
Abstract
For most of our 25,000 genes, the removal of introns by pre-messenger RNA (pre-mRNA) splicing represents an essential step toward the production of functional messenger RNAs (mRNAs). Alternative splicing of a single pre-mRNA results in the production of different mRNAs. Although complex organisms use alternative splicing to expand protein function and phenotypic diversity, patterns of alternative splicing are often altered in cancer cells. Alternative splicing contributes to tumorigenesis by producing splice isoforms that can stimulate cell proliferation and cell migration or induce resistance to apoptosis and anticancer agents. Cancer-specific changes in splicing profiles can occur through mutations that are affecting splice sites and splicing control elements, and also by alterations in the expression of proteins that control splicing decisions. Recent progress in global approaches that interrogate splicing diversity should help to obtain specific splicing signatures for cancer types. The development of innovative approaches for annotating and reprogramming splicing events will more fully establish the essential contribution of alternative splicing to the biology of cancer and will hopefully provide novel targets and anticancer strategies. Metazoan genes are usually made up of several exons interrupted by introns. The introns are removed from the pre-mRNA by RNA splicing. In conjunction with other maturation steps, such as capping and polyadenylation, the spliced mRNA is then transported to the cytoplasm to be translated into a functional protein. The basic mechanism of splicing requires accurate recognition of each extremity of each intron by the spliceosome. Introns are identified by the binding of U1 snRNP to the 5′ splice site and the U2AF65/U2AF35 complex to the 3′ splice site. Following these interactions, other proteins and snRNPs are recruited to generate the complete spliceosomal complex needed to excise the intron. While many introns are constitutively removed by the spliceosome, other splice junctions are not used systematically, generating the phenomenon of alternative splicing. Alternative splicing is therefore the process by which a single species of pre-mRNA can be matured to produce different mRNA molecules (Fig. 1). Depending on the number and types of alternative splicing events, a pre-mRNA can generate from two to several thousands different mRNAs leading to the production of a corresponding number of proteins. It is now believed that the expression of at least 70 % of human genes is subjected to alternative splicing, implying an enormous contribution to proteomic diversity, and by extension, to the development and the evolution of complex animals. Defects in splicing have been associated with human diseases (Caceres and Kornblihtt, Trends Genet 18(4):186–93, 2002, Cartegni et al., Nat Rev Genet 3(4):285–98, 2002, Pagani and Baralle, Nat Rev Genet 5(5):389–96, 2004), including cancer (Brinkman, Clin Biochem 37(7):584–94, 2004, Venables, Bioessays 28(4):378–86, 2006, Srebrow and Kornblihtt, J Cell Sci 119(Pt 13):2635–2641, 2006, Revil et al., Bull Cancer 93(9):909–919, 2006, Venables, Transworld Res Network, 2006, Pajares et al., Lancet Oncol 8(4):349–57, 2007, Skotheim and Nees, Int J Biochem Cell Biol 39:1432–1449, 2007). Numerous studies have now confirmed the existence of specific differences in the alternative splicing profiles between normal and cancer tissues. Although there are a few cases where specific mutations are the primary cause for these changes, global alterations in alternative splicing in cancer cells may be primarily derived from changes in the expression of RNA-binding proteins that control splice site selection. Overall, these cancer-specific differences in alternative splicing offer an immense potential to improve the diagnosis and the prognosis of cancer. This review will focus on the functional impact of cancer-associated alternative splicing variants, the molecular determinants that alter the splicing decisions in cancer cells, and future therapeutic strategies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
For most of our 25,000 genes, the removal of introns by pre-messenger RNA (pre-mRNA) splicing represents an essential step toward the production of functional messenger RNAs (mRNA). Alternative splicing of a single pre-mRNA results in the production of different mRNAs. Although complex organisms use alternative splicing to expand protein function and phenotypic diversity, patterns of alternative splicing are often altered in cancer cells. Alternative splicing contributes to tumorigenesis by producing splice isoforms that can stimulate cell proliferation and cell migration or induce resistance to apoptosis and anticancer agents. Cancer-specific changes in splicing profiles can occur through mutations that are affecting splice sites and splicing control elements, and also by alterations in the expression of proteins that control splicing decisions. Recent progress in global approaches that interrogate splicing diversity should help to obtain specific splicing signatures for cancer types. The development of innovative approaches for annotating and reprogramming splicing events will more fully establish the essential contribution of alternative splicing to the biology of cancer and will hopefully provide novel targets and anticancer strategies.
Metazoan genes are usually made up of several exons interrupted by introns. The introns are removed from the pre-mRNA by RNA splicing. In conjunction with other maturation steps, such as capping and polyadenylation, the spliced mRNA is then transported to the cytoplasm to be translated into a functional protein. The basic mechanism of splicing requires accurate recognition of each extremity of each intron by the spliceosome. Introns are identified by the binding of U1 snRNP to the 5′ splice site and the U2AF65/U2AF35 complex to the 3′ splice site. Following these interactions, other proteins and snRNPs are recruited to generate the complete spliceosomal complex needed to excise the intron.
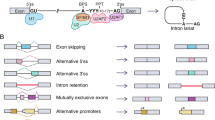
While many introns are constitutively removed by the spliceosome, other splice junctions are not used systematically, generating the phenomenon of alternative splicing. Alternative splicing is therefore the process by which a single species of pre-mRNA can be matured to produce different mRNA molecules (Fig. 1). Depending on the number and types of alternative splicing events, a pre-mRNA can generate from two to several thousands different mRNAs leading to the production of a corresponding number of proteins. It is now believed that the expression of at least 70 % of human genes is subjected to alternative splicing, implying an enormous contribution to proteomic diversity, and by extension, to the development and the evolution of complex animals.

Patterns of alternative splicing. Gray boxes represent exons or exonic fragments that are alternatively spliced. a cassette exon, b mutually exclusive exons, c alternative 5′ splice sites, d alternative 3′ splice sites, e intron retention, f alternative promoters can affect the identity of the first exon, and g alternative polyadenylation sites can impact the structure of the terminal exon
Defects in splicing have been associated with human diseases [1–3], including cancer [4–10]. Numerous studies have now confirmed the existence of specific differences in the alternative splicing profiles between normal and cancer tissues. Although there are a few cases where specific mutations are the primary cause for these changes, global alterations in alternative splicing in cancer cells may be primarily derived from changes in the expression of RNA-binding proteins that control splice site selection. Overall, these cancer-specific differences in alternative splicing offer an immense potential to improve the diagnosis and the prognosis of cancer.
This review will focus on the functional impact of cancer-associated alternative splicing variants, the molecular determinants that alter the splicing decisions in cancer cells, and future therapeutic strategies.
2 Function of Cancer-Associated Splice Variants
Alternative splicing is part of the normal expression program of the majority of human genes. The initial reports describing the importance of alternative splicing in the control of sex determination in Drosophila [11] were followed by several examples linking splicing to the regulation of gene expression in human cells. For example, it was shown that alternative splicing controls the production of membrane-associated or secreted forms of immunoglobulins [12], and the synthesis of hormones with distinct physiological functions [13]. The most striking examples of the effect of splicing on gene expression are found in the nervous system where alternative splicing is used to expand the functional repertoire of receptor molecules. One notable example is the alternative splicing of the Slo gene that leads to the production of different proteins playing a role in the perception of different sound frequencies during audition [14].
The contribution of alternative splicing to gene expression is not always to expand protein diversity. Indeed, alternative splicing can also regulate the level of gene expression by producing mRNA isoforms containing premature stop codons that activate nonsense-mediated RNA decay (NMD). However, a global analysis of the effect of knocking-down a specific NMD component on the abundance of 3126 alternative splicing events suggests that this pathway is not general but rather affects a selected group of pre-mRNAs [15]. Interestingly, splicing factors such as PTB and SC35 autoregulate their own expression by promoting the production of the NMD-sensitive isoforms [16–18]. Recent studies indicate that this mode of regulation is a hallmark of many splicing regulatory factors that belong to the SR and hnRNP family of proteins [17, 19].
Despite the involvement of alternative splicing in the expression and diversity of selective sets of genes, the global impact of the functional diversity imparted by alternative splicing still remains incomplete and controversial [20]. However, some of the best available evidence documenting the breadth and significance of alternative splicing has been provided by the study of cancer cells. Selected examples ordered by cellular functions are presented below.
2.1 Cellular Proliferation
Expression of the fibroblast growth factor receptor (FGFR) family is closely linked to cellular proliferation and cancer. Alternative splicing of FGFR1 and FGFR2 pre-mRNAs produce splice variants that have different affinities for their respective ligands. The FGFR2-IIIc splice isoforms is overexpressed in advanced stages of prostate cancers and transforms human mammary epithelial cells when expressed ectopically [21, 22]. FGFR2-IIIc accumulates in mesenchymal cells while FGFR2-IIIb is preferentially produced in epithelial cells. In a rat model system, prostate cancer cells expressing the FGFR2-IIIc-specific exon in a reporter construct revealed unexpected mesenchymal-epithelial transitions in primary tumors and lung micrometastases, revealing their phenotypic plasticity [23].
Proto-oncogenes and tumor-suppressor genes are essential players in the regulation of cellular proliferation. The majority of these genes express isoforms that are generated by alternative splicing. For example, the pre-mRNA encoding the p53 protein is alternatively spliced to produce isoforms whose abundance vary in breast tumors [24]. Other members of the p53 family such as p73 and p63 are alternatively spliced leading to the inclusion or the exclusion of the transcription transactivation domain. Inclusion of the transactivation domain transforms these tumor-suppressor proteins into oncoproteins [25]. The transcriptional activity of p53 is itself controlled via the alternative splicing of MDM2. Variations in the splicing of MDM2 in cancer tissues influence its accumulation in the nucleus and therefore affect its capacity to repress the transcription of p53 [26, 27]. Other examples of cancer-associated changes in alternative splicing are listed in Table 1.
2.2 Cellular Invasion
Integrins are a family of cell adhesion transmembrane proteins. Their expression modulates the invasive properties of cancer cells. Numerous integrin splice variants that either facilitate or inhibit cellular proliferation have been described [28]. The cell–cell adhesion glycoprotein CD44 whose expression is associated with the metastatic potential of cancer cells [29] is produced in more than 20 splicing isoforms. Isoforms that contain exon v6 induce metastasis in mammary and pancreatic carcinomas in rats [30], while inclusion of exon v10 modifies CD44 adhesion properties and contributes to cancer progression [31]. Integrins often interact with fibronectin, itself produced in various versions. The alternate EDB- and EDA-containing isoforms of fibronectin are involved in cell adhesion and spreading [32]. In breast and colon cancers, overexpression of RonD, the splice variant of the transmembrane receptor MSF (macrophage-stimulating factor) is overexpressed, which enhances the migratory properties of cancer cells [33]. Table 1 lists additional genes whose change in alternative splicing has been shown to affect cell adhesion properties and invasiveness.
2.3 Angiogenesis
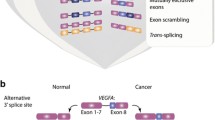
Several splicing changes affect angiogenesis, most notably those of the vascular endothelial growth factor (VEGF) in which an alternative 3′ splice site causes an antiangiogenic form to be produced in normal tissues [34]. Recently, it was also shown that prostate tumors have reduced incorporation of exon 7. Experimental inhibition of exon 7 inclusion reduced the ability of transplanted cells to induce angiogenesis in mice [35]. Exon 7 incorporation is positively regulated by the RNA-binding protein T-STAR (also known as SLM-2) [36]. VEGF expression is itself affected by the alternative splicing products of the Estrogen receptor alpha and the cholecystokin-2/gastrin receptor (CCK2 R) [37, 38]. Another relevant event is the removal of exon 3 of Survivin, which gives it a specific function in promoting angiogenesis [39].
2.4 Resistance to Apoptosis
Alternative splicing has a strong impact on the function of proteins implicated in apoptosis. A comprehensive inventory of isoforms derived by alternative splicing of apoptotic genes and a summary of their known functions appeared recently [40]. The functional consequences of alternative splicing have been demonstrated for the death receptor Fas, as well as the adaptor and regulatory proteins APAF1 and Survivin. The function of apoptotic mediators such as Bcl-x, Bfk, Mcl1, Bim and Bid, and several caspases were also shown to be modulated by alternative splicing [40]. The alternative splicing of these genes produces isoforms that often have different and sometimes opposing activities.
A close association with cancer is particularly well documented for the splicing variants of Bcl-x. Competing 5′ splice sites dictate the expression of the Bcl-xL and Bcl-xS isoforms, two proteins with antagonist regulatory functions [41]. Bcl-xL is an anti-apoptotic protein that protects the integrity of mitochondria, and is overexpressed in many types of cancer cells [42–46]. On the other hand, the pro-apoptotic isoform Bcl-xS can heterodimerize with Bcl-xL to abrogate its anti-apoptotic activity [47]. Furthermore, Bcl-xL overexpression is directly responsible for the resistance of cancer cells to stresses and chemotherapeutic drugs [41, 48–56].
Another example of a splicing event linked to apoptosis is FIR, a known inhibitor of MYC. In this case, a shorter splice isoform of FIR switches protein activity to facilitate the overexpression of MYC, inhibiting apoptosis [57]. The alternative splicing of protein kinase C delta (PKCδ) was also shown to produce an isoform that can protect human teratocarcinoma cells from apoptosis [58]. Other recent examples of splice isoforms affecting apoptosis are presented in Table 1.
2.5 Multidrug Resistance
Drug transporters defend cells against cytotoxic agents. Thus, alterations in their alternative splicing may alter the efficacy of anticancer agents. One major group of transporters are known as the multidrug resistance-associated proteins (MRP; ABCC gene family). Alternative splicing of many genes in this group creates functional variants, as is the case with MRP1 that produces splicing variants conferring resistance to doxorubicin in ovarian tumors [59]. Alternative splicing of MRP4, a drug-efflux pump mediating the efflux of nucleotide analogs, generates a non-functional protein via introduction of premature termination codons [60]. Differential expression of two mRNA isoforms of the ATP-binding cassette transporter gene ABCB 5 was reported in melanoma cells [61]. Several alternatively spliced P-glycoprotein transcripts have been found in multidrug-resistant cells and their expression correlates with drug resistance [62, 63]. Finally, BCRP (ABCG2) splice variants are differentially expressed in human drug-selected breast cancer cell lines [64].
The deregulated expression of dominant negative variants of p63 and p73 lacking the transactivation domain can inhibit the transactivation of target genes and apoptosis, thus contributing to chemoresistance [65]. A splice variant of the spindle checkpoint gene Mad2 abrogates mitotic arrest and adriamycin-induced apoptosis [66]. Finally, a MAP-kinase activating death domain (MADD) splice variant of the IG20 gene suppresses tumor cell survival and enhances susceptibility to apoptosis and anticancer drugs [67].
3 Alternative Splicing Control: Basic Principles
Before discussing specific examples of molecular alterations responsible for cancer-specific splicing patterns, it is important to review some key principles of splice site control and selection. Splice site selection is determined first by the sequence of splice sites. The sequence of constitutive splice sites that are used all the time usually matches the consensus CAG/GUGAGU, for a 5′ splice site («/» indicates the junction between exon and intron), and a polypyrimidine-rich tract followed by CAG/at the 3′ splice site (Fig. 2a). In contrast, alternative splice sites usually display various mismatches to the consensus sequence, which make them intrinsically weaker splice sites. This intrinsic weakness renders them amenable to modulation by control factors that may increase or decrease their use. Hence, the use of alternative splice sites is often influenced by the presence of nearby exonic or intronic auxiliary elements that promote or repress splicing. These sequence elements function by recruiting proteins that either directly modulate splice site recognition, affect the formation of specific splicing complexes or change the conformation of the pre-mRNA to promote the use of certain splice site combinations.

Control of splice site selection. a Schematic representation of a splicing unit (top) and an alternative splicing unit (bottom). The consensus sequences of a 5′ splice site, a branch site and a 3′ splice site are shown. R purine, Y pyrimidine. Elements that control splicing decisions are shown and include positively acting exonic and intronic splicing enhancers (ESEs and ISEs, respectively), as well as negatively acting exonic and intronic splicing silencers (ESSs, ISSs, respectively). b Proteins bound to exonic or intronic enhancers can recruit or stabilize the binding of splicing factors to the 3′ (left) or 5′ (right) splice site. In the illustrated examples, an SR protein bound to an ESE enhances U2AF and U2 snRNP binding to the 3′ splice site/branch site region [102, 217], while binding of TIA1 to ISE can stimulate recognition of an adjacent upstream 5′ splice site by the U1 snRNP [126]. c Splicing inhibition. Steric interference caused by the presence of a protein bound in the proximity of a splice site impedes its recognition. The SR and hnRNP H proteins can, respectively, inhibit the binding of the U2 and U1 snRNPs [218], [219]. d Change in pre-mRNA conformation can be promoted by an interaction between hnRNP A1 proteins bound to introns flanking an alternative exon, provoking repression of the looped out exon, and stimulation of splicing between the distal pair of exons [72]
Direct modulation of splice site recognition is promoted by members of the SR family of proteins, which are frequently associated with stimulatory elements. However, other types of proteins with stimulatory activity have been identified (for example, CELF, hnRNP H, hnRNP L, TIA1, and TIAR) [68]. When these proteins are bound in the proximity of a splice site, they may interact with other components of the splicing machinery (U1 snRNP at the 5′ splice site or U2AF at the 3′ splice site) to improve their binding (Fig. 2b). Alternatively, some sequence elements located near splice sites may repress their use (Fig. 2c) by recruiting proteins that hinder the recognition or utilization of the adjacent splice sites.
In addition to the direct role of proteins recruited in the vicinity of a splice site, changes in the conformation of the pre-mRNA may also influence splicing decisions. These changes sometimes involve the formation of a secondary structure that blocks the utilization of a splice site [69]. In other cases, the changes are induced by an interaction between proteins bound at different locations on a pre-mRNA. Such spatial rearrangement can stimulate the use of a splice site located outside of the loop, and may simultaneously repress splice sites located inside that loop (Fig. 2d). Although this model was initially proposed to explain the mechanism of action of the hnRNP A1 protein in alternative splicing [70–72], it is also relevant to the mechanism of action of hnRNP H, and possibly hnRNP I/PTB and Nova-1 proteins [72–75]. hnRNP A1 may also counteract the activity of certain SR proteins through a different model which proposes that hnRNP A1 can nucleate and spread over regions of a pre-mRNA through cooperative RNA binding [76, 77].
Detailed investigations of selected alternative splicing events indicate that splice site selection is determined by a combination of several layers of positive and negative regulators. Thus, the frequency with which a given alternative splicing pattern is used will be determined by: (1) the intrinsic strength of each splice site involved, (2) the number, identity, and the position of control elements, and (3) the relative concentration and affinity of each RNA-binding protein to its respective binding site. The phosphorylation of specific SR and hnRNP proteins will also have an impact on splice site selection since this modification can affect their activity and cellular distribution [78].
4 Molecular Basis for Splicing Alterations in Cancer
Different types of molecular perturbations can cause alterations in alternative splicing profiles. Point mutations at splice sites have been linked to numerous diseases and have been proposed to account for 15 % of human genetic diseases [2, 79]. This number is likely to be considerably larger since mutations in introns, which constitute the majority of the gene sequence, are rarely considered. A recent study suggests that as much as 60 % of the point mutations that cause genetic diseases affect splicing decisions [80]. Thus, we can anticipate that several cancer-related genes sustain mutations at splice sites or in regions bound by proteins that control the selection of splice sites. Exonic mutations that create a stop codon can also affect the function of a splicing control element, thereby modifying splicing profiles [81]. In other cases, missense mutations, again through changes in splicing, may have a more profound impact on protein structure than the predicted change in amino acids caused by the mutations. Finally, even mutations that are considered to be neutral because they do not change amino acids can have an impact on alternative splicing [2, 3]. The following sections present some examples of mutations (Fig. 3) and changes in the expression of splicing factors (Fig. 4) that affect the splicing of cancer-associated genes.

Alternative splicing of selected cancer-related genes (see main text for details)

Specific examples of mutations reported to affect alternative splicing in cancer. Mutations are organized according to their impact on the inactivation or activation of splice sites by directly changing the structure of the splicing signals or control elements. The name of the gene that is affected, the type of cancer in which it was reported, and relevant references are given
4.1 Mutations at Splice Sites and in Auxiliary Elements
According to the Cancer Genome Project of the Welcome Trust Sanger Institute (http://www.sanger.ac.uk/genetics/CGP/Census/), about 363 human genes have mutations that have been associated with cancer; 90 % of these genes have somatic mutations, approximately 20 % show germline mutations that predispose to cancer, and 10 % show both somatic and germline mutations. Specifically, 42 of these 363 genes have sustained mutations that affect splicing. Splicing mutations are the most prevalent type of mutations found in the NF1 gene, which is implicated in neurofibromatosis, the most common form of autosomal dominant cancer in humans [82]. Mutations in the splice sites of NF2 are used as markers to grade the severity of the disease [83].
Similarly, approximately 30 mutations affecting the splice sites of p53 have been reported in different cancers [84]. Another interesting example concerns the tumor suppressor gene APC, where two mutations have been closely associated with the development of familial adenomatous polyposis [85]. One of these mutations creates a splice site that promotes the deletion of one nucleotide resulting in the production of a truncated APC protein. Other mutations that weaken or create new splice sites in different genes associated with cancers are listed in Fig. 3. This list includes well-known genes such BRCA1, BRCA2, CDKN2, PTEN, KIT, ATM, and XPC.
Mutations in cis-acting splicing regulatory elements can also modify the relative abundance of splicing variants or induce the utilization of new splice sites. For example, a mutation of an exonic element in the BRCA1 gene reduces the binding of ASF/SF2, thereby increasing the exclusion of exon 18 [86]. Many mutations that affect splicing control elements have been described in the BRCA1 gene [87]. A polymorphism in the intron upstream of exon 2 in KLF6 creates a binding site for SRp40 that activates cryptic sites in exon 2 [88]. This mutation is associated with an increased risk of prostate cancer. The altered KLF6 proteins may counteract the tumor-suppressor activity of the wild-type protein. Intronic and exonic mutations in the cadherin CDH17 gene that do not directly affect the splice junctions have been proposed to interfere with splicing decisions [89]. Other examples of this type of mutation have been reported in NF1, APC, and MLH1 genes (Fig. 3).
The diversity of splicing control elements suggests that most intronic or exonic mutations that were initially considered silent mutations may in fact have an important impact on constitutive and alternative splicing. For the same reasons, a large subset of single nucleotide polymorphisms (SNPs) found within the human population may modulate alternative splicing. For example, a SNP in an SRp40 binding site in the APC gene correlates with exon omission and attenuated familial adenomatous polyposis [90]. These differences may predispose some individuals to develop certain types of cancers, while other polymorphisms may increase their resistance. Consistent with this view, a recent study has identified a high frequency of alternative splicing in microsatellite regions linked with human longevity or resistance to anticancer treatments [91].
4.2 Alterations in the Activity of Splicing Proteins
While many cis-acting mutations have been shown to affect splicing, the majority of alterations in alternative splicing profiles appear to proceed from changes in the expression or activity of splicing factors [4]. Because a proof for the direct contribution of such changes to cancer is not always available, only experimentally proven cases will be discussed below.
4.2.1 Chromosomal Translocations
In some cancers, as in Ewing’s sarcomas, chromosomal translocations affect the genes EWS and TLS (FUS or hnRNP P2) that encode RNA-binding proteins. The interaction of these proteins with splicing factors such as SF1, U1C, YB-1 and some SR proteins may explain why their translocation has an impact on splicing control [92–95]. Translocations between these genes and transcription factors of the ETS family result in the production of chimeric proteins with strong transcriptional activity and oncogenic properties, as for example EWS-FLI1, EWS-NOR1, and TLS-ERG [96]. The failure of the hybrid proteins EWS-FLI1 and TLS-ERG to recruit the YB-1 and SR proteins may compromise their role in splicing [95, 97]. The interaction of EWS-FLI1 and TLS-ERG with transcriptionally active form of RNA polymerase II [94, 95, 98] may affect the cotranscriptional selection of splice sites. EWS-NOR1 can interact with the snRNP protein U1C. EWS-NOR1 can also enhance the use of a distal 5′ splice site in a reporter pre-mRNA more efficiently than the EWS protein alone [99]. In leukemia, the well-known Bcr-Abl translocation has been associated with splicing defects in SLP65, Bruton’s tyrosine kinase, Pyk2 and Ikaros. However, the mechanism responsible for these alterations may be indirect since Bcr-Abl enhances the expression of SRPK1, a kinase that modulates the activity of SR proteins [100].
4.2.2 Alterations in the Expression and Localization of Splicing Control Factors
Viral transformation was first associated with the upregulation and increased activity of SR proteins [101], and this change affected fibronectin pre-mRNA splicing [102]. The first case of a deregulation in the expression of hnRNP and SR proteins in cancer cells was observed in colon adenocarcinomas [103]. Since then, alterations in the expression of a variety of RNA-binding proteins have been reported in different types of cancers [104] (see Table 2). For example, expression of the SR protein ASF/SF2 is upregulated in many types of cancers and can elicit the transformation of immortal rodent fibroblasts [105]. Notably, ASF/SF2 shifts the alternative splicing of three pre-mRNAs: BIN1 produces an isoform that lacks tumor–suppressor activity; a MNK2 kinase variant can phosphorylate eiF4E in a MAP kinase-independent manner; the S6K1 kinase is spliced to produce an oncogenic isoform [105]. Since ASF/SF2 overexpression in breast cancer has been linked to cell motility through the production of RonD [33], ASF/SF2 may contribute to early and late steps of carcinogenesis.
While over expression of SR proteins has been linked to cancer, downregulation and altered phosphorylation are relevant to apoptosis, a process that is often defective in cancer cells. A reduction in ASF/SF2 can induce apoptosis. Genomic DNA fragmentation is blocked because the drop in ASF/SF2 changes the alternative splicing profile of the ICAD nuclease to favor the expression of the inactive isoform [106]. Similarly, induction of the apoptotic pathway through activation of the death receptor Fas dephosphorylates SR proteins [107]. Fas and the anticancer drug gemcitabine increase the levels of ceramide that activates protein phosphatase 1 (PP1), which in turn can act on SR proteins. Since ceramide promotes a shift toward the production of the pro-apoptotic Bcl-xS protein, a reduction in phosphorylation of SR proteins may elicit apoptosis.
An increase in the levels of hnRNP I/PTB and SRp20 in ovarian cancers that are resistant to doxorubicin correlates with the overexpression of some isoforms of the multidrug resistance protein MRP1 [59]. Moreover, PTB knockdown suppresses growth and invasiveness [108]. Overexpression of the alternative splicing factor SPF45 is often observed in tumors and may confer resistance to chemotherapeutic agents [109].
Components of the constitutive splicing machinery may also be subject to differential regulation in tumors. The branchpoint binding protein SF1 is downregulated in mouse intestinal tumorigenesis. Expression of SF1 is regulated by β-catenin and it affects alternative splicing of several genes including WISP1, FGFR3 and the Estrogen receptor-beta [110]. In pancreatic tumors, the expression of U2AF35 (part of the other main protein complex that recognizes 3′ splice sites) is also often repressed. This situation promotes the synthesis of a constitutively active isoform of the CCK-B receptor and a resulting stimulation of cellular proliferation [111]. The breast cancer-associated scaffold attachment factor SAFB1 interacts with a plethora of splicing factors and affects splicing [112].
In general, however, while several studies have reported increased expression of specific RNA-binding proteins in cancer tissues, the functional impact of these differences in the alternative splicing of important genes in cancer remains poorly documented. A few of the most important cases where this link has been established are presented below.
FGFRs. Approximately 10 distinct elements control the alternative splicing of the mutually exclusive IIIb and IIIc exons in FGFR2 (Fig. 4a). Fox-2 and hnRNP I/PTB proteins have been implicated in the activity of some of these controlling elements [113, 114]. hnRNP I/PTB also controls the alternative splicing of FGFR1. Interestingly, the high levels of hnRNP I/PTB in glioblastomas may promote the exclusion of exon α, thereby improving its affinity for the ligand [115].
CD44. An increase in the expression of some SR proteins is associated with the progression from the pre-neoplastic to the metastatic status of mammary tumors [103, 116]. This observation correlates with modifications in the alternative splicing of the cell-surface glycoprotein CD44 in human mammary tumors [103]. In ovarian cancers, an increase in the expression of SR proteins and their hyperphosphorylated forms has also been observed [117]. In addition, exon v9 of CD44 contains a stimulatory element that can be bound by low molecular weight SR proteins [118].
Sam68 and hnRNP A1 also contribute to the alternative splicing of CD44 by controlling the inclusion of exon v5 [119, 120]. A protein of the same family as Sam68, T-STAR (SLM2), can also bind to the v5 exon to promote its inclusion and this is antagonized by other RNA-binding proteins such as SRp30c, hnRNP G, and SAF-B, as well as by SIAH-1, a ubiquitin ligase and candidate tumor suppressor [121]. The inclusion of exon v5 is stimulated by the phosphorylation of Sam68 following activation of the Ras signaling pathway. This stimulation requires the participation of the splicing co-activator SRm160, which interacts with Sam68 [122]. As demonstrated in a recent study, the binding of Tra2β to exon v4 of CD44 and its synergic action with the nucleic acid-binding protein YB-1 enhance the inclusion of both exons v4 and v5 [123].
Interestingly, overexpression of Brm, a component of the SWI/SNF chromatin remodeling complex can promote the inclusion of the v5–v9 exons through a process that requires Sam68 [124].
Ron. The binding of the SR protein ASF/SF2 to a sequence of exon 12 on the pre-mRNA of Ron, a tyrosine kinase transmembrane receptor, improves the utilization of its 3′ splice site and enhances the exclusion of the immediate upstream exon 11. Upregulation of ASF/SF2 in cancer cells is associated with enhanced exon 11 skipping and generation of RonD, an isoform that improves the motility and invasive properties of these cells. RonD is upregulated in metastatic breast and colon cancers [33, 125].
Fas. Alternative splicing of the pre-mRNA encoding the cell death receptor Fas generates two isoforms through inclusion or skipping of exon 6. The membrane-bound long isoform activates the extrinsic apoptotic pathway, while the soluble short isoform that lacks the transmembrane domain is an anti-apoptotic factor. The production of the long isoform is facilitated by TIA1 and TIAR that bind to a U-rich sequence situated immediately downstream of the 5′ splice site of alternative exon 6. These proteins stimulate the binding of the U1 snRNP at this 5′ splice site (Fig. 4b) [126]. In contrast, the exclusion of exon 6 is enhanced by the binding of hnRNP I/PTB to exon 6 which interferes with the communication between the downstream U1 snRNP bound to the 5′ splice site and U2AF bound to the upstream 3′ splice site [127].
Bcl-x. The alternative use of two 5′ splice sites produces the pro- and anti-apoptotic isoforms of Bcl-x (Fig. 4c). Several regulatory elements have been identified in the regions flanking these splice sites. Intronic regulatory elements (IRE) may repress the use of the 5′ splice site of Bcl-xL following induction by IL-6 and GM-CSF, or treatment with TPA [128]. In addition, two exonic regions known as CRCE1 and CRCE2 mediate the pro-apoptotic effect conferred by ceramide on the splicing of Bcl-x [129]. Given that the U2 snRNP-associated protein SAP155 binds to CRCE1 and that its genetic depletion by RNA interference also promotes Bcl-xS usage, the signaling pathway activated by ceramide possibly prevents SAP155 binding. More recently, a role for Sam68 has also been uncovered [130]. Although the cis-acting element mediating this function is currently unknown, Sam68 appears to interact with hnRNP A1 to elicit a splicing shift toward Bcl-xS. Three other regions have been identified as important for controlling the use of the alternative 5′ splice sites. One of them (B2) is specifically bound by hnRNP F/H proteins and is required to enforce the use of Bcl-xS 5′ splice site [131]. The B3 region is bound by SRp30c and enforces Bcl-xL usage [132]. Finally, a region (SB1) upstream of the Bcl-xS 5′ splice site distinct from CRCE1 represses the use of the 5′ splice site of Bcl-xS. This SB1-dependent splicing repression is lifted by drugs that inactivate protein kinase C, as well as by a variety of anticancer agents [133, 134]. The pathways that control this aspect of Bcl-xS splicing vary considerably between different cancer cell lines.
Caspase 2. Members of the SR and hnRNP family of proteins participate in the alternative splicing control of caspase 2 [135]. Overexpressing SR proteins enhances the exclusion of exon 9 to favor the production of the pro-apoptotic isoform Casp2L. In contrast, overexpressing hnRNP A1 stimulates the inclusion of exon 9 and the production of the anti-apoptotic isoform Casp2S [136]. The intronic sequence In100 is a control element modulating the alternative splicing of caspase 2 (Fig. 4d). This element is bound by hnRNP I/PTB and it prevents splicing between exons 9 and 10 through formation of a nonproductive complex with splicing factors recruited at the In100 site [137].
4.2.3 Other Perturbations that Impact Alternative Splicing
As discussed above, cancer cells can accumulate genetic changes that directly affect the splicing machinery or splicing regulatory elements in pre-mRNAs to deregulate alternative splicing patterns. In addition, at least four other molecular processes may affect the production of splicing variants to impact on carcinogenesis. These include transcription, RNA editing, NMD, and signal transduction events.
4.2.3.1 Transcription
Simple overexpression of pre-mRNAs may contribute to deregulated alternative splicing. For overexpressed pre-mRNAs, there might not be enough available control factors to modulate their splicing. In addition, the sequestration of splicing factors by this pre-mRNA may affect the splicing of other pre-mRNAs, similar to the situation occurring when triplet repeat expansion in the 3′ untranslated region of the DMPK pre-mRNA sequesters MBNL with an impact on the alternative splicing of cardiac troponin T, tau, insulin receptor, ClC-1, and myotubularin-related 1 pre-mRNAs [138, 139].
The mechanistic coupling of RNA synthesis by RNA polymerase II to alternative splice site choice has been documented [140, 141]. Both enhancer and core promoter elements can modulate the alternative splicing of their nascent pre-mRNA [142, 143]. Promoter identity can control alternative splicing through the differential recruitment of transcription factors and cofactors, such as Brm [124], CoAA [144] and Spi-1 [145], or by recruitment of specific splicing factors, such as SRp20 [146]. Ultimately, promoter-specific transcription complexes can dictate alternative splice site choice by altering the rate of elongation and/or the processivity of RNA polymerase II [147, 148], or through physical interactions with the splicing machinery. Consequently, factors that modify the status of chromatin that change the intrinsic speed of the transcriptional complex or that modify its sensitivity to pausing sites can potentially affect alternative splicing.
In addition to the impact of different complexes at a single promoter on alternative splicing, there is now convincing evidence for the impact of multiple promoters of a single gene on alternative splicing. For Bcl-x [149] and caspase-2 [150], the choice of alternative promoters can define the ratio of splice variant expression. The literature now contains a growing list of genes that contain alternative promoters [151]. In fact, a recent bioinformatics analysis estimated that 52 % of human genes contain alternative promoters [152]. In this context, oncogenic alterations of transcription factors, as well as genetic or epigenetic changes in promoter regions could influence the alternative splicing patterns in tumor cells. We already know that there must be considerable tissue- and cancer-specific variations in the combinatorial assemblies at promoters. What remains to be discovered is the extent of the contribution of these transcriptional aspects to global patterns of alternative splicing in cancer.
4.2.3.2 RNA Editing
RNA editing is a process that directly modifies specific adenosine residues into inosines [153]. RNA editing can influence the secondary structure of specific pre-mRNAs in preparation for splicing. In some types of tumors, editing defects can affect the alternative splicing of the PTPN6 phosphatase and glutamate receptor pre-mRNAs [154, 155]. Because the contribution of RNA editing to the alternative splicing of cancer-related genes has not been investigated systematically, it is likely that more examples of this type exist.
4.2.3.3 NMD
NMD is a surveillance process that eliminates mRNA molecules containing a stop codon localized more than 50 nucleotides upstream of exon/exon junction [156, 157]. Therefore, NMD can potentially neutralize the impact of mutations that create new termination codons [157, 158]. While NMD can affect the accumulation of some splicing variants, its widespread contribution in controlling the expression of splicing isoforms is unlikely [15] but controversial [159]. Interestingly, the non-productive isoforms of splicing factors that are generated from the alternative splicing of exons enriched in ultra conserved sequences form a distinct class of NMD-regulated transcripts [17, 19].
It is possible but yet undocumented that changes in the expression or activity of factors involved in regulation of the NMD may promote the accumulation of the mutated or aberrantly spliced transcripts and therefore may contribute to cancer. Although the number of examples supporting a role for NMD in cancer remains small [160, 161], this pathway appears to contribute to the elimination of mRNA encoding proteins involved in drug-resistance [60]. Such alterations in the NMD pathway may contribute to tumorigenesis.
4.2.3.4 Signal Transduction
Cell signaling impacts alternative splicing [162, 163] and a typical means by which such changes can be mediated is by the phosphorylation of SR proteins, which causes them to accumulate in different subcellular compartments [164]. The upregulation of PKA and the MKK3/6/p38 signaling pathway results in the cytoplasmic localization of hnRNP I/PTB and hnRNP A1, respectively [165, 166]. Another splicing factor displaying this behavior is KSRP, which is implicated in the inclusion a specific exon in the c-src mRNA in neurons [167]. KSRP accumulates in the nucleus when neuroblastoma cells are induced to differentiate [167]. Therefore, the altered subcellular localization of splicing factors via their post-translational modification can potentially play a role in the deregulation of splicing in cancer cells. The impact of splicing factor localization on the cancer phenotype is likely underappreciated, as documenting changes in localization is considerably more laborious than determining changes in total expression levels of the mRNA or protein.
The interface between signal transduction and splice site selection is just beginning to be explored. The Ras/PI 3-kinase/AKT pathway is often activated in human cancer [168]. This pathway can modulate the activity of SR proteins to affect the alternative splicing of fibronectin and PKC βII [169, 170]. The Ras-dependent signaling pathway is also implicated in the alternative splicing of CD44 [171]. The intricate interplay between alternative splicing and cell signaling is illustrated by the fact that just as signaling affects alternative splicing, so alternative splicing can affect signaling. For example, the inclusion of exon v6 in CD44 promoted by Ras helps to sustain late Ras signaling [172]. The SR protein kinase SRPK1 is overexpressed in many types of cancers and its downregulation increases the sensitivity of cells to anticancer drugs [173], and MAP2 K is alternatively spliced in response to activation of SRPK1 [174]. Another recent example of a link between signaling and splicing control involves the Notch3 signaling pathway which regulates the splicing of Ikaros in leukemia by inducing the RNA-binding protein HuD [175–177]. The deregulated expression of CDK12 and cyclin L1/L2 can also affect alternative splicing decisions [178].
Thus, it would not be too surprising if cancer-specific alterations in signaling pathways impact the production of isoforms that contribute to neoplastic transformation. However, the contribution of signal transduction events to splicing decisions that are relevant to cancer remains to be more fully investigated.
Arginine methylation of splicing factors also affects their localization and activity [179, 180]. Recently, the arginine methylase CARM1 was shown to control splicing decisions [181]. Although cancer-specific defects in the arginine methylation of splicing factors are yet to be reported, important contributions of this pathway to various aspects of cell growth can be anticipated.
5 Outlooks and Challenges
5.1 Global Detection of Splicing Variation in Cancer
The existence of cancer-specific signatures made up of individual alternative splicing events was initially investigated by using large-scale compilation of cDNAs and expressed-sequences tags (ESTs). Computational approaches revealed considerable differences between the alternative splicing patterns of normal and cancer prostate tissues [182, 183]. Genome-wide profiling of splicing events became possible with the development of DNA oligonucleotide microarrays. Using this technology, one of the first observations was that the genes displaying tissue-specific alternative splicing were largely different from the genes presenting tissue-specific differences in steady-state expression levels [184]. Indeed, a quarter of the genes that showed alternative splicing between normal and cancerous prostate had no detectable change in overall gene expression level [185]. A similar conclusion was obtained when mining for differences in expression profiles and the detection of splicing isoforms between melanoma and melanocytes [186]. Overall, the classification of tumors was improved when alternative splicing was considered, strongly arguing in favor of mining the wealth of alternative splicing diversity for the purpose of developing a complementary disease-specific signature that could have diagnostic and prognostic value.
Splicing-sensitive arrays were generated using limited sets of alternative splice junction probes and used to detect splicing changes in cancer tissues. For example, changes in splicing profiles and the abundance of splicing factors in Hodgkin lymphoma was observed using an array that measured mRNA levels and 100 splicing events [187]. More recently, a similar design was used to compare alternative splicing of 64 genes in breast cancer cell lines and xenografts [188]. In this case, four genes were found to be differentially spliced between breast cancer and normal cell lines and four other tumor-associated genes were spliced differently according to cell culture conditions.
A new technique called DASL was developed to measure the expression of cancer-related splice isoforms. This technique combines targeted microarray and PCR techniques and treats splicing isoforms as separate genes. DASL therefore estimates changes in global gene expression level, as a normal microarray does, while providing additional information on the ratio of splice isoforms variants [189]. Recently, this approach has been used for the analysis of 1,500 different splice variants from 364 genes in six prostate tumor cell lines and 22 prostate tumors [185, 190]. Fourteen genes had different isoforms whose expression in normal tissues inversely correlated with their expression in tumor samples, implying a likely switch in alternative splicing.
Companies have also designed microarrays for cancer biomarker discovery using known alternatively spliced junction probes identified through bioinformatics mining of EST libraries. The Jivan cancer-specific splice variant array covers 524 putative cancer-specific splices although this has been largely superseded by their total splice form microarray that includes 193,000 specific splice junctions. ExonHit also has pathway-specific microarrays and a ‘genome-wide’ alternative splice junction microarray. The other approach to splicing discovery is to print probes for every exon:exon junction and deduce alternative splicing patterns from the relative junction expression patterns. The first large-scale splicing microarrays using this approach covered about half of the known exon:exon junctions in the human transcriptome and monitored splicing globally in multiple normal and cancer cell lines [191]. Affymetrix now produces a Human Exon GeneChip microarray that detects over 1,000,000 different human exons. This was used to compare splicing between ten matched normal tumor colon cancer pairs and nine high confidence differences were confirmed by RT-PCR [192].
One drawback of most current global approaches is that they rely on previously documented splicing events and are therefore not designed to discover novel splicing events. Because our current collection of existing splicing events is likely incomplete, biased, and poorly validated, there is a need to incorporate strategies that can identify novel splicing events and technologies that can accurately validate the quantitative differences detected between samples. Current microarray technologies may be superseded in the future by high-throughput sequencing technologies of single molecules [193]. However, although all the above approaches are designed to improve the annotation and assessment of alternative splicing events, they all fail to provide a description of the complete structure of splice isoforms. Indeed, pre-mRNA often sustains multiple alternative splicing events, sometimes involving regions that are far apart. Given that splicing decisions taken in one region can affect the splicing outcome of an apparently distinct unit [194], we cannot assume that all combinations of potential isoforms are represented in the mRNA population. The only currently reliable way to obtain this information remains through cDNA cloning and large-scale projects in this direction would provide a useful complement to current high-throughput mapping efforts directed at specific alternative splicing units.
5.2 Depleting Specific Splice Isoforms
We have seen that mutations in splicing elements of key genes and expression defects of splicing control factors can contribute to neoplastic transformation. The alterations in the splicing profiles observed in tumors may lead to the production of novel isoforms. In other cases, the isoforms may correspond to molecules that are normally expressed in other cell types or at other stages of development. Most frequently, splicing alterations will involve differences in the relative abundance of isoforms already expressed in the normal tissue. Although several of these alterations may have a neutral impact, other splicing alterations may confer growth advantages to cancer cells. For example, the signaling pathways that inhibit cell proliferation or induce cell death may be neutralized leading to cell propagation and invasion. Considering the enormous potential for diversity emanating from alternative splicing, the number of isoforms that are known to affect the growth of cancer cells is most probably vastly underestimated. More extensive annotation of the function of different isoforms is a priority that represents a challenge both in terms of commitment and methodology.
To help with the goal of attributing function to splice variants, the RNA interference (RNAi) technology represents a useful approach because it can be used to reduce the expression of specific isoforms. Although this approach has been used successfully in Drosophila cells [195], it is yet to be used in a systematic manner in human cells. If RNA interference-based approaches turn out to promote transcriptional silencing in addition to RNA degradation [196, 197], depletion of specific mRNA isoforms may only be possible with siRNAs targeting exon-exon junctions. This caveat could seriously compromise the use of RNA interference as a tool to assess isoform function.
5.3 Reprogramming Alternative Splicing
A different approach aims at reprogramming alternative splicing decisions. The spectacular physiological improvement obtained by reprogramming the Duchenne muscular dystrophy gene in a mouse model [198] supports applying this strategy to cancer. The original version of the approach employs a complementary oligonucleotide to cover the targeted splice site (Fig. 5a). By blocking the 5′ splice site of the anti-apoptotic Bcl-xL with 2′-O-methyl oligonucleotides, Mercatante et al. [199] could decrease the concentrations of the Bcl-xL isoform and increase the level of the corresponding pro-apoptotic Bcl-xS isoform. This change in the splicing profile of Bcl-x increased the sensitivity of cells to chemotherapeutic agents and even induced apoptosis in some cell lines [200]. Similar approaches have been successfully applied on the FGFR1 and the MYC pre-mRNAs [201, 202].

Reprogramming of alternative splicing through use of oligonucleotides. a An oligonucleotide blocking a splice site favors the use of an alternative site. b Oligonucleotides complementary to exonic or intronic controlling elements can prevent the binding of control factors. If these elements are ESEs and ISEs as shown, the oligos will stimulate splicing to an alternative site. c A complex between hnRNP A1 protein and the tail of an oligonucleotide partially complementary to an exonic sequence can provoke steric interference to reduce the use of a splice site and favor alternative splice site selection. d When the tail of the oligo contains a sequence of high affinity for a SR protein, splicing to the adjacent site can be stimulated. e If the hnRNP A1 binding tail is contained in an oligo complementary to an intron region near the 5′ splice site, splicing stimulation can occur. f A PNA (peptide-nucleic acid) portion covalently linked to an RNA sequence complementary to an exon can stimulate exon inclusion when the PNA tail contains repetitions of the dipeptide arginine-serine to mimic the RS domain of SR proteins
An alternative strategy involves using antisense oligonucleotides that target splicing control elements (Fig. 5b). An oligonucleotide complementary to a sequence upstream of 5′ splice site of Bcl-xL strongly repressed the use of this site [203] probably because its hybridization neutralizes a positive controlling element [131]. In a similar way, inactivating intronic repressor elements can also be used to modulate alternative splicing [202, 204]. Modification of the oligonucleotide with a non-hybridizing tail that contains one or several binding sites for a protein produces a bifunctional oligonucleotide that can recruit factors to a specific region on the pre-mRNA. For example, binding of hnRNP A1 to the tail of a bifunctional oligonucleotide sterically interferes with the efficient utilization of a neighboring 5′ splice site (Fig. 5c) [205]. The architecture of the tail can be modified to provide binding sites for other factors [206]. The general concept is flexible and changing the identity of the recruited protein or modifying the hybridization site can be used to stimulate splicing (Fig. 5d and e) [72, 207]. A PNA (peptide-nucleic acid) version has also been used with success. In this case, the tail is made of alternating serine and arginine amino acids that mimic SR proteins in their ability to promote exon inclusion (Fig. 5f) [208, 209]. The above approaches can be used to document the functions of different splice isoforms and therefore help to define new therapeutic targets. The development of oligonucleotides whose chemistries are compatible with human use may offer new therapeutic means for anticancer treatment. Although trans-acting non-coding nucleic acid molecules to reprogram splicing have been used by researchers for many years, only recently did we realize that this molecular strategy is used normally by cells to modulate splicing decisions. A snoRNA was recently shown to shift the splicing of a serotonin receptor [210]. Likewise, microRNAs (miRNAs) have very recently been shown to regulate alternative splicing during muscle differentiation [211, 212]. Given that miRNA expression is often altered in cancer cells [213], it is likely that additional examples of misregulation of splicing through miRNAs will be found in cancer-associated genes.
Another interesting approach consists in modifying the expression or activity of proteins that affect alternative splicing. This strategy has potential value given that the inhibition of the SRPK1 kinase through RNA interference reduces cellular proliferation and increases apoptosis in cells derived from pancreatic tumors [173, 174]. A more classic pharmacological approach consists in screening libraries for chemical compounds to identify small antagonist molecules with therapeutic value. An application of this strategy has resulted in the discovery of compounds that inhibit other kinases specific for SR proteins and that affect alternative splicing [214, 215]. Recently, screening assays have uncovered molecules that directly target SR proteins and that preferentially inhibit splicing events required for HIV replication [216]. These encouraging results justify that similar approaches be attempted with other regulatory splicing factors.
6 Conclusions
Cancer can arise from alterations in a variety of cellular pathways including signal transduction, cell cycle, and apoptosis. Alternative splicing is an important process that participates in the complex regulation of these cancer-related pathways. Despite incomplete information, it is becoming increasingly evident that defects in alternative splicing imposed by mutations or changes in the levels of splicing factors can generate isoforms whose activities contribute to the initiation and progression of cancer. Given current efforts at documenting the function of splice variants and at cataloguing cancer-specific splicing alterations, we can expect that the role of alternative splicing in cancer will provide many novel anticancer targets. In the meantime, the results of different approaches aimed at reprogramming splice site usage will pave the way for novel therapeutic strategies against cancer.
References
Caceres JF, Kornblihtt AR (2002) Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet 18(4):186–193
Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3(4):285–298
Pagani F, Baralle FE (2004) Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 5(5):389–396
Brinkman BM (2004) Splice variants as cancer biomarkers. Clin Biochem 37(7):584–594
Venables JP (2006) Unbalanced alternative splicing and its significance in cancer. BioEssays 28(4):378–386
Srebrow A, Kornblihtt AR (2006) The connection between splicing and cancer. J Cell Sci 119(Pt 13):2635–2641
Revil T, Shkreta L, Chabot B (2006) Pre-mRNA alternative splicing in cancer: functional impact, molecular mechanisms and therapeutic perspectives. Bull Cancer 93(9):909–919
Venables JP (ed) (2006) Alternative splicing in cancer. Transworld Res Network
Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, Montuenga LM (2007) Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol 8(4):349–357
Skotheim RI, Nees M (2007) Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell Biol 39(7–8):1432–1449
Forch P, Valcarcel J (2003) Splicing regulation in Drosophila sex determination. Prog Mol Subcell Biol 31:127–151
Peterson ML (1994) Regulated immunoglobulin (Ig) RNA processing does not require specific cis-acting sequences: non-Ig RNA can be alternatively processed in B cells and plasma cells. Mol Cell Biol 14(12):7891–7898
Lou H, Gagel RF (1998) Alternative RNA processing–its role in regulating expression of calcitonin/calcitonin gene-related peptide. J Endocrinol 156(3):401–405
Black DL (1998) Splicing in the inner ear: a familiar tune, but what are the instruments? Neuron 20(2):165–168
Pan Q, Saltzman AL, Kim YK et al (2006) Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev 20(2):153–158
Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW (2004) Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell 13(1):91–100
Ni JZ, Grate L, Donohue JP et al (2007) Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21(6):708–718
Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J (2001) SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20(7):1785–1796
Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE (2007) Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446(7138):926–929
Tress ML, Martelli PL, Frankish A et al (2007) The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci U S A 104(13):5495–5500
Carstens RP, Eaton JV, Krigman HR, Walther PJ, Garcia-Blanco MA (1997) Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene 15(25):3059–3065
Moffa AB, Ethier SP (2007) Differential signal transduction of alternatively spliced FGFR2 variants expressed in human mammary epithelial cells. J Cell Physiol 210(3):720–731
Oltean S, Sorg BS, Albrecht T et al (2006) Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci U S A 103(38):14116–14121
Bourdon JC, Fernandes K, Murray-Zmijewski F et al (2005) p53 isoforms can regulate p53 transcriptional activity. Genes Dev 19(18):2122–2137
Ozaki T, Nakagawara A (2005) p73, a sophisticated p53 family member in the cancer world. Cancer Sci 96(11):729–737
Bartel F, Taubert H, Harris LC (2002) Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2(1):9–15
Fridman JS, Hernando E, Hemann MT, de Stanchina E, Cordon-Cardo C, Lowe SW (2003) Tumor promotion by Mdm2 splice variants unable to bind p53. Cancer Res 63(18):5703–5706
Lovecchio M, Maiorano E, Vacca RA et al (2003) beta 1C integrin expression in human endometrial proliferative diseases. Am J Pathol 163(6):2543–2553
Hsieh HF, Yu JC, Ho LI, Chiu SC, Harn HJ (1999) Molecular studies into the role of CD44 variants in metastasis in gastric cancer. Mol Pathol 52(1):25–28
Wallach SB, Friedmann A, Naor D (2000) The CD44 receptor of the mouse LB T-cell lymphoma: analysis of the isoform repertoire and ligand binding properties by reverse-transcriptase polymerase chain reaction and antisense oligonucleotides. Cancer Detect Prev 24(1):33–45
Miyake H, Eto H, Arakawa S, Kamidono S, Hara I (2002) Over expression of CD44V8-10 in urinary exfoliated cells as an independent prognostic predictor in patients with urothelial cancer. J Urol 167(3):1282–1287
Hashimoto-Uoshima M, Yan YZ, Schneider G, Aukhil I (1997) The alternatively spliced domains EIIIB and EIIIA of human fibronectin affect cell adhesion and spreading. J Cell Sci 110(Pt 18):2271–2280
Ghigna C, Giordano S, Shen H et al (2005) Cell motility is controlled by SF2/ASF through alternative splicing of the ron protooncogene. Mol Cell 20(6):881–890
Ladomery MR, Harper SJ, Bates DO (2007) Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett 249(2):133–142
Catena R, Muniz-Medina V, Moralejo B et al (2007) Increased expression of VEGF(121)/VEGF(165–189) ratio results in a significant enhancement of human prostate tumor angiogenesis. Int J Cancer 120(10):2096–2109
Cohen CD, Doran PP, Blattner SM et al (2005) Sam68-like mammalian protein 2, identified by digital differential display as expressed by podocytes, is induced in proteinuria and involved in splice site selection of vascular endothelial growth factor. J Am Soc Nephrol 16(7):1958–1965
Chao C, Goluszko E, Lee YT et al (2007) Constitutively active CCK2 receptor splice variant increases src-dependent HIF-1 alpha expression and tumor growth. Oncogene 26(7):1013–1019
Koduri S, Goldhar AS, Vonderhaar BK (2006) Activation of vascular endothelial growth factor (VEGF) by the ER-alpha variant, ERDelta3. Breast Cancer Res Treat 95(1):37–43
Caldas H, Fangusaro JR, Boue DR, Holloway MP, Altura RA (2007) Dissecting the role of endothelial SURVIVIN deltaEx3 in angiogenesis. Blood 109(4):1479–1489
Schwerk C, Schulze-Osthoff K (2005) Regulation of apoptosis by alternative pre-mRNA splicing. Mol Cell 19(1):1–13
Minn AJ, Boise LH, Thompson CB (1996) Bcl-x(S) anatagonizes the protective effects of Bcl-x(L). J Biol Chem 271(11):6306–6312
Tu Y, Renner S, Xu F et al (1998) BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res 58(2):256–262
Takehara T, Liu X, Fujimoto J, Friedman SL, Takahashi H (2001) Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 34(1):55–61
Olopade OI, Adeyanju MO, Safa AR et al (1997) Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am 3(4):230–237
Yang CC, Lin HP, Chen CS, Yang YT, Tseng PH, Rangnekar VM (2003) Bcl-xL mediates a survival mechanism independent of the phosphoinositide 3-kinase/Akt pathway in prostate cancer cells. J Biol Chem 278(28):25872–25878
Wincewicz A, Sulkowska M, Koda M, Kanczuga-Koda L, Witkowska E, Sulkowski S (2007) Significant coexpression of GLUT-1, Bcl-xL, and bax in colorectal cancer. Ann N Y Acad Sci 1095:53–61
Chang BS, Kelekar A, Harris MH, Harlan JE, Fesik SW, Thompson CB (1999) The BH3 domain of Bcl-x(S) is required for inhibition of the antiapoptotic function of Bcl-x(L). Mol Cell Biol 19(10):6673–6681
Dole MG, Jasty R, Cooper MJ, Thompson CB, Nunez G, Castle VP (1995) Bcl-xL is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res 55(12):2576–2582
Simonian PL, Grillot DA, Nunez G (1997) Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood 90(3):1208–1216
Lebedeva I, Rando R, Ojwang J, Cossum P, Stein CA (2000) Bcl-xL in prostate cancer cells: effects of overexpression and down-regulation on chemosensitivity. Cancer Res 60(21):6052–6060
Williams J, Lucas PC, Griffith KA et al (2005) Expression of Bcl-xL in ovarian carcinoma is associated with chemoresistance and recurrent disease. Gynecol Oncol 96(2):287–295
Cho HJ, Kim JK, Kim KD et al (2006) Upregulation of Bcl-2 is associated with cisplatin-resistance via inhibition of bax translocation in human bladder cancer cells. Cancer Lett 237(1):56–66
Vilenchik M, Raffo AJ, Benimetskaya L, Shames D, Stein CA (2002) Antisense RNA down-regulation of bcl-xL expression in prostate cancer cells leads to diminished rates of cellular proliferation and resistance to cytotoxic chemotherapeutic agents. Cancer Res 62(7):2175–2183
Zhu H, Guo W, Zhang L et al (2005) Bcl-XL small interfering RNA suppresses the proliferation of 5-fluorouracil-resistant human colon cancer cells. Mol Cancer Ther 4(3):451–456
Konishi T, Sasaki S, Watanabe T, Kitayama J, Nagawa H (2006) Overexpression of hRFI inhibits 5-fluorouracil-induced apoptosis in colorectal cancer cells via activation of NF-kappaB and upregulation of BCL-2 and BCL-XL. Oncogene 25(22):3160–3169
Wang P, Song JH, Song DK, Zhang J, Hao C (2006) Role of death receptor and mitochondrial pathways in conventional chemotherapy drug induction of apoptosis. Cell Signal 18(9):1528–1535
Matsushita K, Tomonaga T, Shimada H et al (2006) An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res 66(3):1409–1417
Patel NA, Song SS, Cooper DR (2006) PKCdelta alternatively spliced isoforms modulate cellular apoptosis in retinoic acid-induced differentiation of human NT2 cells and mouse embryonic stem cells. Gene Expr 13(2):73–84
He X, Ee PL, Coon JS, Beck WT (2004) Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin Cancer Res 10(14):4652–4660
Lamba JK, Adachi M, Sun D et al (2003) Nonsense mediated decay downregulates conserved alternatively spliced ABCC4 transcripts bearing nonsense codons. Hum Mol Genet 12(2):99–109
Chen KG, Szakacs G, Annereau JP et al (2005) Principal expression of two mRNA isoforms (ABCB 5alpha and ABCB 5beta) of the ATP-binding cassette transporter gene ABCB 5 in melanoma cells and melanocytes. Pigment Cell Res 18(2):102–112
Devine SE, Hussain A, Davide JP, Melera PW (1991) Full length and alternatively spliced pgp1 transcripts in multidrug-resistant chinese hamster lung cells. J Biol Chem 266(7):4545–4555
Ma JF, Grant G, Staelens B, Howard DL, Melera PW (1999) In vitro translation of a 2.3-kb splicing variant of the hamster pgp1 gene whose presence in transfectants is associated with decreased drug resistance. Cancer Chemother Pharmacol 43(1):19–28
Nakanishi T, Shiozawa K, Hassel BA, Ross DD (2006) Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood 108(2):678–684
Muller M, Schleithoff ES, Stremmel W, Melino G, Krammer PH, Schilling T (2006) One, two, three-p53, p63, p73 and chemosensitivity. Drug Resist Updat 9(6):288–306
Yin F, Du Y, Hu W et al (2006) Mad2beta, an alternative variant of Mad2 reducing mitotic arrest and apoptosis induced by adriamycin in gastric cancer cells. Life Sci 78(12):1277–1286
Efimova EV, Al-Zoubi AM, Martinez O et al (2004) IG20, in contrast to DENN-SV, (MADD splice variants) suppresses tumor cell survival, and enhances their susceptibility to apoptosis and cancer drugs. Oncogene 23(5):1076–1087
Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72:291–336
Blanchette M, Chabot B (1997) A highly stable duplex structure sequesters the 5′ splice site region of hnRNP A1 alternative exon 7B. RNA 3(4):405–419
Blanchette M, Chabot B (1999) Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J 18(7):1939–1952
Nasim FU, Hutchison S, Cordeau M, Chabot B (2002) High-affinity hnRNP A1 binding sites and duplex-forming inverted repeats have similar effects on 5′ splice site selection in support of a common looping out and repression mechanism. RNA 8(8):1078–1089
Martinez-Contreras R, Fisette JF, Nasim FU, Madden R, Cordeau M, Chabot B (2006) Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol 4(2):e21
Wagner EJ, Garcia-Blanco MA (2001) Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol 21(10):3281–3288
Amir-Ahmady B, Boutz PL, Markovtsov V, Phillips ML, Black DL (2005) Exon repression by polypyrimidine tract binding protein. RNA 11(5):699–716
Ule J, Stefani G, Mele A et al (2006) An RNA map predicting Nova-dependent splicing regulation. Nature 444(7119):580–586
Domsic JK, Wang Y, Mayeda A, Krainer AR, Stoltzfus CM (2003) Human immunodeficiency virus type 1 hnRNP A/B-dependent exonic splicing silencer ESSV antagonizes binding of U2AF65 to viral polypyrimidine tracts. Mol Cell Biol 23(23):8762–8772
Zhu J, Mayeda A, Krainer AR (2001) Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell 8(6):1351–1361
Soret J, Gabut M, Tazi J (2006) SR proteins as potential targets for therapy. Prog Mol Subcell Biol 44:65–87
Krawczak M, Reiss J, Cooper DN (1992) The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90(1–2):41–54
Lopez-Bigas N, Blencowe BJ, Ouzounis CA (2006) Highly consistent patterns for inherited human diseases at the molecular level. Bioinformatics 22(3):269–277
Disset A, Bourgeois CF, Benmalek N, Claustres M, Stevenin J, Tuffery-Giraud S (2006) An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum Mol Genet 15(6):999–1013
Serra E, Ars E, Ravella A et al (2001) Somatic NF1 mutational spectrum in benign neurofibromas: mRNA splice defects are common among point mutations. Hum Genet 108(5):416–429
Baser ME, Kuramoto L, Woods R et al (2005) The location of constitutional neurofibromatosis 2 (NF2) splice site mutations is associated with the severity of NF2. J Med Genet 42(7):540–546
Holmila R, Fouquet C, Cadranel J, Zalcman G, Soussi T (2003) Splice mutations in the p53 gene: case report and review of the literature. Hum Mutat 21(1):101–102
Neklason DW, Solomon CH, Dalton AL, Kuwada SK, Burt RW (2004) Intron 4 mutation in APC gene results in splice defect and attenuated FAP phenotype. Fam Cancer 3(1):35–40
Liu HX, Cartegni L, Zhang MQ, Krainer AR (2001) A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet 27(1):55–58
Yang Y, Swaminathan S, Martin BK, Sharan SK (2003) Aberrant splicing induced by missense mutations in BRCA1: clues from a humanized mouse model. Hum Mol Genet 12(17):2121–2131
Narla G, DiFeo A, Yao S et al (2005) Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res 65(13):5761–5768
Wang XQ, Luk JM, Leung PP, Wong BW, Stanbridge EJ, Fan ST (2005) Alternative mRNA splicing of liver intestine-cadherin in hepatocellular carcinoma. Clin Cancer Res 11(2 Pt 1):483–489
Venesio T, Balsamo A, Sfiligoi C et al (2007) Constitutional high expression of an APC mRNA isoform in a subset of attenuated familial adenomatous polyposis patients. J Mol Med 85(3):301–308
Zhuo D, Madden R, Elela SA, Chabot B (2007) Modern origin of numerous alternatively spliced human introns from tandem arrays. Proc Natl Acad Sci U S A 104(3):882–886
Hallier M, Lerga A, Barnache S, Tavitian A, Moreau-Gachelin F (1998) The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J Biol Chem 273(9):4838–4842
Knoop LL, Baker SJ (2000) The splicing factor U1C represses EWS/FLI-mediated transactivation. J Biol Chem 275(32):24865–24871
Yang L, Embree LJ, Hickstein DD (2000) TLS-ERG leukemia fusion protein inhibits RNA splicing mediated by serine-arginine proteins. Mol Cell Biol 20(10):3345–3354
Chansky HA, Hu M, Hickstein DD, Yang L (2001) Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res 61(9):3586–3590
Janknecht R (2005) EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene 363:1–14
Knoop LL, Baker SJ (2001) EWS/FLI alters 5′-splice site selection. J Biol Chem 276(25):22317–22322
Yang L, Chansky HA, Hickstein DD (2000) EWS. Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J Biol Chem 275(48):37612–37618
Ohkura N, Yaguchi H, Tsukada T, Yamaguchi K (2002) The EWS/NOR1 fusion gene product gains a novel activity affecting pre-mRNA splicing. J Biol Chem 277(1):535–543
Salesse S, Dylla SJ, Verfaillie CM (2004) p210BCR/ABL-induced alteration of pre-mRNA splicing in primary human CD34 + hematopoietic progenitor cells. Leukemia 18(4):727–733
Chabot B, Frappier D, La Branche H (1992) Differential ASF/SF2 activity in extracts from normal WI38 and transformed WI38VA13 cells. Nucleic Acids Res 20(19):5197–5204
Lavigueur A, La Branche H, Kornblihtt AR, Chabot B (1993) A splicing enhancer in the human fibronectin alternate ED1 exon interacts with SR proteins and stimulates U2 snRNP binding. Genes Dev 7((12A)):2405–2417
Ghigna C, Moroni M, Porta C, Riva S, Biamonti G (1998) Altered expression of heterogenous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Res 58(24):5818–5824
Pino I, Pio R, Toledo G et al (2003) Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer 41(2):131–143
Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14(3):185–193
Li X, Wang J, Manley JL (2005) Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev 19(22):2705–2714
Chalfant CE, Ogretmen B, Galadari S, Kroesen BJ, Pettus BJ, Hannun YA (2001) FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem 276(48):44848–44855
He X, Pool M, Darcy KM et al (2007) Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene 26(34):4961–4968
Perry WL 3rd, Shepard RL, Sampath J et al (2005) Human splicing factor SPF45 (RBM17) confers broad multidrug resistance to anticancer drugs when overexpressed–a phenotype partially reversed by selective estrogen receptor modulators. Cancer Res 65(15):6593–6600
Shitashige M, Naishiro Y, Idogawa M et al (2007) Involvement of splicing factor-1 in beta-catenin/T-cell factor-4-mediated gene transactivation and pre-mRNA splicing. Gastroenterology 132(3):1039–1054
Ding WQ, Kuntz SM, Miller LJ (2002) A misspliced form of the cholecystokinin-B/gastrin receptor in pancreatic carcinoma: role of reduced sellular U2AF35 and a suboptimal 3′-splicing site leading to retention of the fourth intron. Cancer Res 62(3):947–952
Sergeant KA, Bourgeois CF, Dalgliesh C, Venables JP, Stevenin J, Elliott DJ (2007) Alternative RNA splicing complexes containing the scaffold attachment factor SAFB2. J Cell Sci 120(Pt 2):309–319
Le Guiner C, Plet A, Galiana D, Gesnel MC, Del Gatto-Konczak F, Breathnach R (2001) Polypyrimidine tract-binding protein represses splicing of a fibroblast growth factor receptor-2 gene alternative exon through exon sequences. J Biol Chem 276(47):43677–43687
Baraniak AP, Chen JR, Garcia-Blanco MA (2006) Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Mol Cell Biol 26(4):1209–1222
Jin W, McCutcheon IE, Fuller GN, Huang ES, Cote GJ (2000) Fibroblast growth factor receptor-1 alpha-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res 60(5):1221–1224
Stickeler E, Kittrell F, Medina D, Berget SM (1999) Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene 18(24):3574–3582
Fischer DC, Noack K, Runnebaum IB et al (2004) Expression of splicing factors in human ovarian cancer. Oncol Rep 11(5):1085–1090
Galiana-Arnoux D, Lejeune F, Gesnel MC, Stevenin J, Breathnach R, Del Gatto-Konczak F (2003) The CD44 alternative v9 exon contains a splicing enhancer responsive to the SR proteins 9G8, ASF/SF2, and SRp20. J Biol Chem 278(35):32943–32953
Matter N, Herrlich P, Konig H (2002) Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420(6916):691–695
Matter N, Marx M, Weg-Remers S, Ponta H, Herrlich P, Konig H (2000) Heterogeneous ribonucleoprotein A1 is part of an exon-specific splice-silencing complex controlled by oncogenic signaling pathways. J Biol Chem 275(45):35353–35360
Venables JP, Dalgliesh C, Paronetto MP et al (2004) SIAH1 targets the alternative splicing factor T-STAR for degradation by the proteasome. Hum Mol Genet 13(14):1525–1534
Cheng C, Sharp PA (2006) Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol Cell Biol 26(1):362–370
Watermann DO, Tang Y, Zur Hausen A, Jager M, Stamm S, Stickeler E (2006) Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res 66(9):4774–4780
Batsche E, Yaniv M, Muchardt C (2006) The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol 13(1):22–29
Zhou YQ, He C, Chen YQ, Wang D, Wang MH (2003) Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene 22(2):186–197
Forch P, Puig O, Martinez C, Seraphin B, Valcarcel J (2002) The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J 21(24):6882–6892
Izquierdo JM, Majos N, Bonnal S et al (2005) Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell 19(4):475–484
Li CY, Chu JY, Yu JK et al (2004) Regulation of alternative splicing of Bcl-x by IL-6 GM-CSF and TPA. Cell Res 14(6):473–479
Massiello A, Salas A, Pinkerman RL, Roddy P, Roesser JR, Chalfant CE (2004) Identification of two RNA cis-elements that function to regulate the 5′ splice site selection of Bcl-x pre-mRNA in response to ceramide. J Biol Chem 279(16):15799–15804
Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C (2007) The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol 176(7):929–939
Garneau D, Revil T, Fisette JF, Chabot B (2005) Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem 280(24):22641–22650
Cloutier P, Toutant J, Shkreta L, Goekjian S, Revil T, Chabot B (2008) Antagonistic effects of the SRp30c protein and cryptic 5′ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J Biol Chem 283(31):21315–21324
Revil T, Toutant J, Shkreta L, Garneau D, Cloutier P, Chabot B (2007) Protein kinase C-dependent control of Bcl-x alternative splicing. Mol Cell Biol 27(24):8431–8441
Shkreta L, Froehlich U, Paquet ER, Toutant J, Elela SA, Chabot B (2008) Anticancer drugs affect the alternative splicing of Bcl-x and other human apoptotic genes. Mol Cancer Ther 7(6):1398–1409
Wu JY, Tang H, Havlioglu N (2003) Alternative pre-mRNA splicing and regulation of programmed cell death. Prog Mol Subcell Biol 31:153–185
Jiang ZH, Zhang WJ, Rao Y, Wu JY (1998) Regulation of Ich-1 pre-mRNA alternative splicing and apoptosis by mammalian splicing factors. Proc Natl Acad Sci U S A 95(16):9155–9160
Cote J, Dupuis S, Jiang Z, Wu JY (2001) Caspase-2 pre-mRNA alternative splicing: Identification of an intronic element containing a decoy 3′ acceptor site. Proc Natl Acad Sci U S A 98(3):938–943
Faustino NA, Cooper TA (2003) Pre-mRNA splicing and human disease. Genes Dev 17(4):419–437
Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA (2004) Muscleblind proteins regulate alternative splicing. EMBO J 23(15):3103–3112
Auboeuf D, Batsche E, Dutertre M, Muchardt C, O’Malley BW (2007) Coregulators: transducing signal from transcription to alternative splicing. Trends Endocrinol Metab 18(3):122–129
Kornblihtt AR (2005) Promoter usage and alternative splicing. Curr Opin Cell Biol 17(3):262–268
Cramer P, Pesce CG, Baralle FE, Kornblihtt AR (1997) Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci U S A 94(21):11456–11460
Gendra E, Colgan DF, Meany B, Konarska MM (2007) A sequence motif in the SV40 early core promoter affects alternative splicing of transcribed mRNA. J Biol Chem 282(16):11648–11657
Auboeuf D, Dowhan DH, Li X et al (2004) CoAA, a nuclear receptor coactivator protein at the interface of transcriptional coactivation and RNA splicing. Mol Cell Biol 24(1):442–453
Guillouf C, Gallais I, Moreau-Gachelin F (2006) Spi-1/PU.1 oncoprotein affects splicing decisions in a promoter binding-dependent manner. J Biol Chem 281(28):19145–19155
de la Mata M, Kornblihtt AR (2006) RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol 13(11):973–980
de la Mata M, Alonso CR, Kadener S et al (2003) A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12(2):525–532
Nogues G, Kadener S, Cramer P et al (2003) Control of alternative pre-mRNA splicing by RNA Pol II elongation: faster is not always better. IUBMB Life 55(4–5):235–241
Pecci A, Viegas LR, Baranao JL, Beato M (2001) Promoter choice influences alternative splicing and determines the balance of isoforms expressed from the mouse bcl-X gene. J Biol Chem 276(24):21062–21069
Logette E, Wotawa A, Solier S, Desoche L, Solary E, Corcos L (2003) The human caspase-2 gene: alternative promoters, pre-mRNA splicing and AUG usage direct isoform-specific expression. Oncogene 22(6):935–946
Landry JR, Mager DL, Wilhelm BT (2003) Complex controls: the role of alternative promoters in mammalian genomes. Trends Genet 19(11):640–648
Kimura K, Wakamatsu A, Suzuki Y et al (2006) Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res 16(1):55–65
Bass BL (2002) RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71:817–846
Beghini A, Ripamonti CB, Peterlongo P et al (2000) RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum Mol Genet 9(15):2297–2304
Maas S, Patt S, Schrey M, Rich A (2001) Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A 98(25):14687–14692
Weischenfeldt J, Lykke-Andersen J, Porse B (2005) Messenger RNA surveillance: neutralizing natural nonsense. Curr Biol 15(14):R559–R562
Rossi MR, Hawthorn L, Platt J, Burkhardt T, Cowell JK, Ionov Y (2005) Identification of inactivating mutations in the JAK1, SYNJ2, and CLPTM1 genes in prostate cancer cells using inhibition of nonsense-mediated decay and microarray analysis. Cancer Genet Cytogenet 161(2):97–103
Ware MD, DeSilva D, Sinilnikova OM, Stoppa-Lyonnet D, Tavtigian SV, Mazoyer S (2006) Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene 25(2):323–328
Green RE, Lewis BP, Hillman RT et al (2003) Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics 19(Suppl 1):i118–i121
El-Bchiri J, Buhard O, Penard-Lacronique V, Thomas G, Hamelin R, Duval A (2005) Differential nonsense mediated decay of mutated mRNAs in mismatch repair deficient colorectal cancers. Hum Mol Genet 14(16):2435–2442
De Rosa M, Morelli G, Cesaro E et al (2007) Alternative splicing and nonsense-mediated mRNA decay in the regulation of a new adenomatous polyposis coli transcript. Gene 395(1–2):8–14
Tarn WY (2007) Cellular signals modulate alternative splicing. J Biomed Sci 14(4):517–522
Blaustein M, Pelisch F, Srebrow A (2007) Signals, pathways and splicing regulation. Int J Biochem Cell Biol 39(11):2031–2048
Ding JH, Zhong XY, Hagopian JC et al (2006) Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell 17(2):876–885
Xie J, Lee JA, Kress TL, Mowry KL, Black DL (2003) Protein kinase A phosphorylation modulates transport of the polypyrimidine tract-binding protein. Proc Natl Acad Sci U S A 100(15):8776–8781
Allemand E, Guil S, Myers M, Moscat J, Caceres JF, Krainer AR (2005) Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc Natl Acad Sci U S A 102(10):3605–3610
Hall MP, Huang S, Black DL (2004) Differentiation-induced colocalization of the KH-type splicing regulatory protein with polypyrimidine tract binding protein and the c-src pre-mRNA. Mol Biol Cell 15(2):774–786
Scheid MP, Woodgett JR (2001) PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol 2(10):760–768
Blaustein M, Pelisch F, Coso OA, Bissell MJ, Kornblihtt AR, Srebrow A (2004) Mammary epithelial-mesenchymal interaction regulates fibronectin alternative splicing via phosphatidylinositol 3-kinase. J Biol Chem 279(20):21029–21037
Patel NA, Kaneko S, Apostolatos HS et al (2005) Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CbetaII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J Biol Chem 280(14):14302–14309
Konig H, Ponta H, Herrlich P (1998) Coupling of signal transduction to alternative pre-mRNA splicing by a composite splice regulator. EMBO J 17(10):2904–2913
Cheng C, Yaffe MB, Sharp PA (2006) A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev 20(13):1715–1720
Hayes GM, Carrigan PE, Beck AM, Miller LJ (2006) Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res 66(7):3819–3827
Hayes GM, Carrigan PE, Miller LJ (2007) Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res 67(5):2072–2080
Bellavia D, Mecarozzi M, Campese AF et al (2007) Notch3 and the Notch3-upregulated RNA-binding protein HuD regulate Ikaros alternative splicing. EMBO J 26(6):1670–1680
Shin C, Manley JL (2004) Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol 5(9):727–738
Blaustein M, Pelisch F, Tanos T et al (2005) Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 12(12):1037–1044
Chen HH, Wang YC, Fann MJ (2006) Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol Cell Biol 26(7):2736–2745
Shen EC, Henry MF, Weiss VH, Valentini SR, Silver PA, Lee MS (1998) Arginine methylation facilitates the nuclear export of hnRNP proteins. Genes Dev 12(5):679–691
Yu MC, Bachand F, McBride AE, Komili S, Casolari JM, Silver PA (2004) Arginine methyltransferase affects interactions and recruitment of mRNA processing and export factors. Genes Dev 18(16):2024–2035
Ohkura N, Takahashi M, Yaguchi H, Nagamura Y, Tsukada T (2005) Coactivator-associated arginine methyltransferase 1, CARM1, affects pre-mRNA splicing in an isoform-specific manner. J Biol Chem 280(32):28927–28935
Hui L, Zhang X, Wu X et al (2004) Identification of alternatively spliced mRNA variants related to cancers by genome-wide ESTs alignment. Oncogene 23(17):3013–3023
Xu Q, Lee C (2003) Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res 31(19):5635–5643
Pan Q, Shai O, Misquitta C et al (2004) Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol Cell 16(6):929–941
Zhang C, Li HR, Fan JB et al (2006) Profiling alternatively spliced mRNA isoforms for prostate cancer classification. BMC Bioinformatics 7:202
Watahiki A, Waki K, Hayatsu N et al (2004) Libraries enriched for alternatively spliced exons reveal splicing patterns in melanocytes and melanomas. Nat Methods 1(3):233–239
Relogio A, Ben-Dov C, Baum M et al (2005) Alternative splicing microarrays reveal functional expression of neuron-specific regulators in Hodgkin lymphoma cells. J Biol Chem 280(6):4779–4784
Li C, Kato M, Shiue L, Shively JE, Ares M Jr, Lin RJ (2006) Cell type and culture condition-dependent alternative splicing in human breast cancer cells revealed by splicing-sensitive microarrays. Cancer Res 66(4):1990–1999
Yeakley JM, Fan JB, Doucet D et al (2002) Profiling alternative splicing on fiber-optic arrays. Nat Biotechnol 20(4):353–358
Li HR, Wang-Rodriguez J, Nair TM et al (2006) Two-dimensional transcriptome profiling: identification of messenger RNA isoform signatures in prostate cancer from archived paraffin-embedded cancer specimens. Cancer Res 66(8):4079–4088
Johnson JM, Castle J, Garrett-Engele P et al (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302(5653):2141–2144
Gardina PJ, Clark TA, Shimada B et al (2006) Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics 7:325
Margulies M, Egholm M, Altman WE et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437(7057):376–380
Fededa JP, Petrillo E, Gelfand MS et al (2005) A polar mechanism coordinates different regions of alternative splicing within a single gene. Mol Cell 19(3):393–404
Celotto AM, Graveley BR (2002) Exon-specific RNAi: a tool for dissecting the functional relevance of alternative splicing. RNA 8(6):718–724
Morris KV, Chan SW, Jacobsen SE, Looney DJ (2004) Small interfering RNA-induced transcriptional gene silencing in human cells. Science 305(5688):1289–1292
Kim DH, Villeneuve LM, Morris KV, Rossi JJ (2006) Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol 13(9):793–797
Goyenvalle A, Vulin A, Fougerousse F et al (2004) Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science 306(5702):1796–1799
Mercatante DR, Bortner CD, Cidlowski JA, Kole R (2001) Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. Analysis of apoptosis and cell death. J Biol Chem 276(19):16411–16417
Mercatante DR, Mohler JL, Kole R (2002) Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J Biol Chem 277(51):49374–49382
Giles RV, Spiller DG, Clark RE, Tidd DM (1999) Antisense morpholino oligonucleotide analog induces missplicing of C-myc mRNA. Antisense Nucleic Acid Drug Dev 9(2):213–220
Bruno IG, Jin W, Cote GJ (2004) Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet 13(20):2409–2420
Taylor JK, Zhang QQ, Wyatt JR, Dean NM (1999) Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol 17(11):1097–1100
Singh NK, Singh NN, Androphy EJ, Singh RN (2006) Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 26(4):1333–1346
Villemaire J, Dion I, Elela SA, Chabot B (2003) Reprogramming alternative pre-messenger RNA splicing through the use of protein-binding antisense oligonucleotides. J Biol Chem 278(50):50031–50039
Gendron D, Carriero S, Garneau D et al (2006) Modulation of 5′ splice site selection using tailed oligonucleotides carrying splicing signals. BMC Biotechnol 6:5
Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F (2003) Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci U S A 100(7):4114–4119
Cartegni L, Krainer AR (2003) Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol 10(2):120–125
Wilusz JE, Devanney SC, Caputi M (2005) Chimeric peptide nucleic acid compounds modulate splicing of the bcl-x gene in vitro and in vivo. Nucleic Acids Res 33(20):6547–6554
Kishore S, Stamm S (2006) The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311(5758):230–232
Bland CS, Cooper TA (2007) Micromanaging alternative splicing during muscle differentiation. Dev Cell 12(2):171–172
Boutz PL, Chawla G, Stoilov P, Black DL (2007) MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 21(1):71–84
Calin GA, Croce CM (2006) MicroRNA-cancer connection: the beginning of a new tale. Cancer Res 66(15):7390–7394
Muraki M, Ohkawara B, Hosoya T et al (2004) Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem 279(23):24246–24254
Pilch B, Allemand E, Facompre M et al (2001) Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res 61(18):6876–6884
Soret J, Bakkour N, Maire S et al (2005) Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc Natl Acad Sci U S A 102(24):8764–8769
Wang Z, Hoffmann HM, Grabowski PJ (1995) Intrinsic U2AF binding is modulated by exon enhancer signals in parallel with changes in splicing activity. RNA 1(1):21–35
Kanopka A, Muhlemann O, Akusjarvi G (1996) Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 381(6582):535–538
Caputi M, Zahler AM (2002) SR proteins and hnRNP H regulate the splicing of the HIV-1 tev-specific exon 6D. EMBO J 21(4):845–855
Vickers SM, Huang ZQ, MacMillan-Crow L, Greendorfer JS, Thompson JA (2002) Ligand activation of alternatively spliced fibroblast growth factor receptor-1 modulates pancreatic adenocarcinoma cell malignancy. J Gastrointest Surg 6(4):546–553
Jang JH (2005) Reciprocal relationship in gene expression between FGFR1 and FGFR3: implication for tumorigenesis. Oncogene 24(5):945–948
Kornmann M, Ishiwata T, Matsuda K et al (2002) IIIc isoform of fibroblast growth factor receptor 1 is overexpressed in human pancreatic cancer and enhances tumorigenicity of hamster ductal cells. Gastroenterology 123(1):301–313
Sturla LM, Merrick AE, Burchill SA (2003) FGFR3IIIS: a novel soluble FGFR3 spliced variant that modulates growth is frequently expressed in tumour cells. Br J Cancer 89(7):1276–1284
Jang JH, Shin KH, Park YJ, Lee RJ, McKeehan WL, Park JG (2000) Novel transcripts of fibroblast growth factor receptor 3 reveal aberrant splicing and activation of cryptic splice sequences in colorectal cancer. Cancer Res 60(15):4049–4052
Takaishi S, Sawada M, Morita Y, Seno H, Fukuzawa H, Chiba T (2000) Identification of a novel alternative splicing of human FGF receptor 4: soluble-form splice variant expressed in human gastrointestinal epithelial cells. Biochem Biophys Res Commun 267(2):658–662
Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW (2007) The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 117(5):1370–1380
Nyman U, Sobczak-Pluta A, Vlachos P, Perlmann T, Zhivotovsky B, Joseph B (2005) Full-length p73alpha represses drug-induced apoptosis in small cell lung carcinoma cells. J Biol Chem 280(40):34159–34169
Watson IR, Blanch A, Lin DC, Ohh M, Irwin MS (2006) Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J Biol Chem 281(45):34096–34103
Steinman HA, Burstein E, Lengner C et al (2004) An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis. J Biol Chem 279(6):4877–4886
Chandler DS, Singh RK, Caldwell LC, Bitler JL, Lozano G (2006) Genotoxic stress Induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res 66(19):9502–9508
Bartel F, Schulz J, Bohnke A et al (2005) Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int J Cancer 117(3):469–475
Giglio S, Mancini F, Gentiletti F et al (2005) Identification of an aberrantly spliced form of HDMX in human tumors: a new mechanism for HDM2 stabilization. Cancer Res 65(21):9687–9694
Agrawal S, Eng C (2006) Differential expression of novel naturally occurring splice variants of PTEN and their functional consequences in Cowden syndrome and sporadic breast cancer. Hum Mol Genet 15(5):777–787
Colgin LM, Wilkinson C, Englezou A, Kilian A, Robinson MO, Reddel RR (2000) The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia 2(5):426–432
Burd CJ, Petre CE, Morey LM et al (2006) Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A 103(7):2190–2195
Knudsen KE, Diehl JA, Haiman CA, Knudsen ES (2006) Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 25(11):1620–1628
Leung YK, Lau KM, Mobley J, Jiang Z, Ho SM (2005) Overexpression of cytochrome P450 1A1 and its novel spliced variant in ovarian cancer cells: alternative subcellular enzyme compartmentation may contribute to carcinogenesis. Cancer Res 65(9):3726–3734
Yu Y, Jiang X, Schoch BS, Carroll RS, Black PM, Johnson MD (2007) Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res 67(1):130–138
Di Modugno F, DeMonte L, Balsamo M et al (2007) Molecular cloning of hMena (ENAH) and its splice variant hMena +11a: epidermal growth factor increases their expression and stimulates hMena +11a phosphorylation in breast cancer cell lines. Cancer Res 67(6):2657–2665
Carson DJ, Santoro IM, Groden J (2004) Isoforms of the APC tumor suppressor and their ability to inhibit cell growth and tumorigenicity. Oncogene 23(42):7144–7148
Kawasaki Y, Sato R, Akiyama T (2003) Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol 5(3):211–215
Cooper DL, Dougherty GJ (1995) To metastasize or not? Selection of CD44 splice sites. Nat Med 1(7):635–637
Scotlandi K, Zuntini M, Manara MC, et al. (2007) CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene 26(46):6604–6618
Byun HJ, Hong IK, Kim E et al (2006) A splice variant of CD99 increases motility and MMP-9 expression of human breast cancer cells through the AKT-, ERK-, and JNK-dependent AP-1 activation signaling pathways. J Biol Chem 281(46):34833–34847
Oyama F, Hirohashi S, Sakamoto M, Titani K, Sekiguchi K (1993) Coordinate oncodevelopmental modulation of alternative splicing of fibronectin pre-messenger RNA at ED-A, ED-B, and CS1 regions in human liver tumors. Cancer Res 53(9):2005–2011
Khan ZA, Chan BM, Uniyal S et al (2005) EDB fibronectin and angiogenesis: a novel mechanistic pathway. Angiogenesis 8(3):183–196
Wang L, Lin SH, Wu WG et al (2000) C-CAM1, a candidate tumor suppressor gene, is abnormally expressed in primary lung cancers. Clin Cancer Res 6(8):2988–2993
Luo W, Wood CG, Earley K, Hung MC, Lin SH (1997) Suppression of tumorigenicity of breast cancer cells by an epithelial cell adhesion molecule (C-CAM1): the adhesion and growth suppression are mediated by different domains. Oncogene 14(14):1697–1704
DiFeo A, Narla G, Hirshfeld J et al (2006) Roles of KLF6 and KLF6-SV1 in ovarian cancer progression and intraperitoneal dissemination. Clin Cancer Res 12(12):3730–3739
Camacho-Vanegas O, Narla G, Teixeira MS, et al. (2007) Functional inactivation of the KLF6 tumor suppressor gene by loss of heterozygosity and increased alternative splicing in glioblastoma. Int J Cancer 121(6):1390–1395
Esufali S, Charames GS, Pethe VV, Buongiorno P, Bapat B (2007) Activation of tumor-specific splice variant Rac1b by dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res 67(6):2469–2479
Matos P, Jordan P (2006) Rac1, but not Rac1B, stimulates RelB-mediated gene transcription in colorectal cancer cells. J Biol Chem 281(19):13724–13732
Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW (2007) An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 26(44):6361–6371
Aigner A, Juhl H, Malerczyk C, Tkybusch A, Benz CC, Czubayko F (2001) Expression of a truncated 100 kDa HER2 splice variant acts as an endogenous inhibitor of tumour cell proliferation. Oncogene 20(17):2101–2111
Woolard J, Wang WY, Bevan HS et al (2004) VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res 64(21):7822–7835
Lokeshwar VB, Estrella V, Lopez L et al (2006) HYAL1-v1, an alternatively spliced variant of HYAL1 hyaluronidase: a negative regulator of bladder cancer. Cancer Res 66(23):11219–11227
van Nimwegen MJ, Verkoeijen S, van Buren L, Burg D, van de Water B (2005) Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Res 65(11):4698–4706
Song SW, Fuller GN, Zheng H, Zhang W (2005) Inactivation of the invasion inhibitory gene IIp45 by alternative splicing in gliomas. Cancer Res 65(9):3562–3567
Luther T, Kotzsch M, Meye A et al (2003) Identification of a novel urokinase receptor splice variant and its prognostic relevance in breast cancer. Thromb Haemost 89(4):705–717
Midis GP, Shen Y, Owen-Schaub LB (1996) Elevated soluble Fas (sFas) levels in nonhematopoietic human malignancy. Cancer Res 56(17):3870–3874
Ugurel S, Rappl G, Tilgen W, Reinhold U (2001) Increased soluble CD95 (sFas/CD95) serum level correlates with poor prognosis in melanoma patients. Clin Cancer Res 7(5):1282–1286
Ueno T, Toi M, Tominaga T (1999) Circulating soluble Fas concentration in breast cancer patients. Clin Cancer Res 5(11):3529–3533
Nonomura N, Nishimura K, Ono Y et al (2000) Soluble Fas in serum from patients with renal cell carcinoma. Urology 55(1):151–155
Benedict MA, Hu Y, Inohara N, Nunez G (2000) Expression and functional analysis of Apaf-1 isoforms. Extra Wd-40 repeat is required for cytochrome c binding and regulated activation of procaspase-9. J Biol Chem 275(12):8461–8468
Ogawa T, Shiga K, Hashimoto S, Kobayashi T, Horii A, Furukawa T (2003) APAF-1-ALT, a novel alternative splicing form of APAF-1, potentially causes impeded ability of undergoing DNA damage-induced apoptosis in the LNCaP human prostate cancer cell line. Biochem Biophys Res Commun 306(2):537–543
Li F (2005) Role of survivin and its splice variants in tumorigenesis. Br J Cancer 92(2):212–216
Krieg A, Mahotka C, Krieg T et al (2002) Expression of different survivin variants in gastric carcinomas: first clues to a role of survivin-2B in tumour progression. Br J Cancer 86(5):737–743
Vegran F, Boidot R, Oudin C, Riedinger JM, Lizard-Nacol S (2005) Distinct expression of Survivin splice variants in breast carcinomas. Int J Oncol 27(4):1151–1157
Tsujimoto Y, Croce CM (1986) Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci U S A 83(14):5214–5218
Tanaka S, Saito K, Reed JC (1993) Structure-function analysis of the Bcl-2 oncoprotein. Addition of a heterologous transmembrane domain to portions of the Bcl-2 beta protein restores function as a regulator of cell survival. J Biol Chem 268(15):10920–10926
Boise LH, Gonzalez-Garcia M, Postema CE et al (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74(4):597–608
Marone M, Scambia G, Mozzetti S et al (1998) bcl-2, bax, bcl-XL, and bcl-XS expression in normal and neoplastic ovarian tissues. Clin Cancer Res 4(2):517–524
Wu J, Shao ZM, Shen ZZ et al (2000) Significance of apoptosis and apoptotic-related proteins, Bcl-2, and Bax in primary breast cancer. Breast J 6(1):44–52
Schmitt E, Paquet C, Beauchemin M, Dever-Bertrand J, Bertrand R (2000) Characterization of Bax-sigma, a cell death-inducing isoform of Bax. Biochem Biophys Res Commun 270(3):868–879
Shi B, Triebe D, Kajiji S, Iwata KK, Bruskin A, Mahajna J (1999) Identification and characterization of baxepsilon, a novel bax variant missing the BH2 and the transmembrane domains. Biochem Biophys Res Commun 254(3):779–785
Chang DW, Xing Z, Pan Y et al (2002) c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J 21(14):3704–3714
Longley DB, Wilson TR, McEwan M et al (2006) c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 25(6):838–848
Mulherkar N, Ramaswamy M, Mordi DC, Prabhakar BS (2006) MADD/DENN splice variant of the IG20 gene is necessary and sufficient for cancer cell survival. Oncogene 25(47):6252–6261
Bae J, Leo CP, Hsu SY, Hsueh AJ (2000) MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem 275(33):25255–25261
Liu JW, Chandra D, Tang SH, Chopra D, Tang DG (2002) Identification and characterization of Bimgamma, a novel proapoptotic BH3-only splice variant of Bim. Cancer Res 62(10):2976–2981
Yamaguchi T, Okada T, Takeuchi K et al (2003) Enhancement of thymidine kinase-mediated killing of malignant glioma by BimS, a BH3-only cell death activator. Gene Ther 10(5):375–385
Abrams MT, Robertson NM, Yoon K, Wickstrom E (2004) Inhibition of glucocorticoid-induced apoptosis by targeting the major splice variants of BIM mRNA with small interfering RNA and short hairpin RNA. J Biol Chem 279(53):55809–55817
Wang L, Miura M, Bergeron L, Zhu H, Yuan J (1994) Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 78(5):739–750
Droin N, Beauchemin M, Solary E, Bertrand R (2000) Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res 60(24):7039–7047
Vegran F, Boidot R, Oudin C, Riedinger JM, Bonnetain F, Lizard-Nacol S (2006) Overexpression of caspase-3 s splice variant in locally advanced breast carcinoma is associated with poor response to neoadjuvant chemotherapy. Clin Cancer Res 12(19):5794–5800
Muzio M, Chinnaiyan AM, Kischkel FC et al (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death–inducing signaling complex. Cell 85(6):817–827
Himeji D, Horiuchi T, Tsukamoto H, Hayashi K, Watanabe T, Harada M (2002) Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood 99(11):4070–4078
Waltereit R, Weller M (2002) The role of caspases 9 and 9-short (9S) in death ligand- and drug-induced apoptosis in human astrocytoma cells. Brain Res Mol Brain Res 106(1–2):42–49
Lee SB, Haber DA (2001) Wilms tumor and the WT1 gene. Exp Cell Res 264(1):74–99
Richard DJ, Schumacher V, Royer-Pokora B, Roberts SG (2001) Par4 is a coactivator for a splice isoform-specific transcriptional activation domain in WT1. Genes Dev 15(3):328–339
Tojo Y, Bandoh S, Fujita J et al (2003) Aberrant messenger RNA splicing of the cytokeratin 8 in lung cancer. Lung Cancer 42(2):153–161
Yin F, Hu WH, Qiao TD, Fan DM (2004) Multidrug resistant effect of alternative splicing form of MAD2 gene-MAD2beta on human gastric cancer cell. Zhonghua Zhong Liu Za Zhi 26(4):201–204
Lixia M, Zhijian C, Chao S, Chaojiang G, Congyi Z (2007) Alternative splicing of breast cancer associated gene BRCA1 from breast cancer cell line. J Biochem Mol Biol 40(1):15–21
Orban TI, Olah E (2003) Emerging roles of BRCA1 alternative splicing. Mol Pathol 56(4):191–197
Bieche I, Lidereau R (1999) Increased level of exon 12 alternatively spliced BRCA2 transcripts in tumor breast tissue compared with normal tissue. Cancer Res 59(11):2546–2550
Khan SG, Muniz-Medina V, Shahlavi T et al (2002) The human XPC DNA repair gene: arrangement, splice site information content and influence of a single nucleotide polymorphism in a splice acceptor site on alternative splicing and function. Nucleic Acids Res 30(16):3624–3631
Shiote Y, Ouchida M, Jitsumori Y et al (2006) Multiple splicing variants of Naf1/ABIN-1 transcripts and their alterations in hematopoietic tumors. Int J Mol Med 18(5):917–923
Lee EJ, Jo M, Park J, Zhang W, Lee JH (2006) Alternative splicing variants of IRF-1 lacking exons 7, 8, and 9 in cervical cancer. Biochem Biophys Res Commun 347(4):882–888
Hube F, Guo J, Chooniedass-Kothari S et al (2006) Alternative splicing of the first intron of the steroid receptor RNA activator (SRA) participates in the generation of coding and noncoding RNA isoforms in breast cancer cell lines. DNA Cell Biol 25(7):418–428
Wang L, Duke L, Zhang PS et al (2003) Alternative splicing disrupts a nuclear localization signal in spleen tyrosine kinase that is required for invasion suppression in breast cancer. Cancer Res 63(15):4724–4730
Katoh M, Kirikoshi H, Terasaki H, Shiokawa K (2001) WNT2B2 mRNA, up-regulated in primary gastric cancer, is a positive regulator of the WNT- beta-catenin-TCF signaling pathway. Biochem Biophys Res Commun 289(5):1093–1098
Lee JH, Gao CF, Lee CC, Kim MD (2006) Vande Woude GF. An alternatively spliced form of Met receptor is tumorigenic. Exp Mol Med 38(5):565–573
Treeck O, Pfeiler G, Horn F et al (2007) Novel estrogen receptor beta transcript variants identified in human breast cancer cells affect cell growth and apoptosis of COS-1 cells. Mol Cell Endocrinol 264(1–2):50–60
Pind MT, Watson PH (2003) SR protein expression and CD44 splicing pattern in human breast tumours. Breast Cancer Res Treat 79(1):75–82
Patry C, Bouchard L, Labrecque P et al (2003) Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res 63(22):7679–7688
Zhou J, Nong L, Wloch M, Cantor A, Mulshine JL, Tockman MS (2001) Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 34(3):341–350
Zech VF, Dlaska M, Tzankov A, Hilbe W (2006) Prognostic and diagnostic relevance of hnRNP A2/B1, hnRNP B1 and S100 A2 in non-small cell lung cancer. Cancer Detect Prev 30(5):395–402
Hiraki A, Murakami T, Aoe K et al (2006) Heterogeneous nuclear ribonucleoprotein B1 expression in malignant mesothelioma. Cancer Sci 97(11):1175–1181
Brose MS, Volpe P, Paul K et al (2004) Characterization of two novel BRCA1 germ-line mutations involving splice donor sites. Genet Test 8(2):133–138
Chen X, Truong TT, Weaver J et al (2006) Intronic alterations in BRCA1 and BRCA2: effect on mRNA splicing fidelity and expression. Hum Mutat 27(5):427–435
Bonatti F, Pepe C, Tancredi M et al (2006) RNA-based analysis of BRCA1 and BRCA2 gene alterations. Cancer Genet Cytogenet 170(2):93–101
Humar B, Toro T, Graziano F et al (2002) Novel germline CDH1 mutations in hereditary diffuse gastric cancer families. Hum Mutat 19(5):518–525
Loo JC, Liu L, Hao A et al (2003) Germline splicing mutations of CDKN2A predispose to melanoma. Oncogene 22(41):6387–6394
Wolf M, Hemminki A, Kivioja A et al (1998) A novel splice site mutation of the EXT2 gene in a finnish hereditary multiple exostoses family. Mutations in brief no. 197. Online. Hum Mutat 12(5):362
Tamary H, Dgany O, Toledano H et al (2004) Molecular characterization of three novel Fanconi anemia mutations in Israeli Arabs. Eur J Haematol 72(5):330–335
Hastings ML, Resta N, Traum D, Stella A, Guanti G, Krainer AR (2005) An LKB1 AT-AC intron mutation causes Peutz-Jeghers syndrome via splicing at noncanonical cryptic splice sites. Nat Struct Mol Biol 12(1):54–59
Turner JJ, Leotlela PD, Pannett AA et al (2002) Frequent occurrence of an intron 4 mutation in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 87(6):2688–2693
Baehring J, Sutter C, Kadmon M, Doeberitz MV, Gebert J (2006) A ‘nonsense’ mutation leads to aberrant splicing of hMLH1 in a German hereditary non-polyposis colorectal cancer family. Fam Cancer 5(2):195–199
Nemoto H, Tate G, Schirinzi A et al (2006) Novel NF1 gene mutation in a Japanese patient with neurofibromatosis type 1 and a gastrointestinal stromal tumor. J Gastroenterol 41(4):378–382
Reifenberger J, Rauch L, Beckmann MW, Megahed M, Ruzicka T, Reifenberger G (2003) Cowden’s disease: clinical and molecular genetic findings in a patient with a novel PTEN germline mutation. Br J Dermatol 148(5):1040–1046
Niemann S, Muller U, Engelhardt D, Lohse P (2003) Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum Genet 113(1):92–94
Martella M, Salviati L, Casarin A et al (2006) Molecular analysis of two uncharacterized sequence variants of the VHL gene. J Hum Genet 51(11):964–968
Abu-Amero KK, Owaidah TM, Al Jefri A, Al-Ghonaium A, Fawaz IM, Al-Hamed MH (2004) A novel splice site mutation in the WAS gene causes Wiskott-Aldrich syndrome in two siblings of a Saudi family. Blood Coagul Fibrinolysis 15(7):599–603
Tanioka M, Budiyant A, Ueda T et al (2005) A novel XPA gene mutation and its functional analysis in a Japanese patient with xeroderma pigmentosum group A. J Invest Dermatol 125(2):244–246
Spirio L, Green J, Robertson J et al (1999) The identical 5′ splice-site acceptor mutation in five attenuated APC families from Newfoundland demonstrates a founder effect. Hum Genet 105(5):388–398
Chen LL, Sabripour M, Wu EF, Prieto VG, Fuller GN, Frazier ML (2005) A mutation-created novel intra-exonic pre-mRNA splice site causes constitutive activation of KIT in human gastrointestinal stromal tumors. Oncogene 24(26):4271–4280
Tala HP, Carvajal CA, Gonzalez AA et al (2006) New splicing mutation of MEN1 gene affecting the translocation of menin to the nucleus. J Endocrinol Invest 29(10):888–893
Martinez R, Schackert HK, von Kannen S, Lichter P, Joos S, Schackert G (2003) Independent molecular development of metachronous glioblastomas with extended intervening recurrence-free interval. Brain Pathol 13(4):598–607
Ariga T, Yamada M, Pudua FR, Sakiyama Y (1996) Detection of a novel splice-site mutation that results in skipping exon 3 of the WASP gene in a patient with Wiskott-Aldrich syndrome. Biochim Biophys Acta 1317(3):158–160
Kanemoto K, Ishikura K, Ariyasu D et al (2007) WT1 intron 9 splice acceptor site mutation in a 46, XY male with focal segmental glomerulosclerosis. Pediatr Nephrol 22(3):454–458
Aceto G, Cristina Curia M, Veschi S et al (2005) Mutations of APC and MYH in unrelated Italian patients with adenomatous polyposis coli. Hum Mutat 26(4):394
Rutter JL, Goldstein AM, Davila MR, Tucker MA, Struewing JP (2003) CDKN2A point mutations D153spl(c.457G > T) and IVS2 + 1G > T result in aberrant splice products affecting both p16INK4a and p14ARF. Oncogene 22(28):4444–4448
Pagenstecher C, Wehner M, Friedl W et al (2006) Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet 119(1–2):9–22
Raponi M, Upadhyaya M, Baralle D (2006) Functional splicing assay shows a pathogenic intronic mutation in neurofibromatosis type 1 (NF1) due to intronic sequence exonization. Hum Mutat 27(3):294–295
Trojan J, Plotz G, Brieger A et al (2001) Activation of a cryptic splice site of PTEN and loss of heterozygosity in benign skin lesions in Cowden disease. J Invest Dermatol 117(6):1650–1653
Andreu N, Carreras C, Prieto F, Estivill X, Volpini V, Fillat C (2003) Identification and characterization of a novel splice-site mutation in a patient with Wiskott-Aldrich syndrome. J Hum Genet 48(11):590–593
Heikkinen K, Mansikka V, Karppinen SM, Rapakko K, Winqvist R (2005) Mutation analysis of the ATR gene in breast and ovarian cancer families. Breast Cancer Res 7(4):R495–R501
Mazoyer S, Puget N, Perrin-Vidoz L, Lynch HT, Serova-Sinilnikova OM, Lenoir GM (1998) A BRCA1 nonsense mutation causes exon skipping. Am J Hum Genet 62(3):713–715
Fackenthal JD, Cartegni L, Krainer AR, Olopade OI (2002) BRCA2 T2722R is a deleterious allele that causes exon skipping. Am J Hum Genet 71(3):625–631
McVety S, Li L, Gordon PH, Chong G, Foulkes WD (2006) Disruption of an exon splicing enhancer in exon 3 of MLH1 is the cause of HNPCC in a Quebec family. J Med Genet 43(2):153–156
Baralle M, Skoko N, Knezevich A et al (2006) NF1 mRNA biogenesis: effect of the genomic milieu in splicing regulation of the NF1 exon 37 region. FEBS Lett 580(18):4449–4456
Bottillo I, De Luca A, Schirinzi A et al (2007) Functional analysis of splicing mutations in exon 7 of NF1 gene. BMC Med Genet 8:4
Suphapeetiporn K, Kongkam P, Tantivatana J, Sinthuwiwat T, Tongkobpetch S, Shotelersuk V (2006) PTEN c.511C > T nonsense mutation in a BRRS family disrupts a potential exonic splicing enhancer and causes exon skipping. Jpn J Clin Oncol 36(12):814–821
Gorlov IP, Gorlova OY, Frazier ML, Amos CI (2004) Missense mutations in cancer suppressor gene TP53 are colocalized with exonic splicing enhancers (ESEs). Mutat Res 554(1–2):175–183
Narla G, Difeo A, Reeves HL et al (2005) A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res 65(4):1213–1222
Khan SG, Metin A, Gozukara E et al (2004) Two essential splice lariat branchpoint sequences in one intron in a xeroderma pigmentosum DNA repair gene: mutations result in reduced XPC mRNA levels that correlate with cancer risk. Hum Mol Genet 13(3):343–352
De Klein A, Riegman PH, Bijlsma EK et al (1998) A G– > A transition creates a branch point sequence and activation of a cryptic exon, resulting in the hereditary disorder neurofibromatosis 2. Hum Mol Genet 7(3):393–398
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Shkreta, L. et al. (2013). Cancer-Associated Perturbations in Alternative Pre-messenger RNA Splicing. In: Wu, J. (eds) RNA and Cancer. Cancer Treatment and Research, vol 158. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-31659-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-642-31659-3_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-31658-6
Online ISBN: 978-3-642-31659-3
eBook Packages: MedicineMedicine (R0)