
Abstract
Chronic pain is a widespread problem that plagues an estimated 10 to 30% of the world’s population. The current therapeutic repertoire is inadequate in managing patient pain with narcotic use resulting in a drug overdose epidemic, affirming the need for the development of new therapeutics. Adenosine and its four cognate receptors (A1AR, A2AAR, A2BAR, and A3AR) play essential roles in physiological and pathophysiological states, including chronic pain. For decades, preclinical and clinical studies have revealed that adenosine and A1AR- and to a lesser extent A2AAR-selective agonists have analgesic properties, yet their therapeutic utility has been limited by adverse cardiovascular side effects. There is no evidence that A2BAR plays a role in pain. Recent preclinical studies have demonstrated that selective A3AR agonists result in antinociception in models of acute and chronic pain while lacking unwanted side effects. These exciting preclinical observations of A3AR agonists have been bolstered by clinical trials of A3AR agonists in other disease states including rheumatoid arthritis and psoriasis that suggests a clinical benefit without cardiotoxicity. Our goal herein is to briefly discuss adenosine and its receptors in the context of pathological pain and examine what is known at present regarding A3AR-mediated antinociception. We will highlight recent findings pertaining to A3AR in pain and describe possible pathways by which A3AR may mediate its effects and the current state of selective A3AR agonists used in pain studies. The adenosine-to-A3AR pathway represents an important endogenous system that can be targeted to provide safe, effective pain relief in patients suffering with chronic pain.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
16.1 Introduction
Chronic pain afflicts an estimated 10% of the world’s adult population (Goldberg and McGee 2011) and approximately 30% of American adults with an estimated societal cost in the billions annually (Institute of Medicine 2011). The current therapeutic approaches for chronic pain include but are not limited to the use of NSAIDs, antidepressants, anticonvulsants, and opioid pain relievers; however, these strategies are frequently inadequate and/or are associated with side effects that reduce quality of life. Escalating doses are needed to produce analgesic efficacy, while side effects and potential addiction often result in the discontinuation of therapy (Goldberg and McGee 2011; Pizzo and Clark 2012). There is a desperate need for novel therapeutics that engage molecular targets in the nociceptive and inflammatory pathways that will not result in unwanted side effects nor result in severe analgesic tolerance. Adenosine and two of its associated adenosine receptor (AR) subtypes, A1AR and A2AAR, have been investigated for their ability to inhibit pain with varying degrees of success but lack a useful therapeutic index due to cardiovascular side effects and are not being currently pursued for pain. However, the A3 subtype, A3AR, has resulted in preclinical antinociceptive efficacy in a variety of pain models (Ford et al. 2015; Janes et al. 2014b, 2015; Little et al. 2015; Yoon et al. 2004) and has demonstrated efficacy and safety in trials for non-pain conditions including psoriasis, hepatitis, rheumatoid arthritis, dry eye, and glaucoma. Hence, A3AR agonists have clinically acceptable therapeutic indices that may be suitable for the treatment of chronic pain (Fishman et al. 2012). Importantly, such compounds lack rewarding behavior removing the potential for addiction and show long-term efficacy after sustained used (Little et al. 2015). The aim of this review is to summarize the existing literature on adenosine and its receptors in the context of pain with a particular emphasis on A3AR and its prospect as a novel solution to the problem of chronic pain management.
16.2 Adenosine Production and Metabolism
The endogenous purine nucleoside adenosine through its cognate receptors is a potent regulator of a wide variety of physiological processes affecting nervous (Boison 2013, 2016; Gomes et al. 2011; Wei et al. 2011), cardiovascular (Headrick et al. 2011), renal (Vallon and Osswald 2009), immune (Hasko et al. 1998; Hasko et al. 1996), and cell cycle (Fishman et al. 2009) functions. In the central nervous system (CNS), extracellular adenosine provides neuroprotective, anti-inflammatory, and neuromodulatory effects by regulating glial activity (Cunha 2008; Dias et al. 2013) and glutamatergic, GABAergic, cholinergic, and dopaminergic neurotransmission (Sebastiao and Ribeiro 1996). The extracellular function of adenosine is tightly regulated by homeostatic control of the intracellular/extracellular adenosine gradient and the local adenosine receptor profile (Deussen et al. 1999; Zimmermann 2000).
Adenosine is produced by nearly all cells (Zimmermann 2000) through intracellular and extracellular metabolic pathways (Fig. 16.1). Intracellular adenosine is generated either by the dephosphorylation of AMP via soluble 5′-nucleotidases or S-adenosylhomocysteine (SAH) hydrolysis (Latini and Pedata 2001). In the CNS , soluble 5′-nucleotidases activity appears to be the predominate route of intracellular adenosine production (Engler 1991). Extracellular generation of adenosine arises from the dephosphorylation of ATP by ectonucleotidase activity within the extracellular space. Upon its extracellular release during neurotransmission or in response to cellular injury (Ballarin et al. 1991; Engler 1991; Latini and Pedata 2001), ATP is first dephosphorylated by ectonucleoside triphosphate diphosphohydrolases (CD39 family) to AMP and then to adenosine by ecto-5′-nucleotidase (CD73) (Bonan 2012; Robson et al. 2006). Alternatively, extracellular adenosine can be generated by tissue-nonspecific alkaline phosphatase dephosphorylation of any of the adenosine nucleotides (Sebastian-Serrano et al. 2015).

Adenosine synthesis and metabolism. (a, b) ATP is released from various cell types in response to a number of stimuli. The phosphate groups of ATP can then be sequentially removed giving rise to ADP, AMP, and then adenosine. Ectonucleotidases (CD39, CD73) feed into this pathway by hydrolyzing nucleotides to adenosine for transport back into the cell via equilibrative nucleoside transporters (ENTs) or concentrative nucleoside transporters (CNTs). In the intracellular space, adenosine can be converted to AMP (by adenosine kinase, AdK) which in turn is catalyzed to AMP and then ATP or deaminated to inosine by ADA. Intracellular adenosine can be generated from AMP by 5′-nucleotidase. (c) Extracellular adenosine can act on its cognate receptors (ARs: A1, A2A, A2B, and A3)
Basal levels of extracellular adenosine within the CNS are maintained around 25–250 nM (Dunwiddie and Masino 2001). This is accomplished by sodium-coupled influx of adenosine through concentrative nucleoside transporters (CNT / Slc28) (Bonan 2012; Choi and Berdis 2012) or by passive influx/efflux of adenosine down its gradient through the ubiquitous equilibrative nucleoside transporters (ENT1/Slc29A1 and ENT2/SLC29A2) (Brundege and Dunwiddie 1998; Peng et al. 2005). The adenosine gradient for ENT function is established by the balance of adenosine production as already described and the depletion of its intracellular stores by adenosine kinase (AdK) phosphorylation of adenosine (Spychala et al. 1996) and catabolism by adenosine deaminase (ADA) to inosine (Blackburn and Kellems 1996). AdK and ADA limit the physiological half-life of adenosine to <1 s (Moser et al. 1989) to establish a normally inward driving adenosine gradient. Studies have revealed AdK is a major driving force for both intracellular and extracellular adenosine (Boison 2016). The expression of AdK in neurons during development of the nervous system is necessary for neurite outgrowth and synaptic formation; but during postnatal development, AdK expression shifts primarily to astrocytes where it is involved in maintaining adenosine homeostasis (Studer et al. 2006). Inhibiting AdK activity significantly increases the intracellular concentrations of adenosine, which reverses its gradient and drives it out through the ENT channels into the extracellular space (Keil and DeLander 1992; Zhang et al. 1993). Consequently, efforts have been made to target AdK activity for the treatment of a number of neuropathologies (Boison 2008b, 2013, 2016; Kowaluk et al. 1999). In pain, pharmacological inhibition of AdK in the CNS results increased the ENT-dependent release of adenosine, which in turn attenuated spinal nociceptive transmission (Otsuguro et al. 2015). Moreover, enhancing endogenous adenosine signaling using AdK inhibitors has been shown to be efficacious in rodent models neuropathic pain (Kowaluk et al. 2000; Little et al. 2015; McGaraughty et al. 2005).
16.3 Adenosine Receptors
Adenosinergic signaling is mediated through four cognate G protein-coupled receptors: A1, A2A, A2B, and A3AR. The A1A and A3A receptor subtypes couple to Gαi to inhibit adenylate cyclase formation (Boison et al. 2010; Fredholm et al. 2011). However, there is evidence that A3AR also associates with Gαq/11 to stimulate phospholipase C (Parsons et al. 2000). In contrast, A2A and A2B receptor couple to Gs and stimulate adenylyl cyclase and produce elevations in intracellular cAMP (Fredholm et al. 2001).
In the CNS, A1AR is widely expressed in the brain and superficial laminae of the spinal cord dorsal horn (Gessi et al. 2011). The expression of A1AR is highest in neurons (Cunha 2001, 2005) where it is expressed on both the presynaptic and postsynaptic membrane (Cunha 2001, 2005; Gessi et al. 2011) and associated with the modulation of neurotransmission by reducing the presynaptic release of glutamate and increasing the postsynaptic hyperpolarization (Cunha 2005). In glia, A1AR expression is downregulated in multiple sclerosis (Johnston et al. 2001) and the dorsal horn of the lumbar spinal cord following plantar incision, a model of postoperative pain (Yamaoka et al. 2013). However, A1AR has also been shown increase in the dorsal horn following traumatic nerve injury (Yamaoka et al. 2013). These findings suggest receptor expression is differentially regulated depending on the nature of the injury. This is further supported by findings that in primary mouse microglia, A1AR expression increases in response to ATP but reduced following exposures to endotoxin (Luongo et al. 2014). From a clinical standpoint, it is important to note that A1AR is also highly expressed in cardiovascular tissue, particularly the atrioventricular node, which is associated with A1AR-mediated high-grade atrioventricular block mediated following treatments with A1AR agonists (Kiesman et al. 2009).
A2AAR expression in the brain on striatal postsynaptic neurons, hippocampal and cortical presynaptic neurons, and glial cells (Rebola et al. 2005; Svenningsson et al. 1997). The expression of A2AAR can increase following hypoxia, spinal cord injury, and streptozotocin-induced diabetes (Janes et al. 2014b). In monocytes/microglia, the expression of A2AAR is enhanced by pro-inflammatory mediators such as IL-1β and TNF-α (Morello et al. 2006). In the cardiovascular system , the epithelium of coronary blood vessels express A2AAR and exert vasodilatory effects in response to A2AR agonists (Fredholm et al. 2011; Gao and Jacobson 2007; Jacobson and Gao 2006).
Collectively, A1AR and A2AAR comprise the bulk of the adenosine receptor expression the CNS (Gomes et al. 2011). In contrast, the lower-expressed and lower-affinity A2BAR is found in the neuroimmune cells of the CNS and within the cardiovascular system. A2BAR activity in microglia is associated with IL-6 expression and microglial proliferation (Merighi et al. 2017). However, A2BAR transcript in the cortex of a normal mouse brain has been reported to be expressed mainly in astrocytes and oligodendrocyte progenitor cells (Zhang et al. 2014).
Species-specific differences exist for A3AR structure and distribution. In rats, A3AR expression is highest in testis and mast cells, whereas in humans, A3AR expression is highest in the liver and lung (Borea et al. 2015). High expression of A3AR has been reported in human coronary and carotid arteries (Grandoch et al. 2013; Hinze et al. 2012) and several studies have found A3AR signaling is cardioprotective during ischemic injury (Cross et al. 2002; Harrison et al. 2002; Headrick and Peart 2005; Thourani et al. 1999a, b; Tracey et al. 1997) and doxorubicin-induced cardiotoxicity (Shneyvays et al. 1998, 2001). In the CNS, A3AR is expressed at much lower levels than A1AR and A2AAR. However, A3AR has higher expression on many immune cell types, including glial cells (Abbracchio et al. 1997; Ochaion et al. 2009; Poulsen and Quinn 1998), and can be found on both peripheral (Ru et al. 2011) and central neurons (Giannaccini et al. 2008; Jacobson et al. 1993; Lopes et al. 2003; Zhang et al. 2010) of the brain and spinal cord (Borea et al. 2015; Haeusler et al. 2015). In pain-processing centers, A3AR transcript and protein have been identified in the lumbar spinal cord and rostral ventromedial medulla (RVM) (Little et al. 2015).
The expression and distribution of adenosine receptors throughout the CNS and on cells responsible for pathophysiological changes within the CNS during development and maintenance of pain (Cao and Zhang 2008; Nagata et al. 2009; Obata and Noguchi 2008; Watkins et al. 2001) provide unique advantages to targeting these receptors. However, as will be discussed, activation of many of these receptors provides similar effects in models of pain despite differences in the coupling mechanisms of these receptors. These similarities may be due to their tissue distribution, expression regulation under pain conditions, the components of the microdomains in which they associate, and the endogenous ligands to which they respond such as the partial agonism of inosine at A1AR and A3AR.
16.4 Adenosine and Pain
The analgesic effects of adenosine have been known for many years now. In the clinic, intrathecal adenosine provided sustained relief for several hours to months of chronic neuropathic pain (Hayashida et al. 2005). Adenosine and its analogues have consistently been shown to inhibit pain behavior in a number of neuropathic and inflammatory pain models arising from various etiologies , such as spinal cord injury, spinal nerve ligation, and exposure to mustard oil, formalin, or carrageenan (Dickenson et al. 2000). The beneficial effects of adenosine have been associated with its regulation of excitatory neurotransmission, persistent neuronal signaling, and glial activation and proliferation (Boison 2008a; Boison et al. 2010; Cunha 2005; Daniele et al. 2014; Studer et al. 2006). Despite the promising data from animal pain models and early clinical chronic pain studies, the effectiveness of adenosine therapy for the prevention of postoperative pain has been mixed. Prophylactic intravenous administration of adenosine prior to surgical procedures conferred persistent pain relief in several studies (Gan and Habib 2007; Hayashida et al. 2005), but not in others (Habib et al. 2008). Moreover, intravenous adenosine therapy is associated with serious adverse cardiac side effects (Zylka 2011) limiting its utility. Thus, evaluating the receptor subtypes involved in order to separate the antinociceptive adenosinergic signaling from cardiovascular adenosinergic effects is important in developing adenosine-based therapeutics in pain.
16.4.1 A1AR and A2AAR in Pain
Despite demonstrated preclinical efficacy in several pain models, agonists of A1AR and A2AAR have not been the focus of clinical trials due to their potential cardiotoxicity (Chen et al. 2013; Fredholm et al. 2011; Sawynok 1998; Varani et al. 2017; Zylka 2011). Yet, these receptors have played an important role in evolving our current understanding of adenosine-mediated antinociception. Prior studies attributed the adenosine antinociception to the activation of the A1 and A2A receptor subtypes (Sawynok 2013, 2016; Zylka 2011). For example, genetic knockout of A1ARs elicits thermal hypersensitivity and exacerbates neuropathic behavioral responses to cold and heat (Wu et al. 2005). In contrast, A1AR activation alleviates nerve injury-induced pain (Cui et al. 1997; Gong et al. 2010), perioperative pain (Gan and Habib 2007), inflammatory pain (Sowa et al. 2010), central pain following spinal cord injury (Sjolund et al. 1998), complex regional pain syndrome type I (CRPS-I) (Martins et al. 2013), and painful diabetic neuropathy (Katz et al. 2015; Vincenzi et al. 2014) in preclinical models. Intrathecal administration of an A1AR agonist reduced non-evoked spontaneous pain behaviors resulting from a surgical model of pain (Zahn et al. 2007). Repeated sessions of high-intensity swimming exercise increased endogenous adenosine levels, which played a role in the attenuation of mechanical allodynia in an animal model of CRPS-I (Martins et al. 2013). Intervention with the adenosine deaminase inhibitor erythro-9-(2-hydroxy-3nonyl)adenine (EHNA) , which limits adenosine degradation, enhanced the pain-relieving effects of swimming through mechanisms involving A1AR (Martins et al. 2013). Intravenous infusions of adenosine in humans reduced some aspects of neuropathic pain and were shown to decrease postoperative pain mainly through the A1AR (Gao and Jacobson 2007). The preclinical robustness of A1AR pain relief resulted in clinical trials for multiple A1AR agonists and an A1AR allosteric enhancer; however, these drug trials were discontinued due to limited efficacy, presumably driven by a low therapeutic index (Gessi et al. 2011; Romagnoli et al. 2010). Recently, additional novel allosteric enhancers of the A1AR, including TRR469, have demonstrated antinociceptive efficacy in two preclinical models of acute pain, writhing and formalin tests, and in chronic streptozotocin-induced diabetic neuropathy (Vincenzi et al. 2014). These data suggest that A1AR allosteric enhancers may still be promising candidates to treat acute and chronic pain, with the potential advantages of their unique mechanism of action and lack of side effects. TRR469 dramatically increases adenosine affinity in mouse spinal cord membranes, suggesting the possibility of exploiting the antinociceptive effect of endogenous adenosine in a physiological way (Vincenzi et al. 2014).
Controversy surrounds the role of A2AAR in nociception/antinociception. Depressed responses to acute pain stimuli were observed in mice lacking the A2AAR (Ledent et al. 1997). Similarly, an intracerebroventricular injection of an A2AAR-targeted antibody with agonist-like activity produces antinociceptive effects in naïve mice (By et al. 2011). Peripheral administration of an A2AAR agonist is associated with nociceptive behaviors (Taiwo and Levine 1990), whereas very low doses of A2AAR agonists promote reversal of nerve injury-induced pain in rats for weeks after a single spinal injection (Loram et al. 2009). In models of postsurgical pain (Zahn et al. 2007) and inflammatory pain (Poon and Sawynok 1998), intrathecal administration of A2AAR agonists had limited antinociceptive efficacy. At the clinical level, a phase II trial of an oral A2AAR agonist BVT-115959 in the treatment of diabetic neuropathy was completed in 2008 (Gao and Jacobson 2011); no further data has been provided at this time. The differing observations of A2AAR agonists in pain highlight an apparent dichotomy of peripheral versus central A2AARs in pain signaling .
Unfortunately, a narrow therapeutic focus on only two of the AR subtypes has contributed to a decade of failed preclinical and clinical development efforts. Indeed, a focus on the A1 and A2A receptors has failed to harness adenosine antinociception effectively and without cardiovascular side effects (Boison 2013; Zylka 2011). In response, we anticipate that a greater emphasis on the A3 receptor may provide an answer to the question of whether adenosine antinociception can provide safe, clinical pain relief. Recently, the combination of an A1AR agonist and an A3AR agonist demonstrated highly potent analgesic activity using a preclinical model of formalin-induced flinching (Petrelli et al. 2017). The combining of both A1AR and A3AR agonistic activity in one single molecule may act synergistically reducing the overall dose and therefore reduce the A1AR-induced cardiotoxicity.
16.4.2 A3AR and Pain
The A3AR is a rapidly growing focus in the area of pain. Early literature was confounded by results gleaned from A3AR-targeted compounds with poor specificity (Sawynok et al. 1997, 1999) or from a single study performed in A3AR−/− mice (Wu et al. 2002). To inform the progress of A3AR development, it is important to clarify the findings of these initial studies. In the earliest paper published in 1997 examining the contribution of A3AR in pain, Sawynok and colleagues reported that subcutaneous administration of N6-benzyl–NECA into the hindpaw of rodents produces a dose-related increase in nociceptive flinching behavior (Sawynok et al. 1997). It was found that this behavior was blocked by inhibitors of the histamine H1 receptor and of 5-hydroxytryptamine2 (5-HT2) , but was not modified by A1AR or A2AR antagonists. The authors speculated that A3AR activation was responsible for the pro-nociceptive response, possibly by inducing mast cell degranulation (Sawynok et al. 1997). However, there was no evidence that linked A3AR to the effects of N6-benzyl–NECA. Moreover, N6-benzyl–NECA is not selective for A3AR (Gallo-Rodriguez et al. 1994). In the follow-up studies in 1999, the effects of N6-benzyl–NECA were not influenced by an A3AR antagonist (MRS1191) but rather abrogated by an A2BAR antagonist. These results suggest that the pro-inflammatory , pro-nociceptive effect of N6-benzyl–NECA was likely due to activation of the A2BAR, a subtype previously implicated in inflammation (Feoktistov and Biaggioni 2011). Unfortunately, the notion of A3AR-mediated pro-nociceptive effects remained. The erroneous notion that A3AR activation led to pain and inflammation was further supported by a study in 2002 characterizing the development of carrageenan-induced paw edema and hyperalgesia in the A3AR−/− mouse. This study reported a minimal increase in thermal hyperalgesia compared to wild-type control animals, but no observable differences in the normal (protective) nociceptive response of A3AR−/− animals to indicate that A3AR is not physiologically involved in modulating normal nociception (Wu et al. 2002). However, a year later, another study revealed decreased hot plate but not tail-flick responses of A3AR−/− mice (Fedorova et al. 2003).
The notion of the A3AR-mediated pro-nociceptive effects was challenged when more selective A3AR agonists, such as IB-MECA (N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide), began to be employed in pain models. IB-MECA is 50-fold more selective for A3AR over rat A1AR or A2AAR , whereas N6-benzyl–NECA only displays 14-fold selectivity (Gallo-Rodriguez et al. 1994; Jacobson 1998). A single investigation in 2005 reported that systemic administration of IB-MECA had no effect on normal nociception nor in the first phase of the formalin test but exerted significant antinociceptive effects on the second phase of the formalin test (Yoon et al. 2005). In a follow-up report, it was noted that intrathecal administration of an A3AR antagonist (MRS1220) prevented the antinociceptive actions of adenosine in the second phase of the formalin test, supporting a role for spinal A3ARs in the effect of adenosine (Yoon et al. 2006). No other papers were published between 2006 and 2012 that examined the contribution of A3AR in pain.
In 2012, we revisited the A3AR hypothesis and demonstrated that selective activation of A3AR exerts potent antinociceptive effects in models of neuropathic pain (Chen et al. 2012; Little et al. 2015), validating the observations in models of non-neuropathic pain states (Yoon et al. 2006). Both IB-MECA and Cl-IB-MECA blocked the development of mechano-allodynia following chronic constriction injury (CCI), which was attenuated by an antagonist of A3AR but not of A1AR or A2AAR (Chen et al. 2012). Moreover, low doses of IB-MECA that lacked analgesic effects provided profound increases in the analgesic potency of morphine, gabapentin, and amitriptyline when coadministered (Chen et al. 2012). The antinociceptive effects of IB-MECA and Cl-IB-MECA have since been corroborated with even more selective A3 agonists, such as MRS1898 (>100-fold over A1AR or A2AAR (Gao et al. 2009)) and more recently MRS5698 (>10,000-fold over A1AR or A2AAR (Tosh et al. 2012)), in rodent CCI, spared nerve injury, and spinal nerve ligation neuropathic pain models (Chen et al. 2012; Ford et al. 2015; Little et al. 2015). The loss of MRS5698 antinociception in the A3AR−/− mouse or in the presence of the specific A3AR antagonist, MRS1523, corroborates the specificity of these newer-generation compounds as A3AR antinociceptive agents (Little et al. 2015). Indeed, these pharmacological tools have facilitated a better understanding of the levels at which A3AR functions to attenuate pain: A3AR agonists administered via intradermal (ipsilateral paw) injection (IB-MECA, 3–60 nmol), intrathecal cannula (MRS5698, 3–60 nmol), or RVM cannula (MRS5698, 0.3–3 nmol) dose-dependently attenuate CCI-induced mechanical allodynia (Little et al. 2015). Systemic administration of a peripherally restricted A3AR agonist also reverses CCI-induced peak mechanical allodynia, and the inability of an intrathecal A3AR antagonist to reverse this effect validated its peripheral site of action (Paoletta et al. 2013). Conversely, antinociception conferred via systemic administration of the CNS-permeant MRS5698 is attenuated with intrathecal or intra-RVM delivery of an A3AR antagonist, highlighting the dual peripheral and central roles of A3AR in antinociception (Little et al. 2015). Further studies are warranted to explore the relationship between peripheral and central A3ARs in pain.
The beneficial effects of A3AR agonists extend to number of cancer-related pain states. In models of neuropathic pain associated with the administration of chemotherapeutics (chemotherapy-induced peripheral neuropathy , CIPN), IB-MECA (Chen et al. 2012; Janes et al. 2014b) and MRS5698 (Janes et al. 2015; Little et al. 2015; Wahlman et al. 2018) blocked the development of neuropathic pain. Similar antinociceptive effects were provided by Cl-IB-MECA (Varani et al. 2013) and MRS5698 (Little et al. 2015) in rodent models of pain associated with breast cancer bone metastasis. Interestingly, A3AR agonists do not interfere with antitumor effects (Chen et al. 2012) but instead are in themselves antitumor agents. High expression of A3AR is detected on many malignant cell types and accordingly A3AR agonists have been shown to produce direct anticancer effects on their own and have been documented to enhance the actions of several widely used chemotherapeutics and attenuate the associated myelosuppression (Fishman et al. 2002, 2009, 2012). Cl-IB-MECA was shown to reduce tumor growth in the rat model of breast cancer bone metastasis (Varani et al. 2013). Indeed, Cl-IB-MECA is currently in phase II clinical trials for hepatocellular carcinoma as an anticancer agent. Therefore, the use of A3AR agonists may provide dual benefits in the treatment of a variety of cancer-related pain states.
The antinociceptive effects of A3AR agonists persist even with long-term treatment, such as repeated daily injections for 6 days or continuous infusion for 7 days (Little et al. 2015). These findings suggest that there is no development of antinociceptive tolerance to A3AR agonists, unlike morphine, where tolerance to its antinociceptive effects develops only after 6 days of injections (Muscoli et al. 2010). These findings are curious as all adenosine receptor subtypes exhibit a “desensitization phenomenon” resulting in the diminished response and receptor surface expression after repeated or continuous exposure agonists (Klaasse et al. 2008). However, in animal models of autoimmune disorders and cancer, chronic administration of A3AR agonists maintains anti-inflammatory/anticancer effects even during A3AR downregulation (Madi et al. 2003). It is thought that the functionality of A3AR agonist in inflammation/tumor growth may be dependent on the downregulation of A3AR to inhibit downstream regulatory proteins (Fishman et al. 2006). Whether this mechanism explains the action of IB-MECA and other A3AR agonists in pain requires further investigation.
In preclinical animal models, the antinociceptive effects of A3AR agonists are not dependent upon endogenous opioid or endocannabinoid pathways (Ford et al. 2015; Little et al. 2015), suggesting that A3AR agonists lack inherent reward properties that would heighten the potential risk of abuse and dependence. Emerging data indicates that A3AR agonists, such as MRS5698, produce a preference in nerve-injured rats to the particular chamber in which they received the A3AR agonists termed “conditioned place preference” (CPP) (Little et al. 2015). This suggests that A3AR agonists provided relief of spontaneous pain in these animals. However, sham rats given A3AR agonists did not exhibit any CPP, indicating a lack of inherent reward with these compounds (Little et al. 2015). In contrast, opioids and other drugs of abuse elicit CPP from both naïve and injured animals (Prus et al. 2009). Therefore, A3AR agonists have the potential to selectively modify pathological but not protective pain , while avoiding the tolerance and abuse potential associated with opioid therapy.
16.4.3 Mechanisms of A3AR Antinociception
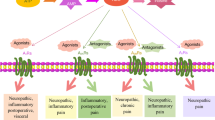
The antinociceptive and regulatory mechanisms and pathways modulated by A3AR agonists in pathological pain states is only now beginning to be explored. However, as already discussed, A3AR agonists act the level of the peripheral afferent, the spinal cord, and the RVM as selective A3AR agonists administered via intradermally, intrathecally, or intra-RVM dose-dependently attenuate neuropathic pain behaviors (Little et al. 2015). The actions of A3AR agonist are independent of opioidergic and cannabinoid systems (Little et al. 2015) and engage serotonergic and noradrenergic bulbospinal circuits in neuropathic pain, suggesting the involvement of A3AR signaling in summary descending inhibition of wide dynamic range spinal neurons (Little et al. 2015). In other disease states, A3AR activation has been shown to alter components that are critically involved in the development of central sensitization and pain, including protein kinase activity , glutamatergic neurotransmission, ion conductance, and neuroinflammation. To inform the potential mechanism(s) of A3AR-mediated antinociception, we have summarized the consequences of A3AR activation as they are relevant to pain (Fig. 16.2).

Potential mechanisms of A3AR-mediated antinociception
The A3AR agonist MRS5698 has been recently shown to reverse traumatic nerve injury-induced pain by maintaining GABAergic signaling (Ford et al. 2015). The GABAergic system is an important inhibitory regulator of nociceptive transmission. GABA is released from interneurons within the CNS and resulting activation of GABA receptors dampens neuronal excitability to reduce nociceptive signaling (Zeilhofer et al. 2012). In the pathological pain state, the GABAergic system becomes dysregulated and the balance of nociceptive signaling shifts toward state hyperexcitability (Zeilhofer et al. 2012). GABAergic dysregulation results from reduced GAD65-dependent GABA synthesis (Eaton et al. 1998; Stiller et al. 1996), increased GABA reuptake transporter GAT-1 expression (Eaton et al. 1998; Moore et al. 2002), and reduction in K+-Cl− cotransporter (KCC2) activity that results in the loss of the anion gradient necessary to drive Cl− through GABAA channels (Coull et al. 2003; Price et al. 2005). In a traumatic nerve injury-induced pain animal model, MRS5698 attenuated the dephosphorylation of GAD65 and GAT-1 and the phosphorylation of KCC2 and maintained appropriate Cl− flux (Ford et al. 2015). Moreover, A3AR agonists attenuated brain-derived neurotrophic factor (BDNF) signaling (Ford et al. 2015), which has been shown to inhibit GABAergic signaling (Biggs et al. 2010; Ferrini and De Koninck 2013; Smith 2014).
A3AR agonists may also exert their effects through the RhoA-phospholipase D (PLD) signaling pathways. In other animal models, A3AR agonists prevent the decrease in PLD activity in response to reactive oxygen species exposure during cardiomyocyte apoptosis (Asemu et al. 2005; Lee et al. 2001). Proper PLD function is necessary for the production of choline in order to activate α7 nicotinic acetylcholine receptors (Lee et al. 1993). Activation of these receptors is both neuroprotective and antinociceptive during chronic neuropathic pain (Feuerbach et al. 2009).
A3AR activation is associated with the attenuation of astrocyte reactivity, neuroinflammatory response (Janes et al. 2015), and reactive microglial chemotaxis (Choi et al. 2011), such that A3AR agonists may reduce BDNF associated with glial hyperactivation and free the GABAergic system to function properly. Glial cells (astrocytes and microglia) are critical to the development and maintenance of many pathological pain states (Cao and Zhang 2008; Nagata et al. 2009; Obata and Noguchi 2008; Watkins et al. 2001). Targeting the glial activity can prevent and attenuate a variety of pain states (Hashizume et al. 2000; Meller et al. 1994; Sweitzer et al. 2001; Watkins et al. 1997, 2001). In pathological pain states, glial cells can release a number of pro-inflammatory cytokines and nitroxidative species that increase neuronal sensitivities in the dorsal horn (Cao and Zhang 2008; Milligan and Watkins 2009) and further increase glial activity to establish an amplification loop that may account for the persistence of hypersensitivities in chronic pain states (Bradesi et al. 2001). Moreover, activation of innate immune receptor toll-like receptor 4 (TLR4) expressed on glial cells has been implicated in the neuroinflammatory response in the development of neuropathic pain (Li et al. 2014; Watkins et al. 2009).
A3AR agonists are anti-inflammatory in autoimmune and inflammatory diseases (Bar Yehuda et al. 2010). Both in vitro and in vivo studies have revealed that A3AR attenuates pro-inflammatory cytokines by inhibiting the p38 MAPK and nuclear factor κB (NFκB) signaling pathways (Janes et al. 2014a; Madi et al. 2007; Varani et al. 2010, 2011). IB-MECA has been documented to decrease the TLR4-induced pro-inflammatory mediators, such as tumor necrosis factor (TNF) and macrophage inflammatory protein 1α (MIP-1α) (Hasko et al. 1998; Hasko et al. 1996; Sajjadi et al. 1996; Szabo et al. 1998). A3AR-mediated suppression of pro-inflammatory mediators following TLR stimulation is lost in A3AR knockout mice (Salvatore et al. 2000). In models of CIPN, IB-MECA reduced the level of reactive astrocytes, NFκB and MAPK activation, and level of pro-inflammatory/neuroexcitatory cytokines (Janes et al. 2014a, 2015; Wahlman et al. 2018). In the oxaliplatin-induced neuropathic pain model, administration of oxaliplatin increased NOD-like receptor with pyrin domain subtype 3 (NLRP3) inflammasome activation of IL-1β in the spinal cord and pharmacological inhibition of NLRP3 activity attenuated pain to suggest the involvement of this pathway in the development of mechano-hypersensitivities (Wahlman et al. 2018). Attenuation of CIPN with intrathecal MRS5698 was associated with reduced expression and activation of NLRP3 in the spinal cord (Wahlman et al. 2018). Interestingly, A3AR activation also enhances formation of the anti-inflammatory cytokine IL-10 (Hasko et al. 1996; Janes et al. 2014a, 2015) and glial-derived neuroprotective substances (Wittendorp et al. 2004). Moreover, inhibition of IL-10 with neutralizing antibodies not only attenuated the beneficial effects of A3AR agonists on pain behavior but also restored the expression and activation of NLRP3 inflammasomes (Wahlman et al. 2018). MRS5698 also lost its beneficial effects on CIPN in IL-10−/− mice (Wahlman et al. 2018). These findings suggest that this shift in the spinal neuroinflammatory environment may be a major contributor to the effects of A3AR in pain. More work is necessary to understand at what point A3AR exerts its effects on neuroinflammation.
In addition to neuroinflammatory mediators, nitroxidative species including superoxide (SO), nitric oxide (NO), and their highly pro-nociceptive reaction product peroxynitrite (PN) (Salvemini and Neumann 2010) are important in the development and maintenance of pain of several etiologies, including acute and chronic inflammation (Ndengele et al. 2008), orofacial pain (Yeo et al. 2008), and opiate-induced hyperalgesia and antinociceptive tolerance (Muscoli et al. 2007), nerve injury-induced pain (Rausaria et al. 2011), and CIPN (Doyle et al. 2012; Janes et al. 2013). In CIPN, IB-MECA attenuated the activation NADPH oxidase, a source of SO as a precursor to PN formation (Janes et al. 2014a; Poderoso et al. 1996), in the spinal cord. Inhibition of NADPH oxidase in prostate cells following IB-MECA is linked to the inhibition of intracellular cyclic AMP/PKA (Jajoo et al. 2009) and reduced expression of NADPH oxidase subunits (Rac1 and p47phox) through inhibition of ERK1/2 activity (Jajoo et al. 2009).
Activation of A3AR may play a critical role in the inhibitory actions of adenosine on excitatory neurotransmission and its neuroprotective effects. A3AR activation in vitro protects against the neurotoxic rises in intracellular Ca2+ and neuronal excitability mediated by P2X7R (Zhang et al. 2006) and NMDAR (Zhang et al. 2010). Dysregulated glutamatergic neurotransmission and increased neuronal excitability are hallmarks of chronic pain (Amadesi et al. 2006; Chen et al. 2010; Doyle et al. 2012; Elliott et al. 1994; Mayer et al. 1999; Muscoli et al. 2007; Xu et al. 2010; Zhang et al. 2012). Treatment with A3AR agonists attenuates posttranslational nitration of glutamate transporter GLT-1 and glutamate synthase (Janes et al. 2014a) in the spinal cord. Nitration of these proteins leads to a loss in their activity that consequently reduces the capacity to remove glutamate from the synapse and terminate glutamatergic signaling (Mao et al. 2002).
16.4.4 Pharmacological Probes for the Study of A3AR in Pain
A toolbox of selective A3AR modulators is now accessible, which includes high affinity directly acting agonists 1-8 (Table 16.1, Fig. 16.3) and antagonists 9-12, as well as indirect modulators of A3AR activity. Indirect modulators include inhibitors of adenosine degrading enzymes, adenosine deaminase (ADA; 13), and adenosine kinase (ADK; 14, 15). Furthermore, there are selective allosteric enhancers of the action of endogenous adenosine at the A3AR (16, 17). Although these positive allosteric modulators are selective for the human A3AR and do not act at other AR subtypes, there is a large species dependence such that their activity is only subtle in rodent species.

Pharmacological agents useful for the study of A3AR-mediated antinociception
At the 5′ position, an amide in place of the CH2OH, as for all agonists shown in Fig. 16.3, favors affinity and efficacy at the A3AR. At the N6 position, either small hydrophobic groups, e.g., methyl 7 and ethyl 8, or large hydrophobic groups, e.g., m-substituted benzyl rings in 5 and 6, are tolerated when bound to the receptor . The A3AR affinity of N6-benzyl analogues is often better preserved in rodent species than in compounds with small N6 groups. For example, MRS5698 (5) is of the same affinity (Ki ~3 nM) at human and mouse A3ARs with high selectivity. At 10 μM (close to its solubility limit), 5 displays a low percent inhibition of binding (less than 50%) at the A1AR and A2AAR.
Among widely used A3AR agonists, IB-MECA (1) and Cl-IB-MECA (2) have varying degrees of AR subtype selectivity, as shown in a comparison of affinities at human, mouse, and rat ARs (Table 16.1). The affinity (Ki value) of IB-MECA at the mouse A3AR is an impressive 87 pM, and its Ki value at the human A3AR is 20-fold higher. These two agonists are moderately selective for the A3AR, which is often sufficient to achieve a dose window of selectivity depending on the pharmacological model and species being studied. Compounds 3 and 4 contain a ring constraint in the ribose-like moiety, known as the (North)-methancarba modification of nucleosides, which maintains a conformation preferred at the A3AR. Native ribose can freely twist to achieve a range of conformations, but if a favored conformation is pre-installed in the nucleoside, there is an advantage for binding to that subtype. In general, this ribose modification tends to increase affinity and selectivity, because the other AR subtypes are either adversely affected by this ribose substitution (A2AAR) or favor the substitution (A1AR) to a lesser degree than the A3AR. The more highly derivatized agonists containing a rigid C2 extension consisting of an arylethynyl group, in addition to the (North)-methancarba modification, are even more A3AR selective. Thus, the combination of these two substituents , as present in compounds 5-8, achieves 10,000-fold selectivity or greater for the A3AR. These particularly potent and specific A3AR agonists are especially useful in pharmacological studies of this receptor in pain models (Tosh et al. 2012, 2014, 2015). Compounds 5, 7, and 8 have been shown to be orally active in a dose-dependent manner in reducing or completely suppressing mechanoallodynia in the CCI model. The terminal C2 aryl group in this series of agonists may be substituted with a wide range of chemical functionality and still retain A3AR selectivity. Compound 5 contains a 3,4-difluorophenyl ring at the terminal position, while compounds 7 and 8 contain a 5-chlorothienyl ring that is associated with long duration of action (3 h or greater) in the mouse CCI model following oral administration. Compound 6 is not intended for oral administration, because it contains a fully negatively charged aryl sulfonate group that prevents its diffusion across biological membranes . It displayed no permeability in the PAMPA model of membrane permeability, indicating that it likely does not diffuse across the blood brain barrier. Due to this property, compound 6 was used to separate central from peripheral effects of A3AR agonists in the mouse CCI model, depending on the site of administration.
The use of selective A3AR antagonists or mice in which A3AR is genetically knocked out in conjunction with agonists or enhancers is important to delineate A3AR-mediated effects, especially with agonists that are only moderately selective. Nonnucleoside A3AR antagonists (e.g., 1,4-dihydropyridines 9 and 10 and pyridine 11) have varying degrees of AR subtype selectivity, depending on species. By progressively truncating the structure of nucleosides that are selective for the A3AR, it is possible to shift the activity from full agonist to partial agonist to antagonists. Thus, a truncated nucleoside 12 was shown to have considerable affinity at both human and mouse A3AR with selectivity, but the efficacy of this compound in A3AR activation was greatly diminished, such that it can serve as an antagonist of a full A3AR agonist. Thus, more potent and selective A3AR antagonists for application to a range of species are still needed.
An indirect means of pharmacologically enhancing activation of the A3AR, and potentially other ARs, is to enhance levels of extracellular adenosine by inhibiting ADA (e.g., 13 Pentostatin) or ADK (e.g., 14 5-iodotubercidin and 15 ABT-702). ADK inhibitor 15 was found to reduce both chronic and acute pain through action at both peripheral or central sites (Kowaluk et al. 2000). Recently, Little et al. used 15 to reveal an effect of endogenous adenosine acting through the A3AR to reduce chronic neuropathic pain.
16.5 Concluding Remarks
The development of selective pharmacological tools targeting A3AR has uncovered the exciting, robust antinociceptive properties of A3ARs agonists in a variety of pathological pain states. Emerging evidence suggests that harnessing the endogenous antinociceptive A3AR pathway yields effective pain relief without altering normal protective nociception and without producing inherent reward that is associated with abuse potential. As selective A3AR agonists in ongoing phase II/III clinical trials for non-pain conditions display good safety profile, we propose that A3AR agonists may be a safe and successful strategy for exploiting the potent analgesic actions of adenosine to provide a breakthrough non-opioid treatment for patients suffering from chronic pain.
References
Abbracchio MP, Rainaldi G, Giammarioli AM et al (1997) The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl-XL:studies in human astroglioma cells. Biochem Biophys Res Commun 241:297–304
Amadesi S, Cottrell GS, Divino L et al (2006) Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 575:555–571
Asemu G, Dent MR, Singal T et al (2005) Differential changes in phospholipase D and phosphatidate phosphohydrolase activities in ischemia-reperfusion of rat heart. Arch Biochem Biophys 436:136–144
Ballarin M, Fredholm BB, Ambrosio S et al (1991) Extracellular levels of adenosine and its metabolites in the striatum of awake rats:inhibition of uptake and metabolism. Acta Physiol Scand 142:97–103
Bar Yehuda S, Fishman P, Stemmer S et al (2010) CF102 exerts a differential effect in various pathological liver conditions:protection from inflammation damage and anti-tumor activity. Purinergic Signal 6:88
Biggs JE, Lu VB, Stebbing MJ et al (2010) Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol Pain 6:44
Blackburn MR, Kellems RE (1996) Regulation and function of adenosine deaminase in mice. Prog Nucleic Acid Res Mol Biol 55:195–226
Boison D (2008a) Adenosine as a neuromodulator in neurological diseases. Curr Opin Pharmacol 8:2–7
Boison D (2008b) The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol 84:249–262
Boison D (2013) Adenosine kinase:exploitation for therapeutic gain. Pharmacol Rev 65:906–943
Boison D (2016) Adenosinergic signaling in epilepsy. Neuropharmacology 104:131–139
Boison D, Chen JF, Fredholm BB (2010) Adenosine signaling and function in glial cells. Cell Death Differ 17:1071–1082
Bonan CD (2012) Ectonucleotidases and nucleotide/nucleoside transporters as pharmacological targets for neurological disorders. CNS Neurol Disord Drug Targets 11:739–750
Borea PA, Varani K, Vincenzi F et al (2015) The A3 adenosine receptor:history and perspectives. Pharmacol Rev 67:74–102
Bradesi S, Eutamene H, Theodorou V et al (2001) Effect of ovarian hormones on intestinal mast cell reactivity to substance P. Life Sci 68:1047–1056
Brundege JM, Dunwiddie TV (1998) Metabolic regulation of endogenous adenosine release from single neurons. Neuroreport 9:3007–3011
By Y, Condo J, Durand-Gorde JM et al (2011) Intracerebroventricular injection of an agonist-like monoclonal antibody to adenosine A(2A) receptor has antinociceptive effects in mice. J Neuroimmunol 230:178–182
Cao H, Zhang YQ (2008) Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev 32:972–983
Chen Z, Muscoli C, Doyle T et al (2010) NMDA-receptor activation and nitroxidative regulation of the glutamatergic pathway during nociceptive processing. Pain 149:100–106
Chen Z, Janes K, Chen C et al (2012) Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J 26:1855–1865
Chen JF, Eltzschig HK, Fredholm BB (2013) Adenosine receptors as drug targets--what are the challenges? Nat Rev 12:265–286
Choi JS, Berdis AJ (2012) Nucleoside transporters: biological insights and therapeutic applications. Future Med Chem 4:1461–1478
Choi IY, Lee JC, Ju C et al (2011) A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am J Pathol 179:2042–2052
Coull JA, Boudreau D, Bachand K et al (2003) Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424:938–942
Cross HR, Murphy E, Black RG et al (2002) Overexpression of A(3) adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am J Phys Heart Circ Phys 283:H1562–H1568
Cui JG, Sollevi A, Linderoth B et al (1997) Adenosine receptor activation suppresses tactile hypersensitivity and potentiates spinal cord stimulation in mononeuropathic rats. Neurosci Lett 223:173–176
Cunha RA (2001) Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system:different roles, different sources and different receptors. Neurochem Int 38:107–125
Cunha RA (2005) Neuroprotection by adenosine in the brain:from A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal 1:111–134
Cunha RA (2008) Different cellular sources and different roles of adenosine:A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int 52:65–72
Daniele S, Zappelli E, Natali L et al (2014) Modulation of A1 and A2B adenosine receptor activity:a new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis 5:e1539
Deussen A, Stappert M, Schafer S et al (1999) Quantification of extracellular and intracellular adenosine production: understanding the transmembranous concentration gradient. Circulation 99:2041–2047
Dias RB, Rombo DM, Ribeiro JA et al (2013) Adenosine: setting the stage for plasticity. Trends Neurosci 36:248–257
Dickenson AH, Suzuki R, Reeve AJ (2000) Adenosine as a potential analgesic target in inflammatory and neuropathic pains. CNS Drugs 13:77–85
Doyle T, Chen Z, Muscoli C et al (2012) Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci 32:6149–6160
Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55
Eaton MJ, Plunkett JA, Karmally S et al (1998) Changes in GAD- and GABA- immunoreactivity in the spinal dorsal horn after peripheral nerve injury and promotion of recovery by lumbar transplant of immortalized serotonergic precursors. J Chem Neuroanat 16:57–72
Elliott K, Minami N, Kolesnikov YA et al (1994) The NMDA receptor antagonists, LY274614 and MK-801, and the nitric oxide synthase inhibitor, NG-nitro-L-arginine, attenuate analgesic tolerance to the mu-opioid morphine but not to kappa opioids. Pain 56:69–75
Engler RL (1991) Adenosine. The signal of life? Circulation 84:951–954
Fedorova IM, Jacobson MA, Basile A et al (2003) Behavioral characterization of mice lacking the A3 adenosine receptor:sensitivity to hypoxic neurodegeneration. Cell Mol Neurobiol 23:431–447
Feoktistov I, Biaggioni I (2011) Role of adenosine A(2B) receptors in inflammation. Adv Pharmacol 61:115–144
Ferrini F, De Koninck Y (2013) Microglia control neuronal network excitability via BDNF signalling. Neural Plast 2013:429815
Feuerbach D, Lingenhoehl K, Olpe HR et al (2009) The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology 56:254–263
Fishman P, Bar-Yehuda S, Madi L et al (2002) A3 adenosine receptor as a target for cancer therapy. Anti-Cancer Drugs 13:437–443
Fishman P, Bar-Yehuda S, Madi L et al (2006) The PI3K-NF-kappaB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res Ther 8:R33
Fishman P, Bar-Yehuda S, Synowitz M et al (2009) Adenosine receptors and cancer. Handb Exp Pharmacol 193:399–441
Fishman P, Bar-Yehuda S, Liang BT et al (2012) Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov Today 17:359–366
Ford A, Castonguay A, Cottet M et al (2015) Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J Neurosci 35:6057–6067
Fredholm BB, AP IJ, Jacobson KA et al (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552
Fredholm BB, AP IJ, Jacobson KA et al (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol Rev 63:1–34
Gallo-Rodriguez C, Ji XD, Melman N et al (1994) Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem 37:636–646
Gan TJ, Habib AS (2007) Adenosine as a non-opioid analgesic in the perioperative setting. Anesth Analg 105:487–494
Gao ZG, Jacobson KA (2007) Emerging adenosine receptor agonists. Expert Opin Emerg Drugs 12:479–492
Gao ZG, Jacobson KA (2011) Emerging adenosine receptor agonists:an update. Expert Opin Emerg Drugs 16:597–602
Gao ZG, Teng B, Wu H et al (2009) Synthesis and pharmacological characterization of [(125)I]MRS1898, a high-affinity, selective radioligand for the rat A(3) adenosine receptor. Purinergic Signal 5:31–37
Gessi S, Merighi S, Varani K et al (2011) Adenosine receptors in health and disease. Adv Pharmacol 61:41–75
Giannaccini G, Betti L, Palego L et al (2008) Species comparison of adenosine receptor subtypes in brain and testis. Neurochem Res 33:852–860
Goldberg DS, McGee SJ (2011) Pain as a global public health priority. BMC Public Health 11:770
Gomes CV, Kaster MP, Tomé AR et al (2011) Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta Biomembr 1808:1380–1399
Gong QJ, Li YY, Xin WJ et al (2010) Differential effects of adenosine A1 receptor on pain-related behavior in normal and nerve-injured rats. Brain Res 1361:23–30
Grandoch M, Hoffmann J, Rock K et al (2013) Novel effects of adenosine receptors on pericellular hyaluronan matrix: implications for human smooth muscle cell phenotype and interactions with monocytes during atherosclerosis. Basic Res Cardiol 108:340
Habib AS, Minkowitz H, Osborn T et al (2008) Phase 2, double-blind, placebo-controlled, dose-response trial of intravenous adenosine for perioperative analgesia. Anesthesiology 109:1085–1091
Haeusler D, Grassinger L, Fuchshuber F et al (2015) Hide and seek:a comparative autoradiographic in vitro investigation of the adenosine A3 receptor. Eur J Nucl Med Mol Imaging 42:928–939
Harrison GJ, Cerniway RJ, Peart J et al (2002) Effects of A(3) adenosine receptor activation and gene knock-out in ischemic-reperfused mouse heart. Cardiovasc Res 53:147–155
Hashizume H, DeLeo JA, Colburn RW et al (2000) Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine (Phila Pa 1976) 25:1206–1217
Hasko G, Szabo C, Nemeth ZH et al (1996) Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 2647 macrophages and in endotoxemic mice. J Immunol 157:4634–4640
Hasko G, Nemeth ZH, Vizi ES et al (1998) An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur J Pharmacol 358:261–268
Hayashida M, Fukuda K, Fukunaga A (2005) Clinical application of adenosine and ATP for pain control. J Anesth 19:225–235
Headrick JP, Peart J (2005) A3 adenosine receptor-mediated protection of the ischemic heart. Vasc Pharmacol 42:271–279
Headrick JP, Peart JN, Reichelt ME et al (2011) Adenosine and its receptors in the heart:regulation, retaliation and adaptation. Biochim Biophys Acta 1808:1413–1428
Hinze AV, Mayer P, Harst A et al (2012) Adenosine A(3) receptor-induced proliferation of primary human coronary smooth muscle cells involving the induction of early growth response genes. J Mol Cell Cardiol 53:639–645
Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education (2011) Relieving pain in America: a blueprint for transforming prevention, care, education, and research. National Academies Press, Washington, DC
Jacobson KA (1998) Adenosine A3 receptors:novel ligands and paradoxical effects. Trends Pharmacol Sci 19:184–191
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5:247–264
Jacobson KA, Nikodijevic O, Shi D et al (1993) A role for central A3-adenosine receptors. Mediation of behavioral depressant effects. FEBS Lett 336:57–60
Jajoo S, Mukherjea D, Watabe K et al (2009) Adenosine A(3) receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia 11:1132–1145
Janes K, Doyle T, Bryant L et al (2013) Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain 154:2432–2440
Janes K, Esposito E, Doyle T et al (2014a) A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain 155:2560–2567
Janes K, Little JW, Li C et al (2014b) The development and maintenance of paclitaxel-induced neuropathic pain require activation of the sphingosine 1-phosphate receptor subtype 1. J Biol Chem 289:21082–21097
Janes K, Wahlman C, Little JW et al (2015) Spinal neuroimmmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun 44:91–99
Johnston JB, Silva C, Gonzalez G et al (2001) Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol 49:650–658
Katz NK, Ryals JM, Wright DE (2015) Central or peripheral delivery of an adenosine A1 receptor agonist improves mechanical allodynia in a mouse model of painful diabetic neuropathy. Neuroscience 285:312–323
Keil GJ 2nd, DeLander GE (1992) Spinally-mediated antinociception is induced in mice by an adenosine kinase-, but not by an adenosine deaminase-, inhibitor. Life Sci 51:PL171–PL176
Kiesman WF, Elzein E, Zablocki J (2009) A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handb Exp Pharmacol 193:25–58
Klaasse EC, Ijzerman AP, de Grip WJ et al (2008) Internalization and desensitization of adenosine receptors. Purinergic Signal 4:21–37
Kowaluk EA, Kohlhaas KL, Bannon A et al (1999) Characterization of the effects of adenosine kinase inhibitors on acute thermal nociception in mice. Pharmacol Biochem Behav 63:83–91
Kowaluk EA, Mikusa J, Wismer CT et al (2000) ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin- 3-yl)pyrido[2,3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties. II. In vivo characterization in the rat. J Pharmacol Exp Ther 295:1165–1174
Latini S, Pedata F (2001) Adenosine in the central nervous system:release mechanisms and extracellular concentrations. J Neurochem 79:463–484
Ledent C, Vaugeois JM, Schiffmann SN et al (1997) Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 388:674–678
Lee HC, Fellenz-Maloney MP, Liscovitch M et al (1993) Phospholipase D-catalyzed hydrolysis of phosphatidylcholine provides the choline precursor for acetylcholine synthesis in a human neuronal cell line. Proc Natl Acad Sci U S A 90:10086–10090
Lee JE, Bokoch G, Liang BT (2001) A novel cardioprotective role of RhoA: new signaling mechanism for adenosine. FASEB J 15:1886–1894
Li Y, Zhang H, Kosturakis AK et al (2014) Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain 15:712–725
Little JW, Ford A, Symons-Liguori AM et al (2015) Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138:28–35
Lopes LV, Rebola N, Pinheiro PC et al (2003) Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport 14:1645–1648
Loram LC, Harrison JA, Sloane EM et al (2009) Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists:a novel therapy for neuropathic pain. J Neurosci 29:14015–14025
Luongo L, Guida F, Imperatore R et al (2014) The A1 adenosine receptor as a new player in microglia physiology. Glia 62:122–132
Madi L, Bar-Yehuda S, Barer F et al (2003) A3 adenosine receptor activation in melanoma cells:association between receptor fate and tumor growth inhibition. J Biol Chem 278:42121–42130
Madi L, Cohen S, Ochayin A et al (2007) Overexpression of A3 adenosine receptor in peripheral blood mononuclear cells in rheumatoid arthritis:involvement of nuclear factor-kappaB in mediating receptor level. J Rheumatol 34:20–26
Mao J, Sung B, Ji RR et al (2002) Chronic morphine induces downregulation of spinal glutamate transporters:implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22:8312–8323
Martins DF, Mazzardo-Martins L, Soldi F et al (2013) High-intensity swimming exercise reduces neuropathic pain in an animal model of complex regional pain syndrome type I:evidence for a role of the adenosinergic system. Neuroscience 234:69–76
Mayer DJ, Mao J, Holt J et al (1999) Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A 96:7731–7736
McGaraughty S, Cowart M, Jarvis MF et al (2005) Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem 5:43–58
Meller ST, Dykstra C, Grzybycki D et al (1994) The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 33:1471–1478
Merighi S, Bencivenni S, Vincenzi F et al (2017) A2B adenosine receptors stimulate IL-6 production in primary murine microglia through p38 MAPK kinase pathway. Pharmacol Res 117:9–19
Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36
Moore KA, Kohno T, Karchewski LA et al (2002) Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22:6724–6731
Morello S, Ito K, Yamamura S et al (2006) IL-1 beta and TNF-alpha regulation of the adenosine receptor (A2A) expression:differential requirement for NF-kappa B binding to the proximal promoter. J Immunol 177:7173–7183
Moser GH, Schrader J, Deussen A (1989) Turnover of adenosine in plasma of human and dog blood. Am J Phys 256:C799–C806
Muscoli C, Cuzzocrea S, Ndengele MM et al (2007) Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest 117:3530–3539
Muscoli C, Doyle T, Dagostino C et al (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neurosci 30:15400–15408
Nagata K, Imai T, Yamashita T et al (2009) Antidepressants inhibit P2X4 receptor function: a possible involvement in neuropathic pain relief. Mol Pain 5:20
Ndengele MM, Cuzzocrea S, Esposito E et al (2008) Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J 22:3154–3164
Obata K, Noguchi K (2008) Contribution of primary sensory neurons and spinal glial cells to pathomechanisms of neuropathic pain. Brain Nerve 60:483–492
Ochaion A, Bar-Yehuda S, Cohen S et al (2009) The anti-inflammatory target A(3) adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn's disease. Cell Immunol 258:115–122
Otsuguro KI, Tomonari Y, Otsuka S et al (2015) An adenosine kinase inhibitor, ABT-702, inhibits spinal nociceptive transmission by adenosine release via equilibrative nucleoside transporters in rat. Neuropharmacology 97:160–170
Paoletta S, Tosh DK, Finley A et al (2013) Rational design of sulfonated A3 adenosine receptor-selective nucleosides as pharmacological tools to study chronic neuropathic pain. J Med Chem 56:5949–5963
Parsons M, Young L, Lee JE et al (2000) Distinct cardioprotective effects of adenosine mediated by differential coupling of receptor subtypes to phospholipases C and D. FASEB J 14:1423–1431
Peng L, Huang R, Yu AC et al (2005) Nucleoside transporter expression and function in cultured mouse astrocytes. Glia 52:25–35
Petrelli R, Scortichini M, Kachler S et al (2017) Exploring the role of N(6)-substituents in potent dual acting 5′-C-Ethyltetrazolyladenosine derivatives:synthesis, binding, functional assays, and antinociceptive effects in mice nabla. J Med Chem 60:4327–4341
Pizzo PA, Clark NM (2012) Alleviating suffering 101--pain relief in the United States. N Engl J Med 366:197–199
Poderoso JJ, Carreras MC, Lisdero C et al (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92
Poon A, Sawynok J (1998) Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain 74:235–245
Poulsen SA, Quinn RJ (1998) Adenosine receptors:new opportunities for future drugs. Bioorg Med Chem 6:619–641
Price TJ, Cervero F, de Koninck Y (2005) Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr Top Med Chem 5:547–555
Prus AJ, James JR, Rosecrans JA (2009) Conditioned place preference. In: Buccafusco JJ (ed) Methods of behavior analysis in neuroscience, 2nd edn. CRC Press/Taylor Francis, Boca Raton
Rausaria S, Ghaffari MM, Kamadulski A et al (2011) Retooling manganese(III) porphyrin-based peroxynitrite decomposition catalysts for selectivity and oral activity:a potential new strategy for treating chronic pain. J Med Chem 54:8658–8669
Rebola N, Canas PM, Oliveira CR et al (2005) Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 132:893–903
Robson SC, Sevigny J, Zimmermann H (2006) The E-NTPDase family of ectonucleotidases:structure function relationships and pathophysiological significance. Purinergic Signal 2:409–430
Romagnoli R, Baraldi PG, Tabrizi MA et al (2010) Allosteric enhancers of A1 adenosine receptors:state of the art and new horizons for drug development. Curr Med Chem 17:3488–3502
Ru F, Surdenikova L, Brozmanova M et al (2011) Adenosine-induced activation of esophageal nociceptors. Am J Physiol Gastrointest Liver Physiol 300:G485–G493
Sajjadi FG, Takabayashi K, Foster AC et al (1996) Inhibition of TNF-alpha expression by adenosine:role of A3 adenosine receptors. J Immunol 156:3435–3442
Salvatore CA, Tilley SL, Latour AM et al (2000) Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem 275:4429–4434
Salvemini D, Neumann W (2010) Targeting peroxynitrite driven nitroxidative stress with synzymes:a novel therapeutic approach in chronic pain management. Life Sci 86:604–614
Sawynok J (1998) Adenosine receptor activation and nociception. Eur J Pharmacol 347:1–11
Sawynok J (2013) Adenosine and pain. In: Boison D, Masino SA (eds) Adenosine: a key link between metabolism and brain activity. Springer, Berlin, pp 343–360
Sawynok J (2016) Adenosine receptor targets for pain. Neuroscience 338:1–18
Sawynok J, Zarrindast MR, Reid AR et al (1997) Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5-hydroxytryptamine. Eur J Pharmacol 333:1–7
Sawynok J, Reid A, Liu XJ (1999) Acute paw oedema induced by local injection of adenosine A(1), A(2) and A(3) receptor agonists. Eur J Pharmacol 386:253–261
Sebastian-Serrano A, de Diego-Garcia L, Martinez-Frailes C et al (2015) Tissue-nonspecific alkaline phosphatase regulates purinergic transmission in the central nervous system during development and disease. Comput Struct Biotechnol J 13:95–100
Sebastiao AM, Ribeiro JA (1996) Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog Neurobiol 48:167–189
Shneyvays V, Nawrath H, Jacobson KA et al (1998) Induction of apoptosis in cardiac myocytes by an A3 adenosine receptor agonist. Exp Cell Res 243:383–397
Shneyvays V, Mamedova L, Zinman T et al (2001) Activation of A(3) adenosine receptor protects against doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol 33:1249–1261
Sjolund KF, von Heijne M, Hao JX et al (1998) Intrathecal administration of the adenosine A1 receptor agonist R-phenylisopropyl adenosine reduces presumed pain behaviour in a rat model of central pain. Neurosci Lett 243:89–92
Smith PA (2014) BDNF:no gain without pain? Neuroscience 283C:107–123
Sowa NA, Street SE, Vihko P et al (2010) Prostatic acid phosphatase reduces thermal sensitivity and chronic pain sensitization by depleting phosphatidylinositol 4,5-bisphosphate. J Neurosci 30:10282–10293
Spychala J, Datta NS, Takabayashi K et al (1996) Cloning of human adenosine kinase cDNA:sequence similarity to microbial ribokinases and fructokinases. Proc Natl Acad Sci U S A 93:1232–1237
Stiller CO, Cui JG, O’Connor WT et al (1996) Release of gamma-aminobutyric acid in the dorsal horn and suppression of tactile allodynia by spinal cord stimulation in mononeuropathic rats. Neurosurgery 39:367–374
Studer FE, Fedele DE, Marowsky A et al (2006) Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 142:125–137
Svenningsson P, Hall H, Sedvall G et al (1997) Distribution of adenosine receptors in the postmortem human brain:an extended autoradiographic study. Synapse 27:322–335
Sweitzer SM, Schubert P, DeLeo JA (2001) Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther 297:1210–1217
Szabo C, Scott GS, Virag L et al (1998) Suppression of macrophage inflammatory protein (MIP)-1alpha production and collagen-induced arthritis by adenosine receptor agonists. Br J Pharmacol 125:379–387
Taiwo YO, Levine JD (1990) Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 38:757–762
Thourani VH, Nakamura M, Ronson RS et al (1999a) Adenosine A(3)-receptor stimulation attenuates postischemic dysfunction through K(ATP) channels. Am J Phys 277:H228–H235
Thourani VH, Ronson RS, Jordan JE et al (1999b) Adenosine A3 pretreatment before cardioplegic arrest attenuates postischemic cardiac dysfunction. Ann Thorac Surg 67:1732–1737
Tosh DK, Deflorian F, Phan K et al (2012) Structure-guided design of A(3) adenosine receptor-selective nucleosides:combination of 2-arylethynyl and bicyclo[ 3 10]hexane substitutions. J Med Chem 55:4847–4860
Tosh DK, Finley A, Paoletta S et al (2014) In vivo phenotypic screening for treating chronic neuropathic pain:modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J Med Chem 57:9901–9914
Tosh DK, Paoletta S, Chen Z et al (2015) Structure-based design, synthesis by click chemistry and in vivo activity of highly selective A3 adenosine receptor agonists. Med Chem Commun 6:555–563
Tracey WR, Magee W, Masamune H et al (1997) Selective adenosine A3 receptor stimulation reduces ischemic myocardial injury in the rabbit heart. Cardiovasc Res 33:410–415
Vallon V, Osswald H (2009) Adenosine receptors and the kidney. Handb Exp Pharmacol 193:443–470
Varani K, Vincenzi F, Tosi A et al (2010) Expression and functional role of adenosine receptors in regulating inflammatory responses in human synoviocytes. Br J Pharmacol 160:101–115
Varani K, Padovan M, Vincenzi F et al (2011) A2A and A3 adenosine receptor expression in rheumatoid arthritis:upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther 13:R197
Varani K, Vincenzi F, Targa M et al (2013) The stimulation of A(3) adenosine receptors reduces bone-residing breast cancer in a rat preclinical model. Eur J Cancer 49:482–491
Varani K, Vincenzi F, Merighi S et al (2017) Biochemical and pharmacological role of A1 adenosine receptors and their modulation as novel therapeutic strategy. Adv Exp Med Biol 1051:193–232
Vincenzi F, Targa M, Romagnoli R et al (2014) TRR469, a potent A(1) adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 81:6–14
Wahlman C, Doyle TM, Little JW et al (2018) Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels via astrocyte-dependent mechanisms. Pain, in press. https://doi.org/10.1097/j.pain.0000000000001177
Watkins LR, Martin D, Ulrich P et al (1997) Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain 71:225–235
Watkins LR, Milligan ED, Maier SF (2001) Glial activation: a driving force for pathological pain. Trends Neurosci 24:450–455
Watkins LR, Hutchinson MR, Rice KC et al (2009) The “toll” of opioid-induced glial activation:improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30:581–591
Wei CJ, Li W, Chen JF (2011) Normal and abnormal functions of adenosine receptors in the central nervous system revealed by genetic knockout studies. Biochim Biophys Acta 1808:1358–1379
Wittendorp MC, Boddeke HW, Biber K (2004) Adenosine A3 receptor-induced CCL2 synthesis in cultured mouse astrocytes. Glia 46:410–418
Wu WP, Hao JX, Halldner-Henriksson L et al (2002) Decreased inflammatory pain due to reduced carrageenan-induced inflammation in mice lacking adenosine A3 receptors. Neuroscience 114:523–527
Wu WP, Hao JX, Halldner L et al (2005) Increased nociceptive response in mice lacking the adenosine A1 receptor. Pain 113:395–404
Xu X, Wang P, Zou X et al (2010) The effects of sympathetic outflow on upregulation of vanilloid receptors TRPV(1) in primary afferent neurons evoked by intradermal capsaicin. Exp Neurol 222:93–107
Yamaoka G, Horiuchi H, Morino T et al (2013) Different analgesic effects of adenosine between postoperative and neuropathic pain. J Orthop Sci 18:130–136
Yeo JF, Ling SF, Tang N et al (2008) Antinociceptive effect of CNS peroxynitrite scavenger in a mouse model of orofacial pain. Exp Brain Res 184:435–438
Yoon MH, Choi JI, Park HC et al (2004) Interaction between intrathecal gabapentin and adenosine in the formalin test of rats. J Korean Med Sci 19:581–585
Yoon MH, Bae HB, Choi JI (2005) Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth Analg 101:1417–1421
Yoon MH, Bae HB, Choi JI et al (2006) Roles of adenosine receptor subtypes in the antinociceptive effect of intrathecal adenosine in a rat formalin test. Pharmacology 78:21–26
Zahn PK, Straub H, Wenk M et al (2007) Adenosine A1 but not A2a receptor agonist reduces hyperalgesia caused by a surgical incision in rats:a pertussis toxin-sensitive G protein-dependent process. Anesthesiology 107:797–806
Zeilhofer HU, Wildner H, Yevenes GE (2012) Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev 92:193–235
Zhang G, Franklin PH, Murray TF (1993) Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J Pharmacol Exp Ther 264:1415–1424
Zhang X, Zhang M, Laties AM et al (2006) Balance of purines may determine life or death of retinal ganglion cells as A3 adenosine receptors prevent loss following P2X7 receptor stimulation. J Neurochem 98:566–575
Zhang M, Hu H, Zhang X et al (2010) The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem Int 56:35–41
Zhang H, Yoon SY, Dougherty PM (2012) Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. J Pain 13:293–303
Zhang Y, Chen K, Sloan SA et al (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947
Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedeberg's Arch Pharmacol 362:299–309
Zylka MJ (2011) Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol Med 17:188–196
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Salvemini, D., Doyle, T.M., Largent-Milnes, T.M., Vanderah, T.W. (2018). The Adenosine-Receptor Axis in Chronic Pain. In: Borea, P., Varani, K., Gessi, S., Merighi, S., Vincenzi, F. (eds) The Adenosine Receptors. The Receptors, vol 34. Humana Press, Cham. https://doi.org/10.1007/978-3-319-90808-3_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-90808-3_16
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-90807-6
Online ISBN: 978-3-319-90808-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)