminomas and nonseminomas is the presence of increased copies of 12p, usually as an isochromosome, (i[12p]) [45].
Debate Over the Cell of Origin of GCT
Although it is widely agreed that GCTs arise from malignant transformation of germ cells along their development, there is disagreement over the precise time point at which this occurs. Two models have been proposed. One, proposed by Skakkebaek and colleagues [46], postulates that PGCs or gonocytes, while still in utero, but after reaching the genital ridge, initiate abnormal cell proliferation under the direction of KIT pathway activation, leading to ITGCNU. This premalignant lesion remains dormant until puberty when, under stimulation from gonadotrophins, undergoes further transformation, acquires extra copies of 12p, and evolves to an invasive GCT. As such, this theory supposes that ITGCNU precedes acquisition of i[12p] and is supported by common expression patterns between PGCs/gonocytes and ITGCNU, of genes such as PRDM1/BLIMP1 and PRMT5, as well as the observation that not all ITGCNU may contain extra copies of 12p [47, 48]. In addition, epidemiologic data and the characteristics of germ cells in developmental abnormality syndromes that predispose patients to GCT such as testicular feminization and testicular dysgenesis, are cited in support this hypothesis. Another model, proposed by Chaganti and Houldsworth [49], suggests that the zygotene/pachytene spermatocyte with a 4n DNA content is the cell of origin. The error-prone homologous recombination at this stage of germ cell development allows acquisition of increased 12p copy number, which leads to aberrant gene expression, increased mitosis, re-establishment of pluripotentiality, and genomic instability that support malignant transformation to GCT. Evidence supporting this theory includes the shared chromosomal aneuploidy between GCTs and zygotene/pachytene spermatocytes and the abundant expression of wild-type p53, a hallmark of germ cells and GCTs. However, neither hypothesis has been experimentally validated.
Debate Over the Origin of Extragonadal GCTs
Adult GCTs and testicular cancer have become synonymous since more than 95 % of testicular cancers are GCTs and more than 90 % of GCTs originate in the testis. Nevertheless, 5–10 % of GCTs arise from extragonadal locations, with the most common sites including the mediastinum, retroperitoneum, and the pineal gland. With the exception of tumors that arise in the pineal gland and show a predominance of seminomatous histology (often referred to as germinomas), the majority of extragonadal GCTs are nonseminomas [5]. The concept of retroperitoneal primary tumor remains controversial as many believe these cases to represent metastatic lesions from testicular tumors that were not able to be identified by ultrasound or at orchiectomy. Changes in the testicular parenchyma where a tumor has undergone spontaneous regression are referred to as having a “burnt out” appearance and could explain the failure to identify a gonadal primary tumor in some cases of solitary retroperitoneal GCT masses [50, 51].
Two mechanisms have been proposed to explain how GCTs of extragonadal primary sites other than the retroperitoneum arise. The conventional hypothesis suggests that PGCs or gonocytes get “left behind” while migrating through the embryo to the genital ridges and eventually transform. While this model is easy to conceptualize, misplaced germ cells at the PGC or gonocyte stage have never been identified in developing human embryos. Such cells have been observed in mouse embryos but are not viable due to a predilection to rapid apoptosis [52]. Finally, extragonadal GCTs have been identified to have chromosomal changes highly similar to gonadal GCTs with increased 12p copy number and aneuploidy [45]. These alterations are thought to be acquired later in GC development (meiosis of primary spermatocytes) than during the migratory stage of gonocytes, raising doubt to this theory and instead supporting a common cell of origin for gonadal and extragonadal GCT. An alternative explanation involves the potential of transformed germ cells to undergo reverse migration to the mediastinum or pineal gland, where stromal environments left over from embryological development could remain fertile for transformed germ cell proliferation. At present, this remains an open area of controversy.
Genetics of Germ Cell Tumors
Gain of Chromosome 12p
GCTs are one of only a handful of malignancies (e.g., CML, GIST) that contain a pathognomonic genetic abnormality, present in nearly all cases. In GCT, this abnormality is the i(12p), which was first described in 1982 by Atkin and Baker during karyotyping of metaphase chromosomes from cases of GCTs [53]. Subsequently, several studies have documented approximately 85 % of GCTs to contain this chromosomal abnormality, and in cases where i(12p) was absent, extra copies of part or all of 12p occur as tandem duplications in situ or within other chromosomes [54]. As such, this assay has provided diagnostic utility for poorly differentiated midline tumors of unknown histogenesis, allowing the diagnosis of GCT to be made, and permitting administration of potentially curative chemotherapy [55]. Several studies have indicated that i(12p) is evident as early in GCT neoplasm development as ITGCNU, yet others have indicated that the appearance of this marker is associated with tumor invasion out of the tubules [48, 49, 56, 57].
Regardless of whether or not chromosome 12p gain is present only in fully malignant GCT or also ITGCNU, its omnipresence in invasive disease strongly suggests a role in the pathogenesis of GCT. Initially, it was thought that aberration of a single gene within 12p would be found responsible for GCT pathogenesis/progression. However, with more than 400 genes located on chromosome 12p and no overwhelming evidence to support one gene in particular, conventional wisdom now asserts that multiple genes on 12p, possibly in conjunction with other chromosomal anomalies, enable invasive GCT development. One gene of particular interest is cyclin D2 (CCND2), whose protein product is involved in regulation of DNA replication at the G1/S transition of the cell cycle. Overexpression of this protein leads to increased cell cycling and has been identified in ITGCNU, seminoma, and EC [58]. In contrast, normal spermatogonial cells in the adult testis rarely express cyclinD2, although expression of this protein has been observed in neonatal spermatogonial cells of the mouse.
Additional genes of interest have been identified through gene expression profiling, including a group of stem cell associated genes all mapping to a 200 Kb region at 12p13.31. These genes include STELLAR, NANOG, and GDF3, all of which demonstrate elevated expression in seminomas and embryonal carcinomas [44]. The overexpression of these genes through gain of 12p may be responsible for the undifferentiated phenotype observed in these two GCT histologies. Furthermore, exposure of EC cell lines to differentiating agents such as all-trans retinoic acid (ATRA) or bone morphogenic protein 2 (BMP2) lead to downregulation of these genes and resultant loss of pluripotency [44, 59, 60].
Evaluations of chromosomal changes within GCTs, primarily seminomas, by comparative genomic hybridization (CGH) revealed the frequent presence of a high-level amplification of the 12p11-12.2 region in addition to gain of the entire short arm of chromosome 12 [61, 62]. However, attempts to identify the specific target gene within this amplicon using molecular cytogenetic studies and global genomic screening have not been conclusive [63–65].
Chromosomal Changes Other Than 12p
In addition to i[12p], conventional karyotype analyses have demonstrated that GCTs are aneuploid in DNA content, typically hypertriploid or tetraploid. Specific chromosomal abnormalities have been identified as recurrent across GCTs, some correlating with particular histologic subtypes [66, 67]. For example, breakpoints at 1p32-36 and 7q11.2 have been associated with teratoma whereas breakpoints at 1p22 correlated with yolk sac tumor histology [67]. Deletion or rearrangement of 12q and deletion of 6q13-25 constitute other frequently observed chromosomal changes in GCTs [66].
Interrogation of ITGCNU demonstrated frequent gain of portions of chromosomes 1, 5, 7, 8, 12p, and X and loss of DNA content from chromosome 18 [48, 56]. Adjacent invasive tumors also exhibited many of these changes but in addition, frequently had gains of portions of chromosomes 2, 3, 4, 6,13q, 14q, 17q, 18q, 20, and 21 and losses of portions of chromosomes 1p, 4, 6q, 9, 11, 13q, and 19 [48]. In the case of 17q gain, GRB7 and JUP were identified as potential target genes through microarray analysis [46].
Potential tumor suppressor genes involved in GCT pathogenesis have also been identified, primarily through loss of heterozygosity (LOH) studies [68, 69]. These studies demonstrated GCTs frequently contain loss of regions including the known tumor suppressor genes, RB1, DCC, and NME. In addition, loss of heterozygosity was demonstrated in regions where other proposed tumor suppressor genes are located (1p, 3p, 5q, 10q, 11p, 11q, and 17p) as well as new sites not previously identified as containing tumor suppressor genes (1q, 2q, 3q, 5p, 9q, 12q, 18p, and 20p). Epigenetic modifications such as promoter methylation might also contribute to loss of heterozygosity for tumor suppressor genes involved in GCT histopathogenesis. For example, seminomas have been demonstrated to contain lower levels of promoter methylation than nonseminomas. In addition, methylation of MGMT correlated with loss of its expression [70, 71]. However, methylation changes and expression of other genes have not correlated well in other studies [72].
Mutations
In contrast to most malignancies, GCTs are believed to contain relatively few driver mutations. However, mutations in KRAS [73], KIT [74, 75], and SMAD4 [76] have been identified in some GCTs and have been proposed as important in germ cell transformation. KIT is perhaps the most well studied of these genes. In one series, activating KIT mutations were found in a large proportion of bilateral GCTs, particularly bilateral seminomas [74]. However, other studies did not support this claim [77, 78]. As discussed earlier, aberration of KIT signaling was also recently identified as increasing susceptibility to GCT development [25, 26]. More recent efforts by our group and others have identified additional mutations within a subset of GCTs, particularly those that demonstrate cisplatin resistance [111].
Genetics and Pathobiology of Chemosensitivity and Resistance
The introduction of cisplatin in the late 1970s radically changed the outlook for post-pubertal men with advanced GCTs, increasing the complete remission rate from approximately 25 % to nearly 80 % [79]. Subsequently, GCTs have become a model for the curable malignancy and for investigations into platinum sensitivity. With the activity of cisplatin, albeit to a lesser extent, against a number of malignancies, there has been great interest in understanding the biological basis of the platinum sensitivity of GCTs as well as the mechanisms of resistance.
The transformation from a platinum-sensitive to a platinum-resistant phenotype likely depends on changes in several intracellular pathways including those involved in cellular response to DNA damage, apoptosis, differentiation, and cell growth (Table 26.2). Several studies have demonstrated differences in immunohistochemical staining of markers of cell proliferation and apoptosis between different GCT histologies [80–82]. For example, in one study, ECs were demonstrated to have the highest rate of apoptosis and negative staining for BCL2 in contrast to mature Ts which had very low levels of apoptosis and positive staining for BCL2 [82]. However, these investigations did not identify any markers specific to platinum resistance. In order to more specifically identify resistance markers, studies were carried out within pure EC specimens, demonstrating improved outcomes for ECs with higher rates of proliferation (Ki-67) and lower rates of spontaneous apoptosis [83].
Differentiation and Resistance
Several pieces of evidence suggest an association between differentiation and development of chemotherapy resistance. For example, Ts, which represent somatic differentiation of malignant germ cells, are highly chemoresistant with surgical resection comprising the mainstay of treatment [84]. While Ts are thought to lack the ability to metastasize, they can nonetheless be problematic through local uncontrolled growth leading to compression of important structures, or differentiation along the lines of a secondary somatic malignancy [85]. In vitro studies support the concept of differentiation leading to chemotherapy resistance. For instance, the pluripotent and undifferentiated EC cell line NT2/D1 undergoes rapid apoptosis upon exposure to cisplatin. In contrast, upon exposure to the differentiating agent ATRA, NT2/D1 cells become relatively resistant to cisplatin with a marked attenuation in apoptosis [86]. Other studies have demonstrated that loss of expression of POU5F1, a stem cell marker involved in maintenance of pluripotency, also correlates with development of cisplatin resistance [87].
DNA Repair Proficiency and Resistance
Since the mechanism of action of cisplatin involves the formation of DNA adducts which lead to apoptosis, a long held belief has been that a decreased proficiency in DNA repair underlies the unique sensitivity of GCT to platinum-based chemotherapy. One study suggested that certain high mobility group (HMG) domain proteins specific to germ cells could allow shielding of cisplatin-DNA adducts, preventing effective DNA repair from taking place and increasing cisplatin sensitivity [88]. GCTs have also been demonstrated to contain decreased levels of the xeroderma pigmentosum complementation group A (XPA) protein, which is involved in repair of DNA adducts and UV radiation-induced photoproducts, providing another possible explanation for the platinum sensitivity of this tumor type [89, 90]. However, another study found conflicting results, demonstrating that sensitive GCT cell lines containing low levels of XPA remained capable of efficient DNA repair. In addition, overexpression of XPA did not confer cisplatin resistance [91]. As such, there remains no convincing evidence to support the defective DNA repair hypothesis as the underlying Achilles heel of GCT exposed to cisplatin.
Mutations and Resistance
As previously mentioned, GCTs have historically been regarded as a malignancy associated with relatively few driver mutations and instead, characterized by more global changes in chromosomal content (aneuploidy, and large region chromosomal gains and losses). However, inactivating mutation or deletion of the tumor suppressor gene, TP53, a critical regulator of the cellular response to DNA damage and induction of apoptosis, has been demonstrated to correlate with GCT platinum-resistance [92]. Some investigators have proposed that the rare (<5 %) incidence of TP53 loss in GCT as compared to the more frequent (≈50 %) aberrations in other malignancies [93, 94] may explain why the vast majority of GCTs are sensitive to cisplatin. However, not all TP53 mutant GCT cell lines have been found to be platinum-resistant [95], suggesting that TP53 mutation might not be sufficient by itself to induce resistance in some cases.
More recently, the V600E mutation in BRAF was reported to be associated with resistant GCT. Honecker and colleagues found that 9 (26 %) of 35 resistant GCTs harbored V600E BRAF mutations as compared to only 1 (1 %) of 100 unselected cases. These authors also reported an increase in microsatellite instability among resistant GCT tumors as compared to unselected cases [96]. However, these results contrast with those of other groups [97, 98] and have not been confirmed. Our group recently attempted to validate the findings of Honnecker within 46 cisplatin-resistant and 24 cisplatin-sensitive GCTs. Using a Sequenom approach, no BRAF mutations were identified in any of the tumors. However, mutations in KRAS, HRAS, PIK3CA, or AKT1 were observed in tumors from 9/46 (20%) patients with resistant GCT as compared 0/24 patients with sensitive GCT [111].
It is possible that with more sensitive next generation sequencing techniques, an increasing number of mutations may be identified within GCT that correlate with resistance.
Apoptotic Pathway Proficiency and Resistance
In addition to mutations in TP53, aberrations in other parts of apoptotic signaling could also lead to cisplatin resistance [99]. For example, a cell line with inability to activate caspase-9 maintained cisplatin resistance, independent of whether TP53 was wild-type or mutant [100]. Another study found that upon exposure to cisplatin, sensitive GCT cell lines displayed an increase in expression of FAS and recruitment of FADD and caspase-8 to FAS, whereas resistant GCT cell lines did not [101].
Recent work has further implicated aberrations in the p21-CDK2 pathway to also lead to cisplatin resistance. Koster and colleagues found sensitive GCT cell lines to have decreased cytoplasmic staining for p21 as compared to resistant cell lines. Furthermore, silencing of p21 or manipulations to increase p21 shuttling from the cytoplasm to the nucleus increased apoptosis and restored the cisplatin sensitivity of the resistant GCT cell lines. Finally, these authors demonstrated that phospho-AKT is responsible for phosphorylation of p21 that prevents shuttling to the nucleus; inhibition of AKT led to decreased cytoplasmic AKT and increased apoptosis upon cisplatin exposure of cisplatin-resistant cell lines, an effect that was reversed by silencing of CDK2 [102, 103]. Our recent work demonstrating the presence of PIK3CA and AKT1 mutations in cisplatin-resistant but not sensitive GCT [111], lends support to activation of this pathway as a possible mechanism of escape from cisplatin-induced apoptosis.
Apart from the pluripotency and differentiation hypothesis, the common premise to all of the aforementioned mechanisms underlying cisplatin sensitivity and resistance is that GCTs appear to have an innate response to DNA-damaging agents that causes rapid initiation of multiple cell death pathways. In many but not all cases, this response appears to be dependent upon TP53. In contrast, exposure of other malignant cell types to cisplatin results in halting of the cell cycle, allowing DNA repair and avoidance of apoptosis, followed by re-entry into the cell cycle and active proliferation. An improved understanding of the factors that lead to the rapid upregulation of apoptosis in GCT could have major implications to identifying and overcoming the mechanisms of cisplatin resistance in other neoplasms.
Utility of GCT Genetics to Predict Patient Outcome
Investigations into GCT biology and molecular pathogenesis offer not only the potential to increase our understanding of these tumors but also to enhance patient outcome prediction beyond current systems which rely solely on clinicohistologic factors. For example, following orchiectomy, patients with Stage I seminoma, the most commonly encountered GCT stage-histology combination, have an approximately 20 % risk of recurrence. In one study, the presence of specific pathologic factors such as tumor size >4 cm and rete testis involvement increased the recurrence rate from 12 % (for patients with neither factor) to 32 % (for patients with both factors) [104]. Yet, even in the highest risk cases, the majority of patients remain relapse-free without any further treatment. As such, based on the emerging appreciation of the long-term risks of radiation (e.g., secondary malignancies) as well as the nearly 100 % survival for patients treated at recurrence, adjuvant radiation therapy, once the universal treatment for Stage I seminoma, has fallen out of favor over the last several years. It would be highly beneficial if intratumoral molecular markers that predict recurrence could be identified, aiding in the selection of patients for adjuvant therapy as well as intensity of follow-up.
For advanced disease, a clinicopathologic prognostic system is currently in use, known as the International Germ Cell Cancer Collaborative Group (IGCCG) risk model [105]. This classification system takes into account histology (seminoma vs. nonseminoma), primary tumor site (mediastinal vs. gonadal/retroperitoneal), tumor marker (HCG, AFP, and LDH) levels, and sites of metastases (non-pulmonary visceral metastases). Based on these factors, patients are classified into good-, intermediate-, and poor-risk groups, with 5-year survival rates of approximately 90, 75, and 50 % respectively [105]. Therefore, for patients in the poor-risk group, the probability of survival is predicted no better than the flip of a coin. It would certainly be of great value to be able to predict which of these patients will be cured with standard chemotherapy so that more intensive or novel strategies could be applied to the remaining patients. Similarly, it would be very helpful if we could predict which 10 and 25 % of patients with good- and intermediate-risk disease, respectively, are destined to fail conventional therapy.
In general, the molecular markers of cisplatin resistance discussed above could all represent potential prognostic factors for patients with advanced GCT, since cisplatin sensitivity is crucial to the efficacy of first-line therapy in advanced GCT. However, most of these markers have not been validated. Furthermore, even those that are well established, such as mutations in TP53, occur only rarely among resistant cases and are not universally predictive of poor outcome, limiting their clinical utility [92, 93, 95]. As discussed above, BRAF mutations and microsatellite instability were recently reported in a retrospective study to be found in more than 25 % of cisplatin-resistant GCT as compared to only 1 % of unselected cases [96]. While these results are interesting, they have not yet been independently validated, or studied in a prospective manner.
Several studies have focused on the association of DNA copy number changes with outcome in GCT. Early chromosomal comparative genomic hybridization (CGH) studies revealed gains of genetic material at multiple sites other than 12p in 5 of 17 cisplatin-resistant cases as compared to none of the cisplatin-sensitive cases [62]. More recently, another CGH study demonstrated gains of an 8.7 Mb region in chromosome 6q and loss of 0.3 Mb region in chromosome 10q to be present in 3 cisplatin-resistant GCT cell lines that were derived from repeated cisplatin exposure of their cisplatin-sensitive parental clones [106]. However, these findings have not been duplicated among additional GCT samples taken from patients with cisplatin-resistant disease.
Our group performed an array-based CGH (aCGH) on a larger set of tumor specimens (n = 53) with known outcome to cisplatin-based chemotherapy and identified 16 regions of DNA gain or loss that were associated with 5-year overall survival. Using expression data from this same cohort, we identified 75 probe sets within these 16 regions that demonstrated >2-fold differences in expression as compared to tumors with normal copy numbers or >3 fold differences in expression as compared to normal testis tissue. These data were used to build a model predictive of outcome based on expression of these 75 probe sets, which was then applied to an independent set of 54 tumors for which we had gene expression profile data but not aCGH data. The model predicted 5yOS with 75 % accuracy and this could be increased to 80 % when including probes from regions that were predictive of 2-year disease-specific survival. Importantly, on multivariate analysis, the model’s prognostic capability was independent of the IGCCCG risk classification (Unpublished data).
Tumor gene expression patterns have recently been studied in multiple malignancies as predictors of outcome. In particular, the OncotypeDx® (Genomic Health) assay [107] has been commercialized for predicting the likelihood of recurrence in hormone-receptor positive breast cancer patients with localized lymph node negative disease treated with adjuvant hormonal therapy. Patients with a high risk of recurrence on the OncotypeDx assay are offered adjuvant chemotherapy prior to starting hormonal therapy whereas those with a low risk of recurrence are treated with adjuvant hormone therapy.
Based on the success of this approach in other tumor types, we have recently completed an evaluation of differential gene expression as a predictor of outcome in GCT. We conducted whole genome microarray analysis on fresh frozen tumor specimens from 74 NSGCTs and used the prediction analysis for microarray (PAM) software to identify genes associated with favorable and unfavorable outcomes. In total, 170 probe sets corresponding to 135 genes had a significant association with 5-years OS. When the PAM classifier was applied to an independent validation set of 34 NSGCTs, it correctly predicted 5-years OS with 90 % accuracy. On multivariate analysis, the PAM classifier was independent of the IGCCCG classification model, serving as proof of concept that in GCTs, like other malignancies, gene expression-based modeling can enhance outcome prediction [108]. We further categorized the prognostic genes into defined groups using the GOMINER algorithm. Interestingly, overexpression of genes with an immune function (immunoglobulin and T-cell related genes) was associated with a favorable outcome, whereas overexpression of genes involved in differentiation, particularly into a neural lineage was associated with a poor outcome [108].
Conclusions
GCTs encompass a fascinating group of neoplasms with diverse clinical features, histologic appearance, protein and gene expression patterns, and differentiating capability. This vast biologic spectrum relates to the derivation of GCT from malignant transformation of a developing germ cell, existing in an undifferentiated state and with capability to acquire pluripotentiality. Despite their diversity, these tumors are uniquely sensitive to the DNA damaging agent, cisplatin, and therefore, in advanced disease, have among the most favorable prognosis of any metastatic neoplasm. An improved understanding of the biology of these tumors likely will provide invaluable insight into gametogenesis, embryology, stem cell biology, and mechanisms of cisplatin sensitivity and resistance.
References
Biggs ML, Schwartz SM. Cancer of the testis. In: Ries LAG et al., editors. SEER survival monograph: cancer survival among adults: U.S. SEER program, 1988–2001, patient and tumor characteristics. Bethesda: National Cancer Institute; 2007. p. 165–70.
Feldman DR, et al. Medical treatment of advanced testicular cancer. JAMA. 2008;299(6): 672–84.
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29.
Risk MC, Porter CR. Management of non-germinal testicular tumors. World J Urol. 2009;27(4):507–12.
Stang A, et al. Gonadal and extragonadal germ cell tumours in the United States, 1973–2007. Int J Androl. 2012;35(4):616–25.
Chia VM, et al. International trends in the incidence of testicular cancer, 1973–2002. Cancer Epidemiol Biomarkers Prev. 2010;19(5):1151–9.
Howlader N, et al. SEER cancer statistics review, 1975–2008. 2011 [cited 2012; Available from: http://seer.cancer.gov/csr/1975_2008/. Last Accessed on Apr 15th 2014.
McGlynn KA, et al. Trends in the incidence of testicular germ cell tumors in the United States. Cancer. 2003;97(1):63–70.
Townsend JS, Richardson LC, German RR. Incidence of testicular cancer in the United States, 1999–2004. Am J Mens Health. 2010;4(4):353–60.
Wood HM, Elder JS. Cryptorchidism and testicular cancer: separating fact from fiction. J Urol. 2009;181(2):452–61.
Myrup C, Schnack TH, Wohlfahrt J. Correction of cryptorchidism and testicular cancer. N Engl J Med. 2007;357(8):825–7; author reply 825–7.
Schnack TH, et al. Familial coaggregation of cryptorchidism, hypospadias, and testicular germ cell cancer: a nationwide cohort study. J Natl Cancer Inst. 2010;102(3):187–92.
Raman JD, Nobert CF, Goldstein M. Increased incidence of testicular cancer in men presenting with infertility and abnormal semen analysis. J Urol. 2005;174(5):1819–22; discussion 1822.
Cools M, et al. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006;27(5):468–84.
Fossa SD, et al. Risk of contralateral testicular cancer: a population-based study of 29,515 U.S. men. J Natl Cancer Inst. 2005;97(14):1056–66.
Andreassen KE, et al. Risk of metachronous contralateral testicular germ cell tumors: a population-based study of 7,102 Norwegian patients (1953–2007). Int J Cancer. 2011;129(12):2867–74.
Osterlind A, et al. Incidence of bilateral testicular germ cell cancer in Denmark, 1960–84: preliminary findings. Int J Androl. 1987;10(1):203–8.
Skakkebaek NE. Possible carcinoma-in-situ of the testis. Lancet. 1972;2(7776):516–7.
von der Maase H, et al. Carcinoma in situ of contralateral testis in patients with testicular germ cell cancer: study of 27 cases in 500 patients. Br Med J (Clin Res Ed). 1986;293(6559):1398–401.
Hartmann JT, et al. The relative risk of second nongerminal malignancies in patients with extragonadal germ cell tumors. Cancer. 2000;88(11):2629–35.
Heimdal K, et al. Familial testicular cancer in Norway and Southern Sweden. Br J Cancer. 1996;73(7):964–9.
Sonneveld DJ, et al. Familial testicular cancer in a single-centre population. Eur J Cancer. 1999;35(9):1368–73.
Spermon JR, et al. Cancer incidence in relatives of patients with testicular cancer in the eastern part of The Netherlands. Urology. 2001;57(4):747–52.
Westergaard T, et al. Cancer risk in fathers and brothers of testicular cancer patients in Denmark. A population-based study. Int J Cancer. 1996;66(5):627–31.
Kanetsky PA, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41(7):811–5.
Rapley EA, et al. Somatic mutations of KIT in familial testicular germ cell tumours. Br J Cancer. 2004;90(12):2397–401.
Blume-Jensen P, et al. Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3’-kinase is essential for male fertility. Nat Genet. 2000;24(2):157–62.
Gu Y, et al. Steel factor controls primordial germ cell survival and motility from the time of their specification in the allantois, and provides a continuous niche throughout their migration. Development. 2009;136(8):1295–303.
Daling JR, et al. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer. 2009;115(6):1215–23.
Trabert B, et al. Marijuana use and testicular germ cell tumors. Cancer. 2011;117(4):848–53.
Hardell L, et al. Increased concentrations of polychlorinated biphenyls, hexachlorobenzene, and chlordanes in mothers of men with testicular cancer. Environ Health Perspect. 2003;111(7):930–4.
McGlynn KA, et al. Persistent organochlorine pesticides and risk of testicular germ cell tumors. J Natl Cancer Inst. 2008;100(9):663–71.
Nichols CR, et al. Klinefelter’s syndrome associated with mediastinal germ cell neoplasms. J Clin Oncol. 1987;5(8):1290–4.
Ginsburg M, Snow MH, McLaren A. Primordial germ cells in the mouse embryo during gastrulation. Development. 1990;110(2):521–8.
Skakkebaek NE. Carcinoma in situ of the testis: frequency and relationship to invasive germ cell tumours in infertile men. Histopathology. 1978;2(3):157–70.
de Jong J, et al. Diagnostic value of OCT3/4 for pre-invasive and invasive testicular germ cell tumours. J Pathol. 2005;206(2):242–9.
Jones TD, et al. OCT4 staining in testicular tumors: a sensitive and specific marker for seminoma and embryonal carcinoma. Am J Surg Pathol. 2004;28(7):935–40.
Koshida K, Uchibayashi T, Hisazumi H. Characterization of seminoma-derived placental-like alkaline phosphatase. Urol Int. 1991;47 Suppl 1:96–9.
Donovan PJ, de Miguel MP. Turning germ cells into stem cells. Curr Opin Genet Dev. 2003;13(5):463–71.
Durcova-Hills G, et al. Reprogramming primordial germ cells into pluripotent stem cells. PLoS One. 2008;3(10):e3531.
Looijenga LH, Stoop H, de Leeuw HP, et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors.Cancer Res 2003;63(9):2244–2250.
Sperger JM, et al. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci U S A. 2003;100(23):13350–5.
Brivanlou AH, et al. Stem cells. Setting standards for human embryonic stem cells. Science. 2003;300(5621):913–6.
Korkola JE, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66(2):820–7.
Samaniego F, et al. Cytogenetic and molecular analysis of human male germ cell tumors: chromosome 12 abnormalities and gene amplification. Genes Chromosomes Cancer. 1990;1(4):289–300.
Skakkebaek NE, et al. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl. 1987;10(1):19–28.
Ottesen AM, et al. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer. 2003;38(2):117–25.
Summersgill B, et al. Chromosomal imbalances associated with carcinoma in situ and associated testicular germ cell tumours of adolescents and adults. Br J Cancer. 2001;85(2):213–20.
Chaganti RS, Houldsworth J. The cytogenetic theory of the pathogenesis of human adult male germ cell tumors. Review article APMIS. 1998;106(1):80–3; discussion 83–4.
Angulo JC, et al. Clinicopathological study of regressed testicular tumors (apparent extragonadal germ cell neoplasms). J Urol. 2009;182(5):2303–10.
Balzer BL, Ulbright TM. Spontaneous regression of testicular germ cell tumors: an analysis of 42 cases. Am J Surg Pathol. 2006;30(7):858–65.
Chaganti RS, Houldsworth J. Genetics and biology of adult human male germ cell tumors. Cancer Res. 2000;60(6):1475–82.
Atkin NB, Baker MC. Specific chromosome change, i(12p), in testicular tumours? Lancet. 1982;2(8311):1349.
Rodriguez E, et al. Molecular cytogenetic analysis of i(12p)-negative human male germ cell tumors. Genes Chromosomes Cancer. 1993;8(4):230–6.
Motzer RJ, et al. Molecular and cytogenetic studies in the diagnosis of patients with poorly differentiated carcinomas of unknown primary site. J Clin Oncol. 1995;13(1):274–82.
Rosenberg C, et al. Overrepresentation of the short arm of chromosome 12 is related to invasive growth of human testicular seminomas and nonseminomas. Oncogene. 2000;19(51):5858–62.
Vos A, et al. Cytogenetics of carcinoma in situ of the testis. Cancer Genet Cytogenet. 1990;46(1):75–81.
Houldsworth J, et al. Aberrant expression of cyclin D2 is an early event in human male germ cell tumorigenesis. Cell Growth Differ. 1997;8(3):293–9.
Clark AT, et al. Human STELLAR, NANOG, and GDF3 genes are expressed in pluripotent cells and map to chromosome 12p13, a hotspot for teratocarcinoma. Stem Cells. 2004;22(2):169–79.
Giuliano CJ, et al. Retinoic acid represses a cassette of candidate pluripotency chromosome 12p genes during induced loss of human embryonal carcinoma tumorigenicity. Biochim Biophys Acta. 2005;1731(1):48–56.
Mostert MM, et al. Comparative genomic hybridization of germ cell tumors of the adult testis: confirmation of karyotypic findings and identification of a 12p-amplicon. Cancer Genet Cytogenet. 1996;89(2):146–52.
Rao PH, et al. Chromosomal amplification is associated with cisplatin resistance of human male germ cell tumors. Cancer Res. 1998;58(19):4260–3.
Bourdon V, et al. Genomic and expression analysis of the 12p11-p12 amplicon using EST arrays identifies two novel amplified and overexpressed genes. Cancer Res. 2002;62(21):6218–23.
Rodriguez S, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. 2003;22(12):1880–91.
Zafarana G, et al. 12p-amplicon structure analysis in testicular germ cell tumors of adolescents and adults by array CGH. Oncogene. 2003;22(48):7695–701.
Murty VV, Chaganti RS. A genetic perspective of male germ cell tumors. Semin Oncol. 1998;25(2):133–44.
Rodriguez E, et al. Cytogenetic analysis of 124 prospectively ascertained male germ cell tumors. Cancer Res. 1992;52(8):2285–91.
Murty VV, et al. Allelic loss and somatic differentiation in human male germ cell tumors. Oncogene. 1994;9(8):2245–51.
Mathew S, et al. Loss of heterozygosity identifies multiple sites of allelic deletions on chromosome 1 in human male germ cell tumors. Cancer Res. 1994;54(23):6265–9.
Koul S, et al. Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol Cancer. 2002;1:8.
Smith-Sorensen B, et al. Frequent promoter hypermethylation of the O6-Methylguanine-DNA Methyltransferase (MGMT) gene in testicular cancer. Oncogene. 2002;21(57):8878–84.
Honorio S, et al. Frequent epigenetic inactivation of the RASSF1A tumour suppressor gene in testicular tumours and distinct methylation profiles of seminoma and nonseminoma testicular germ cell tumours. Oncogene. 2003;22(3):461–6.
Roelofs H, et al. Restricted 12p amplification and RAS mutation in human germ cell tumors of the adult testis. Am J Pathol. 2000;157(4):1155–66.
Looijenga LH, et al. Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res. 2003;63(22):7674–8.
Tian Q, et al. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999;154(6):1643–7.
Bouras M, et al. A novel SMAD4 gene mutation in seminoma germ cell tumors. Cancer Res. 2000;60(4):922–8.
Coffey J, et al. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes Chromosomes Cancer. 2008;47(1):34–42.
Sakuma Y, et al. Mutations of c-kit gene in bilateral testicular germ cell tumours in Japan. Cancer Lett. 2008;259(1):119–26.
Einhorn LH, Donohue J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann Intern Med. 1977;87(3):293–8.
Grobholz R, et al. Bax, Bcl-2, fas and Fas-L antigen expression in human seminoma: correlation with the apoptotic index. APMIS. 2002;110(10):724–32.
Mayer F, et al. Molecular determinants of treatment response in human germ cell tumors. Clin Cancer Res. 2003;9(2):767–73.
Soini Y, Paakko P. Extent of apoptosis in relation to p53 and bcl-2 expression in germ cell tumors. Hum Pathol. 1996;27(11):1221–6.
Mazumdar M, et al. Cluster analysis of p53 and Ki67 expression, apoptosis, alpha-fetoprotein, and human chorionic gonadotrophin indicates a favorable prognostic subgroup within the embryonal carcinoma germ cell tumor. J Clin Oncol. 2003;21(14):2679–88.
Logothetis CJ, et al. The growing teratoma syndrome. Cancer. 1982;50(8):1629–35.
Donadio AC, et al. Chemotherapy for teratoma with malignant transformation. J Clin Oncol. 2003;21(23):4285–91.
Timmer-Bosscha H, et al. Differential effects of all-trans-retinoic acid, docosahexaenoic acid, and hexadecylphosphocholine on cisplatin-induced cytotoxicity and apoptosis in a cisplantin-sensitive and resistant human embryonal carcinoma cell line. Cancer Chemother Pharmacol. 1998;41(6):469–76.
Mueller T, et al. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumour Biol. 2006;27(2):71–83.
Zamble DB, et al. Testis-specific HMG-domain protein alters the responses of cells to cisplatin. J Inorg Biochem. 2002;91(3):451–62.
Welsh C, et al. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 2004;110(3):352–61.
Koberle B, et al. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol. 1999;9(5):273–6.
Koberle B, et al. Elevation of XPA protein level in testis tumor cells without increasing resistance to cisplatin or UV radiation. Mol Carcinog. 2008;47(8):580–6.
Houldsworth J, et al. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene. 1998;16(18):2345–9.
Heimdal K, et al. No germline TP53 mutations detected in familial and bilateral testicular cancer. Genes Chromosomes Cancer. 1993;6(2):92–7.
Peng HQ, et al. Mutations of the p53 gene do not occur in testis cancer. Cancer Res. 1993;53(15):3574–8.
Burger H, et al. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int J Cancer. 1997;73(4):592–9.
Honecker F, et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol. 2009;27(13):2129–36.
McIntyre A, et al. Activating mutations and/or expression levels of tyrosine kinase receptors GRB7, RAS, and BRAF in testicular germ cell tumors. Neoplasia. 2005;7(12):1047–52.
Sommerer F, et al. Mutations of BRAF and RAS are rare events in germ cell tumours. Int J Cancer. 2005;113(2):329–35.
Spierings DC, et al. The attractive Achilles heel of germ cell tumours: an inherent sensitivity to apoptosis-inducing stimuli. J Pathol. 2003;200(2):137–48.
Mueller T, et al. Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer Res. 2003;63(2):513–21.
Spierings DC, et al. Loss of drug-induced activation of the CD95 apoptotic pathway in a cisplatin-resistant testicular germ cell tumor cell line. Cell Death Differ. 2003;10(7):808–22.
Koster R, de Jong S. Lessons learned from testicular cancer: identification of cytoplasmic p21 as an Achilles’ heel of cisplatin resistance. Cell Cycle. 2010;9(24):4776–7.
Koster R, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010;120(10):3594–605.
Warde P, et al. Prognostic factors for relapse in stage I seminoma managed by surveillance: a pooled analysis. J Clin Oncol. 2002;20(22):4448–52.
International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. J Clin Oncol. 1997;15(2):594–603.
Noel EE, et al. Identification of genomic changes associated with cisplatin resistance in testicular germ cell tumor cell lines. Genes Chromosomes Cancer. 2008;47(7):604–13.
Paik S, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–26.
Korkola JE, et al. Identification and validation of a gene expression signature that predicts outcome in adult men with germ cell tumors. J Clin Oncol. 2009;27(31):5240–7.
Ramani P, Yeung CK, Habeebu SS. Testicular intratubular germ cell neoplasia in children and adolescents with intersex. Am J Surg Pathol. 1993;17(11):1124–33.
Rapley EA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41(7):807–10.
Feldman DR, Iyer G, Van Alstine L, Patil S, Al-Ahmadie HA, Reuter VE, Bosl GJ, Chaganti RS, Solit DB. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant Germ Cell Tumors. Clin Cancer Res. [Epub May 8, 2014].
Acknowledgments
The reported studies were supported by the Byrne Fund and the Lance Armstrong Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag London
About this chapter
Cite this chapter
Feldman, D.R., Chaganti, R.S.K. (2015). Epidemiology, Biology, and Genetics of Adult Male Germ Cell Tumors. In: Nargund, V., Raghavan, D., Sandler, H. (eds) Urological Oncology. Springer, London. https://doi.org/10.1007/978-0-85729-482-1_26
Download citation
DOI: https://doi.org/10.1007/978-0-85729-482-1_26
Published:
Publisher Name: Springer, London
Print ISBN: 978-0-85729-481-4
Online ISBN: 978-0-85729-482-1
eBook Packages: MedicineMedicine (R0)