
Abstract
Tuberculosis (TB) is the world’s second leading infectious killer. Cases of multidrug-resistant (MDR-TB) and extremely drug-resistant (XDR-TB) have increased globally. Therapeutic drug monitoring (TDM) remains a standard clinical technique for using plasma drug concentrations to determine dose. For TB patients, TDM provides objective information for the clinician to make informed dosing decisions. Some patients are slow to respond to treatment, and TDM can shorten the time to response and to treatment completion. Normal plasma concentration ranges for the TB drugs have been defined. For practical reasons, only one or two samples are collected post-dose. A 2-h post-dose sample approximates the peak serum drug concentration (Cmax) for most TB drugs. Adding a 6-h sample allows the clinician to distinguish between delayed absorption and malabsorption. TDM requires that samples are promptly centrifuged, and that the serum is promptly harvested and frozen. Isoniazid and ethionamide, in particular, are not stable in human serum at room temperature. Rifampicin is stable for more than 6 h under these conditions. Since our 2002 review, several papers regarding TB drug pharmacokinetics, pharmacodynamics, and TDM have been published. Thus, we have better information regarding the concentrations required for effective TB therapy. In vitro and animal model data clearly show concentration responses for most TB drugs. Recent studies emphasize the importance of rifamycins and pyrazinamide as sterilizing agents. A strong argument can be made for maximizing patient exposure to these drugs, short of toxicity. Further, the very concept behind ‘minimal inhibitory concentration’ (MIC) implies that one should achieve concentrations above the minimum in order to maximize response. Some, but not all clinical data are consistent with the utility of this approach. The low ends of the TB drug normal ranges set reasonable ‘floors’ above which plasma concentrations should be maintained. Patients with diabetes and those infected with HIV have a particular risk for poor drug absorption, and for drug–drug interactions. Published guidelines typically describe interactions between two drugs, whereas the clinical situation often is considerably more complex. Under ‘real–life’ circumstances, TDM often is the best available tool for sorting out these multi-drug interactions, and for providing the patient safe and adequate doses. Plasma concentrations cannot explain all of the variability in patient responses to TB treatment, and cannot guarantee patient outcomes. However, combined with clinical and bacteriological data, TDM can be a decisive tool, allowing clinicians to successfully treat even the most complicated TB patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Tuberculosis (TB) will not be going away any time soon. The World Health Organization (WHO) estimates that 8.6 million people developed TB and 1.3 million died from it in 2012 [1]. Since the publication of an earlier edition of therapeutic drug monitoring (TDM) in the treatment of TB in Drugs [2] cases of multidrug-resistant TB (MDR-TB) have increased globally, and cases of extremely drug-resistant TB (XDR-TB) are common in countries already burdened with MDR-TB [3]. Therefore, we are updating that prior review. If the current regimen and current doses were completely adequate, this should not be happening. ‘Something’ is interfering with TB elimination. To be sure, the current regimens can work, and clinical trials have shown success rates at approximately 95 %, based primarily upon ‘per protocol’ analyses [4–8]. However, as with many other diseases, TB treatment results in general practice often fall short of clinical trial results. The reasons for this shortfall may include a more diverse patient population in general practice, and co-morbid conditions that either were excluded from the trials, or were not as prevalent when the original clinical trials took place (examples: diabetes mellitus and HIV). Further, trial clinicians may be better equipped to ensure patient adherence, and they may be particularly attentive to patient follow-up.
Mycobacterium tuberculosis is never considered a colonizer, nor is it considered part of the normal human flora on any surface, or in any organ or secretion. Latent TB infection (LTBI) is an infection that has been contained by the immune system, but one that can reactivate given the right conditions [9]. LTBI can be treated with isoniazid alone, or the new combination of isoniazid and rifapentine [10]. In contrast, active TB disease always requires treatment with multiple drugs [11]. In comparison with drugs used for bacterial infections, the mechanisms of action for several of the TB drugs are poorly understood, although this field continues to advance [12–15]. Traditionally, the treatment of TB disease can be divided into two parts. The initial, intensive phase typically uses at least four drugs, and is designed to rapidly reduce the total body burden of TB [8, 11, 16]. It appears that many of these organisms are extracellular (that is, not within macrophages) and are multiplying approximately once daily, making them more accessible to bactericidal drugs. Next, generally after 2 months of intensive treatment, the continuation phase is designed to eliminate the ‘persisters’. These are organisms that survive the initial phase of treatment, for reasons that are poorly understood. Persisters are thought to be the primary source of relapses following treatment regimens of insufficient duration (i.e., <6 months for most patients). It is not known how similar TB persisters are to organisms found during LTBI; they may represent distinct physiological states [17–21]. Because so little is known about these persisters, it is difficult to design optimal treatment regimens for them.
The majority of patients who receive 6 or more months of treatment for drug-susceptible TB will respond completely [11]. Cure rates vary by country, and as noted, often differ in general practice as compared with clinical trials. Most regimens include isoniazid, rifampicin, pyrazinamide, and ethambutol [11]. Isoniazid is good at reducing the bacillary load rapidly (bactericidal activity evident within 2 days) and rifampicin and pyrazinamide are the best sterilizing agents (i.e., preventing post-treatment relapses) [8, 16]. The typical adult rifampicin dose of 600 mg appears to be at the low end of the effective range, and new clinical trials are showing better responses with higher daily doses of rifampicin or rifapentine (also known as cyclopentyl-rifampicin) [16, 22–27]. According to the US Centers for Disease Control and Prevention (CDC), the proportion of patients who completed therapy within 1 year increased from 64 % in 1993 to 88 % in 2010 (the latest year for which complete outcome data are available). However, these completion rates address patients who are given what should be a 6-month treatment regimen [28]. Thus, statements that “6-month treatment regimens are 95–98 % effective” do not reflect what actually happens in general practice, even in countries with excellent TB control infrastructures. The reasons for these disparities are multi-factorial, but at their core, revolve around the drug therapy itself. This simply must be the case, since very few TB patients in any country are cured surgically.
Given the above, we think TDM has a role and can improve the treatment of TB. TDM remains a standard clinical technique for using plasma drug concentrations to determine dose. TDM provides objective information for the clinician to make informed dosing decisions. Some patients are slow to respond to treatment, and TDM can shorten the time to response and to treatment completion. Patients with drug-resistant TB are treated with weak drugs, and TDM can determine that adequate concentrations have been achieved.
Since our 2002 review, several papers regarding TB drug pharmacokinetics, pharmacodynamics, and TDM have been published. Thus, we have better information regarding the concentrations required for effective TB therapy, and this field of research continues to grow. In this paper, we review these recent advances, and how they apply clinically.
2 Therapeutic Drug Monitoring (TDM) Studies Since 2002
Over the past decade, several studies have been published describing the application of TDM and its benefits in TB clinical practice [29–36]. The search engine used to find these papers was PubMed, and the medical terms used were therapeutic drug monitoring, tuberculosis, and treatment. The search was limited to 2003–2013. Most papers focused on drug concentrations for the first-line TB drugs in the treatment of drug-susceptible TB. The normal ranges from our 2002 review are widely accepted, and were used in these published studies. We highlight a few of those studies below.
Heysell et al. [29] performed a retrospective study to assess the prevalence of low TB drug concentrations in patients with slow responses to TB treatment in the state of Virginia, USA. Patients (n = 42) with slow responses were selected for routine TDM. Drug concentrations were measured at 2 h post-dose and were used as estimated peak serum drug concentration (Cmax) concentrations. Only rifampicin drug concentrations were available for all 42 patients, and were below the expected range in 22 patients (52 %). Isoniazid drug concentrations were low in 23 of 39 patients tested (59 %). For pyrazinamide, all 20 patients tested had normal concentrations, and, for ethambutol, concentrations were low in eight of 26 patients (31 %). In summary, most patients with slow responses to TB treatment had low 2-h concentrations of rifampicin and isoniazid. The authors concluded that TDM could help to identify the cause of slow responses to TB treatment. It is important to note that 2-h drug concentrations alone will not capture delayed Cmax values [29].
Another study looked at TDM in a selected group of patients [30]. The authors describe how TDM was used to manage difficult TB cases in their clinic. The first case described a patient with a relapse of TB. The patient was initially treated with the standard four-drug regimen under directly observed treatment (DOT). Nine months after treatment completion, he presented with a relapse. The patient restarted the standard regimen, but there was no clinical improvement after 1 month of treatment. Therefore, drug concentrations were measured for isoniazid, rifampicin, and pyrazinamide at 2 h. Isoniazid was undetectable, the rifampicin concentration was low at 5.6 mg/l, and pyrazinamide was low at 8.3 mg/l. Doses for all three drugs were increased, and drug concentrations were measured again. Isoniazid remained trace, rifampicin increased to 15.2 mg/l, and pyrazinamide increased to 12.2 mg/l, which is still low. More importantly, the patient’s clinical signs improved, and he was culture negative by week 6. The patient completed treatment and remained culture negative until his last follow-up visit 7 months after treatment completion. The authors also discussed three other difficult cases with relapse TB who had low drug concentrations. They used TDM to adjust the doses for these patients, and achieved good clinical responses [30].
A recent study by Babalik et al. [31] described the application of TDM in their practice. Indications to perform TDM in their center were a slow clinical response to TB treatment, or having other comorbid conditions like HIV. TDM was performed for 20 patients in their center. Low drug concentrations of any TB drug were present in 17 of the 20 patients. Isoniazid concentrations were low in 13 of 15 patients (87 %), rifampicin concentrations were low in 8 of 12 patients (67 %), and pyrazinamide concentration was low in only 2 of 13 patients (15 %). Rifabutin concentrations also were measured, and were low in four of five patients (80 %). Time to culture conversion was higher in the 17 patients with low drug concentrations than in the three patients with normal concentrations. Overall, most patients had low concentrations of the TB drugs. The authors also found that it was difficult to achieve adequate concentrations for rifabutin in patients with HIV. The authors considered TDM useful in the treatment of TB, especially in the presence of other comorbidities like HIV [31].
A study by Holland et al. [32] looked at the application of TDM in patients with TB and advanced HIV. They measured the concentrations of both isoniazid and the rifamycins (rifampicin or rifabutin). Of 21 patients included in the study, 18 had a low concentration of isoniazid and/or one of the rifamycins at 2 h (rifabutin was measured at 3 h) (86 %). The authors were able to increase the concentrations into the normal range in their patients by increasing the doses. This suggests that higher initial doses may be needed in patients with HIV. Other published data thus far are not definitive on this point.
At the 2013 Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) meeting, Prahl et al. [37] from Denmark presented the paper, Clinical Significance of 2-Hour Plasma Concentrations of First-line Tuberculosis Drugs. A 2-h post-dose blood sample was collected from 35 patients. Plasma concentrations below the normal range were observed for isoniazid in 25 of 35 patients (71 %), for rifampicin in 19 of 33 patients (58 %), for ethambutol in 13 of 28 patients (46 %), and for pyrazinamide in 3 of 29 patients (10 %). Treatment failure occurred more frequently in patients with low values of either isoniazid or rifampicin or both [37].
From these abstracted papers, and in the additional references cited, it is clear that the authors took advantage of the flexibility offered by TDM. Although two or more sampling times post-dose would be preferred, some authors were only able to collect 2-h or ‘peak’ concentrations. Still, useful data were obtained. The alternative to TDM is to watch, wait, and hope for the best. It is common for patients to have their treatment regimens empirically extended when responses to treatment are delayed. However, there are consequences to this strategy, both clinically and financially. Clinically, patients may remain infectious longer, and extended durations of treatment provide many more months during which adverse drug reactions may occur. Finally, some of these patients fail treatment or relapse. On the financial side, prolonged treatment means prolonged expenditure, and failures and relapses mean considerably larger expenditures. Thus, when considering that TDM ‘is expensive’, one has to ask ‘compared with what?’. Formal pharmacoeconomic analyses have not been performed in this area. ‘Back of the envelope’ calculations suggest that, in the USA, checking 2- and 6-h post-dose samples for four drugs would cost approximately $US600. If we assume that TB treatment costs $US10,000–12,000 per patient for a 6-month course, it certainly costs an additional $US3,000 to extend the treatment to 9 months, and $US6,000 to extend it to 12 months. The CDC data cited above suggest that prolonged treatment occurs in at least 10 % of the patients in the USA. Using rounded numbers, 900 patients × $US3,000 = $US2,700,000—the low-end estimate for the cost of extend durations of treatment in the USA, and it could be much higher. One set of TDM for 900 patients would cost roughly $US540,000. Not all of the extended durations could be prevented by TDM, but it seems likely, based on the available reports, that many of them could be prevented, and at a considerable cost savings. Prospective studies are needed to confirm or modify these estimates.
3 Otherwise Healthy Patients with Tuberculosis (TB)
Patients with uncomplicated pulmonary TB usually respond to standard treatment, although as noted, often with extended durations of treatment [11]. DOT is a key component of TB treatment, with a member of the healthcare team confirming that the patient swallowed the tablets and capsules. Because even ‘short-course’ TB treatment is 6 months, the potential for patient default is high. Healthcare providers must recognize and plan for this problem.
With successfully delivered four-drug TB treatment, roughly 80 % of patients with fully susceptible isolates will be sputum smear and culture negative by 2 months [6, 38]. Of course, the culture results take 2–6 weeks from the time of collection to become available, so there is a ‘blind period’ while waiting for those results, which can last from the 2-month point until the 3.5-month point. Given that culture positivity at 4 months is considered treatment failure, this process can leave only a small window of time within which to ‘salvage’ the situation. Given these delays with cultures, smears provide the first clues to the success of TB treatment. Most patients are sputum smear negative by 2 weeks, and certainly 3 weeks of treatment, when they receive the standard regimen. Patients who are slower to respond may be at greater risk for poor outcomes. Such slow responders may be candidates for TDM, as discussed by several authors [27, 29–36, 39–41]. Some clinicians prefer to observe the patient and continue with the standard doses of the TB drugs. Others prefer to check serum drug concentrations early during treatment, hoping to head off prolonged treatment durations and the potential development of drug resistance. Since TB drugs are known to show variable pharmacokinetics, it should be anticipated that some patients are under-dosed even with standard doses [29, 42–44]. This is a correctable problem.
A key component of the decision-making process is the clinical status of the patient. In this section, otherwise healthy patients are described. If the patient is making steady progress (fevers have ended, weight is stable or increasing, no more night sweats, diminished cough), then careful observation of slow converters may be reasonable. However, the authors advocate checking serum concentrations in patients who are slow to respond clinically, or in those whose condition is worsening. TDM allows you to identify and control one of the potential causes of slow clinical responses.
4 Performing TDM
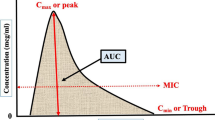
The general approach for TDM in most patient populations is the same. Additional details are provided in further sections for specific types of patients, and for specific drugs. Since the trough concentrations for many of the TB drugs are below the limit of detection for the assays, and because the peak concentrations may be more important for several of these drugs, 2-h post-dose samples can be collected to estimate the peak concentrations. For certain drugs, such as rifabutin, 3-h samples approximate the peak concentrations better. Given the variability of oral drug absorption, single time points may miss the actual peak concentrations. Therefore, second samples, typically 6-h post-dose (7 h for rifabutin), allow one to capture information on the rate and completeness of drug absorption. The second samples also provide information regarding the elimination of drugs that have short half-lives, such as isoniazid and rifampicin, provided that absorption was nearly completed 2-h post-dose.
The normal pattern for TB drug serum concentrations shows the 2-h values as substantially higher than the 6-h values. Should the 2- and 6-h values be similar, often somewhat below the expected 2-h ranges, or should the 6-h values be higher than the 2-h values, delayed absorption is likely occurring. In these situations, it is possible that the peak concentrations occurred between the two blood draws. One may recommend that the patient take the drugs on an empty stomach, especially for isoniazid and rifampicin. Finally, if both values are well below the expected ranges, malabsorption is likely occurring. With malabsorption, protein-free drug exposures may be lower than the minimal inhibitory concentrations (MICs), and higher doses of the drugs may be used.
For most TB drugs, the area under the concentration versus time curve (AUC)/MIC is the most important pharmacodynamic parameter. However, it is not practical to estimate the AUC in patients because it requires several blood samples (minimum of six or seven samples). Another approach to dose the patient is to rely on limited sampling strategies (LSS). The purpose of LSS is to determine what sampling time is most informative of the AUC. This is usually done using the AUC as the dependent variable and time concentrations as predictors, then using linear regression to determine the time points best correlated with AUC [45]. This principle has been applied to some TB drugs like linezolid and moxifloxacin [46, 47]. Another approach is to use a maximum a posteriori estimation (MAP) Bayesian approach to optimize the dosing regimen. This approach combines two sets of information to estimate the individual patient’s pharmacokinetic parameters. Results from previous population pharmacokinetic studies are used as prior information. Then, the patient’s drug concentrations are combined with the prior information to estimate the patient’s pharmacokinetic parameters. From the estimated pharmacokinetic parameters, one can dose the patient to achieve a specific therapeutic target. Potential drawbacks for the Bayesian approach are the need to purchase, learn, and use sophisticated software, and the fact that it is somewhat time consuming.
Generally, blood samples should be collected in plain red top vacuum tubes. For most of the TB drugs, except the injectable drugs, heparin-containing green top vacuum tubes also can be used. The latter are helpful when the location of blood collection is distant from the location of blood processing. Under such conditions, the green top tubes can be gently inverted several times to mix the heparin with the blood. After labeling the tubes with indelible ink, the tubes can be placed in a cup of ice chips. This will keep the samples cold during the period of transportation to the lab.
Samples should be promptly centrifuged, and the serum harvested and frozen as soon as possible after the blood draws. This is critical for isoniazid and ethionamide, since these two drugs are not stable at room temperature in whole blood or in serum. Rifampicin is somewhat more stable, and most of the other TB drugs are stable for 24 h at room temperature in whole blood or in serum. Assays should be very specific for the drugs of interest. In general, high-performance liquid chromatography (HPLC) and gas chromatography (GC) are preferred. Commonly used detection systems (ultraviolet, fluorescence, or mass spectrometry detection for HPLC, mass spectrometry for GC) work well, provided that extensive interference checks are performed while validating the assays. Guidance is available from the College of American Pathologists (CAP), and from the US FDA, regarding proper laboratory techniques.
Sometimes conventional blood sampling cannot be performed, especially in areas with limited resources. A possible alternative is to use dried blood spots, where blood samples are collected and dried before storage and transportation. Dried blood spots are easier to transport and offer cost savings [48]. However, this area still is evolving and requires further research and validation. Also, when using dried blood spots, drug concentrations are being measured in whole blood, and not in plasma [49]. This technique has been applied successfully for some of the TB drugs, like rifampicin and linezolid [50–52].
5 Interpreting the Results from TDM
Better information regarding the concentrations required for effective therapy has emerged over the past decade [43, 53–57]. We do know that, for every drug with a proven mechanism of action, a physical interaction with an intra-bacillary target must take place. This binding to a target produces either an inhibitory or a lethal effect on the bacilli. We also know that if you do not administer the drugs, they do not work. So, as concentrations for a drug recede towards zero within a patient, that drug loses its ability to produce the desired response. These facts have clearly been demonstrated in vitro and in animal models [58–64]. There is no reason to believe that these principles suddenly cease to attain once the drugs are given to people—that would be magical thinking. The difficulty in defining these endpoints for humans comes from the fact that human disease is much more complex that the in vitro situation, and far more variable than disease produced in animal models. In humans, many different strains of TB cause infection. There is re-infection, latency and relapse, varying host immune responses, widely varying pathological presentations, ages of lesions, calcification of lesions, co-morbidities, and so forth. Further, there is variability in drug absorption, protein binding, distribution, metabolism, and elimination. Thus, all of the variables that are tightly controlled with in vitro systems or in animal models are ‘wild cards’ in human disease, and often cannot be quantified. It therefore comes as no surprise that the responses to treatment in humans cover a wide spectrum.
Detailed pharmacokinetic-pharmacodynamic data from human studies are beginning to emerge [43, 53–57]. Better targets for the free drug Cmax (fCmax) relative to the MIC (fCmax /MIC), free drug AUC (fAUC/MIC), or percent of time within a dosing interval with free drug serum concentrations above MIC (f %T > MIC) are being defined [63, 65–69]. In specific cases such as rifabutin reasonable therapeutic ranges have emerged [70]. In other cases, normal ranges, derived from clinical studies of both healthy volunteers and patients with TB, are predictably tolerated and should be expected after standard doses of the TB drugs. It is well known that the standard doses generally are effective, so these standard concentrations also should be effective. Although lower serum concentrations can be effective therapy in some patients, there is no clear guidance on ‘how low can you go’. As described above, ‘how low you can go’ likely varies from patient to patient, and cannot be defined at the outset of therapy. However, one can define, a priori, ‘floor’ values for each drug. Each clinician can choose to keep the concentrations above these ‘floor’ values, in much the same way a pilot achieves a minimum altitude for his airplane. Just as with an airplane, it is not advisable to fly below these ‘floor’ values—bad things can happen. The recent TDM study findings summarized above consistently make this clear.
Once the decision is made to use TDM, it seems logical to achieve ‘normal’ serum concentrations in patients. If patients can achieve ‘normal’ serum concentrations, then poor drug absorption is not the reason for the poor clinical responses. A search for other causes can be continued with greater confidence. In rare situations, with desperately ill patients, concentrations higher than ‘normal’ may be warranted. Because there are no published data to guide these rare decisions, clinical judgment is the only guide available.
For most of the TB drugs, relationships between the dose or serum concentrations and toxicity are not well established. Exceptions include daily pyrazinamide and hepatotoxicity (more common with daily doses above 40 mg/kg, although this has been recently challenged) [71], ethambutol and ocular toxicity (more common with daily doses above 30 mg/kg and in patients with renal dysfunction), and cycloserine (more common with serum concentrations above 35 μg/mL) [72, 73]. It is important to bear in mind these relationships when increasing doses. However, in the face of malabsorption, systemic toxicity is unlikely with higher doses, because significant portions of the doses are not reaching the systemic circulation. It is quite reasonable to use higher than standard doses in the face of documented malabsorption. Serum concentrations can be rechecked after the dose increases to verify achievement of the desired concentrations. In some patients with early malabsorption, once the TB drugs begin to work, absorption also improves. This is particularly true of isoniazid in our experience, with concentrations rebounding later in treatment. This issue of malabsorption is consistent with warnings in older medical textbooks not to treat serious pneumonias with oral medications, because oral drug delivery may be less reliable. If we view TB as ‘a pneumonia with signs of systemic consumption’, the warning applies well to TB. Since intravenous treatment of TB is challenging, expensive, and usually unwarranted, verifying drug delivery and adjusting oral doses appears to be simpler.
6 More Complicated Patients
US Public Health Service (USPHS) TB Study 22 showed that patients with bilateral pulmonary disease, cavitary lesions, and low bodyweight were more likely to have poor outcomes. With once-weekly isoniazid plus rifapentine, this was especially true of HIV-positive patients [55]. Low serum concentrations of isoniazid were associated with treatment failures in patients treated with once-weekly isoniazid plus rifapentine [55]. In TB Study 23, both low isoniazid and rifabutin AUCs were associated with treatment failure or relapse in patients with HIV [56]. A study in Botswana showed an association between pyrazinamide Cmax <35 μg/mL and poor treatment outcome [43]. Another interesting article by Pasipanodya et al. [54] looked at TB drug concentrations and outcome in 142 patients from South Africa. The best predictor of outcome was pyrazinamide AUC, followed by rifampicin AUC and isoniazid AUC. Also, in all three patients who developed drug resistance, rifampicin Cmax concentrations were low. These findings illustrate the importance of drug concentrations as predictors of clinical outcome. A few other studies have found similar associations between TB drug serum concentrations, AUCs and outcomes [53, 54, 57]. A review by Chang et al. [54] described the relationship between dose frequency and treatment outcomes. In general, less frequent dosing, especially early in treatment, was associated with worse outcomes [74]. Thus, drug exposure affects treatment outcomes in two ways, the frequency of exposure, and the absolute exposure per dose. Also, TB drugs show high inter-individual variability, and this variability has been proposed as a possible cause of drug resistance. This is based on the recent study by Srivastava et al. [75], which showed that non-adherence alone is not sufficient to cause drug resistance. These findings strengthen the need to measure drug concentrations, as they are the only way to assure that patients have adequate drug concentrations.
Again, we emphasize that TB treatment outcomes are multi-factorial, and that serum concentrations represent only one of several factors that need to be considered [27, 76, 77]. Clinicians need to consider the conditions of the patients, the extent of disease, the susceptibility of the organisms (once it becomes available), and the rapidity of the clinical and bacteriological responses. TDM offers direct control over the drug exposure component. Given the discussion above, sicker patients, and those who are immunocompromised, certainly should receive more frequent dosing.
Special situations can affect treatment outcomes. TB inside the brain, within the eye, or inside the bone, may limit drug delivery and reduce treatment responses [27, 78, 79]. Abscesses and empyemas also present adverse circumstances for drug treatment, and, if accessible, surgical drainage should be considered. Treatment durations may be extended to 9 months (bone) or 12 months (meningitis) [11].
6.1 Patients with Diabetes Mellitus
It is estimated that the number of patients with TB and diabetes will continue to increase in the future [80]. Patients with diabetes are more likely to progress to active disease if exposed to TB. Also, diabetes might negatively impact disease severity and treatment outcomes in patients with active TB [29, 80–82]. Patients with diabetes are more likely to experience gastrointestinal problems, including gastroparesis [83]. This can produce either delayed drug absorption or malabsorption. Studies of the effects of diabetes on TB drug absorption have produced conflicting results. Some have shown rifampicin absorption is reduced in patients with diabetes and TB, while other studies have not [84]. If diabetes does affect the absorption of TB drugs, and rifampicin in particular, that might contribute to poor treatment outcome in patients with diabetes. Additionally, patients with diabetes frequently have other medical conditions, including coronary artery disease and chronic renal failure; some patients with diabetes are particularly fragile. For these complicated patients, it may be prudent to check serum concentrations early to ensure adequate therapy [29, 36, 84, 85]. A recent study by Heysell et al. [36] describes how early TDM at 2 weeks of treatment was applied to patients with both TB and diabetes and its potential benefits.
6.2 Patients Co-Infected with HIV
HIV-positive patients are more likely to progress to active disease if exposed to, or previously infected with, TB [86]. Active TB is an AIDS-defining illness. Patients with HIV infection represent a continuum from relatively healthy to seriously ill patients. They may have other concurrent opportunistic infections, they may be taking a large number of other drugs, and they may have various forms of malabsorption [87, 88]. Therefore, it is not surprising that some patients with AIDS malabsorb the TB drugs.
There have been a number of studies, with variable results [89–98]. Some of the studies used single samples per patient, so it is possible that delayed absorption was misidentified as malabsorption in some cases. Data from North America have tended to show a greater difference in TB drug serum concentrations between HIV-positive and HIV-negative patients than studies conducted in Asia or Africa. As noted above, the combination of intermittent dosing and low drug concentrations can be particularly dangerous in HIV-positive patients. Low concentrations of rifabutin, in particular, have been associated with failure, relapse, and acquired rifamycin resistance (ARR) [70]. Rifabutin has biphasic decay and a long terminal elimination half-life. Companion drugs, such as isoniazid, pyrazinamide, and ethambutol, have shorter elimination half-lives. Thus, intermittent dosing of these four drugs leaves sub-MIC rifabutin concentrations present as monotherapy in the patient 3–4 days per week. It is easy to see that this could lead to ARR, since active TB disease should never be treated with monotherapy. New guidelines recommend daily dosing of HIV-positive TB patients, particularly early on in treatment [99].
AIDS patients with TB also are at significant risk for drug–drug interactions [70, 100–104]. Rifamycins are potent inducers of hepatic metabolizing enzyme, including but not limited to cytochrome P450 (CYP) enzymes. Many HIV drugs are mixed inhibitors and inducers of hepatic metabolizing enzymes. Most published interaction guidelines address interactions between only two drugs. They are not designed to address the multi-way interactions common in TB/HIV co-infected patients. TDM allows one to verify the adequacy of both the TB drug doses and the HIV drug doses early in treatment, before adverse drug reactions or the selection of drug-resistant TB or HIV occurs [103, 105–107].
6.3 Patients with Renal Failure
Patients receiving dialysis are far more likely to go on to have active TB disease than those with normal renal function, whether they are latently infected or newly exposed to TB [108–111]. Patients with renal failure comprise three groups: those with poor renal function who are not on dialysis, those receiving hemodialysis, and those receiving peritoneal dialysis. All of these patients are at risk of accumulating some of the TB drugs, possibly leading to adverse drug reactions (ADRs) [112, 113].
Ethambutol, cycloserine, and the injectable TB drugs (aminoglycosides and polypeptides) require renal elimination, and their accumulation should be anticipated in patients with poor renal function. Reducing the frequency of dosing may reduce the risk of toxicities (details below). Care must be taken to not over-compensate—giving so little drug as to make it placebo.
Certain drug metabolites also require renal clearance, including pyrazinoic acid, 5-hydroxypyrazinoic acid, and acetyl-para-aminosalicylic acid [78, 79]. The toxicities of these metabolites are not known. In general, for patients with renal failure, we recommend giving standard doses (those typically given daily) no more than three times weekly for ethambutol, aminoglycosides, and pyrazinamide. p-Aminosalicylic acid (PAS) granules can be given twice daily for most patients. TDM can be helpful in this patient population. Blood samples at 2 and 6 h can be used to evaluate the rate and extent of drug absorption, which may be low in these patients. As needed, a pre- or post-dialysis sample can be used to assess residual pre-dose drug.
6.4 Patients with Hepatic Dysfunction
The liver is primarily responsible for the clearance of isoniazid, rifampicin, pyrazinamide, ethionamide, and PAS [78]. Unlike the use of blood urea nitrogen (BUN) and serum creatinine to assess renal function, hepatic drug clearance cannot be predicted based only on the serum tests for liver enzymes (aspartate aminotransferase [AST], alanine aminotransferase [ALT]) or bilirubin. Furthermore, patients with either hepatic or renal dysfunction often experience nausea and vomiting. These patients may have both malabsorption and reduced drug clearance. Individualized dosing seems appropriate for such patients.
7 Specific Drugs
Table 1 shows the ‘normal’ serum concentrations for standard doses of the TB drugs. Additional details for each drug can be found below, and in the references cited.
7.1 Isoniazid
Isoniazid is generally absorbed quickly from the gastrointestinal tract, with Cmax occurring 1–2 h post-dose when given on an empty stomach [78, 114, 115]. Food delays and reduces the absorption of isoniazid. In particular, high-fat meals cause a 51 % drop in Cmax. Therefore, isoniazid should be given on an empty stomach. If only isoniazid is measured, 1- and 4-h concentrations may effectively capture Cmax and most cases of delayed absorption. However, since isoniazid is routinely given with other agents that are somewhat more slowly absorbed, 2- and 6-h post-dose samples are preferred. In USPHS TB Studies 22 and 23, low isoniazid plasma concentrations were associated with treatment failures and relapses [55, 56]. It may be desirable to confirm isoniazid concentrations for patients on intermittent therapy, since fewer total doses are given, and each dose has greater importance. We try to achieve peak concentrations from 3 to 5 or 6 μg/mL after 300-mg daily doses, and from 9 to 15 μg/mL after 900-mg biweekly doses of isoniazid.
Isoniazid is hepatically cleared to a number of inactive compounds, mostly by acetylation through N-acetyl transferase 2 (NAT 2). This enzyme is subject to genetic polymorphisms and is tri-modally distributed into slow, intermediate, and rapid acetylation. Historically, aside from once-weekly regimens, acetylator status has not been correlated with treatment efficacy, but this has been challenged recently [116]. Slow acetylator status might be a risk factor for hepatotoxicity, while rapid acetylators are at risk of treatment failure [116–120]. A recent study showed that genotype-guided dosing can help prevent early treatment failure and unwanted side effects [120].
Generally, isoniazid dose adjustment is not required in patients with renal failure. Some renal failure patients may experience central nervous system (CNS) or peripheral neuropathies, and in these selected patients, less frequent doses may be appropriate. Patients with renal failure should receive pyridoxine 10–50 mg every day to prevent these toxicities.
7.2 Rifampicin (Rifampin)
Rifampicin absorption may be the most variable among the TB drugs [78, 114, 121]. High-fat meals reduce Cmax and delay the time to Cmax (Tmax). Rifampicin should be given on an empty stomach, or with a light snack if necessary. Fixed-drug combinations (FDCs) with isoniazid and pyrazinamide modestly reduce rifampicin absorption. The Sanofi FDC product compensates by increasing the rifampicin dose per tablet. Other formulations have been more problematic, prompting a WHO testing program for these FDC products [122, 123].
As noted above, rifampicin clearly exhibits concentration-dependent killing, and higher doses (1,200 mg or more daily) might be tolerable and more efficacious [16, 22–25, 124–126]. These higher doses are currently being evaluated in large clinical trials (NCT01408914, NCT01785186, NCT01392911). For TDM, 2- and 6-h rifampicin samples are recommended, targeting Cmax values ≥8 μg/mL. Like isoniazid, rifampicin is predominantly cleared by the liver. Dose adjustment is generally not needed for patients with renal failure.
7.3 Rifabutin
Rifabutin can be used instead of rifampicin in situations where drug interactions are especially problematic [26, 100, 127]. These include patients with AIDS, seizure disorders or cardiovascular diseases, or patients receiving poly-pharmacy. Concentrations for many hepatically cleared drugs are substantially reduced by rifampicin. Rifabutin is approximately 40 % as potent an inducer of drug clearance, which may be more manageable in many situations.
Rifabutin is partially cleared by CYP 3A4, and the partially active 25-desacetyl metabolite is significantly cleared by this enzyme [26]. Therefore, rifabutin also is subject to induction or, more commonly, inhibition of clearance caused by co-administered drugs. Potential inducers of rifabutin include efavirenz, and potential inhibitors include clarithromycin, fluconazole, and voriconazole. Because rifabutin toxicities appear to be concentration dependent, increased serum rifabutin concentrations can result in leucopenia, skin discoloration, arthralgias, and anterior uveitis. In HIV-positive patients, poor rifabutin absorption is well documented [70]. Therefore, TDM for rifabutin is highly recommended, using 3- and 7-h post-dose samples. Although we have long considered the normal range to be 0.30–0.90 μg/mL, TB trial 23 and subsequent experience suggests that 0.45 μg/mL would be a better low-end for the range [56]. A dose decrease may be indicated when peak concentrations (or the combination of the parent plus 25-desacetyl-rifatbutin concentrations) exceed 1.0 μg/mL, a value estimated based upon the limited available data [128]. The decision to change the dose should take into account the clinical status of the patient, their need for drug, and any evidence of adverse reactions, in addition to the serum concentrations. TDM for the concurrent medications will facilitate their dosage adjustments.
7.4 Rifapentine
The serum concentrations of rifapentine are slightly higher than those for rifampicin, and its partially active 25-desacetyl metabolite also produces relatively high concentrations [26, 129–131]. The half-life of rifapentine is about 15 h, intermediate between the very short half-life of rifampicin and the very long half-life of rifabutin. Rifapentine typically has a Tmax of around 5 h post-dose, later than either rifampicin or rifabutin.
Rifapentine is roughly 99 % protein bound, and this was not taken into account in early clinical trials. Daily dosing and higher mg doses of rifapentine are showing promise against TB [132, 133]. Once-weekly rifapentine, along with isoniazid, can reduce the treatment of LTBI to only 12 doses [10]. Rifapentine is similar to rifampicin as an inducer of hepatic microsomal enzymes (about 85 to >100 % as potent as rifampicin, depending on rifapentine dose and frequency) [26]. Therefore, it offers no advantage over rifampicin for patients at risk of serious drug interactions. We target 5- to 6-h Cmax values of 8–30 μg/mL after a 600-mg dose of rifapentine, similar to the 2-h Cmax of rifampicin.
7.5 Pyrazinamide
Pyrazinamide is the most reliably absorbed of the TB drugs [114, 134]. Pyrazinamide usually reaches Cmax 1–2 h post-dose and, with its long half-life (approximately 9 h), is present in the serum for many hours. Therefore, 2- and 6-h samples work well for pyrazinamide TDM. Pyrazinamide Cmax are generally 20–50 μg/mL with 25-mg/kg daily doses, proportionally higher with 35-mg/kg daily doses, and typically 60–90 μg/mL after a 50-mg/kg biweekly dose. In vitro and animal studies show pyrazinamide activity is dose dependent and is mainly driven by the AUC/MIC ratio. Also, a couple of recent clinical studies suggested higher Cmax concentrations should be targeted. The first, by Chideya et al. [43], where patients with Cmax concentrations <35 μg/mL were at risk of treatment failure. The second, by Pasipanodya et al. [54], where patients with pyrazinamide Cmax concentrations below 58 μg/mL had lower 2-month sputum culture conversion. Therefore, it might be reasonable to target a higher Cmax concentration. However, this will require using higher doses of pyrazinamide, which might increase its hepatotoxicity. Pyrazinamide elevates serum uric acid concentrations in virtually all patients, and it typically has measurable a 24-h serum concentrations. If pyrazinamide is not present in the serum, and if the patient’s uric acid is normal, then it is very unlikely that the patient is taking the medications. Pyrazinamide and its metabolites inhibit the secretion of uric acid in the kidneys, and possibly the secretion of other drugs [135]. Pyrazinamide was poorly tolerated when given with ofloxacin and levofloxacin treatment for latent MDR-TB [136–138].
7.6 Ethambutol
Ethambutol usually reaches Cmax 2- to 3-h post-dose. However, ethambutol absorption is highly variable and sometimes incomplete. Therefore, 2- and 6-h samples are required to detect delayed absorption or malabsorption [139–141]. Should the patient also be taking rifabutin and, in the case of Mycobacterium avium infection, clarithromycin, 3- and 7-h samples may be preferred [142]. The reliance of ethambutol on renal elimination was described above, so the use of ethambutol in patients with renal failure is best accompanied by TDM. Also, frequent documentation of visual acuity (Snellen chart) and red-green color discrimination (Ishihara plates) is highly recommended. Patients should be instructed to stop taking ethambutol and to call their primary physicians should they notice any change in vision, such as difficulty reading the newspaper. For ethambutol, we target a Cmax of 2–6 μg/mL after daily doses of 15–25 mg/kg, and 4–12 μg/mL after biweekly doses of 50 mg/kg.
7.7 Aminoglycosides and Polypeptides
Streptomycin is given intramuscularly or intravenously, so malabsorption generally is not a concern. Standard doses of 15 mg/kg intramuscularly produce calculated Cmax values of 35–45 μg/mL [79, 143]. Repeated intramuscular injections may toughen the tissue and alter the absorption of streptomycin over time, and this may blunt the Cmax. Renal elimination is responsible for the removal of streptomycin from the body. The adequacy of the Cmax, and potential delays in elimination, can be detected using the 2- and 6-h sampling strategy (post-intramuscular injection, or post-end of intravenous infusion). Sampling too soon after an intravenous infusion of large aminoglycoside doses has been shown to falsely elevate the calculated Cmax values when a one-compartment model is used [144].
Other aminoglycosides, including kanamycin and amikacin, display pharmacokinetics and toxicities similar to those of streptomycin [79]. Capreomycin is a polypeptide, not an aminoglycoside, but it resembles the aminoglycosides in the above two characteristics. For streptomycin and the other injectable anti-TB drugs, we target a calculated Cmax of 35–45 μg/mL after 15-mg/kg daily doses and 65–80 μg/mL after 25-mg/kg biweekly doses. Again, at least 1 h should pass from the end of an intravenous infusion until collection of the peak concentration sample in order to avoid falsely elevated concentrations during the distribution phase.
7.8 p-Aminosalicylic Acid
PAS was a first-line TB drug into the 1960s, when it was replaced by ethambutol [8]. Because of limited use over the past 3 decades, most isolates of TB remain susceptible to PAS, making it useful for patients with MDR-TB. The granule form of PAS is the only form available in several countries, including the USA. PAS granules are enteric coated and sustained release. Therefore, samples for Cmax should be collected approximately 6 h post-dose.
Because only PAS, and not the acetyl-PAS metabolite, has anti-mycobacterial activity, we try to administer PAS in a way that keeps inhibitory concentrations in the blood for most of the dose-administration interval [145]. On the basis of our review of the literature, there appears to be no good reason to avoid PAS in patients with renal failure who require the drug [113]. For PAS, we target Cmax values of 20–60 μg/mL after a 4-g dose. Tmax usually is 4–6 h post-dose for the granules. With regular PAS tablets, Tmax occurs by 2 h in most patients. We generally recommend a dose increase if the Cmax is less than 10 μg/mL. Patients should be monitored for hypothyroidism. [146]
7.9 Cycloserine
Cycloserine remains a ‘second-line’ TB drug because of its frequent CNS effects [79]. Most commonly, patients complain of an inability to concentrate, or lethargy. These complaints appear even with serum concentrations at the low end of the normal range (20–35 μg/mL). More serious CNS toxicity may be associated with elevated serum concentrations, although published literature is hard to find. In the experience of the authors and colleagues, seizures are rare, though this can vary by location [147, 148].
Food modestly decreases the absorption of cycloserine, so it is best to give this drug on an empty stomach [149]. Antacids and orange juice have little effect on cycloserine and the Tmax is about 2 h post-dose. Delayed absorption can be detected with a 6-h sample. For cycloserine, we target Cmax values of 20–35 μg/mL after 250–500 mg doses. Given the longer half-life of cycloserine, it may be best to allow 3–4 days of administration before collecting blood samples. This will allow for the achievement of steady state prior to sampling.
7.10 Ethionamide
Ethionamide is a very difficult drug to take. It frequently causes significant gastrointestinal upset, sometimes with vomiting. Although 2 h is the typical Tmax, delayed absorption is fairly common [150]. The 2- and 6-h samples work reasonably well for this drug. We target Cmax values of 1–5 μg/mL after a 250–500 mg dose. As with PAS, and especially if the two drugs are used together, patients should be monitored for hypothyroidism [146].
7.11 Fluoroquinolones
Levofloxacin, moxifloxacin (and gatifloxacin, where available) are currently used for TB [151–156]. Clinical trials continue in order to better define the roles of these drugs (NCT00864383, NCT01918397) [155, 157–160]. They display concentration-dependent activity against most organisms. Dose increases of moxifloxacin must be carried out very cautiously due to toxicity concerns [161]. It is hoped that higher doses of levofloxacin will be safe and even more effective against TB (NCT01918397). Levofloxacin is currently administered at 750–1,000 mg daily. The associated 2-h Cmax is 8–12 μg/mL, producing a Cmax/MIC of approximately 10 [162]. Clinical trials are needed to validate and improve upon this approach. Samples at 2 and 6 h are typical for fluoroquinolone TDM. Levofloxacin relies on renal elimination, so patients with renal dysfunction should receive the drug either daily, or less often, based on TDM [163].
Moxifloxacin is generally administered at 400 mg daily for TB. Some have suggested that higher doses in the 600–800 mg range should be used; this is based on a previous in vitro study [63]. One concern with using higher doses of moxifloxacin is the potential to cause QT prolongation. A high-dose moxifloxacin study is planned (NCT01329250). When given with rifampicin or rifapentine, serum concentrations are reduced by 20–30 % [164–166]. Thus, a 600-mg dose can be considered in order to maintain the 2-h Cmax in the range of 3–5 μg/mL. Moxifloxacin is both hepatically and renally cleared, and may be easier to administer in patients with renal dysfunction.
As a class, fluoroquinolones can cause CNS excitation, including caffeine-like effects and insomnia. Tendonitis is seen in some patients and, rarely, Achilles tendon rupture has occurred [167]. However, compared with the frequency of ADRs seen with other ‘second-line’ agents, such as cycloserine and ethionamide, the fluoroquinolones are well tolerated.
7.12 Linezolid
Linezolid has good in vitro and in vivo activity against TB, with an MIC of 0.5 μg/mL against M. tuberculosis [168–170]. Data from small clinical studies and case series suggest linezolid contributes to sputum culture conversion in MDR and XDR-TB. However, chronic administration of linezolid is associated with toxicities such as myelosuppression and neuropathy [170, 171]. One approach is to use linezolid 600 mg once daily, 300 mg twice daily, and even 300 mg once daily to limit its toxicity, instead of using the standard 600-mg twice daily dose for other bacterial infections [172, 173]. How this dosage would affect linezolid activity or toxicity is not clear. Linezolid activity against gram-positive infections is mainly driven by the fAUC/MIC ratio and to some extent by the time above MIC [174, 175]. Therefore, it is reasonable to target trough concentrations at or above the MIC of the organism. Additionally, trough concentrations can be as a used a surrogate for the AUC, as linezolid trough concentrations are highly correlated with its AUC [47].
7.13 Clofazimine
Clofazimine was thought to be a weak anti-TB agent, and it was used sparingly in the treatment of MDR-TB. More recent experience, both in animals and in humans, may suggest a larger role in the treatment of MDR-TB [176]. A 2-h post-dose sample can verify that the drug is being absorbed, with most patients showing 0.5–4.0 μg/mL in the serum. Clofazimine is better absorbed when given with food. Given its very long tissue half-life, second samples tend to say more about distribution than elimination of clofazimine [79, 177]. Over time, dry skin and skin discoloration become general clinical clues regarding drug absorption, but are not quantitative.
8 Conclusions
TDM is never a substitute for sound clinical judgment, nor is it a substitute for DOT. However, TDM is a useful tool in a variety of clinical situations, including patients at high risk of treatment failure, or patients with a delayed response. TDM allows one to achieve the desired serum concentrations, increasing the chances of successful clinical and bacteriological outcomes. Several of the TB drugs show high inter-individual variability. TDM also can resolve complicated drug–drug interactions before the patient experiences failure, relapse, or toxicity. In these ways, TDM is a very powerful ally in the management of complex clinical cases. Additional studies are needed to further define therapeutic concentrations, especially for the second-line TB drugs.
References
World Health Organization. Global TB report. Geneva: World Health Organization; 2013.
Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs. 2002;62(15):2169–83.
Multidrug-resistant TB (MDR-TB): 2013 update. Geneva: World Health Organization; 2013.
East African-British Medical Research Councils. Controlled clinical trial of four short-course (6-month) regimens of chemotherapy for treatment of pulmonary tuberculosis. Third report. Lancet. 1974;2(7875):237–40.
Singapore Tuberculosis Service-British Medical Research Council. Clinical trial of six-month and four-month regimens of chemotherapy in the treatment of pulmonary tuberculosis: the results up to 30 months. Tubercle. 1981;62(2):95–102.
British Thoracic Association. A controlled trial of six months chemotherapy in pulmonary tuberculosis. Second report: results during the 24 months after the end of chemotherapy. Am Rev Respir Dis. 1982;126(3):460–2.
Singapore Tuberculosis Service/British Medical Research Council. Long-term follow-up of a clinical trial of six-month and four-month regimens of chemotherapy in the treatment of pulmonary tuberculosis. Am Rev Respir Dis. 1986;133(5):779–83.
Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946–1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3(10 Suppl 2):S231–79.
American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. MMWR Recomm Rep. 2000;49(RR-6):1–51.
Sterling TR, Villarino ME, Borisov AS, et al. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365(23):2155–66.
Blumberg HM, Burman WJ, Chaisson RE, et al. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167(4):603–62.
Somoskovi A, Parsons LM, Salfinger M. The molecular basis of resistance to isoniazid, rifampin, and pyrazinamide in Mycobacterium tuberculosis. Respir Res. 2001;2(3):164–8.
Zhang Y, Mitchison D. The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis. 2003;7(1):6–21.
Zimhony O, Cox JS, Welch JT, et al. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat Med. 2000;6(9):1043–7.
Zhang Y, Wade MM, Scorpio A, et al. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J Antimicrob Chemother. 2003;52(5):790–5.
Mitchison DA. Role of individual drugs in the chemotherapy of tuberculosis. Int J Tuberc Lung Dis. 2000;4(9):796–806.
Hernandez-Pando R, Jeyanathan M, Mengistu G, et al. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet. 2000;356(9248):2133–8.
Bishai WR. Rekindling old controversy on elusive lair of latent tuberculosis. Lancet. 2000;356(9248):2113–4.
Flynn JL, Chan J. Tuberculosis: latency and reactivation. Infect Immun. 2001;69(7):4195–201.
Lenaerts AJ, Hoff D, Aly S, et al. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother. 2007;51(9):3338–45.
Gomez JE, McKinney JD. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb). 2004;84(1–2):29–44.
Ruslami R, Nijland H, Aarnoutse R, et al. Evaluation of high- versus standard-dose rifampin in Indonesian patients with pulmonary tuberculosis. Antimicrob Agents Chemother. 2006;50(2):822–3.
Kreis B, Pretet S, Birenbaum J, et al. Two three-month treatment regimens for pulmonary tuberculosis. Bull Int Union Tuberc. 1976;51(1):71–5.
Diacon AH, Patientia RF, Venter A, et al. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob Agents Chemother. 2007;51(8):2994–6.
Steingart KR, Jotblad S, Robsky K, et al. Higher-dose rifampin for the treatment of pulmonary tuberculosis: a systematic review. Int J Tuberc Lung Dis. 2011;15(3):305–16.
Burman WJ, Gallicano K, Peloquin C. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin Pharmacokinet. 2001;40(5):327–41.
Peloquin CA. Using therapeutic drug monitoring to dose the antimycobacterial drugs. Clin Chest Med. 1997;18(1):79–87.
Reported Tuberculosis in the United States, 2012. 2013; Available from http://www.cdc.gov/tb/statistics/reports/2012/pdf/report2012.pdf.
Heysell SK, Moore JL, Keller SJ, Houpt ER. Therapeutic drug monitoring for slow response to tuberculosis treatment in a state control program, Virginia, USA. Emerg Infect Dis. 2010;16(10):1546–53.
Magis-Escurra C, van den Boogaard J, Ijdema D, et al. Therapeutic drug monitoring in the treatment of tuberculosis patients. Pulm Pharmacol Ther. 2012;25(1):83–6.
Babalik A, Mannix S, Francis D, Menzies D. Therapeutic drug monitoring in the treatment of active tuberculosis. Can Respir J. 2011;18(4):225–9.
Holland DP, Hamilton CD, Weintrob AC, et al. Therapeutic drug monitoring of antimycobacterial drugs in patients with both tuberculosis and advanced human immunodeficiency virus infection. Pharmacotherapy. 2009;29(5):503–10.
Van Tongeren L, Nolan S, Cook VJ, et al. Therapeutic drug monitoring in the treatment of tuberculosis: a retrospective analysis. Int J Tuberc Lung Dis. 2013;17(2):221–4.
Li J, Burzynski JN, Lee YA, et al. Use of therapeutic drug monitoring for multidrug-resistant tuberculosis patients. Chest. 2004;126(6):1770–6.
Ray J, Gardiner I, Marriott D. Managing antituberculosis drug therapy by therapeutic drug monitoring of rifampicin and isoniazid. Intern Med J. 2003;33(5–6):229–34.
Heysell SK, Moore JL, Staley D, et al. Early therapeutic drug monitoring for isoniazid and rifampin among diabetics with newly diagnosed tuberculosis in Virginia, USA. Tuberc Res Treat. 2013;2013.
Prahl JB, Johansen IS, Frimodt-Møller N, Andersen AB. Clinical significance of 2-hour plasma concentrations of first-line tuberculosis drugs (abstract). Interscience Conference on Antimicrobial Agents and Chemotherapy; Denver, CO2013.
A controlled trial of six months chemotherapy in pulmonary tuberculosis. First Report: results during chemotherapy. British Thoracic Association. Br J Dis Chest. 1981;75(2):141–53.
Mehta JB, Shantaveerapa H, Byrd RP Jr, et al. Utility of rifampin blood levels in the treatment and follow-up of active pulmonary tuberculosis in patients who were slow to respond to routine directly observed therapy. Chest. 2001;120(5):1520–4.
Kimerling ME, Phillips P, Patterson P, et al. Low serum antimycobacterial drug levels in non-HIV-infected tuberculosis patients. Chest. 1998;113(5):1178–83.
Yew WW. Therapeutic drug monitoring in antituberculosis chemotherapy. Ther Drug Monit. 1998;20(5):469–72.
McIlleron H, Wash P, Burger A, et al. Determinants of rifampin, isoniazid, pyrazinamide, and ethambutol pharmacokinetics in a cohort of tuberculosis patients. Antimicrob Agents Chemother. 2006;50(4):1170–7.
Chideya S, Winston CA, Peloquin CA, et al. Isoniazid, rifampin, ethambutol, and pyrazinamide pharmacokinetics and treatment outcomes among a predominantly HIV-infected cohort of adults with tuberculosis from Botswana. Clin Infect Dis. 2009;48(12):1685–94.
Babalik A, Ulus IH, Bakirci N, et al. Pharmacokinetics and serum concentrations of antimycobacterial drugs in adult Turkish patients. Int J Tuberc Lung Dis. 2013;17(11):1442–7.
Sprague DA, Ensom MH. Limited-sampling strategies for anti-infective agents: systematic review. Can J Hosp Pharm. 2009;62(5):392–401.
Pranger AD, Kosterink JG, van Altena R, et al. Limited-sampling strategies for therapeutic drug monitoring of moxifloxacin in patients with tuberculosis. Ther Drug Monit. 2011;33(3):350–4.
Alffenaar JW, Kosterink JG, van Altena R, et al. Limited sampling strategies for therapeutic drug monitoring of linezolid in patients with multidrug-resistant tuberculosis. Ther Drug Monit. 2010;32(1):97–101.
Parker SP, Cubitt WD. The use of the dried blood spot sample in epidemiological studies. J Clin Pathol. 1999;52(9):633–9.
Edelbroek PM, van der Heijden J, Stolk LM. Dried blood spot methods in therapeutic drug monitoring: methods, assays, and pitfalls. Ther Drug Monit. 2009;31(3):327–36.
Vu DH, Bolhuis MS, Koster RA, et al. Dried blood spot analysis for therapeutic drug monitoring of linezolid in patients with multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2012;56(11):5758–63.
Vu DH, Koster RA, Alffenaar JW, et al. Determination of moxifloxacin in dried blood spots using LC-MS/MS and the impact of the hematocrit and blood volume. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(15–16):1063–70.
Vu DH, Koster RA, Bolhuis MS, et al. Simultaneous determination of rifampicin, clarithromycin and their metabolites in dried blood spots using LC-MS/MS. Talanta. 2014;121:9–17.
Burhan E, Ruesen C, Ruslami R, et al. Isoniazid, rifampin, and pyrazinamide plasma concentrations in relation to treatment response in Indonesian pulmonary tuberculosis patients. Antimicrob Agents Chemother. 2013;57(8):3614–9.
Pasipanodya JG, McIlleron H, Burger A, et al. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis. 2013;208(9):1464–73.
Weiner M, Burman W, Vernon A, et al. Low isoniazid concentrations and outcome of tuberculosis treatment with once-weekly isoniazid and rifapentine. Am J Respir Crit Care Med. 2003;167(10):1341–7.
Weiner M, Benator D, Burman W, et al. Association between acquired rifamycin resistance and the pharmacokinetics of rifabutin and isoniazid among patients with HIV and tuberculosis. Clin Infect Dis. 2005;40(10):1481–91.
Chigutsa E, Pasipanodya J, Visser ME, et al. Multivariate adaptive regression splines analysis of the effect of drug concentration and MIC on sterilizing activity in patients on multidrug therapy (abstract). Clinical Pharmacology of Tuberculosis Drugs; Denver, CO2013.
Srivastava S, Gumbo T. In vitro and in vivo modeling of tuberculosis drugs and its impact on optimization of doses and regimens. Curr Pharm Des. 2011;17(27):2881–8.
Pasipanodya J, Gumbo T. An oracle: antituberculosis pharmacokinetics-pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother. 2011;55(1):24–34.
Nuermberger E, Grosset J. Pharmacokinetic and pharmacodynamic issues in the treatment of mycobacterial infections. Eur J Clin Microbiol Infect Dis. 2004;23(4):243–55.
Davies GR, Nuermberger EL. Pharmacokinetics and pharmacodynamics in the development of anti-tuberculosis drugs. Tuberculosis (Edinb). 2008;88(Suppl 1):S65–74.
Ahmad Z, Fraig MM, Bisson GP, et al. Dose-dependent activity of pyrazinamide in animal models of intracellular and extracellular tuberculosis infections. Antimicrob Agents Chemother. 2011;55(4):1527–32.
Gumbo T, Louie A, Deziel MR, et al. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190(9):1642–51.
Nuermberger E. Using animal models to develop new treatments for tuberculosis. Semin Respir Crit Care Med. 2008;29(5):542–51.
Pasipanodya J, Srivastava S, Gumbo T. New susceptibility breakpoints and the regional variability of MIC distribution in Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2012;56(10):5428.
Gumbo T, Louie A, Deziel MR, et al. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother. 2007;51(11):3781–8.
Gumbo T, Dona CS, Meek C, Leff R. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother. 2009;53(8):3197–204.
Jayaram R, Gaonkar S, Kaur P, et al. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 2003;47(7):2118–24.
Jayaram R, Shandil RK, Gaonkar S, et al. Isoniazid pharmacokinetics-pharmacodynamics in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 2004;48(8):2951–7.
Boulanger C, Hollender E, Farrell K, et al. Pharmacokinetic evaluation of rifabutin in combination with lopinavir-ritonavir in patients with HIV infection and active tuberculosis. Clin Infect Dis. 2009;49(9):1305–11.
Pasipanodya JG, Gumbo T. Clinical and toxicodynamic evidence that high-dose pyrazinamide is not more hepatotoxic than the low doses currently used. Antimicrob Agents Chemother. 2010;54(7):2847–54.
Talbert Estlin KA, Sadun AA. Risk factors for ethambutol optic toxicity. Int Ophthalmol. 2010;30(1):63–72.
Hasenbosch RE, Alffenaar JW, Koopmans SA, et al. Ethambutol-induced optical neuropathy: risk of overdosing in obese subjects. Int J Tuberc Lung Dis. 2008;12(8):967–71.
Chang KC, Leung CC, Grosset J, Yew WW. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66(11):997–1007.
Srivastava S, Pasipanodya JG, Meek C, et al. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis. 2011;204(12):1951–9.
Peloquin CA. Tuberculosis drug serum levels. Clin Infect Dis. 2001;33(4):584–5.
Peloquin CA. Pharmacological issues in the treatment of tuberculosis. Ann NY Acad Sci. 2001;953:157–64.
Holdiness MR. Clinical pharmacokinetics of the antituberculosis drugs. Clin Pharmacokinet. 1984;9(6):511–44.
Peloquin C. Antituberculosis drugs: pharmacokinetics. In: LB H, editor. Drug susceptibility in the chemotherapy of mycobacterial infections. Boca Raton, FL: CRC Press; 1991. p. 89–122.
Dooley KE, Chaisson RE. Tuberculosis and diabetes mellitus: convergence of two epidemics. Lancet Infect Dis. 2009;9(12):737–46.
Dooley KE, Tang T, Golub JE, et al. Impact of diabetes mellitus on treatment outcomes of patients with active tuberculosis. Am J Trop Med Hyg. 2009;80(4):634–9.
Alisjahbana B, Sahiratmadja E, Nelwan EJ, et al. The effect of type 2 diabetes mellitus on the presentation and treatment response of pulmonary tuberculosis. Clin Infect Dis. 2007;45(4):428–35.
Dostalek M, Akhlaghi F, Puzanovova M. Effect of diabetes mellitus on pharmacokinetic and pharmacodynamic properties of drugs. Clin Pharmacokinet. 2012;51(8):481–99.
Ruslami R, Nijland HM, Adhiarta IG, et al. Pharmacokinetics of antituberculosis drugs in pulmonary tuberculosis patients with type 2 diabetes. Antimicrob Agents Chemother. 2010;54(3):1068–74.
Nijland HM, Ruslami R, Stalenhoef JE, et al. Exposure to rifampicin is strongly reduced in patients with tuberculosis and type 2 diabetes. Clin Infect Dis. 2006;43(7):848–54.
Diagnostic Standards and Classification of Tuberculosis in Adults and Children. This official statement of the American Thoracic Society and the Centers for Disease Control and Prevention was adopted by the ATS Board of Directors, July 1999. This statement was endorsed by the Council of the Infectious Disease Society of America, September 1999. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1376–95.
Kotler DP, Gaetz HP, Lange M, et al. Enteropathy associated with the acquired immunodeficiency syndrome. Ann Intern Med. 1984;101(4):421–8.
Gillin JS, Shike M, Alcock N, et al. Malabsorption and mucosal abnormalities of the small intestine in the acquired immunodeficiency syndrome. Ann Intern Med. 1985;102(5):619–22.
Peloquin CA, MacPhee AA, Berning SE. Malabsorption of antimycobacterial medications. N Engl J Med. 1993;329(15):1122–3.
Gordon SM, Horsburgh CR, Peloquin CA, et al. Low serum levels of oral antimycobacterial agents in patients with disseminated Mycobacterium avium complex disease. J Infect Dis. 1993;168(6):1559–62.
Peloquin CA, Nitta AT, Burman WJ, et al. Low antituberculosis drug concentrations in patients with AIDS. Ann Pharmacother. 1996;30(9):919–25.
Sahai J, Gallicano K, Swick L, et al. Reduced plasma concentrations of antituberculosis drugs in patients with HIV infection. Ann Intern Med. 1997;127(4):289–93.
Gurumurthy P, Ramachandran G, Hemanth Kumar AK, et al. Decreased bioavailability of rifampin and other antituberculosis drugs in patients with advanced human immunodeficiency virus disease. Antimicrob Agents Chemother. 2004;48(11):4473–5.
Gurumurthy P, Ramachandran G, Hemanth Kumar AK, et al. Malabsorption of rifampin and isoniazid in HIV-infected patients with and without tuberculosis. Clin Infect Dis. 2004;38(2):280–3.
Taylor B, Smith PJ. Does AIDS impair the absorption of antituberculosis agents? Int J Tuberc Lung Dis. 1998;2(8):670–5.
Peloquin CA, Berning SE, Huitt GA, Iseman MD. AIDS and TB drug absorption. Int J Tuberc Lung Dis. 1999;3(12):1143–4.
McIlleron H, Rustomjee R, Vahedi M, et al. Reduced antituberculosis drug concentrations in HIV-infected patients who are men or have low weight: implications for international dosing guidelines. Antimicrob Agents Chemother. 2012;56(6):3232–8.
Narita M, Hisada M, Thimmappa B, et al. Tuberculosis recurrence: multivariate analysis of serum levels of tuberculosis drugs, human immunodeficiency virus status, and other risk factors. Clin Infect Dis. 2001;32(3):515–7.
Kaplan JE, Benson C, Holmes KK, et al. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep. 2009;58(Rr-4):1–207.
Burman WJ, Gallicano K, Peloquin C. Therapeutic implications of drug interactions in the treatment of human immunodeficiency virus-related tuberculosis. Clin Infect Dis. 1999;28(3):419–29.
Narita M, Stambaugh JJ, Hollender ES, et al. Use of rifabutin with protease inhibitors for human immunodeficiency virus-infected patients with tuberculosis. Clin Infect Dis. 2000;30(5):779–83.
Weiner M, Benator D, Peloquin CA, et al. Evaluation of the drug interaction between rifabutin and efavirenz in patients with HIV infection and tuberculosis. Clin Infect Dis. 2005;41(9):1343–9.
Schwiesow JN, Iseman MD, Peloquin CA. Concomitant use of voriconazole and rifabutin in a patient with multiple infections. Pharmacotherapy. 2008;28(8):1076–80.
Benator DA, Weiner MH, Burman WJ, et al. Clinical evaluation of the nelfinavir–rifabutin interaction in patients with tuberculosis and human immunodeficiency virus infection. Pharmacotherapy. 2007;27(6):793–800.
Durant J, Clevenbergh P, Garraffo R, et al. Importance of protease inhibitor plasma levels in HIV-infected patients treated with genotypic-guided therapy: pharmacological data from the Viradapt Study. AIDS. 2000;14(10):1333–9.
Angel JB, Khaliq Y, Monpetit ML, et al. An argument for routine therapeutic drug monitoring of HIV-1 protease inhibitors during pregnancy. AIDS. 2001;15(3):417–9.
Back D, Gatti G, Fletcher C, et al. Therapeutic drug monitoring in HIV infection: current status and future directions. AIDS. 2002;16(Suppl 1):S5–37.
Cengiz K. Increased incidence of tuberculosis in patients undergoing hemodialysis. Nephron. 1996;73(3):421–4.
Hu HY, Wu CY, Huang N, et al. Increased risk of tuberculosis in patients with end-stage renal disease: a population-based cohort study in Taiwan, a country of high incidence of end-stage renal disease. Epidemiol Infect. 2014:142(1):191–9.
Chia S, Karim M, Elwood RK, FitzGerald JM. Risk of tuberculosis in dialysis patients: a population-based study. Int J Tuberc Lung Dis. 1998;2(12):989–91.
Cuss FM, Carmichael DJ, Linington A, Hulme B. Tuberculosis in renal failure: a high incidence in patients born in the Third World. Clin Nephrol. 1986;25(3):129–33.
Malone RS, Fish DN, Spiegel DM, et al. The effect of hemodialysis on isoniazid, rifampin, pyrazinamide, and ethambutol. Am J Respir Crit Care Med. 1999;159(5 Pt 1):1580–4.
Malone RS, Fish DN, Spiegel DM, et al. The effect of hemodialysis on cycloserine, ethionamide, para-aminosalicylate, and clofazimine. Chest. 1999;116(4):984–90.
Peloquin CA, Jaresko GS, Yong CL, et al. Population pharmacokinetic modeling of isoniazid, rifampin, and pyrazinamide. Antimicrob Agents Chemother. 1997;41(12):2670–9.
Peloquin CA, Namdar R, Dodge AA, Nix DE. Pharmacokinetics of isoniazid under fasting conditions, with food, and with antacids. Int J Tuberc Lung Dis. 1999;3(8):703–10.
Pasipanodya JG, Srivastava S, Gumbo T. Meta-analysis of clinical studies supports the pharmacokinetic variability hypothesis for acquired drug resistance and failure of antituberculosis therapy. Clin Infect Dis. 2012;55(2):169–77.
Cho HJ, Koh WJ, Ryu YJ, et al. Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis (Edinb). 2007;87(6):551–6.
Ben Mahmoud L, Ghozzi H, Kamoun A, et al. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatotoxicity in Tunisian patients with tuberculosis. Pathol Biol (Paris). 2012;60(5):324–30.
Ohno M, Yamaguchi I, Yamamoto I, et al. Slow N-acetyltransferase 2 genotype affects the incidence of isoniazid and rifampicin-induced hepatotoxicity. Int J Tuberc Lung Dis. 2000;4(3):256–61.
Azuma J, Ohno M, Kubota R, et al. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: a randomized controlled trial for pharmacogenetics-based therapy. Eur J Clin Pharmacol. 2013;69(5):1091–101.
Peloquin CA, Namdar R, Singleton MD, Nix DE. Pharmacokinetics of rifampin under fasting conditions, with food, and with antacids. Chest. 1999;115(1):12–8.
Ellard GA, Fourie PB. Rifampicin bioavailability: a review of its pharmacology and the chemotherapeutic necessity for ensuring optimal absorption. Int J Tuberc Lung Dis. 1999;3(11 Suppl 3):S301–8 discussion S17–21.
Ellard GA. The evaluation of rifampicin bioavailabilities of fixed-dose combinations of anti-tuberculosis drugs: procedures for ensuring laboratory proficiency. Int J Tuberc Lung Dis. 1999;3(11 Suppl 3):S322–4 discussion S51–2.
Acocella G, Bertrand A, Beytout J, et al. Comparison of three different regimens in the treatment of acute brucellosis: a multicenter multinational study. J Antimicrob Chemother. 1989;23(3):433–9.
Peloquin C. What is the ‘right’ dose of rifampin? Int J Tuberc Lung Dis. 2003;7(1):3–5.
Boeree M, Diacon A, Dawson R, et al. What Is the “Right” Dose of Rifampin? (abstract) The annual Conference on retroviruses and opportunistic infections; Atlanta, GA2013.
(CDC) CfDCaP. Updated guidelines for the use of rifabutin or rifampin for the treatment and prevention of tuberculosis among HIV-infected patients taking protease inhibitors or nonnucleoside reverse transcriptase inhibitors. MMWR Morb Mortal Wkly Rep. 2000;49(9):185–9.
Hafner R, Bethel J, Power M, et al. Tolerance and pharmacokinetic interactions of rifabutin and clarithromycin in human immunodeficiency virus-infected volunteers. Antimicrob Agents Chemother. 1998;42(3):631–9.
Temple ME, Nahata MC. Rifapentine: its role in the treatment of tuberculosis. Ann Pharmacother. 1999;33(11):1203–10.
Gao XF, Li J, Yang ZW, Li YP. Rifapentine vs. rifampicin for the treatment of pulmonary tuberculosis: a systematic review. Int J Tuberc Lung Dis. 2009;13(7):810–9.
Tam CM, Chan SL, Kam KM, et al. Rifapentine and isoniazid in the continuation phase of a 6-month regimen. Final report at 5 years: prognostic value of various measures. Int J Tuberc Lung Dis. 2002;6(1):3–10.
Dorman SE, Goldberg S, Stout JE, et al. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: Study 29 of the tuberculosis trials consortium. J Infect Dis. 2012;206(7):1030–40.
Savic R, Weiner M, Mac Kenzie W, et al. PKPD analysis of rifapentine in patients during intensive phase treatment for tuberculosis from Tuberculosis Trial Consortium Studies 29 and 29X (abstract). Clinical Pharmacology of Tuberculosis Drugs; Denver, CO2013.
Peloquin CA, Bulpitt AE, Jaresko GS, et al. Pharmacokinetics of pyrazinamide under fasting conditions, with food, and with antacids. Pharmacotherapy. 1998;18(6):1205–11.
Weiner IM, Tinker JP. Pharmacology of pyrazinamide: metabolic and renal function studies related to the mechanism of drug-induced urate retention. J Pharmacol Exp Ther. 1972;180(2):411–34.
Horn DL, Hewlett D Jr, Alfalla C, et al. Limited tolerance of ofloxacin and pyrazinamide prophylaxis against tuberculosis. N Engl J Med. 1994;330(17):1241.
Lou HX, Shullo MA, McKaveney TP. Limited tolerability of levofloxacin and pyrazinamide for multidrug-resistant tuberculosis prophylaxis in a solid organ transplant population. Pharmacotherapy. 2002;22(6):701–4.
Papastavros T, Dolovich LR, Holbrook A, et al. Adverse events associated with pyrazinamide and levofloxacin in the treatment of latent multidrug-resistant tuberculosis. CMAJ. 2002;167(2):131–6.
Peloquin CA, Bulpitt AE, Jaresko GS, et al. Pharmacokinetics of ethambutol under fasting conditions, with food, and with antacids. Antimicrob Agents Chemother. 1999;43(3):568–72.
Tappero JW, Bradford WZ, Agerton TB, et al. Serum concentrations of antimycobacterial drugs in patients with pulmonary tuberculosis in Botswana. Clin Infect Dis. 2005;41(4):461–9.
Zhu M, Burman WJ, Starke JR, et al. Pharmacokinetics of ethambutol in children and adults with tuberculosis. Int J Tuberc Lung Dis. 2004;8(11):1360–7.
Peloquin CA. Mycobacterium avium complex infection. Pharmacokinetic and pharmacodynamic considerations that may improve clinical outcomes. Clin Pharmacokinet. 1997;32(2):132–44.
Zhu M, Burman WJ, Jaresko GS, et al. Population pharmacokinetics of intravenous and intramuscular streptomycin in patients with tuberculosis. Pharmacotherapy. 2001;21(9):1037–45.
Demczar DJ, Nafziger AN, Bertino JS Jr. Pharmacokinetics of gentamicin at traditional versus high doses: implications for once-daily aminoglycoside dosing. Antimicrob Agents Chemother. 1997;41(5):1115–9.
Peloquin CA, Berning SE, Huitt GA, et al. Once-daily and twice-daily dosing of p-aminosalicylic acid granules. Am J Respir Crit Care Med. 1999;159(3):932–4.
Andries A, Isaakidis P, Das M, et al. High rate of hypothyroidism in multidrug-resistant tuberculosis patients co-infected with HIV in Mumbai, India. PLoS One. 2013;8(10):e78313.
Hwang TJ, Wares DF, Jafarov A, et al. Safety of cycloserine and terizidone for the treatment of drug-resistant tuberculosis: a meta-analysis. Int J Tuberc Lung Dis. 2013;17(10):1257–66.
Carroll MW, Lee M, Cai Y, et al. Frequency of adverse reactions to first- and second-line anti-tuberculosis chemotherapy in a Korean cohort. Int J Tuberc Lung Dis. 2012;16(7):961–6.
Zhu M, Nix DE, Adam RD, et al. Pharmacokinetics of cycloserine under fasting conditions and with high-fat meal, orange juice, and antacids. Pharmacotherapy. 2001;21(8):891–7.
Auclair B, Nix DE, Adam RD, et al. Pharmacokinetics of ethionamide administered under fasting conditions or with orange juice, food, or antacids. Antimicrob Agents Chemother. 2001;45(3):810–4.
Ziganshina LE, Titarenko AF, Davies GR. Fluoroquinolones for treating tuberculosis (presumed drug-sensitive). Cochrane Database Syst Rev. 2013;6:Cd004795.
Moadebi S, Harder CK, Fitzgerald MJ, et al. Fluoroquinolones for the treatment of pulmonary tuberculosis. Drugs. 2007;67(14):2077–99.
Berning SE. The role of fluoroquinolones in tuberculosis today. Drugs. 2001;61(1):9–18.
Pranger AD, van Altena R, Aarnoutse RE, et al. Evaluation of moxifloxacin for the treatment of tuberculosis: 3 years of experience. Eur Respir J. 2011;38(4):888–94.
Rustomjee R, Lienhardt C, Kanyok T, et al. A Phase II study of the sterilising activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2008;12(2):128–38.
Wang JY, Wang JT, Tsai TH, et al. Adding moxifloxacin is associated with a shorter time to culture conversion in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2010;14(1):65–71.
Jawahar MS, Banurekha VV, Paramasivan CN, et al. Randomized clinical trial of thrice-weekly 4-month moxifloxacin or gatifloxacin containing regimens in the treatment of new sputum positive pulmonary tuberculosis patients. PLoS One. 2013;8(7):e67030.
Dawson R, Diacon A. PA-824, moxifloxacin and pyrazinamide combination therapy for tuberculosis. Expert Opin Investig Drugs. 2013;22(7):927–32.
Dorman SE, Johnson JL, Goldberg S, et al. Substitution of moxifloxacin for isoniazid during intensive phase treatment of pulmonary tuberculosis. Am J Respir Crit Care Med. 2009;180(3):273–80.
Koh WJ, Lee SH, Kang YA, et al. Comparison of levofloxacin versus moxifloxacin for multidrug-resistant tuberculosis. Am J Respir Crit Care Med. 2013;188(7):858–64.
Demolis JL, Kubitza D, Tenneze L, Funck-Brentano C. Effect of a single oral dose of moxifloxacin (400 mg and 800 mg) on ventricular repolarization in healthy subjects. Clin Pharmacol Ther. 2000;68(6):658–66.
Peloquin CA, Hadad DJ, Molino LP, et al. Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52(3):852–7.
Fish DN, Chow AT. The clinical pharmacokinetics of levofloxacin. Clin Pharmacokinet. 1997;32(2):101–19.
Nijland HM, Ruslami R, Suroto AJ, et al. Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin Infect Dis. 2007;45(8):1001–7.
Weiner M, Burman W, Luo CC, et al. Effects of rifampin and multidrug resistance gene polymorphism on concentrations of moxifloxacin. Antimicrob Agents Chemother. 2007;51(8):2861–6.
Dooley K, Flexner C, Hackman J, et al. Repeated administration of high-dose intermittent rifapentine reduces rifapentine and moxifloxacin plasma concentrations. Antimicrob Agents Chemother. 2008;52(11):4037–42.
Corrao G, Zambon A, Bertu L, et al. Evidence of tendinitis provoked by fluoroquinolone treatment: a case-control study. Drug Saf. 2006;29(10):889–96.
Lauzardo M, Peloquin CA. Antituberculosis therapy for 2012 and beyond. Expert Opin Pharmacother. 2012;13(4):511–26.
Garcia-Tapia A, Rodriguez JC, Ruiz M, Royo G. Action of fluoroquinolones and Linezolid on logarithmic- and stationary-phase culture of Mycobacterium tuberculosis. Chemotherapy. 2004;50(5):211–3.
Rodriguez JC, Ruiz M, Lopez M, Royo G. In vitro activity of moxifloxacin, levofloxacin, gatifloxacin and linezolid against Mycobacterium tuberculosis. Int J Antimicrob Agents. 2002;20(6):464–7.
Sotgiu G, Centis R, D’Ambrosio L, et al. Efficacy, safety and tolerability of linezolid containing regimens in treating MDR-TB and XDR-TB: systematic review and meta-analysis. Eur Respir J. 2012;40(6):1430–42.
Alffenaar JW, van Altena R, Harmelink IM, et al. Comparison of the pharmacokinetics of two dosage regimens of linezolid in multidrug-resistant and extensively drug-resistant tuberculosis patients. Clin Pharmacokinet. 2010;49(8):559–65.
Park IN, Hong SB, Oh YM, et al. Efficacy and tolerability of daily-half dose linezolid in patients with intractable multidrug-resistant tuberculosis. J Antimicrob Chemother. 2006;58(3):701–4.
Andes D, van Ogtrop ML, Peng J, Craig WA. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob Agents Chemother. 2002;46(11):3484–9.
Rayner CR, Forrest A, Meagher AK, et al. Clinical pharmacodynamics of linezolid in seriously ill patients treated in a compassionate use programme. Clin Pharmacokinet. 2003;42(15):1411–23.
Gopal M, Padayatchi N, Metcalfe JZ, O’Donnell MR. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int J Tuberc Lung Dis. 2013;17(8):1001–7.
Garrelts JC. Clofazimine: a review of its use in leprosy and Mycobacterium avium complex infection. DICP. 1991;25(5):525–31.
Acknowledgments
Charles Peloquin does not have funding or conflicts of interest relevant to the content of this review. Abdullah Alsultan does not have funding or conflict of interest relevant to the content of this review. The authors acknowledge academic support from King Saud University for Dr. Sultan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Alsultan, A., Peloquin, C.A. Therapeutic Drug Monitoring in the Treatment of Tuberculosis: An Update. Drugs 74, 839–854 (2014). https://doi.org/10.1007/s40265-014-0222-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0222-8