
Abstract
The heart involvement in systemic autoimmune diseases represents a growing burden for patients and health systems. Cardiac function can be impaired as a consequence of systemic conditions and manifests with threatening clinical pictures or chronic myocardial damage. Direct injuries are mediated by the presence of inflammatory infiltrate which, even though unusual, is one of the most danger manifestations requiring prompt recognition and treatment. On the other hand, a not well-managed inflammatory status leads to accelerated atherosclerosis that precipitates ischemic disease. All cardiac structures may be damaged with different grades of intensity; moreover, lesions can appear simultaneously or more frequently at a short distance from each other leading to the onset of varied clinical pictures. The pathogenesis of heart damages in systemic autoimmune conditions is not yet completely understood for the great part of situations, even if several mechanisms have been investigated. The principal biochemical circuits refer to the damaging role of autoantibodies on cardiac tissues and the precipitation of immune complexes on endocardium. These events are finally responsible of inflammatory infiltration which leads to subsequent worsening of the previous damage. For these reasons, it appears of paramount importance a regular and deepened cardiovascular assessment to prevent a progressive evolution toward heart failure in patient affected by autoimmune diseases.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Immune-mediated injury
- Chronic inflammation
- Autoimmunity
- Atherosclerosis
- Connective tissue diseases
- Arthritis
- Antiphospholipid syndrome
1 Introduction
Autoimmune diseases represent a broad range of related disorders affecting nearly 5% of the European population [1]. These conditions are cumulatively characterized by more or less defined clinical pictures sometimes overlapping within the same patient. These include connective tissue diseases such as systemic lupus erythematosus (SLE) , systemic sclerosis (SSc) , mixed connective tissue disease (MCTD) , Sjögren’s syndrome (SS) , rheumatoid arthritis (RA) , myositis , and antiphospholipid syndrome (APS) . The common pathogenic mark is the breakdown of tolerance against self-peptide antigens which derives from a failure of peripheral tolerance under genetic predisposing factors and environmental triggering agents [2, 3]. One of the most threatening complications is the cardiovascular (CV) involvement, which represents a significant cause of morbidity and mortality [4,5,6,7,8,9,10].
1.1 Mechanisms of CV Damage
The hypothesized pathogenesis encompasses different mechanisms, but only some of these have been clarified, especially in their cellular and biochemical pathways, but ultimately remaining elusive. The heart can be primarily involved by the localization of an autoimmune process or can be indirectly damaged by chronic inflammation, by other injured organs, and by drugs used to manage the primary disease. In the case of a primary involvement, the impairment of different cardiac structures derives from the inflammatory infiltrate, the deposition of immune complexes, leading to the activation of the complement cascade (Fig. 8.1). Alternatively, the cardiac function is threatened by the general inflammatory status, strictly dependent on disease activity, or the heart can be severely damaged in end stages of a systemic autoimmune process. This last condition is principally determined by the impaired function of other organs, such as the kidneys or the lungs, which compromise hemodynamic. It is now well established that atherosclerosis appears to be significantly accelerated in systemic autoimmune diseases compared to general population. This condition is one of the most powerful causes of CV diseases due to thromboembolic phenomena following plaque rupture ; moreover, chronic inflammation activates the endothelial cells which express different surface molecule patterns acquiring a prothrombotic phenotype.

Pathogenic mechanisms of heart damages in autoimmune diseases. LDL light-density lipoprotein, oxLDLs oxidized light-density lipoproteins
The mechanisms of the accelerated atherosclerosis are not well defined, but chronic inflammation has been suggested as a contributing factor with pro-inflammatory cytokines (i.e., TNFα and IL-6) leading to endothelial dysfunction and activation and subsequent rise of low-density lipoprotein (LDL) uptake and deposition under endothelial surface. Specifically, TNFα can alter the endothelial structure mediating the inflammatory vascular injury in both acute and chronic conditions, ranging from the remodeling by smooth muscle cells to a direct role in rupture of atherosclerotic plaques [11]. Similarly, IL-6 induces an overall change of surface molecules of the endothelium that develops a pro-inflammatory status. This enhances the migration of lipoproteins, especially LDLs, from bloodstream to subendothelial space at higher concentrations than the cellular metabolic needs and when in contact with high amount of free oxygen radicals produced by activated inflammatory cells undergoing oxidation (oxLDLs). High oxLDL levels stimulate macrophages through the interaction with scavenger receptors and lead to massive phagocytosis which, after a gradual engulfment of cytoplasm, generates the so-called foam cells. The reaction of the immune system against oxLDLs is of paramount importance because it represents the first pathological mechanism leading to the atheromatous plaque . OxLDLs stimulate not only innate immunity but also the adaptive compartment. Studies have demonstrated the production of antibodies against oxLDLs which seem to be related to cardiovascular events [12, 13], even though some authors have reported, conversely, a protective role [14].
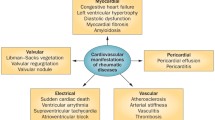
Chronic inflammation stimulates the expression of adhesion molecules, such as VCAM-1, by the endothelium and smooth muscle cells. This pathological activation promotes the recruitment of leukocytes into the subendothelial space which ultimately lead to the perpetuation of inflammation [15]. T cell activation can also be involved, and activated T cells are present in atherosclerotic plaques, especially in unstable plaques accounting for the increased CV risk and higher mortality. The clinical manifestations of heart involvement can be strikingly diverse because all the anatomical heart structures can be affected simultaneously or not, with damages of various seriousness [16] (Fig. 8.2). Recognized since the beginning of the twentieth century, cardiac involvement related to autoimmunity conditions has been carefully investigated in the last decades. The following findings led to recognize new clinical entities but also to introduce different available treatments. We will focus on cardiac manifestations operating in systemic autoimmune diseases, with particular interest on pathogenic mechanisms .

Pathological involvement of the heart by different systemic autoimmune diseases. SLE systemic lupus erythematosus, APS antiphospholipid syndrome, IIM idiopathic inflammatory myositis, MCTD mixed connective tissue diseases, RA rheumatoid arthritis, SSc systemic sclerosis, SS Sjögren’s syndrome
1.2 The Impact of Medical Treatments
Medications used to treat the autoimmune diseases can have, sometimes, a negative impact on cardiac function by a direct toxicity (antimalarials, nonsteroidal anti-inflammatory drugs (NSAIDs)) or worsening a pre-existing cardiac disease (Fig. 8.1). On the other hand, several drugs used in chronic inflammatory diseases represent paradigms of potential treatments for CV comorbidities, but effects are poorly defined.
First, antimalarials, such as chloroquine and hydroxychloroquine, in chronic use rarely cause conduction abnormalities, arrhythmias, and infiltrative cardiomyopathy with heart failure (HF) due to impairment of diastolic, systolic, or both cardiac functions [17, 18]. Second, biologics were ideal candidates as anti-TNFα biologics are used for RA treatment, but data from clinical trials reported no beneficial effects and a possible worsening of heart function leading to a contraindication of anti-TNFα use in patients with HF New York Heart Association (NYHA) classes III–IV (moderate and severe) [19]. To further investigate the HF risk with anti-TNFα, longer observational studies were conducted on almost all TNFα inhibitors, including etanercept, which, due to its peculiar structure, has lower avidity for TNFα compared to monoclonal antibodies, but results were inconsistent [20, 21]. Other biologics, with different mechanisms of action, have also been investigated: rituximab (anti-CD20), tocilizumab (anti-IL-6), and abatacept (anti-CTLA4) showed no overall increased CV risk in the RA population; however, no reduction of the risk of developing HF and other CV events was found in comparison with the RA population treated on standard therapy or anti-TNFα biologic agents [22]. Third, NSAIDs are frequently used in rheumatic conditions and are associated with an increased CV risk, and a recent meta-analysis of various clinical, preclinical, meta-analysis and observational studies showed that coxibs and NSAIDs increase the risk of cardiotoxicity in a dose-dependent manner. The cardiotoxicity associated with the use of NSAIDs might be due to inhibition of prostacyclin synthesis, oxidative stress, increase in blood pressure, and impaired endothelial function [23].
2 Systemic Lupus Erythematosus
SLE is frequently associated with CV manifestations, and all three layers of the heart (i.e., pericardium, myocardium, and endocardium) can be affected with the parietal sheet of cardiac serous being the most frequently involved, as suggested by its inclusion in the American College of Rheumatology (ACR) classification criteria [24]. The valvular apparatus, the conducting system , and the coronary vessels may be other noteworthy sites of pathological involvement in SLE.
2.1 Pericardium
Clinically, pericarditis manifests with variable symptoms ranging from precordial or substernal positional chest pain to dyspnea. At physical examination, patients may have fever, tachycardia, decreased heart sounds, and pericardial rubs, a specific sign which, however, is rarely hearable. The diagnosis may be sustained by an electrocardiogram (ECG) showing elevated ST segments and peaked T waves (although slight T-wave changes and transient elevation of ST segments are most characteristic) [25]. Transthoracic echocardiography and chest X-ray are the standard methods to investigate pericarditis [26, 27]. One of the most fearsome complications is cardiac tamponade, although constrictive and purulent processes are equally challenging. Pericarditis may be either characterized by an acute or chronic process which leads to the appearance of clinical symptoms in only 25% of the SLE population; however it is reported in as many as >50% of patients undergoing echocardiography or up to 62% during autoptic examination [28,29,30,31,32,33,34,35,36,37,38,39]. Pericardial involvement appears more frequently at SLE onset or during disease relapses, although it can occur at any time [40]. Common pathological findings include fibrinous or fibrous reactions, which, in chronic conditions, can lead to adhesive processes both focal and diffuse. Microscopically, the pericardium appears infiltrated by plasma cells and lymphocytes with fibroblastic proliferation, findings that are not unique for SLE and which resemble tubercular pericarditis [35]. The identification of hematoxylin bodies is a more specific finding, but particles of denatured nuclei are rarely seen [41]. Immunofluorescence may be helpful in characterizing SLE pericarditis; indeed i t is possible to demonstrate a granular deposition of immunoglobulin, C1q, and C3, in the pericardial vessels, which ultimately suggests a pathogenic role of immune complexes [42].
2.2 Myocardium
Clinically, patients manifest with symptoms resembling ischemic heart disease such as chest pain dyspnea, tachycardia, and arrhythmias [40]. Diagnosis may be challenging, with ECG not showing any typical findings and cardiac enzymes within limits. Echocardiographic imaging cannot definitely diagnose myocarditis , but the finding of global hypokinesis or pericardial effusion, in the absence of other known causes, is strongly suggestive [43]. More recently, cardiac magnetic resonance imaging (MRI), the gold standard for measuring ventricular volumes and ejection fraction, can be used to detect acute myocarditis, myocardial fibrosis, and even subtle tissue changes [43]. Endomyocardial biopsy is the gold standard for diagnosis; however this challenging procedure can be aggravated by complications both during and after execution, and for these reasons, it should be reserved only for selected situations [6, 44]. One of the most threatening consequences of chronic myocarditis is represented by dilated cardiomyopathy , which ultimately can evolve toward HF. Myocarditis represents one of the most important manifestations of heart involvement in SLE, even if myocardial disease may be determined or worsened by other mechanisms, such as drug toxicity (i.e., antimalarials). The inflammation of the myocardium, largely ignited by immune complex deposition and complement activation, is the pathological condition determining tissue injuries. Myocarditis prevalence is reported to be as high as 80% in SLE autoptic series [30]; however, recent findings suggest a rate of 6%, probably due to the introduction of effective treatments. From a clinical standpoint, this process can be detected only in 3–15% of patients with SLE due to the paucity of symptoms [45]. Pathologically, the findings are nonspecific, with a perivascular and interstitial infiltrate of mononuclear cells, constituted by lymphocytes, plasma cells, macrophages, and myocardial fibrosis. Immunohistochemistry is helpful to identify aggregates of lymphocytes and macrophages. The presence of myocyte vacuolization at optical microscopy, defined as a dilatation of sarcoplasmic reticulum that contains laminar amorphous material, is a characteristic of antimalarial-induced cardiomyopathy , even if the detection of myeloid bodies by electron microscope is the only pathognomonic sign [35, 46].
2.3 Valvular Disease
From a clinical standpoint, most of the valvular findings are asymptomatic, but extended lesions which cause hemodynamic dysfunctions become clinically evident and require, in about 3–4% of cases, valve replacement [6]. Valvular involvement in SLE is one of the most important and prevalent forms of carditis, and nearly 60% of patients who undergo echocardiography manifest abnormalities, with postmortem studies showing a prevalence between 11 and 74% [47]. The most common pathological findings derive from regurgitation and stenosis even if milder defects such as thickening and vegetations can be found. Among valve alterations, the Libman-Sacks endocarditis is a histopathological picture significantly associated with SLE and APS, albeit currently is rare thanks to the better treatment of the underlying disease. The persistent inflammation of endocardium generates sterile verrucous lesions affecting all structures with a remarkable prevalence for the mitral valve [48]. Histologically, Libman-Sacks vegetations consist of platelets, fibrin, degenerating blood products, chronic fibrosis, active fibroblasts, and neovascularization. These lesions may be active or healed [35].
2.4 Arrhythmias
Congenital heart block (CHB) is one the most representative cardiac rhythm disturbances associated with autoimmune diseases. This condition, among endocardial fibroelastosis and dilated cardiomyopathy , is a threatening manifestation of neonatal lupus, characterized by cardiac and skin involvement in newborns from mothers with SLE or Sjögren’s syndrome positive for anti-Ro/SSA and/or anti-La/SSB antibodies [49]. CHB arises by definition during uterine life or in the neonatal period (<28 days of life) [50, 51], but the entity of the problem remains elusive. Two studies, performed, respectively, in Finland and the USA on large case series, have estimated an incidence of ~1 case on 20,000 live births; but it was impossible to evaluate what is the real impact of maternal autoimmunity, given that no autoantibody status was reported [50, 51]. Nonetheless, CHB prevalence was recently determined to be less than 1% in anti-SSA-positive women [52, 53], and recurrence rate is defined at 19% [53]. The pathogenic pathway is represented by the transfer of maternal autoantibodies through the placenta into fetal bloodstream, even though the mechanism by which anti-SSA/Ro and anti-SSB/La act on the heart conduction system is not yet understood. Evidence of complete atrioventricular (AV) block in mice pups passively injected with anti-SSA/Ro 52 antibodies supports data from earlier reports [54]. Furthermore, studies in vitro demonstrate how anti-SSA/Ro IgG inhibits L-type CA2þ channel in rat and rabbit heart [55, 56]. Researches on humans have confirmed the potential role of what previously described. The presence of maternal autoantibodies in fetal circulation and the elution of anti-SSA/Ro antibodies from cardiac tissues of affected fetuses support animal models, even if the rarity o f CHB and discordance rate in twins remain unsolved problems [57]. National database analyses reveal a CHB-associated mortality rate of 20% with nearly 80% of deaths occurring in the uterus [58]. The great majority of pregnancies results in live births (81%), with a prematurity rate (birth <37 weeks) of 38% [58]. CHB can present with various grades of rhythm alterations in live births, and challenge refers to therapeutic strategies because most of the babies require pacing for the first 10 days of life and about two thirds have to maintain this support for 1 year; in fact, a quarter of total deaths related to autoimmune CHB are reported during this span of life [50]. Other rare rhythm disturbances have been described in association with anti-SSA/Ro antibodies, and the most significant include bradycardia, AV blocks of various degrees, and prolongation of the QT interval that usually resolve within the first year of life when maternal autoantibodies disappear [57].
Limited prevention strategies are available for women who are anti-SSA/SSB positive and are considered at high risk for offspring CHB as those with a previous history of neonatal lupus syndrome. Fetal monitoring with echocardiography is a safe and noninvasive method for screening CHB that offers an accurate assessment of the fetal heart rate, rhythm, and ventricular function. This procedure should be performed weekly between 16th and 24th week of gestation. At birth neonates should be observed during the first month of life [50]. If rhythm alterations are detected prenatally, therapeutic options to reduce the risk of CHB are represented by fluorinated steroids, which should diminish the inflammatory component in the fetal heart reducing tissue injury. Other strategies to treat intrauterine heart blocks include plasma exchange and/or intravenous immunoglobulin used in various combinations [59].
3 Systemic Sclerosis
SSc is a connective tissue disorder of unknown etiology characterized by the damage and fibrosis of multiple organs. The main pathogenic treats are represented by microvascular damages but also dysregulation of innate and adaptive immunity which lead to fibrosis in multiple organs. The prevalence ranges from 7 to 700 cases per million depending on geographic regions, ethnic differences, and gender; in fact women are more frequently affected than men [60, 61]. SSc can be classified into limited and diffuse variant, according to the extension of skin fibrosis [62]. Patients can develop heterogeneous clinical manifestations on the basis of variable involvement of different organs. Heart damage can derive from indirect chronic injuries which are principally dependent on lung and kidney pathology. Pulmonary arterial hypertension (PAH) and interstitial lung disease (ILD) are the main causes of right ventricular dysfunction. These conditions determine a persistent increased outflow resistance which causes right ventricular remodeling . The myocardial response to this chronic excessive work brings the heart to adopt counterproductive mechanisms, such as hypertrophy, generating various grades of HF. The kidney involvement is represented by scleroderma renal crisis , a rare (4–6%) but life-threatening manifestation, defined by malignant hypertension and acute renal failure which can be sometimes associated with congestive HF [63].
Primary cardiac involvement represents a frequent cause of morbidity and mortality in limited and diffuse SSc, as demonstrated by several studies [64, 65]. All heart structures can be affected, resulting in different impairments, which are often subclinical [66]. The overall hazard of early disease is higher in diffuse pattern, especially during the first year after the onset of the disease [67], and in patients with anti-RNA polymerases (I–III) and anti-fibrillarin antibodies [68] (Table 8.1). The etiology of cardiac damages is likely dependent from microvascular ischemic lesions and from fibrous tissue deposition. These events could impair regional microcirculation causing areas of localized myocardial hypokinesis which could evolve toward global heart dysfunction in later stages of disease [69, 70]. Interestingly, nailfold capillaroscopy could help in identifying patients more prone to develop cardiac involvement through the identification of peripheral microangiopathy, which has been shown to correlate with internal organ involvement [71].
3.1 Pericardium
The clinical picture of pericardial SSc manifestations may cover a wide range of conditions, including acute or constrictive pericardial effusion and the most threatening cardiac tamponade. The pericardial involvement is relatively common in SSc, even though it is frequently asymptomatic. Autoptic studies reported pathological findings in about 78% of specimens, but only 5–16% of patients present symptoms [72]. The cause of pericardial pathology in SSc is not well understood, even if studies performed on pericardial biopsies have often found diffuse fibrosis as in other tissues [73]. Clinically, the inflammation of pericardium is associated with chest and substernal pain, dyspnea, pericardial effusion, and symptoms of HF if tamponade is present. Echocardiography with Doppler imaging is demonstrated to be a precious instrument in diagnostic process of pericardial disease; indeed, it can precociously detect effusion. Treatment is usually unnecessary, even if pericardiocentesis should be considered to discern diagnostic doubts and for patients who develop hemodynamic impairment. Constrictive pericarditis may be difficult to identify; moreover its differentiation from restrictive cardiomyopathy can be a hard challenge for the clinician, especially when both processes coexist. Two echocardiographic signs favor a diagnosis of pericardium pathology; in fact, abnormal interventricular motion and preserved mitral parameters focus the attention on the serous sack. Cardiac MRI and computed tomography (CT) may be of additional help [72]. Intrinsic myocardial disease in SSc manifests as myocardial fibrosis due to microvascular ischemia and ultimately determines left ventricular dysfunction.
3.2 Myocardium
Myocardial fibrosis has been reported to be an important manifestation of SSc, being found in as many as 80% of autoptic cases, because symptomatic conditions are less frequent. The pathogenic mechanisms are still matter of discussion even if several factors have been identified as possible actors in myocardial injuries which lead to fibrotic process. Some of these are strikingly dependent on recurrent vasospasms and poor vasodilator reserve which determine, respectively, recurrent ischemia-reperfusion damages and areas of focal ischemia. Histology of SSc myocardium demonstrates focal myocardial fibrosis and necrosis of the contraction bands, findings that are extremely similar to non-SSc-related ischemia-reperfusion injury. From a clinical standpoint, myocardial fibrosis becomes manifest only in late stages, when the heart stiffness is so important to produce a function deficit. The tissue alterations can impair the systolic (reduced ejection fraction) or diastolic (preserved ejection fraction) function, leading to various degrees of heart chamber dysfunction. Right ventricular dysfunction has been frequently described in SSc, independently of PAH. The main clinical sign is a reduced right ventricle ejection fraction due to myocardial fibrosis as proved by a recent study which demonstrates altered ejection fractions in about one fifth of SSc patients using cardiac MRI [74]. A clinical suspect of heart fibrosis can be solved performing cardiac MRI, even if echocardiography and natriuretic peptide levels are a useful instrument to monitor cardiac function [75].
3.3 Arrhythmias
Arrhythmias are common in SSc and may be considered a complication of the fibrotic process of the conduction system and myocardium. Patients will develop different symptoms such as palpitations, vertigo, dizziness, syncope, and even sudden death; for these reasons, a precocious recognition and an accurate identification are essential. Atrial and ventricular tachyarrhythmias derive from myocardial fibrosis, whereas the conduction system involvement could give rise to brady-arrhythmias [66, 76]. Supraventricular rhythm disturbances are more common and manifest in about two thirds of SSc patients, compared to other conditions which have a lower prevalence [77]. ECG represents the first diagnostic tool even if up to 50% of patients have not any alterations at rest [78]. Some studies demonstrate that ECG abnormalities are predictive of survival, although it is unclear whether these changes contribute directly to morbidity and mortality or reflect the overall disease burden [77]. Symptomatic patients, who have a normal ECG, should undergo ECG Holter monitoring to better characterize cardiac rhythm changes; moreover, treadmill exercise can be performed to identify exertional arrhythmias. Echocardiography should be performed as second-line test. Treatment of arrhythmias should be according to general guidelines [76].
Early assessment of the overall CV involvement represents a mandatory step in a proper management of SSc patients, since its development increases morbidity and mortality. A good organ involvement screening should be sought in every patient, with special attention to those who have complaints which refer to cardiac involvement.
4 Inflammatory Muscle Disease
Idiopathic inflammatory myositis (IIM) includes a group of diseases characterized by proximal muscle weakness due to chronic inflammation of striated muscles mirrored by elevation of serum muscle enzymes, particularly creatine kinase [79]. Heart involvement is uncommon in IIM, especially in polymyositis (PM) and dermatomyositis (DM), but represents a poor prognostic factor responsible approximately of 10–20% of deaths [80]. Cardiac abnormality prevalence ranges from 6 to 72% in PM/DM. These results are strongly dependent on selection criteria; in fact, the broad interval suggests an underestimation of the problem, as demonstrated by autoptic studies which report alterations in about 30% of cases [81]. There is no clear relationship between IIM clinical patterns and specific heart diseases [81], and most studies reveal the occurrence of several cardiac pathologies during the course of both active and under remission myositis [2], even if a recent study shows significant correlations with disease onset [82]. Clinically, about 3–6% of patients with PM/DM present with symptoms such as dyspnea, chest pain, palpitations, or less common peripheral edema and syncope [83]. All these manifestations are mainly due to end-stage heart disease which ultimately evolves toward pump failure.
4.1 Pericardium
Pericardial disease is generally asymptomatic and sporadic, being reported in less than 10% of patients. The most frequent finding is pericardial effusion which is often revealed by investigations made for other clinical needs [81].
4.2 Myocardium
Myocarditis has been linked to IIM and is found in approximately 8% of patients. Cardiac MRI is a helpful tool to detect inflammatory infiltrates in the tissue [84,85,86,87], making endomyocardial biopsy less necessary. The histopathology resembles the picture found in skeletal muscles, with focal fibrosis, vasculitis, intimal proliferation, and medial sclerosis of blood vessels [88]. Clinically, it manifests with chest pain and dyspnea [83], but it can produce advanced signs of heart failure in widespread involvement of myocardial muscle. This group of patient is related with a worse prognosis, and for this reason, it could benefit from immunosuppressant therapies , but available data are lacking [83] in spite of some case reports which show a benefit of steroid [89].
4.3 Conduction System
Arrhythmias are a common feature observed in IIM. The incidence of EKG alterations is reported between 25 and 85% of patients with PM/DM. Branch block and supraventricular arrhythmias are the most frequent findings which range from 13.6% to 2.4%, in prospective and retrospective cohorts, respectively [83]. Other disturbances include atrial or ventricular premature beats, tachycardia, atrial fibrillation, conduction blocks, and abnormal Q-waves as nonspecific ST-T wave changes. Histopathologically, abnormalities in the conducting system include lymphocytic infiltration, sinoatrial node fibrosis, and contraction band necrosis [81].
4.4 Valves, Coronary, and Heart Function
Valvular abnormalities are relatively uncommon, as clinically significant valve diseases may be observed in 7–23% of patients [90].
Coronary heart disease is reported with uncertain incidence; in fact a recent study investigating CV risk factors in IIM suggests a prevalence of 26%. Sporadic reports of inflammatory arteritis of coronaries without obstruction have been described. Vasospastic angina (Prinzmetal’s angina) is occasionally reported in DM with Raynaud’s phenomenon [91]. HF develops as a consequence of left ventricular diastolic dysfunction that is observed in 12–42% of cases. Traditional echocardiography associated with tissue Doppler imaging can detect this pump defect in about 14–62% of circumstances [81] and should be carefully performed in high-risk patients, such as female sex and late onset and long course of disease [92].
5 Mixed Connective Tissue Disease
Overlap autoimmune features of SLE, PM, SSc, and RA associated with antibodies directed toward U1 nuclear ribonucleoprotein (RNP) configure MCTD [93]. CV involvement is rather frequent in MCTD. It has been reported from 13 to 65% depending on patient selection criteria and to diagnostic procedures performed; in fact, in symptomatic patients, cardiac disease ranges from 24 to 63% [94].
5.1 Pericardium
Pericarditis is the most common cardiac manifestation with a prevalence of 10–40% [95], when echocardiography is used. This condition can be related to the rheumatoid arthritis-like and/or the lupus-like spectra of MCTD [94]. The treatment is based on NSAIDs or steroids (0.25–1.0 mg/kg). Rare cases of large effusion [96] which evolve toward cardiac tamponade [97] may need percutaneous and surgical drainage.
5.2 Myocardium
Myocarditis has been rarely reported, although it could be underestimated as demonstrated in postmortem studies. This pathological entity might be responsible for conduction abnormalities and diastolic dysfunction [94]. Symptoms include dyspnea, chest pain, arrhythmias, and elevated cardiac enzymes. When an endomyocardial biopsy is performed, the most common findings include interstitial lymphocytic infiltrate, myocardial fiber, and interstitial necrosis [98, 99].
5.3 Valve, Conduction System, and Heart Function
Valvular abnormalities are reported with high frequency, especially verrucous thickening alterations or mitral valve prolapse (MVP), which is detectable by echocardiography in about 12–32% of patients [100, 101]. MVP might be due to focal degeneration of the valve leaflets resulting in reduced capacity to support systolic stress, as seen in SLE [8]. Conduction disorder prevalence rates are reported as high as 20%, and the most common condition observed is a deviation of QRS axis, which is related to anterior hemiblock [102]. The presence of ST-T abnormalities is detected with prevalence around 29% on ECG [103, 104]. Diastolic dysfunction is a common defect in MCTD, and echocardiography demonstrates abnormalities of the diastolic filling indexes due to atrial contraction, such as prolongation of isovolumetric relaxation time, reduction of peak early diastolic flow velocity, and an increase of peak late diastolic flow velocity [105, 106]. Systolic function is usually conserved. Histologically sections reveal inflammatory cell infiltrate and/or proliferative vasculopathy of the epicardial and intramural arteries. Autoptic studies have proven the presence of lymphocytic and polymorphonuclear infiltrates in the perivascular space but also intimal proliferation with an increased number of acellular elements [103].
6 Antiphospholipid Syndrome
APS is a rare autoimmune disease characterized by a high tendency of developing thrombotic events . It is diagnosed on the basis of defined clinical criteria and specific laboratory findings [107]. The former are represented by vascular thrombosis and/or pregnancy morbidity, while the latter are elevated levels of serum antiphospholipid antibodies (aPL). These include lupus anticoagulant (LA), anticardiolipin (aCL), and/or anti-beta2-glycoprotein I (anti-β2GPI) antibodies. The syndrome can be a primary disorder or it can be secondary to an underlying condition, most commonly SLE. The heart involvement is a frequent finding that is supposed to be mediated by direct aPL actions on cardiac structures or through vessel thrombosis which lead to myocardial ischemia.
6.1 Valve Involvement
Valvular damage is the most frequent cardiac manifestation of APS, being detected in one third of patients. The major alterations include vegetations or thickening of valve leaflets as Libman-Sacks endocarditis which was first described in SLE [108,109,110]. The pathogenic role of aPL seems to have a close link with the valvular damage, as supported by the evidence of deformed heart valves in patients with aPL positivity and no clinical signs or symptoms of disease [108, 111]. The prevalence of valvular defects in primary APS is reported to be between 32 and 38%, based on echocardiographic studies [112]. The most frequent altered valve is the mitral, followed by aortic; on the contrary, Libman-Sacks endocarditis involves often the tricuspid valve. These abnormalities are usually asymptomatic from a clinical standpoint; in fact, only 4–6% of APS patients develop severe disease that requires surgical treatment [40, 113]. The diagnosis can be made with traditional echocardiography, even if transesophageal approach is more sensitive [114, 115]. The importance of this pathological consequence of the syndrome goes beyond heart damage; indeed, some studies prove how it may be considered as a major risk factor for cerebrovascular accidents, particularly in primary APS [116, 117]. The pathogenesis of the valvular damage is referred to micro-injuries of some hemodynamically vulnerable sites which undergo repetitive mechanical stress [118]. These conditions should induce the exposition of negative phospholipids on both valve surface and endothelial cells of intra-valve capillaries leading to the interaction with aPL [118]. Histologically, some studies prove the coexistence of new and old lesions characterized by the presence of superficial or intravalvular fibrin deposits. This pathological feature triggers subsequent vascular proliferation, fibrosis, and calcification which lead to valve thickening, fusion of commissures, and rigidity of valvular apparatus [119]. Inflammation seems to be not a prominent feature, even in the presence of a linear subendothelial deposition of immunoglobulins and complement components [119]. The same pattern and location of staining were observed with anti-idiotypic antibody to aCL; moreover, a significant amount of IgG immunoglobulins that bound to cardiolipin was eluted from valves of patient with secondary APS [119]. Such deposits may be probably involved in the pathogenesis of valvular lesions [119].
6.2 Atherosclerosis
Atherosclerosis is significantly accelerated and more prevalent in patients with APS than in the general population, despite similar CV risk factors [120]. From a clinical standpoint, this process manifests with syndromes of different severity, from angina to sudden death, passing through myocardial infarction. Increasingly data demonstrate how this process is driven by direct immunological effects of aPL, especially anti-β2GPI [121], but also through autoantibodies cross-reactions and increased oxidative stress [122]. Histological studies on atherosclerotic plaques prove the contiguity of beta-2 glycoprotein I to T-CD4+ lymphocytes areas in the subendothelial regions [123]; moreover, in vitro experiments demonstrate how aPL accelerate plaque formation, enhance macrophage transformation in foam cells, and reduce the activity of paraoxonase which increases oxLDLs [124]. The binding of beta-2 glycoprotein I to oxLDLs generates a molecular complex that interacts with aPL leading to an increased uptake by macrophages via Fcγ receptors [125]. The proof of an increased rate of CV events in APS patients is represented by the prevalence of myocardial infarction which is 5.5% [108], 2.8% as first manifestation of the disease, compared to 1.4–3.2% of general population [126]; moreover, some studies correlate the levels of aPL with the incidence of severe CV events [127].
6.3 Myocardium
Chronic damages to heart tissue can manifest also with a less acute myocardial dysfunction . The pathogenesis is hypothesized to be derived from both direct antibody effects and repeated micro-embolism phenomena which ultimately impair pump function. Histologically, there is evidence of inflammation, immune deposits, and thrombosis in intramyocardial arteriolar with microinfarction of surrounding areas [122]. Cardiac MRI is the most sensible test to detect microvascular damages; in fact, a study shed on light that APS patients with low pretest probability of coronary heart diseases (CHD) have ischemic lesions in 11% of cases, while only 3.7% of the controls show the same alterations [128]. For all these reasons, the assessment of patient atherosclerotic status is crucial to estimate the overall CV risk of thromboembolic complications. Carotid intima-media thickness is considered an early marker of generalized atherosclerosis [122] and can be easily explored with echocardiography, a llowing a noninvasive but accurate evaluation of the situation.
7 Sjögren’s Syndrome
Sjögren’s syndrome is an autoimmune condition characterized by a chronic inflammation of exocrine glands which leads to their functional impairment and manifests with sicca syndrome.
The extraglandular signs are represented mainly by arthritis, even though it is described a wide range of organ involvement such as pulmonary and renal abnormalities but also vascular and gastrointestinal alterations. Heart injuries are rarely reported, even though echocardiographic studies suggest that subclinical abnormalities are relatively common [8].
7.1 Pericardium
Data demonstrate that approximately 33% of patients affected by SS present ultrasound signs of pericarditis , but only half of them complaint some symptoms [129]. Pericardium wall thickening or echodense signals may be consequences of subclinical conditions which can be derived from either the underlying disease or other causes. Some studies suggest that pericardial inflammation may be more frequent in older patients or in those that present a shorter disease duration and ANA positivity but also high levels of orosomucoid and haptoglobin [130].
7.2 Heart Function
Left ventricle diastolic dysfunction is an important cause of morbidity and may be an early sign of myocardial damage in various diseases. It has been shown that it is detectable in a significant number of patients. The pathogenesis underlying this heart abnormality remains still unclear, even if it has been suggested that myocardial Raynaud’s phenomenon may mediate the damage. Other mechanisms refer to stable microcircle alterations such as small intramyocardial vessel vasculitis or vasa vasorum impairment, but these conditions can be confirmed only by myocardial biopsy [130]. Only a limited number of HF cases have been reported and no histological diagnosis was made. The suspicion of autoimmune etiology was derived from the exclusion of other possible causes and from the rapid response to immunosuppressive therapy [131, 132].
8 Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic disease characterized by a chronic inflammatory condition affecting primarily the joints. Extra-articular manifestations include frequently and can directly involve several organs such as the lungs, skin, and eyes. Heart involvement is frequently found and substantially increases the risk of mortality, accounting for about a 50% excess compared to the general population [133, 134]. The pathogenesis is mainly related to the pro-inflammatory status of the patients which drive augmented oxidative stress of lipoprotein leading to accelerated atherosclerosis. For this reason, RA should be considered as a major CV risk factor . European League Against Rheumatism (EULAR) guidelines for CV risk recommend an annual assessment [135]; however it is important to note that traditional scores may underestimate the real hazard in RA patients [136].
8.1 Pericardium
Pericarditis is believed to be the most common cardiac manifestation of RA [137], and clinical symptoms are present in less than 5% of patients, while asymptomatic finding is seen in 20–50% by echocardiography. Pericarditis is more frequent in men, especially if autoantibodies are positive and in case of severe or active disease [138]. It can include both exudative and constrictive manifestations. Studies on pericardial fluid have demonstrated the presence of high level of proteins and lactate dehydrogenase but low levels of glucose [139]. Symptomatic patients usually manifest chest pain and dyspnea, but some of these develop nonspecific clinical pictures; therefore, echocardiography is being revealed an essential procedure to clarify undefined clinical situations detecting pericardial effusion. It is important to point out that sometimes pericarditis may develop after biologic initiation; in this case infections and tumors should be excluded, as well as SLE after anti-TNFα treatment [140].
8.2 Valvular Disease
Valvular heart disease is not considered a major extra-articular manifestation of RA, even if echocardiographic studies demonstrate the presence of different valve defects which affect from 24 to 39% of patients. Mitral valve, the most frequent site of pathological involvement, can be aggravated by both stenosis and regurgitation of various grades. This is the result of structure thickening generated by localized fibrosis or nodules [138].
8.3 Atherosclerosis
Atherosclerosis is considered as a collateral but not secondary process related to RA; in fact, chronic systemic inflammation is known to increase the CV burden inducing endothelial dysfunction that leads to accelerated atherogenesis [141, 142]. Several studies prove how inflammatory molecules, such as CRP or cytokines, are common key players in the pathogenesis of both diseases [11]. TNFα can alter the endothelial metabolism, and similarly, IL-6 induce an overall change of surface molecules of the endothelium that develops a pro-inflammatory status; moreover a study demonstrates how IL-6 overproduction reduces lipid concentrations in RA patient and enhances the expression of very low-density lipoprotein receptors in mice [143]. The link between inflammation and atherosclerosis in RA patients is further underlined by the evidence that the association between carotid plaques and inflammatory markers is independent of classical CV risk factors [144]. The major complex of histocompatibility DRB1 is significantly associated with RA susceptibility and also can potentially confer an increased risk of coronary heart disease [145]. This finding adds evidence to the not completely clarified role of adaptive immunity in the pathogenesis in atherosclerosis [145]. Nonetheless, additional factors (i.e., smoke habit, obesity, hypertension, dyslipidemia) alter the fibrinolytic pathway and coagulation status, raising the rate of plaque rupture. For these reasons, EULAR recommends using a modified Systematic Coronary Risk Evaluation (SCORE) to determine the 10-year risk of fatal CVD in RA patients [146].
8.4 Heart Function
Ischemic heart disease is a frequent complication in RA. The risk of acute myocardial infarction is about double compared to the general population, and the presence of unrecognized coronary heart disease is not rare in RA. Sudden death prevalence in RA patients is almost twofold than the general population [10]. HF has been reported in about 4–11% of RA cases; however, some studies demonstrate how prevalence can rise to 24% in selected RA populations which are characterized by high disease activity, glucocorticoid treatments, and positivity of rheumatoid factor [147]. HF derives from a progressively reduced organ reserve that ultimately leads to HF resulting from a detrimental remodeling of myocardial tissue driven by acute or chronic insults. The pathological injury can be the result of ischemic heart events as well as valvular disease, cardiomyopathies, and rhythm abnormalities [138]. Echocardiography and MRI are helpful to recognize potential etiologies and also to establish cardiac function, which, in RA patient, is usually depressed in its diastolic function. Studies on myocardial damage demonstrate how innate immunity cells are critical players in HF remodeling. The production of pro-inflammatory cytokines by injured tissue is the first step in the activation of acute inflammation [138] which aggravates the damage. The amplification of inflammatory pathways determines harmful effects on healthy cell metabolism by increased oxidative stress; moreover, these redundant molecular circuits recall additional immune cells in situ which worsen the process. The strong relation between inflammation and heart disease is nowadays widely proved by several data; some of these demonstrate the active roles played by TNFα and other pro-inflammatory cytokines (i.e., IL-6 and IL-1β) on the progression of obstructive coronary artery disease [138] and left ventricle remodeling [148,149,150]. Interestingly, Giles et al. have observed a robust association among myocardial dysfunction and disease activity and age but also interstitial fibrosis in RA compared to non-RA groups [151]; these findings are strengthened by histological analysis of myocardial samples which manifest higher levels of interstitial citrullination and fibrosis [151].
9 Conclusions
Cardiac involvement is a very common comorbidity of systemic autoimmune diseases which greatly contributes to a general deterioration of patient health status and to a higher mortality rate in comparison to general population. Traditional risk factors do not explain entirely a so significant CV risk, as reported in different case series and national databases; in fact, increasingly evidences link the mediators involved in chronic inflammation and in other immune mechanisms to the harmful action on heart tissue, as previously described. The cornerstone for a comprehensive management of autoimmune diseases lies thus in an aggressive control of disease activity associated with an accurate modification of traditional CV risk factors.
References
Cooper GS, Bynum ML, Somers EC. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun. 2009;33:197–207.
Cavazzana I, Fredi M, Selmi C, Tincani A, Franceschini F. The clinical and histological spectrum of idiopathic inflammatory myopathies. Clin Rev Allergy Immunol. 2015;52(1):88–98.
Meroni PL, Penatti AE. Epigenetics and systemic lupus erythematosus: unmet needs. Clin Rev Allergy Immunol. 2015;50(3):367–76.
Agmon-Levin N, Selmi C. The autoimmune side of heart and lung diseases. Clin Rev Allergy Immunol. 2013;44:1–5.
Mason JC, Libby P. Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J. 2015;36:482–9c.
Miner JJ, Kim AH. Cardiac manifestations of systemic lupus erythematosus. Rheum Dis Clin N Am. 2014;40:51–60.
Muangchan C, Canadian Scleroderma Research Group, Baron M, Pope J. The 15% rule in scleroderma: the frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol. 2013;40:1545–56.
Riboldi P, Gerosa M, Luzzana C, Catelli L. Cardiac involvement in systemic autoimmune diseases. Clin Rev Allergy Immunol. 2002;23:247–61.
Van Gelder H, Charles-Schoeman C. The heart in inflammatory myopathies. Rheum Dis Clin N Am. 2014;40:1–10.
Wright K, Crowson CS, Gabriel SE. Cardiovascular comorbidity in rheumatic diseases: a focus on heart failure. Heart Fail Clin. 2014;10:339–52.
Sattar N, McCarey DW, Capell H, McInnes IB. Explaining how “high-grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003;108:2957–63.
Inoue T, Uchida T, Kamishirado H, Takayanagi K, Hayashi T, Morooka S. Clinical significance of antibody against oxidized low density lipoprotein in patients with atherosclerotic coronary artery disease. J Am Coll Cardiol. 2001;37:775–9.
Nowak B, Szmyrka-Kaczmarek M, Durazinska A, Plaksej R, Borysewicz K, Korman L, Wiland P. Anti-ox-LDL antibodies and anti-ox-LDL-B2GPI antibodies in patients with systemic lupus erythematosus. Adv Clin Exp Med. 2012;21:331–5.
Karvonen J, Paivansalo M, Kesaniemi YA, Horkko S. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation. 2003;108:2107–12.
Bartoloni E, Shoenfeld Y, Gerli R. Inflammatory and autoimmune mechanisms in the induction of atherosclerotic damage in systemic rheumatic diseases: two faces of the same coin. Arthritis Care Res (Hoboken). 2011;63:178–83.
Hollan I, Meroni PL, Ahearn JM, Cohen Tervaert JW, Curran S, Goodyear CS, Hestad KA, Kahaleh B, Riggio M, Shields K, Wasko MC. Cardiovascular disease in autoimmune rheumatic diseases. Autoimmun Rev. 2013;12:1004–15.
Costedoat-Chalumeau N, Hulot JS, Amoura Z, Delcourt A, Maisonobe T, Dorent R, Bonnet N, Sable R, Lechat P, Wechsler B, Piette JC. Cardiomyopathy related to antimalarial therapy with illustrative case report. Cardiology. 2007;107:73–80.
White NJ. Cardiotoxicity of antimalarial drugs. Lancet Infect Dis. 2007;7:549–58.
Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, Vaysbrot E, McNaughton C, Osani M, Shmerling RH, Curtis JR, Furst DE, Parks D, Kavanaugh A, O’dell J, King C, Leong A, Matteson EL, Schousboe JT, Drevlow B, Ginsberg S, Grober J, St Clair EW, Tindall E, Miller AS, McAlindon T. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26.
Diamantopoulos AP, Larsen AI, Omdal R. Is it safe to use TNF-alpha blockers for systemic inflammatory disease in patients with heart failure? Importance of dosage and receptor specificity. Int J Cardiol. 2013;167:1719–23.
Javed Q, Murtaza I. Therapeutic potential of tumour necrosis factor-alpha antagonists in patients with chronic heart failure. Heart Lung Circ. 2013;22:323–7.
Rubbert-Roth A. Assessing the safety of biologic agents in patients with rheumatoid arthritis. Rheumatology (Oxford). 2012;51(Suppl 5):V38–47.
Singh BK, Haque SE, Pillai KK. Assessment of nonsteroidal anti-inflammatory drug-induced cardiotoxicity. Expert Opin Drug Metab Toxicol. 2014;10:143–56.
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
Yoneda S, Koyama M, Matsubara T, Toyama S. Electrocardiographic studies in acute pericarditis with specific reference to ventricular involvement of non-specific pericarditis. Acta Cardiol. 1977;32:337–52.
Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus. 2005;14:683–6.
Turiel M, Peretti R, Sarzi-Puttini P, Atzeni F, Doria A. Cardiac imaging techniques in systemic autoimmune diseases. Lupus. 2005;14:727–31.
Brigden W, Bywaters EG, Lessof MH, Ross IP. The heart in systemic lupus erythematosus. Br Heart J. 1960;22:1–16.
Bulkley BH, Roberts WC. The heart in systemic lupus erythematosus and the changes induced in it by corticosteroid therapy. A study of 36 necropsy patients. Am J Med. 1975;58:243–64.
Griffith GC, Vural IL. Acute and subacute disseminated lupus erythematosus; a correlation of clinical and postmortem findings in eighteen cases. Circulation. 1951;3:492–500.
Gross L. The cardiac lesions in Libman-Sacks disease: with a consideration of its relationship to acute diffuse lupus erythematosus. Am J Pathol. 1940;16:375–408. 11
Harvey AM, Shulman LE, Tumulty PA, Conley CL, Schoenrich EH. Systemic lupus erythematosus: review of the literature and clinical analysis of 138 cases. Medicine (Baltimore). 1954;33:291–437.
Hejtmancik MR, Wright JC, Quint R, Jennings FL. The cardiovascular manifestations of systemic lupus erythematosus. Am Heart J. 1964;68:119–30.
Humphreys EM. The cardiac lesions of acute disseminated lupus erythematosus. Ann Intern Med. 1948;28:12–4.
Jain D, Halushka MK. Cardiac pathology of systemic lupus erythematosus. J Clin Pathol. 2009;62:584–92.
Jessar RA, Lamont-Havers RW, Ragan C. Natural history of lupus erythematosus disseminatus. Ann Intern Med. 1953;38:717–31.
Kong TQ, Kellum RE, Haserick JR. Clinical diagnosis of cardiac involvement in systemic lupus erythematosus. A correlation of clinical and autopsy findings in thirty patients. Circulation. 1962;26:7–11.
Panchal L, Divate S, Vaideeswar P, Pandit SP. Cardiovascular involvement in systemic lupus erythematosus: an autopsy study of 27 patients in India. J Postgrad Med. 2006;52:5–10. Discussion 10
Shearn MA. The heart in systemic lupus erythematosus. Am Heart J. 1959;58:452–66.
Tincani A, Rebaioli CB, Taglietti M, Shoenfeld Y. Heart involvement in systemic lupus erythematosus, anti-phospholipid syndrome and neonatal lupus. Rheumatology (Oxford). 2006;45(Suppl 4):iv8–13.
Ansari A, Larson PH, Bates HD. Cardiovascular manifestations of systemic lupus erythematosus: current perspective. Prog Cardiovasc Dis. 1985;27:421–34.
Bidani AK, Roberts JL, Schwartz MM, Lewis EJ. Immunopathology of cardiac lesions in fatal systemic lupus erythematosus. Am J Med. 1980;69:849–58.
Lin K, Lloyd-Jones DM, Li D, Liu Y, Yang J, Markl M, Carr JC. Imaging of cardiovascular complications in patients with systemic lupus erythematosus. Lupus. 2015;24:1126–34.
Yilmaz A, Ferreira V, Klingel K, Kandolf R, Neubauer S, Sechtem U. Role of cardiovascular magnetic resonance imaging (CMR) in the diagnosis of acute and chronic myocarditis. Heart Fail Rev. 2013;18:747–60.
Wijetunga M, Rockson S. Myocarditis in systemic lupus erythematosus. Am J Med. 2002;113:419–23.
Yogasundaram H, Putko BN, Tien J, Paterson DI, Cujec B, Ringrose J, Oudit GY. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol. 2014;30:1706–15.
Moyssakis I, Tektonidou MG, Vasilliou VA, Samarkos M, Votteas V, Moutsopoulos HM. Libman-Sacks endocarditis in systemic lupus erythematosus: prevalence, associations, and evolution. Am J Med. 2007;120:636–42.
Roldan CA, Tolstrup K, Macias L, Qualls CR, Maynard D, Charlton G, Sibbitt WL Jr. Libman-Sacks endocarditis: detection, characterization, and clinical correlates by three-dimensional transesophageal echocardiography. J Am Soc Echocardiogr. 2015;28:770–9.
Brucato A, Cimaz R, Caporali R, Ramoni V, Buyon J. Pregnancy outcomes in patients with autoimmune diseases and anti-Ro/SSA antibodies. Clin Rev Allergy Immunol. 2011;40:27–41.
Brito-Zeron P, Izmirly PM, Ramos-Casals M, Buyon JP, Khamashta MA. The clinical spectrum of autoimmune congenital heart block. Nat Rev Rheumatol. 2015;11:301–12.
Landtman B, Linder E, Hjelt L, Tuuteri L. Congenital complete heart block. I. A clinical study of 27 cases. Ann Paediatr Fenn. 1964;10:99–104.
Costedoat-Chalumeau N, Amoura Z, Lupoglazoff JM, Huong DL, Denjoy I, Vauthier D, Sebbouh D, Fain O, Georgin-Lavialle S, Ghillani P, Musset L, Wechsler B, Duhaut P, Piette JC. Outcome of pregnancies in patients with anti-SSA/Ro antibodies: a study of 165 pregnancies, with special focus on electrocardiographic variations in the children and comparison with a control group. Arthritis Rheum. 2004;50:3187–94.
Izmirly PM, Costedoat-Chalumeau N, Pisoni CN, Khamashta MA, Kim MY, Saxena A, Friedman D, Llanos C, Piette JC, Buyon JP. Maternal use of hydroxychloroquine is associated with a reduced risk of recurrent anti-SSA/Ro-antibody-associated cardiac manifestations of neonatal lupus. Circulation. 2012;126:76–82.
Buyon JP, Clancy RM. Neonatal lupus: basic research and clinical perspectives. Rheum Dis Clin N Am. 2005;31:299–313. vii
Garcia S, Nascimento JH, Bonfa E, Levy R, Oliveira SF, Tavares AV, De Carvalho AC. Cellular mechanism of the conduction abnormalities induced by serum from anti-Ro/SSA-positive patients in rabbit hearts. J Clin Invest. 1994;93:718–24.
Xiao GQ, Hu K, Boutjdir M. Direct inhibition of expressed cardiac L- and T-type calcium channels by igg from mothers whose children have congenital heart block. Circulation. 2001;103:1599–604.
Tincani A, Biasini-Rebaioli C, Cattaneo R, Riboldi P. Nonorgan specific autoantibodies and heart damage. Lupus. 2005;14:656–9.
Brito-Zeron P, Izmirly PM, Ramos-Casals M, Buyon JP, Khamashta MA. Autoimmune congenital heart block: complex and unusual situations. Lupus. 2016;25:116–28.
Tonello M, Ruffatti A, Marson P, Tison T, Marozio L, Hoxha A, De Silvestro G, Punzi L. Plasma exchange effectively removes 52- and 60-kDa anti-Ro/SSA and anti-La/SSB antibodies in pregnant women with congenital heart block. Transfusion. 2015;55:1782–6.
Elhai M, Avouac J, Walker UA, Matucci-Cerinic M, Riemekasten G, Airo P, Hachulla E, Valentini G, Carreira PE, Cozzi F, Balbir Gurman A, Braun-Moscovici Y, Damjanov N, Ananieva LP, Scorza R, Jimenez S, Busquets J, Li M, Muller-Ladner U, Kahan A, Distler O, Allanore Y. A gender gap in primary and secondary heart dysfunctions in systemic sclerosis: a EUSTAR prospective study. Ann Rheum Dis. 2016;75:163–9.
Ranque B, Mouthon L. Geoepidemiology of systemic sclerosis. Autoimmun Rev. 2010;9:A311–8.
Van Den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Matucci-Cerinic M, Naden RP, Medsger TA Jr, Carreira PE, Riemekasten G, Clements PJ, Denton CP, Distler O, Allanore Y, Furst DE, Gabrielli A, Mayes MD, Van Laar JM, Seibold JR, Czirjak L, Steen VD, Inanc M, Kowal-Bielecka O, Muller-Ladner U, Valentini G, Veale DJ, Vonk MC, Walker UA, Chung L, Collier DH, Csuka ME, Fessler BJ, Guiducci S, Herrick A, Hsu VM, Jimenez S, Kahaleh B, Merkel PA, Sierakowski S, Silver RM, Simms RW, Varga J, Pope JE. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737–47.
Mouthon L, Bussone G, Berezne A, Noel LH, Guillevin L. Scleroderma renal crisis. J Rheumatol. 2014;41:1040–8.
Elhai M, Meune C, Avouac J, Kahan A, Allanore Y. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2012;51:1017–26.
Ferri C, Sebastiani M, Lo Monaco A, Iudici M, Giuggioli D, Furini F, Manfredi A, Cuomo G, Spinella A, Colaci M, Govoni M, Valentini G. Systemic sclerosis evolution of disease pathomorphosis and survival. Our experience on Italian patients’ population and review of the literature. Autoimmun Rev. 2014;13:1026–34.
Vacca A, Meune C, Gordon J, Chung L, Proudman S, Assassi S, Nikpour M, Rodriguez-Reyna TS, Khanna D, Lafyatis R, Matucci-Cerinic M, Distler O, Allanore Y, Scleroderma Clinical Trial Consortium Cardiac Subcommittee. Cardiac arrhythmias and conduction defects in systemic sclerosis. Rheumatology (Oxford). 2014;53:1172–7.
Fernandez-Codina A, Simeon-Aznar CP, Pinal-Fernandez I, Rodriguez-Palomares J, Pizzi MN, Hidalgo CE, Del Castillo AG, Prado-Galbarro FJ, Sarria-Santamera A, Fonollosa-Pla V, Vilardell-Tarres M. Cardiac involvement in systemic sclerosis: differences between clinical subsets and influence on survival. Rheumatol Int. 2015;37(1):75–84.
Harvey GR, Butts S, Rands AL, Patel Y, McHugh NJ. Clinical and serological associations with anti-RNA polymerase antibodies in systemic sclerosis. Clin Exp Immunol. 1999;117:395–402.
Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol. 2010;28:S48–53.
Kahan A, Allanore Y. Primary myocardial involvement in systemic sclerosis. Rheumatology (Oxford). 2006;45(Suppl 4):Iv14–7.
Cutolo M, Sulli A, Secchi ME, Paolino S, Pizzorni C. Nailfold capillaroscopy is useful for the diagnosis and follow-up of autoimmune rheumatic diseases. A future tool for the analysis of microvascular heart involvement? Rheumatology (Oxford). 2006;45(Suppl 4):Iv43–6.
Desai CS, Lee DC, Shah SJ. Systemic sclerosis and the heart: current diagnosis and management. Curr Opin Rheumatol. 2011;23:545–54.
Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol. 2013;31:314–9.
Hachulla AL, Launay D, Gaxotte V, De Groote P, Lamblin N, Devos P, Hatron PY, Beregi JP, Hachulla E. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis. 2009;68:1878–84.
Allanore Y, Meune C. N-terminal pro brain natriuretic peptide: the new cornerstone of cardiovascular assessment in systemic sclerosis. Clin Exp Rheumatol. 2009;27:59–63.
Lambova S. Cardiac manifestations in systemic sclerosis. World J Cardiol. 2014;6:993–1005.
Clements PJ, Furst DE, Cabeen W, Tashkin D, Paulus HE, Roberts N. The relationship arrhythmias and conduction disturbances to other manifestations of cardiopulmonary disease in progressive systemic sclerosis (PSS). Am J Med. 1981;71:38–46.
Follansbee WP, Curtiss EI, Rahko PS, Medsger TA Jr, Lavine SJ, Owens GR, Steen VD. The electrocardiogram in systemic sclerosis (Scleroderma). Study of 102 consecutive cases with functional correlations and review of the literature. Am J Med. 1985;79:183–92.
Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–82.
Danieli MG, Gambini S, Pettinari L, Logullo F, Veronesi G, Gabrielli A. Impact of treatment on survival in polymyositis and dermatomyositis. A single-centre long-term follow-up study. Autoimmun Rev. 2014;13:1048–54.
Zhang L, Wang GC, Ma L, Zu N. Cardiac involvement in adult polymyositis or dermatomyositis: a systematic review. Clin Cardiol. 2012;35:686–91.
Diederichsen LP, Simonsen JA, Diederichsen AC, Kim WY, Hvidsten S, Hougaard M, Junker P, Lundberg IE, Petersen H, Hansen ES, Eskerud KS, Kay SD, Jacobsen S. Cardiac abnormalities assessed by non-invasive techniques in patients with newly diagnosed idiopathic inflammatory myopathies. Clin Exp Rheumatol. 2015;33:706–14.
Gupta R, Wayangankar SA, Targoff IN, Hennebry TA. Clinical cardiac involvement in idiopathic inflammatory myopathies: a systematic review. Int J Cardiol. 2011;148:261–70.
Mavrogeni S, Douskou M, Manoussakis MN. Contrast-enhanced CMR imaging reveals myocardial involvement in idiopathic inflammatory myopathy without cardiac manifestations. JACC Cardiovasc Imaging. 2011;4:1324–5.
Mavrogeni S, Sfikakis PP, Dimitroulas T, Kolovou G, Kitas GD. Cardiac and muscular involvement in idiopathic inflammatory myopathies: noninvasive diagnostic assessment and the role of cardiovascular and skeletal magnetic resonance imaging. Inflamm Allergy Drug Targets. 2014;13:206–16.
Rosenbohm A, Buckert D, Gerischer N, Walcher T, Kassubek J, Rottbauer W, Ludolph AC, Bernhardt P. Early diagnosis of cardiac involvement in idiopathic inflammatory myopathy by cardiac magnetic resonance tomography. J Neurol. 2015;262:949–56.
Toong C, Puranik R, Adelstein S. Use of cardiac MR imaging to evaluate the presence of myocarditis in autoimmune myositis: three cases. Rheumatol Int. 2012;32:779–82.
Haupt HM, Hutchins GM. The heart and cardiac conduction system in polymyositis-dermatomyositis: a clinicopathologic study of 16 autopsied patients. Am J Cardiol. 1982;50:998–1006.
Matsumoto K, Tanaka H, Yamana S, Kaneko A, Tsuji T, Ryo K, Sekiguchi K, Kawakami F, Kawai H, Hirata K. Successful steroid therapy for heart failure due to myocarditis associated with primary biliary cirrhosis. Can J Cardiol. 2012;28:515.e3–6.
Gonzalez-Lopez L, Gamez-Nava JI, Sanchez L, Rosas E, Suarez-Almazor M, Cardona-Munoz C, Ramos-Remus C. Cardiac manifestations in dermato-polymyositis. Clin Exp Rheumatol. 1996;14:373–9.
Bazzani C, Cavazzana I, Ceribelli A, Vizzardi E, Dei Cas L, Franceschini F. Cardiological features in idiopathic inflammatory myopathies. J Cardiovasc Med (Hagerstown). 2010;11:906–11.
Lu Z, Wei Q, Ning Z, Qian-Zi Z, Xiao-Ming S, Guo-Chun W. Left ventricular diastolic dysfunction—early cardiac impairment in patients with polymyositis/dermatomyositis: a tissue Doppler imaging study. J Rheumatol. 2013;40:1572–7.
Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an Extractable Nuclear Antigen (ENA). Am J Med. 1972;52:148–59.
Ungprasert P, Wannarong T, Panichsillapakit T, Cheungpasitporn W, Thongprayoon C, Ahmed S, Raddatz DA. Cardiac involvement in mixed connective tissue disease: a systematic review. Int J Cardiol. 2014;171:326–30.
Hajas A, Szodoray P, Nakken B, Gaal J, Zold E, Laczik R, Demeter N, Nagy G, Szekanecz Z, Zeher M, Szegedi G, Bodolay E. Clinical course, prognosis, and causes of death in mixed connective tissue disease. J Rheumatol. 2013;40:1134–42.
Kim P, Grossman JM. Treatment of mixed connective tissue disease. Rheum Dis Clin N Am. 2005;31:549–65. Viii
Arroyo-Avila M, Vila LM. Cardiac tamponade in a patient with mixed connective tissue disease. J Clin Rheumatol. 2015;21:42–5.
Hammann C, Genton CY, Delabays A, Bischoff Delaloye A, Bogousslavsky J, Spertini F. Myocarditis of mixed connective tissue disease: favourable outcome after intravenous pulsed cyclophosphamide. Clin Rheumatol. 1999;18:85–7.
Lash AD, Wittman AL, Quismorio FP Jr. Myocarditis in mixed connective tissue disease: clinical and pathologic study of three cases and review of the literature. Semin Arthritis Rheum. 1986;15:288–96.
Comens SM, Alpert MA, Sharp GC, Pressly TA, Kelly DL, Hazelwood SE, Mukerji V. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol. 1989;63:369–70.
Leung WH, Wong KL, Lau CP, Wong CK, Cheng CH, Tai YT. Echocardiographic identification of mitral valvular abnormalities in patients with mixed connective tissue disease. J Rheumatol. 1990;17:485–8.
Rebollar-Gonzalez V, Torre-Delgadillo A, Orea-Tejeda A, Ochoa-Perez V, Navarrete-Gaona R, Asensio-Lafuente E, Dorantes-Garcia J, Narvaez R, Rangel-Pena AM, Hernandez-Reyes P, Oseguera-Moguel J. Cardiac conduction disturbances in mixed connective tissue disease. Rev Investig Clin. 2001;53:330–4.
Alpert MA, Goldberg SH, Singsen BH, Durham JB, Sharp GC, Ahmad M, Madigan NP, Hurst DP, Sullivan WD. Cardiovascular manifestations of mixed connective tissue disease in adults. Circulation. 1983;68:1182–93.
Oetgen WJ, Mutter ML, Lawless OJ, Davia JE. Cardiac abnormalities in mixed connective tissue disease. Chest. 1983;83:185–8.
Leung WH, Wong KL, Lau CP, Wong CK, Cheng CH, Tai YT. Doppler-echo evaluation of left ventricular diastolic filling in patient with mixed connective tissue disease. Cardiology. 1990;77:93–100.
Vegh J, Hegedus I, Szegedi G, Zeher M, Bodolay E. Diastolic function of the heart in mixed connective tissue disease. Clin Rheumatol. 2007;26:176–81.
Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, Pg DEG, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA. International consensus statement on an update of the classification criteria for definite Antiphospholipid Syndrome (APS). J Thromb Haemost. 2006;4:295–306.
Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, Jacobsen S, Lakos G, Tincani A, Kontopoulou-Griva I, Galeazzi M, Meroni PL, Derksen RH, De Groot PG, Gromnica-Ihle E, Baleva M, Mosca M, Bombardieri S, Houssiau F, Gris JC, Quere I, Hachulla E, Vasconcelos C, Roch B, Fernandez-Nebro A, Boffa MC, Hughes GR, Ingelmo M, Euro-Phospholipid Project G. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46:1019–27.
Long BR, Leya F. The role of antiphospholipid syndrome in cardiovascular disease. Hematol Oncol Clin North Am. 2008;22:79–94. vi–vii
Roldan CA. Valvular and coronary heart disease in systemic inflammatory diseases: systemic disorders in heart disease. Heart. 2008;94:1089–101.
Hojnik M, George J, Ziporen L, Shoenfeld Y. Heart valve involvement (Libman-Sacks endocarditis) in the antiphospholipid syndrome. Circulation. 1996;93:1579–87.
Vianna JL, Khamashta MA, Ordi-Ros J, Font J, Cervera R, Lopez-Soto A, Tolosa C, Franz J, Selva A, Ingelmo M, et al. Comparison of the primary and secondary antiphospholipid syndrome: a European Multicenter Study of 114 patients. Am J Med. 1994;96:3–9.
Nesher G, Ilany J, Rosenmann D, Abraham AS. Valvular dysfunction in antiphospholipid syndrome: prevalence, clinical features, and treatment. Semin Arthritis Rheum. 1997;27:27–35.
Espinola-Zavaleta N, Vargas-Barron J, Colmenares-Galvis T, Cruz-Cruz F, Romero-Cardenas A, Keirns C, Amigo MC. Echocardiographic evaluation of patients with primary antiphospholipid syndrome. Am Heart J. 1999;137:973–8.
Zavaleta NE, Montes RM, Soto ME, Vanzzini NA, Amigo MC. Primary antiphospholipid syndrome: a 5-year transesophageal echocardiographic followup study. J Rheumatol. 2004;31:2402–7.
Erdogan D, Goren MT, Diz-Kucukkaya R, Inanc M. Assessment of cardiac structure and left atrial appendage functions in primary antiphospholipid syndrome: a transesophageal echocardiographic study. Stroke. 2005;36:592–6.
Krause I, Lev S, Fraser A, Blank M, Lorber M, Stojanovich L, Rovensky J, Chapman J, Shoenfeld Y. Close association between valvular heart disease and central nervous system manifestations in the antiphospholipid syndrome. Ann Rheum Dis. 2005;64:1490–3.
Garcia-Torres R, Amigo MC, De La Rosa A, Moron A, Reyes PA. Valvular heart disease in primary antiphospholipid syndrome (PAPS): clinical and morphological findings. Lupus. 1996;5:56–61.
Ziporen L, Goldberg I, Arad M, Hojnik M, Ordi-Ros J, Afek A, Blank M, Sandbank Y, Vilardell-Tarres M, De Torres I, Weinberger A, Asherson RA, Kopolovic Y, Shoenfeld Y. Libman-Sacks endocarditis in the antiphospholipid syndrome: immunopathologic findings in deformed heart valves. Lupus. 1996;5:196–205.
Cervera R, Boffa MC, Khamashta MA, Hughes GR. The Euro-phospholipid project: epidemiology of the antiphospholipid syndrome in Europe. Lupus. 2009;18:889–93.
Artenjak A, Lakota K, Frank M, Cucnik S, Rozman B, Bozic B, Shoenfeld Y, Sodin-Semrl S. Antiphospholipid antibodies as non-traditional risk factors in atherosclerosis based cardiovascular diseases without overt autoimmunity. A critical updated review. Autoimmun Rev. 2012;11:873–82.
Denas G, Jose SP, Bracco A, Zoppellaro G, Pengo V. Antiphospholipid syndrome and the heart: a case series and literature review. Autoimmun Rev. 2015;14:214–22.
George J, Shoenfeld Y, Harats D. The involvement of beta2-glycoprotein I (beta2-GPI) in human and murine atherosclerosis. J Autoimmun. 1999;13:57–60.
Delgado Alves J, Ames PR, Donohue S, Stanyer L, Nourooz-Zadeh J, Ravirajan C, Isenberg DA. Antibodies to high-density lipoprotein and beta2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum. 2002;46:2686–94.
Matsuura E, Kobayashi K, Koike T, Shoenfeld Y. Autoantibody-mediated atherosclerosis. Autoimmun Rev. 2002;1:348–53.
Daly CA, De Stavola B, Sendon JL, Tavazzi L, Boersma E, Clemens F, Danchin N, Delahaye F, Gitt A, Julian D, Mulcahy D, Ruzyllo W, Thygesen K, Verheugt F, Fox KM. Predicting prognosis in stable angina—results from the Euro heart survey of stable angina: prospective observational study. BMJ. 2006;332:262–7.
Veres K, Lakos G, Kerenyi A, Szekanecz Z, Szegedi G, Shoenfeld Y, Soltesz P. Antiphospholipid antibodies in acute coronary syndrome. Lupus. 2004;13:423–7.
Mavrogeni SI, Sfikakis PP, Kitas GD, Kolovou G, Tektonidou MG. Cardiac involvement in antiphospholipid syndrome: the diagnostic role of noninvasive cardiac imaging. Semin Arthritis Rheum. 2015;45(5):611–6.
Rantapaa-Dahlqvist S, Backman C, Sandgren H, Ostberg Y. Echocardiographic findings in patients with primary Sjogren’s syndrome. Clin Rheumatol. 1993;12:214–8.
Gyongyosi M, Pokorny G, Jambrik Z, Kovacs L, Kovacs A, Makula E, Csanady M. Cardiac manifestations in primary Sjogren’s syndrome. Ann Rheum Dis. 1996;55:450–4.
Golan TD, Keren D, Elias N, Naschitz JE, Toubi E, Misselevich I, Yeshurun D. Severe reversible cardiomyopathy associated with systemic vasculitis in primary Sjogren’s syndrome. Lupus. 1997;6:505–8.
Levin MD, Zoet-Nugteren SK, Markusse HM. Myocarditis and primary Sjogren’s syndrome. Lancet. 1999;354:128–9.
Del Rincon ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737–45.
Nurmohamed MT, Heslinga M, Kitas GD. Cardiovascular comorbidity in rheumatic diseases. Nat Rev Rheumatol. 2015;11:693–704.
Ikdahl E, Rollefstad S, Olsen IC, Kvien TK, Hansen IJ, Soldal DM, Haugeberg G, Semb AG. Eular task force recommendations on annual cardiovascular risk assessment for patients with rheumatoid arthritis: an audit of the success of implementation in a rheumatology outpatient clinic. Biomed Res Int. 2015;2015:515280.
Arts EE, Popa C, Den Broeder AA, Semb AG, Toms T, Kitas GD, Van Riel PL, Fransen J. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann Rheum Dis. 2015;74:668–74.
Voskuyl AE. The heart and cardiovascular manifestations in rheumatoid arthritis. Rheumatology (Oxford). 2006;45(Suppl 4):iv4–7.
Sen D, Gonzalez-Mayda M, Brasington RD Jr. Cardiovascular disease in rheumatoid arthritis. Rheum Dis Clin N Am. 2014;40:27–49.
Barcin C, Yalcinkaya E, Kabul HK. Cholesterol pericarditis associated with rheumatoid arthritis: a rare cause of pericardial effusion. Int J Cardiol. 2013;166:E56–8.
Ambrose NL, O’connell PG. Anti-TNF alpha therapy does not always protect rheumatoid arthritis patients against developing pericarditis. Clin Exp Rheumatol. 2007;25:660.
Choy E, Ganeshalingam K, Semb AG, Szekanecz Z, Nurmohamed M. Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology (Oxford). 2014;53:2143–54.
Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52:402–11.
Hashizume M, Yoshida H, Koike N, Suzuki M, Mihara M. Overproduced interleukin 6 decreases blood lipid levels via upregulation of very-low-density lipoprotein receptor. Ann Rheum Dis. 2010;69:741–6.
Park YB, Ahn CW, Choi HK, Lee SH, In BH, Lee HC, Nam CM, Lee SK. Atherosclerosis in rheumatoid arthritis: morphologic evidence obtained by carotid ultrasound. Arthritis Rheum. 2002;46:1714–9.
Paakkanen R, Lokki ML, Seppanen M, Tierala I, Nieminen MS, Sinisalo J. Proinflammatory HLA-DRB1*01-haplotype predisposes to ST-elevation myocardial infarction. Atherosclerosis. 2012;221:461–6.
Peters MJ, Symmons DP, McCarey D, Dijkmans BA, Nicola P, Kvien TK, McInnes IB, Haentzschel H, Gonzalez-Gay MA, Provan S, Semb A, Sidiropoulos P, Kitas G, Smulders YM, Soubrier M, Szekanecz Z, Sattar N, Nurmohamed MT. Eular evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325–31.
Myasoedova E, Crowson CS, Nicola PJ, Maradit-Kremers H, Davis JM 3rd, Roger VL, Therneau TM, Gabriel SE. The influence of rheumatoid arthritis disease characteristics on heart failure. J Rheumatol. 2011;38:1601–6.
Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103:2055–9.
Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41.
Myasoedova E, Davis JM 3rd, Crowson CS, Roger VL, Karon BL, Borgeson DD, Therneau TM, Matteson EL, Rodeheffer RJ, Gabriel SE. Brief report: rheumatoid arthritis is associated with left ventricular concentric remodeling: results of a population-based cross-sectional study. Arthritis Rheum. 2013;65:1713–8.
Giles JT, Fert-Bober J, Park JK, Bingham CO 3rd, Andrade F, Fox-Talbot K, Pappas D, Rosen A, Van Eyk J, Bathon JM, Halushka MK. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther. 2012;14:R39.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Generali, E., Folci, M., Selmi, C., Riboldi, P. (2017). Immune-Mediated Heart Disease. In: Sattler, S., Kennedy-Lydon, T. (eds) The Immunology of Cardiovascular Homeostasis and Pathology. Advances in Experimental Medicine and Biology, vol 1003. Springer, Cham. https://doi.org/10.1007/978-3-319-57613-8_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-57613-8_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57611-4
Online ISBN: 978-3-319-57613-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)