
Abstract
Allergies to metals are common, with nickel allergy being the most frequent form of contact allergy. Contact allergies are classified as delayed-type hypersensitivity reactions in which T cells are the key players. In this chapter, we describe how metals are presented to T cells and discuss the current literature on the role of CD4+ and CD8+ T cells in contact allergies to metals.
The original version of this chapter was revised. An erratum to this chapter can be found at https://doi.org/10.1007/978-3-319-58503-1_44
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
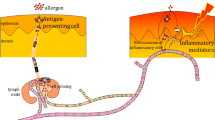
Metal ions are common triggers of allergic contact dermatitis (ACD) [1]. ACD is a T cell-mediated inflammatory response classified as a type IV delayed-type hypersensitivity reaction. ACD can be divided into two phases—the sensitization and the elicitation phase (Fig. 9.1) [2]. Sensitization occurs after contact with a specific allergen and requires activation of the innate immune system in the skin, eventually leading to the activation and migration of dendritic cells (DC) to the draining lymph nodes [3]. In the lymph nodes, the DC presents the allergen in association with a major histocompatibility complex (MHC)-bound peptide to naïve T cells expressing an allergen-specific T cell receptor (TCR) [4]. These events lead to clonal expansion of the allergen-specific T cells and generation of allergen-specific memory T cells that can be found in both skin and blood. During re-exposure to the specific allergen in the elicitation phase, allergen-specific memory T cells are recruited and activated, resulting in the cellular damage and inflammation responsible for the clinically apparent eczematous skin reaction [2]. In this chapter, we focus on T cell responses to metals with special emphasis on the response to nickel, as nickel allergy is the most common and thoroughly studied of the metal allergies.

The immunological mechanisms of contact allergy. In the sensitization phase, allergens (e.g., metal ions) penetrate the skin and trigger activation of an innate inflammatory response with the production of pro-inflammatory cytokines, eventually leading to activation of dendritic cells (Langerhans cells in the epidermis and dermal dendritic cells in the dermis). The DC migrate to the draining lymph nodes and present the allergen to naïve allergen-specific T cells, which leads to clonal expansion and generation of allergen-specific memory T cells. Upon re-exposure to the same allergen in the elicitation phase, memory T cells are recruited and activated, mediating skin inflammation and cellular damage
2 Presentation and T Cell Recognition of Metal Ions
T cells recognize antigens in the form of MHC-peptide complexes via their TCR. The TCR is composed of the variable, antigen-recognizing TCRα and β chains in the majority of circulating T cells and the TCRγ and δ chain in a minority of circulating T cells. Together with the invariable chains CD3γ, CD3δ, CD3ε, and ζ, which are responsible for signaling, the antigen-recognizing chains make up the complete multimeric TCR (Fig. 9.2a). To be recognized by the TCR, an antigen must be processed into peptides and presented on MHC molecules. Two major classes of MHC molecules present peptides to the TCR, namely, MHC class I and MHC class II molecules. CD8+ and CD4+ T cells recognize MHC class I-peptide and MHC class II-peptide complexes, respectively (Fig. 9.2b). Like most other contact allergens, metal ions are low molecular weight chemicals (< 500 Da) referred to as haptens, which have to bind proteins or peptides to become immunogenic. Most of our knowledge about the way nickel is presented to T cells comes from studies using isolated nickel-reactive T cell clones from patients with nickel allergy [5]. Stimulation of these clones with nickel ions (Ni2+) could induce strong proliferative responses, showing that T cells are involved in the pathogenesis of nickel allergy [6,7,8,9].

Composition of (a) the T cell receptor (TCR) and (b) major histocompatibility complex (MHC) classes I and II
Metal ions are proposed to be presented to T cells much like classical haptens by forming complexes with amino acid residues on the MHC molecules and/or on the MHC-bound peptides [10] (Fig. 9.3a). However, unlike classical haptens, metal ions do not form stable covalent bonds to proteins. They rather interact with nitrogen or oxygen in amino acid side chains to produce non-covalent, reversible coordination protein-metal complexes [2, 5]. The first study indicating that nickel could bind an MHC-bound peptide was published in 1991. In this study, it was found that T cell clones reactive to a malarial peptide were inhibited from recognizing the peptide by treatment of the peptide-pulsed, antigen-presenting cells (APC) with Ni2+ [11]. A histidine (His) residue of the malaria peptide was found to be responsible for the binding of Ni2+. Similar findings were later reported for gold [12, 13].

Models of metal presentation to CD4+ T cells. Several different molecular interactions between metal ions, the TCR, and MHC class II-peptide complexes have been proposed. (a) Metal ions can directly bind to the MHC molecule and the associated peptide, or it can bind to either the peptide or the MHC molecule only (not shown). (b) Similarly to superantigens, metal ions might directly bridge TCR with MHC, independent of the MHC-associated peptide. (c) Some MHC-peptide-metal complexes may be formed by cellular processing of metal-modified proteins, and (d) in other instances, metal ions may alter the processing of self-proteins and trigger activation of T cells with metal-free cryptic self-peptides. (e) Some metal ions may create neo-antigens by altering the conformation of preexisting self-MHC-peptide complexes. The TCR subsequently recognizes the neo-antigen but does not directly interact with the metal ion
In 2003, Lu et al. studied the components of the ligand for a human nickel-reactive CD4+ T cell clone (ANi-2.3) that had been isolated from a patient with nickel allergy [14]. ANi-2.3 belongs to a group of TCR-Vβ17-expressing T cells that are found to be overrepresented among nickel-specific CD4+ T cells in patients suffering from particularly severe nickel allergy [15, 16]. Lu et al. identified the MHC restriction element for ANi-2.3 as HLA-DR52c [14]. They found that the functional ligand for this T cell clone was a complex of Ni2+ bound to the combination of HLA-DR52c and an unknown peptide produced in B cells. Furthermore, Ni2+ could be presented to ANi-2.3 by paraformaldehyde-fixed autologous APC, which indicated that Ni2+ can be presented on preformed MHC-peptide complexes [14]. In addition, it was shown that ANi-2.3 recognition of the Ni2+-MHC-peptide complex was dependent on His81 of the HLA-DR52c β chain—a residue that is conserved in the β chain of many MHC class II molecules. It was proposed that Ni2+ was coordinated by His81 and by two amino acid side chains of the bound peptide. As ANi-2.3 is cross-reactive to copper and gold cations, it was speculated that these could be coordinated similarly to Ni2+ [14]. Unfortunately, a crystal structure of the complete complex of TCR, MHC class II, peptide, and Ni2+ does not exist [17]. However, recently a screening of libraries of DR52c-bound peptides with ANi-2.3 resulted in the identification of so-called mimotope peptides that could substitute for Ni2+ and the self-peptide in the natural TCR ligand [18]. By solving the structure of two of these mimotopes, insight was offered into the binding site for Ni2+ in the natural ligand and how it interacts with the TCR [18]. Thus, structural analyses revealed that ANi-2.3 TCR docked on the mimotope peptide-DR52c complex in a typical diagonal orientation, and it was suggested that a conserved lysine residue at the p7 position of the DR52c-bound peptide mimicked Ni2+ in the natural TCR ligand [18]. Supporting that the ANi-2.3 TCR interacts with Ni2+ complexed to a self-peptide, this study further showed that Ni2+ and maybe also other metal cations were accommodated by an acidic pocket formed by the α1 and β1 chains of DR52c in the peripheral region of the peptide-binding groove where the conserved p7 lysine of the peptide mimotope was located [18, 19]. Future structural studies similar to this might provide a more complete understanding of metal recognition by T cells [17].
Gamerdinger et al., who studied another CD4+, TCR-Vβ17-expressing nickel-reactive T cell clone (SE9), identified a second and quite different type of Ni2+ recognition. As shown for ANi-2.3, nickel reactivity of SE9 did not depend on antigen processing as it could be activated in the presence of fixed APC, but in this case Ni2+ recognition was also found to be independent of the nature of the MHC-associated peptides. On the other hand, SE9 cells required permanent availability of Ni2+ in the medium for activation, arguing against the existence of preformed Ni2+-MHC determinants for the SE9 TCR [20]. Furthermore, SE9 activation crucially depended on the conserved His81 in the HLA-DR β chain, as well as on two tyrosine residues in the CDR1 and CDR3 region of the TCRα chain (Fig. 9.2a). Thus, it was proposed that Ni2+, in analogy to superantigens, directly links and stabilizes intramolecular bridges between the TCR and MHC independently of MHC-associated peptides [20] (Fig. 9.3b). Whether this proposed model also applies to other metals remains unknown, as the SE9 clone was not cross-reactive to copper, palladium, cobalt or chromium [20]. In addition, it has been suggested that particular proteins such as human serum albumin could be responsible for transferring Ni2+ to the high-affinity coordination sites within the contact zone between certain TCR and MHC molecules [5, 21].
Activation of ANi-2.3 and SE9 by Ni2+ was not dependent on active antigen processing. However, this does not account for all isolated nickel-reactive clones. A study on 42 independent T cell clones showed that 40% of the clones could not be activated by glutaraldehyde-fixed APC, meaning that these were strictly dependent on active antigen processing [8]. Thus some MHC-peptide-metal complexes may be formed by cellular processing of metal-modified proteins [9, 10] (Fig. 9.3c). In this context, it has been suggested that metal ions in some instances alter the processing of self-proteins and trigger activation of T cells with metal-free cryptic self-peptides [22, 23] (Fig. 9.3d). Moreover, it has been shown that certain noble metals, including palladium and gold, are able to destabilize peptide binding to MHC class II complexes, whereby the metal-bound MHC molecule adopts a stable, “peptide-empty” conformation that resembles the transition state of peptide loading [24]. This metal-induced peptide stripping may also be involved in the formation of new antigenic epitopes. Hence, although not tested in the study, the peptide-empty MHC molecules could be recognized by certain T cells as neo-antigens [17, 24]. Some metal ions may also create neo-antigens by indirect modification of preexisting self-MHC-peptide complexes as shown for beryllium ions (Be2+) in a study on chronic beryllium disease [25]. Here, it was shown that the TCR did not interact directly with Be2+. Instead, Be2+ was buried in an MHC-peptide complex, altering the charge and conformation of the surface of the complex, which was then recognized by the TCR [25] (Fig. 9.3e).
The studies described above reveal that several different molecular interactions between metal ions, the TCR and MHC class II-peptide complexes must be considered in CD4+ T cell responses to metals (Fig. 9.3). How nickel and other metals are presented to CD8+ cells by MHC class I molecules is still not known, but similar mechanisms as described for CD4+ T cells probably apply. It is known that the strength whereby a TCR is triggered affects the resulting effector T cell response [26]. How metals are presented to T cells therefore most likely has a great impact on the T cell response and thereby the development of ACD. Specifically, modulation of metal presentation could potentially be useful for the development of more specific treatments of metal allergies.
3 CD4+ Versus CD8+ T Cells in Metal Allergy
T cells involved in metal allergy seem to be a very heterogeneous group with regard to their cytokine profile and function. Early studies on blood-derived nickel-specific T cell clones suggested that nickel allergy was a CD4+ Th1-dominated response mainly mediated by IFN-γ [6, 27]. The important role of CD4+ cells in the response have been confirmed by several studies showing the involvement of both Th1 and Th2 components in nickel allergy [28,29,30,31] as well as in allergies to other metals, including palladium, cobalt, chromium, and gold [32, 33]. However, Cavani et al. compared the characteristics of the T cell responses to nickel in allergic patients and healthy individuals [34]. They found that both allergic and nonallergic individuals carried CD4+ memory T cells responsive to nickel. In contrast, they found that nickel-specific CD8+ T cell responses were restricted to the nickel-allergic patients. Nickel-specific CD8+ T cell clones expressed the skin-homing marker cutaneous lymphocyte-associated antigen (CLA) and released high amounts of IFN-γ and no IL-4, thus belonging to the T cytotoxic 1 (Tc1) subset [34]. In contrast, both allergic and nonallergic individuals carried CD4+ memory T cells responsive to nickel. The nickel-specific CD4+CLA+ T cell clones from the nonallergic donors secreted higher amounts of IL-10 and lower amounts of IFN-γ compared to the T cell clones from the allergic patients. From these results, it was concluded that CD8+ T cells are crucial for the induction of nickel allergy, whereas CD4+ T cells may predominantly have a regulatory role [34]. In the same year, another study also demonstrated the presence of nickel-specific CD8+CLA+ T cells in the peripheral blood of nickel-allergic individuals [35]. Although the CD8+ T cells only constituted a minor subpopulation of the nickel-specific T cells, which had also been shown by others [28], they clearly had effector functions in the presence of nickel, involving cytokine production (IFN-γ and some IL-4 in addition) and cytotoxic activity [35]. Traidl et al. further demonstrated an important role of CD8+ T cells in nickel allergy, as nickel-loaded keratinocytes were found to be highly susceptible to nickel-specific cytotoxicity induced by skin-infiltrating CD8+ Tc1 and Tc2 cells [36].
However, the role of CD8+ cells has been questioned [37]. By using T cells isolated directly from peripheral blood mononuclear cells (PBMC), Moed et al. showed that only CD4+CLA+CD45RO+ and not CD8+ T cells proliferate and produce both Th1- (IFN-γ) and Th2-type (IL-5) cytokines in response to nickel [37]. In line with this, Minang et al. examined the role of CD4+ and CD8+ T cells on nickel-induced Th1- and Th2-type cytokine production. By depleting CD4+, CD3+, or CD8+ T cell populations from PBMC, they showed that CD4+ T cells were responsible for the nickel-specific production of both IFN-γ and IL-4 [38]. Thus, these studies suggested that CD4+ T cells are the most important effector cells in nickel allergy. In accordance, we have demonstrated a massive cellular infiltration in nickel-challenged skin from nickel-allergic patients with CD4+ T cells being the predominant cells [39]. However, CD8+ T cells were also found in the infiltrate [39], suggesting that at least small numbers of CD8+ nickel-specific T cells contribute to nickel-induced allergic skin reactions.
The varying results presented above might be ascribed to the use of different experimental models, e.g., the use of long-term cultured T cell clones or freshly isolated T cells from blood or skin, as well as patient-related differences [37]. Thus, although there is some inconsistency concerning the roles of CD4+ versus CD8+ T cells during the response to nickel, we find it likely that both subsets play important roles as effector cells probably in different phases of the response. Accordingly, we have recently shown that exposure to nickel in nickel-allergic individuals results in localization of epidermal-resident memory CD8+ T cells in the specific skin area exposed to nickel [40]. Even though the epidermal-resident memory CD8+ T cells appear to be a minor subset during the peak of the ACD response, they probably boost the innate immune response and thereby are important initiators and accelerators of the early inflammatory response [39, 40].
4 Th17 Cells and Nickel Allergy
Although the studies described above point toward metal allergy being a Th1/Tc1-dominated or a mixed Th1/Th2 response, evidence for the involvement of other T cell subsets has emerged. Thus, Albanesi et al. found that IL-17 mRNA was expressed in skin biopsies from positive patch tests to nickel but not in normal skin [41]. In addition, IL-17 was found to be produced by approximately 50% of activated blood- and skin-derived nickel-specific CD4+ T cell clones, although the authors did not find IL-17 production to segregate into a distinct Th subset [42]. Rather, IL-17 was produced together with either IFN-γ (Th1), IL-4 (Th2), or both (Th0), and it was demonstrated that IL-17 modulated various pro-inflammatory functions of keratinocytes, especially when acting together with IFN-γ and IL-4 [41, 42]. We have identified nickel-specific Th1 and Th17 cells in the circulating memory T cell compartment from nickel-allergic patients but not from healthy controls following restimulation with autologous nickel-pulsed DC [43]. Furthermore, we showed infiltration of cells expressing IL-17, IL-22, CCR6, as well as the IL-22 receptor in an inflamed skin of nickel-challenged allergic individuals [43]. These findings have been confirmed by a study showing that IL-17-producing CD4+ T cells infiltrate the skin during ACD elicited by nickel, cobalt, fragrances, or thiuram [44]. One way IL-17 seems to amplify the allergic skin reaction is by acting synergistically with IFN-γ to increase T cell-keratinocyte adhesiveness via ICAM-1, which promotes the Fas-FasL-mediated T cell killing of keratinocytes [44]. In addition to IFN-γ and IL-17, IL-22 seems to be involved in the response as increased serum levels of IL-22 have been found in nickel-allergic individuals [45]. In correlation with this, we have shown that the number of IL-17-, IFN-γ-, and IL-22-producing CD4+ T cells is increased in skin-derived T cell pools from nickel-exposed skin compared to vehicle-exposed skin from nickel-allergic patients [39]. Thus, these studies suggest that Th17 and Th22 cells are also important effector cells in nickel allergy and presumably in other metal allergies as well. Accordingly, IL-17 and IL-22 stimulate keratinocytes to release IL-1β, a cytokine that is indeed associated with ACD and nickel allergy [46,47,48]. Furthermore, IL-1β may provide positive feedback for further IL-17 and IL-22 production [49, 50].
The involvement of Th17 and Th22 cells in metal allergy was recently substantiated by Dhingra et al. who performed extensive molecular and cellular profiling of skin samples from patients sensitized to several common haptens, including nickel and other metal allergens, compared to petrolatum-occluded skin. Using RT-PCR, gene arrays, and immunohistochemistry, nickel was found to induce potent innate immune responses and Th1/Th17 polarization along with a Th22 component. Other metals, including cobalt and chromium, generally showed weaker immune activation predominantly characterized by induction of Th17 markers [51].
5 Induction of Regulatory T Cells in Nickel Allergy
As mentioned above, nickel allergy is the most frequent form of ACD, affecting approximately 14.5% of the general European population [52]. However, as nickel is ubiquitously present in our everyday life, one would expect that even more people developed nickel allergy or that different types of immune responses (pro- or anti-inflammatory) could develop depending on the route and dose of the primary nickel exposure. The development of nickel tolerance was suggested based on epidemiological studies and was later confirmed in both human studies and in animal models [34, 53,54,55,56,57,58]. Interestingly, it was shown that oral contact with nickel in the form of dental braces prior to ear piercing reduced the risk of developing nickel allergy by approximately 50% [53]. Later, it was shown that oral tolerance correlated with the induction of nickel-specific IL-10 production by PBMC [30]. The mechanisms for oral tolerance to metal have been further studied using different animal models. It was shown that it is possible to induce long-lasting (> 2 years) nickel-specific tolerance in a dose-dependent manner by feeding guinea pigs NiSO4 [55]. Similar results were obtained by feeding animals with chromium [55]. The mechanism mediating this tolerance seems to be the induction of suppressor cells as the tolerance could be transferred to naïve animals by transferring spleen and lymph node cells from nickel-fed animals [55]. Studies in mice confirmed the induction of oral tolerance by giving mice NiSO4 in the drinking water, and it was shown that the cells mediating the suppression were CD8+ T cells [56, 57]. In contrast, in a series of studies, Gleichmann and co-workers showed that both CD4+ and CD8+ T cells are required to obtain oral tolerance to nickel and that the T cells work together with both tolerogenic APC and CD4+ invariant NKT cells [58,59,60,61,62]. The different results obtained in various studies might be due to the use of different mice strains and/or doses of nickel [56,57,58]. Interestingly, when comparing the effector function of nickel-responsive T cell clones isolated from nickel-allergic and healthy controls, it was shown that in healthy controls only CD4+ T cells proliferated following nickel stimulation, whereas the proliferation of both CD4+ T cells and CD8+ T cells was seen in allergic individuals [34]. The CD4+ T cell clones isolated from healthy controls were characterized by high production of IL-10 compared to the CD4+ T cell clones isolated from nickel-allergic individuals [34]. The nickel-specific IL-10-producing CD4+ T cells (Tr1 cells) were further analyzed and they were identified both in blood and skin from nickel-allergic individuals and in blood from healthy controls [63]. The nickel-specific Tr1 cells were shown to inhibit the maturation of dendritic cells and monocytes via an IL-10-dependent mechanism, thereby inhibiting the activation of Th1 and Tc1 cells [63]. The presence of anti-inflammatory mechanisms in individuals with negative patch tests was confirmed by Rustemeyer et al., who found a higher number of individuals with blood-derived, nickel-specific IL-10- and TGF-β-producing T cells among a group of individuals with negative patch tests compared to a positive patch test group [30]. In addition to Tr1 cells, CD4+CD25+ T cells (Treg cells) with an anti-inflammatory function have been isolated from nickel-patch test negative skin [54]. Interestingly, Treg cells could inhibit both primary and secondary nickel-specific responses in a cell-cell contact-dependent and IL-10- and TGF-β-independent way [54]. The induction of tolerance in already allergic individuals has been studied using a mouse model for oral nickel desensitization [58]. Unfortunately, even though it was possible to desensitize allergic mice, this required constitutive oral exposure to nickel [58].
6 Conclusion
T cells play a central role in immune responses to metals, with Tc1, Th1, and Th17 cells being the major effector cells and Tr1 and Treg cells having an anti-inflammatory role (Fig. 9.4). However, additional studies are clearly required to define the exact role of the different T cell subsets in metal allergy. For example, it has been suggested that Th2 responses to metals and other contact allergens are elicited to counterbalance the detrimental Th1/Tc1 responses [1, 38, 64]. In addition, a study recently found that nickel increases IL-9 production in human PBMC from nickel-allergic patients but not from healthy donors. It was suggested that Th9 cells exert a regulatory role in nickel allergy by modulating the Th1 response both directly and via its ability to promote secretion of the Th2 cytokine IL-4 [65]. Finally, it would be highly valuable to understand the immunological mechanisms underlying the fact that only temporary desensitization can be induced in individuals with metal allergy.

T cells involved in metal allergy. Metal allergy is a T cell-mediated reaction with Tc1, Th1, and Th17 cells being the major effector cells and Tr1 and Treg cells having an anti-inflammatory role
Change history
28 August 2018
Correction to: Chapter 9 in: J.K. Chen, J.P. Thyssen (eds.), Metal Allergy, https://doi.org/10.1007/978-3-319-58503-1_9
References
Schmidt M, Goebeler M. Immunology of metal allergies. J Dtsch Dermatol Ges. 2015;13:653–9.
Vocanson M, Hennino A, Rozières A, Poyet G, Nicolas JF. Effector and regulatory mechanisms in allergic contact dermatitis. Allergy. 2009;64:1699–714.
Martin S, Esser P, Weber F. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy. 2011;66:1152–63.
Kaplan DH, Igyártó BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. 2012;12:114–24.
Thierse H-J, Gamerdinger K, Junkes C, Guerreiro N, Weltzien HU. T cell receptor (TCR) interaction with haptens: metal ions as non-classical haptens. Toxicology. 2005;209:101–7.
Sinigaglia F, Scheidegger D, Garotta G, Scheper R, Pletscher M, Lanzavecchia A. Isolation and characterization of Ni-specific T cell clones from patients with Ni-contact dermatitis. J Immunol. 1985;135:3929–32.
Kapsenberg ML, Res P, Bos JD, Schootemijer A, Teunissen MB, Van Schooten W. Nickel-specific T lymphocyte clones derived from allergic nickel-contact dermatitis lesions in man: heterogeneity based on requirement of dendritic antigen-presenting cell subsets. Eur J Immunol. 1987;17:861–5.
Moulon C, Vollmer J, Weltzien HU. Characterization of processing requirements and metal cross-reactivities in T cell clones from patients with allergic contact dermatitis to nickel. Eur J Immunol. 1995;25:3308–15.
Büdinger L, Hertl M. Immunologic mechanisms in hypersensitivity reactions to metal ions: an overview. Allergy. 2000;55:108–15.
Sinigaglia F. The molecular basis of metal recognition by T cells. J Invest Dermatol. 1994;102:398–401.
Romagnoli P, Labhardt AM, Sinigaglia F. Selective interaction of Ni with an MHC-bound peptide. EMBO J. 1991;10:1303–6.
Romagnoli P, Spinas GA, Sinigaglia F. Gold-specific T cells in rheumatoid arthritis patients treated with gold. J Clin Invest. 1992;89:254–8.
Griem P. The antirheumatic drug disodium aurothiomalate inhibits CD4+ T cell recognition of peptides containing two or more cysteine residues. J Immunol. 1995;155(3):1575–87.
Lu L, Vollmer J, Moulon C, Weltzien HU, Marrack P, Kappler J. Components of the ligand for a Ni++ reactive human T cell clone. J Exp Med. 2003;197:567–74.
Vollmer J, Fritz M, Dormoy A, Weltzien HU, Moulon C. Dominance of the BV17 element in nickel-specific human T cell receptors relates to severity of contact sensitivity. Eur J Immunol. 1997;27:1865–74.
Büdinger L, Neuser N, Totzke U, Merk HF, Hertl M. Preferential usage of TCR-Vbeta17 by peripheral and cutaneous T cells in nickel-induced contact dermatitis. J Immunol. 2001;167:6038–44.
Wang Y, Dai S. Structural basis of metal hypersensitivity. Immunol Res. 2013;55:83–90.
Yin L, Crawford F, Marrack P, Kappler JW, Dai S. T-cell receptor (TCR) interaction with peptides that mimic nickel offers insight into nickel contact allergy. Proc Natl Acad Sci U S A. 2012;109:18517–22.
Weltzien HU, Martin SF, Nicolas J-F. T cell responses to contact allergens. In: Martin FS, editor. T lymphocytes as tools in diagnostics and immunotoxicology. Basel: Springer; 2014. p. 41–9.
Gamerdinger K, Moulon C, Karp DR, Van Bergen J, Koning F, Wild D, Pflugfelder U, Weltzien HU. A new type of metal recognition by human T cells: contact residues for peptide-independent bridging of T cell receptor and major histocompatibility complex by nickel. J Exp Med. 2003;197:1345–53.
Thierse H-J, Moulon C, Allespach Y, Zimmermann B, Doetze A, Kuppig S, Wild D, Herberg F, Weltzien HU. Metal-protein complex-mediated transport and delivery of Ni2+ to TCR/MHC contact sites in nickel-specific human T cell activation. J Immunol. 2004;172:1926–34.
Griem P, Panthel K, Kalbacher H. Alteration of a model antigen by Au(III) leads to T cell sensitization to cryptic peptides. Eur J Immunol. 1996;26(2):279–87.
Griem P, Von Vultée C, Panthel K, Best SL, Sadler PJ, Shaw CF. T cell cross-reactivity to heavy metals: identical cryptic peptides may be presented from protein exposed to different metals. Eur J Immunol. 1998;28:1941–7.
De Wall SL, Painter C, Stone JD, Bandaranayake R, Wiley DC, Mitchison TJ, Stern LJ, DeDecker BS. Noble metals strip peptides from class II MHC proteins. Nat Chem Biol. 2006;2:197–201.
Clayton GM, Wang Y, Crawford F, Novikov A, Wimberly BT, Kieft JS, Falta MT, Bowerman NA, Marrack P, Fontenot AP, Dai S, Kappler JW. Structural basis of chronic beryllium disease: linking allergic hypersensitivity and autoimmunity. Cell. 2014;158:132–42.
Rogers PR, Croft M. Peptide dose, affinity, and time of differentiation can contribute to the Th1/Th2 cytokine balance. J Immunol. 1999;163:1205–13.
Kapsenberg ML, Wierenga EA, Stiekema FE, Tiggelman AM, Bos JD. Th1 lymphokine production profiles of nickel-specific CD4+T-lymphocyte clones from nickel contact allergic and non-allergic individuals. J Invest Dermatol. 1992;98:59–63.
Werfel T, Hentschel M, Kapp A, Renz H. Dichotomy of blood- and skin derived IL-4-producing allergen-specific T cells and restricted Vb repertoire in nickel-mediated contact dermatitis. J Immunol. 1997;158:2500–5.
Borg L, Molin J, Kristiansen J, Henrik N. Nickel-induced cytokine production from mononuclear cells in nickel-sensitive individuals and controls with nickel allergy-related hand eczema before and after nickel challenge. Arch Dermatol Res. 2000;292(6):285–91.
Rustemeyer T, von Blomberg BME, van Hoogstraten IMW, Bruynzeel DP, Scheper RJ. Analysis of effector and regulatory immune reactivity to nickel. Clin Exp Allergy. 2004;34:1458–66.
Minang JT, Troye-Blomberg M, Lundeberg L, Ahlborg N. Nickel elicits concomitant and correlated in vitro production of Th1-, Th2-type and regulatory cytokines in subjects with contact allergy to nickel. Scand J Immunol. 2005;62:289–96.
Minang JT, Areström I, Troye-Blomberg M, Lundeberg L, Ahlborg N. Nickel, cobalt, chromium, palladium and gold induce a mixed Th1- and Th2-type cytokine response in vitro in subjects with contact allergy to the respective metals. Clin Exp Immunol. 2006;146:417–26.
Muris J, Feilzer AJ, Kleverlaan CJ, Rustemeyer T, Van Hoogstraten IMW, Scheper RJ, Von Blomberg BME. Palladium-induced Th2 cytokine responses reflect skin test reactivity. Allergy. 2012;67:1605–8.
Cavani A, Mei D, Guerra E, Corinti S. Patients with allergic contact dermatitis to nickel and nonallergic individuals display different nickel-specific T cell responses. Evidence for the presence of effector. J Invest Dermatol. 1998;111:621–8.
Moulon C, Wild D, Dormoy A, Weltzien HU. MHC-dependent and -independent activation of human nickel-specific CD8+ cytotoxic T cells from allergic donors. J Invest Dermatol. 1998;111:360–6.
Traidl C, Sebastiani S, Albanesi C, Merk HF, Puddu P, Girolomoni G, Cavani A. Disparate cytotoxic activity of nickel-specific CD8+ and CD4+ T cell subsets against keratinocytes. J Immunol. 2000;165:3058–64.
Moed H, Boorsma DM, Stoof TJ, Von Blomberg BME, Bruynzeel DP, Scheper RJ, Gibbs S, Rustemeyer T. Nickel-responding T cells are CD4+ CLA+ CB45RO+ and express chemokine receptors CXCR3, CCR4 and CCR10. Br J Dermatol. 2004;151:32–41.
Minang JT, Arestrom I, Zuber B, Jonsson G, Troye-Blomberg M, Ahlborg N. Nickel-induced IL-10 down-regulates Th1- but not Th2-type cytokine responses to the contact allergen nickel. Clin Exp Immunol. 2006;143:494–502.
Dyring-Andersen B, Skov L, Løvendorf MB, Bzorek M, Søndergaard K, Lauritsen J-PH, Dabelsteen S, Geisler C, Bonefeld CM. CD4(+) T cells producing interleukin (IL)-17, IL-22 and interferon-γ are major effector T cells in nickel allergy. Contact Dermatitis. 2013;68:339–47.
Schmidt JD, Ahlström MG, Johansen JD, Dyring-Andersen B, Agerbeck C, Nielsen MM, Poulsen SS, Woetmann A, Ødum N, Thomsen AR, Geisler C, Bonefeld CM. Rapid allergen-induced IL-17 and IFN-gamma secretion by skin-resident memory CD8+ T cells. Contact Dermatitis. 2016;76(4):218–27.
Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. J Immunol. 1999;162:494–502.
Albanesi C, Scarponi C, Cavani A, Federici M, Nasorri F, Girolomoni G. Interleukin-17 is produced by both Th1 and Th2 lymphocytes, and modulates interferon-gamma- and interleukin-4-induced activation of human keratinocytes. J Invest Dermatol. 2000;115:81–7.
Larsen JM, Bonefeld CM, Poulsen SS, Geisler C, Skov L. IL-23 and T(H)17-mediated inflammation in human allergic contact dermatitis. J Allergy Clin Immunol. 2009;123:486–92.
Pennino D, Eyerich K, Scarponi C, Carbone T, Eyerich S, Nasorri F, Garcovich S, Traidl-Hoffmann C, Albanesi C, Cavani A. IL-17 amplifies human contact hypersensitivity by licensing hapten nonspecific Th1 cells to kill autologous keratinocytes. J Immunol. 2010;184:4880–8.
Ricciardi L, Minciullo PL, Saitta S, Trombetta D, Saija A, Gangemi S. Increased serum levels of IL-22 in patients with nickel contact dermatitis. Contact Dermatitis. 2009;60:57–8.
Watanabe H, Gaide O, Pétrilli V, Martinon F, Contassot E, Roques S, Kummer JA, Tschopp J, French LE. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956–63.
Li X, Zhong F. Nickel induces interleukin-1β secretion via the NLRP3-ASC-caspase-1 pathway. Inflammation. 2014;37:457–66.
Vennegaard MT, Dyring-Andersen B, Skov L, Nielsen MM, Schmidt JD, Bzorek M, Poulsen SS, Thomsen AR, Woetmann A, Thyssen JP, Johansen JD, Odum N, Menné T, Geisler C, Bonefeld CM. Epicutaneous exposure to nickel induces nickel allergy in mice via a MyD88-dependent and interleukin-1-dependent pathway. Contact Dermatitis. 2014;71:224–32.
Cho K-A, Suh JW, Lee KH, Kang JL, Woo S-Y. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1beta by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int Immunol. 2012;24:147–58.
Nielsen MM, Lovato P, MacLeod AS, Witherden DA, Skov L, Dyring-Andersen B, Dabelsteen S, Woetmann A, Ødum N, Havran WL, Geisler C, Bonefeld CM. IL-1β-dependent activation of dendritic epidermal T cells in contact hypersensitivity. J Immunol. 2014;192:2975–83.
Dhingra N, Shemer A, Correa Da Rosa J, Rozenblit M, Fuentes-Duculan J, Gittler JK, Finney R, Czarnowicki T, Zheng X, Xu H, Estrada YD, Cardinale I, Suárez-Fariñas M, Krueger JG, Guttman-Yassky E. Molecular profiling of contact dermatitis skin identifies allergen-dependent differences in immune response. J Allergy Clin Immunol. 2014;134:362–72.
Diepgen TL, Ofenloch RF, Bruze M, Bertuccio P, Cazzaniga S, Coenraads P-J, Elsner P, Goncalo M, Svensson Å, Naldi L. Prevalence of contact allergy in the general population in different European regions. Br J Dermatol. 2016;174:319–29.
Van Hoogstraten IM, Andersen KE, Von Blomberg BM, Boden D, Bruynzeel DP, Burrows D, Camarasa JG, Dooms-Goossens A, Kraal G, Lahti A. Reduced frequency of nickel allergy upon oral nickel contact at an early age. Clin Exp Immunol. 1991;85:441–5.
Cavani A, Nasorri F, Ottaviani C, Sebastiani S, De Pita O, Girolomoni G. Human CD25+ regulatory T cells maintain immune tolerance to nickel in healthy, nonallergic individuals. J Immunol. 2003;171:5760–8.
van Hoogstraten IM, Boden D, von Blomberg ME, Kraal G, Scheper RJ. Persistent immune tolerance to nickel and chromium by oral administration prior to cutaneous sensitization. J Invest Dermatol. 1992;99:608–16.
Van Hoogstraten IM, Boos C, Boden D, Von Blomberg ME, Scheper RJ, Kraal G. Oral induction of tolerance to nickel sensitization in mice. J Invest Dermatol. 1993;101:26–31.
Ishii N, Moriguchi N, Nakajima H, Tanaka S, Amemiya F. Nickel sulfate-specific suppressor T cells induced by nickel sulfate in drinking water. J Dermatol Sci. 1993;6:159–64.
Artik S, Haarhuis K, Wu X, Begerow J, Gleichmann E. Tolerance to nickel: oral nickel administration induces a high frequency of anergic T cells with persistent suppressor activity. J Immunol. 2001;167:6794–803.
Roelofs-Haarhuis K, Wu X, Nowak M, Fang M, Artik S, Gleichmann E. Infectious nickel tolerance: a reciprocal interplay of tolerogenic APCs and T suppressor cells that is driven by immunization. J Immunol. 2003;171:2863–72.
Roelofs-Haarhuis K, Wu X, Gleichmann E. Oral tolerance to nickel requires CD4+ invariant NKT cells for the infectious spread of tolerance and the induction of specific regulatory T cells. J Immunol. 2004;173:1043–50.
Nowak M, Kopp F, Roelofs-Haarhuis K, Wu X, Gleichmann E. Oral nickel tolerance: Fas ligand-expressing invariant NK T cells promote tolerance induction by eliciting apoptotic death of antigen-carrying, effete B cells. J Immunol. 2006;176:4581–9.
Wu X, Roelofs-Haarhuis K, Zhang J, Nowak M, Layland L, Jermann E, Gleichmann E. Dose dependence of oral tolerance to nickel. Int Immunol. 2007;19:965–75.
Cavani A, Nasorri F, Prezzi C, Sebastiani S, Albanesi C, Girolomoni G. Human CD4+ T lymphocytes with remarkable regulatory functions on dendritic cells and nickel-specific Th1 immune responses. J Invest Dermatol. 2000;114:295–302.
Xu H, DiIulio N, Fairchild R. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon gamma-producing (Tc1). J Exp Med. 1996;183:1001–12.
Liu J, Harberts E, Tammaro A, Girardi N, Filler RB, Fishelevich R, Temann A, Licona-Limón P, Girardi M, Flavell RA, Gaspari AA. IL-9 regulates allergen-specific Th1 responses in allergic contact dermatitis. J Invest Dermatol. 2014;134:1903–11.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Petersen, T.H., Geisler, C., Bonefeld, C.M. (2018). Acquired Immunity in Metal Allergy: T Cell Responses. In: Chen, J., Thyssen, J. (eds) Metal Allergy. Springer, Cham. https://doi.org/10.1007/978-3-319-58503-1_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-58503-1_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58502-4
Online ISBN: 978-3-319-58503-1
eBook Packages: MedicineMedicine (R0)