
Abstract
Understanding the basic mechanisms of drug interactions allows researchers and clinicians to best interpret and apply drug interaction data and make predictions about patient-specific interactions. Drug interactions can occur during the absorption, distribution, metabolism, and excretion phases of drug distribution (pharmacokinetic interactions) and at the site of action (pharmacodynamic interactions). The consequences of unintended interactions can be extremely harmful and potentially fatal, such as those leading to cardiac conduction abnormalities. Knowledge of the mechanisms of drug interactions has also identified useful interactions with therapeutic benefits, such as in the development of feasible dosing regimens for protease inhibitors in the treatment of HIV and hepatitis C infection. This chapter describes the mechanisms of drug interactions for each of the aforementioned pharmacokinetic processes. The cytochrome P450 family of enzymes, the P-glycoprotein drug transporter, and their mechanisms for inhibition, induction, and suppression are reviewed. Preclinical and clinical methods used to study cytochrome P450 are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Pharmacokinetics
- Pharmacodynamics
- HIV
- Hepatitis C
- CYP450
- UGT
- P-glycoprotein
- OATP
- Gastric pH
- Ritonavir
- Cobicistat
- Pharmacogenetics
- Induction
- Inhibition
- Tubular secretion
- Phenotyping
2.1 Introduction
It is difficult to assess the overall clinical importance of many drug interactions. Often, drug interaction reports are based on anecdotal or case reports, and the involved interaction mechanisms are not always clearly defined. In addition, determining clinical significance requires an assessment of the severity of potential harm. This makes an unequivocal determination of “clinically significant” difficult.
Drug interactions can be pharmacokinetic or pharmacodynamic in nature. Pharmacokinetic interactions result from alterations in a drug’s absorption, distribution, metabolism, and/or excretion characteristics. Pharmacodynamic interactions are a result of the influence of combined treatment at a site of biological activity, and yield altered pharmacologic actions at standard plasma concentrations. Although drug interactions occur through a variety of mechanisms, the effects are the same: the potentiation or antagonism of the effects of drugs.
The mechanisms by which changes in absorption, distribution, metabolism, and excretion occur have been understood for decades. However, more recently developed technology has allowed for a more thorough understanding of drug-metabolizing isoforms and influences thereon. Much information has been published regarding drug interactions involving the cytochrome P450 (CYP450) enzyme system [1,2,3]. This will be an important focus of this chapter, since the majority of currently available anti-infectives are metabolized by, or influence the activity of, the CYP450 system. This chapter provides a detailed review of the mechanisms by which clinically significant pharmacokinetic drug interactions occur. Drug transporter-based interactions will be mentioned where appropriate, but for a more detailed description, the reader is referred to Chap. 3.
2.2 Drug Interactions Affecting Absorption
Mechanisms of absorption include passive diffusion, convective transport, active transport, facilitated transport, ion-pair transport, and endocytosis. Certain drug combinations can affect the rate or extent of absorption of anti-infectives by interfering with one or more of these mechanisms. Generally, a change in the extent of a medication’s absorption of greater than 20% may be considered clinically significant in case of drugs with a relatively narrow therapeutic index. The most common mechanisms of drug interactions affecting absorption are shown in Table 2.1.
2.2.1 Changes in pH
The rate of drug absorption by passive diffusion is limited by the solubility, or dissolution, of a compound in gastric fluid. Basic drugs are more soluble in acidic fluids and acidic drugs are more soluble in basic fluids. Therefore, compounds that create an environment with a specific pH may decrease (or increase) the solubility of compounds with pH-dependent absorption. However, drug solubility does not completely ensure absorption, since only un-ionized molecules are absorbed. Although acidic drugs are soluble in basic fluids, basic environments can also decrease the proportion of solubilized acidic molecules that are in an un-ionized state. Therefore, weak acids (pKa = 3–8) may have limited absorption in an alkaline environment and weak bases (pKa = 5–11) have limited absorption in an acidic environment.
Antacids, histamine receptor antagonists, and proton-pump inhibitors all raise gastric pH to varying degrees. Antacids transiently (0.5–2 h) raise gastric pH by 1–2 units [4], H2-antagonists dose-dependently maintain gastric pH > 5 for many hours, and proton-pump inhibitors dose-dependently raise gastric pH > 5 for up to 19 h [5]. The concomitant administration of these compounds leads to significant alterations in the extent of absorption of basic compounds [6].
These interactions can also be clinically significant. For example, when patients in the Hepatitis C Virus (HCV) Target study used a proton-pump inhibitor while starting HCV treatment with a ledipasvir-containing regimen, lower rates of sustained virological response were observed [7]. Ledipasvir is an NS5A-inhibitor of HCV replication that has poor solubility at pH >3.0. Similar effects have been seen for the HIV protease inhibitors indinavir and atazanavir [8] and the non-nucleoside reverse transcriptase inhibitor rilpivirine [9]. When combined, plasma concentrations of the antiretroviral agents may become subtherapeutic, and virological failure may occur [10]. Other examples of anti-infective agents known to require an acidic environment for dissolution are ketoconazole [11], itraconazole [12,13,14,15], posaconazole [16, 17], and dapsone [18, 19]. Because of large interindividual variability in the extent of altered gastric pH , significant interactions may not occur in all patients.
It must be noted here that pH-dependent effects may vary between different formulations of some of the abovementioned anti-infectives. For instance, posaconazole absorption is negatively influenced when the oral suspension is taken with acid-reducing agents, but this does not occur with posaconazole tablet formulation [20]. Likewise, itraconazole dissolution is affected by omeprazole when taken as capsules but not as oral solution which contains itraconazole already dissolved in cyclodextrins [21].
2.2.2 Chelation and Adsorption
Drugs may form insoluble complexes by chelation in the gastrointestinal tract. Chelation involves the formation of a ring structure between a metal ion (e.g., aluminum, magnesium, iron, and to a lesser degree calcium ions) and an organic molecule (e.g., anti-infective medication), which results in an insoluble compound that is unable to permeate the intestinal mucosa due to the lack of drug dissolution. High concentrations of cations are present in food supplements, including many multivitamin preparations, but also in some antacids. The latter can be confusing as both a pH effect and a chelation effect may occur after simultaneous intake with an organic molecule.
A number of examples of the influence on anti-infective exposure by this mechanism exist in the literature including the quinolone antibiotics in combination with magnesium and aluminum-containing antacids, sucralfate, ferrous sulfate, or certain buffers. These di- and trivalent cations complex with the 4-oxo and 3-carboxyl groups of the quinolones, resulting in clinically significant decreases in the quinolone area under the concentration–time curve (AUC) by 30–50% [22,23,24]. A second well-documented, clinically significant example of this type of interaction involves the complexation of tetracycline and iron. By this mechanism, tetracycline antibiotic AUCs are decreased by up to 80% [25]. More recently, the absorption of members of the group of HIV-integrase inhibitors also appears to be harmed by concomitant intake of divalent cations, as has been demonstrated for raltegravir [26], elvitegravir [27], and dolutegravir [28].
Cations present in enteral feeding formulations do not appear to interfere significantly with the absorption of these compounds [29, 30].
Adsorption is the process of ion binding or hydrogen binding and may occur between anti-infectives such as penicillin G, cephalexin, sulfamethoxazole, or tetracycline and adsorbents such as cholestyramine. Since this process can significantly decrease antibiotic exposure, the concomitant administration of adsorbents and antibiotics should be avoided.
2.2.3 Changes in Gastric Emptying and Intestinal Motility
The presence or absence of food can affect the absorption of anti-infectives by a variety of mechanisms. High-fat meals can significantly increase the extent of absorption of fat soluble compounds such as griseofulvin, cefpodoxime, cefuroxime axetil, saquinavir, and rilpivirine. Prolonged stomach retention can cause excessive degradation of acid-labile compounds such as penicillin and erythromycin.
Since the primary location of drug absorption is the small intestine, changes in gastric emptying and gastrointestinal motility may have significant effects on drug exposure. Rapid gastrointestinal transit effected by prokinetic agents such as cisapride, metoclopramide, and domperidone may decrease the extent of absorption of poorly soluble drugs or drugs that are absorbed in a limited area of the intestine [31]. However, clinically significant effects on anti-infectives have not been documented.
2.2.4 Effects of Intestinal Blood Flow
Intestinal blood flow can be modulated by vasoactive agents and theoretically can affect the absorption of lipophilic compounds. However, there is no evidence to date that this results in clinically significant drug interactions.
2.2.5 Changes in Presystemic Clearance
The drug-metabolizing cytochromes P450 (CYP) 3A4 and 5 are expressed at high concentrations in the intestine and contribute to drug inactivation. P-glycoprotein is expressed at the lumenal surface of the intestinal epithelium and serves to extrude unchanged drug from the enterocyte into the lumen. Both CYP3A4/5 and P-glycoprotein share a significant overlap in substrate specificity [32, 33], although there is no correlation between affinities [34]. Determining the relative contributions of intestinal CYP3A4/5 and P-glycoprotein activity to drug bioavailability and interactions is an active area of investigation. Potential drug interactions involving these mechanisms are discussed in detail below.
2.2.6 Cytochrome P450 Isozymes
Gastrointestinal cytochrome P450 isozymes , responsible for Phase I oxidative metabolism (for a more detailed discussion of CYP isoforms, see Sect. 2.4.1 Phase I Drug Metabolism), are most highly concentrated in the proximal two-thirds of the small intestine [35]. Two intestinal CYP isoforms, CYP3A4 and CYP3A5 (CYP3A4/5), account for approximately 70% of total intestinal P450 protein and are a major determinant of the systemic bioavailability of orally administered drugs [36,37,38,39].
For example, the benzodiazepine midazolam is a specific CYP3A4/5 substrate with no affinity for P-glycoprotein . An investigation of oral and intravenous midazolam plasma clearance in 20 healthy young volunteers [40] revealed an incomplete correlation between the two measures (r = 0.70). The large variability in midazolam oral clearance not accounted for by hepatic metabolism most likely represents the contribution of intestinal CYP3A4/5. Therefore, it appears that at least 30–40% of the clearance of many CYP3A metabolized compounds may be significantly influenced by CYP3A4/5 located in enterocytes. Since the activity of intestinal CYP3A4/5 can also be influenced by a variety of environmental factors, the potential for drug interactions to occur during drug absorption is great.
A good example of the significant effects of drug interactions occurring at the intestinal isozyme level involve the inhibition of CYP3A4/5 with grapefruit juice [41, 42]. Generally, this interaction results in a minimum threefold increase in the extent of absorption and toxicity of the concomitantly administered agent, but can also result in decreased efficacy of prodrugs needing CYP3A for conversion to active moieties. The concern of this interaction is strictly limited to orally administered agents, since the active components of grapefruit juice are either inactivated in the gut or are present in such minute quantities in the portal circulation that no effect on hepatic metabolism occurs. Clinical data available for anti-infective–grapefruit juice interactions include the protease inhibitor saquinavir [43], the antifungal agent itraconazole [44], and the macrolide clarithromycin [45], and there are also indications for effects on anthelmintics and antimalarials [42]. Whereas saquinavir AUC increases twofold with a single 400-mL dose of commercially available grapefruit juice, itraconazole and clarithromycin AUCs do not change significantly. The absence of an effect of grapefruit juice on the oral clearance of these latter two compounds suggests that their first-pass metabolism does not rely significantly on intestinal CYP3A4/5.
Anti-infectives can also inhibit intestinal CYP isozyme activity themselves. For example, the protease inhibitor ritonavir is a potent inhibitor of CYP3A4 activity [46, 47]. This characteristic can be clinically useful, as demonstrated by the increased bioavailability of several HIV protease inhibitors including saquinavir, lopinavir, atazanavir, and darunavir when given in combination with low-dose ritonavir [48]. This application is called “pharmaco-enhancement” or “boosting ” and has now also been introduced in HCV therapy by the development of the HCV protease inhibitor paritaprevir that needs low-dose ritonavir to boost its plasma exposure and activity [49, 50].
Whereas the discovery of (low-dose) ritonavir as a pharmaco-enhancer can be seen as the direct consequence of the observed high drug interaction potential of this agent at its therapeutic dose, it is not a surprise that pharmaceutical companies have searched for non-therapeutic agents with similar pharmaco-enhancement profiles as ritonavir . Cobicistat is an agent chemically related to ritonavir but without its anti-HIV activity; its affinity for CYP3A is similar to ritonavir [51, 52].
Other CYP isozymes present in enterocytes may also influence drug absorption. Environmental factors may influence their activity as well, and drug–environment interactions may result in significantly altered absorption. However, further research is needed to better characterize these influences before specific interactions can be predicted.
2.2.7 Changes in Active and Passive Transport: P-Glycoprotein
A rapidly expanding field of research is that of intestinal transcellular transport. Over the past 20 years, multiple intestinal transporters located on the brush-border and basolateral membrane of the enterocyte have been identified [53,54,55]. The potential for competitive inhibition of these transporters with quinolone antibiotics, antiretroviral agents, and directly acting antivirals for HCV infection among others has been documented in many studies [56, 57]. This contributes an additional mechanism by which anti-infective drug interactions may occur.
The Caco-2 cell model is a human colonic cell line sharing similarities with enterocytes and is widely used as a model for oral absorption [58,59,60]. Investigations using this cell line have demonstrated that certain compounds can modulate the tight junctions of the intestinal epithelia and alter paracellular drug absorption. There is still incomplete understanding of the structure and function of tight junctions, which has limited the development of such modulating compounds to enhance paracellular absorption [61, 62].
Of the intestinal transporters, P-glycoprotein is probably the most relevant. This transporter is the product of the multidrug resistance 1 (MDR1) gene found in a variety of human tissues including the gastrointestinal epithelium [63, 64]. This efflux pump is expressed at the lumenal surface of the intestinal epithelium and opposes the absorption of unchanged drug by transporting lipophilic compounds out of enterocytes back into the gastrointestinal lumen. P-glycoprotein has demonstrated up to tenfold variability in activity between subjects [65] and has a significant role in oral drug absorption. Decreased bioavailability occurs because intact drug molecules are pumped back into the gastrointestinal tract lumen and exposed multiple times to enterocyte metabolism.
P-glycoprotein has broad substrate specificity, and inhibiting or inducing the activity of this protein can lead to significant alterations in drug exposure. P-glycoprotein genotype has also been associated with basal expression and induction of CYP3A4 [66]. However, because many drugs have affinities for both P-glycoprotein and CYP3A4/5, it is difficult to determine by what specific mechanism drug interactions occur. For some compounds, inhibition of both P-glycoprotein function and CYP3A4/5 activity may be required to produce clinically significant interactions.
Many anti-infectives have binding affinity for P-glycoprotein. These include erythromycin, clarithromycin [67], ketoconazole, sparfloxacin [68], almost all HIV-1 protease inhibitors [69], tenofovir disoproxil fumarate [70], posaconazole [71], and sofosbuvir [72]. Since drugs that have affinity for P-glycoprotein are not necessarily removed from the enterocyte by this efflux pump, anti-infectives may participate in, but are not necessarily influenced by, drug interactions involving P-glycoprotein . This concept is illustrated by an in vitro investigation of ketoconazole and erythromycin [73]. Both drugs demonstrate significant affinity for P-glycoprotein. However, in combination with verapamil (a classic P-glycoprotein inhibitor), significantly decreased P-glycoprotein-mediated efflux occurred only with erythromycin. Therefore, although ketoconazole exhibits binding affinity for P-glycoprotein, it can be concluded that P-glycoprotein does not contribute significantly to the process of first-pass effect of ketoconazole.
2.3 Drug Interactions Affecting Distribution
2.3.1 Protein Binding and Displacement
Drug interactions affecting distribution are in general those that alter protein binding (Table 2.1). Initially, the importance of drug displacement interactions has been overestimated, with the extrapolation of data from in vitro investigations without consideration for subsequent physiologic phenomena. The lack of well-designed studies has prevented precise quantification of the influence of protein binding on (anti-infective) therapeutic efficacy in vivo. The main reason for the general lack of clinical relevance of protein displacement effects is that redistribution and excretion of drugs generally occurs quickly after displacement, and hence the effects of any transient rise in unbound concentration of the object drug are rarely clinically important [74].
Albumin constitutes the main protein fraction (~5%) in blood plasma. As albumin contains both basic and acidic groups, it can bind basic as well as acidic drugs. Acidic drugs (e.g., penicillins, sulfonamides, doxycycline, and clindamycin) are strongly bound to albumin at a small number of binding sites, and basic drugs (e.g., erythromycin) are weakly bound to albumin at a larger number of sites [75, 76]. Basic drugs such as most HIV protease inhibitors [77] may also preferentially bind to α-1-acid glycoprotein .
Depending on relative plasma concentrations and protein-binding affinities, one drug may displace another with temporary clinically significant results. This interaction is much more likely to occur with drugs that are at least 80–90% bound to plasma proteins, with small changes in protein binding leading to large relative changes in free drug concentration. Drugs that are poorly bound to plasma proteins may also be displaced, but the relative increase in free drug concentration is generally of less consequence. When a protein displacement interaction occurs, the increased free drug in plasma quickly distributes throughout the body and will localize in tissues if the volume of distribution is large. An increase in unbound drug concentrations at metabolism and elimination sites will also lead to increased rates of elimination.
Generally, interactions between basic drugs and albumin are not clinically significant. In subjects with normal concentrations of albumin and anti-infective concentrations of less than 100 μg/mL, the degree of protein binding will be relatively constant. At higher anti-infective concentrations, available binding sites may theoretically become saturated, and the extent of binding subsequently decreased. Clinically significant displacement interactions for α-1-acid glycoprotein have not been described.
Before it is concluded that protein displacement interactions are never clinically relevant, one should keep this mechanism in mind in case unexpected acute toxicity occurs when (novel) drugs with high protein binding are combined. One such example is the recent occurrence of severe symptomatic bradycardia when sofosbuvir-containing HCV therapy was initiated in patients concomitantly taking amiodarone. Although the mechanism of this interaction has not yet been fully discovered, protein-binding displacement of amiodarone by anti-HCV agents is one of the hypotheses [78].
In summary, drug interactions involving albumin binding displacement may potentially be clinically significant if the compound is greater than 80% protein bound, has a high hepatic extraction ratio, a narrow therapeutic index, and a small volume of distribution. Although temporary increase in drug concentrations may be clinically significant with such drugs as warfarin and phenytoin, mean steady-state free drug concentrations will remain unaltered [79,80,81,82].
2.4 Drug Interactions Affecting Drug Metabolism
The principal site of drug metabolism is the liver . Metabolism generally converts lipophilic compounds into ionized metabolites for renal elimination. Drug-metabolizing activity can be classified according to nonsynthetic (Phase I) and synthetic (Phase II) reactions. Phase I reactions include oxidation, reduction, and hydrolysis and occur in the membrane of hepatocyte endoplasmic reticula. Phase II reactions result in conjugation (i.e., glucuronidation, sulfation) and occur in the cytosol of the hepatocyte.
2.4.1 Phase I Drug Metabolism
The majority of oxidative reactions are catalyzed by a superfamily of mixed-function mono-oxygenases called the cytochrome P450 enzyme system. Although cytochrome P450 (CYP) isozymes are located in numerous tissues throughout the body, the liver is the largest source of CYP protein. Many significant pharmacokinetic drug interactions involve the hepatic cytochrome P450 isozymes (Table 2.2).
Nomenclature for this superfamily is based on amino acid sequence homology and groups enzymes and genes into families and subfamilies [58, 83]. To designate the cytochrome P450 enzymes, the “CYP” prefix is used. All isozymes having at least 40% amino acid sequence homology are members of an enzyme family, as designated by an Arabic number (e.g., CYP3). All isozymes that have at least 55% amino acid sequence homology are members of an enzyme subfamily, as designated by a capital letter (e.g., CYP3A). An Arabic number is used to represent an individual enzyme (e.g., CYP3A4). Italicized nomenclature represents the gene coding for a specific enzyme (e.g., CYP3A4).
To date, at least 14 human families, 22 human subfamilies, and 36 human CYP enzymes have been identified [1, 84]. However, the CYP1, 2, and 3 families account for 70% of the total hepatic P450 content [85, 86]. Approximately 95% of all therapeutic drug oxidation can be accounted for by the activities of CYP1A2, CYP2C8/9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5. Drug interactions involving these isozymes result from enzyme inhibition or induction, although genetic polymorphisms can attenuate these interactions.
2.4.1.1 Genetic Polymorphisms
Polymorphisms are generated by nonrandom genetic mutations that occur in at least 1% of a population and give rise to distinct subgroups within that population that differ in their ability to metabolize xenobiotics [87, 88]. Clinically significant polymorphisms in CYP enzymes have been documented for CYP2B6, CYP2D6, CYP2C9, and CYP2C19 [87, 89, 90]. Extensive or rapid metabolizers (generally the largest proportion of a population) have heterozygous or homozygous dominant alleles, poor metabolizers possess variant homozygous autosomal recessive alleles, and ultraextensive metabolizers exhibit gene amplification of autosomal dominant alleles.
Poor-metabolizer phenotypes can be at high risk for toxicity from drugs that require CYP inactivation and at high risk for therapeutic inefficacy from prodrugs that need CYP activation.
Two recent examples of the importance of genetic polymorphisms in evaluating the outcome of drug–drug interactions with anti-infectives are related to efavirenz (CYP2B6) and voriconazole (CYP2C19, CYP3A). The non-nucleoside reverse transcriptase inhibitor efavirenz is primarily metabolized by CYP2B6, but many patients possess a 516G > T variant in this enzyme (defined as CYP2B6*6 haplotype) that has almost no enzyme activity [91]. The prevalence of this polymorphism varies among ethnic groups: African Americans and sub-Saharan Africans, 45%; Hispanics and Caucasians, 21–27%; and Japanese and Asians, 18% [91]. Not only do these patients have a higher risk of discontinuation of efavirenz because of adverse effects (associated with higher efavirenz plasma concentrations), but they are also less prone to a drug–drug interaction with the enzyme inducer rifampin [92]. This has led to unexpected clinical observations of patients on efavirenz treated with rifampin that need a lower dose of efavirenz; there was no drug interaction, but the genetic polymorphism in CYP2B6 determined the therapeutic dose of efavirenz in such an individual [93].
The antifungal agent voriconazole is extensively metabolized by CYP2C19 and to a lesser extent by CYP2C9 and CYP3A. The antiretroviral combination atazanavir/ritonavir is an inhibitor of CYP3A but also an in vivo inducer of CYP2C19 and CYP2C9. It has been demonstrated [94] that when atazanavir/ritonavir is added to voriconazole in CYP2C19 extensive metabolizers, a moderate (10–40%) reduction in voriconazole exposure can be seen; this is explained by CYP2C9/19 induction by ritonavir . However, when atazanavir/ritonavir is added to voriconazole in CYP2C19 poor metabolizers, the net effect is 4.4–7.7-fold increase in voriconazole exposure. Here, atazanavir-/ritonavir-mediated CYP3A inhibition becomes dominant in the absence of CYP2C19 activity. Ideally, drug–drug interactions with (anti-infective) agents that are metabolized by polymorphic CYP enzymes should be studied in both extensive and poor metabolizers.
2.4.1.2 Mechanisms of Inhibition
Enzyme inhibition can result in sudden catastrophic drug interactions. Several mechanisms of inhibition exist, and many drugs can interact by multiple mechanisms [85, 86].
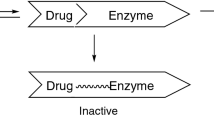
Reversible inhibition is the most common mechanism. Reversible inhibition occurs when compounds quickly form weak bonds with CYP isozymes without permanently disabling them [95]. This can occur both competitively (competition for the same binding site between inhibitor and substrate) and noncompetitively (inhibitor binds at a site on the enzyme distinct from the substrate).The magnitude of this type of inhibition depends both on the affinity of substrate and inhibitor for the enzyme, and on the concentration of the inhibitor at the enzyme site. Affinity is represented by an inhibitor constant (Ki), which is the concentration of inhibitor required to decrease the maximal rate of the reaction to half of the uninhibited value [96, 97]. For example, potent reversible CYP3A inhibitors generally have Ki values below 1 μM (e.g., ketoconazole, itraconazole, ritonavir, and cobicistat), although drugs with Ki values in the low micromolar range can also demonstrate competitive inhibition (e.g., erythromycin). Compounds with Ki’s greater than 100 μM for the CYP3A subfamily tend not to produce clinically significant inhibition [98, 99].
CYP inhibition can also occur as a result of a slowly reversible reaction. When an inhibitor binds to a CYP isozyme and undergoes oxidation to a nitrosoalkane species, it can form a slowly reversible complex with the reduced heme in the CYP isozyme. This interaction has been documented between the macrolide antibiotics and CYP3A [100] and explains why clinically significant interactions (i.e., erythromycin and terfenadine) can occur with compounds that have modest Ki values.
A second, distinct type of enzyme inhibition is called mechanism-based inhibition (or suicide inhibition). This type of interaction is usually irreversible and generally occurs with the CYP-mediated formation of a reactive metabolite [95, 101]. This metabolite can covalently and irreversibly bind to the catalytic site residue and permanently inactivate the enzyme for subsequent reactions. The extent of the clinical importance of this reaction depends on the total amount of CYP isozyme present, the total amount of inhibitor to which the isozyme is exposed, and the rate of new isozyme synthesis. Examples of anti-infectives that display mechanism-based enzyme inhibition include isoniazid, ritonavir , and also macrolide antibiotics (which thus combine different mechanisms of enzyme inhibition).
2.4.1.3 Mechanisms of Suppression of Inflammation-Induced Enzyme Inhibition
As early as the 1960s, inflammation and infection were demonstrated to decrease Phase I metabolism of drugs and toxins in animals, thereby modulating pharmacologic and toxicologic effects. One of the earliest reports of infection altering human drug-metabolizing enzyme activity occurred a decade later, with quinidine concentrations consistently elevated in subjects experimentally infected with plasmodium falciparum malaria [102]. Since that time, numerous reports have described alterations in drug metabolism with viral and bacterial infections [103,104,105], in addition to complex events such as surgery and bone marrow transplantation.
The effects of inflammation and infection on CYP activity are ascribed to stimulation of the cellular immune response [104]. Although many different mediators may be involved, there has been particular focus on the major proinflammatory cytokines interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α. Generally, IL-1, IL-6, and TNFα demonstrate a suppressive effect on CYP isozymes by decreasing mRNA up to 80%. However correlations between mRNA , enzyme protein content, and enzyme activity are incomplete both within and between investigations [104].
A number of clinical investigations have also documented decreased drug-metabolizing enzyme activity during the administration of therapeutic interferons and interleukins. These studies demonstrate variable and conflicting results with respect to the magnitude of drug–cytokine interactions. With the increasing use of cytokines as therapeutic agents for a variety of disease states, further investigation is required to elucidate the mechanisms of drug–cytokine interactions in order to optimize anti-infective therapeutic regimens.
2.4.1.4 Mechanisms of Induction
An increase in cytochrome P450 activity through induction is less of an immediate concern than inhibition, since induction occurs gradually rather than rapidly and generally leads to compromised therapeutic goals rather than profound toxicity. Since the time course of enzyme induction is determined by the half-life of the substrate as well as the rate of isozyme turnover [99], it is often difficult to predict this time course specifically [106, 107]. Clinically significant induction results from a > 50-fold increase in the number of enzyme molecules. This generally occurs through an increase in P450 synthesis by either receptor-mediated transcriptional activation or mRNA stabilization. However, protein stabilization leading to decreased rates of P450 degradation has also been observed. It should be noted that enzyme induction also persists for days to weeks after stopping the inducing drug.
Induction of the CYP1 family by cigarette smoke, charcoal-broiled foods, indoles (found in broccoli, cauliflower, cabbage, Brussels sprouts, kale, watercress), and omeprazole occurs primarily by substrate binding to the aryl hydrocarbon receptor (AhR/dioxin receptor). This complex subsequently binds with a receptor nuclear translocator, enters the hepatocyte nucleus, and binds with regulatory DNA sequences to enhance gene transcription and stabilize mRNA.
The CYP2 and CYP3 families are induced by a variety of structurally diverse compounds. Activation of CYP2C genes is regulated by constitutive androstane receptor (CAR) and pregnane X receptor (PXR) in addition to multiple co-activators [98, 108,109,110]. Both PXR and CAR can regulate CYP2B6 and CYP3A expression; however, induction by efavirenz and nevirapine of these enzymes is mediated by specifically activating CAR [111]. PXR is activated by a range of drugs known to induce CYP3A4/5 expression (e.g., rifampicin, clotrimazole) [98]. PXR is expressed most abundantly in the liver, but is also present in the small intestine and colon. Transcriptional factors not directly activated by xenobiotics have also been shown to be critical for enzyme induction.
CYP3A can also be induced by posttranscriptional message stabilization and protein stabilization with the following anti-infectives: macrolides, imidazole antifungal agents, and rifampicin. A proposed mechanism for posttranscriptional protein stabilization is proteasome inhibition by NF kappaB activation [112], and message stabilization may involve a similar phosphorylation process.
2.4.2 Phase II Drug Metabolism
The term “Phase II” metabolism was developed originally to represent synthetic reactions occurring after “Phase I” processes. It is now known that many xenobiotics do not require Phase I metabolism before undergoing conjugation reactions [113]. The group of Phase II isozymes consists of UDP-glucuronosyltransferases, sulfotransferases, acetyltransferases, glutathione S-transferase, and methyltransferases. Many of these families of enzymes are still growing in complexity, and drug interactions involving these isozymes continue to be under investigation [114,115,116,117,118].
2.4.2.1 Genetic Polymorphisms
Many of the Phase II enzymes exhibit polymorphism [119, 120]. Although these polymorphisms have been implicated in selected anti-infective-associated adverse drug reactions (e.g., dapsone, isoniazid, sulfonamides [121]), influences of these polymorphisms on anti-infective drug interactions have not been documented.
2.4.2.2 Inhibition
Phase II drug-metabolizing enzymes do not currently appear to play as prominent a role in clinical drug interactions with anti-infectives as the cytochrome P450 enzyme system. This may be due to the large capacity of the conjugation system, in which only profound disturbances result in clinically significant alterations in drug pharmacokinetics.
UDP-glucuronosyltransferase represents the most common conjugation reaction in drug metabolism. Many drugs have been characterized as competitive inhibitors of UDP-glucuronosyltransferases [122], but the roles of these interactions in practical drug metabolism issues are currently only partly explored.
The HIV protease inhibitor atazanavir is a strong inhibitor of UGT1A1 [117]; the pharmacokinetic booster ritonavir is a moderate inducer of UGT1A1. When atazanavir is combined with ritonavir, the net inhibition effect is smaller than when atazanavir is given unboosted [117]. The HIV integrase inhibitors raltegravir and dolutegravir are UGT1A1 substrates, and their metabolism is thus inhibited by atazanavir [123, 124]. A well-known characteristic of UGT1A1 inhibitors is that hyperbilirubinemia occurs as bilirubin is an endogenous substrate of UGT1A1.
2.4.2.3 Induction
Far less is known about the potential for induction of Phase II enzymes than the cytochrome P450 enzyme system. The UDP-glucuronosyltransferases can be induced, but the clinical significance of this is not fully understood. However, the increased clearance of zidovudine that has been documented with the coadministration of rifampicin suggests that induction of these enzymes may be clinically significant [125]. Glutathione S-transferase is also known to be inducible, although these activities rarely exceed two- to threefold times baseline and are not involved in anti-infective metabolism [126]. Another example involves the induction of the sulfotransferase enzyme . Exposure to moxifloxacin is decreased by circa 30% upon coadministration of rifampicin [127, 128]. As moxifloxacin does not undergo Phase I metabolism , this interaction is probably due to induction of sulfation (and possibly glucuronidation) of moxifloxacin by rifampicin [128].
2.5 Drug Interactions Affecting Excretion
Renal elimination of drugs involves glomerular filtration, tubular secretion, and tubular reabsorption. Five mechanisms of drug–drug interactions can occur at the site of renal elimination. The most common mechanisms are discussed below (Table 2.3).
2.5.1 Glomerular Filtration
Rates of glomerular filtration can be affected by changes in renal blood flow, cardiac output, and extent of protein binding. With highly protein-bound drugs (e.g., >80%), a significant increase in the unbound fraction can lead to an increase in glomerular filtration and subsequent increased drug elimination [129, 130]. Conversely, if saturation of tubular secretion transporters occurs, and renal elimination is at a maximal, elimination rates may decrease significantly with increased free drug .
2.5.2 Tubular Secretion
The most common renal drug interactions occur at the transport site of tubular secretion. Many organic anionic and cationic drugs and metabolites compete with each other for secretion, as they share the same proximal tubular active transport system [54, 131]. A classic example of this interaction, used long ago intentionally for therapeutic benefit, is the combination of probenecid and penicillin to decrease the secretion of penicillin and increase its serum concentrations [132]. Examples of other anti-infectives that may exhibit interactions by this mechanism include the sulfonamides, penicillins, and zidovudine. Also a range of antiretrovirals are subjected to tubular secretion and/or interact with the renal transport system [130].
P-glycoprotein has been identified in the apical membrane of the proximal tubule and can transport a large variety of drugs into the lumen [54]. A number of experimental drug interaction investigations have implicated the inhibition of renal p-glycoprotein to result in an increase in plasma drug concentrations. Quinolones [133], macrolides [134], and azole antifungals [135] demonstrate affinity for renal p-glycoprotein and can potentially be subjected to or cause significant drug interactions.
Besides p-glycoprotein many other renal transporters have been identified in the last 20 years [54]. For more detailed description, see Chap. 3.
2.5.3 Tubular Reabsorption
Reabsorption of drugs from the tubular lumen involves both passive diffusion and active transport processes. Only nonionized compounds are passively reabsorbed from the renal tubule, and thus manipulating urinary pH can alter the reabsorption of weak organic acids and bases. Renal clearance of weak organic bases (pKa = 7–10) is increased with urine acidification (i.e., by salicylates and ascorbic acid) and decreased with urine alkalinization (i.e., by antacids, calcium carbonate, thiazide diuretics, and sodium bicarbonate). Likewise, renal elimination of weak organic acids (pKa = 3–7; nitrofurantoin, sulfonamides, aminoglycosides, and vancomycin) is increased with urine alkalinization and decreased with urine acidification. Generally, these interactions are not clinically significant, since few drugs can have altered urinary excretion to a large enough extent to affect plasma half-life. The role of active transport reabsorption in anti-infective drug interactions remains largely unknown.
2.6 Pharmacodynamic Drug Interactions
Drug interactions are not limited to mechanisms of absorption, distribution, metabolism, and elimination, but can also result from pharmacodynamic interactions. Pharmacodynamic interactions may occur at the intended site of biological activity, i.e., on the same receptors or physiological systems, and they occur irrespective of drug concentrations in the blood or plasma. This type of interaction is fairly common, but is not always recognized or denoted as an interaction. For example, many antibiotics and antiviral drugs are applied in combination for their additive or synergistic effect to achieve improved efficacy or prevent the emergence of resistance.
Pharmacodynamic interactions may also have detrimental effects. Examples of such interactions include the potential for seizures with quinolones when combined with NSAIDs or other medications that lower seizure thresholds and the increased risk of serotonin syndrome after coadministration of linezolid with other medications with serotonergic activity such as antidepressants and opioids [136]. Another example is QT-interval prolongation by combination of anti-infectives and other agents including macrolides, quinolones, antimalarials, and azole antifungals [137]. A third example is the overlapping adverse effect profiles of antiretroviral and anti-TB drugs. Understanding drug mechanisms and side-effect profiles of the antimicrobial agent and concomitant therapy can prevent these complications.
2.7 Significance of Drug Interactions
Many drug interactions are primarily assessed in vitro (see Sect. 2.8 Preclinical Methods for Predicting Drug Interactions). However, absolute in vitro/in vivo correlations are infrequent. Even when assessed in a clinical trial, not all statistically significant drug interactions are of clinical significance. For example, interactions that involve drugs with wide therapeutic indices that demonstrate even more than 20% changes in exposure when combined with a second agent will most likely be of little, if any, clinical significance.
The greatest risk of documented clinically significant pharmacokinetic drug interactions involving anti-infective-induced altered protein binding, drug-metabolizing enzyme inhibition, and altered renal elimination include combinations of anti-infectives with anticoagulants, antidepressants, and cardiovascular agents. The most clinically significant anti-infective drug interactions involving enzyme induction are subtherapeutic concentrations resulting from the combination of rifampicin with various co-medications including anticoagulants, immunosuppressants, antiretrovirals, and oral contraceptives [125, 138, 139].
Conversely, the reduction of AUC and/or Cmax of anti-infectives by other drugs or environmental influences can result in a much greater chance of failure of therapy and possibly an increase in the development of resistance. This now also includes the novel class of direct-acting antivirals against HCV where resistance may develop associated with low plasma concentrations of these agents [140, 141].
Again, not all pharmacokinetic drug interactions involving anti-infectives are detrimental, however. Ketoconazole has been used for a number of years to inhibit the metabolism of oral cyclosporine by approximately 80%, thereby reducing the cost of therapy as well as the rates of rejection and infection. As mentioned previously, the administration of ritonavir or cobicistat to enhance the oral absorption of antiretrovirals is a well-known component of potent antiretroviral combination regimens [142].
Beneficial and detrimental pharmacodynamic antimicrobial drug interactions also exist. The use of lower concentrations of two synergistic antibacterials to reduce the toxicity of each while having the same pharmacologic effect has been advocated, although the clinical data supporting superior efficacy is weak. Synergistic combinations of antimicrobials may produce better results in the treatment of Pseudomonas aeruginosa and Enterococcus species. Clinical data are largely lacking for detrimental effects of potentially antagonistic combinations of antimicrobials (e.g., a bacteriostatic drug combined with a bactericidal agent). However, these combinations are best avoided unless clinically warranted for the treatment of multiple pathogens.
2.8 Preclinical Methods for Predicting Drug Interactions
Although understanding and anticipating pharmacokinetic drug interactions are important components of rational therapeutics, there is a limit to the number and scope of clinical studies that can reasonably be performed. The development of human in vitro models allows information to be obtained without the expense and potential risks involved in conducting human trials. However, scaling of in vitro data to the clinical situation is not always accurate, and the results of these methods may not be definitive. A primary focus of preclinical screening methods for assessing drug–drug interactions is the identification of isozymes responsible for the metabolism of these compounds and the relative contribution of an inhibited pathway to a compound’s overall elimination.
To account for variability in individual enzyme expression, positive controls for inhibition and induction should always be used (e.g., troleandomycin or ketoconazole for CYP3A inhibition, quinidine for CYP2D6 inhibition, and rifampicin for CYP3A induction). Modern technology has allowed in vitro screening techniques to become widely available, and much of these data are currently included in package inserts.
In addition, there is now guidance from FDA on how to select in vitro and in vivo systems for evaluating drug–drug interactions [143]. The following briefly summarizes the strengths and weaknesses of currently available in vitro human methodologies for assessing cytochrome P450 drug interactions and predicting their clinical significance (Table 2.4).
2.8.1 Purified P450 Isozymes
In an attempt to identify specific isozymes responsible for the metabolism of compounds, investigators have tried to isolate human cytochrome P450 enzymes and purify them from hepatic tissue. However, only small amounts of protein can be isolated at any one time, and specific isozymes from certain subfamilies often cannot be separated (e.g., CYP2C9 vs CYP2C19 vs CYP2C10). To ensure correct interpretation of the results obtained from this method, it is most critical to examine the isozyme purification methods and quality control procedures. This method has now been superceded by the use of recombinant human cytochrome P450 isozymes.
2.8.2 Recombinant Human P450 Isozymes
Complementary DNA expression has been used to produce recombinant human cytochrome P450 isozymes in yeast, insects, bacteria, and mammalian cells, to be used in in vitro interaction experiments [115, 144]. An advantage of these systems is the ability to identify specific isozymes of a subfamily that are responsible for the metabolism of a compound and to confirm interaction of a compound with suspected isozyme-selective inhibitors. However, this remains an artificial system, and discrepancies can exist between results obtained by complementary DNA methods and other in vitro systems.
2.8.3 Microsomes
Microsomes isolated from human hepatocytes have become the “gold standard” of in vitro experimentation for drug interactions [145,146,147]. Microsomes are isolated membranes of hepatocyte endoplasmic reticula and contain the cytochromes P450 in proportion to their in vivo representation. Given the large interindividual variability in CYP expression, using microsomes from a single individual may produce distorted results. To circumvent this, pooling microsomes from multiple sources in order to obtain an average representation of activity is advocated. Human microsomes are widely available at relatively low cost, but they can only be used to determine direct inhibition of metabolism. Investigations of drug–drug interactions involving induction or suppression of CYP isozymes require intact cellular machinery [110, 148].
2.8.4 Immortalized Cell Lines
An ideal in vitro model for studying drug–drug interactions involving inhibition, suppression, and induction would be a validated, immortalized, readily available cell line, the results from which could be extrapolated directly to the clinical environment. However, no such model currently exists. All available immortalized human cell lines do not maintain a full complement of cytochrome P450 enzyme activities, nor do they maintain other potentially important physiologic processes, including membrane transporters. One commonly used immortalized cell line is derived from a human hepatoma (HepG2 cells). This model has been investigated for CYP1A1 induction, but does not significantly express other cytochrome P450s [149, 150].
2.8.5 Liver Slices
Human liver slices have been used with moderate success in determining the hepatic metabolism of certain compounds. Liver slices are relatively easy to prepare, and they maintain the hepatic ultrastructure [151,152,153,154]. However, up to half of constitutive (baseline) cytochrome P450 activity is lost within the first 24 h after isolation, and all constitutive cytochrome P450 activity is lost by 96 h. This makes investigations of induction and suppression of drug-metabolizing enzyme activity difficult. In addition, a distribution equilibrium is not achieved between all hepatocytes within the slice and the incubation media, resulting in decreased rates of metabolism compared to a hepatocyte monolayer culture system.
2.8.6 Human Hepatocyte Cultures
Primary human hepatocyte culture systems are ideal for studying drug interactions, as they maintain both Phase I and Phase II activity and form and maintain physiologic processes such as biliary canaliculi and transporters [153, 155]. Determining drug interactions in this system often allows for the closest prediction of potential drug interactions. Although this system does not mimic the pharmacokinetic alterations in drug concentrations seen clinically, it does allow quantitation of “best” and “worst” scenarios that may be extrapolated to the clinical setting. Inhibition, suppression, and induction interactions can all be performed with this model. Although maintaining constitutive levels of cytochrome P450 activity has been challenging, currently available enriched media and improved culture conditions allow for maintenance of control activity for at least 72–96 h after isolation. Challenges encountered with this system are primarily in obtaining fresh hepatic tissue for digestion and the specialized technique of perfusion for isolation of the hepatocytes. In addition, with the wide variability in enzyme activity seen clinically, investigations in a limited number of hepatocyte preparations will not be able to definitively reflect the occurrence of drug interactions in an entire population, but only suggest the potential for interactions to occur. These limitations (availability and reproducibility) can be partially overcome with cryopreserved human hepatocytes.
2.9 In Vitro/In Vivo Scaling of Drug Interactions
Extrapolating in vitro results to an in vivo situation is often complicated. The process of using in vitro models to predict drug interactions in vivo, preferably in humans, is still under development, and extensive validation of this approach is needed. In vitro models predictive of drug interactions are essential for rapid, cost-effective screening of pharmaceutical compounds and are important for reducing risks to patient safety. Currently these models are constructed from a combination of laboratory and theoretical components [150, 156,157,158]. In addition, preclinical screening of promising compounds frequently include the study of nonhuman mammalian species, although interspecies differences in expression and regulation of transporters and enzymes are well documented [159,160,161]. These differences limit the translation of preclinical animal data to the human situation.
Ideally, in a valid model, the clinical decrease in clearance caused by coadministration of an inhibitor would be specifically predicted by the decrease in reaction velocity (e.g., formation rate of a metabolite) for the same compound in vitro when the inhibitor is present in the same concentration. However, presently available models contain a number of weaknesses and assumptions that make scaling of in vitro data to the clinical situation complicated and not always accurate. Poor predictions occur with compounds that have flow-dependent hepatic clearance, with mechanism-based inhibition , and with compounds that concurrently induce and inhibit enzyme activity. In addition, inhibitor and substrate plasma concentrations are not always proportional to the inhibitor and substrate concentrations to which the enzyme is exposed in vitro. For example, supratherapeutic, as opposed to clinically relevant, concentrations of inhibitors and substrates may be utilized. Furthermore, experimental conditions such as enzyme protein concentration and buffers can critically affect specific results and confound in vitro/in vivo correlations [158]. For example, in vitro and cell culture models can demonstrate extensive partitioning of lipophilic compounds into cells, with uptake not restricted by plasma protein binding.
In order to establish the feasibility of in vitro to in vivo scaling, most currently reported predictions of inhibitory drug interactions are retrospective. Presently available methods allow a general assessment of what may occur (i.e., an unlikely interaction versus a probable interaction). However, to be most useful, in vitro data should not only indicate the possibility of an interaction but also predict its magnitude and clinical importance. Until such a time, the clinical study remains the ultimate means by which a drug interaction and its importance can be assessed.
2.10 Overview of Clinical Methods for Predicting Drug Interactions
The primary cause of clinically significant drug interactions is the involvement of drug-metabolizing enzymes. An overview of relevant substrates, inhibitors, and inducers of CYP450 enzymes is given in Table 2.5. Because great variability exists in drug-metabolizing enzyme activity among subjects, and drug interactions may not achieve clinical significance in all patients, interactions may be better clinically predicted by the knowledge of individual patient isozyme activities. However, there is currently a need for the development of reliable, accurate, and noninvasive methods to monitor drug-metabolizing enzyme expression in humans in order to guide drug dosage, reduce toxicity, and predict potential drug interactions.
Genotyping involves identification of mutant genes causing poor or ultra-extensive metabolizer activity. Genotyping has been demonstrated to predict the clinical outcome of drug interactions involving both Phase I and Phase II metabolism. However, drug-metabolizing enzyme activity can be exquisitely sensitive to other non-genetic factors, i.e., environmental and physiologic influences. Therefore, genotyping allows for the determination of an individual’s genetic predisposition to a specific enzyme activity, but may not reflect true phenotype at any one point in time.
Phenotyping for drug-metabolizing enzymes or transporters is defined as measuring its actual in vivo activity in an individual [162]. This is performed by administration of a selective substrate (“probe”) for this enzyme and subsequent determination of appropriate pharmacokinetic parameters. The metric used may be systemic clearance of a drug eliminated exclusively by the respective enzyme, partial clearance for a metabolic pathway, or absorption rate in the case of a transporter. Other parameters such as single-point concentrations or ratios of metabolite over parent concentrations in plasma, saliva, and/or urine are also often used [162, 163]. Specific methods have been developed to phenotype CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A, and N-acetyltransferase activities [162]. Multiple substrates can be studied in a combination using a “cocktail approach,” which involves the administration of more than one probe drug simultaneously [162, 164].
Phenotyping offers the primary advantage of quantitating enzyme activity and accounts for combined genetic, environmental, and endogenous influences on drug-metabolizing or drug-transporting enzyme activity. However, a number of currently available phenotyping methods are invasive (requiring pharmacokinetic sampling of blood) and impractical (requiring multiple samples), and analytical methods are not readily available. With a simplification of phenotyping methods, and an increase in the availability of analytical procedures [163], it may be possible to use these methods to determine correlations between enzyme activity and the risk of significant drug interactions in individual patients.
More details can be found in Chap. 23.
The practice of therapeutic drug monitoring (TDM) , i.e., the measurement of drug concentrations and subsequent individualization of doses, is also a means to detect and monitor drug interactions in clinical practice. Currently, TDM is available for a range of antibiotics, among others for HIV drugs [165], anti-TB agents [166], antifungals [167], aminoglycosides [168], and vancomycin [169].
2.11 Conclusions and Future Directions
It is difficult to assess the true incidence and clinical significance of drug interactions. Understanding the mechanisms underlying drug interactions is important for the prediction and avoidance of drug toxicity when initiating combination therapy. Although multiple in vitro methods are currently in use to assess drug interactions, not all have allowed the prediction of clinically significant events. As drug interactions most commonly result from influences on drug-metabolizing enzymes, future research defining the origins of enzyme activity variability and characterizing individual patient activity will certainly improve our ability to predict these interactions and improve drug therapy.
References
Guengerich FP, Waterman MR, Egli M (2016) Recent structural insights into cytochrome P450 function. Trends Pharmacol Sci 37(8):625–640
Ong CE, Pan Y, Mak JW, Ismail R (2013) In vitro approaches to investigate cytochrome P450 activities: update on current status and their applicability. Expert Opin Drug Metab Toxicol 9(9):1097–1113
Rendic S, Guengerich FP (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem Res Toxicol 28(1):38–42
Ogawa R, Echizen H (2011) Clinically significant drug interactions with antacids: an update. Drugs 71(14):1839–1864
Ogawa R, Echizen H (2010) Drug-drug interaction profiles of proton pump inhibitors. Clin Pharmacokinet 49(8):509–533
Zhang L, Wu F, Lee SC, Zhao H, Zhang L (2014) pH-dependent drug-drug interactions for weak base drugs: potential implications for new drug development. Clin Pharmacol Ther 96(2):266–277
Terrault NA, Zeuzem S, Di Bisceglie AM, Lim JK, Pockros PJ, Frazier LM et al (2016) Effectiveness of Ledipasvir-Sofosbuvir combination in patients with hepatitis C virus infection and factors associated of sustained virologic response. Gastroenterology 151:1131
Falcon RW, Kakuda TN (2008) Drug interactions between HIV protease inhibitors and acid-reducing agents. Clin Pharmacokinet 47(2):75–89
Crauwels H, van Heeswijk RP, Stevens M, Buelens A, Vanveggel S, Boven K et al (2013) Clinical perspective on drug-drug interactions with the non-nucleoside reverse transcriptase inhibitor rilpivirine. AIDS Rev 15(2):87–101
Burger DM, Hugen PW, Kroon FP, Groeneveld P, Brinkman K, Foudraine NA et al (1998) Pharmacokinetic interaction between the proton pump inhibitor omeprazole and the HIV protease inhibitor indinavir. AIDS (London, England) 12(15):2080–2082
Chin TW, Loeb M, Fong IW (1995) Effects of an acidic beverage (Coca-Cola) on absorption of ketoconazole. Antimicrob Agents Chemother 39(8):1671–1675
Kanda Y, Kami M, Matsuyama T, Mitani K, Chiba S, Yazaki Y et al (1998) Plasma concentration of itraconazole in patients receiving chemotherapy for hematological malignancies: the effect of famotidine on the absorption of itraconazole. Hematol Oncol 16(1):33–37
Jaruratanasirikul S, Sriwiriyajan S (1998) Effect of omeprazole on the pharmacokinetics of itraconazole. Eur J Clin Pharmacol 54(2):159–161
Jaruratanasirikul S, Kleepkaew A (1997) Influence of an acidic beverage (Coca-Cola) on the absorption of itraconazole. Eur J Clin Pharmacol 52(3):235–237
Moreno F, Hardin TC, Rinaldi MG, Graybill JR (1993) Itraconazole-didanosine excipient interaction. JAMA 269(12):1508
Alffenaar JW, van Assen S, van der Werf TS, Kosterink JG, Uges DR (2009) Omeprazole significantly reduces posaconazole serum trough level. Clin Infect Dis Off Publ Infect Dis Soc Am 48(6):839
Krishna G, Moton A, Ma L, Medlock MM, McLeod J (2009) Pharmacokinetics and absorption of posaconazole oral suspension under various gastric conditions in healthy volunteers. Antimicrob Agents Chemother 53(3):958–966
Horowitz HW, Jorde UP, Wormser GP (1992) Drug interactions in use of dapsone for pneumocystis carinii prophylaxis. Lancet (London, England) 339(8795):747
Metroka CE, McMechan MF, Andrada R, Laubenstein LJ, Jacobus DP (1991) Failure of prophylaxis with dapsone in patients taking dideoxyinosine. N Engl J Med 325(10):737
Kraft WK, Chang PS, van Iersel ML, Waskin H, Krishna G, Kersemaekers WM (2014) Posaconazole tablet pharmacokinetics: lack of effect of concomitant medications altering gastric pH and gastric motility in healthy subjects. Antimicrob Agents Chemother 58(7):4020–4025
Johnson MD, Hamilton CD, Drew RH, Sanders LL, Pennick GJ, Perfect JR (2003) A randomized comparative study to determine the effect of omeprazole on the peak serum concentration of itraconazole oral solution. J Antimicrob Chemother 51(2):453–457
Knupp CA, Barbhaiya RH (1997) A multiple-dose pharmacokinetic interaction study between didanosine (Videx) and ciprofloxacin (Cipro) in male subjects seropositive for HIV but asymptomatic. Biopharm Drug Dispos 18(1):65–77
Polk RE (1989) Drug-drug interactions with ciprofloxacin and other fluoroquinolones. Am J Med 87(5A):76S–81S
Sahai J, Gallicano K, Oliveras L, Khaliq S, Hawley-Foss N, Garber G (1993) Cations in the didanosine tablet reduce ciprofloxacin bioavailability. Clin Pharmacol Ther 53(3):292–297
Campbell NR, Hasinoff BB (1991) Iron supplements: a common cause of drug interactions. Br J Clin Pharmacol 31(3):251–255
Moss DM, Siccardi M, Murphy M, Piperakis MM, Khoo SH, Back DJ et al (2012) Divalent metals and pH alter raltegravir disposition in vitro. Antimicrob Agents Chemother 56(6):3020–3026
Ramanathan S, Mathias A, Wei X, Shen G, Koziara J, Cheng A et al (2013) Pharmacokinetics of once-daily boosted elvitegravir when administered in combination with acid-reducing agents. J Acquir Immune Defic Syndr 64(1):45–50
Song I, Borland J, Arya N, Wynne B, Piscitelli S (2015) Pharmacokinetics of dolutegravir when administered with mineral supplements in healthy adult subjects. J Clin Pharmacol 55(5):490–496
Yuk JH, Nightingale CH, Quintiliani R, Yeston NS, Orlando R 3rd, Dobkin ED et al (1990) Absorption of ciprofloxacin administered through a nasogastric or a nasoduodenal tube in volunteers and patients receiving enteral nutrition. Diagn Microbiol Infect Dis 13(2):99–102
Yuk JH, Nightingale CH, Sweeney KR, Quintiliani R, Lettieri JT, Frost RW (1989) Relative bioavailability in healthy volunteers of ciprofloxacin administered through a nasogastric tube with and without enteral feeding. Antimicrob Agents Chemother 33(7):1118–1120
Greiff JM1, Rowbotham D (1994) Pharmacokinetic drug interactions with gastrointestinal motility modifying agents. 27(6):447—61
van Waterschoot RA, Schinkel AH (2011) A critical analysis of the interplay between cytochrome P450 3A and P-glycoprotein: recent insights from knockout and transgenic mice. Pharmacol Rev 63(2):390–410
Knight B, Troutman M, Thakker DR (2006) Deconvoluting the effects of P-glycoprotein on intestinal CYP3A: a major challenge. Curr Opin Pharmacol 6(5):528–532
Bertz RJ, Granneman GR (1997) Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet 32(3):210–258
Peters SA et al. (2016) Predicting drug extraction in the Human Gut Wall: Assessing contributions from drug metabolizing enzymes and transporter proteins using preclinical models. Clin Pharmacokinet 55:673–696
Thummel KE, Wilkinson GR (1998) In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol 38:389–430
Komura H, Iwaki M (2011) In vitro and in vivo small intestinal metabolism of CYP3A and UGT substrates in preclinical animals species and humans: species differences. Drug Metab Rev 43(4):476–498
Thelen K, Dressman JB (2009) Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol 61(5):541–558
Zanger UM, Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138(1):103–141
Thummel KE, O'Shea D, Paine MF, Shen DD, Kunze KL, Perkins JD et al (1996) Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther 59(5):491–502
Hanley MJ, Cancalon P, Widmer WW, Greenblatt DJ (2011) The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol 7(3):267–286
Seden K, Dickinson L, Khoo S, Back D (2010) Grapefruit-drug interactions. Drugs 70(18):2373–2407
Kupferschmidt HH, Fattinger KE, Ha HR, Follath F, Krahenbuhl S (1998) Grapefruit juice enhances the bioavailability of the HIV protease inhibitor saquinavir in man. Br J Clin Pharmacol 45(4):355–359
Kawakami M, Suzuki K, Ishizuka T, Hidaka T, Matsuki Y, Nakamura H (1998) Effect of grapefruit juice on pharmacokinetics of itraconazole in healthy subjects. Int J Clin Pharmacol Ther 36(6):306–308
Cheng KL, Nafziger AN, Peloquin CA, Amsden GW (1998) Effect of grapefruit juice on clarithromycin pharmacokinetics. Antimicrob Agents Chemother 42(4):927–929
Kempf DJ, Marsh KC, Kumar G, Rodrigues AD, Denissen JF, McDonald E et al (1997) Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob Agents Chemother 41(3):654–660
Hsu A, Granneman GR, Bertz RJ (1998) Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin Pharmacokinet 35(4):275–291
Hill A, van der Lugt J, Sawyer W, Boffito M (2009) How much ritonavir is needed to boost protease inhibitors? Systematic review of 17 dose-ranging pharmacokinetic trials. AIDS (London, England) 23(17):2237–2245
Menon RM, Klein CE, Podsadecki TJ, Chiu YL, Dutta S, Awni WM (2016) Pharmacokinetics and tolerability of paritaprevir, a direct acting antiviral agent for hepatitis C virus treatment, with and without ritonavir in healthy volunteers. Br J Clin Pharmacol 81(5):929–940
Brayer SW, Reddy KR (2015) Ritonavir-boosted protease inhibitor based therapy: a new strategy in chronic hepatitis C therapy. Expert Rev Gastroenterol Hepatol 9(5):547–558
Shah BM, Schafer JJ, Priano J, Squires KE (2013) Cobicistat: a new boost for the treatment of human immunodeficiency virus infection. Pharmacotherapy 33(10):1107–1116
Nathan B, Bayley J, Waters L, Post FA (2013) Cobicistat: a novel pharmacoenhancer for co-formulation with HIV protease and integrase inhibitors. Infectious diseases and therapy 2(2):111–122
Silva R, Vilas-Boas V, Carmo H, Dinis-Oliveira RJ, Carvalho F, de Lourdes BM et al (2015) Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther 149:1–123
Konig J, Muller F, Fromm MF (2013) Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev 65(3):944–966
Estudante M, Morais JG, Soveral G, Benet LZ (2013) Intestinal drug transporters: an overview. Adv Drug Deliv Rev 65(10):1340–1356
Mulgaonkar A, Venitz J, Sweet DH (2012) Fluoroquinolone disposition: identification of the contribution of renal secretory and reabsorptive drug transporters. Expert Opin Drug Metab Toxicol 8(5):553–569
Pal D, Kwatra D, Minocha M, Paturi DK, Budda B, Mitra AK (2011) Efflux transporters- and cytochrome P-450-mediated interactions between drugs of abuse and antiretrovirals. Life Sci 88(21–22):959–971
Sevrioukova IF, Poulos TL (2013) Understanding the mechanism of cytochrome P450 3A4: recent advances and remaining problems. Dalton Trans (Cambridge, England : 2003) 42(9):3116–3126
Feng B, Varma MV, Costales C, Zhang H, Tremaine L (2014) In vitro and in vivo approaches to characterize transporter-mediated disposition in drug discovery. Expert Opin Drug Discovery 9(8):873–890
Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X et al (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9(3):215–236
Salama NN, Eddington ND, Fasano A (2006) Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev 58(1):15–28
Assimakopoulos SF, Dimitropoulou D, Marangos M, Gogos CA (2014) Intestinal barrier dysfunction in HIV infection: pathophysiology, clinical implications and potential therapies. Infection 42(6):951–959
Zakeri-Milani P, Valizadeh H (2014) Intestinal transporters: enhanced absorption through P-glycoprotein-related drug interactions. Expert Opin Drug Metab Toxicol 10(6):859–871
Glaeser H (2011) Importance of P-glycoprotein for drug-drug interactions. Handb Exp Pharmacol 201:285–297
Lown KS, Fontana RJ, Schmiedlin-Ren P et al (1995) Interindividual variation in intestinal mdr1:lack of short diet effects. Gastroenterology 108:A737
Lamba J, Strom S, Venkataramanan R, Thummel KE, Lin YS, Liu W et al (2006) MDR1 genotype is associated with hepatic cytochrome P450 3A4 basal and induction phenotype. Clin Pharmacol Ther 79(4):325–338
Wakasugi H, Yano I, Ito T, Hashida T, Futami T, Nohara R et al (1998) Effect of clarithromycin on renal excretion of digoxin: interaction with P-glycoprotein. Clin Pharmacol Ther 64(1):123–128
Cormet-Boyaka E, Huneau JF, Mordrelle A, Boyaka PN, Carbon C, Rubinstein E et al (1998) Secretion of sparfloxacin from the human intestinal Caco-2 cell line is altered by P-glycoprotein inhibitors. Antimicrob Agents Chemother 42(10):2607–2611
Srinivas RV, Middlemas D, Flynn P, Fridland A (1998) Human immunodeficiency virus protease inhibitors serve as substrates for multidrug transporter proteins MDR1 and MRP1 but retain antiviral efficacy in cell lines expressing these transporters. Antimicrob Agents Chemother 42(12):3157–3162
Neumanova Z, Cerveny L, Ceckova M, Staud F (2014) Interactions of tenofovir and tenofovir disoproxil fumarate with drug efflux transporters ABCB1, ABCG2, and ABCC2; role in transport across the placenta. AIDS (London, England) 28(1):9–17
Lempers VJ, van den Heuvel JJ, Russel FG, Aarnoutse RE, Burger DM, Bruggemann RJ et al (2016) Inhibitory potential of antifungal drugs on ATP-binding cassette transporters P-glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob Agents Chemother 60(6):3372–3379
Kirby BJ, Symonds WT, Kearney BP, Mathias AA (2015) Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor Sofosbuvir. Clin Pharmacokinet 54(7):677–690
Takano M, Hasegawa R, Fukuda T, Yumoto R, Nagai J, Murakami T (1998) Interaction with P-glycoprotein and transport of erythromycin, midazolam and ketoconazole in Caco-2 cells. Eur J Pharmacol 358(3):289–294
Sansom LN, Evans AM (1995) What is the true clinical significance of plasma protein binding displacement interactions? Drug Saf 12(4):227–233
Onufrak NJ, Forrest A, Gonzalez D (2016) Pharmacokinetic and pharmacodynamic principles of anti-infective dosing. Clin Ther 38:1930
Barbour A, Scaglione F, Derendorf H (2010) Class-dependent relevance of tissue distribution in the interpretation of anti-infective pharmacokinetic/pharmacodynamic indices. Int J Antimicrob Agents 35(5):431–438
Boffito M, Back DJ, Blaschke TF, Rowland M, Bertz RJ, Gerber JG et al (2003) Protein binding in antiretroviral therapies. AIDS Res Hum Retrovir 19(9):825–835
Back DJ, Burger DM (2015) Interaction between amiodarone and sofosbuvir-based treatment for hepatitis C virus infection: potential mechanisms and lessons to be learned. Gastroenterology 149(6):1315–1317
Rolan PE (1994) Plasma protein binding displacement interactions--why are they still regarded as clinically important? Br J Clin Pharmacol 37(2):125–128
Schmidt S, Gonzalez D, Derendorf H (2010) Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci 99(3):1107–1122
Benet LZ, Hoener BA (2002) Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther 71(3):115–121
Schmidt S, Barbour A, Sahre M, Rand KH, Derendorf H (2008) PK/PD: new insights for antibacterial and antiviral applications. Curr Opin Pharmacol 8(5):549–556
Nelson DR (2006) Cytochrome P450 nomenclature, 2004. Methods Mol Biol 320:1–10
Martiny VY, Miteva MA (2013) Advances in molecular modeling of human cytochrome P450 polymorphism. J Mol Biol 425(21):3978–3992
Murray M (1997) Drug-mediated inactivation of cytochrome P450. Clin Exp Pharmacol Physiol 24(7):465–470
Vanden Bossche H, Koymans L, Moereels H (1995) P450 inhibitors of use in medical treatment: focus on mechanisms of action. Pharmacol Ther 67(1):79–100
Ma Q, Lu AY (2011) Pharmacogenetics pharmacogenomics, and individualized medicine. Pharmacol Rev 63(2):437–459
Michaud V, Bar-Magen T, Turgeon J, Flockhart D, Desta Z, Wainberg MA (2012) The dual role of pharmacogenetics in HIV treatment: mutations and polymorphisms regulating antiretroviral drug resistance and disposition. Pharmacol Rev 64(3):803–833
Backman JT, Filppula AM, Niemi M, Neuvonen PJ (2016) Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharmacol Rev 68(1):168–241
Werk AN, Cascorbi I (2014) Functional gene variants of CYP3A4. Clin Pharmacol Ther 96(3):340–348
Naidoo P, Chetty VV, Chetty M (2014) Impact of CYP polymorphisms, ethnicity and sex differences in metabolism on dosing strategies: the case of efavirenz. Eur J Clin Pharmacol 70(4):379–389
Semvua HH, Mtabho CM, Fillekes Q, van den Boogaard J, Kisonga RM, Mleoh L et al (2013) Efavirenz, tenofovir and emtricitabine combined with first-line tuberculosis treatment in tuberculosis-HIV-coinfected Tanzanian patients: a pharmacokinetic and safety study. Antivir Ther 18(1):105–113
van Luin M, Brouwer AM, van der Ven A, de Lange W, van Schaik RH, Burger DM (2009) Efavirenz dose reduction to 200 mg once daily in a patient treated with rifampicin. AIDS (London, England) 23(6):742–744
Zhu L, Bruggemann RJ, Uy J, Colbers A, Hruska MW, Chung E et al (2016) CYP2C19 genotype-dependent pharmacokinetic drug interaction between voriconazole and ritonavir-boosted atazanavir in healthy subjects. J Clin Pharmacol
Kamel A, Harriman S (2013) Inhibition of cytochrome P450 enzymes and biochemical aspects of mechanism-based inactivation (MBI). Drug Discov Today Technol 10(1):e177–e189
Greenblatt DJ (2014) In vitro prediction of clinical drug interactions with CYP3A substrates: we are not there yet. Clin Pharmacol Ther 95(2):133–135
Brown HS, Galetin A, Hallifax D, Houston JB (2006) Prediction of in vivo drug-drug interactions from in vitro data : factors affecting prototypic drug-drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin Pharmacokinet 45(10):1035–1050
Hisaka A, Ohno Y, Yamamoto T, Suzuki H (2010) Prediction of pharmacokinetic drug-drug interaction caused by changes in cytochrome P450 activity using in vivo information. Pharmacol Ther 125(2):230–248
Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H (2008) Inhibition and induction of human cytochrome P450 enzymes: current status. Arch Toxicol 82(10):667–715
Ke AB, Zamek-Gliszczynski MJ, Higgins JW, Hall SD (2014) Itraconazole and clarithromycin as ketoconazole alternatives for clinical CYP3A inhibition studies. Clin Pharmacol Ther 95(5):473–476
Zhou S, Yung Chan S, Cher Goh B, Chan E, Duan W, Huang M et al (2005) Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet 44(3):279–304
Trenholme GM, Williams RL, Rieckmann KH, Frischer H, Carson PE (1976) Quinine disposition during malaria and during induced fever. Clin Pharmacol Ther 19(4):459–467
Lee JI, Zhang L, Men AY, Kenna LA, Huang SM (2010) CYP-mediated therapeutic protein-drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet 49(5):295–310
Morgan ET (2009) Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther 85(4):434–438
Aitken AE, Richardson TA, Morgan ET (2006) Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol 46:123–149
Zhu M, Kaul S, Nandy P, Grasela DM, Pfister M (2009) Model-based approach to characterize efavirenz autoinduction and concurrent enzyme induction with carbamazepine. Antimicrob Agents Chemother 53(6):2346–2353
Smythe W, Khandelwal A, Merle C, Rustomjee R, Gninafon M, Bocar Lo M et al (2012) A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob Agents Chemother 56(4):2091–2098
Rana R, Chen Y, Ferguson SS, Kissling GE, Surapureddi S, Goldstein JA (2010) Hepatocyte nuclear factor 4{alpha} regulates rifampicin-mediated induction of CYP2C genes in primary cultures of human hepatocytes. Drug Metab Dispos 38(4):591–599
Fahmi OA, Ripp SL (2010) Evaluation of models for predicting drug-drug interactions due to induction. Expert Opin Drug Metab Toxicol 6(11):1399–1416
Sinz M, Wallace G, Sahi J (2008) Current industrial practices in assessing CYP450 enzyme induction: preclinical and clinical. AAPS J 10(2):391–400
Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL et al (2007) Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther 320(1):72–80
Zangar RC, Bollinger N, Verma S, Karin NJ, Lu Y (2008) The nuclear factor-kappa B pathway regulates cytochrome P450 3A4 protein stability. Mol Pharmacol 73(6):1652–1658
Wu B, Kulkarni K, Basu S, Zhang S, Hu M (2011) First-pass metabolism via UDP-glucuronosyltransferase: a barrier to oral bioavailability of phenolics. J Pharm Sci 100(9):3655–3681
Devineni D, Vaccaro N, Murphy J, Curtin C, Mamidi RN, Weiner S et al (2015) Effects of rifampin, cyclosporine A, and probenecid on the pharmacokinetic profile of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in healthy participants. Int J Clin Pharmacol Ther 53(2):115–128
Walsky RL, Bauman JN, Bourcier K, Giddens G, Lapham K, Negahban A et al (2012) Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab Dispos 40(5):1051–1065
Belanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C (2009) Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine. Drug Metab Dispos 37(9):1793–1796
Burger DM, Huisman A, Van Ewijk N, Neisingh H, Van Uden P, Rongen GA et al (2008) The effect of atazanavir and atazanavir/ritonavir on UDP-glucuronosyltransferase using lamotrigine as a phenotypic probe. Clin Pharmacol Ther 84(6):698–703
van der Lee MJ, Dawood L, ter Hofstede HJ, de Graaff-Teulen MJ, van Ewijk-Beneken Kolmer EW, Caliskan-Yassen N et al (2006) Lopinavir/ritonavir reduces lamotrigine plasma concentrations in healthy subjects. Clin Pharmacol Ther 80(2):159–168
Sim SC, Kacevska M, Ingelman-Sundberg M (2013) Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J 13(1):1–11
Yiannakopoulou E (2013) Pharmacogenomics of phase II metabolizing enzymes and drug transporters: clinical implications. Pharmacogenomics J 13(2):105–109
Perwitasari DA, Atthobari J, Wilffert B (2015) Pharmacogenetics of isoniazid-induced hepatotoxicity. Drug Metab Rev 47(2):222–228
Knights KM, Rowland A, Miners JO (2013) Renal drug metabolism in humans: the potential for drug-endobiotic interactions involving cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT). Br J Clin Pharmacol 76(4):587–602
Jansen A, Colbers EP, van der Ven AJ, Richter C, Rockstroh JK, Wasmuth JC et al (2013) Pharmacokinetics of the combination raltegravir/atazanavir in HIV-1-infected patients. HIV Med 14(7):449–452
Bollen P, Reiss P, Schapiro J, Burger D (2015) Clinical pharmacokinetics and pharmacodynamics of dolutegravir used as a single tablet regimen for the treatment of HIV-1 infection. Expert Opin Drug Saf 14(9):1457–1472
Regazzi M, Carvalho AC, Villani P, Matteelli A (2014) Treatment optimization in patients co-infected with HIV and mycobacterium tuberculosis infections: focus on drug-drug interactions with rifamycins. Clin Pharmacokinet 53(6):489–507
Higgins LG, Hayes JD (2011) Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev 43(2):92–137
Nijland HM, Ruslami R, Suroto AJ, Burger DM, Alisjahbana B, van Crevel R et al (2007) Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin Infect Dis Off Publ Infect Dis Soc Am 45(8):1001–1007
Weiner M, Burman W, Luo CC, Peloquin CA, Engle M, Goldberg S et al (2007) Effects of rifampin and multidrug resistance gene polymorphism on concentrations of moxifloxacin. Antimicrob Agents Chemother 51(8):2861–2866
Feng B, LaPerle JL, Chang G, Varma MV (2010) Renal clearance in drug discovery and development: molecular descriptors, drug transporters and disease state. Expert Opin Drug Metab Toxicol 6(8):939–952
Yombi JC, Pozniak A, Boffito M, Jones R, Khoo S, Levy J et al (2014) Antiretrovirals and the kidney in current clinical practice: renal pharmacokinetics, alterations of renal function and renal toxicity. AIDS (London, England) 28(5):621–632
Morrissey KM, Stocker SL, Wittwer MB, Xu L, Giacomini KM (2013) Renal transporters in drug development. Annu Rev Pharmacol Toxicol 53:503–529
Kampmann J, Hansen JM, Siersboek-Nielsen K, Laursen H (1972) Effect of some drugs on penicillin half-life in blood. Clin Pharmacol Ther 13(4):516–519
Douros A, Grabowski K, Stahlmann R (2015) Safety issues and drug-drug interactions with commonly used quinolones. Expert Opin Drug Metab Toxicol 11(1):25–39
Cohen O, Locketz G, Hershko AY, Gorshtein A, Levy Y (2015) Colchicine-clarithromycin-induced rhabdomyolysis in familial mediterranean fever patients under treatment for helicobacter pylori. Rheumatol Int 35(11):1937–1941
Bruggemann RJ, Alffenaar JW, Blijlevens NM, Billaud EM, Kosterink JG, Verweij PE et al (2009) Clinical relevance of the pharmacokinetic interactions of azole antifungal drugs with other coadministered agents. Clin Infect Dis Off Publ Infect Dis Soc Am 48(10):1441–1458
Woytowish MR, Maynor LM (2013) Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 47(3):388–397
Owens RC Jr, Nolin TD (2006) Antimicrobial-associated QT interval prolongation: pointes of interest. Clin Infect Dis Off Publ Infect Dis Soc Am 43(12):1603–1611
Chen J, Raymond K (2006) Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob 5:3
Semvua HH, Kibiki GS, Kisanga ER, Boeree MJ, Burger DM, Aarnoutse R (2015) Pharmacological interactions between rifampicin and antiretroviral drugs: challenges and research priorities for resource-limited settings. Ther Drug Monit 37(1):22–32
Langness JA, Everson GT (2016) Viral hepatitis: drug-drug interactions in HCV treatment--the good, the bad and the ugly. Nat Rev Gastroenterol Hepatol 13(4):194–195
Burger D, Back D, Buggisch P, Buti M, Craxi A, Foster G et al (2013) Clinical management of drug-drug interactions in HCV therapy: challenges and solutions. J Hepatol 58(4):792–800
Larson KB, Wang K, Delille C, Otofokun I, Acosta EP (2014) Pharmacokinetic enhancers in HIV therapeutics. Clin Pharmacokinet 53(10):865–872
FDA. Guidance for Industry. Drug interaction studies — study design, data analysis, implications for dosing, and labeling recommendations 2012 [September 13, 2016]. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
Stringer RA, Strain-Damerell C, Nicklin P, Houston JB (2009) Evaluation of recombinant cytochrome P450 enzymes as an in vitro system for metabolic clearance predictions. Drug Metab Dispos Biol Fate Chem 37(5):1025–1034
Spaggiari D, Geiser L, Daali Y, Rudaz S (2014) A cocktail approach for assessing the in vitro activity of human cytochrome P450s: an overview of current methodologies. J Pharm Biomed Anal 101:221–237
Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AY (2011) Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re-evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 36(1):1–16
Parkinson A, Kazmi F, Buckley DB, Yerino P, Ogilvie BW, Paris BL (2010) System-dependent outcomes during the evaluation of drug candidates as inhibitors of cytochrome P450 (CYP) and uridine diphosphate glucuronosyltransferase (UGT) enzymes: human hepatocytes versus liver microsomes versus recombinant enzymes. Drug Metab Pharmacokinet 25(1):16–27
Donato MT, Lahoz A, Castell JV, Gomez-Lechon MJ (2008) Cell lines: a tool for in vitro drug metabolism studies. Curr Drug Metab 9(1):1–11
Alqahtani S, Mohamed LA, Kaddoumi A (2013) Experimental models for predicting drug absorption and metabolism. Expert Opin Drug Metab Toxicol 9(10):1241–1254
Scotcher D, Jones C, Posada M, Rostami-Hodjegan A, Galetin A (2016) Key to opening kidney for in vitro-in vivo extrapolation entrance in health and disease: part I: in vitro systems and physiological data. AAPS J
Graaf IA, Groothuis GM, Olinga P (2007) Precision-cut tissue slices as a tool to predict metabolism of novel drugs. Expert Opin Drug Metab Toxicol 3(6):879–898
de Graaf IA, Olinga P, de Jager MH, Merema MT, de Kanter R, van de Kerkhof EG et al (2010) Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc 5(9):1540–1551
Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S et al (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87(8):1315–1530
Olinga P, Schuppan D (2013) Precision-cut liver slices: a tool to model the liver ex vivo. J Hepatol 58(6):1252–1253
Lash LH, Putt DA, Cai H (2008) Drug metabolism enzyme expression and activity in primary cultures of human proximal tubular cells. Toxicology 244(1):56–65
Peters SA, Jones CR, Ungell AL, Hatley OJ (2016) Predicting drug extraction in the human gut wall: assessing contributions from drug metabolizing enzymes and transporter proteins using preclinical models. Clin Pharmacokinet 55(6):673–696
Moss DM, Marzolini C, Rajoli RK, Siccardi M (2015) Applications of physiologically based pharmacokinetic modeling for the optimization of anti-infective therapies. Expert Opin Drug Metab Toxicol 11(8):1203–1217
Rostami-Hodjegan A (2012) Physiologically based pharmacokinetics joined with in vitro-in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther 92(1):50–61
Chu X, Bleasby K, Evers R (2013) Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin Drug Metab Toxicol 9(3):237–252
Graham MJ, Lake BG (2008) Induction of drug metabolism: species differences and toxicological relevance. Toxicology 254(3):184–191
Martignoni M, Groothuis GM, de Kanter R (2006) Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2(6):875–894
Fuhr U, Jetter A, Kirchheiner J (2007) Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the “cocktail” approach. Clin Pharmacol Ther 81(2):270–283
De Kesel PM, Lambert WE, Stove CP (2016) Alternative sampling strategies for cytochrome P450 phenotyping. Clin Pharmacokinet 55(2):169–184
Breimer DD, Schellens JH (1990) A ‘cocktail’ strategy to assess in vivo oxidative drug metabolism in humans. Trends Pharmacol Sci 11(6):223–225
Pretorius E, Klinker H, Rosenkranz B (2011) The role of therapeutic drug monitoring in the management of patients with human immunodeficiency virus infection. Ther Drug Monit 33(3):265–274
Alsultan A, Peloquin CA (2014) Therapeutic drug monitoring in the treatment of tuberculosis: an update. Drugs 74(8):839–854
Bruggemann RJ, Aarnoutse RE (2015) Fundament and prerequisites for the application of an antifungal TDM service. Curr Fungal Infect Rep 9(2):122–129
Croes S, Koop AH, van Gils SA, Neef C (2012) Efficacy, nephrotoxicity and ototoxicity of aminoglycosides, mathematically modelled for modelling-supported therapeutic drug monitoring. Eur J Pharm Sci Off J Eur Fed Pharm Sci 45(1–2):90–100
Ye ZK, Li C, Zhai SD (2014) Guidelines for therapeutic drug monitoring of vancomycin: a systematic review. PLoS One 9(6):e99044
Acknowledgments
We would like to thank Kevin C. Brown and Angela D. M. Kashuba for providing us with the text files of this chapter in the previous edition.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Burger, D.M., te Brake, L.H.M., Aarnoutse, R.E. (2018). Mechanisms of Drug Interactions I: Absorption, Metabolism, and Excretion. In: Pai, M., Kiser, J., Gubbins, P., Rodvold, K. (eds) Drug Interactions in Infectious Diseases: Mechanisms and Models of Drug Interactions. Infectious Disease. Humana Press, Cham. https://doi.org/10.1007/978-3-319-72422-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-72422-5_2
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-72421-8
Online ISBN: 978-3-319-72422-5
eBook Packages: MedicineMedicine (R0)