
Abstract
The gut possesses a diverse microbial community comprising bacteria, eukaryotes, archaea, and viruses, all of which make up the gut microbiome. The interactions between these various organisms are complex and difficult to model; however, they greatly influence our human health in a variety of ways. Commensals form the majority of this community and have a great impact on our immunity and resistance to disease. Consequently, the genetic pool or “metagenome” of the gut microbiome is a valuable resource into studies on human health. Metagenomic studies have revealed the presence of several genes contributing to drug resistance in the microbiome. These may have arisen either as a by-product of an essential survival pathway for the microbe or through spontaneous mutations. Another possible mode of entry is through pathogens carrying drug-resistant genes that may be introduced into the gut environment in a variety of ways, food being a significant point of entry. Consequently, all of the above factors contribute to an increasing number of drug-resistant genes in the gut microbiome. To add to this phenomenon, transmission of these genes through members of the microbiome may occur by horizontal gene transfer mechanisms adding to the diversity of organisms exhibiting resistance. Moreover, the administration of antibiotics for routine treatments has been found to further exacerbate this by deleting the beneficial commensal pool. Thus, it is of utmost importance to investigate and impede the emergence of resistance in the gut microbiome to benefit long-term human health.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Resistance Gene
- Antibiotic Resistance
- Horizontal Gene Transfer
- Clostridium Difficile Infection
- Terminal Restriction Fragment Length Polymorphism
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The human gut possesses a vast microbial community consisting of several trillion cells far outnumbering the number of human cells by a considerable margin. This community of microbes resident in the gut is termed the gut microbiome and includes bacteria, archaea, eukarya, and viruses. The human microbiome refers to the entire set of genetic elements of all the organisms comprising the microbiota. The term metagenomics refers to a study of the functions and interactions of all of these organisms inferred from their genomic data. The gut microbiome plays several essential functions, namely, the development of both adaptive and innate immunity, maintaining the integrity of the intestinal lining, energy production, synthesis of several vitamins, and factors and protection from colonization of invasive pathogens. To this effect, the Human Microbiome Project was constituted by the National Institute of Health to gain a deeper understanding of this diverse, complex microbial community with the following points of focus:
-
1.
Complete characterization of the resident communities.
-
2.
Is there a core community shared by all?
-
3.
To study the effects of changes to this community on human health (The NIHHMPWG et al. 2009).
The use of antibiotics affects not only a specific target but also a broader population, some of which are beneficial to the host. Such is the effect exerted by antibiotics on the gut microbiome. The use of antibiotics or consumption of plant, animal, and dairy products containing antibiotics exerts an impact on these microbiota resulting in their destabilization or emergence of resistance. Destabilization has consequences on the host resulting in diarrhea and other opportunistic infections, while emergence of resistance leads to further complication for much longer periods of time. Thus, a study of the gut microbiome and the effects of drugs on its constituent communities are essential in determining the benefits and consequences to human health.
2 The Normal Gut Microbiota
The terms “normal” and “healthy” in context of the human gut microbiome are somewhat difficult to define primarily due to the significant interindividual differences that are prevalent. Thus, defining a “core microbiota” poses a greater challenge as opposed to defining “core functions” as a more unifying term. Several factors have been attributed to differences in the human gut microbiome, namely, age, gender, ethnic background, and environmental factors (diet, medication, stress, smoking, and infections) (Mueller et al. 2006).
A large proportion of gut bacteria (≈80 %) cannot be cultured in vitro by standard microbiological techniques, primarily due to their stringent nutrient requirements, anaerobic nature of their niche, and interdependence on one other (Dethlefsen et al. 2007). Consequently, several molecular techniques such as 16S rRNA sequencing, terminal restriction fragment length polymorphism (TRFLP), denaturing gradient gel electrophoresis (DGGE), and fluorescent in situ hybridization have proved useful in this regard for the identification and characterization of constituent members of the human microbiome. Studies have revealed large, diverse microbial communities to reside in the oral cavity and distal gastrointestinal (GI) tract with simpler groups residing in the esophagus, stomach, and small intestine. There have been very few studies on the latter group, and thus identifying them as resident bacteria or just transient travelers presents a significant challenge. Predominant members of the oral cavity include Firmicutes, Bacteroidetes, Proteobacteria, Fusobacteria, and Actinobacteria constituting about 99 % of the total members present. The remainder is composed of Cyanobacteria, Spirochaetes, TM7, and several others. The distal esophagus harbors a microbial community that is similar to the oral cavity with exception that most of the members could be cultured microbiologically (Ahn et al. 2011). Earlier, it was believed that the stomach with its extremely low pH possessed a transient microbial community with a simpler communal structure; however, gastric biopsies of 23 human patients presented a 16S rRNA library comprising diverse microbes, including members of Actinobacteria, Proteobacteria, Fusobacteria, and more (Bik et al. 2006). This community was found to differ considerably from communities of the mouth and esophagus. The microbial constitution of the small intestine was found to be rich in facultative anaerobic species; however, there are relatively few studies, and thus much more work needs to be done in this area. The colon on the other hand presented the greatest concentration of microbes with approximately 1011 to 1012 cells per gram of feces and a twofold greater number of microbial genes as compared to the human genome itself (Ley et al. 2006). The dominant phyla in the colon include Firmicutes and Bacteroides with seven other groups (Fusobacteria, Actinobacteria, Verrucomicrobia, Proteobacteria, Spirochaetes, Cyanobacteria, and VadinBE97) also serving as residents (Backhed et al. 2005).
The Metagenome of the Human Intestinal Tract consortium studied the metagenomes of 124 fecal specimens utilizing the Illumina Genome Analyzer. Specimens were isolated from healthy, overweight, and obese individuals and those suffering from inflammatory bowel disease (IBD) residing in Spain or Denmark (Arumugam et al. 2011). Data from the study revealed the human gut microbiota to comprise of about 1150 different species (Arumugam et al. 2011). Additionally, they identified 536,112 unique genes of which 99 % were found to be bacterial. At least 40 % of the bacterial genes from each specimen were found to be present within about half of the remaining specimens. These conserved genes were found to cluster to pathways involved in digestion and degradation of complex sugars, short-chain fatty acid production, and vitamin biosynthesis. The study also revealed the presence of a set of core functions that the microbiome performs, rather than a core group of organisms.
The establishment of the gut microbiome occurs at birth and continues to evolve and develop throughout the lifetime of the host. Studies have found variations in the gut microbiome of infants depending on the mode of delivery. 16S rRNA pyrosequencing revealed that infants delivered naturally possessed bacterial communities that resembled the vaginal microbiota of their mothers (Lactobacillus, Prevotella, etc.), while those delivered by Caesarian section possessed communities resembling those on the surface of the skin (Staphylococcus, Propionibacterium, etc.). These initial species termed as “founder species” undergo evolution within each host under both extrinsic and intrinsic factors that may be genetic, physiological, and environmental and ultimately constitute the gut microbiome.
Gut microbiota dysbiosis could lead to several pathological conditions. Diarrheal infections pose a significant challenge worldwide with a large proportion of them being food-borne. The most common bacterial infections are caused by Salmonella, Shigella, Campylobacter, Vibrio cholerae, etc., while viral pathogens include Norovirus, Rotavirus, etc. and Giardia, Cryptosporidia, etc. constitute the parasites. Antibiotic-associated diarrheal infections such as those caused by Clostridium difficile are due to a dysbiosis induced by the use of antibiotics. This leads to a loss of colonization resistance normally provided by the microbiota that subsequently allows overgrowth of Clostridium difficile and production of toxins A and B that bring about colonic damage, diarrhea, and, in severe cases, even death. Short bowel syndrome is a disorder that causes malabsorption due to either dysfunction of a large portion of the intestine or surgical removal of part of the intestine, or in some cases, it may be congenital. In such patients, the colon serves as an extremely important organ for energy salvage; however, in some cases, excess bacteria from the colon may lead to overgrowth, causing small intestinal bacterial overgrowth (SIBO). Studies have revealed a link between interaction of the host and microbiota in inflammatory bowel syndrome (IBS) pathology. Thus, the focus is now underway as to what antigens are present in the normal microbiota that may drive chronic inflammation in predisposed individuals.
3 Factors Affecting Drug Resistance
The spread of bacteria and genes responsible for the emergence of antibiotic resistance in the microbiota of the gut is dependent on a variety of factors, the most important of which is antibiotic use. Additional factors that play a role in the emergence and persistence of drug-resistant strains include their rate of adaptability, frequency of mutations that confer resistant traits, and ability to undergo various mechanisms of horizontal gene transfer (HGT) such as transformation, transduction, and conjugation. When exposed to a particular antibiotic, the acquisition of resistance by the bacterium endows it with an advantage in that particular environment. On the flipside, if the particular selective pressure, in this case an antibiotic, is removed, the resistant bacterium may not survive as well as its susceptible other.
A number of studies investigating the effects of antibiotic use on gut flora have shown resistant genes to be detected in members of the microbiota several years after treatment. Thus, bacteria are able to persist and transfer resistance traits for a considerable amount of time even after antibiotic use has been discontinued. For example, a study by Sjolund et al. revealed the ermB gene responsible for conferring clarithromycin resistance to be present in enterococci up to 1 year posttreatment (Sjolund et al. 2003). Interestingly, one particular patient was detected to have a resistant clone 3 years posttreatment. Such results reveal crucial clinical consequences since there remains an extended window during which resistance genes may be transferred to other species as well.
The intestine presents a highly suitable environment for the transfer of resistant genes. It provides a warm moisture-laden environment having an abundance of nutrients and a diverse bacterial population that could serve as a source and targets for acquiring resistance. Once particular resistance genes have been selected in the commensals, they may then be transferred to more pathogenic bacteria, thus posing a severe risk to health. The selection pressure provides a double blow with an increase in the number of bacteria possessing the resistant gene that could in turn further transfer it through the microbiota. Several bacteria are not residents rather passing through the intestine along with food. These may well serve as additional sources of resistant genes that could be transferred to commensal bacteria. Another potential source of selective pressure could be from agriculture. The use of certain compounds may impose selective pressure leading to the emergence of resistant genes in the microbiota when foods containing them are consumed.
4 External and Environmental Factors
4.1 Antibiotics
There is a delicate ecological balance that exists between the gut-associated microorganisms and their human host. This balance is however quite easily disturbed by a number of factors, the most important of which is antibiotic use. Major consequences of this are a decreased ability of commensal bacteria to resist colonization of an invasive species and the emergence of drug-resistant strains. While there are several studies investigating the short-term effects of the use of such antibiotics, their long-term effects are still relatively less studied. The effects induced on the microbiota by an antibiotic depend on a number of factors such as the dosage, administration route, time course, spectrum (broad or narrow), and mode of action. Antibiotic use that results in discharge into the gut may gravely affect the resident microbiota and consequently have an adverse impact on health by creating unforeseen complication such as Clostridium difficile infections (Sullivan et al. 2001).
Intestinal microbiota are dominated by anaerobic and facultative anaerobic species which play a major role in the production of volatile fatty acids, however many of which serve as specific targets for antibiotics resulting in destabilizing of the microbiota. As an example, the broad-spectrum antibiotic, clindamycin, has been found in high levels in the bile and fecal matter of treated individuals. However, it has also been associated in decreasing the colonization resistance of the resident microbiota resulting in C. difficile infections (Sullivan et al. 2001). This organism is normally present in relatively few numbers in healthy individuals; however, its numbers rapidly increase as a result of antibiotic-induced microbiota disturbances. Other side effects of this antibiotic include diarrhea, gastritis, intestinal pain, and swelling. Another drug of interest is amoxicillin, the use of which has been associated with an increase of antibiotic-resistant bacteria as well as a reduction in the number of gram-positive cocci. Several metagenomic techniques including DGGE have revealed a change in the microbiota constitution with the use of this antibiotic (Brugere et al. 2009). The Helicobacter pylori infection treatment protocol consists of the use of a combination of three drugs, namely, omeprazole, metronidazole, and clarithromycin. Dramatic disturbances in the gut microflora have been reported as a result of this treatment regimen with effects lasting for several years after treatment.
4.2 Food Animals
For a long time, the practice of administering antibiotics to animals reared for food, as a means to promote growth or for treatment, has been the norm. However, several studies have highlighted this as a potential source for the transfer of pathogens harboring antibiotic-resistant genes to humans. For example, a study by Levy et al. demonstrated that the use of the antibiotic oxytetracycline to enhance growth in chickens favored the selection of tetracycline-resistant Escherichia coli in them, which were subsequently detected in people that consumed them (Levy et al. 1976a, b). Another more recent example relates to the use of avoparcin in animals and its link to the emergence of vancomycin-resistant enterococci (VRE) which are highly pathogenic to humans. Thus, animals used for food, as well as pets and wild animals, can serve as sources of antibiotic-resistant genes. Additionally, several metals and compounds used in the veterinary industry have also been found to induce cross-resistance to various antibiotics. Not limited to animals but animal products such as milk and other dairy items were found to contain lactobacilli and other bacteria that possessed antibiotic-resistant genes.
4.3 Aquaculture
The fairly recent field of aquaculture is another example of the indiscriminate use of antibiotics by direct addition to water or direct administration, to promote growth or treatment of fish. This has resulted in antibiotic-resistant gene pools emerging in fish that are spread throughout the aquatic environment and ultimately to us. Several classes of antibiotics used are related or identical to those used to treat human infections, as a result of which cross-resistance has emerged in intestinal gut microbes. This has been well documented in several Aeromonas strains of fish origin that were able to transfer their antibiotic-resistant plasmids to human pathogens in the gut, such as E. coli and Salmonella (Cabello 2006). Another recent example is the emergence of Salmonella enterica serotype Typhimurium DT104 as responsible for outbreaks in the United States and Europe most likely originated from an aquaculture setup (Cabello 2006). The gene encoding a resistance to florfenicol which is widely used in aquaculture was isolated from these pathogens. Another source for the acquisition of antibiotic-resistant genes is through direct consumption of water or fish products that were contaminated.
4.4 Raw Fruits and Fresh Vegetables
Fruits and vegetables could also serve as a source of antibiotic-resistant genes if not washed or cooked adequately before consumption. As an example, Pseudomonas aeruginosa strains that were resistant to several commonly used antibiotics such as chloramphenicol, sulfamethoxazole, and ampicillin were isolated from several sources such as lettuce, tomatoes, cucumbers, and carrots (Allydice-Francis and Brown 2012). Another frequently reported source occurs through the consumption of salads and other meals that utilize uncooked vegetable and fruit products.
5 Methods to Study the Gut Resistome
One of the major limitations to study the human gut resistome is the unavailability of techniques to fully characterize resistance genes. In addition, determination and functional characterization of gene sequences providing resistance to antibiotics are challenging. Thus, in order to characterize a gut resistome completely, a combination of methods should be employed.
The simplest method to detect resistance genes present in gut commensals is to isolate strains and characterize them under laboratory conditions. These techniques were used frequently during the 1970s and 1980s to determine antibiotic resistance in gut commensals belonging to the Bacteroidales and Clostridiales phyla. However, owing to a large number of bacteria present in the gut microbiome, it is practically impossible to culture every gut commensal under laboratory conditions. Therefore, culture-based techniques cannot reveal the complete resistance profile of microbes present in the gut. Furthermore, this approach does not provide information about the mobility of resistance genes. Recent advances in laboratory culture-based techniques have made it possible to perform comparative genome analysis for Bacteroidales (Coyne et al. 2014) and Enterococcaceae (Palmer et al. 2012; Lebreton et al. 2013). However, this approach is very extensive, and it is unclear whether it can capture the entire gut resistome.
Culture-independent techniques have been found to be fairly useful for the analysis of the gut resistome. Often, PCR and microarray hybridization techniques are used to determine resistance gene reservoirs in the human gut. DNA can be isolated from fecal samples and analyzed for the presence of antibiotic-resistant genes by PCR, using gene-specific primers. Alternatively, isolated DNA samples can be hybridized with known antibiotic-resistant gene sequences (probes) and detected through a technique known as DNA microarray. With the help of quantitative PCR, the relative abundance of specific antibiotic resistance can also be determined (Jernberg et al. 2007; Buelow et al. 2014). The advantage of DNA microarray over PCR-based technique is that large numbers of resistance genes can be detected within a short time period. PCR on the other hand is comparatively slower and yields less information. A major limitation of the above techniques is the detection of known resistance markers as opposed to detection of novel resistance genes (Card et al. 2014; Lu et al. 2014). Furthermore, these techniques do not provide information about the bacterium harboring the resistance gene.
Metagenomic sequencing has proved to be a useful technique for the genetic and functional characterization of the human gut resistome. In this approach, DNA samples are purified from feces and sequenced. With the advent of next-generation sequencing platforms such as the Roche 454 sequencer, the Genome Analyzer of Illumina, and the SOLiD system of Applied Biosystems, sequencing costs have reduced dramatically with considerable increases in throughput. The sequence data sets thus obtained are assembled to form large contiguous DNA fragments and analyzed through bioinformatics tools. This technique not only allows the detection of resistance genes in any sample but also reveals the phylogenetic composition of the microbiota. Additionally, antibiotic-resistant genes in any microbiome can be quantified through these techniques (Forslund et al. 2014). However, even this approach cannot be used to discover new antibiotic resentence genes. On the other hand, functional metagenomics has proved useful in this regard, in which random fragments of metagenomic DNA are cloned in E. coli vectors and screened for antibiotic-resistant clones. From this library of antibiotic-resistant clones, gene sequences conferring resistance are determined. Although functional metagenomics is labor intensive, the major advantage of this approach is the identification of novel antibiotic-resistant genes.
6 The Gut Resistome as the Epicenter of Drug-Resistant Genes
Recent advances in metagenomic approaches have dramatically enhanced our knowledge about the gut microbiome (The NIHHMPWG et al. 2009). HGT is a common biological phenomenon in Enterobacteriaceae and has also been found to occur between pathogens and gut microflora particularly when the intestinal barrier is altered (Schjorring and Krogfelt 2011; Cremet et al. 2012; Stecher et al. 2012). The gut receives a wide number of bacteria from different sources such as hands, pharyngeal, food, water, beverages, nasal secretions, etc. Neonates acquire environmental microflora rapidly, and in some cases, sepsis occurs due to translocation of new microflora (Tezuka and Ohteki 2010; Das et al. 2011). Surprisingly, in healthy individuals, the gut microflora is stable, and pathogens ingested through food and water are cleared from the gut due to the presence of commensals.
Thus, it is essential to explore the gut microbiota for the presence of antibiotic-resistant genes and subsequently expand our knowledge on mechanism of emergence of multidrug-resistant pathogens. All resistance genes contributed by the gut microbiota have been termed as the “gut resistome.” Antibiotic resistance may arise by two mechanisms, namely, “by chance” or “by mutation.” The former mechanism describes a gene having a specific role in the host and accidentally neutralizes an antibiotic due to substrate or target similarity. Mutations, on the other hand, generate phenotypic resistance such as target modification, antibiotic inactivation, etc. Thus, antibiotic resistance may naturally be present or can be acquired through the accumulation of mutation. Resistance acquired through any method may be transferred to phylogenetically distant microorganisms sharing a similar ecological niche.
Antibiotics have been the cornerstone of defense against pathogenic infections, particularly after the Second World War. In natural environments, antibiotics provide selective advantage to the producing bacteria and prevent invasive competitors from establishing themselves. Besides, antibiotics may also function as signaling molecules, triggering developmental processes such as biofilm formation (Aminov 2009). Resistance to antibiotics is acquired in a population of susceptible bacteria that accumulate mutations. One such example is the occurrence of point mutation in DNA gyrase which confers resistance to quinolones. Besides this, many bacteria acquire antibiotic-resistant genes that protect microbial cells from the lethal action of antibiotics. Antibiotic-resistant genes confer phenotypic resistance against antibiotics through a variety of mechanisms which include enzymatic inactivation of antibiotics, modification of targets of antibiotics, and pumping antibiotics out of the cells through efflux pumps, thereby preventing accumulation of lethal doses (Martinez 2008, 2014; Allen et al. 2010). Antibiotic-resistant genes have been present in the environment for millennia. For instance, the beta-lactamases have been found to originate about 2 billion years ago (Hall and Barlow 2004). This is evident from the finding that OXA-type beta-lactamases carried on plasmids have been moved between different bacterial phyla for millions of years (Barlow and Hall 2002). Of note, genes conferring antibiotic resistance may have entirely different functions in the original host. For example, 2′-N-acetyltransferase encoded by a gene in a Gammaproteobacterium, namely, Providencia stuartii, is involved in peptidoglycan modification; however, aminoglycosides are structurally similar to the natural substrate of 2′-N-acetyltransferase and hence are inactivated by the enzyme. Therefore, Providencia stuartii are naturally resistant to aminoglycosides. Such genes can be termed as “accidental resistance genes,” and when acquired by any pathogen, these genes can provide resistance against aminoglycosides making them antibiotic resistant (Martinez 2008). Emergence of antibiotic resistance among human pathogens has become a major threat to modern medicine. Therefore, identification of niches where microbes acquire antibiotic resistance is of great importance. Further, the mechanisms by which this antibiotic resistance is mobilized to pathogens are also equally important.
There is adequate evidence to suggest that the gut can serve as a reservoir for opportunistic pathogens, which under immunocompromised conditions may cause severe infections. These findings raise a concern particularly for hospitalized patients who undergo antibiotic therapy to prevent infections. Treatment of immunocompromised patients with various antibiotics may provide selective advantage to multidrug-resistant opportunistic pathogens present in the gut.
Since commensals and pathogens present in the gut share similar ecology, therefore, gene transfer is more likely to occur between them (Smillie et al. 2011). Consequently, it becomes very important to understand the nature of the human gut resistome and its role in the spread of antibiotic-resistant genes among members of the gut microflora, particularly between commensals and opportunistic pathogens (Penders et al. 2013).
As previously mentioned, the human gut presents an environment where millions of phylogenetically distinct bacteria may colonize and interact with each other which increases the possibility for the existence of ample antibiotic-resistant genes contributing to the genetic pool. Available facts about the human gut resistome raise concern in light of the emergence of multidrug-resistant opportunistic pathogens. A metagenomic study of 252 fecal samples performed by Forslund et al. revealed the presence of resistance genes for 50 classes of antibiotics accounting for 21 antibiotic-resistant genes per sample collected from different countries (Forslund et al. 2013). In a similar study, 1093 antibiotic-resistant genes were identified in 162 individuals from Denmark, China, and Spain (Hu et al. 2013). The most commonly found resistance genes are tet32, tet40, tetO, tetQ, and tetW which provide resistance against tetracycline and are found to be present in gut microflora of almost all individuals. Several such genes are ubiquitously present in bacterial genomes such as ant(6)-Ia, bacitracin (bacA), and glycopeptide vancomycin (vanRA and vanRG). These genes are thought to confer resistance to aminoglycosides. Similarly, metagenomic screening of gut resistomes of hospitalized patients also revealed many antibiotic-resistant genes in the gut microbiota of patients, which appeared to increase with increasing antibiotic therapy (Perez-Cobas et al. 2013; Buelow et al. 2014). For instance, Buelow et al. reported that genes conferring resistance to aminoglycosides expanded during a hospital stay, especially in intensive care units (Buelow et al. 2014). The observed expansion of the gut resistome was linked with the use of tobramycin which is an aminoglycoside used for selective decontamination of the digestive tract (de Smet et al. 2009). This treatment regime is generally used as a prophylactic measure for patients admitted to intensive care units in order to lower the risk of infection of opportunistic pathogens. Notably, antibiotic use is not always associated with expansion of the gut resistome in patients; rather, in certain cases, resistant genes may be lost during antibiotic treatment (Perez-Cobas et al. 2013; Buelow et al. 2014). This is generally observed during combination therapy in which bacteria carrying resistance against one antibiotic are still susceptible to another one and are thus lost from the microbial population.
7 From the Gut Resistome to Multidrug-Resistant Pathogens
Pathogenic bacterial loads in the gut are generally lower than that of commensal bacteria. Interestingly, the majority of antibiotic-resistant genes are found on the genome of commensals. Despite this, multidrug-resistant pathogens are emerging at an alarming rate posing a serious threat to the future of medicine. Thus, an understanding of the mechanism of transfer of antibiotic resistance from commensals to pathogens is of particular interest. There are two basic mechanisms by which antibiotic-resistant pathogens can emerge: one by opportunity and another by horizontal gene transfer (HGT). In the former, a small population of drug-resistant pathogenic strains exists naturally which, under suitable conditions such as an immunocompromised state of the host, multiply and increase in number. In the second mechanism, a pathogen acquires antibiotic-resistant genes from commensals by HGT.
8 Mobility of Antibiotic-Resistant Genes
Since antibiotic-resistant genes provide acute fitness in various environments, they are often encoded on mobile elements such as conjugative elements, extra chromosomal DNAs, and viral particles. An estimated 1–3 % of people from the developed world undergo antibiotic treatment daily making the gut environment a dynamic niche (Costello et al. 2012). Conjugative elements with resistance genes have been found to be associated with a large number of single-nucleotide polymorphism (SNP) (Schloissnig et al. 2013). Besides point mutation, HGT is also one of the sources of variability. Mobility of resistance genes is further augmented by the tendency of several antibiotics to facilitate gene transfer. For instance, antibiotics which inhibit DNA synthesis are known to induce interspecific transfer of integrating conjugative elements containing multidrug-resistant genes (Beaber et al. 2004). In the mice gut, tetracycline increases the rate of conjugation between Enterococcus faecalis and Listeria monocytogenes (Doucet-Populaire et al. 1991). Besides conjugation, antibiotics also enhance phage mobility by activating bacterial DNA damage response having cross talk with phage regulation. Treatment with antibiotics has been shown to increase the connectivity between phage and bacterial networks, increasing the accessibility of the microbial genome to phages (Modi et al. 2013). Gut commensals play an important role in host defense against pathogenic microbes by outcompeting for space and nutrients (Brandl et al. 2008; Fukuda et al. 2011; Costello et al. 2012) and inducing host immune responses (Brandl et al. 2008; Fukuda et al. 2011). Antibiotic treatment disrupts the highly organized microbial population structure of the gut, thus exposing a new niche for pathogens and increasing the availability and mobility of resistance genes to virulent species. For instance, methicillin-resistant Staphylococcus aureus (MRSA) emerged by obtaining a gene cluster from Staphylococcus epidermis which is a skin commensal. This gene cluster improved the colonization of host sites (Diep et al. 2006). There are several other examples of gene cluster movement between different bacteria. For example, a specialized polysaccharide degradation gene cluster which is typically present in marine bacteria was found to be present in the gut microbiota of Japanese individuals. Most likely, the gene cluster was transferred to the gut microbiome due to consumption of seaweeds over a long period of time. Besides this, there is evidence for the exchange of antibiotic-resistant genes between soil bacteria and human pathogens (Forsberg et al. 2012).
Generally, exchange of antibiotic-resistant genes between bacteria from different environments is more frequent than within the same environment, probably because of adaptive advantage provided by the resistance genes (Smillie et al. 2011). However, exchange of antibiotic-resistant genes between gut microbiota and the pathogenic gene pool is yet to be confirmed. Metagenomic analysis of the human gut resistome has revealed that functional sequences of antibiotic-resistant genes present in the gut microbial gene pool share very low homology to those present on known pathogens. Possible uncharacterized barriers hinder the transfer of resistance genes between commensals and pathogens resulting in the compartmentalization of the resistome of gut commensals and pathogens. Although antibiotic treatment has been shown to have significant implications, the mechanisms which govern the flow of genes in vivo have yet to be elucidated. For instance, antibiotic-induced HGT may improve the ability of gut microbiota to withstand a particular stress. One such example is the transfer of carbohydrate-active enzymes across phylogenetically distinct commensal bacteria which in turn helps diverse communities withstand shared challenges encountered in a dynamic gut environment. Antibiotic treatment in mice has been reported to enrich a carbohydrate-active enzyme encoded by phages. This finding suggests that increased gene transfer during antibiotic treatment helps the gut microbiota store and access functional genes that facilitate niche recolonization (Modi et al. 2013).
9 Acquisition of Genes by Pathogens
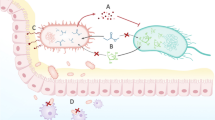
Human pathogens may acquire antibiotic-resistant genes from the gut reservoir by HGT which can occur by several mechanisms such as transformation, conjugation, and transduction (Fig. 1). In transformation, naked DNA is taken up by competent bacteria. Thus, if the DNA carries an antibiotic-resistant gene, it will be transferred to the recipient bacterium. Transformation is a common phenomenon, and many bacteria have been reported to be naturally competent to take up DNA from different species. Conjugation is a mating process in which exchange of genetic material takes place through a conjugative bridge. During this process, resistance genes can spread from donor cells to recipients. Transduction is mediated by bacteriophages. Several bacteriophages encode antibiotic-resistant genes which can be transferred and integrated into the chromosome or transferred from one cell to another (Furuya and Lowy 2006). Among the three processes, transformation does not contribute significantly to HGT in the human gut (Nordgard et al. 2012). However, conjugation and transduction play an important role in the exchange of antibiotic genes among gut microflora. Most commonly, gene transfer in the human gut occurs through conjugative plasmids or transposons (Coyne et al. 2014). Genome and plasmid sequence analyses indicate that though conjugation between closely related bacteria is more frequent than remotely related ones, it plays a significant role in the dissemination of antibiotic-resistant genes (Jones et al. 2010; Tamminen et al. 2012). Conjugative transfer of antibiotic resistance from gut commensals to pathogens is evidenced from the finding that the vanB gene responsible for vancomycin resistance is present on a transposon of a commensal belonging to the phylum Firmicutes which serves as the source of vancomycin resistance in the nosocomial pathogen E. faecium (Stinear et al. 2001; Graham et al. 2008; Howden et al. 2013). Inside the gut, gene transfer takes place in both directions, i.e., from commensals to pathogens and from pathogens to commensals. The most common gram-negative bacteria that play an important role in gene transfer in the gut belong to the phylum Bacteroidetes as they are capable of acquiring DNA from a wide range of bacteria including the gram-positives E. faecalis and Clostridium perfringens. Thus, Bacteroidetes can serve as a source of resistance genes for other bacteria within the gut (Coyne et al. 2014). These groups of bacteria contain a conjugative transposon CTnDOT which plays an active role in the spread of erythromycin and tetracycline resistance inside the gut (Waters and Salyers 2013). Besides obligate anaerobes, facultative anaerobic commensals of the gut such as lactic acid bacteria are also involved in channeling gene transfer in the intestinal tract (Ogilvie et al. 2012). Among others, enterococci are exceptionally efficient in trafficking drug-resistant genes in human gut (Werner et al. 2013).

Spread of drug-resistant genes in the gut microbiome. The figure depicts the mechanisms at play that contribute to the emergence of drug resistance in the gut microbiome. Drug resistance may arise as the mutation of a gene to prevent it from serving as a drug target or an unrelated enzyme whose secondary function serves to inactivate the effect of a drug. Conjugation, transformation, and transduction serve as mechanisms that aid the spread of drug resistance in a population either from pathogens to commensals and vice versa or between the commensals themselves
Furthermore, pathogens of Enterobacteriaceae can also easily exchange plasmid-bearing antibiotic-resistant and virulent genes during gut colonization (Goren et al. 2010). Conjugative exchange of genetic material inside the gut is not only determined by bacterial factors but also affected by host factors. Human epithelial cells produce several proteinaceous compounds which can lower the conjugation efficiency of resistance-encoding plasmids in E. coli strains (Machado and Sommer 2014). On the contrary, gut inflammation facilitates conjugation between pathogenic bacteria and gut commensals (Stecher et al. 2012). Besides bacteria, phages also play an important role in gene transfer in the intestinal tract. The prophages carrying antibiotic-resistant genes that remain integrated into the genome of commensals often enter into the lytic cycle and can thereby transfer antibiotic-resistant genes to other bacteria (Quiros et al. 2014; Waller et al. 2014). In a recent study involving metagenomic analysis of phage DNA from the gut, it was found that 70 % of the samples contained antibiotic-resistant genes such as beta-lactamase resistance gene blaTEM and the quinolone resistance gene qnrA. The role of bacteriophages in HGT in the gut has been proved experimentally as well where antibiotic-resistant genes increased considerably in the phage metagenome after treatment with ampicillin or ciprofloxacin (Modi et al. 2013). Phages isolated from mice treated with antibiotics show higher gene transfer rates as compared to those isolated from untreated mice (Modi et al. 2013). Thus, the gut presents a highly dynamic niche where constant gene transfer takes place among different communities by several methods.
10 Geographical Signature of Antibiotic-Resistant Genes
Although the human gut has been found to harbor a large number of resistance genes, there are country-specific variations (although small, approximately 1.5- to twofold) in the gut resistome of individuals. This could be due to differential use of antibiotics in different countries because individuals from countries such as Denmark with restricted use of antibiotics have been found to contain lower levels of antibiotic-resistant genes than the countries like Spain and China where the use of antibiotic is relatively higher. However, the relation between variation of resistome and incidences of antibiotic-resistant infections in different countries still remains to be determined.
Interestingly, analysis of SNPs in the antibiotic-resistant genes isolated from different geographical locations indicates that there is a specific geographical signature present in antibiotic-resistant genes. For example, sequences of antibiotic resistance isolated from Chinese individuals form a distinct cluster than those isolated from Danish and Spanish individuals (Hu et al. 2013). Similar to geographic-level variation, regional-level differences were also observed when resistance genes from different populations of China were analyzed.
11 Conclusion
The human gut environment presents a dynamic niche for microbes where millions of bacteria belonging to different phyla can colonize and exchange chemical and genetic information. In addition, gut microbes can exchange genetic material with microbes from the external environment through food, water, and beverages. This phenomenon has the result of accumulation of resistance genes in the gut commensals. Hence, the gut commensals have been depicted as the epicenter of antibiotic resistance. Antibiotic treatment has become an integral part of modern-day medicine. However, the disruptions to gut bacteria caused by antibiotic treatment are raising concerns, highlighting the need for new antibacterial therapies. A major concern from the extensive use of antibiotics is the emergence of multidrug-resistant bacterial strains and the widespread transfer of their resistance genes to surrounding nonpathogenic bacteria such as gut commensals (Fig. 2). Thus, the extensive use of antibiotics is shaping our gut microbiota contributing significantly to the emergence of multidrug-resistant pathogens.

A summary of the emergence and modes of transmission of drug-resistant genes in the gut microbiome. The figure illustrates the major external and environmental factors contributing to the emergence of drug resistance in the gut microbiome, namely, antibiotic use and the spread of antibiotic-resistant pathogens from various sources such as fruits, vegetables, animals, and fish. The effects of antibiotic use are the destabilization of the gut and selection of drug-resistant strains that in turn serve as sources of drug resistance, thus contributing to the pool of resistance genes in the population
References
Ahn J, Yang L, Paster BJ, Ganly I, Morris L, Pei Z, Hayes RB (2011) Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS One 6, e22788. doi:10.1371/journal.pone.0022788
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J (2010) Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 8:251–259. doi:10.1038/nrmicro2312
Allydice-Francis K, Brown PD (2012) Diversity of antimicrobial resistance and virulence determinants in Pseudomonas aeruginosa associated with fresh vegetables. Int J Microbiol 2012:426241. doi:10.1155/2012/426241
Aminov RI (2009) The role of antibiotics and antibiotic resistance in nature. Environ Microbiol 11:2970–2988. doi:10.1111/j.1462-2920.2009.01972.x
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, Weissenbach J, Ehrlich SD, Bork P (2011) Enterotypes of the human gut microbiome. Nature 473:174–180. doi:10.1038/nature09944(supplementary-information)
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI (2005) Host-bacterial mutualism in the human intestine. Science 307:1915–1920. doi:10.1126/science.1104816
Barlow M, Hall BG (2002) Phylogenetic analysis shows that the OXA beta-lactamase genes have been on plasmids for millions of years. J Mol Evol 55:314–321. doi:10.1007/s00239-002-2328-y
Beaber JW, Hochhut B, Waldor MK (2004) SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72–74. doi:10.1038/nature02241
Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA (2006) Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A 103:732–737. doi:10.1073/pnas.0506655103
Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, DeMatteo RP, Pamer EG (2008) Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455:804–807. doi:10.1038/nature07250
Brugere JF, Mihajlovski A, Missaoui M, Peyret P (2009) Tools for stools: the challenge of assessing human intestinal microbiota using molecular diagnostics. Expert Rev Mol Diagn 9:353–365. doi:10.1586/erm.09.16
Buelow E, Gonzalez TB, Versluis D, Oostdijk EA, Ogilvie LA, van Mourik MS, Oosterink E, van Passel MW, Smidt H, D’Andrea MM, de Been M, Jones BV, Willems RJ, Bonten MJ, van Schaik W (2014) Effects of selective digestive decontamination (SDD) on the gut resistome. J Antimicrob Chemother 69:2215–2223. doi:10.1093/jac/dku092
Cabello FC (2006) Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol 8:1137–1144. doi:10.1111/j.1462-2920.2006.01054.x
Card RM, Warburton PJ, MacLaren N, Mullany P, Allan E, Anjum MF (2014) Application of microarray and functional-based screening methods for the detection of antimicrobial resistance genes in the microbiomes of healthy humans. PLoS One 9, e86428. doi:10.1371/journal.pone.0086428
Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA (2012) The application of ecological theory toward an understanding of the human microbiome. Science 336:1255–1262. doi:10.1126/science.1224203
Coyne MJ, Zitomersky NL, McGuire AM, Earl AM, Comstock LE (2014) Evidence of extensive DNA transfer between bacteroidales species within the human gut. mBio 5:e01305–e01314. doi:10.1128/mBio.01305-14
Cremet L, Bourigault C, Lepelletier D, Guillouzouic A, Juvin ME, Reynaud A, Corvec S, Caroff N (2012) Nosocomial outbreak of carbapenem-resistant Enterobacter cloacae highlighting the interspecies transferability of the blaOXA-48 gene in the gut flora. J Antimicrob Chemother 67:1041–1043. doi:10.1093/jac/dkr547
Das P, Singh AK, Pal T, Dasgupta S, Ramamurthy T, Basu S (2011) Colonization of the gut with Gram-negative bacilli, its association with neonatal sepsis and its clinical relevance in a developing country. J Med Microbiol 60:1651–1660. doi:10.1099/jmm.0.033803-0
de Smet AM, Kluytmans JA, Cooper B, Mascini EM, Benus RF, van der Werf TS, van der Hoeven JG, Pickkers P, Bogaers-Hofman D, van der Meer NJ, Bernards AT, Kuijper EJ, Joore JC, Leverstein-van Hall MA, Bindels AJ, Jansz AR, Wesselink RM, de Jongh BM, Dennesen PJ, van Asselt GJ, te Velde LF, Frenay IH, Kaasjager K, Bosch FH, van Iterson M, Thijsen SF, Kluge GH, Pauw W, de Vries JW, Kaan JA, Arends JP, Aarts LP, Sturm PD, Harinck HI, Voss A, Uijtendaal EV, Blok HE, Thieme Groen ES, Pouw ME, Kalkman CJ, Bonten MJ (2009) Decontamination of the digestive tract and oropharynx in ICU patients. N Engl J Med 360:20–31. doi:10.1056/NEJMoa0800394
Dethlefsen L, McFall-Ngai M, Relman DA (2007) An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811–818. doi:10.1038/nature06245
Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F (2006) Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi:10.1016/s0140-6736(06)68231-7
Doucet-Populaire F, Trieu-Cuot P, Dosbaa I, Andremont A, Courvalin P (1991) Inducible transfer of conjugative transposon Tn1545 from Enterococcus faecalis to Listeria monocytogenes in the digestive tracts of gnotobiotic mice. Antimicrob Agents Chemother 35:185–187. doi:10.1128/AAC.35.1.185
Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G (2012) The shared antibiotic resistome of soil bacteria and human pathogens. Science 337:1107–1111. doi:10.1126/science.1220761
Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, Bork P (2013) Country-specific antibiotic use practices impact the human gut resistome. Genome Res 23:1163–1169. doi:10.1101/gr.155465.113
Forslund K, Sunagawa S, Coelho LP, Bork P (2014) Metagenomic insights into the human gut resistome and the forces that shape it. Bioessays 36:316–329. doi:10.1002/bies.201300143
Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H (2011) Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547. doi:10.1038/nature09646
Furuya EY, Lowy FD (2006) Antimicrobial-resistant bacteria in the community setting. Nat Rev Microbiol 4:36–45. doi:10.1038/nrmicro1325
Goren MG, Carmeli Y, Schwaber MJ, Chmelnitsky I, Schechner V, Navon-Venezia S (2010) Transfer of carbapenem-resistant plasmid from Klebsiella pneumoniae ST258 to Escherichia coli in patient. Emerg Infect Dis 16:1014–1017. doi:10.3201/eid1606.091671
Graham M, Ballard SA, Grabsch EA, Johnson PD, Grayson ML (2008) High rates of fecal carriage of nonenterococcal vanB in both children and adults. Antimicrob Agents Chemother 52:1195–1197. doi:10.1128/aac.00531-07
Hall BG, Barlow M (2004) Evolution of the serine beta-lactamases: past, present and future. Drug Resist Updat 7:111–123. doi:10.1016/j.drup.2004.02.003
Howden BP, Holt KE, Lam MM, Seemann T, Ballard S, Coombs GW, Tong SY, Grayson ML, Johnson PD, Stinear TP (2013) Genomic insights to control the emergence of vancomycin-resistant enterococci. mBio 4:e00412-13. doi:10.1128/mBio.00412-13
Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X, Meng Z, Zhao F, Liu D, Ma J, Qin N, Xiang C, Xiao Y, Li L, Yang H, Wang J, Yang R, Gao GF, Wang J, Zhu B (2013) Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 4:2151. doi:10.1038/ncomms3151
Jernberg C, Lofmark S, Edlund C, Jansson JK (2007) Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J 1:56–66. doi:10.1038/ismej.2007.3
Jones BV, Sun F, Marchesi JR (2010) Comparative metagenomic analysis of plasmid encoded functions in the human gut microbiome. BMC Genomics 11:46. doi:10.1186/1471-2164-11-46
Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, Corander J, Cheng L, Saif S, Young S, Zeng Q, Wortman J, Birren B, Willems RJ, Earl AM, Gilmore MS (2013) Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 4:e00534-13. doi:10.1128/mBio.00534-13
Levy SB, FitzGerald GB, Macone AB (1976a) Changes in intestinal flora of farm personnel after introduction of a tetracycline-supplemented feed on a farm. N Engl J Med 295:583–588. doi:10.1056/nejm197609092951103
Levy SB, FitzGerald GB, Macone AB (1976b) Spread of antibiotic-resistant plasmids from chicken to chicken and from chicken to man. Nature 260:40–42. doi:10.1038/260040a0
Ley RE, Peterson DA, Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837–848. doi:10.1016/j.cell.2006.02.017
Lu N, Hu Y, Zhu L, Yang X, Yin Y, Lei F, Zhu Y, Du Q, Wang X, Meng Z, Zhu B (2014) DNA microarray analysis reveals that antibiotic resistance-gene diversity in human gut microbiota is age related. Sci Rep 4:4302. doi:10.1038/srep04302
Machado AM, Sommer MO (2014) Human intestinal cells modulate conjugational transfer of multidrug resistance plasmids between clinical Escherichia coli isolates. PLoS One 9, e100739. doi:10.1371/journal.pone.0100739
Martinez JL (2008) Antibiotics and antibiotic resistance genes in natural environments. Science 321:365–367. doi:10.1126/science.1159483
Martinez JL (2014) General principles of antibiotic resistance in bacteria. Drug Discov Today Technol 11:33–39. doi:10.1016/j.ddtec.2014.02.001
Modi SR, Lee HH, Spina CS, Collins JJ (2013) Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 499:219–222. doi:10.1038/nature12212
Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, Cresci A, Silvi S, Orpianesi C, Verdenelli MC, Clavel T, Koebnick C, Zunft HJ, Dore J, Blaut M (2006) Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol 72:1027–1033. doi:10.1128/aem.72.2.1027-1033.2006
Nordgard L, Brusetti L, Raddadi N, Traavik T, Averhoff B, Nielsen KM (2012) An investigation of horizontal transfer of feed introduced DNA to the aerobic microbiota of the gastrointestinal tract of rats. BMC Res Notes 5:170. doi:10.1186/1756-0500-5-170
Ogilvie LA, Firouzmand S, Jones BV (2012) Evolutionary, ecological and biotechnological perspectives on plasmids resident in the human gut mobile metagenome. Bioeng Bugs 3:13–31. doi:10.4161/bbug.3.1.17883
Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS (2012) Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 3:e00318-11. doi:10.1128/mBio.00318-11
Penders J, Stobberingh EE, Savelkoul PH, Wolffs PF (2013) The human microbiome as a reservoir of antimicrobial resistance. Front Microbiol 4:87. doi:10.3389/fmicb.2013.00087
Perez-Cobas AE, Artacho A, Knecht H, Ferrus ML, Friedrichs A, Ott SJ, Moya A, Latorre A, Gosalbes MJ (2013) Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS One 8, e80201. doi:10.1371/journal.pone.0080201
Quiros P, Colomer-Lluch M, Martinez-Castillo A, Miro E, Argente M, Jofre J, Navarro F, Muniesa M (2014) Antibiotic resistance genes in the bacteriophage DNA fraction of human fecal samples. Antimicrob Agents Chemother 58:606–609. doi:10.1128/aac.01684-13
Schjorring S, Krogfelt KA (2011) Assessment of bacterial antibiotic resistance transfer in the gut. Int J Microbiol 2011:312956. doi:10.1155/2011/312956
Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, Kota K, Sunyaev SR, Weinstock GM, Bork P (2013) Genomic variation landscape of the human gut microbiome. Nature 493:45–50. doi:10.1038/nature11711
Sjolund M, Wreiber K, Andersson DI, Blaser MJ, Engstrand L (2003) Long-term persistence of resistant Enterococcus species after antibiotics to eradicate Helicobacter pylori. Ann Intern Med 139:483–487. doi:10.7326/0003-4819-139-6-200309160-00011
Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ (2011) Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480:241–244. doi:10.1038/nature10571
Stecher B, Denzler R, Maier L, Bernet F, Sanders MJ, Pickard DJ, Barthel M, Westendorf AM, Krogfelt KA, Walker AW, Ackermann M, Dobrindt U, Thomson NR, Hardt WD (2012) Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc Natl Acad Sci U S A 109:1269–1274. doi:10.1073/pnas.1113246109
Stinear TP, Olden DC, Johnson PD, Davies JK, Grayson ML (2001) Enterococcal vanB resistance locus in anaerobic bacteria in human faeces. Lancet 357:855–856. doi:10.1016/s0140-6736(00)04206-9
Sullivan A, Edlund C, Nord CE (2001) Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect Dis 1:101–114. doi:10.1016/s1473-3099(01)00066-4
Tamminen M, Virta M, Fani R, Fondi M (2012) Large-scale analysis of plasmid relationships through gene-sharing networks. Mol Biol Evol 29:1225–1240. doi:10.1093/molbev/msr292
Tezuka H, Ohteki T (2010) Regulation of intestinal homeostasis by dendritic cells. Immunol Rev 234:247–258. doi:10.1111/j.0105-2896.2009.00872.x
The NIHHMPWG, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M (2009) The NIH human microbiome project. Genome Res 19:2317–2323. doi:10.1101/gr.096651.109
Waller AS, Yamada T, Kristensen DM, Kultima JR, Sunagawa S, Koonin EV, Bork P (2014) Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J 8:1391–1402. doi:10.1038/ismej.2014.30
Waters JL, Salyers AA (2013) Regulation of CTnDOT conjugative transfer is a complex and highly coordinated series of events. MBio 4:e00569-13. doi:10.1128/mBio.00569-13
Werner G, Coque TM, Franz CM, Grohmann E, Hegstad K, Jensen L, van Schaik W, Weaver K (2013) Antibiotic resistant enterococci-tales of a drug resistance gene trafficker. IJMM 303:360–379. doi:10.1016/j.ijmm.2013.03.001
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ryan, D., Jaiswal, S., Suar, M. (2017). Role of External and Environmental Factors in Drug Resistance Emergence: Gut Microbiota. In: Arora, G., Sajid, A., Kalia, V. (eds) Drug Resistance in Bacteria, Fungi, Malaria, and Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-48683-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-48683-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48682-6
Online ISBN: 978-3-319-48683-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)