
Abstract
Neuroinflammation is a common pathological feature in almost all neurological diseases and is a response triggered as a consequence of the chronic activation of the innate immune response in the CNS against a variety of stimuli, including infection, traumatic brain injury, toxic metabolites, aggregated proteins, or autoimmunity. Crucial mediators of this neurinflammatory process are the intracellular protein complexes known as inflammasomes which can be triggered by pathogens as well as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). However, chronic inflammasome activation can eventually result in cellular death and tissue damage, leading to the release of DAMPs that can reactivate the inflammasome, thereby propagating a vicious cycle of inflammation. The primary cells involved in CNS inflammasome activation are the immunocompetent microglia and the infiltrating macrophages into the CNS. However, astrocytes and neurons also express inflammasomes, and the understanding of how they are engaged in the pathogenesis of a variety of neurological diseases is crucial to develop effective therapeutic approaches for CNS pathologies that are propagated by chronic inflammasome activation. This chapter covers the activation mechanisms of relevant inflammasomes in the brain and summarizes their roles in the pathogenesis and progression of different neurological conditions.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Introduction
Inflammation of the central nervous system (CNS) or neuroinflammation is a common underlying pathological feature of most neurological disorders. Chronic neuroinflammation is evident in progressive neurodegenerative diseases like Alzheimer’s and Parkinson’s disease (Heneka et al. 2014) and in autoimmune disorders such as multiple sclerosis (Barclay and Shinohara 2017). Neuroinflammation is also present in psychiatric illnesses such as depression (Alcocer-Gomez et al. 2014) and a consequence of direct damage to the CNS in the form of brain injuries such as stroke (Barrington et al. 2017) or traumatic brain injury (Wallisch et al. 2017). Innate immune activation is also evident as an acute response against viral or bacterial infections in the CNS (Klein et al. 2017). The neuroinflammatory process is a response to a variety of pathogenic signals, and its primary role is to maintain homeostasis in the brain conferring neuroprotection and promoting remyelination and even axonal regeneration (Wee Yong 2010). Nevertheless, the delicate balance between the benefit and harm that is conferred by activation of the innate immune system is directly related to length, reactivation, and spread of the inflammatory response. This mechanism of defense in the innate immune response occurs through pattern recognition receptors (PRRs) that can recognize infections through pathogen-specific proteins (PAMPs) and internal cellular disturbances through damage-associated proteins (DAMPs) (Kigerl et al. 2014). Primarily expressed in the CNS by glial cells (microglia and astrocytes) and also in neurons, these receptors can be located on extracellular membranes or within the cytosol such as the NOD-like receptors (NLRs). NLR activation leads to the oligomerization of cytosolic protein complex known as inflammasomes that regulate the activation of caspase-1 (Martinon et al. 2002). This active caspase cleaves the precursor forms of the pro-inflammatory cytokines of the IL-1 family, IL-1β, IL-18, and IL-33, into their active forms that exert their biological effects. The expression and release of the IL-1 family cytokines in the normal brain is normally at low levels and is dramatically upregulated in response to local or systemic disease and injury (Rothwell and Luheshi 2000). In the CNS, multiple cell types express receptors for IL-1β and IL-18 making them particularly sensitive to these cytokines. (Allan et al. 2005; Alboni et al. 2010). The primary role of the inflammasome activation in the healthy brain is to limit and clear any pathogenic or metaboloic damage, but when this activation becomes persistent, it entails other associated process that can propagate and spread the inflammatory response. These include pyroptotic cell death and the release of ASC specks, NLRP3, and DAMPs that can propagate the inflammatory response leading to tissue damage (Franklin et al. 2014; Baroja-Mazo et al. 2014).
Recent advances in our understanding of inflammasome triggers and activation mechanisms in the CNS, as well as its regulation by endogenous inhibitors such as dopamine and the ketone body β-hydroxybutyrate (Youm et al. 2015) could have therapeutic applications for CNS diseases. Indeed, the development of highly specific NLRP3 inhibitors such as MCC950 (Coll et al. 2015) and caspase-1/ICE inhibitors have opened up an exciting new area of translational research with the potential for disease modification and therapeutic targeting of progressive neurodegenerative diseases (Lipinska et al. 2014; Boxer et al. 2010; Ross et al. 2007).
3.2 Inflammasomes in the Healthy Brain
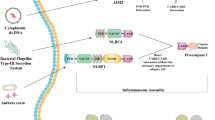
Until recently, the CNS was considered an immune-privileged site, protected by the blood-brain barrier (BBB), creating an isolated environment between the CNS and the peripheral immune system (Carson et al. 2006). However, the recent paradigm shifts in our understanding has revealed the far-reaching scope and intricate communication between the innate immune response in the CNS and the peripheral immune system in maintaining brain homeostasis (Louveau et al. 2015a, b). The immunocompetent microglia are primarily responsible for sensing a broad range of exogenous pathogenic stimuli, such as fungal, bacterial, and viral components, or endogenous such as aggregated and misfolded proteins, extracellular ATP, and reactive oxygen species (ROS) among others (Walsh et al. 2014). The microglia express and can activate NLRC4 and NLRP3 inflammasomes, the best characterized of which is NLRP3, considered in the recent years as a key pathway in the development of neuroinflammation and neurodegeneration (Song et al. 2017). NLRP3 activation requires two signals: the first is a priming step (such as TLR4 activation) which drives the transcription and translation of inflammasome components including IL-1β, caspase-1, NLRP3, and ASC. A secondary signal or activation step (i.e., pathogen infiltration or aggregated proteins) is then required to trigger the formation of the inflammasome complex (NRLP3-ASC-caspase-1) leading to cluster-dependent caspase-1 activation and the cleavage and release of IL1-β (Guo et al. 2015). There is believed to be an association of this pro-inflammatory phenotype or M1-like microglia with the expression of IL-1β, IL-6, IL-18, tumor necrosis factor-α (TNFα), the production of superoxide, reactive oxygen species (ROS), and nitric oxide (NO), and an impaired phagocytic capacity (Tang and Le 2016). The microglia also expresses NLRC4 a key sensor for bacterial infection, sensing bacterial flagellin related with the cause of meningitis in the CNS (Wu et al. 2010). Unlike NLRP3, NLRC4 can be activated without the ASC adapter protein (Latz et al. 2013), and bacterial activation can lead to a dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex (Man et al. 2014). In contrast, in the healthy brain, the microglia interact with the neighboring neurons, remodeling synapses and secreting soluble neurotrophic factors, such as brain-derived neurotrophic factor (BDNF) and transforming growing factor-β (TGF-β). Microglial cells in the healthy brain are involved in regulation of synaptic strength, neuronal pruning, protease secretion to maintain extracellular matrices, and building and maintaining proper neuronal network functions and phagocytic activity to remove accumulating cell debris and misfolded proteins to maintain brain homeostasis (Fig. 3.1) (Waisman et al. 2015). This neuroprotective phenotype or M2-like is associated with the production of anti-inflammatory cytokines such as IL-4, IL-13, and IL-10 (Kabba et al. 2018). The balance between microglial phenotypes (M1/M2) depends on the disease stage and severity, and the type of response may determine whether microglial activation can be beneficial or leads to chronic neuroinflammation (Fig. 3.1) (Tang and Le 2016). The astrocytes also play a significant role in the brain homeostasis, including regulating neurotransmitter and growth factor release, forming the BBB, and regulating the immune response (Dong and Benveniste 2001). The role of astrocytes in innate immunity is crucial; these cells express an array of receptors including Toll-like receptors (TLR), double-stranded RNA-dependent protein kinase, scavenger receptors, components of the complement system, and NOD-like receptors like NLRP3 (Farina et al. 2007; Johann et al. 2015). Recently, it was shown that human astrocytes also express NLRP2 inflammasome (Minkiewicz et al. 2013). ATP can activate the inflammasome forming a multiprotein complex with ASC and caspase-1 and also interacts with the P2X7 receptor and pannexin 1 channel, leading to the release of IL-1β (Minkiewicz et al. 2013). Furthermore, increasing evidence has shown the expression of inflammasome-forming NLRs in non-myeloid cells such as neurons; the most characterized and implicated in some pathologies is NRLP1 (Kaushal et al. 2015). Human NLRP1 is unique compared to the rest of the inflammasomes due to the complexity of its domain structure, possessing two interaction domains, an N-terminal PYD and a C-terminal CARD, and interestingly the adaptor protein ASC is not necessary for activating the pathway, but it might enhance NLRP1 activation (Faustin et al. 2007). Furthermore, cerebral pericytes express inflammasomes, and Toll-like receptors, which are involved in controlling key neurovascular functions and BBB permeability and peripheral leukocyte trafficking (Nyul-Toth et al. 2017). Emerging evidence suggests that inflammasomes could have previously unknown roles in normal brain function beyond neuronal protection and maintaining homeostasis. It can therefore be expected that new roles for the various inflammasome components will emerge as these pathways are studied in more detail in the context of the central nervous system.

Overview of inflammasome activation in neurological diseases. Microglial inflammasomes can be chronically activated in progressive neurodegenerative diseases such as Alzheimer and Parkinson’s disease where they drive neuropathology (1). Inflammasome activation has also been documented in autoimmune diseases such as Multiple Sclerosis where it contributes to T cell dysregulation and nerve damage (2). Emerging evidence also indicates a pathogenic role for inflammasome activation in Traumatic Brain Injury (TBI) and stroke (3), as well as CNS infection by bacteria and viruses (4). The pathological contributions of systemic versus CNS inflammasome activation remains to be determined in these diseases and is an active area of research
3.3 Inflammasomes in Neurodegenerative Diseases
There is extensive accumulating evidence for the pathogenic role of chronic inflammasome activation in propagating neuroinflammation, a key underlying feature of neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease (Heneka 2017; Mao et al. 2017). Despite the different factors and mediators that can trigger inflammasome activation in this sterile environment, aging is a common factor in most progressive neurodegenerative diseases (Wyss-Coray 2016). Recent studies correlate microglia and enhanced sensitivity to inflammatory stimuli with aging, demonstrating in senescence-accelerated mice a “primed microglial” phenotype, characterized by increased production of pro-inflammatory cytokines and ROS, leading to elevated basal inflammasome activation that can influence neurodegeneration in more elderly populations (Luo et al. 2010; Spittau 2017). Furthermore, it has recently been demonstrated that the expression of specific inflammasome gene modules can accurately classify older populations in two groups, one with constitutive expression of IL-1β and the other without this expression, showing a correlation in the first group with an increased mortality and related inflammatory disease state, a common hallmark of neurodegenerative diseases (Furman et al. 2017). We describe below, the role of the inflammasome and their therapeutic approaches in these two most common neurodegenerative diseases.
3.3.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is the most prevalent chronic and progressive neurodegenerative disease. The first symptoms are short-term memory loss and worsen over time leading to progressive cognitive impairment and dementia in 70% of the cases (Tarawneh and Holtzman 2012). Currently, there are no treatments to stop the progression of the disease, only to ameliorate some symptoms (Yiannopoulou and Papageorgiou 2013). The primary hallmark of the development of AD is the deposition of amyloid β (Aβ) aggregates in the hippocampus (Friedrich et al. 2010), correlated with a chronic activation of the innate immune system and microglial activation (Doens and Fernandez 2014). The microglia can bind soluble Aβ oligomers and fibrils via cell-surface receptors including CD14, CD36, CD47, and TLRs among others (Bamberger et al. 2003). In AD, NLRP3 inflammasome activation proceeds via phagosomal disruption or cell surface K+ channels (Salminen et al. 2009). Increasing evidence from AD mouse models such as APP/PS1 transgenic model and clinical studies have shown the relationship between innate immune mechanisms and neurodegenerative process involving the persistent activation of NLRP3 in microglia and peripheral macrophages and NLRP1 inflammasome in neurons (Saresella et al. 2016). NLRP3 activation has been shown to enhance AD pathology and may be involved in synaptic dysfunction, cognitive impairments, and the restriction of microglial clearance functions (Heneka et al. 2013). Aβ fibrils can activate NLRP3 in mouse microglia, and higher levels of active caspase-1 and IL-1β in the brains of patients with AD and APP/PS1 mice have been detected compared to healthy patients and wild-type (WT) control mice. Furthermore, there is a protective phenotype in caspase-1 or NLRP3 knockout (KO) in terms of spatial memory impairments and loss of hippocampal neurons, associated with behavioral disturbances present in AD, showing a neuroprotective M2-like phenotype (Heneka et al. 2013; Heneka 2017). In neurons NLRP1 levels are upregulated in APP/PS1 mice, and in vitro results have shown that silencing NLRP1 reduces Aβ-induced neuronal pyroptotic cell death, positioning NLRP1 activation in neurons as a new factor relevant to neurodegeneration in AD (Tan et al. 2014; Kaushal et al. 2015). Recently the NLRC4 inflammasome was shown to be upregulated in AD brains; it has been shown that NLRC4 can be activated in response to fatty acid palmitate in astrocytes (Kaushal et al. 2015); this is correlated with a higher fatty acid content in AD brains compared with healthy brains (Cutler et al. 2004). Based on these results, introducing pharmacologic treatments targeting the NLRP3 inflammasome at different levels in the pathway may have beneficial effects in patients with AD (White et al. 2017). The first successful study targeting NLRP3 pharmacologically in a transgenic mouse model of AD was recently published (Daniels et al. 2016). This study has shown that several clinically approved and widely used nonsteroidal anti-inflammatory drugs (NSAIDs) of the fenamate class are efficient and selective inhibitors of the NLRP3 inflammasome via reversible blockade of volume-regulated anion channel (VRAC) in the plasma membrane of macrophages. Inhibiting cognitive impairments in 3 × TgAD transgenic mice (Daniels et al. 2016), this treatment completely abated AD-related neuroinflammation with reduced levels of microglial activation and IL-1β expression compared to WT mice, opening an exciting new translational field to repurposition these drugs as Alzheimer’s disease therapeutics (Venegas et al. 2017). A potentially key paradigm shift in our understanding of inflammasome pathology in driving AD is that microglial ASC specks can cross-seed amyloid-β during the course of the disease and thereby contribute to the spread of amyloid-β pathology.
3.3.2 Parkinson’s Disease
Parkinson’s disease (PD) is the most prevalent synucleinopathy and the second most common neurodegenerative disorder worldwide after AD (Mhyre et al. 2012). PD is a chronic neurodegenerative disease of the CNS, and its pathological hallmark is a profound loss of nigrostriatal dopaminergic neurons that is preceded by the accumulation and spread of characteristic Lewy bodies, consisting primarily of misfolded fibrillar α-synuclein (Syn) (Obeso et al. 2010). There is a correlation between lack of dopamine in the CNS with debilitating motor symptoms including tremor, rigidity, and slowness of the movements and in advanced-stage dementia (Lotharius and Brundin 2002). The current treatments target the symptoms but similar to AD do not stop the progression of the disease due to the lack of treatments to prevent chronic neuroinflammation that leads to neurodegeneration (Schapira et al. 2006). In recent years, accumulating evidence suggests that the innate immune system specifically NLRP3 is involved in the prominent neuroinflammatory response observed in PD (Codolo et al. 2013; Walsh et al. 2014; Guo et al. 2015; Mao et al. 2017). It has been shown that Syn fibrils can activate NLRP3 inflammasome in macrophages and microglia, acting as an endogenous trigger of the inflammasome in PD (Codolo et al. 2013; Guo et al. 2015; Gustot et al. 2015). The mechanism of Syn activation of NLRP3 is not fully elucidated, but recent evidence suggests that it could be through microglial endocytosis and subsequent lysosomal cathepsin B release (Zhou et al. 2016) and deficiency of caspase-1, significantly inhibited Syn-induced microglia activation, and IL-1β production in vitro (Zhou et al. 2016). Clinical studies have found higher levels of IL-1β and an upregulation of NLRP3 in PD patients (Zhou et al. 2016; Zhang et al. 2016). Additionally, it has been demonstrated that dopamine can inhibit systemic NLRP3 activation through dopamine D1 receptor (DDR1) via a second messenger cyclic adenosine monophosphate (cAMP), which binds to NLRP3 and promote its ubiquitination and degradation via the E3 ubiquitin ligase MARCH7 (Yan et al. 2015), suggesting an important novel endogenous regulatory role for dopamine that correlates with an increased NLRP3 activation in PD patients who inevitably have reduced levels of dopamine over the course of the disease. Although the etiology of PD is unknown, pesticide exposure is well recognized as an environmental risk factor to acquire this disease (Hancock et al. 2008). In fact chronic infusion of the pesticide, rotenone, has been used as a rodent model of PD, since this model reproduces many relevant features of the disease (Panov et al. 2005). In this model it has also been demonstrated that microglial NLRP3 inflammasome can be activated by rotenone exposure (Liang et al. 2015). Microglial NLRP3 can be activated with rotenone via the ROS/c-abl/NLRP3 signaling pathway, affecting the auto-lysosomal system. In this study it was demonstrated that targeting oxidative stress-induced c-Abl activation in the microglia can diminish microglial activation associated with neurodegeneration in PD (Lawana et al. 2017). The effect of rotenone also was observed in neurons in an animal model showing that pathological pesticide exposure could activate neuronal inflammasome in the substantia nigra and promote the expression of NLRP3, ASC, and caspase-1 and the secretion of IL-1β and IL-18 in a dose- and time-dependent manner (Zhang et al. 2016). Moreover, inflammasome activation components were detected in cerebrospinal fluid of PD patients, showing that cyclin-dependent kinase 5 (Cdk5) is necessary for NLRP3 activation in neurons and its pharmacological inhibition with roscovitine a Cdk5 inhibitor or Cdk5-targeted deletion could efficiently block neuronal inflammasome activation in 1,2,3,6-methyl-phenyl-tetrahydropyridine (MPTP) and Syn transgenic mouse PD models (Zhang et al. 2016). Besides, a central component of the inflammasome, caspase-1, causes truncation and aggregation of Syn, and this was recently demonstrated in a neuronal cell model of PD, dmeonstrating another potentially pathogenic role of the NLRP3 inflammasome in PD etiology (Wang et al. 2016). NLRP3 upregulation also was observed in a rat model of PD, the 6-hydroxydopamine (6-OHDA) model, showing high levels of mRNA and protein expression of NLRP3 components in the 6-OHDA injected side. In this study, microinjections of different doses of caspase-1 inhibitor (Ac-YVAD-CMK) in the striatum were performed. This treatment showed an inhibition of the mRNA and protein expression of NLRP3 components and an improvement in the rotational behavior and the number of dopamine neurons in the substantia nigra, indicating that NLRP3 inflammasome participates in the pathogenesis of PD and downstream inhibition of the inflammasome can alleviate the occurrence of PD symptoms (Mao et al. 2017). Astrocytes also play an important role in neurodegeneration, regulating ROS production through uncoupling protein 2 (UCP2), maintaining the proper levels of oxidative stress in the brain (Lu et al. 2014). It has been shown that UCP2 knockout mice exhibit exacerbated dopaminergic neuron loss in MPTP mouse model, with NLRP3 inflammasome activation in astrocytes (Lu et al. 2014). Moreover, targeting NLRP3 to inhibit the inflammatory process also has been approached with gene therapy. In a recent study, it was shown that NLRP3 is a target gene of microRNA-7 (miR-7) and microglial transfection with miR-7 produces an inhibition of microglial NLRP3 inflammasome activation, whereas anti-miR-7 aggravated inflammasome activation in vitro (Zhou et al. 2016). Also, this was demonstrated in vivo with stereotaxic injections of miR-7 into mouse striatum, decreasing dopaminergic neuron loss accompanied by the amelioration of microglial activation in MPTP mouse model (Zhou et al. 2016). Collectively, these exciting new findings place NLRP3 inflammasome as a central pathway in the progression of the neuroinflammatory process in PD and an attractive disease-modifying therapeutic target.
3.4 Inflammasome in CNS Autoimmune Disease: Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune inflammatory demyelinating disease of the CNS that is thought to be mediated by myelin-specific autoreactive T cells, characterized by an increased microglial activation associated with extensive and chronic neurodegeneration (Compston and Coles 2008). This neuroinflammatory damage disrupts the proper transmission of the nerve impulse, resulting in a range of debilitating signs and symptoms including fatigue, ataxia, cognitive impairment, and depression among others (Bruck 2005). Although the etiology of the disease remains unknown, several clinical studies have reported the association of the elevated expression of caspase-1, IL-1β, and IL-18 with the susceptibility, progression, and severity of MS patients (Losy and Niezgoda 2001; Ming et al. 2002; Huang et al. 2004; Heidary et al. 2014). This finding is linked with an increasing number of reports that strongly suggest the involvement of NLRP3 inflammasome in the pathophysiology of MS (Gris et al. 2010; Inoue and Shinohara 2013; Barclay and Shinohara 2017; Guo et al. 2017). Recent studies have shown that NLRP3 plays a critical role in the induction and progression of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. This is through direct effects on caspase-1-dependent cytokines which influence Th1 and Th17 responses (Gris et al. 2010), showing a protective phenotype in NLRP3 KO mice with EAE, due to a reduction in the severity of the disease with a significant decrease of the inflammatory infiltrates including macrophages, dendritic cells, CD4, and CD8+ T cells in the spinal cord and with a reduction in the destruction of myelin and astrogliosis (Gris et al. 2010). Additionally, high levels of cytoplasmic caspase-1 in resident oligodendrocytes of MS lesions have been reported. In this study oligodendrocytes were exposed to a cytokine challenge, observing a blockage in cell death induction by the caspase-1inhibitor Z-YVAD-FMK, suggesting that caspase-1 may play a key role in the inflammatory and pyroptotic processes associated with MS pathogenesis (Ming et al. 2002). Furthermore, evidence in patients with MS suggests that inflammasome activation occurs during MS progression finding caspase-1 and IL-1β in MS plaques and an increase of IL-1β and IL-18 in peripheral blood mononuclear cells (PBMCs) of MS patients (Huang et al. 2004; Inoue and Shinohara 2013). Moreover, pharmacological studies with the specific NLRP3 inhibitor, the small molecule MCC950, were performed investigating the possibility that MCC950 may suppress the T-cell response that mediates autoimmune diseases (Coll et al. 2015). Pretreatment of EAE mice with MCC950 delayed the onset and reduced the severity of EAE, with a reduction in serum concentration of IL-1β and IL-6. Furthermore, analysis of brain mononuclear cells from mice culled on day 22 after treatment showed modestly reduced frequencies of IL-17- and IFN-γ-producing CD3+ T cells in MCC950-treated mice in comparison with vehicle-treated mice, showing an attenuation in the severity and progression of the disease, positioning this drug as a potential therapeutic for NLRP3-associated syndromes including auto-inflammatory and autoimmune diseases (Coll et al. 2015). Similar results were observed with a novel small molecule, a hydroxyl sulfonamide analog JC-171, delaying the progression and severity of EAE in prophylactic and therapeutic settings and blocking IL-1β production and Th17 response (Guo et al. 2017). Collectively, all these novel therapeutic approaches suggest that sustained NLRP3 inflammasome activation is an important mechanism in MS pathophysiology and a potential therapeutic target for the treatment.
3.5 Inflammasomes in Brain Injury
The leading causes of brain injury related to mortality and morbidity are acute ischemic stroke and traumatic brain injury (Feigin et al. 2014; Levin and Diaz-Arrastia 2015). The first is due to an insufficient supply of blood to regions of the brain producing damage and death of tissue (Donnan et al. 2008). The second, TBI, is a consequence of a mechanical trauma to the CNS such as head or spinal cord injury (Ghajar 2000). Both forms of injury result in an acute necrotic and apoptotic loss of neuronal and some glial populations driven by inflammatory cascades leading to the overactivation of innate immune responses. Below we describe the role of the inflammasome activation in stroke and TBI.
3.5.1 Stroke
Stroke is one of the most frequent causes of death and disability worldwide (Donnan et al. 2008; Kuklina et al. 2012). The most common form is ischemic stroke and is caused when a blood clot slows or interrupts the normal blood flow to a region of the brain leading to inflammation and tissue damage (Feigin et al. 2003). Increasing evidence indicates that NLRP3 inflammasome plays a significant role in the pathogenesis and progression of stroke (Barrington et al. 2017; Ye et al. 2017). Increased levels of NLRP3 protein has been found after experimental ischemic stroke, accompanied by elevated levels of IL-1β and IL-18 and extensive neuronal and glial cell death (Lammerding et al. 2016). Preclinical studies in animal models have also found an increase in NLRP3 and NLRP1 expression and activation in primary cortical neurons and cerebral tissue under in vitro and in vivo ischemic conditions. This activation is through NF-κB and MAPK signaling pathways, showing high levels of IL-1β, IL-18, and caspase-3 triggering neuronal apoptosis (Fann et al. 2018). Furthermore, in the same study, treatment with intravenous immunoglobulin (IVIg) was reported to reduce the activation of the NF-κB and MAPK pathways resulting in decreased expression and activation of NLRP1 and NLRP3 in primary cortical neurons under ischemic conditions, suggesting that therapeutic interventions targeting inflammasome activation in neurons may provide new treatments for ischemic stroke. Moreover, NLRP3 deficiency ameliorated cerebral injury in mice after ischemic stroke by reducing infarcts and BBB damage through NOX2-mediated oxidative stress (Yang et al. 2014). Also, the specific selective inhibition of NLRP3 with MCC950 or with P2X7R antagonist BBG post stroke has shown a reduction in neuronal apoptosis, infarction volume, and neurological impairment demonstrating that P2X7R/NLRP3 pathway plays a critical role in caspase-3-dependent neuronal apoptosis after ischemic stroke (Ye et al. 2017). The increase of caspase-1 expression has also been described in neurons and astrocytes after thromboembolic stroke and observed 24 h later in microglia (Abulafia et al. 2009). Therapeutic caspase-1 inhibition has been evaluated pharmacologically and with transgenic mice, showing a reduction of brain damage in knockout mice compared with WT controls after experimental stroke (Friedlander et al. 1997), as well as a protective effect with the intracerebroventricular administration of the caspase-1 inhibitors Ac-YVAD-cmk or VRT-018858 in experimental stroke models (Rabuffetti et al. 2000; Ross et al. 2007). Collectively, these findings suggest that the NLRP3 inflammasome could be a potential novel therapeutic target for stroke.
3.5.2 Traumatic Brain Injury
Traumatic brain injury (TBI), or mechanical trauma to the CNS, results in the disruption of the cellular microenvironment leading to massive necrotic and apoptotic loss of neuronal and glial populations. This loss is accompanied by an acute production of ROS and activation of the innate immune system triggering inflammation with the release of pro-inflammatory cytokines leading to neuron damage and death (Werner and Engelhard 2007). Growing evidence indicates that TBI could activate the inflammasome, specifically NLRP3 with increased levels of ASC, activation of caspase-1, and release of IL-1β and IL-18 in humans and murine models of TBI (Adamczak et al. 2012; Liu et al. 2013). The first study in human patients has shown increased levels of NLRP1 and NLRP3 in cerebrospinal fluid (CFS) after severe TBI in children correlating NLRP3 as a marker of TBI severity (Adamczak et al. 2012). Also, neuronal NLRP1 constitutes an important component of the innate immune response after TBI, demonstrated in a rat model neutralizing ASC with anti-ASC antibodies, showing a reduced caspase-1 activation and processing of IL-1β resulting in a significant decrease in contusion volume after injury (de Rivero Vaccari et al. 2009). To study if NLRP3 is involved in the outcome of TBI, NLRP3 activation was targeted pharmacologically in a rat model of blast-induced TBI (bTBI) with the administration of propofol. Propofol is a lipid-soluble intravenous anesthetic, which has been shown to possess therapeutic benefit during neuroinflammation on various brain injury models, indicating an inhibition of the inflammatory response and a reduction in brain injury by inhibiting ROS. This inhibition leads to a decrease in NLRP3 activation and pro-inflammatory cytokines release in the cerebral cortex of bTBI rats with an amelioration of cerebral cortex damage (Ma et al. 2016). Moreover, similar results were obtained with the Chinese medicine mangiferin, showing a neuroprotective effect in rats treated with mangiferin in a bTBI model, suppressing the activation and expression of NLRP3 through the inhibition of oxidative stress and pro-inflammatory cytokines production in the cerebral cortex, alleviating brain damage, and positioning this approach as a potential therapeutic strategy for bTBI (Fan et al. 2017). Another therapeutic approach has been achieved, targeting NLRP3 in TBI with the use of omega-3 fatty acids (ω−3 FAs). This treatment prevents NLRP3 mitochondrial localization and caspase-1 cleavage through G protein-coupled receptor 40 (GPR40) reducing the release of IL-1β ameliorating neuronal death and behavioral deficits after TBI in rats (Lin et al. 2017), positioning the innate immune response, specifically NLRP3, as a major contributor to the neuroinflammatory process that leads to the severity of TBI and as an important novel therapeutic target for the treatment of this debilitating neurological disease.
3.6 Inflammasome Activation in CNS Infections
Infections of the CNS can be caused primarily by viruses, bacteria, and fungi as well as pathogenic prions (Koyuncu et al. 2013; Coureuil et al. 2017; Shi and Mody 2016). Some acute infections can lead to meningitis or encephalitis and have the potential to contribute to massive acute inflammasome activation. This response against a particular pathogen can be distinct in the CNS compared to systemic responses (Walsh et al. 2014). Therefore it is important to understand how the inflammasome can be distinctly activated in the CNS to develop novel and effective therapeutic strategies against CNS infections; below we outline the role of the inflammasomes in bacterial and viral infections in the CNS based on recent evidence.
3.6.1 Bacterial Infection
Streptococcus pneumoniae is a common cause of bacterial meningitis which occurs when these bacteria invade the CSF leading to an inflammatory process and brain damage (Geldhoff et al. 2013). This process is orchestrated by a variety of innate immune responses, including through acute NLRP3 inflammasome activation. As recently demonstrated in murine models and human patients, NLRP3 could play an important role in the pathologic progression of pneumococcal meningitis infection (Hoegen et al. 2011; Geldhoff et al. 2013). In a murine model of pneumococcal meningitis, ASC and NLRP3 were shown to be involved in modulating the severity of the disease through the release of IL-1β and IL-18. In this study using differentiated human macrophages, THP-1 cells were utilized to confirm that the pneumococcal pore-forming toxin pneumolysin is an essential inducer of IL-1β expression and inflammasome activation upon pneumococcal challenge, through the release of ATP, lysosomal destabilization, and cathepsin B activation (Hoegen et al. 2011). In murine microglia pneumolysin can induce caspase-1-dependent pyroptotic cell death with NLRP3 being critical for caspase-1 activation during the process, and the induction of autophagy could transiently protect microglia from pyroptosis boosting the infective mechanism (Kim et al. 2015). In patients with bacterial meningitis, CSF levels of IL-1β and IL-18 were correlated with severity of the disease. Inversely in ASC and NLRP3 KO mice, a decreased systemic inflammatory response and bacterial outgrowth is observed in blood and brain homogenates compared to WT mice. Moreover, the NLRP3 deficiency was associated with an increase in cerebral neutrophil infiltration and cerebral hemorrhages in comparison to WT controls (Geldhoff et al. 2013). Another common etiological agent of brain abscesses is Staphylococcus aureus (S. aureus) which is characterized by widespread inflammation and necrosis. S. aureus also induces NLRP3 inflammasome activation in microglia in an ATP and cathepsin B-dependent manner, with significantly reduced IL-1β production in NLRP3 and ASC KO microglia following exposure to S. aureus (Hanamsagar et al. 2011). Listeria monocytogenes (LM) infection, or listeriosis, is a common foodborne disease that can lead to severe and potentially fatal cases of bacteremia and meningitis (Thonnings et al. 2016). Infection with LM can activate caspase-1 and the processing of IL-1β and IL-18 and pyroptosis through the activation of multiple inflammasomes such as AIM2, NLRC4, and NLRP3 and collectively orchestrate a robust pro-inflammatory response (Wu et al. 2010).
3.6.2 Viral Infections
Viral infections typically begin in the peripheral tissues and rarely invade the CNS; however, some viruses can infect the CNS triggering the innate immune response (Koyuncu et al. 2013). Japanese encephalitis virus (JEV) represents a common cause of acute viral encephalitis; JEV can invade the CNS and consequently induce acute neuroinflammation, which is characterized by neurodegeneration, astrogliosis. JEV infection has recently been linked with microglial NLRP3 activation resulting in the production of IL-1β and IL-18 (Kaushik et al. 2012). JEV activates NLRP3 through K+ efflux and ROS production in mouse microglia, and depletion of NLRP3 results in the reduction of caspase-1 activity and cytokine release (Kaushik et al. 2012). Another related flavivirus that causes viral encephalitis; West Nile virus (WNV) has been shown to interact with the innate immune response. In contrast with JEV, NLRP3 inflammasome activation has shown to be a protective response during encephalitis caused with WNV. IL-1β production is a key host restriction factor involved in WNV control, as shown in animals lacking the IL-1 receptor or components involved in inflammasome pathway a higher susceptibility to WNV pathogenesis. In this study it was demonstrated that IL-1β production is essential for the development of an effective host immunity against WNV, revealing a novel role for IL-1β in antiviral action that restricts virus replication in neurons (Ramos et al. 2012). Furthermore, ASC-deficient mice exhibited increased susceptibility to WNV infection, associated with a reduced survival with enhanced virus replication in the peripheral tissues and CNS. However, brains from ASC KO mice displayed unrestrained inflammation, including elevated levels of pro-inflammatory cytokines and chemokines, correlated with astrogliosis and enhanced infiltration of peripheral immune cells in the CNS (Kumar et al. 2013). Recently neurological complications related to Zika virus (ZIKV) infection have emerged as a significant threat to public health worldwide (Russo et al. 2017). Several studies have identified microglial nodules, gliosis, neuronal and glial cell degeneration, and necrosis in the brain of ZIKV-infected infants. This suggests that ZIKV could play a role in these neurological disorders through neuroinflammation and microglial dysfunction (Tricarico et al. 2017). ZIKV has been shown to replicate in infective cells causing increased ROS leading to NLRP3 activation and IL-1β release in the human primary glioblastoma cell line U87-MG process that culminates in cell death, implicating inflammasome as a novel and relevant pathogenic factor in ZIKV infection and associated neuropathology (Tricarico et al. 2017).
3.7 Concluding Remarks
In the past decade, crucial advances have been made in our understanding of functional roles of inflammasomes in the healthy CNS and more significantly, in understading the pathological consequences of chronic inflammasome activation during neurological disease. Indeed, sustained innate immune activation more broadly has emerged as a key pathological mechanism during progressive neurodegeneration. While insufficient activation causes the host to become vulnerable to PAMPs and DAMPs, on the contrary chronic and sustained inflammasome activation can drive unfavorable outcomes in almost all progressive neurological diseases that have been studied (Singhal et al. 2014; Song et al. 2017). The current understanding of the specific pathological mechanisms driving inflammasome activation remains insufficient to develop effective therapeutic strategies for complex and challening CNS diseases with multifactorial etioligies. Specifically, the mechanisms by which inflammasome activation is terminated and CNS homeostasis is restored remain to be defined. Likewise the contribution of systemic inflammasome activation to CNS pathology has not been elucidated. Therefore, a more in-depth understanding of the regulation of this key pathway in different neurological contexts could lead to novel and more effective disease-modifying therapeutic strategies that could slow or halt disease progression and potentially restore homeostasis in the CNS.
References
Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD (2009) Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab 29:534–544
Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW (2012) Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg 117:1119–1125
Alboni S, Cervia D, Sugama S, Conti B (2010) Interleukin 18 in the CNS. J Neuroinflammation 7:9
Alcocer-Gomez E, de Miguel M, Casas-Barquero N, Nunez-Vasco J, Sanchez-Alcazar JA, Fernandez-Rodriguez A, Cordero MD (2014) NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun 36:111–117
Allan SM, Tyrrell PJ, Rothwell NJ (2005) Interleukin-1 and neuronal injury. Nat Rev Immunol 5:629–640
Bamberger ME, Harris ME, Mcdonald DR, Husemann J, Landreth GE (2003) A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci 23:2665–2674
Barclay W, Shinohara ML (2017) Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol 27:213–219
Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, Barbera-Cremades M, Yague J, Ruiz-Ortiz E, Anton J, Bujan S, Couillin I, Brough D, Arostegui JI, Pelegrin P (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15:738–748
Barrington J, Lemarchand E, Allan SM (2017) A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol 27:205–212
Boxer MB, Quinn AM, Shen M, Jadhav A, Leister W, Simeonov A, Auld DS, Thomas CJ (2010) A highly potent and selective caspase 1 inhibitor that utilizes a key 3-cyanopropanoic acid moiety. ChemMedChem 5:730–738
Bruck W (2005) Clinical implications of neuropathological findings in multiple sclerosis. J Neurol 252(Suppl 3):iii10–iii14
Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC (2006) CNS immune privilege: hiding in plain sight. Immunol Rev 213:48–65
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M (2013) Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One 8:e55375
Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:248–255
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372:1502–1517
Coureuil M, Lecuyer H, Bourdoulous S, Nassif X (2017) A journey into the brain: insight into how bacterial pathogens cross blood-brain barriers. Nat Rev Microbiol 15:149–159
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101:2070–2075
Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS, Baldwin AG, Freeman S, Wong R, Latta C, Yu S, Jackson J, Fischer N, Koziel V, Pillot T, Bagnall J, Allan SM, Paszek P, Galea J, Harte MK, Eder C, Lawrence CB, Brough D (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun 7:12504
de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29:1251–1261
Doens D, Fernandez PL (2014) Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J Neuroinflammation 11:48
Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–190
Donnan GA, Fisher M, Macleod M, Davis SM (2008) Stroke. Lancet 371:1612–1623
Fan K, Ma J, Xiao W, Chen J, Wu J, Ren J, Hou J, Hu Y, Gu J, Yu B (2017) Mangiferin attenuates blast-induced traumatic brain injury via inhibiting NLRP3 inflammasome. Chem Biol Interact 271:15–23
Fann DY, Lim YA, Cheng YL, Lok KZ, Chunduri P, Baik SH, Drummond GR, Dheen ST, Sobey CG, Jo DG, Chen CL, Arumugam TV (2018) Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol Neurobiol 55(2):1082–1096
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145
Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25:713–724
Feigin VL, Lawes CM, Bennett DA, Anderson CS (2003) Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol 2:43–53
Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, Moran AE, Sacco RL, Anderson L, Truelsen T, O’Donnell M, Venketasubramanian N, Barker-Collo S, Lawes CM, Wang W, Shinohara Y, Witt E, Ezzati M, Naghavi M, Murray C, Global Burden of Diseases I, Risk Factors S, The, G. B. D. S. E. G (2014) Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet 383:245–254
Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, Mangan MS, Zimmer S, Monks BG, Fricke M, Schmidt RE, Espevik T, Jones B, Jarnicki AG, Hansbro PM, Busto P, Marshak-Rothstein A, Hornemann S, Aguzzi A, Kastenmuller W, Latz E (2014) The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15:727–737
Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, Macdonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J (1997) Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med 185:933–940
Friedrich RP, Tepper K, Ronicke R, Soom M, Westermann M, Reymann K, Kaether C, Fandrich M (2010) Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A 107:1942–1947
Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, Ganio EA, Fragiadakis GK, Spitzer MH, Douchet I, Daburon S, Moreau JF, Nolan GP, Blanco P, Dechanet-Merville J, Dekker CL, Jojic V, Kuo CJ, Davis MM, Faustin B (2017) Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med 23:174–184
Geldhoff M, Mook-Kanamori BB, Brouwer MC, Troost D, Leemans JC, Flavell RA, van der Ende A, van der Poll T, van de Beek D (2013) Inflammasome activation mediates inflammation and outcome in humans and mice with pneumococcal meningitis. BMC Infect Dis 13:358
Ghajar J (2000) Traumatic brain injury. Lancet 356:923–929
Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP (2010) NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185:974–981
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687
Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, Wang XY, Zhang S (2017) Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci 8(10):2194–2201
Gustot A, Gallea JI, Sarroukh R, Celej MS, Ruysschaert JM, Raussens V (2015) Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem J 471:323–333
Hanamsagar R, Torres V, Kielian T (2011) Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem 119:736–748
Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, Stacy MA, Scott BL, Vance JM, Scott WK (2008) Pesticide exposure and risk of Parkinson’s disease: a family-based case-control study. BMC Neurol 8:6
Heidary M, Rakhshi N, Pahlevan Kakhki M, Behmanesh M, Sanati MH, Sanadgol N, Kamaladini H, Nikravesh A (2014) The analysis of correlation between IL-1B gene expression and genotyping in multiple sclerosis patients. J Neurol Sci 343:41–45
Heneka MT (2017) Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol 27:220–222
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477
Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, Pfister HW, Fontana A, Hammerschmidt S, Koedel U (2011) The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol 187:5440–5451
Huang WX, Huang P, Hillert J (2004) Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler 10:482–487
Inoue M, Shinohara ML (2013) NLRP3 inflammasome and MS/EAE. Autoimmune Dis 2013:859145
Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63:2260–2273
Kabba JA, Xu Y, Christian H, Ruan W, Chenai K, Xiang Y, Zhang L, Saavedra JM, Pang T (2018) Microglia: housekeeper of the central nervous system. Cell Mol Neurobiol 38(1):53–71
Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, Leblanc AC (2015) Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ 22:1676–1686
Kaushik DK, Gupta M, Kumawat KL, Basu A (2012) NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS One 7:e32270
Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW (2014) Pattern recognition receptors and central nervous system repair. Exp Neurol 258:5–16
Kim JY, Paton JC, Briles DE, Rhee DK, Pyo S (2015) Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget 6:44161–44178
Klein RS, Garber C, Howard N (2017) Infectious immunity in the central nervous system and brain function. Nat Immunol 18:132–141
Koyuncu OO, Hogue IB, Enquist LW (2013) Virus infections in the nervous system. Cell Host Microbe 13:379–393
Kuklina EV, Tong X, George MG, Bansil P (2012) Epidemiology and prevention of stroke: a worldwide perspective. Expert Rev Neurother 12:199–208
Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M Jr, Verma S (2013) Inflammasome adaptor protein apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J Virol 87:3655–3667
Lammerding L, Slowik A, Johann S, Beyer C, Zendedel A (2016) Poststroke inflammasome expression and regulation in the peri-infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103:460–475
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411
Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A (2017) Involvement of c-Abl kinase in microglial activation of NLRP3 inflammasome and impairment in autolysosomal system. J Neuroimmune Pharmacol 12(4):624–660
Levin HS, Diaz-Arrastia RR (2015) Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol 14:506–517
Liang Y, Jing X, Zeng Z, Bi W, Chen Y, Wu X, Yang L, Liu J, Xiao S, Liu S, Lin D, Tao E (2015) Rifampicin attenuates rotenone-induced inflammation via suppressing NLRP3 inflammasome activation in microglia. Brain Res 1622:43–50
Lin C, Chao H, Li Z, Xu X, Liu Y, Bao Z, Hou L, Liu Y, Wang X, You Y, Liu N, Ji J (2017) Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp Neurol 290:115–122
Lipinska K, Malone KE, Moerland M, Kluft C (2014) Applying caspase-1 inhibitors for inflammasome assays in human whole blood. J Immunol Methods 411:66–69
Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L, Hang CH (2013) Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38:2072–2083
Losy J, Niezgoda A (2001) IL-18 in patients with multiple sclerosis. Acta Neurol Scand 104:171–173
Lotharius J, Brundin P (2002) Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3:932–942
Louveau A, Harris TH, Kipnis J (2015a) Revisiting the mechanisms of CNS immune privilege. Trends Immunol 36:569–577
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J (2015b) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341
Lu M, Sun XL, Qiao C, Liu Y, Ding JH, Hu G (2014) Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol Aging 35:421–430
Luo XG, Ding JQ, Chen SD (2010) Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener 5:12
Ma J, Xiao W, Wang J, Wu J, Ren J, Hou J, Gu J, Fan K, Yu B (2016) Propofol inhibits NLRP3 inflammasome and attenuates blast-induced traumatic brain injury in rats. Inflammation 39:2094–2103
Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, Bryant CE (2014) Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A 111:7403–7408
Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, Xue Z (2017) The NLRP3 inflammasome is involved in the pathogenesis of Parkinson’s disease in rats. Neurochem Res 42:1104–1115
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426
Mhyre TR, Boyd JT, Hamill RW, Maguire-Zeiss KA (2012) Parkinson’s disease. Subcell Biochem 65:389–455
Ming X, Li W, Maeda Y, Blumberg B, Raval S, Cook SD, Dowling PC (2002) Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci 197:9–18
Minkiewicz J, De Rivero Vaccari JP, Keane RW (2013) Human astrocytes express a novel NLRP2 inflammasome. Glia 61:1113–1121
Nyul-Toth A, Kozma M, Nagyoszi P, Nagy K, Fazakas C, Hasko J, Molnar K, Farkas AE, Vegh AG, Varo G, Galajda P, Wilhelm I, Krizbai IA (2017) Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav Immun 64:220–231
Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G (2010) Missing pieces in the Parkinson’s disease puzzle. Nat Med 16:653–661
Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT (2005) Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem 280:42026–42035
Rabuffetti M, Sciorati C, Tarozzo G, Clementi E, Manfredi AA, Beltramo M (2000) Inhibition of caspase-1-like activity by Ac-Tyr-Val-Ala-Asp-chloromethyl ketone induces long-lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J Neurosci 20:4398–4404
Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M Jr (2012) IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 8:e1003039
Ross J, Brough D, Gibson RM, Loddick SA, Rothwell NJ (2007) A selective, non-peptide caspase-1 inhibitor, VRT-018858, markedly reduces brain damage induced by transient ischemia in the rat. Neuropharmacology 53:638–642
Rothwell NJ, Luheshi GN (2000) Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci 23:618–625
Russo FB, Jungmann P, Beltrao-Braga PCB (2017) Zika infection and the development of neurological defects. Cell Microbiol 19:e12744
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009) Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87:181–194
Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R, Mancuso R, Clerici M (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11:23
Schapira AH, Bezard E, Brotchie J, Calon F, Collingridge GL, Ferger B, Hengerer B, Hirsch E, Jenner P, Le Novere N, Obeso JA, Schwarzschild MA, Spampinato U, Davidai G (2006) Novel pharmacological targets for the treatment of Parkinson’s disease. Nat Rev Drug Discov 5:845–854
Shi M, Mody CH (2016) Fungal infection in the brain: what we learned from intravital imaging. Front Immunol 7:292
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT (2014) Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci 8:315
Song L, Pei L, Yao S, Wu Y, Shang Y (2017) NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci 11:63
Spittau B (2017) Aging microglia-phenotypes, functions and implications for age-related neurodegenerative diseases. Front Aging Neurosci 9:194
Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, Yu JT (2014) Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis e1382:5
Tang Y, Le W (2016) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53:1181–1194
Tarawneh R, Holtzman DM (2012) The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med 2:a006148
Thonnings S, Knudsen JD, Schonheyder HC, Sogaard M, Arpi M, Gradel KO, Ostergaard C, Danish Collaborative Bacteraemia, N (2016) Antibiotic treatment and mortality in patients with Listeria monocytogenes meningitis or bacteraemia. Clin Microbiol Infect 22:725–730
Tricarico PM, Caracciolo I, Crovella S, D’Agaro P (2017) Zika virus induces inflammasome activation in the glial cell line U87-MG. Biochem Biophys Res Commun 492(4):597–602
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT (2017) Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552(7685):355–361. https://doi.org/10.1038/nature25158
Waisman A, Ginhoux F, Greter M, Bruttger J (2015) Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol 36:625–636
Wallisch JS, Simon DW, Bayir H, Bell MJ, Kochanek PM, Clark RS (2017) Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care 27(1):44–50
Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97
Wang W, Nguyen LT, Burlak C, Chegini F, Guo F, Chataway T, Ju S, Fisher OS, Miller DW, Datta D, Wu F, Wu CX, Landeru A, Wells JA, Cookson MR, Boxer MB, Thomas CJ, Gai WP, Ringe D, Petsko GA, Hoang QQ (2016) Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc Natl Acad Sci U S A 113:9587–9592
Wee Yong V (2010) Inflammation in neurological disorders: a help or a hindrance? Neuroscientist 16:408–420
Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth 99:4–9
White CS, Lawrence CB, Brough D, Rivers-Auty J (2017) Inflammasomes as therapeutic targets for Alzheimer’s disease. Brain Pathol 27:223–234
Wu J, Fernandes-Alnemri T, Alnemri ES (2010) Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol 30:693–702
Wyss-Coray T (2016) Ageing, neurodegeneration and brain rejuvenation. Nature 539:180–186
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160:62–73
Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, Shi W, Li F, Xin T, Pang Q, Yi F (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab 34:660–667
Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao L, Cui C, Jin G, Zan K, Zhang Z, Yang X, Shi H, Zu J, Yu M, Song C, Wang Y, Qi S, Cui G (2017) Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol 292:46–55
Yiannopoulou KG, Papageorgiou SG (2013) Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord 6:19–33
Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, Dixit VD (2015) The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21:263–269
Zhang P, Shao XY, Qi GJ, Chen Q, Bu LL, Chen LJ, Shi J, Ming J, Tian B (2016) Cdk5-dependent activation of neuronal inflammasomes in Parkinson’s disease. Mov Disord 31:366–376
Zhou Y, Lu M, Du RH, Qiao C, Jiang CY, Zhang KZ, Ding JH, Hu G (2016) MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener 11:28
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Albornoz, E.A., Woodruff, T.M., Gordon, R. (2018). Inflammasomes in CNS Diseases. In: Cordero, M., Alcocer-Gómez, E. (eds) Inflammasomes: Clinical and Therapeutic Implications. Experientia Supplementum, vol 108. Springer, Cham. https://doi.org/10.1007/978-3-319-89390-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-89390-7_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89389-1
Online ISBN: 978-3-319-89390-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)