
Abstract
Tuberculosis (TB) remains a global health concern, despite availability of antituberculosis drugs. Drug-resistant Mycobacterium tuberculosis strains were identified shortly after the discovery and introduction of streptomycin for the treatment of this disease. Subsequently, multidrug therapy was implemented for TB treatment; however, this was soon followed by reports of multi-, extensively, and totally drug-resistant tuberculosis cases globally. The amplification of this drug resistance is due to the sequential accumulation of chromosomal alterations in target genes in the Mycobacterium tuberculosis genome. It is also evident that the presence of mutations that confer drug resistance results in the emergence of compensatory mechanisms which restore bacterial fitness. The recent approval by the Food and Drug Administration for bedaquiline as an antituberculosis drug provided some hope. However, clinical resistance to this new drug has already been reported. This underscores that it is imperative to understand drug resistance and its associated mechanisms in order to direct research efforts to the development of antituberculosis regimens with novel mechanisms of actions.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
In 2015 the World Health Organization (WHO) reported 9.6 million new cases of tuberculosis (TB), with 3.3 % of these and 20 % of previously treated cases infected with a multidrug-resistant (MDR) strain of Mycobacterium tuberculosis (WHO 2015). More alarmingly, the average proportion of MDR-TB cases with extensively drug-resistant TB (XDR-TB) is 9.7 % (WHO 2015). Resistance to the first effective anti-TB drug, streptomycin (STR), was observed shortly after its introduction in 1944 (Nachega and Chaisson 2003; Keshavjee and Farmer 2012), and this trend had continued for many other TB drugs (Fig. 1). Numerous MDR-TB outbreaks were identified in the early 1990s, emphasizing TB as a global health problem (Nachega and Chaisson 2003) (Fig. 1). MDR-TB is characterized by a mycobacterial infection with M. tuberculosis strains that are resistant to rifampicin (RIF) and isoniazid (INH) (Gupta et al. 2003). Outbreaks of XDR-TB have been reported globally (Gandhi et al. 2006; Migliori et al. 2007b; Masjedi et al. 2010; Klopper et al. 2013; Cohen et al. 2015), with XDR-TB defined as an infection with an MDR-TB strain with further resistance to a fluoroquinolone (FQ) and one injectable drug, amikacin (AMI), kanamycin (KANA), and capreomycin (CAP) (Holtz 2007; Holtz and Cegielski 2007; Louw et al. 2009). Recently, M. tuberculosis strains resistant to all available anti-TB drugs have been identified globally and have been named totally drug-resistant TB (TDR-TB) (Migliori et al. 2007a; Velayati et al. 2009; Udwadia 2012; Udwadia et al. 2012; Klopper et al. 2013; Udwadia and Vendoti 2013) (Fig. 1). Although this term is somewhat controversial, TDR-TB has been defined as M. tuberculosis strains with in vitro resistance to all available first- and second-line drugs tested (INH, RIF, STR, EMB, PZA, ETH, PAS, DCS, OFL, AMI, CIP, CAP, KANA) (Parida et al. 2015). Factors fueling the drug-resistant TB epidemic include the inadequacies of TB control in combination with HIV coinfection.

Illustration of the tuberculosis drug discovery timeline and drug resistance development reports: STR streptomycin, PAS para-aminosalicylic acid, INH isoniazid, PZA pyrazinamide, DCS d-cycloserine, KANA kanamycin, EMB ethambutol, ETH ethionamide, RIF rifampicin, CAP capreomycin, AMI amikacin, OFL ofloxacin, LEVO levofloxacin, BDQ bedaquiline, MDR-TB multidrug-resistant tuberculosis, WHO World Health Organization, XDR-TB extensively drug-resistant tuberculosis, TDR-TB totally drug-resistant tuberculosis. aFirst report of BDQ resistance identified in a TB patient (Bloemberg et al. 2015)
The WHO recommends that new patients with pulmonary TB receive intensive phase treatment (2 months duration) which consists of INH, RIF, PZA, and EMB. Subsequently, a patient infected with a drug-sensitive M. tuberculosis strain is treated with INH and RIF during the 4-month continuation phase treatment (WHO 1997). Patient noncompliance is a consequence of the long treatment duration, and these factors fuel the development of drug resistance. An 8-month retreatment regimen with first-line anti-TB drugs for previously treated patients awaiting DST results consists of 2 months with INH, RIF, PZA, EMB, and STR; 1 month with INH, RIF, PZA, and EMB; and 5 months with INH, RIF, and EMB. Treatment of MDR-TB requires a regimen with second-line drugs administered over 18–24 months (Mukherjee et al. 2004). Recommendations for the treatment of various forms of drug-resistant TB are tabulated in Table 1. Current drugs used for TB treatment have limited efficacy against drug-resistant M. tuberculosis strains. However, new anti-TB drugs in development, specifically drugs with different modes of actions than the current drugs, could be effective against both drug-sensitive and drug-resistant TB.
2 Mode of Action and Mycobacterial Drug Resistance Mechanism
2.1 Cell Wall Synthesis Inhibitors
2.1.1 Isoniazid
INH is a prodrug that inhibits mycolic acid biosynthesis (Vilcheze and Jacobs 2007). This inhibition occurs via multiple mechanisms and results in the loss of trehalose monomycolate, trehalose dimycolate, and mycolates (Vilcheze and Jacobs 2007). INH is activated by KatG, which is a catalase-peroxidase, encoded by the katG gene. Upon activation, INH forms an adduct with NAD (Rozwarski et al. 1998) and binds and inhibits inhA, encoded by the enoyl-acyl carrier protein reductase InhA (NADH dependent), which is part of the fatty acid synthase type II system (Marrakchi et al. 2000). The INH-NAD adducts inhibit the activity of InhA, thereby resulting in intracellular accumulation of long-chain fatty acids, decreased mycolic acid biosynthesis, and subsequent cell death.
The loss of activation of INH by KatG is one of the mechanisms of INH resistance in mycobacteria. Mutations in the katG gene lead to a reduction in catalase activity. This results in a decrease in activated INH and a decreased capacity to form the INH-NAD adduct to inhibit InhA and subsequent high-level INH resistance (Heym et al. 1999; Ramaswamy et al. 2003). The Ser315Thr mutation in the katG gene is reported to be the most frequent mutation found in clinical M. tuberculosis strains resistant to INH (Seifert et al. 2015). Mutations within the inhA promoter (−15T and −8A loci) result in overexpression or modification of inhA and subsequently confer low-level INH resistance and ETH cross-resistance (Banerjee et al. 1994). Mutations in the structural gene are less frequent, but the Ser94Ala inhA mutation has been reported to be associated with low-level INH resistance (Quemard et al. 1995). Approximately 10 % of INH resistance is not attributed to mutations in katG and inhA, suggesting that additional resistance mechanisms contribute to INH resistance in mycobacteria. Additional genes (kasA, ahpC, ndh, and the ahpC-oxyR intergenic region) have been implicated in INH resistance; however, their direct impact on clinical INH resistance is not fully understood (Vilcheze et al. 2005; Vilcheze and Jacobs 2007; Campbell et al. 2011).
2.1.2 Ethionamide
The second-line drug, ETH, has a common molecular target to INH, namely, InhA of the FAS II system (Banerjee et al. 1994; Marrakchi et al. 2000). ETH is a prodrug and INH structural analog, which also inhibits mycolic acid biosynthesis. It was shown that M. tuberculosis strains with low-level INH resistance also exhibit resistance to ETH (Banerjee et al. 1994). ETH is activated by the monooxygenase, ethA, with subsequent formation of an ETH-NAD adduct. Even though the ETH-NAD adduct inhibits InhA, in the same manner as the INH-NAD adduct, the activating enzymes of the different compounds are distinct.
Numerous mutations in the ethA gene, resulting in a failure to activate ETH, have been reported to contribute to ETH resistance (Morlock et al. 2003; Brossier et al. 2011). The TetR-like repressor, EthR, negatively regulates the expression of ethA and interacts directly with the ethA promoter region, and EthR overexpression leads to ETH resistance (Baulard et al. 2000; DeBarber et al. 2000). Intragenic inhA mutations (Ser94Ala, Ser94Trp, Leu11Val) in addition to inhA promoter mutations (−102A and −47C) have also been identified in ETH-resistant M. tuberculosis isolates (Morlock et al. 2003; Brossier et al. 2011).
Approximately 50 % of ETH-resistant M. tuberculosis strains exhibit an absence of mutations in inhA or ethA, suggesting an alternative resistance mechanism (Boonaiam et al. 2010). Recently, mutations in the mshA gene (including a Val171Gly-Ala187Val double mutation) were identified in ETH-resistant isolates (Vilcheze et al. 2008; Brossier et al. 2011). MshA is a glycosyltransferase that is involved in mycothiol biosynthesis, and mutations in mshA have been proposed to result in the failure to activate ETH (Vilcheze et al. 2008). Interestingly, it was also observed that mutations in ndh resulted in defects in NdhII activity, subsequently leading to increased intracellular NADH/NAD+ ratio (Vilcheze et al. 2005). The increase in the NADH levels protects against InhA inhibition by either the INH-NAD or ETH-NAD formed when INH and ETH is activated, subsequently leading to ETH and INH co-resistance (Vilcheze et al. 2005). Even with the identification of the additional gene mutations, it is evident that additional resistance mechanisms exist that could contribute to ETH resistance.
2.1.3 Ethambutol
EMB is a bacteriostatic agent that targets the integral membrane arabinosyltransferases involved in polymerizing arabinose into arabinan components of arabinogalactan (Takayama and Kilburn 1989; Zhu et al. 2004; Wolucka 2008; Xu et al. 2015). Resistance to EMB is primarily attributed to mutations in the arabinosyltransferases encoded by embB, with 60 % of EMB-resistant isolates carrying a mutation at embB306 (Ramaswamy et al. 2000; Zhang and Yew 2009; Safi et al. 2013; Xu et al. 2015). However, several studies report discordance between genotypic and phenotypic resistance testing; this could be due to inaccurate diagnostic tests that are dependent on the medium used (Sreevatsan et al. 1997; Johnson et al. 2006a; Plinke et al. 2010; Xu et al. 2015).
Mutations in the embC, embA, and embR genes have also been implicated in EMB resistance, with alterations located in the embC-embA intergenic region conferring high-level EMB resistance (Cui et al. 2014; Xu et al. 2015). embR has been reported to modulate the level of arabinosyltransferase activity in vitro in a phosphorylation-dependent manner, acting downstream of the Ser/Thr-kinase PknH (Belanger et al. 1996). Interestingly, mutations were identified in the ubiA gene in EMB-resistant XDR-TB isolates lacking shared embB mutations (Motiwala et al. 2010; He et al. 2015), and these mutations were associated with high-level EMB resistance (Safi et al. 2013). The ubiA gene is essential for growth of M. tuberculosis and is involved in the synthesis of decaprenylphosphoryl-d-arabinose (Huang et al. 2005). It was recently reported that overexpression of wild-type ubiA gene resulted in an increase in EMB resistance in M. tuberculosis (He et al. 2015). This indicates that multiple mechanisms could result in the EMB resistance phenotype in mycobacteria.
2.1.4 SQ109
One of the newer anti-TB drugs, SQ109, was identified by screening a library of EMB derivatives based on the upregulation of the iniBAC operon promoter (Lee et al. 2003; Protopopova et al. 2005). Exposure of mycobacteria to SQ109 leads to the inhibition of trehalose dimycolate production and concomitant upregulation of trehalose monomycolate levels (Li et al. 2014b). This results in failure to attach mycolic acids to the cell wall arabinogalactan (Grzegorzewicz et al. 2012; Tahlan et al. 2012). The MIC for SQ109 ranges from 0.16 to 0.78 μg/ml for all M. tuberculosis strains tested (Jia et al. 2005), and synergy was observed between INH/RIF and SQ109 in in vitro and in vivo analysis (Nikonenko et al. 2007). M. tuberculosis has a low spontaneous mutation rate of 2.55 × 10−11 for SQ109 resistance (Sacksteder et al. 2012).
The mycobacterial transport protein responsible for trehalose dimycolate transport, MmpL3, has been identified as the target of SQ109 (Sacksteder et al. 2012; Tahlan et al. 2012). Attempts to generate mutants against SQ109 have been unsuccessful. However, whole genome sequencing of in vitro mutants generated against analogs of SQ109 revealed that mutations in the mmpL3 gene led to SQ109 and SQ109 analog resistance without cross-resistance to EMB (Tahlan et al. 2012). Mmpl3 mutations (Ala700Thr, Gln40Arg, and Leu567Pro) were reported to result in a greater than fourfold increase in SQ109 resistance level (Tahlan et al. 2012), with cross-resistance being observed between other MmpL3 inhibitors (Li et al. 2014b). Recently, it was observed that SQ109 inhibits enzymes involved in menaquinone synthesis, respiration, and therefore ATP synthesis (Li et al. 2014a). Additionally, SQ109 disrupts the proton motive force, thereby acting as an uncoupler (Li et al. 2014b). This effect on the proton motive force may also impact MmpL proteins, since it is suggested that the resistance-nodulation-division transporters catalyze the export of substrates via a proton anti-port mechanism (Li et al. 2014b).
2.1.5 d-Cycloserine
DCS is recommended by the WHO for the treatment of drug-resistant TB, despite severe side effects (WHO 2000). Resistance to DCS is attributed to overexpression of alrA in M. smegmatis (Caceres et al. 1997). AlrA encodes for d-alanine racemase that is involved in d-alanine synthesis. d-Alanine is an integral component of peptidoglycan which is an essential component of the cell wall. l-Alanine is converted to d-alanine by the catalytic activity of AlrA (Chacon et al. 2002). Subsequently, the d-alanine/d-alanine ligase (Ddl) catalyzes the dimerization of d-alanine into d-alanyl-d-alanine (Chacon et al. 2002). Studies indicate that alrA overexpression is a result of a G→T transversion in the alrA promoter (Caceres et al. 1997). These reports also show that M. smegmatis alrA null mutants have the ability to grow in the absence of d-alanine, suggesting the presence of another pathway of d-alanine biosynthesis (Chacon et al. 2002). Moreover, these alrA null mutants were more susceptible to DCS. It was also observed that a mutation (Gly122Ala) in the cycA gene, which encodes a d-serine/alanine/glycine transporter, partially contributes to the DCS resistance phenotype in M. bovis BCG vaccine strains (Chen et al. 2012). From these reports it is evident that more research needs to be done on DCS in order to elucidate and understand its resistance mechanisms fully.
2.2 Inhibitors of DNA Replication
2.2.1 Fluoroquinolones
Quinolones are synthetic compounds active on the enzymes essential for DNA replication, the DNA gyrases (Ginsburg et al. 2003). By interfering with DNA gyrase activity, the FQs disrupt DNA supercoiling, thereby inhibiting cell division and gene expression. DNA gyrase is comprised of two alpha and two beta subunits, encoded by the gyrA and gyrB genes, respectively (Takiff et al. 1994). Scientific reports indicate that spontaneous mutations develop at a frequency of 2 × 10−6 to 10−8 (Alangaden et al. 1995).
Approximately 90 % of FQ resistance in M. tuberculosis is attributed to mutations in a region named the quinolone-resistance-determining region (QRDR) in the gyrA and the gyrB gene (Takiff et al. 1994; Aubry et al. 2006). Mutations at codons 90 and 94 in the gyrA gene are most commonly observed among clinical isolates (Aubry et al. 2006), along with a Ser95Thr polymorphism in gyrA that is also present in FQ-sensitive clinical isolates (Maruri et al. 2012). Double mutations in gyrA and gyrB have been reported to exhibit high-level OFL resistance (Isaeva et al. 2013; Nosova et al. 2013). Mutations in gyrA (e.g., Ser91Pro, Asp94Ala, Ala90Val) also result in OFL, MOXI, and LFX cross-resistance with MIC90 > 4 μg/ml (Kambli et al. 2015; Willby et al. 2015). Although the majority of clinical FQ resistance is attributed to mutations in the gyrA and gyrB genes, additional mechanisms that can contribute to FQ resistance include efflux and DNA mimicry (Pasca et al. 2004). The clinical significance of these mechanisms has not been extensively investigated yet.
2.3 Inhibitors of Transcription
2.3.1 Rifampicin
RIF is a highly effective rifamycin that interferes with transcription by inhibiting the DNA-dependent RNA polymerase (RNAP) enzyme (McClure and Cech 1978). The majority of RIF-resistant M. tuberculosis strains harbor mutations in an 81 bp RIF resistance-determining region (RRDR) of the rpoβ gene, which encodes the β-subunit of RNAP (Telenti et al. 1993). Mutations at different loci in the RRDR of the rpoβ gene result in different RIF resistance levels (Louw et al. 2011), with His526Arg, His526Asp, His526Pro, His526Tyr, and Ser531Leu mutations being among the most common among RIF-resistant M. tuberculosis isolates (Telenti et al. 1993; Bodmer et al. 1995). Mutations in the RRDR are not the sole contributors to RIF resistance; mutations outside of the RRDR (Heep et al. 2001; Siu et al. 2011), along with the significant upregulation of efflux pumps upon RIF exposure (Louw et al. 2011), have been associated with RIF resistance. In 2011, the WHO endorsed the implementation of an automated test, Xpert® MTB/RIF assay, to rapidly detect TB and RIF-resistant TB (Friedrich et al. 2013). Assessments of the assay indicates that despite the cost limitations, it does provide rapid results, and it significantly increases detection of TB and RIF resistance in culture-confirmed cases, compared to smear microscopy (Steingart et al. 2014).
2.4 Inhibitors of Translation
2.4.1 Aminoglycosides
2.4.1.1 Streptomycin, Amikacin, and Kanamycin
The aminoglycosides inhibit protein synthesis by binding to the 30S subunit of the mycobacterial ribosome (Ramaswamy and Musser 1998), with mutations in the rpsL, rrs, gidB, and eis genes implicated in aminoglycoside resistance (Maus et al. 2005a; Zaunbrecher et al. 2009; Georghiou et al. 2012; Reeves et al. 2013). Mutations in the essential rpsL gene, which encodes the 12S protein, result in resistance to STR, with the most common rpsL mutations being K43R and K88R (Ali et al. 2015). Mutations in the rrs gene, encoding for 16S rRNA, result in high-level resistance to STR, AMI, and KANA, with the A1401G mutation being the most frequently observed in AMI and KANA co-resistance (Campbell et al. 2011). Various different mutations in the gidB gene, which encodes a 7-methylguanosine methyltransferase that specifically modifies residues on 16S rRNA, have been identified in STR-resistant M. tuberculosis strains. These mutations result in the failure to methylate specific residues on the 16S rRNA molecule, thereby leading to resistance conferred by loss-of-function mutations (Ali et al. 2015). It was reported that promoter mutations in the 5′ untranslated region of the eis gene, encoding an aminoglycoside acetyltransferase, confer clinical low-level resistance to KANA. This acetyltransferase acetylates KANA, thereby leading to its inactivation, which subsequently prevents the drug from binding to the 30S ribosome (Zaunbrecher et al. 2009). To date, these mutations have been relatively selective for KANA resistance; therefore many strains with eis mutations would be classified as AMI susceptible. Interestingly, it has recently been reported that mutations in the 5′ untranslated region of the eis transcriptional activator, whiB7, also results in KANA resistance. These mutations in whiB7 lead to an upregulation of eis, thereby resulting in KANA degradation and subsequent resistance (Reeves et al. 2013).
2.4.2 Cyclic Peptides
2.4.2.1 Capreomycin and Viomycin
CAP and VIO are cyclic peptides that inhibit protein synthesis. VIO has been shown to bind both the 30S and 50S ribosome subunits and to inhibit ribosomal translocation by interference with the peptidyl tRNA acceptor site (Yamada et al. 1978). VIO and CAP cross-resistance occurs in M. tuberculosis. Cross-resistance between CAP and AMI/KANA has been reported, but cross-resistance between CAP and STR is rare (Maus et al. 2005a). Mutations at A1401G, C1402T, and G1484T are associated with CAP resistance, with additional mutations at various positions in the tlyA gene, an rRNA methyltransferase reported to exhibit VIO and CAP resistance (Maus et al. 2005a, b).
2.4.3 Oxazolidinones
2.4.3.1 Linezolid
Linezolid (LIN) was first introduced to treat gram-positive infections, including staphylococcal and streptococcal infections (Perry and Jarvis 2001). In vitro linezolid MICs for susceptible M. tuberculosis strains ranged from 0.25 to 1 μg/ml with an MIC90 of 0.5 μg/ml. Development of resistance against linezolid was considered to be rare (Richter et al. 2007). Reported in vitro frequencies for linezolid resistant mutants were 2 × 10−8 to 5 × 10−9 (Hillemann et al. 2008). Sequencing of the 23S rRNA gene in linezolid resistant mutants revealed the presence of a G to T nucleotide substitution at either position 2061 or position 2576 (Richter et al. 2007). The level of resistance for LIN mutants with the nucleotide substitution at position 2061 was 32 μg/ml, whereas those with a nucleotide substitution at position 2576 had a resistance level of 16 μg/ml (Richter et al. 2007). Interestingly, the predominant mutation identified in clinical and in vitro selected LIN mutants was in the rplC gene, encoding the L3 ribosomal protein, at T460C (Beckert et al. 2012).
2.5 Anti-TB Drugs That Target Energy Metabolism
2.5.1 Pyrazinamide
Pyrazinamide (PZA) susceptibility testing is technically difficult due to the acidic medium required for DST tests (Hoffner et al. 2013). PZA-resistant M. tuberculosis strains emerge due to a lack of pyrazinamidase (PZase) activity. PZase is required to convert PZA to its active form pyrazinoic acid (POA) (Konno et al. 1967). The protonated form, HPOA, enters the cell, accumulates, and eventually kills the cell (Zhang and Mitchison 2003). The PZA MIC of M. tuberculosis ranges from 6.25 to 50 μg/ml at pH 5.5 (Stottmeier et al. 1967). However, a PZA MIC > 2000 μg/ml has been reported for M. avium and M. smegmatis due to intrinsic PZA resistance as a result of efflux. M. bovis is also naturally resistant to PZA due to C→Gnt169 in pncA, whereas M. kansasii has weak PZase activity and exhibits an MIC of 250 μg/ml (Ramirez-Busby and Valafar 2015). PZA resistance in M. tuberculosis is mostly due to mutations in the pncA gene (Whitfield et al. 2015a); however, pncA polymorphisms that do not confer the PZA-resistant phenotype have also been identified (Whitfield et al. 2015b). Mutations in rspA, involved in trans-translation, have also been identified in PZA-resistant strains (Louw et al. 2006; Shi et al. 2011; Feuerriegel et al. 2013; Simons et al. 2013b; Tan et al. 2014). Interestingly, M. canetti is naturally resistant to PZA due to a mutation (Met117Thr) in panD (Zhang et al. 2013). Subsequently, panD mutations in PZA-resistant M. tuberculosis strains lacking rpsA or pncA mutations have also been identified (Shi et al. 2014). POA inhibits enzymatic activity of panD, and it was observed that anti-TB activity of POA could be antagonized by B-alanine or pantothenate (Dillon et al. 2014).
2.5.2 Bedaquiline
Bedaquiline (BDQ) (Sirturo or TMC207) is the first anti-TB drug in 40 years to be FDA approved for treatment of sensitive and MDR-TB. The use of BDQ in addition to the standard TB therapy in the murine model accelerated the bactericidal effect (Andries et al. 2005; Lounis et al. 2006; Ibrahim et al. 2007). The minimum inhibitory concentrations of BDQ for M. tuberculosis H37Rv and drug-susceptible strains ranged from 0.03 to 0.12 μg/ml (Table 1) (Andries et al. 2005). Computational models suggest that BDQ restricts the rotational activity of ATP synthase, thereby inhibiting ATP production (deJonge et al. 2007). Spontaneous mutant selection and subsequent whole genome sequence analysis of the resistant M. tuberculosis and M. smegmatis mutants identified mutations (Ala63Pro and Asp32Val) in the c-subunit of ATP synthase encoded by the atpE gene (Andries et al. 2005; Koul et al. 2007). Mutations in atpE partially account for the BDQ resistance phenotype, with the report of spontaneous mutants without atpE gene mutations (Andries et al. 2005; Huitric et al. 2007, 2010). Recently, clofazimine (CFZ)-BDQ cross-resistance was observed in CFZ-resistant in vitro mutants. In the absence of atpE mutations, these mutants harbored mutations in the transcriptional repressor, Rv0678, which subsequently resulted in the upregulation of the Rv0678 and the mmpL5-mmpS5 efflux system (Milano et al. 2009; Hartkoorn et al. 2014). This upregulation led to a four- to eightfold increase in the level of resistance for CFZ and BDQ, which could be reversed with the addition of verapamil and reserpine (Andries et al. 2014; Hartkoorn et al. 2014).
2.6 Multi-target Drugs
2.6.1 PA-824/Pretomanid
PA-824 is a member of the nitroimidazole family containing a nitroimidazopyran nucleus. The MIC for PA-824 ranges from 0.039 to 0.25 μg/ml for sensitive strains compared to 0.015–0.513 μg/ml for drug-resistant strains, with a mutation frequency of 1.9 × 10−5 to 6.38 × 10−7 (Stover et al. 2000). PA-824 is a prodrug that is activated to its toxic form, by the mycobacterial membrane-bound nitroreductase Ddn, a deazaflavin F420-dependent enzyme. This activation leads to the inhibition of mycolic acid synthesis, resulting in cell death (Singh et al. 2008). Investigation on the modes of action of PA-824 has shown that intermediate metabolites of PA-824 act as intracellular nitric oxide donors, therefore encouraging intracellular killing of M. tuberculosis in anaerobic conditions (Singh et al. 2008; Manjunatha et al. 2009). When bacteria are in a hypoxic nonreplicating state, PA-824 kills as a nitrous donor (Manjunatha et al. 2009). Interestingly, M. leprae is intrinsically resistant to PA-824 due to the lack of the ddn gene (Manjunatha et al. 2006).
Another mode of action for PA-824 is suggested by the observation that an fbiC knockout mutant in H37Rv, which is deficient for F420 production, is hypersensitive to oxidative stress and INH, moxifloxacin, and CFZ (Gurumurthy et al. 2013). By isolating PA-824-resistant mutants from the H37Rv M. tuberculosis background, it was observed that 29 % of isolates harbored mutations in the ddn gene and 26 % (fbiC), 19 % (fbiA), 7 % (fgd1), and 2 % in the fbiA gene. The mutation Ser11STOP in ddn gene conferred high-level PA-824 resistance; however, approximately 17 % of mutants lacked mutations in target genes screened, suggesting a different resistance mechanism (Haver et al. 2015).
2.6.2 OPC67683/Delamanid
Delamanid belongs to the nitro-dihydro-imidazooxazole class of antibiotics that inhibit mycolic acid synthesis (Barry and O’Connor 2007). Delamanid has an MIC90 of 0.006–0.05 μg/ml (Diacon et al. 2011), with an in vitro mutation frequency of 6.44 × 10−6 to 4.19 × 10−5 (Szumowski and Lynch 2015). Mutations in F420 biosynthetic genes also result in PA-824-delamanid cross-resistance.
2.6.3 Clofazimine
CFZ is lipophilic riminophenazine developed in 1957 for the treatment of MR-TB (Van Deun et al. 2010). It is a prodrug that is reduced by NADH dehydrogenase (Ndh2), and subsequently re-oxidized by O2, to release reactive oxygen species (ROS). The production of ROS and subsequent cell death have been reported in M. smegmatis treated with CFZ and CFZ analogs (Yano et al. 2011). In vitro isolation of CFZ mutants reported cross-resistance to BDQ due to the presence of mutations in the transcriptional repressor, Rv0678, and subsequent upregulation of efflux pumps mmpL5-mmpS5 (Hartkoorn et al. 2014). Recently, whole genome sequence analysis of spontaneous CFZ mutants revealed mutations in two additional genes that conferred the CFZ-resistant phenotype. These mutations were Glu89STOP in the putative peptidase, PepQ, resulting in the inactivation of this protein (Zhang et al. 2015a). The authors suggest that PepQ could be involved in CFZ activation. The additional mutation, Val351Ala, was identified in a possible permease, Rv1979c, which is involved in amino acid transport (Zhang et al. 2015a). Although it is suggested that this protein could be involved in CFZ uptake and transport, it is evident that the direct effect of these two additional genes on CFZ resistance should be investigated further.
2.7 Anti-TB Drugs That Target Pathways
2.7.1 Para-aminosalicylic Acid
PAS is used as a second-line drug that targets the mycobacterial folate pathway (WHO 2000; Chakraborty et al. 2013). This prodrug is a structural analog of para-aminobenzoic acid (PABA) that is the substrate of the dihydropteroate synthase, encoded by folP1/folP2. The condensation of PABA and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate to 7,8-dihydropteroate is catalyzed by dihydropteroate synthase. This is subsequently converted to dihydrofolate and reduced by dihydrofolate reductase, encoded by dfrA, to produce tetrahydrofolate (Table 2).
Rengarajan and colleagues showed that PAS resistance is attributed to mutations in the thyA gene, encoded for by thymidylate synthase A, which is essential for thymine synthesis. In addition, thyA gene mutations were also present in clinical M. tuberculosis isolates resistant to PAS, indicating that PAS functions as a folate antagonist (Rengarajan et al. 2004; Fivian-Hughes et al. 2012). The dihydrofolate synthase, FolC, is essential for the activation of PAS, and mutations in folC have been reported to result in the PAS-resistant phenotype (Zhao et al. 2014). In addition, mutations in ribD, encoded for by the alternate dihydrofolate reductase, have been reported to result in its overexpression, thereby leading to PAS resistance (Zheng et al. 2013; Zhao et al. 2014; Zhang et al. 2015b). It was suggested that overexpression of ribD confers resistance by compensating for the inhibition of DfrA function.
3 Drug Resistance Mechanisms Other Than Chromosomal Mutations
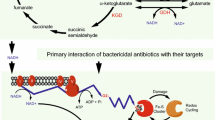
Drug resistance in M. tuberculosis is not attributed to horizontal gene transfer, due to the lack of plasmids in this bacillus (Zainuddin and Dale 1990). Alternative mechanisms that contribute to mycobacterial drug resistance include (a) the production of drug-modifying enzymes, (b) the production of enzymes that inactivated the drug, (c) low cell wall permeability resulting in a decrease in drug influx, and (d) efflux-related mechanisms leading to a reduction in intracellular drug concentration (Davies and Courvalin 1977; Dabbs et al. 1995; Liu et al. 1996; Takiff et al. 1996; Davies and Wright 1997; Quan et al. 1997; Imai et al. 1999; Nikaido 2001; Brennan 2003; Draker et al. 2003; Li et al. 2004; Ashenafi et al. 2014) (Fig. 2).

Mycobacterial drug resistance mechanisms other than chromosomal alterations. Mechanisms such as the activation of efflux pumps and limited drug influx due to the decreased drug permeability lead to a reduction in the intracellular drug concentration and subsequent intrinsic resistance. The production of drug-inactivating and drug-modifying enzymes also results in drug resistance
3.1 Permeability Barrier and Activation of Efflux Pumps
Certain mycobacterial species exhibit an intrinsic drug-resistant phenotype that is not the result of antibiotic exposure (Fajardo et al. 2008). Intrinsic drug resistance is attributed to the activation of efflux pumps and an inherently low permeability of the mycobacterial cell wall (Nikaido 2001; Borges-Walmsley et al. 2003; Louw et al. 2009). Recently, knockdown of Rv1026 (ppx2), an exopolyphosphatase, was shown to result in increased bacterial cell wall thickness and decreased INH permeability (Nikaido 2001; Brennan 2003; Chuang et al. 2015). This indicated a molecular basis contributing to decreased permeability and intrinsic drug resistance.
Whole genome sequencing of M. tuberculosis revealed the presence of various efflux pumps that may enable the bacilli to evade the antimycobacterial killing action. Efflux pumps export various toxic compounds including antibiotics and metabolites, resulting in a decrease in intracellular concentration (Pages et al. 2005; Gupta et al. 2006). This phenomenon has been extensively studied in mycobacteria recently (Li et al. 2004; Morris et al. 2005; Buroni et al. 2006; Zechini and Versace 2009; Adams et al. 2011; Louw et al. 2011; Rodrigues et al. 2011, 2012; Balganesh et al. 2012; Hartkoorn et al. 2014).
In vivo and in vitro studies have revealed that antibiotic exposure of mycobacterial cells resulted in the significant upregulation of efflux pumps. It was shown that exposure to RIF resulted in an increase in expression of Rv1258c, which is a tap-like efflux pump (Adams et al. 2011, 2014), and arise in the RIF resistance level. Treatment with efflux pump inhibitors, verapamil, reserpine, and tetrandrine, along with RIF, INH, and EMB, could reverse the resistance phenotype of these anti-TB drugs (Adams et al. 2011, 2014; Louw et al. 2011). Studies have also shown that the exposure of M. tuberculosis to anti-TB drugs such as EMB, INH, RIF, OFL, STR, and PAS results in the upregulation of efflux pumps like drrA, drrB, efpA, mmr, jefA, Rv1634, whiB7, Rv1456c-Rv1457c-Rv1458c, Rv1258c, and pstB (Morris et al. 2005; Ramon-Garcia et al. 2012; Gupta et al. 2014; Hartkoorn et al. 2014; Garima et al. 2015; Li et al. 2015; Zhang et al. 2015c). The upregulation of the efflux pumps results in an MDR phenotype. Interestingly, the organosilicon compound, SILA-421 and thioradazine, both shown to have efflux pump inhibitory activity, demonstrated time- and concentration-dependent activity against M. tuberculosis as well as the enhanced killing of intracellular XDR-TB (Martins et al. 2009; Simons et al. 2013a; de Knegt et al. 2014, 2015). These compounds also enhanced the activity of INH and RIF in vitro and prevented the emergence of INH- and RIF-resistant mutants. However, they did not show in vivo activity enhancement of INH and RIF in M. tuberculosis-infected mice treated with INH-RIF-PZA for 13 weeks (de Knegt et al. 2014, 2015).
3.2 Production of Drug-Modifying and Inactivating Enzymes
M. smegmatis has been confirmed to be naturally resistant to RIF due the rifampin ADP-ribosyltransferase (Arr-ms), encoded by the chromosome, which assists in covalently adding a ribose group to RIF. This addition modifies and inactivates RIF, thus resulting in intrinsic resistance in M. smegmatis to RIF (Dabbs et al. 1995; Imai et al. 1999; Quan et al. 1997; Baysarowich et al. 2008).
The production of inactivating enzymes, e.g., the acetyltransferase AAC (2′)—Ic and the phosphotransferase encoded by the Rv3225c gene, APH (6)-la and APH (6)-ld from producer strain Streptomyces griseus, has been associated with STR resistance (Davies and Courvalin 1977; Davies and Wright 1997; Draker et al. 2003; Ashenafi et al. 2014). Similarly, the lack of antimicrobial activity in M. abscessus of aminoglycosides could be reversed by disruption of the chromosomally encoded aac(2′) gene (Maurer et al. 2014, 2015) (Fig. 2). By using M. smegmatis, it was shown that the activity of acetyltransferase was significantly induced in response to aminoglycoside, thereby resulting in the inhibition of protein synthesis.
4 Compensatory Mechanisms, Fitness, and Drug Resistance
Some resistance-causing mutations have been found to incur a fitness cost (Gagneux et al. 2006). The fitness cost may be compensated for by the acquisition of secondary mutations at a different site during the evolution of resistant bacteria (Bjorkman et al. 2000). The mutant carrying the chromosomal alteration can become extinct, or the mutations might be fixed in the population by means of compensatory evolution (Bottger and Springer 2008). These compensatory mechanisms can reduce the cost by restoring physiological functions impaired by the resistance mutations without altering the level of bacterial resistance (Schrag and Perrot 1996).
Recently, whole genome sequencing of RIF-resistant M. tuberculosis strains with rpoB mutations revealed novel mutations in rpoA and rpoC that emerged over time. Strains with these mutations exhibit high competitive fitness in vitro and in vivo and lead to MDR strains with high fitness (Comas et al. 2012). Previously, it was shown by in vitro pair-wise competition experiments that the wild-type rpoB M. tuberculosis strains outcompeted strains harboring the Ser522Leu, His526Tyr, and Ser531Trp mutations (Billington et al. 1999; Mariam et al. 2004). The extent of fitness loss was dependent on the specific rpoB mutation, with the Ser531Leu rpoB mutation only exhibiting a minor fitness defect compared to other mutations (Billington et al. 1999; Gagneux et al. 2006; Mariam et al. 2004). Additionally, mutations in rpoC illustrated that epistatic interactions between mutations that confer drug resistance, compensatory mutations, and diverse strain genetic background might influence compensatory evolution (de Vos et al. 2012).
In INH resistance, mutations in katG eliminate catalase-peroxidase activity, thereby preventing the activation of INH (Heym et al. 1999). It was shown that the expression of KatG or the alkyl hydroperoxidase, AhpC, exhibited a protective effect against organic peroxides in bacilli. The overexpression of AhpC, due to the presence of a mutation in ahpC, enabled INH-resistant katG mutants to survive during infection (Sherman et al. 1996).
These alternative mechanisms compensating for the loss of fitness caused by genetic mutations are difficult to detect using PCR-based methods as these methods only target mutation hotspots associated with drug resistance. Thus, it is imperative to also consider these compensatory mechanisms upon designing and developing new drugs and treatment regimens.
5 Perspectives
The history of TB drug development and use provides numerous examples of chromosomally encoded resistance, which often emerges very rapidly after the introduction of new drugs. This highlights the need for a diverse product portfolio entering the TB drug development pipeline. Fortunately, there are several promising new drugs at various stages within the TB drug development pipeline. These include bactericidal compounds in the benzothiazinone class, targeting the enzyme decaprenylphosphoryl-β-d-ribose 2′-oxidase (DprE1), which is essential for cell wall synthesis (Makarov et al. 2015). However, as with all TB drugs, there is a need for a better understanding of mechanisms of drug resistance and consequences of mutations that confer drug resistance. The emergence of compensatory mechanisms following the evolution of drug resistance-conferring mutations, after selective pressure, is an additional factor to consider upon rational drug design. Recently, bacterial collateral resistance and sensitivity to various combinations of anti-TB drugs have been reported. However, it is evident that the collateral sensitivity and resistance networks are complex, thereby complicating tailoring specific treatment regimens based on existing drug treatments. It would be desirable to explore alternative approaches to treatment, including the inclusion of efflux pump inhibitors or immunomodulators. Ideal treatment regimens would eliminate the formation of bacterial persisters, reduce the selection of resistant mutants, and ultimately offer a much-reduced treatment regime, to increase compliance.
References
Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L (2011) Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. doi:10.1016/j.cell.2011.02.022
Adams KN, Szumowski JD, Ramakrishnan L (2014) Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J Infect Dis 210:456–466. doi:10.1093/infdis/jiu095
Alangaden GJ, Manavathu EK, Vakulenko SB, Zvonok NM, Lerner SA (1995) Characterization of fluoroquinolone-resistant mutant strains of Mycobacterium tuberculosis selected in the laboratory and isolated from patients. Antimicrob Agents Chemother 39:1700–1703. doi:10.1128/AAC.39.8.1700
Ali A, Hasan Z, McNerney R, Mallard K, Hill-Cawthorne G, Coll F, Nair M, Pain A, Clark TG, Hasan R (2015) Whole genome sequencing based characterization of extensively drug-resistant Mycobacterium tuberculosis isolates from Pakistan. PLoS One 10, e0117771. doi:10.1371/journal.pone.0117771
Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi:10.1126/science.1106753
Andries K, Villellas C, Coeck N, Thys K, Gevers T, Vranckx L, Lounis N, de Jong BC, Koul A (2014) Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9, e102135. doi:10.1371/journal.pone.0102135
Ashenafi M, Ammosova T, Nekhai S, Byrnes WM (2014) Purification and characterization of aminoglycoside phosphotransferase APH(6)-Id, a streptomycin-inactivating enzyme. Mol Cell Biochem 387:207–216. doi:10.1007/s11010-013-1886-1
Aubry A, Veziris N, Cambau E, Truffot-Pernot C, Jarlier V, Fisher LM (2006) Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: functional analysis of mutant enzymes. Antimicrob Agents Chemother 50:104–112. doi:10.1128/AAC.50.1.104-112.2006
Avalos E, Catanzaro D, Catanzaro A, Ganiats T, Brodine S, Alcaraz J, Rodwell T (2015) Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: a systematic review. PLoS One 10, e0120470. doi:10.1371/journal.pone.0120470
Balganesh M, Dinesh N, Sharma S, Kuruppath S, Nair AV, Sharma U (2012) Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob Agents Chemother 56:2643–2651. doi:10.1128/AAC.06003-11
Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR Jr (1994) inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263:227–230. doi:10.1126/science.8284673
Barry PJ, O’Connor TM (2007) Novel agents in the management of Mycobacterium tuberculosis disease. Curr Med Chem 14:2000–2008. doi:10.2174/092986707781368496
Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, Locht C, Besra GS (2000) Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem 275:28326–28331
Baysarowich J, Koteva K, Hughes DW, Ejim L, Griffiths E, Zhang K, Junop M, Wright GD (2008) Rifamycin antibiotic resistance by ADP-ribosylation: structure and diversity of Arr. Proc Natl Acad Sci U S A 105:4886–4891. doi:10.1073/pnas.0711939105
Beckert P, Hillemann D, Kohl TA, Kalinowski J, Richter E, Niemann S, Feuerriegel S (2012) rplC T460C identified as a dominant mutation in linezolid-resistant Mycobacterium tuberculosis strains. Antimicrob Agents Chemother 56:2743–2745. doi:10.1128/AAC.06227-11
Belanger AE, Besra GS, Ford ME, Mikusova K, Belisle JT, Brennan PJ, Inamine JM (1996) The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci U S A 93:11919–11924
Billington OJ, McHugh TD, Gillespie SH (1999) Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother 43:1866–1869
Bjorkman J, Nagaev I, Berg OG, Hughes D, Andersson DI (2000) Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287:1479–1482. doi:10.1126/science.287.5457.1479
Bloemberg GV, Keller PM, Stuckia D, Trauner A, Borrell S, Latshang T, Coscolla M, Rothe T, Homke R, Ritter C, Feldmann J, Schulthess B, Gagneux S, Bottger EC (2015) Acquired resistance to bedaquiline and delamanid in therapy for tuberculosis. N Engl J Med 373:1986–1988. doi:10.1056/NEJMc1505196
Bodmer T, Zurcher G, Imboden P, Telenti A (1995) Mutation position and type of substitution in the beta-subunit of the RNA polymerase influence in-vitro activity of rifamycins in rifampicin-resistant Mycobacterium tuberculosis. J Antimicrob Chemother 35:345–348. doi:10.1093/jac/35.2.345
Boonaiam S, Chaiprasert A, Prammananan T, Leechawengwongs M (2010) Genotypic analysis of genes associated with isoniazid and ethionamide resistance in MDR-TB isolates from Thailand. Clin Microbiol Infect 16:396–399. doi:10.1111/j.1469-0691.2009.02838.x
Borges-Walmsley MI, McKeegan KS, Walmsley AR (2003) Structure and function of efflux pumps that confer resistance to drugs. Biochem J 376:313–338. doi:10.1042/bj20020957
Bottger EC, Springer B (2008) Tuberculosis: drug resistance, fitness, and strategies for global control. Eur J Pediatr 167:141–148
Brennan PJ (2003) Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb) 83:91–97
Brossier F, Veziris N, Truffot-Pernot C, Jarlier V, Sougakoff W (2011) Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 55:355–360. doi:10.1128/AAC.01030-10
Buroni S, Manina G, Guglierame P, Pasca MR, Riccardi G, De Rossi E (2006) LfrR is a repressor that regulates expression of the efflux pump LfrA in Mycobacterium smegmatis. Antimicrob Agents Chemother 50:4044–4052. doi:10.1128/AAC.00656-06
Caceres NE, Harris NB, Wellehan JF, Feng Z, Kapur V, Barletta RG (1997) Overexpression of the D-alanine racemase gene confers resistance to D-cycloserine in Mycobacterium smegmatis. J Bacteriol 179:5046–5055
Campbell PJ, Morlock GP, Sikes RD, Dalton TL, Metchock B, Starks AM, Hooks DP, Cowan LS, Plikaytis BB, Posey JE (2011) Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob Agents Chemother 55:2032–2041. doi:10.1128/AAC.01550-10
Chacon O, Feng Z, Harris NB, Caceres NE, Adams LG, Barletta RG (2002) Mycobacterium smegmatis D-Alanine Racemase mutants are not dependent on D-Alanine for growth. Antimicrob Agents Chemother 46:47–54. doi:10.1128/AAC.46.2.47-54.2002
Chakraborty S, Gruber T, Barry CE III, Boshoff HI, Rhee KY (2013) Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339:88–91. doi:10.1126/science.1228980
Chen JM, Uplekar S, Gordon SV, Cole ST (2012) A point mutation in cycA partially contributes to the D-cycloserine resistance trait of Mycobacterium bovis BCG vaccine strains. PLoS One 7, e43467. doi:10.1371/journal.pone.0043467
Chuang YM, Bandyopadhyay N, Rifat D, Rubin H, Bader JS, Karakousis PC (2015) Deficiency of the novel exopolyphosphatase Rv1026/PPX2 leads to metabolic downshift and altered cell wall permeability in Mycobacterium tuberculosis. mBio 6, e02428. doi:10.1128/mBio.02428-14
Cohen KA, Abeel T, Manson McGuire A, Desjardins CA, Munsamy V, Shea TP, Walker BJ, Bantubani N, Almeida DV, Alvarado L, Chapman SB, Mvelase NR, Duffy EY, Fitzgerald MG, Govender P, Gujja S, Hamilton S, Howarth C, Larimer JD, Maharaj K, Pearson MD, Priest ME, Zeng Q, Padayatchi N, Grosset J, Young SK, Wortman J, Mlisana KP, O’Donnell MR, Birren BW, Bishai WR, Pym AS, Earl AM (2015) Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med 12, e1001880. doi:10.1371/journal.pmed.1001880
Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S (2012) Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44:106–110. doi:10.1038/ng.1038
Cui Z, Li Y, Cheng S, Yang H, Lu J, Hu Z, Ge B (2014) Mutations in the embC-embA intergenic region contribute to Mycobacterium tuberculosis resistance to ethambutol. Antimicrob Agents Chemother 58:6837–6843. doi:10.1128/AAC.03285-14
Dabbs ER, Yazawa K, Mikami Y, Miyaji M, Morisaki N, Iwasaki S, Furihata K (1995) Ribosylation by mycobacterial strains as a new mechanism of rifampin inactivation. Antimicrob Agents Chemother 39:1007–1009
Davies J, Courvalin P (1977) Mechanisms of resistance to aminoglycosides. Am J Med 62:868–872
Davies J, Wright GD (1997) Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:234–240. doi:10.1016/S0966-842X(97)01033-0
de Knegt GJ, Ten Kate MT, van Soolingen D, Aarnoutse R, Boeree MJ, Bakker-Woudenberg IA, de Steenwinkel JE (2014) Enhancement of in vitro activity of tuberculosis drugs by addition of thioridazine is not reflected by improved in vivo therapeutic efficacy. Tuberculosis (Edinb). doi:10.1016/j.tube.2014.09.002
de Knegt GJ, Bakker-Woudenberg IA, van Soolingen D, Aarnoutse R, Boeree MJ, de Steenwinkel JE (2015) SILA-421 activity in vitro against rifampicin-susceptible and rifampicin-resistant Mycobacterium tuberculosis, and in vivo in a murine tuberculosis model. Int J Antimicrob Agents 46:66–72. doi:10.1016/j.ijantimicag.2015.02.025
de Vos M, Muller B, Borrell S, Black P, van Helden P, Warren R, Gagneux S, Victor T (2012) Putative compensatory mutations in the rpoC gene of rifampicin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother 57:827–832. doi:10.1128/AAC.01541-12
DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE III (2000) Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 97:9677–9682. doi:10.1073/pnas.97.17.9677
deJonge MR, Koymans LH, Guillemont JE, Koul A, Andries K (2007) A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910. Proteins 67:971–980. doi:10.1002/prot.21376
Diacon AH, Dawson R, Hanekom M, Narunsky K, Venter A, Hittel N, Geiter LJ, Wells CD, Paccaly AJ, Donald PR (2011) Early bactericidal activity of delamanid (OPC-67683) in smear-positive pulmonary tuberculosis patients. Int J Tuberc Lung Dis 15:949–954. doi:10.5588/ijtld.10.0616
Dillon NA, Peterson ND, Rosen BC, Baughn AD (2014) Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob Agents Chemother 58:7258–7263. doi:10.1128/AAC.04028-14
Draker KA, Boehr DD, Elowe NH, Noga TJ, Wright GD (2003) Functional annotation of putative aminoglycoside antibiotic modifying proteins in Mycobacterium tuberculosis H37Rv. J Antibiot (Tokyo) 56:135–142
Fajardo A, Martinez-Martin N, Mercadillo M, Galan JC, Ghysels B, Matthijs S, Cornelis P, Wiehlmann L, Tummler B, Baquero F, Martinez JL (2008) The neglected intrinsic resistome of bacterial pathogens. PLoS One 3, e1619. doi:10.1371/journal.pone.0001619
Feuerriegel S, Koser CU, Richter E, Niemann S (2013) Mycobacterium canettii is intrinsically resistant to both pyrazinamide and pyrazinoic acid. J Antimicrob Chemother 68:1439–1440. doi:10.1093/jac/dkt042
Fivian-Hughes AS, Houghton J, Davis EO (2012) Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology 158:308–318. doi:10.1099/mic.0.053983-0
Friedrich SO, Rachow A, Saathoff E, Singh K, Mangu CD, Dawson R, Phillips PP, Venter A, Bateson A, Boehme CC, Heinrich N, Hunt RD, Boeree MJ, Zumla A, McHugh TD, Gillespie SH, Diacon AH, Hoelscher M, Pan African Consortium for the Evaluation of Anti-tuberculosis A (2013) Assessment of the sensitivity and specificity of Xpert MTB/RIF assay as an early sputum biomarker of response to tuberculosis treatment. Lancet Respir Med 1:462–470. doi:10.1016/S2213-2600(13)70119-X
Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ (2006) The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946. doi:10.1126/science.1124410
Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G (2006) Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 368:1575–1580. doi:10.1016/S0140-6736(06)69573-1
Garima K, Pathak R, Tandon R, Rathor N, Sinha R, Bose M, Varma-Basil M (2015) Differential expression of efflux pump genes of Mycobacterium tuberculosis in response to varied subinhibitory concentrations of antituberculosis agents. Tuberculosis (Edinb) 95:155–161. doi:10.1016/j.tube.2015.01.005
Georghiou SB, Magana M, Garfein RS, Catanzaro DG, Catanzaro A, Rodwell TC (2012) Evaluation of genetic mutations associated with Mycobacterium tuberculosis resistance to amikacin, kanamycin and capreomycin: a systematic review. PLoS One 7, e33275. doi:10.1371/journal.pone.0033275
Ginsburg AS, Grosset JH, Bishai WR (2003) Fluoroquinolones, tuberculosis, and resistance. Lancet Infect Dis 3:432–442. doi:10.1016/S1473-3099(03)00671-6
Grzegorzewicz AE, Pham H, Gundi VA, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SE, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M (2012) Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol 8:334–341. doi:10.1038/nchembio.794
Gupta R, Espinal M, Stop TBWGoD-PfMDRTB (2003) A prioritised research agenda for DOTS-Plus for multidrug-resistant tuberculosis (MDR-TB). Int J Tuberc Lung Dis 7:410–414
Gupta AK, Chauhan DS, Srivastava K, Das R, Batra S, Mittal M, Goswami P, Singhal N, Sharma VD, Venkatesan K, Hasnain SE, Katoch VM (2006) Estimation of efflux mediated multi-drug resistance and its correlation with expression levels of two major efflux pumps in mycobacteria. J Commun Dis 38:246–254
Gupta S, Cohen KA, Winglee K, Maiga M, Diarra B, Bishai WR (2014) Efflux inhibition with verapamil potentiates bedaquiline in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:574–576. doi:10.1128/AAC.01462-13
Gurumurthy M, Rao M, Mukherjee T, Rao SP, Boshoff HI, Dick T, Barry CE III, Manjunatha UH (2013) A novel F(420)-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol 87:744–755. doi:10.1111/mmi.12127
Hartkoorn RC, Uplekar S, Cole ST (2014) Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2979–2981. doi:10.1128/AAC.00037-14
Haver HL, Chua A, Ghode P, Lakshminarayana SB, Singhal A, Mathema B, Wintjens R, Bifani P (2015) Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:5316–5323. doi:10.1128/AAC.00308-15
He L, Wang X, Cui P, Jin J, Chen J, Zhang W, Zhang Y (2015) ubiA (Rv3806c) encoding DPPR synthase involved in cell wall synthesis is associated with ethambutol resistance in Mycobacterium tuberculosis. Tuberculosis (Edinb) 95:149–154. doi:10.1016/j.tube.2014.12.002
Heep M, Brandstatter B, Rieger U, Lehn N, Richter E, Rusch-Gerdes S, Niemann S (2001) Frequency of rpoB mutations inside and outside the cluster I region in rifampin-resistant clinical Mycobacterium tuberculosis isolates. J Clin Microbiol 39:107–110. doi:10.1128/JCM.39.1.107-110.2001
Heym B, Saint-Joanis B, Cole ST (1999) The molecular basis of isoniazid resistance in Mycobacterium tuberculosis. Int J Tuberc Lung Dis 79:267–271
Hillemann D, Rusch-Gerdes S, Richter E (2008) In vitro-selected linezolid-resistant Mycobacterium tuberculosis mutants. Antimicrob Agents Chemother 52:800–801. doi:10.1128/AAC.01189-07
Hoffner S, Angeby K, Sturegard E, Jonsson B, Johansson A, Sellin M, Werngren J (2013) Proficiency of drug susceptibility testing of Mycobacterium tuberculosis against pyrazinamide: the Swedish experience. Int J Tuberc Lung Dis 17:1486–1490. doi:10.5588/ijtld.13.0195
Holtz TH (2007) XDR-TB in South Africa: revised definition. PLoS Med 4, e161. doi:10.1371/journal.pmed.0040161
Holtz TH, Cegielski JP (2007) Origin of the term XDR-TB. Eur Respir J 30:396. doi:10.1183/09031936.00042607
Huang H, Scherman MS, D’Haeze W, Vereecke D, Holsters M, Crick DC, McNeil MR (2005) Identification and active expression of the Mycobacterium tuberculosis gene encoding 5-phospho-{alpha}-d-ribose-1-diphosphate:decaprenyl-phosphate5-phosphoribosyltransferase, the first enzyme committed to decaprenylphosphoryl-d-arabinose synthesis. J Biol Chem 280:24539–24543. doi:10.1074/jbc.M504068200
Huitric E, Verhasselt P, Andries K, Hoffner SE (2007) In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrob Agents Chemother 51:4202–4204. doi:10.1128/AAC.00181-07
Huitric E, Verhasselt P, Koul A, Andries K, Hoffner S, Andersson DI (2010) Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob Agents Chemother 54:1022–1028. doi:10.1128/AAC.01611-09
Ibrahim M, Andries K, Lounis N, Chauffour A, Truffot-Pernot C, Jarlier V, Veziris N (2007) Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob Agents Chemother 51:1011–1015. doi:10.1128/AAC.00898-06
Imai T, Watanabe K, Mikami Y, Yazawa K, Ando A, Nagata Y, Morisaki N, Hashimoto Y, Furihata K, Dabbs ER (1999) Identification and characterization of a new intermediate in the ribosylative inactivation pathway of rifampin by Mycobacterium smegmatis. Microb Drug Resist 5:259–264. doi:10.1089/mdr.1999.5.259
Isaeva Y, Bukatina A, Krylova L, Nosova E, Makarova M, Moroz A (2013) Determination of critical concentrations of moxifloxacin and gatifloxacin for drug susceptibility testing of Mycobacterium tuberculosis in the BACTEC MGIT 960 system. J Antimicrob Chemother 68:2274–2281. doi:10.1093/jac/dkt202
Jia L, Tomaszewski JE, Hanrahan C, Coward L, Noker P, Gorman G, Nikonenko B, Protopopova M (2005) Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br J Pharmacol 144:80–87. doi:10.1038/sj.bjp.0705984
Johnson R, Jordaan AM, Pretorius L, Engelke E, van der Spuy G, Kewley C, Bosman M, van Helden PD, Warren R, Victor TC (2006a) Ethambutol resistance testing by mutation detection. Int J Tuberc Lung Dis 10:68–73
Johnson R, Streicher EM, Louw GE, Warren RM, van Helden PD, Victor TC (2006b) Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol 8:97–111
Kambli P, Ajbani K, Sadani M, Nikam C, Shetty A, Udwadia Z, Rodwell TC, Catanzaro A, Rodrigues C (2015) Correlating Minimum Inhibitory Concentrations of ofloxacin and moxifloxacin with gyrA mutations using the genotype MTBDRsl assay. Tuberculosis (Edinb) 95:137–141. doi:10.1016/j.tube.2014
Keshavjee S, Farmer PE (2012) Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med 367:931–936. doi:10.1056/NEJMra1205429
Klopper M, Warren RM, Hayes C, Gey van Pittius NC, Streicher EM, Muller B, Sirgel FA, Chabula-Nxiweni M, Hoosain E, Coetzee G, David van Helden P, Victor TC, Trollip AP (2013) Emergence and spread of extensively and totally drug-resistant tuberculosis, South Africa. Emerg Infect Dis 19:449–455. doi:10.3201/EID1903.120246
Konno K, Feldmann FM, McDermott W (1967) Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis 95:461–469
Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K (2007) Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324. doi:10.1038/nchembio884
Lee RE, Protopopova M, Crooks E, Slayden RA, Terrot M, Barry CE III (2003) Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J Comb Chem 5:172–187. doi:10.1021/cc020071p
Li XZ, Zhang L, Nikaido H (2004) Efflux pump-mediated intrinsic drug resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 48:2415–2423. doi:10.1128/AAC.48.7.2415-2423.2004
Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, Oldfield E (2014a) Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem 57:3126–3139. doi:10.1021/jm500131s
Li W, Upadhyay A, Fontes FL, North EJ, Wang Y, Crans DC, Grzegorzewicz AE, Jones V, Franzblau SG, Lee RE, Crick DC, Jackson M (2014b) Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:6413–6423. doi:10.1128/AAC.03229-14
Li G, Zhang J, Guo Q, Jiang Y, Wei J, Zhao LL, Zhao X, Lu J, Wan K (2015) Efflux pump gene expression in multidrug-resistant Mycobacterium tuberculosis clinical isolates. PLoS One 10, e0119013. doi:10.1371/journal.pone.0119013
Liu J, Takiff HE, Nikaido H (1996) Active efflux of fluoroquinolones in Mycobacterium smegmatis mediated by LfrA, a multidrug efflux pump. J Bacteriol 178:3791–3795
Lounis N, Veziris N, Chauffour A, Truffot-Pernot C, Andries K, Jarlier V (2006) Combinations of R207910 with drugs used to treat multidrug-resistant tuberculosis have the potential to shorten treatment duration. Antimicrob Agents Chemother 50:3543–3547. doi:10.1128/AAC.00766-06
Louw GE, Warren RM, Donald PR, Murray MB, Bosman M, Van Helden PD, Young DB, Victor TC (2006) Frequency and implications of pyrazinamide resistance in managing previously treated tuberculosis patients. Int J Tuberc Lung Dis 10:802–807
Louw GE, Warren RM, Gey van Pittius NC, McEvoy CR, Van Helden PD, Victor TC (2009) A balancing act: efflux/influx in mycobacterial drug resistance. Antimicrob Agents Chemother 53:3181–3189. doi:10.1128/AAC.01577-08
Louw GE, Warren RM, Gey van Pittius NC, Leon R, Jimenez A, Hernandez-Pando R, McEvoy CR, Grobbelaar M, Murray M, van Helden PD, Victor TC (2011) Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am J Respir Crit Care Med 184:269–276. doi:10.1164/rccm.201011-1924OC
Lu Y, Zheng M, Wang B, Fu L, Zhao W, Li P, Xu J, Zhu H, Jin H, Yin D, Huang H, Upton AM, Ma Z (2011) Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob Agents Chemother 55:5185–5193. doi:10.1128/AAC.00699-11
Makarov V, Neres J, Hartkoorn RC, Ryabova OB, Kazakova E, Sarkan M, Huszar S, Piton J, Kolly GS, Vocat A, Conroy TM, Mikusova K, Cole ST (2015) The 8-pyrrole-benzothiazinones are noncovalent inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:4446–4452. doi:10.1128/AAC.00778-15
Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE III (2006) Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103:431–436. doi:10.1073/pnas.0508392103
Manjunatha U, Boshoff HI, Barry CE (2009) The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun Integr Biol 2:215–218
Mariam DH, Mengistu Y, Hoffner SE, Andersson DI (2004) Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob Agents Chemother 48:1289–1294. doi:10.1128/AAC.48.4.1289-1294.2004
Marrakchi H, Laneelle G, Quemard A (2000) InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 146:289–296. doi:10.1099/00221287-146-2-289
Martins M, Viveiros M, Ramos J, Couto I, Molnar J, Boeree M, Amaral L (2009) SILA 421, an inhibitor of efflux pumps of cancer cells, enhances the killing of intracellular extensively drug-resistant tuberculosis (XDR-TB). Int J Antimicrob Agents 33:479–482. doi:10.1016/j.ijantimicag.2008.10.028
Maruri F, Sterling TR, Kaiga AW, Blackman A, van der Heijden YF, Mayer C, Cambau E, Aubry A (2012) A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J Antimicrob Chemother 67:819–831. doi:10.1093/jac/dkr566
Masjedi MR, Tabarsi P, Baghaei P, Jalali S, Farnia P, Chitsaz E, Amiri M, Mansouri D, Velayati AA (2010) Extensively drug-resistant tuberculosis treatment outcome in Iran: a case series of seven patients. Int J Infect Dis 14:e399–e402. doi:10.1371/journal.pmed.0030466
Maurer FP, Bruderer VL, Ritter C, Castelberg C, Bloemberg GV, Bottger EC (2014) Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob Agents Chemother 58:3828–3836. doi:10.1128/AAC.02448-14
Maurer FP, Bruderer VL, Castelberg C, Ritter C, Scherbakov D, Bloemberg GV, Bottger EC (2015) Aminoglycoside-modifying enzymes determine the innate susceptibility to aminoglycoside antibiotics in rapidly growing mycobacteria. J Antimicrob Chemother 70:1412–1419. doi:10.1093/jac/dku550
Maus CE, Plikaytis BB, Shinnick TM (2005a) Molecular analysis of cross-resistance to capreomycin, kanamycin, amikacin, and viomycin in Mycobacterium tuberculosis. Antimicrob Agents Chemother 49:3192–3197. doi:10.1128/AAC.49.8.3192-3197.2005
Maus CE, Plikaytis BB, Shinnick TM (2005b) Mutation of tlyA confers capreomycin resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 49:571–577. doi:10.1128/AAC.49.2.571-577.2005
McClure WR, Cech CL (1978) On the mechanism of rifampicin inhibition of RNA synthesis. J Biol Chem 253:8949–8956
Migliori GB, De Iaco G, Besozzi G, Centis R, Cirillo DM (2007a) First tuberculosis cases in Italy resistant to all tested drugs. Euro Surveill 12:E070517 070511
Migliori GB, Ortmann J, Girardi E, Besozzi G, Lange C, Cirillo DM, Ferrarese M, De Iaco G, Gori A, Raviglione MC, Group STS (2007b) Extensively drug-resistant tuberculosis, Italy and Germany. Emerg Infect Dis 13:780–782. doi:10.3201/eid1305.060200
Milano A, Pasca MR, Provvedi R, Lucarelli AP, Manina G, Ribeiro AL, Manganelli R, Riccardi G (2009) Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberculosis (Edinb) 89:84–90. doi:10.1016/j.tube.2008.08.003
Minato Y, Thiede JM, Kordus SL, McKlveen EJ, Turman BJ, Baughn AD (2015) Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob Agents Chemother 59:5097–5106. doi:10.1128/AAC.00647-15
Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC (2003) ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 47:3799–3805. doi:10.1128/AAC.47.12.3799-3805.2003
Morris RP, Nguyen L, Gatfield J, Visconti K, Nguyen K, Schnappinger D, Ehrt S, Liu Y, Heifets L, Pieters J, Schoolnik G, Thompson CJ (2005) Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 102:12200–12205. doi:10.1073/pnas.0505446102
Motiwala AS, Dai Y, Jones-Lopez EC, Hwang SH, Lee JS, Cho SN, Via LE, Barry CE, Alland D (2010) Mutations in extensively drug-resistant Mycobacterium tuberculosis that do not code for known drug-resistance mechanisms. J Infect Dis 201:881–888. doi:10.1086/650999
Mukherjee JS, Rich ML, Socci AR, Joseph JK, Viru FA, Shin SS, Furin JJ, Becerra MC, Barry DJ, Kim JY, Bayona J, Farmer P, Smith Fawzi MC, Seung KJ (2004) Programmes and principles in treatment of multidrug-resistant tuberculosis. Lancet 363:474–481. doi:10.1016/S0140-6736(04)15496-2
Nachega JB, Chaisson RE (2003) Tuberculosis drug resistance: a global threat. Clin Infect Dis 36(Suppl 1):S24–S30
Nikaido H (2001) Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol 12:215–223
Nikonenko BV, Protopopova M, Samala R, Einck L, Nacy CA (2007) Drug therapy of experimental tuberculosis (TB): improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob Agents Chemother 51:1563–1565. doi:10.1128/AAC.01326-06
Nosova EY, Bukatina AA, Isaeva YD, Makarova MV, Galkina KY, Moroz AM (2013) Analysis of mutations in the gyrA and gyrB genes and their association with the resistance of Mycobacterium tuberculosis to levofloxacin, moxifloxacin and gatifloxacin. J Med Microbiol 62:108–113. doi:10.1099/jmm.0.046821-0
Pages JM, Masi M, Barbe J (2005) Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol Med 11:382–389. doi:10.1016/j.molmed.2005.06.006
Parida SK, Axelsson-Robertson R, Rao MV, Singh N, Master I, Lutckii A, Keshavjee S, Andersson J, Zumla A, Maeurer M (2015) Totally drug-resistant tuberculosis and adjunct therapies. J Intern Med 277:388–405. doi:10.1111/joim.12264
Pasca MR, Guglierame P, Arcesi F, Bellinzoni M, De Rossi E, Riccardi G (2004) Rv2686c-Rv2687c-Rv2688c, an ABC fluoroquinolone efflux pump in Mycobacterium tuberculosis. Antimicrob Agents Chemother 48:3175–3178. doi:10.1128/AAC.48.8.3175-3178.2004
Perry CM, Jarvis B (2001) Linezolid: a review of its use in the management of serious gram-positive infections. Drugs 61:525–551
Petrella S, Cambau E, Chauffour A, Andries K, Jarlier V, Sougakoff W (2006) Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob Agents Chemother 50:2853–2856. doi:10.1128/AAC.00244-06
Plinke C, Cox HS, Zarkua N, Karimovich HA, Braker K, Diel R, Rusch-Gerdes S, Feuerriegel S, Niemann S (2010) embCAB sequence variation among ethambutol-resistant Mycobacterium tuberculosis isolates without embB306 mutation. J Antimicrob Chemother 65:1359–1367. doi:10.1093/jac/dkq120
Protopopova M, Hanrahan C, Nikonenko B, Samala R, Chen P, Gearhart J, Einck L, Nacy CA (2005) Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother 56:968–974. doi:10.1093/jac/dki319
Quan S, Venter H, Dabbs ER (1997) Ribosylative inactivation of rifampin by Mycobacterium smegmatis is a principal contributor to its low susceptibility to this antibiotic. Antimicrob Agents Chemother 41:2456–2460
Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs WR Jr, Blanchard JS (1995) Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 34:8235–8241
Ramaswamy S, Musser JM (1998) Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Int J Tuberc Lung Dis 79:3–29
Ramaswamy SV, Amin AG, Goksel S, Stager CE, Dou SJ, El Sahly H, Moghazeh SL, Kreiswirth BN, Musser JM (2000) Molecular genetic analysis of nucleotide polymorphisms associated with ethambutol resistance in human isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 44:326–336. doi:10.1128/AAC.44.2.326-336.2000
Ramaswamy SV, Reich R, Dou SJ, Jasperse L, Pan X, Wanger A, Quitugua T, Graviss EA (2003) Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 47:1241–1250. doi:10.1128/AAC.47.4.1241-1250.2003
Ramirez-Busby SM, Valafar F (2015) Systematic review of mutations in pyrazinamidase associated with pyrazinamide resistance in Mycobacterium tuberculosis clinical isolates. Antimicrob Agents Chemother 59:5267–5277. doi:10.1128/AAC.00204-15
Ramon-Garcia S, Mick V, Dainese E, Martin C, Thompson CJ, De Rossi E, Manganelli R, Ainsa JA (2012) Functional and genetic characterization of the tap efflux pump in Mycobacterium bovis BCG. Antimicrob Agents Chemother 56:2074–2083. doi:10.1128/AAC.05946-11
Rastogi N, Labrousse V, Goh KS (1996) In vitro activities of fourteen antimicrobial agents against drug susceptible and resistant clinical isolates of Mycobacterium tuberculosis and comparative intracellular activities against the virulent H37Rv strain in human macrophages. Curr Microbiol 33:167–175. doi:10.1007/s002849900095
Reddy VM, O’Sullivan JF, Gangadharam PR (1999) Antimycobacterial activities of riminophenazines. J Antimicrob Chemother 43:615–623. doi:10.1093/jac/43.5.615
Reeves AZ, Campbell PJ, Sultana R, Malik S, Murray M, Plikaytis BB, Shinnick TM, Posey JE (2013) Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7. Antimicrob Agents Chemother 57:1857–1865. doi:10.1128/AAC.02191-12
Rengarajan J, Sassetti CM, Naroditskaya V, Sloutsky A, Bloom BR, Rubin EJ (2004) The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria. Mol Microbiol 53:275–282. doi:10.1111/j.1365-2958.2004.04120.x
Richter E, Rusch-Gerdes S, Hillemann D (2007) First linezolid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1534–1536. doi:10.1128/AAC.01113-06
Rodrigues L, Ainsa JA, Amaral L, Viveiros M (2011) Inhibition of drug efflux in mycobacteria with phenothiazines and other putative efflux inhibitors. Recent Pat Antiinfect Drug Discov 6:118–127. doi:10.2174/157489111796064579
Rodrigues L, Machado D, Couto I, Amaral L, Viveiros M (2012) Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infect Genet Evol 12:695–700. doi:10.1016/j.meegid.2011.08.009
Rozwarski DA, Grant GA, Barton DH, Jacobs WR Jr, Sacchettini JC (1998) Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279:98–102. doi:10.1126/science.279.5347.98
Sacksteder KA, Protopopova M, Barry CE III, Andries K, Nacy CA (2012) Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol 7:823–837. doi:10.2217/fmb.12.56
Safi H, Lingaraju S, Amin A, Kim S, Jones M, Holmes M, McNeil M, Peterson SN, Chatterjee D, Fleischmann R, Alland D (2013) Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat Genet 45:1190–1197. doi:10.1038/ng.2743
Schrag SJ, Perrot V (1996) Reducing antibiotic resistance. Nature 381:120–121. doi:10.1038/381120b0
Seifert M, Catanzaro D, Catanzaro A, Rodwell TC (2015) Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: a systematic review. PLoS One 10, e0119628. doi:10.1371/journal.pone.0119628
Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE III, Stover CK (1996) Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272:1641–1643. doi:10.1126/science.272.5268.1641
Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE III, Wang H, Zhang W, Zhang Y (2011) Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. doi:10.1126/science.1208813
Shi W, Chen J, Feng J, Cui P, Zhang S, Weng X, Zhang W, Zhang Y (2014) Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg Microbes Infect 3, e58. doi:10.1038/emi.2014.61
Siddiqi S, Takhar P, Baldeviano C, Glover W, Zhang Y (2007) Isoniazid induces its own resistance in nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:2100–2104. doi:10.1128/AAC.00086-07
Simons SO, Kristiansen JE, Hajos G, van der Laan T, Molnar J, Boeree MJ, van Ingen J, Christensen JB, Viveiros M, Riedl Z, Amaral L, van Soolingen D (2013a) Activity of the efflux pump inhibitor SILA 421 against drug-resistant tuberculosis. Int J Antimicrob Agents 41:488–489. doi:10.1016/j.ijantimicag.2013.01.001
Simons SO, Mulder A, van Ingen J, Boeree MJ, van Soolingen D (2013b) Role of rpsA gene sequencing in diagnosis of pyrazinamide resistance. J Clin Microbiol 51:382. doi:10.1128/JCM.02739-12
Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE III (2008) PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322:1392–1395. doi:10.1126/science.1164571
Singh P, Jain A, Dixit P, Prakash S, Jaiswal I, Venkatesh V, Singh M (2015) Prevalence of gyrA and B gene mutations in fluoroquinolone-resistant and -sensitive clinical isolates of Mycobacterium tuberculosis and their relationship with MIC of ofloxacin. J Antibiot (Tokyo) 68:63–66. doi:10.1038/ja.2014.95
Siu GK, Zhang Y, Lau TC, Lau RW, Ho PL, Yew WW, Tsui SK, Cheng VC, Yuen KY, Yam WC (2011) Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 66:730–733. doi:10.1093/jac/dkq519
Sreevatsan S, Stockbauer KE, Pan X, Kreiswirth BN, Moghazeh SL, Jacobs WR Jr, Telenti A, Musser JM (1997) Ethambutol resistance in Mycobacterium tuberculosis: critical role of embB mutations. Antimicrob Agents Chemother 41:1677–1681
Steingart KR, Schiller I, Horne DJ, Pai M, Boehme CC, Dendukuri N (2014) Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev 1, CD009593. doi:10.1002/14651858.CD009593.pub3
Stottmeier KD, Beam RE, Kubica GP (1967) Determination of drug susceptibility of mycobacteria to pyrazinamide in 7H10 agar. Am Rev Respir Dis 96:1072–1075
Stover CK, Warrener P, Van Devanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405:962–966. doi:10.1038/35016103
Szumowski JD, Lynch JB (2015) Profile of delamanid for the treatment of multidrug-resistant tuberculosis. Drug Des Devel Ther 9:677–682. doi:10.2147/DDDT.S60923
Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE III, Boshoff HI (2012) SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:1797–1809. doi:10.1128/AAC.05708-11
Takayama K, Kilburn JO (1989) Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob Agents Chemother 33:1493–1499. doi:10.1128/AAC.33.9.1493
Takiff HE, Salazar L, Guerrero C, Philipp W, Huang WM, Kreiswirth B, Cole ST, Jacobs WR Jr, Telenti A (1994) Cloning and nucleotide sequence of Mycobacterium tuberculosisgyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob Agents Chemother 38:773–780. doi:10.1128/AAC.38.4.773
Takiff HE, Cimino M, Musso MC, Weisbrod T, Martinez R, Delgado MB, Salazar L, Bloom BR, Jacobs WR Jr (1996) Efflux pump of the proton antiporter family confers low-level fluoroquinolone resistance in Mycobacterium smegmatis. Proc Natl Acad Sci U S A 93:362–366
Tan Y, Hu Z, Zhang T, Cai X, Kuang H, Liu Y, Chen J, Yang F, Zhang K, Tan S, Zhao Y (2014) Role of pncA and rpsA gene sequencing in detection of pyrazinamide resistance in Mycobacterium tuberculosis isolates from southern China. J Clin Microbiol 52:291–297. doi:10.1128/JCM.01903-13
Telenti A, Imboden P, Marchesi F, Lowrie D, Cole S, Colston MJ, Matter L, Schopfer K, Bodmer T (1993) Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 341:647–650. doi:10.1016/0140-6736(93)90417-F
Udwadia ZF (2012) Totally drug-resistant tuberculosis in India: who let the djinn out? Respirology 17:741–742. doi:10.1111/j.1440-1843.2012.02192.x
Udwadia Z, Vendoti D (2013) Totally drug-resistant tuberculosis (TDR-TB) in India: every dark cloud has a silver lining. J Epidemiol Community Health 67:471–472. doi:10.1136/jech-2012-201640
Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C (2012) Totally drug-resistant tuberculosis in India. Clin Infect Dis 54:579–581. doi:10.1093/cid/cir889
Van Deun A, Maug AK, Salim MA, Das PK, Sarker MR, Daru P, Rieder HL (2010) Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med 182:684–692. doi:10.1164/rccm.201001-0077OC
Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, Ziazarifi AH, Hoffner SE (2009) Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 136:420–425. doi:10.1378/chest.08-2427
Vilcheze C, Jacobs WR Jr (2007) The mechanism of isoniazid killing: clarity through the scope of genetics. Annu Rev Microbiol 61:35–50. doi:10.1146/annurev.micro.61.111606.122346
Vilcheze C, Weisbrod TR, Chen B, Kremer L, Hazbon MH, Wang F, Alland D, Sacchettini JC, Jacobs WR Jr (2005) Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother 49:708–720. doi:10.1128/AAC.49.2.708-720.2005
Vilcheze C, Av-Gay Y, Attarian R, Liu Z, Hazbon MH, Colangeli R, Chen B, Liu W, Alland D, Sacchettini JC, Jacobs WR Jr (2008) Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol 69:1316–1329. doi:10.1111/j.1365-2958.2008.06365.x
Whitfield MG, Soeters HM, Warren RM, York T, Sampson SL, Streicher EM, van Helden PD, van Rie A (2015a) A global perspective on pyrazinamide resistance: systematic review and meta-analysis. PLoS One 10, e0133869. doi:10.1371/journal.pone.0133869
Whitfield MG, Warren RM, Streicher EM, Sampson SL, Sirgel FA, van Helden PD, Mercante A, Willby M, Hughes K, Birkness K, Morlock G, van Rie A, Posey JE (2015b) Mycobacterium tuberculosis pncA polymorphisms that do not confer pyrazinamide resistance at a breakpoint concentration of 100 Micrograms per Milliliter in MGIT. J Clin Microbiol 53:3633–3635. doi:10.1128/JCM.01001-15
WHO (1997) Treatment of tuberculosis: guidelines for national programmes. WHO/TB/97.220. Accessed 26 Oct 2015
WHO (2000) Guidelines for establishing DOTS-PLUS pilot projects for the management of multidrug-resitant tuberculosis (MDR-TB). WHO/CDS/TB/2000.279. Accessed 3 Nov 2015
WHO (2008) Management of MDR-TB: a field guide: a companion document to guidelines for programmatic management of drug-resistant TB: integrated management of adolescent and adult illness (IMAI). WHO/HTM/TB/2008.402a. Accessed 3 Nov 2015
WHO (2015) Global tuberculosis report. WHO/HTM/TB/2015.22. Accessed 5 Nov 2015
Willby M, Sikes RD, Malik S, Metchock B, Posey JE (2015) Correlation between GyrA substitutions and ofloxacin, levofloxacin, and moxifloxacin cross-resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:5427–5434. doi:10.1128/AAC.00662-15
Wolucka BA (2008) Biosynthesis of D-arabinose in mycobacteria – a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J 275:2691–2711. doi:10.1111/j.1742-4658.2008.06395.x
Xu Y, Jia H, Huang H, Sun Z, Zhang Z (2015) Mutations found in embCAB, embR, and ubiA genes of ethambutol-sensitive and -resistant Mycobacterium tuberculosis clinical isolates from China. Biomed Res Int 2015:951706. doi:10.1155/2015/951706
Yamada T, Mizugichi Y, Nierhaus KH, Wittmann HG (1978) Resistance to viomycin conferred by RNA of either ribosomal subunit. Nature 275:460–461. doi:10.1038/275460a0
Yano T, Kassovska-Bratinova S, Teh JS, Winkler J, Sullivan K, Isaacs A, Schechter NM, Rubin H (2011) Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem 286:10276–10287. doi:10.1074/jbc.M110.200501
Zainuddin ZF, Dale JW (1990) Does Mycobacterium tuberculosis have plasmids? Tubercle 71:43–49
Zaunbrecher MA, Sikes RD Jr, Metchock B, Shinnick TM, Posey JE (2009) Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 106:20004–20009. doi:10.1073/pnas.0907925106
Zechini B, Versace I (2009) Inhibitors of multidrug resistant efflux systems in bacteria. Recent Pat Antiinfect Drug Discov 4:37–50. doi:10.2174/157489109787236256
Zhang Y, Mitchison D (2003) The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis 7:6–21
Zhang Y, Yew WW (2009) Mechanisms of drug resistance in Mycobacterium tuberculosis. Int J Tuberc Lung Dis 13:1320–1330
Zhang S, Chen J, Shi W, Liu W, Zhang W, Zhang Y (2013) Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg Microbes Infect 2, e34. doi:10.1038/emi.2013.38
Zhang S, Chen J, Cui P, Shi W, Zhang W, Zhang Y (2015a) Identification of novel mutations associated with clofazimine resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 70:2507–2510. doi:10.1093/jac/dkv150
Zhang X, Liu L, Zhang Y, Dai G, Huang H, Jin Q (2015b) Genetic determinants involved in p-aminosalicylic acid resistance in clinical isolates from tuberculosis patients in northern China from 2006 to 2012. Antimicrob Agents Chemother 59:1320–1324. doi:10.1128/AAC.03695-14
Zhang Z, Yan J, Xu K, Ji Z, Li L (2015c) Tetrandrine reverses drug resistance in isoniazid and ethambutol dual drug-resistant Mycobacterium tuberculosis clinical isolates. BMC Infect Dis 15:153. doi:10.1186/s12879-015-0905-0
Zhao F, Wang XD, Erber LN, Luo M, Guo AZ, Yang SS, Gu J, Turman BJ, Gao YR, Li DF, Cui ZQ, Zhang ZP, Bi LJ, Baughn AD, Zhang XE, Deng JY (2014) Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:1479–1487. doi:10.1128/AAC.01775-13
Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, Jang J, Nam J, Dick T, Walker JR, Pethe K, Camacho LR (2013) para-Aminosalicylic acid is a prodrug targeting dihydrofolatereductase in Mycobacterium tuberculosis. J Biol Chem 288:23447–23456. doi:10.1074/jbc.M113.475798
Zhu M, Burman WJ, Starke JR, Stambaugh JJ, Steiner P, Bulpitt AE, Ashkin D, Auclair B, Berning SE, Jelliffe RW, Jaresko GS, Peloquin CA (2004) Pharmacokinetics of ethambutol in children and adults with tuberculosis. Int J Tuberc Lung Dis 8:1360–1367
Acknowledgements
This work was funded, in part, by the Intramural Research Program of the NIAID and by grants from the Foundation for the National Institutes of Health. SLS is funded by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (NRF) of South Africa, award number UID 86539. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NRF.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Louw, G.E., Sampson, S.L. (2017). Implications of Chromosomal Mutations for Mycobacterial Drug Resistance. In: Arora, G., Sajid, A., Kalia, V. (eds) Drug Resistance in Bacteria, Fungi, Malaria, and Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-48683-3_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-48683-3_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48682-6
Online ISBN: 978-3-319-48683-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)