
Abstract
Tuberculosis (TB) has become a curable disease, thanks to the discovery of antibiotics. However, it has remained one of the most difficult infections to treat. Most current TB regimens consist of 6–9 months of daily doses of four drugs that are highly toxic to patients. The purpose of these lengthy treatments is to completely eradicate Mycobacterium tuberculosis, notorious for its ability to resist most antibacterial agents, thereby preventing the formation of drug resistant mutants. On the contrary, the prolonged therapies have led to poor patient adherence. This, together with a severe limit of drug choices, has resulted in the emergence of strains that are increasingly resistant to the few available antibiotics. Here, we review our current understanding of molecular mechanisms underlying the profound drug resistance of M. tuberculosis. This knowledge is essential for the development of more effective antibiotics, which are not only potent against drug resistant M. tuberculosis strains but also help shorten the current treatment courses required for drug susceptible TB.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Drug Tolerance
- Tuberculosis Strain
- Mycobacterial Species
- Mycobacterial Cell Wall
- Bactericidal Antibiotic
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Tuberculosis (TB) has plagued humans since antiquity. During the pre-antibiotic era, patients diagnosed with this so-called “consumption” disease would have felt hopeless, much like how people feel about many cancers today. TB was treated with gold, arsenic, cod liver oil, herbs, bed rest, sunshine and fresh air, etc. (Birnbaum et al. 1891), but none of these therapies were really effective. It was therefore one of the most deadly diseases during this long period of time. The recent discovery of antibiotics, dubbed “magic bullets” because of their powerful potency against pathogenic bacteria, has brought about a real revolution in TB chemotherapy. Starting with streptomycin in 1943, series of potent TB drugs were introduced to clinical practice during this “golden age of antibiotics” (1940s–1960s). The implementation of these drugs to TB treatment immediately resulted in a sharp decline of TB incidence throughout the world. In fact, the euphoria of the great success brought a sense of total control over this disease. It was commonly thought that TB was no longer a public health concern in many developed countries. Some even went further to predict that TB would be soon eradicated from the world (Myers 1963).
Yet, the disease came back in the 1980s with outbreaks of multidrug resistant (MDR) strains, often associated with the spreading epidemic of the acquired immune deficiency syndrome (AIDS). It is currently estimated that M. tuberculosis is infecting around 2 billion people worldwide, equivalent to one-third of the world population (Corbett et al. 2003). Besides an ability to persist in the face of host defense mechanisms, allowing its wide spread, M. tuberculosis is capable of resistance to most antimicrobial agents available (Nguyen and Pieters 2009). As a result, the existing options for TB chemotherapy are severely restricted. The repeated use of the same drugs, together with prolonged regimens that often lead to poor patient compliance, has resulted in the emergence of strains that are increasingly resistant to the available drugs. From the first isolated strains that were resistant to single drugs, sequential accumulation of resistance mutations has led to the emergence of MDR, extensively drug resistant (XDR), and most recently totally drug resistant (TDR) M. tuberculosis strains (Ormerod 2005; Dorman and Chaisson 2007; Udwadia 2012). Infections with some of the latter strains are essentially incurable by the current TB drugs. Therefore, these resistant strains of M. tuberculosis pose a serious threat to worldwide TB control programs. To tackle the current epidemic of drug resistant TB, novel therapeutic interventions are urgently needed. Besides the efforts to develop completely new antibiotics that are not affected by the existing resistance mechanisms, other nontraditional approaches such as targeting resistance mechanisms or repurposing old drugs need to be further investigated. For these approaches to be successful, drug resistance mechanisms in M. tuberculosis should be thoroughly studied and well understood.
2 Acquired Antibiotic Resistance Mechanisms
Pathogenic bacteria including M. tuberculosis are able to acquire resistance to a particular antibiotic to which they were previously susceptible. The concept here referred to as “acquired antibiotic resistance” is in contrast with the intrinsic resistance discussed later in this chapter. Acquired resistances might occur through either mutation or horizontal gene transfer. In M. tuberculosis, horizontal transfer of resistance genes via plasmids or transposon elements has not been reported. By contrast, all currently known acquired resistances are mediated through chromosomal mutations that arise under selective pressure of antibiotic use. A summary of M. tuberculosis genes to which mutations confer TB drug resistance is presented in Table 1.
2.1 Genetic Mutation
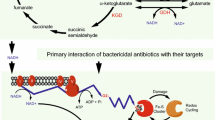
Darwin’s theory of evolution is perfectly epitomized by the progression of drug resistance in M. tuberculosis. Like any new traits arising during selective evolution, antibiotic resistant strains only become predominant in M. tuberculosis populations if the resistance phenotypes provide the mutants with survival advantages over their susceptible counterparts. The prolonged drug exposure due to lengthy regimens might have greatly contributed to the continued progression of the selective evolution of resistant strains that otherwise would hardly predominate the population because of their reduced fitness compared to susceptible strains. The ever-increasing drug use in response to growing TB incidences has resulted in a steady evolution of M. tuberculosis strains that are progressively resistant to the available drugs. Besides the matter of selective survival, recent studies show that exposure of bacterial cells to sublethal levels of bactericidal antibiotics promotes cellular mutagenesis leading to increased mutation rates in other drug resistance genes (Kohanski et al. 2010a). This phenomenon, possibly mediated through the increased production of reactive oxygen species triggered by bactericidal antibiotics (Kohanski et al. 2007), might play a key role in the rapid emergence of multidrug resistance phenotypes in pathogenic bacteria such as M. tuberculosis. In light of this knowledge, many current TB drugs might well represent this double-edged sword. Both isoniazid and ethionamide require activation by redox enzymes in the mycobacterial cytoplasm to become inhibitory. This process produces reactive oxygen and/or radicals that exert the mycobactericidal activity (Fig. 1) (Ito et al. 1992; Wang et al. 1998). But once a mutant survives the killing action of reactive oxygen and radicals, these same chemical matters would enhance its mutability leading to the acquisition of additional drug resistance mutations. Whether or not, and to what level, reactive oxygen and radical species contribute to the rise of resistance mutations remains to be understood in M. tuberculosis.

Possible interconnections of the TCA cycle, cellular redox homeostasis, and action of bactericidal antibiotics in M. tuberculosis. The primary interactions between bactericidal antibiotics and their targets trigger oxidation of NADH, produced in the TCA cycle, through the electron transport chain. This leads to increased production of superoxide that destroys iron–sulfur clusters yielding iron for oxidation of the Fenton reaction. The Fenton reaction results in the formation of hydroxyl radicals that damage nucleic acids, proteins, and lipids, eventually leading to cell death. However, if a cell survives, these hydroxyl radicals increase its mutagenesis thus promoting the emergence of MDR strains. The current TB drugs isoniazid (INH) and ethionamide (ETH) kill mycobacteria via direct conversion to free radicals that may contribute to the formation of MDR M. tuberculosis strains. The recently discovered MDR determinant PknG might regulate activity of many enzymes of the TCA cycle via its phosphorylation of GarA. These enzymes (KGD, GS, and GDH, in red) might affect the cellular NADH pool required for the downstream electron transport chain triggered by bactericidal antibiotics thus leading to cell death. MD: malate dehydrogenase, ICD: isocitrate dehydrogenase. Redrawn with modifications from (Kohanski et al. 2007)
2.1.1 New Insights into Pyrazinamide Action and Resistance
Pyrazinamide is a first-line TB antibiotic that is commonly used in combination with other drugs to shorten treatment regimens. Despite its widespread applications in TB chemotherapy, the precise mechanism underlying its action, as well as the mycobacterial mechanisms conferring pyrazinamide resistance had been elusive until recently. Similar to isoniazid and ethionamide, pyrazinamide is a prodrug and conversion to its active form pyrazinoic acid is catalyzed by the mycobacterial enzyme pyrazinamidase, encoded by the pncA gene (Scorpio and Zhang 1996). Loss of pyrazinamidase activity leads to pyrazinamide resistance, whereas overexpression confers increased susceptibility (Boshoff and Mizrahi 2000; Bamaga et al. 2002). Mapping of pyrazinamide resistance in M. tuberculosis clinical isolates found most mutations associated with pncA (Scorpio et al. 1997). However, there is a small subset of low-level pyrazinamide resistant strains that have no mutation in pncA, suggesting that these strains might carry mutations in genes encoding targets of pyrazinamide. In a search for M. tuberculosis proteins that bind the active molecule pyrazinoic acid, the 30S ribosomal protein S1 (RpsA) was identified (Shi et al. 2011). Overexpression of rpsA results in increased pyrazinamide resistance (Shi et al. 2011). Sequencing of a non-pncA pyrazinamide resistant strain revealed a 3-base pair in-frame deletion that leads to loss of Alanine 438 at the C terminus of RpsA (Shi et al. 2011). These observations suggested that RpsA is involved in M. tuberculosis pyrazinamide resistance.
Besides its essential ribosomal function required for protein translation, RpsA was previously found to be involved in trans-translation through its specific binding to transfer-messenger RNA (tmRNA) (Wower et al. 2000; Saguy et al. 2007). Trans-translation is a cellular process involved in rescuing ribosomes that are stalled during translation as well as in the degradation of the incomplete polypeptide chain and its messenger RNA (Keiler 2008). In this process, stalled mRNA is displaced by tmRNA that encodes a short peptide tagging the stalled protein for subsequent degradation.
In vitro binding experiments confirmed that pyrazinoic acid, but not pyrazinamide prevents interaction between RpsA and tmRNA (Shi et al. 2011). In addition, the 3-base pair deletion found in rpsA of the non-pncA pyrazinamide resistant strain mentioned above abolishes pyrazinoic acid binding (Shi et al. 2011). Also, pyrazinoic acid was shown to inhibit trans-translation of the reporter protein dihydrofolate reductase (Shi et al. 2011). These results suggested that the molecular target of pyrazinamide is the trans-translation process that might be essential for stress survival and recovery from nutrient starvation (Keiler 2008). Large-scale sequencing needs to be done to evaluate the significance of rpsA mutations in clinical pyrazinamide resistant M. tuberculosis strains.
2.2 Fitness Compensation and the Inevitable Evolution to the Totally Drug Resistant Phenotype
Acquired antibiotic resistance mechanisms are often associated with a reduced fitness of the resistant mutants (Andersson and Levin 1999; Andersson 2006; Andersson and Hughes 2010). This “fitness cost” affects the development, as well as the stability and domination of the resistance phenotypes. It has been generally observed that epidemics of drug resistant M. tuberculosis strains mainly circulate among HIV positive patients, raising the hope that these mutants might never spread widely among healthy people. However, first studies using mathematical modeling, as well as those carried out in other organisms, predict that the fitness cost of M. tuberculosis drug resistant strains could be reduced by the subsequent appearance of “compensatory mutations”, i.e., mutations that correct the fitness cost due to earlier mutations (Reynolds 2000; Andersson and Hughes 2010; Sergeev et al. 2012). This compensatory evolution, restoring fitness of resistant strains, might stabilize the epidemic of resistance phenotypes after they emerge. Indeed, in vitro studies suggest that resistant M. tuberculosis isolates are able to restore their fitness after prolonged exposure to antibiotics (Gillespie et al. 2002; Gagneux et al. 2006), although the identity of those mutations was not known. A recent study using whole-genome sequencing identified a set of compensatory mutations in the RNA polymerase genes of rifampicin resistant M. tuberculosis strains (Comas et al. 2012). In vitro growth competition assays confirmed that these mutations restore fitness of the rifampicin resistant M. tuberculosis strains compared to their susceptible counterparts. More convincingly, these mutations were mapped in 30 % of MDR M. tuberculosis strains isolated from regions of MDR TB prevalence (Comas et al. 2012).
TDR M. tuberculosis strains have been confirmed in India this year (Ormerod 2005; Dorman and Chaisson 2007; Udwadia 2012). Like MDR and XDR M. tuberculosis strains, the appearance of these TDR isolates is possibly inevitable with the increasing use of TB antibiotics. It is probably just a matter of time before these deadly bacteria appear more frequently in other places. The crucial question is whether compensatory evolution might allow stabilization of these drug resistance phenotypes, thus enhancing transmission rates of the resistant strains. The coevolution of drug resistance and fitness traits in M. tuberculosis therefore represents a deadly menace to humans.
3 Intrinsic Antibiotic Resistance Mechanisms
Besides the ability to acquire new resistance through the acquisition of chromosomal mutations, M. tuberculosis is endowed with an array of intrinsic resistance mechanisms that allow active neutralization of antibiotic actions. These resistance mechanisms provide a high resistance background that not only limits the application of available antibiotics to TB treatment, but also hampers the development of new drugs. M. tuberculosis intrinsic drug resistance can be divided into two categories: passive resistance and specialized resistance mechanisms.
3.1 Passive Resistance Mechanisms
Similar to the problems encountered in drug development and chemotherapy of Gram-negative bacteria, the impermeable cell wall of mycobacteria functions as an effective barrier for the penetration of antibiotics. Although mycobacteria are classified as Gram-positive bacteria, their cell wall is extremely thick and multilayered with varied hydrophobicity. These layers create an interlayer space similar to the periplasm of the Gram-negative bacterial cell wall (Hoffmann et al. 2008; Zuber et al. 2008). The peptidoglycan sacculus is covered by an arabinogalactan layer, both of which are hydrophilic, thus preventing the transport of hydrophobic molecules (Brennan and Nikaido 1995). These two layers are covalently linked to an outward layer of mycolic acids, long chain fatty acids that form a waxy, nonfluid barrier restricting penetration of both hydrophobic and hydrophilic molecules (Liu et al. 1995). For example, diffusion of β-lactams through the mycobacterial cell wall is hundreds of fold slower than the penetration through that of Escherichia coli (Kasik and Peacham 1968; Chambers et al. 1995).
The role of the mycobacterial cell wall in intrinsic antibiotic resistance is well demonstrated by the studies of mutants defective in cell wall biosynthesis. Pioneering work by the Nikaido laboratory identified a M. smegmatis mutant defective in mycolate biosynthesis. This mutant exhibited increased uptake of and sensitivity to erythromycin, chloramphenicol, novobiocin, and rifampicin (Liu and Nikaido 1999). Also, transposon mutagenesis studies confirmed the role of cell wall integrity in mycobacterial intrinsic drug resistance (Gao et al. 2003; Philalay et al. 2004). For example, transposon insertions in kasB or the virS-mymA operon (rv3082 to rv3089), genes that were defined to be involved in mycolic acid biosynthesis, lead to increased chemical penetration and sensitivity to various antibiotics (rifampicin, ciprofloxacin, isoniazid and pyrazinamide) (Gao et al. 2003; Singh et al. 2003, 2005). The ligation of mycolic acids to sugar moieties (arabinogalactan or trehalose) in the cell wall is catalyzed by a family of redundant mycolyltransferase enzymes initially known as “the antigen 85 complex” (Belisle et al. 1997). Deletion of the fbpA gene encoding one of the mycolyltransferase results in reduced levels of trehalose dimycolates and increased sensitivity to antibiotics widely used for antibacterial chemotherapy (Nguyen et al. 2005). These observations confirm that the mycobacterial cell wall plays an important role in mycobacterial intrinsic resistance against antibiotics. However, because the doubling time of M. tuberculosis is extremely long, the slow penetration rate might in some cases be high enough to allow antibiotics to accumulate to inhibitory levels well before cell division occurs, thus making cell wall permeability an important but not a decisive determinant of drug resistance (Brennan and Nikaido 1995; Chambers et al. 1995; Quinting et al. 1997).
Similar to the cell wall of Gram-negative bacteria, porins are mounted to outer layers of the mycobacterial cell wall, thus allowing import of nutrients and small molecules required for growth (Niederweis 2003). These porins might also play a role in the import of antibiotics through the outer layer of the mycobacterial cell wall (Danilchanka et al. 2008). In trans expression of the major porin MspA from M. smegmatis increases susceptibility of M. tuberculosis and M. bovis to β-lactams, isoniazid, ethambutol, and streptomycin (Stephan et al. 2004). By contrast, deletion of mspA or mspC in M. smegmatis leads to increased resistance to not only hydrophilic, but also hydrophobic and large antibiotics such as vancomycin, erythromycin, and rifampicin (Stephan et al. 2004; Danilchanka et al. 2008). Although M. tuberculosis encodes at least two porin-like proteins, OmpA and Rv1698 (Senaratne et al. 1998; Siroy et al. 2008), and in trans expression of rv1698 restores antibiotic susceptibility to a M. smegmatis mspA mutant (Siroy et al. 2008), the role of porins in antibiotic uptake and susceptibility has not been demonstrated directly in M. tuberculosis.
3.2 Specialized Resistance Mechanisms
Besides the cell wall barrier that helps slow down the penetration of antibiotics (Nikaido 1994), M. tuberculosis and other mycobacterial species operate multiple specialized resistance machineries that allow active detoxification of drugs once they reach the cytoplasmic space.
3.2.1 Modification of Drug Targets
Pathogenic bacteria are able to avoid antibacterial activity of antibiotics through structural modifications of their targets, thereby reducing antibiotic binding affinity. The mechanism that confers the intrinsic resistance of M. tuberculosis to macrolide and lincosamide antibiotics represents a typical example for this type of resistance. M. tuberculosis and other mycobacterial species are naturally resistant to macrolides and lincosamides. These antibiotics stop the growth of bacterial cells through inhibitory action on the protein synthetic machinery. They bind reversibly to a specific site of the ribosomal RNA in the 50S subunit of bacterial ribosomes, thus inhibiting translocation of peptidyl-tRNA (Buriankova et al. 2004). It has been observed that the Pasteur vaccine strain Bacillus of Calmette and Guérin (BCG) is uniquely susceptible to many macrolides and lincosamides, whereas its parental strain M. bovis and other vaccine strains remain resistant to the antibiotics. Through comparative genomics, it was realized that the sensitivity of BCG to macrolides and lincosamides is due to its chromosomal deletion of the erm37 gene encoding a ribosomal RNA methyltransferase (Buriankova et al. 2004). erm37 is located within a larger chromosomal locus known as Region of Difference 2 (RD2) which was deleted in BCG during its culture passage. Genetic experiments showed that the macrolide–lincosamide resistance of BCG could be restored to the level of its parental M. bovis strain by in trans expression of the erm37 gene from M. tuberculosis (Buriankova et al. 2004). Chemical analyses showed that this enzyme alters structures of the M. tuberculosis ribosomes via methylation of 23S ribosomal RNA (Madsen et al. 2005). In vitro macrolide binding assays also confirmed that Erm37 activity reduces affinity of macrolides to the ribosomes, thus lowering the inhibitory activity of macrolides on protein synthesis (Buriankova et al. 2004). Other erm genes conferring macrolide and lincosamide resistance were also identified in M. smegmatis and M. fortuitum (Nash 2003; Nash et al. 2005). Interestingly, expression of erm genes in mycobacteria is inducible by exposure to macrolides and lincosamide antibiotics (Nash 2003; Nash et al. 2005; Andini and Nash 2006), suggesting that their function is specialized for macrolide and lincosamide resistance. This inducible expression of Erm37 is most likely controlled by the MDR transcription regulator WhiB7 (Morris et al. 2005; Burian et al. 2012), which is discussed elsewhere in this chapter.
Another example demonstrating the ability of M. tuberculosis to neutralize drug action through enzymatic modification of its target is the methylation of ribosomal RNA that mediates resistance to cyclic peptide antibiotics. Capreomycin and viomycin are commonly used to treat MDR TB, but clinical strains resistant to these drugs have been isolated. Genetic studies carried out with M. smegmatis and M. tuberculosis found that mutations associated with capreomycin resistance are mapped to the tlyA gene that encodes a 2’-O-methyltransferase (Maus et al. 2005). TlyA methylates both 16S and 23S ribosomal RNA at nucleotide C1409 and C1920, respectively (Johansen et al. 2006). These methylations render mycobacterial ribosomes susceptible to the binding of capreomycin and viomycin (Maus et al. 2005; Johansen et al. 2006). Inactivation of tlyA led to increased resistance to these cyclic peptides, whereas in trans complementation restored capreomycin susceptibility (Maus et al. 2005).
3.2.2 Chemical Modification of Drugs
Mycobacteria are also able to inactivate antibiotics via direct chemical modifications. Recent studies have revealed the importance of acetylation on mycobacterial resistance to aminoglycosides. Aminoglycosides are broad-spectrum antibiotics that can act either as bactericidal or bacteriostatic drugs depending on concentration. This group of antibiotics constitutes an important position in the history of TB chemotherapy. Whereas streptomycin was the first effective remedy for TB, kanamycin and amikacin are currently used as last resorts to treat MDR TB cases. In addition, resistance to these drugs in MDR strains is the hallmark to define XDR TB. Aminoglycosides may have multiple potential mechanisms of action. Although some of them have been shown to act as inhibitors of protein biosynthesis, the precise mechanism of action of aminoglycosides remains to be understood. Early studies in M. smegmatis and M. fortuitum identified homologs of aminoglycoside 2’-N-acetyltransferase (aac) that confer resistance to gentamicin, dibekacin, tobramycin, and netilmicin (Ainsa et al. 1996). Although a homolog of AAC is apparently present in M. tuberculosis (Vetting et al. 2003), its function in aminoglycoside resistance has not been demonstrated. Surprisingly, the intrinsic resistance of M. tuberculosis to aminoglycosides has been recently attributed to a different acetyltransferase (Zaunbrecher et al. 2009). The protein termed Enhanced Intracellular Survival (EIS) was first discovered as a determinant of mycobacterial survival in host macrophages (Wei et al. 2000).
High-level kanamycin resistance in M. tuberculosis isolates has been mapped to the 16S ribosomal RNA gene rrs. However, the majority of isolates that display lower levels of kanamycin resistance have no mutation in rrs. To study mechanisms underlying kanamycin resistance in these isolates, a cosmid library was constructed using genomic DNA from one of these kanamycin resistant M. tuberculosis strains (Zaunbrecher et al. 2009). The cosmid library was transformed into a kanamycin susceptible M. tuberculosis strain and kanamycin resistant transformants were obtained. Further mapping of the cosmid, which conferred kanamycin resistance, identified mutations within the eis promoter region, which increased transcription of eis by 180-fold (Zaunbrecher et al. 2009). Importantly, these mutations were found in 80 % of low-level kanamycin resistant clinical isolates (Zaunbrecher et al. 2009; Campbell et al. 2011; Engstrom et al. 2011), as well as in MDR M. tuberculosis isolates (Huang et al. 2011). In addition, in vitro studies showed that Eis acetylates multiple amine groups of aminoglycosides using acetyl-coenzyme A as an acetyl donor (Chen et al. 2011), thereby inactivating the antibiotics. The dual function of EIS in the protection of M. tuberculosis against mycobactericidal mechanisms of both host immunity and antibiotics indicates a sinister coevolution of these two traits under the pressure of the recent antibiotic use.
3.2.3 Enzymatic Degradation of Drugs
Another pathway that pathogenic bacteria commonly use to subvert antibiotics is to directly degrade them using hydrolases. This mechanism is best studied in the case of β-lactams. These antibiotics bind and inhibit the activity of penicillin binding proteins (PBPs) involved in the assembly of the peptidoglycan network, thereby disrupting cell wall biosynthesis and leading to cell death. There are at least four major PBPs encoded in the genome of M. tuberculosis. These proteins bind β-lactams at clinically achievable concentrations (Chambers et al. 1995), indicating that target affinity is not an important determinant of β-lactam resistance in mycobacteria. The cell wall of mycobacterial species clearly contributes to the reduced accessibility of β-lactams to their targets. In this regard, the slow growth rate of M. tuberculosis serves both as a negative and positive factor in contributing to β-lactam resistance. For example, carbapenems are relatively unstable, and therefore lose activity much faster than the mycobacterial growth rate. However, daily uptake could lead to lethal concentrations that are sufficient to inhibit the slow cell division machinery of mycobacteria (Watt et al. 1992). This makes the cell wall barrier an important but not the principal determinant of β-lactam resistance.
β-lactamases, which hydrolyze the β-lactam ring of β-lactams, have been proven to provide the paramount β-lactam resistance in mycobacteria (Chambers et al. 1995). This conclusion is most supported by studies in M. fallax, the only Mycobacterium species highly susceptible to β-lactams (Kasik 1979; Quinting et al. 1997). Permeability assays first showed that β-lactam penetration through the cell wall of M. fallax is similar to those observed in other mycobacteria. This barrier apparently slows down accessibility of β-lactams to their targets, but half equilibration times should allow accumulation of the drugs at concentrations lethal to the bacterium (Quinting et al. 1997). However, when the β-lactamase from M. fortuitum was in trans expressed in M. fallax, its resistance was elevated to levels comparable to other mycobacterial species (Quinting et al. 1997). It is important to note that mycobacterial β-lactamases are generally considered less active than those of other pathogenic bacteria. However, the slow penetration of β-lactams across the thick cell wall of mycobacteria renders this low β-lactamase activity effective enough to protect mycobacteria from β-lactam action (Jarlier et al. 1991).
The most important β-lactamase in M. tuberculosis is BlaC, which belongs to the Ambler class-A β-lactamases (Voladri et al. 1998; Wang et al. 2006), whose enzymology and structures have been thoroughly characterized (Voladri et al. 1998; Wang et al. 2006). BlaC exhibits broad substrate specificity, possibly due to its large and flexible substrate-binding site (Wang et al. 2006). In addition, M. tuberculosis BlaC displays measurable activity to carbapenems, which are generally resistant to β-lactamases of other pathogenic bacteria (Hugonnet and Blanchard 2007; Tremblay et al. 2010). Also, β-lactamase inhibitors such as clavulanic acid are less effective against BlaC compared to other class A enzymes. Besides BlaC, M. tuberculosis encodes at least three more β-lactamase genes: blaS, rv0406c, and rv3677c, which were shown to provide M. tuberculosis with lower β-lactamase activities (Flores et al. 2005; Nampoothiri et al. 2008). Expression of BlaC in M. tuberculosis is inducible by β-lactams (Sala et al. 2009), indicating that this system has been specialized for β-lactam resistance. The induction of BlaC was recently shown to be mediated through BlaI, a winged helix regulator that functions as a transcriptional repressor of blaC expression. In the absence of β-lactams, BlaI forms homodimers that bind the promoter of the blaC gene, thus inhibiting blaC transcription (Sala et al. 2009). When M. tuberculosis is exposed to β-lactams, BlaI is dissociated from its DNA binding site, thereby derepressing the transcription of blaC that confers increased β-lactam resistance (Sala et al. 2009).
3.2.4 Molecular Mimicry of Drug Targets
Molecular mimicry represents a fascinating mechanism that M. tuberculosis might use to neutralize action of fluoroquinolones. These synthetic antibiotics have recently become important drugs for the treatment of drug resistant TB cases (Duncan and Barry 2004). Fluoroquinolones are bactericidal drugs that kill bacteria through inhibition of DNA replication, transcription, and repair. These antibiotics interact with DNA gyrase or topoisomerase in complexes with DNA, thereby stabilizing DNA breaks while inhibiting resealing of DNA strands. These events eventually result in DNA degradation and cell death (Andriole 2005).
Similar to other bacteria, acquired fluoroquinolone resistance in M. tuberculosis is commonly mapped to mutations in the genes that encode DNA gyrases, gyrA, and gyrB (Table 1). However, the molecular mechanisms responsible for the intrinsic fluoroquinolone resistance in mycobacteria have not been well understood. A M. smegmatis protein (MfpA) was first identified to confer low-level resistance to fluoroquinolones. Overexpression of mfpA from a multicopy plasmid results in increased resistance to ciprofloxacin and sparfloxacin in M. smegmatis and M. bovis (Montero et al. 2001). By contrast, deletion of mfpA leads to reduced fluoroquinolone resistance, indicating that the resistance level is dependent on mfpA expression. The sequence of MfpA has highest homology to pentapeptide repeat proteins in which every fifth amino acid is either leucine or phenylalanine. Interestingly, structure of the M. tuberculosis MfpA resembles the 3D structure of the DNA double helix (Ferber 2005; Hegde et al. 2005), with the tandem of pentapeptide repeats coiling around in a right-handed helix of the same width as DNA (Morais Cabral et al. 1997; Hegde et al. 2005). It was suggested that MfpA mimics DNA structure to sequester fluoroquinolones in the cytoplasm, thus setting DNA free from the drug attack (Ferber 2005). However, the physiological function of MfpA and how MfpA contributes to fluoroquinolone resistance in the clinic remain to be established. Nevertheless, this finding reveals a fascinating capability of pathogenic bacteria in evolving novel resistance mechanisms against antibiotics.
3.2.5 Drug Deportation By Efflux Pumps
An active mechanism that commonly provides protection against antibiotics in pathogenic bacteria is to expel the reagents using efflux pumps. Most of these membrane-spanning proteins play roles in the physiology or metabolism of bacteria; for example, transporting of nutrients, toxins, wastes, or signaling molecules through the cell wall. Therefore, the functions in antibiotic resistance of many transporters might be secondary and due to nonspecific transportation. Indeed, 20 out of the total of 36 genes encoding membrane transport proteins in the E. coli genome confer some levels of resistance to one or more antibiotics (Nishino and Yamaguchi 2001). However, these transporters are commonly controlled by regulatory systems that might have evolved to respond to antibiotics, thus making transport activities specialized for antibiotic resistance. For example, a major MDR determinant in E. coli is AcrB, a transporter of a broad substrate specificity. However, activity of AcrB has evolved as its expression is controlled by three antibiotic-responsive regulatory systems: Mar, Sox, and Rob (Alekshun and Levy 1997).
There are at least 18 transporters thus far characterized to be involved in antibiotic susceptibility of mycobacterial species (Viveirosa et al. 2012). For more specific information related to these transporters and their role in antibiotic resistance, readers are referred to many recent excellent reviews (da Silva et al. 2011; Viveirosa et al. 2012). Most of these transporters confer low-level antibiotic resistance to mycobacteria. Similar to E. coli, activities of some of these transporters are coupled to regulatory systems that are responsive to antibiotics. For example, two mycobacterial transporters, encoded by the iniBAC and efpA genes, are negatively controlled by the transcription regulator Lsr2 (Colangeli et al. 2007). Whereas IniBAC confers resistance to isoniazid and ethambutol, EfpA is rather a nonspecific MDR transporter (Colangeli et al. 2005, 2007). Importantly, the transcriptional control of IniBAC and EfpA by Lsr2 is inducible by isoniazid or ethambutol (Colangeli et al. 2007), indicating a functional evolution toward antibiotic resistance. Another example is the positive regulation of Tap, a transporter of aminoglycosides and tetracycline, by the MDR regulatory protein WhiB7 (Ainsa et al. 1998; Morris et al. 2005). Expression of WhiB7 and its regulon including Tap is induced when M. tuberculosis is exposed to tetracycline, streptomycin, or erythromycin (Morris et al. 2005; Burian et al. 2012). Interestingly, a recent study showed that expression of multiple efflux pumps including Tap is induced in mycobacteria residing within host granulomas, indicating that these molecular pumps may contribute to the drug tolerance of M. tuberculosis in latent TB (Adams et al. 2011).
3.3 Redox Homeostasis and the Mycobactericidal Activity of Drugs
Pioneering work in E. coli and Salmonella established the relationship of antibiotic resistance and oxidative stress responses (Demple 2005). These two phenotypes are controlled by common regulatory proteins such as SoxRS, MarRAB, or Rob. Although a clear reason for this regulatory overlap has not been established, it is conserved in both E. coli and Salmonella and believed to provide evolutionary advantages to these bacteria in the defense against general toxic environments (Demple 2005). Growing evidence recently revealed a tight relationship of killing mechanisms of antibiotics and cellular redox homeostasis in bacteria (Kohanski et al. 2007, 2010a, b), which could help explain the co-evolution of regulatory systems to control these two phenotypes. In E. coli, exposure to bactericidal antibiotics stimulates the production of hydroxyl radicals that ultimately lead to cell death (Kohanski et al. 2007). The terminal production of hydroxyl radicals triggered by bactericidal antibiotics is mediated through complex sequential events starting with the tricarboxylic acid (TCA) cycle. First, NADH produced in the TCA cycle is oxidized via complex electron transport chains, which results in the production of superoxide. Superoxide then damages iron–sulfur clusters, thereby donating ferrous iron to the oxidation of the Fenton reaction that produces hydroxyl radicals. Finally, hydroxyl radicals damage DNA, proteins, and lipids, resulting in cell death (Fig. 1) (Kohanski et al. 2007).
In mycobacteria, the relationship between oxidative stress responses and antibiotic resistance has also been implicated through the studies of the prodrugs isoniazid and ethionamide. Oxidative stress response proteins such as KatG and AhpC were found to be important for the oxidative activation of these antibiotics (Zhang et al. 1992, 1996; Sherman et al. 1996). In addition, expression of the stress responsive sigma factor F (SigF) is induced by antibiotics (Michele et al. 1999). Also, mycothiol, the actinobacterial specific thiol molecule involved in mycobacterial protection against oxygen toxicity, is also required for antibiotic resistance (Rawat et al. 2002; Buchmeier et al. 2003; Vilcheze et al. 2008). More recently, the relationship between redox homeostasis and antibiotic resistance has become clearer from the studies of the MDR systems Lsr2 and WhiB7 (Morris et al. 2005; Colangeli et al. 2009; Burian et al. 2012). Lsr2 was previously reported to control expression of the drug efflux pump IniBAC discussed above (Colangeli et al. 2005, 2007). Interestingly, a recent paper suggested that Lsr2 protects M. tuberculosis from reactive oxygen intermediates, possibly through its histone-like activity that protects DNA from the attacks of hydroxyl radicals (Colangeli et al. 2009). WhiB7 is a MDR determinant found only in actinobacteria including streptomycetes and mycobacteria. Deletion of whiB7 in mycobacteria leads to increased susceptibility to multiple antibiotics, whereas overexpression results in elevated resistance levels, indicating that WhiB7 activity is directly linked to the multidrug resistance phenotype (Morris et al. 2005). The M. tuberculosis WhiB7 protein is a 122-amio acid iron–sulfur cluster carrying protein. Transcriptomic analysis showed that WhiB7 affects transcription of multiple structural antibiotic resistance genes including eis, erm37, and tap that are discussed elsewhere in this chapter. Interestingly, expression of whiB7 could be induced by exposure to not only antibiotics such as erythromycin and tetracycline but also the reducing reagent dithiothreitol (Burian et al. 2012), indicating its function in antibiotic resistance is responsive to cellular redox fluctuations. Combination of antibiotics and dithiothreitol further elevates whiB7 expression. By contrast, an oxidative reagent diamide inhibits whiB7 induction (Burian et al. 2012). These experiments indicate the importance of a reductive environment for WhiB7 activity. Besides WhiB7 and Lsr2, another MDR protein whose function might relate to oxidative stress and the TCA cycle was recently reported in mycobacteria. The eukaryotic-type protein kinase G (PknG) is required for the intrinsic multidrug resistance in M. smegmatis and M. tuberculosis (Wolff et al. 2009). Either genetic deletion or chemical inhibition of PknG kinase activity results in increased susceptibility of mycobacteria to multiple antibiotics (Wolff et al. 2009). Interestingly, PknG was previously reported to mediate survival of pathogenic mycobacteria in host macrophages (Walburger et al. 2004). These findings indicate that traits related to antibiotic resistance and virulence might be interconnected in M. tuberculosis. Although the precise molecular mechanism underlying the function of PknG in mycobacterial antibiotic resistance has remained unknown, the possible involvement of this kinase in the control of the TCA cycle might provide an explanation (Fig. 1). Together with some other kinases, PknG was found to phosphorylate OdhI, an inhibitor of the 2-oxoglutarate dehydrogenase (ODH) complex of the TCA cycle, in Corynebacterium glutamicum (Niebisch et al. 2006). ODH catalyzes the NAD+-dependent conversion of 2-oxoglutarate (or α-ketoglutarate) to succinyl CoA. Phosphorylated OdhI is no longer able to inhibit ODH (Niebisch et al. 2006). In M. tuberculosis, the TCA cycle lacks ODH activity. Instead, α-ketoglutarate is first converted into succinic semialdehyde by α-ketoglutarate decarboxylase (KGD), before further conversion to succinate (Fig. 1) (Tian et al. 2005). The M. tuberculosis OdhI homolog, termed GarA, is also phosphorylated by multiple mycobacterial kinases including PknG. Unphosphorylated GarA was suggested to act as an inhibitor of KGD and NAD+-dependent glutamate dehydrogenase (GDH), yet as an enhancer of glutamate synthase (GS) activity (Nott et al. 2009; Chao et al. 2010). These enzymes are required for the interconversion of NADH and NAD+ (Fig. 1). Therefore, PknG kinase activity may affect the cellular NADH pool that is required for the superoxide production triggered by bactericidal antibiotics leading to eventual cell death (Kohanski et al. 2007, 2010b). It remains to be established whether this model is correct and how PknG affects this NADH-mediated mycobactericidal action of TB antibiotics.
4 Epigenetic Drug Tolerance
Besides the acquired and intrinsic resistance mechanisms that are defined by genetic determinants, epigenetic drug tolerance has been observed in M. tuberculosis, often associated with latent or relapsed TB cases. This phenotypic drug tolerance is associated with the phenomenon of persisters, a small subpopulation of cells with distinct but as-yet-unknown metabolic or physiological states. Persisters are genetically identical with susceptible counterparts and able to convert into susceptible cells upon restoration of normal environments (Lewis 2008; Zhang 2012).
Whereas drug resistance mechanisms result in reduced access of an antibiotic to its target, drug tolerance displayed by persisters might be associated with low metabolic activities or cell division during dormancy. This leads to a reduction of the cellular requirement for the proteins or machineries that are targeted by the antibiotic (Lewis 2008). Therefore, binding and inhibition of antibiotics to targets might occur normally, but the inhibition is no longer lethal to the dormant bacterium (Lewis 2008). In light of the recent knowledge of how bactericidal antibiotics kill bacterial cells (Kohanski et al. 2007, 2010b), inactive metabolic pathways such as a stalled TCA cycle might prevent cell death caused by those antibiotics. Once a persister re-enters active growth, cellular requirement for the targeted proteins is resumed, thus resensitizing the metabolically active cell to antibiotics. M. tuberculosis enters a dormant state during latent infection that is characterized by a shutdown of most of its metabolism, leading to increased antibiotic tolerance (Gomez and McKinney 2004; Gengenbacher and Kaufmann 2012). Indeed, latent infections possibly contain persister cells that are more difficult to treat than active TB normally caused by actively dividing bacilli (Gomez and McKinney 2004; Gengenbacher and Kaufmann 2012). M. tuberculosis isolates obtained from TB patients that had shown prolonged persistence or treatment relapse showed significantly increased tolerance to isoniazid, ethambutol, and rifampicin compared to isolates from regular patients, but this tolerance was strictly phenotypic (Wallis et al. 1999). Also, persister-like bacilli that replicate very slowly or completely stop growth have been isolated from TB sputum (Garton et al. 2008). Transcriptome analysis of these cells indicated that drug tolerance is due to low metabolic rates rather than resistance mutations (Garton et al. 2008).
A dormant-like state could be induced in vitro by mimicking environmental aspects of M. tuberculosis persisters surviving during latent TB. Probably, the best known of such methods is the Wayne model in which gradual oxygen depletion results in a stepwise transition to a dormant cell population that exhibits very low or absent metabolic activities and increased drug tolerance (Wayne and Hayes 1996). Another system uses nutrient starvation to obtain nonreplicating M. tuberculosis cells, which display down-regulated respiration and global metabolism but high levels of multidrug tolerance (Betts et al. 2002; Xie et al. 2005). In addition, exposure of M. tuberculosis to antibiotics such as D-cycloserine allows isolation of drug tolerant persisters whose transcriptome displays a downregulation of metabolic pathways characteristic of bacterial persisters (Keren et al. 2011). While all of these models share certain features, such as reduced intracellular ATP level and increased lipid metabolism, it is not surprising that other markers vary, possibly because of different inducing conditions (Keren et al. 2011; Gengenbacher and Kaufmann 2012). Nevertheless, these studies confirmed the relationship of phenotypic drug resistance and low metabolic activity during M. tuberculosis dormancy in the host.
When bacteria are growing in vitro in the absence of antibiotics, persisters can be steadily isolated with frequencies increasing in stationary phase (Keren et al. 2004, 2011; Hansen et al. 2008; Lewis 2008). Thus, persister formation is likely an intrinsic characteristic of a bacterial population that is related to community heterogeneity. It is thought that persisters function as cells that sacrifice active growth in order to ensure survival of the population in disastrous events such as antibiotic exposure (Keren et al. 2004; Lewis 2008). A recent study demonstrated culture heterogeneity in M. smegmatis arisen from asymmetric cell division and cell aging (Fig. 2) (Aldridge et al. 2012). After a cell division event, only one of the daughter cells contains the pre-existing division pole. and thus quickly progresses through another cycle of division, whereas the second daughter cell must first form a new growing pole of its own (Aldridge et al. 2012). The latter cell also elongates more slowly than the former, but it is more tolerant to antibiotics until the next division event (Fig. 2) (Aldridge et al. 2012). Distinct subpopulations of the diversified cultures thereby display varied levels of antibiotic susceptibility (Aldridge et al. 2012) that might be related to the formation of persisters triggered by antibiotic exposures.

Asymmetric cell division introduces population heterogeneity in growth rate and drug tolerance levels. A microfluidic chamber, which allows culturing mycobacterial cells for live-cell imaging, was developed. Growth from single cells (microcolonies) up to five generations were individually followed and analyzed. a Schematic model of growth and division starting from a single mycobacterial cell. Elongation occurs at one pole (red arrow) that is inherited by only one daughter cell, termed accelerator (Acc). The second daughter cell, or alternator (Alt), has to form a new pole before elongation and cell division can occur again. This requirement for the formation of a new growing pole results in a downshift of the cell’s elongation rate compared to accelerators that divide at unchanged speed due to the inherited pole. b The elongation rate of cells within 10 microcolonies is grouped dependent on cell pole ages (see a). Means of elongation rates are indicated by the larger ovals, distributions are indicated by smaller ovals. c and d Microcolonies in which alternators outnumber accelerators (green area, right) exhibit increased drug persistence compared to those in which accelerators dominate (orange area, left). This is likely due to decreased elongation rates and metabolic activity of alternator cells. Microcolonies were treated with either meropenem (c) or isoniazid (d) at concentrations lethal for actively growing cells. Survival was determined by measuring the number of cells in each microcolony that could regrow after the termination of antibiotic treatment. Reproduced with permission from (Aldridge et al. 2012)
The precise molecular mechanisms underlying the formation of drug tolerant persisters in vivo remain largely unknown. While it is extremely difficult to study the M. tuberculosis persisters residing in granulomas of human TB infections, animal studies and in vitro dormancy systems might help to advance our understanding. In the Cornell mouse model originally described in the 1950s, infected mice are treated with isoniazid and pyrazinamide until mice show no sign of active disease and no detectable bacilli by organ culture (McCune et al. 1956; McCune and Tompsett 1956). Reactivation of the disease occurs spontaneously following cessation of the treatment or triggered by immunosuppression, indicating the existence of drug tolerant persisters that are not cleared by the regimens (McCune et al. 1956; McCune and Tompsett 1956; Scanga et al. 1999). A similar mouse model infected with transposon insertion M. tuberculosis mutants was recently used to screen for mycobacterial genes involved with isoniazid persistence (Dhar and McKinney 2010). Using this animal infection model, genes required for isoniazid persistence have been identified. For example, cydC, encoding a putative ATP-binding subunit of an ABC transporter, which is upregulated during chronic infections in mice (Shi et al. 2005), was found to be required for survival in isoniazid treatment (Dhar and McKinney 2010). Importantly, survival of cydC mutant in the absence of isoniazid was identical to that of wild-type M. tuberculosis, indicating the function of the CydC is specialized for antibiotic persistence.
In other bacteria, toxin–antitoxin (TA) systems have been identified as the most obvious mechanism likely involved in persister formation. Interestingly, the M. tuberculosis genome encodes for over 65 such TA pairs, while E. coli encodes for roughly 20 (Keren et al. 2011). The principle behind these gene pairs consists of a sensitive, quickly modulated method to influence large numbers of genes and pathways by employing two components. While the “toxin” component inhibits critical cellular functions such as DNA replication or translation, thus leading to metabolic downshifts, the matching “antitoxin” neutralizes the activity of the toxin, thereby resulting in upshifts of metabolism (Lewis 2008). The production and degradation of antitoxin molecules therefore allows regulation of dormancy triggers. TA modules are thought to be redundant (Keren et al. 2004, 2011), and the high number contained by M. tuberculosis may indicate the great importance for TA-mediated persister formation during latent infection. In fact, many of the 65 TA pairs encoded in the M. tuberculosis genome have indeed been shown to play a role in drug tolerance and dormancy. When M. tuberculosis was exposed to D-cycloserine to induce persister formation, 10 TA modules were shown to be upregulated (Keren et al. 2004, 2011). Among those induced was rv2866 that encodes the toxin component of the TA pair Rv2865–Rv2866. Rv2866 is a homolog of the mRNA endonuclease RelE, which was shown to induce dormancy by shutting down translation in E. coli (Keren et al. 2004, 2011). Another study showed that VapC, the toxin component of the TA module VapC–VapBC, acts through specific binding and cleavage of RNA, thus downregulating metabolism (McKenzie et al. 2012; Sharp et al. 2012). Similarly, the toxin Rv1102c of the TA module Rv1102c–Rv1103c acts through its ribonuclease activity, which can be neutralized by Rv1103c that forms a complex with the toxin (Han et al. 2010). When Rv1102c was expressed without its antitoxin in M. smegmatis, growth was arrested and the formation of persisters tolerant to kanamycin and gentamycin was increased (Han et al. 2010).
Phosphates were recently implicated in M. tuberculosis drug persistence. Specifically, PhoY2, the M. tuberculosis homolog of E. coli PhoU that regulates the pst operon, acts as a negative regulator of phosphate uptake (Li and Zhang 2007; Shi and Zhang 2010). Deletion of phoY2 led to decreased tolerance of pyrazinamide and rifampicin in M. tuberculosis (Shi and Zhang 2010). How phosphate uptake directly impacts persister formation and drug tolerance remains largely unknown. However, recent studies suggested that inorganic polyphosphate might play a role in the transition from vegetative growth to dormancy and drug tolerance in M. tuberculosis (Thayil et al. 2011). Polyphosphate might act by inducing expression of the sigma factor RpoS that regulates expression of some 50 genes responsible for downregulation of metabolism and cell division (Shiba et al. 1997; Hengge-Aronis 2002).
In an interesting departure from the common theme of drug tolerance-metabolism correlation, a recent study suggested the involvement of efflux pumps in drug tolerance of M. tuberculosis (Adams et al. 2011). This study using an in vivo model of zebrafish infected with M. marinum followed by treatment with TB drugs reported the emergence and enrichment of a multidrug tolerant M. marinum population that was later disseminated into granulomas (Adams et al. 2011). Interestingly, drug tolerance in these bacilli could be reduced by co-treatment with efflux pump inhibitors, indicating that the observed drug tolerance was at least partially mediated by activity of efflux pumps (Adams et al. 2011).
5 Conclusions and Perspectives
It has only been less than 100 years since antibiotics were first used to treat bacterial infections. This time period is very short considering the pre-antibiotic era dated back thousands of years during which infectious diseases might have acted as selective forces of human evolution (Wang et al. 2012). For a long time, humans and bacteria had co-evolved in duels. However, everything has changed since antibiotics were discovered and applied in mass amounts to modern medicine: bacteria now must evolve under the additional selective pressure of these killing molecules. Evidence thus far indicates that pathogenic bacteria such as M. tuberculosis are well able to cope with this pressure and that they have evolved to become progressively resistant to antibiotics. Acquired resistances due to mutations in genes encoding target proteins or genes required for drug activities have allowed rapid evolution of mutants that become newly resistant to the antibiotics used. Accumulation of these resistance mutations has led to the emergence of M. tuberculosis strains that are more and more resistant to the available antibiotics. More alarmingly, recent studies indicate that these drug resistant mutants are able to evolve to regain fitness via compensatory mutations, thus enhancing their transmissibility and/or virulence. In addition to the acquired resistances caused by chromosomal mutations, M. tuberculosis is naturally resistant to most antibiotics. The profound intrinsic drug resistance in M. tuberculosis includes both passive and specialized mechanisms, the latter of which are able to respond to the presence of antibiotics. In fact, structural proteins capable of antibiotic resistance might have existed long before the clinical applications of these molecules (D’Costa et al. 2006, 2011). While these “drug resistance” proteins might still play roles in the physiology or metabolism of M. tuberculosis and other bacteria, inducible expression upon antibiotic exposure allows activities of these proteins important for antibiotic resistance. In this regard, the evolution of regulatory systems toward antibiotic responses might play a key role in specializing the function of those drug resistance systems. In addition to these two types of drug resistance, transition of M. tuberculosis from active growth into a dormant state confers increased phenotypic drug tolerance, which reflects the therapeutic recalcitrance of latent TB. These resistance and tolerance mechanisms hamper not only the clinical application of available antibiotics but also the development of new drugs. Nevertheless, understanding the resistance mechanisms could aid in the discovery of novel chemotherapeutic methods. For example, a recently emerging strategy termed “targeting resistance” uses inhibitors of resistance mechanisms to (re)sensitize resistant bacteria to the inactivated antibiotics. This strategy might help not only to recharge the inactivated drugs but also to synergize drugs that are less active because of intrinsic resistance mechanisms (Nguyen 2012).
References
Adams KN, Takaki K, Connolly LE et al (2011) Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53
Ainsa JA, Blokpoel MC, Otal I, Young DB, De Smet KA, Martin C (1998) Molecular cloning and characterization of Tap, a putative multidrug efflux pump present in Mycobacterium fortuitum and Mycobacterium tuberculosis. J Bacteriol 180:5836–5843
Ainsa JA, Martin C, Gicquel B, Gomez-Lus R (1996) Characterization of the chromosomal aminoglycoside 2’-N-acetyltransferase gene from Mycobacterium fortuitum. Antimicrob Agents Chemother 40:2350–2355
Aldridge BB, Fernandez-Suarez M, Heller D, Ambravaneswaran V, Irimia D, Toner M, Fortune SM (2012) Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335:100–104
Alekshun MN, Levy SB (1997) Regulation of chromosomally mediated multiple antibiotic resistance: the mar regulon. Antimicrob Agents Chemother 41:2067–2075
Andersson DI (2006) The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol 9:461–465
Andersson DI, Hughes D (2010) Antibiotic resistance and its cost: is it possible to reverse resistance? Nature reviews. Microbiology 8:260–271
Andersson DI, Levin BR (1999) The biological cost of antibiotic resistance. Curr Opin Microbiol 2:489–493
Andini N, Nash KA (2006) Intrinsic macrolide resistance of the Mycobacterium tuberculosis complex is inducible. Antimicrob Agents Chemother 50:2560–2562
Andriole VT (2005) The quinolones: past, present, and future. Clin Infect Dis 41(Suppl 2):S113–S119
Bamaga M, Wright DJ, Zhang H (2002) Selection of in vitro mutants of pyrazinamide-resistant Mycobacterium tuberculosis. Int J Antimicrob Agents 20:275–281
Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS (1997) Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276:1420–1422
Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K (2002) Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43:717–731
Birnbaum M, Koch R, Brendecke F (1891) Prof. Koch’s method to cure tuberculosis popularly treated. H.E. Haferkorn, Milwaukee
Boshoff HI, Mizrahi V (2000) Expression of Mycobacterium smegmatis pyrazinamidase in Mycobacterium tuberculosis confers hypersensitivity to pyrazinamide and related amides. J Bacteriol 182:5479–5485
Brennan PJ, Nikaido H (1995) The envelope of mycobacteria. Annu Rev Biochem 64:29–63
Buchmeier NA, Newton GL, Koledin T, Fahey RC (2003) Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol 47:1723–1732
Burian J, Ramon-Garcia S, Sweet G, Gomez-Velasco A, Av-Gay Y, Thompson CJ (2012) The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J Biol Chem 287:299–310
Buriankova K, Doucet-Populaire F, Dorson O, Gondran A, Ghnassia JC, Weiser J, Pernodet JL (2004) Molecular basis of intrinsic macrolide resistance in the Mycobacterium tuberculosis complex. Antimicrob Agents Chemother 48:143–150
Campbell PJ, Morlock GP, Sikes RD et al (2011) Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob Agents Chemother 55:2032–2041
Chambers HF, Moreau D, Yajko D et al (1995) Can penicillins and other beta-lactam antibiotics be used to treat tuberculosis? Antimicrob Agents Chemother 39:2620–2624
Chao J, Wong D, Zheng X, Poirier V, Bach H, Hmama Z, Av-Gay Y (2010) Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim Biophys Acta 1804:620–627
Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S (2011) Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci USA 108:9804–9808
Colangeli R, Haq A, Arcus VL et al (2009) The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc Natl Acad Sci USA 106:4414–4418
Colangeli R, Helb D, Sridharan S et al (2005) The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol Microbiol 55:1829–1840
Colangeli R, Helb D, Vilcheze C et al (2007) Transcriptional regulation of multi-drug tolerance and antibiotic-induced responses by the histone-like protein Lsr2 in M. tuberculosis. PLoS Pathog 3:e87
Comas I, Borrell S, Roetzer A et al (2012) Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44:106–110
Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, Dye C (2003) The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med 163:1009–1021
D’Costa VM, King CE, Kalan L et al (2011) Antibiotic resistance is ancient. Nature 477:457–461
D’Costa VM, McGrann KM, Hughes DW, Wright GD (2006) Sampling the antibiotic resistome. Science 311:374–377
da Silva PE, Von Groll A, Martin A, Palomino JC (2011) Efflux as a mechanism for drug resistance in Mycobacterium tuberculosis. FEMS Immunol Med Microbiol 63:1–9
Danilchanka O, Pavlenok M, Niederweis M (2008) Role of porins for uptake of antibiotics by Mycobacterium smegmatis. Antimicrobial Agents Chemother 52:3127–3134
Demple B (2005) The Nexus of oxidative stress responses and antibiotic resistance mechanisms in Escherichia coli and Salmonella. In: White DG, Alekshun MN, McDermott PF, Levy SB (eds) Frontiers in antimicrobial resistance : a tribute to Stuart B. Levy. American Society for Microbiology, Washington, DC, pp 191–197
Dhar N, McKinney JD (2010) Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid-treated mice. Proc National Acad Sci USA 107:12275–12280
Dorman SE, Chaisson RE (2007) From magic bullets back to the magic mountain: the rise of extensively drug-resistant tuberculosis. Nat Med 13:295–298
Duncan K, Barry CE 3rd (2004) Prospects for new antitubercular drugs. Curr Opin Microbiol 7:460–465
Engstrom A, Perskvist N, Werngren J, Hoffner SE, Jureen P (2011) Comparison of clinical isolates and in vitro selected mutants reveals that tlyA is not a sensitive genetic marker for capreomycin resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 66:1247–1254
Ferber D (2005) Biochemistry. Protein that mimics DNA helps tuberculosis bacteria resist antibiotics. Science 308:1393
Flores AR, Parsons LM, Pavelka MS Jr (2005) Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology 151:521–532
Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ (2006) The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946
Gao LY, Laval F, Lawson EH et al (2003) Requirement for kasB in Mycobacterium mycolic acid biosynthesis, cell wall impermeability and intracellular survival: implications for therapy. Mol Microbiol 49:1547–1563
Garton NJ, Waddell SJ, Sherratt AL et al (2008) Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS medicine 5:e75
Gengenbacher M, Kaufmann SH (2012) Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev 36:514–532
Gillespie SH, Billington OJ, Breathnach A, McHugh TD (2002) Multiple drug-resistant Mycobacterium tuberculosis: evidence for changing fitness following passage through human hosts. Microbial Drug Resist 8:273–279
Gomez JE, McKinney JD (2004) M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis 84:29–44
Han JS, Lee JJ, Anandan T et al (2010) Characterization of a chromosomal toxin-antitoxin, Rv1102c-Rv1103c system in Mycobacterium tuberculosis. Biochemical Biophys Res Commun 400:293–298
Hansen S, Lewis K, Vulic M (2008) Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrobial Agents Chemother 52:2718–2726
Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, Blanchard JS (2005) A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308:1480–1483
Hengge-Aronis R (2002) Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev: MMBR 66:373–395
Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H (2008) Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci USA 105:3963–3967
Huang WL, Chi TL, Wu MH, Jou R (2011) Performance assessment of the GenoType MTBDRsl test and DNA sequencing for detection of second-line and ethambutol drug resistance among patients infected with multidrug-resistant Mycobacterium tuberculosis. J Clin Microbiol 49:2502–2508
Hugonnet JE, Blanchard JS (2007) Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46:11998–12004
Ito K, Yamamoto K, Kawanishi S (1992) Manganese-mediated oxidative damage of cellular and isolated DNA by isoniazid and related hydrazines: non-Fenton-type hydroxyl radical formation. Biochemistry 31:11606–11613
Jarlier V, Gutmann L, Nikaido H (1991) Interplay of cell wall barrier and beta-lactamase activity determines high resistance to beta-lactam antibiotics in Mycobacterium chelonae. Antimicrob Agents Chemother 35:1937–1939
Johansen SK, Maus CE, Plikaytis BB, Douthwaite S (2006) Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2’-O-methylations in 16S and 23S rRNAs. Mol Cell 23:173–182
Kasik JE (1979) Mycobacterial Beta-Lactamases. In: Hamilton-Miller JMT, Smith JT (eds) Beta-Lactamases. Academic Press, London, p 500
Kasik JE, Peacham L (1968) Properties of beta-lactamases produced by three species of mycobacteria. Biochem J 107:675–682
Keiler KC (2008) Biology of trans-translation. Annual Rev Microbiol 62:133–151
Keren I, Minami S, Rubin E, Lewis K (2011) Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2:e00100-00111
Keren I, Shah D, Spoering A, Kaldalu N, Lewis K (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:8172–8180
Kohanski MA, DePristo MA, Collins JJ (2010a) Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37:311–320
Kohanski MA, Dwyer DJ, Collins JJ (2010b) How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810
Lewis K (2008) Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131
Li Y, Zhang Y (2007) PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrobial Agents Chemother 51:2092–2099
Liu J, Nikaido H (1999) A mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids accumulates meromycolates. Proc Natl Acad Sci U S A 96:4011–4016
Liu J, Rosenberg EY, Nikaido H (1995) Fluidity of the lipid domain of cell wall from Mycobacterium chelonae. Proc Natl Acad Sci USA 92:11254–11258
Madsen CT, Jakobsen L, Buriankova K, Doucet-Populaire F, Pernodet JL, Douthwaite S (2005) Methyltransferase Erm(37) slips on rRNA to confer atypical resistance in Mycobacterium tuberculosis. J Biol Chem 280:38942–38947
Maus CE, Plikaytis BB, Shinnick TM (2005) Mutation of tlyA confers capreomycin resistance in Mycobacterium tuberculosis. Antimicrobial Agents Chemother 49:571–577
McCune RM Jr, McDermott W, Tompsett R (1956) The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J Exp Med 104:763–802
McCune RM Jr, Tompsett R (1956) Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. I. The persistence of drug-susceptible tubercle bacilli in the tissues despite prolonged antimicrobial therapy. J Exp Med 104:737–762
McKenzie JL, Robson J, Berney M et al (2012) A VapBC toxin-antitoxin module is a posttranscriptional regulator of metabolic flux in mycobacteria. J Bacteriol 194:2189–2204
Michele TM, Ko C, Bishai WR (1999) Exposure to antibiotics induces expression of the Mycobacterium tuberculosis sigF gene: implications for chemotherapy against mycobacterial persistors. Antimicrobial Agents Chemother 43:218–225
Montero C, Mateu G, Rodriguez R, Takiff H (2001) Intrinsic resistance of Mycobacterium smegmatis to fluoroquinolones may be influenced by new pentapeptide protein MfpA. Antimicrob Agents Chemother 45:3387–3392
Morais Cabral JH, Jackson AP, Smith CV, Shikotra N, Maxwell A, Liddington RC (1997) Crystal structure of the breakage-reunion domain of DNA gyrase. Nature 388:903–906
Morris RP, Nguyen L, Gatfield J et al (2005) Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 102:12200–12205
Myers A (1963) Can tuberculosis be eradicated? Chest 43:327–329
Nampoothiri KM, Rubex R, Patel AK, Narayanan SS, Krishna S, Das SM, Pandey A (2008) Molecular cloning, overexpression and biochemical characterization of hypothetical beta-lactamases of Mycobacterium tuberculosis H37Rv. J Appl Microbiol 105:59–67
Nash KA (2003) Intrinsic macrolide resistance in Mycobacterium smegmatis is conferred by a novel erm gene, erm(38). Antimicrob Agents Chemother 47:3053–3060
Nash KA, Zhang Y, Brown-Elliott BA, Wallace RJ Jr (2005) Molecular basis of intrinsic macrolide resistance in clinical isolates of Mycobacterium fortuitum. J Antimicrob Chemother 55:170–177
Nguyen L (2012) Targeting antibiotic resistance mechanisms in Mycobacterium tuberculosis: recharging the old magic bullets. Expert Rev Anti Infect Ther 10:963–965
Nguyen L, Chinnapapagari S, Thompson CJ (2005) FbpA-Dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J Bacteriol 187:6603–6611
Nguyen L, Pieters J (2009) Mycobacterial subversion of chemotherapeutic reagents and host defense tactics: challenges in tuberculosis drug development. Ann Rev Pharmacol Toxicol 49:427–453
Niebisch A, Kabus A, Schultz C, Weil B, Bott M (2006) Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J Biol Chem 281:12300–12307
Niederweis M (2003) Mycobacterial porins-new channel proteins in unique outer membranes. Mol Microbiol 49:1167–1177
Nikaido H (1994) Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264:382–388
Nishino K, Yamaguchi A (2001) Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol 183:5803–5812
Nott TJ, Kelly G, Stach L, et al (2009) An intramolecular switch regulates phosphoindependent FHA domain interactions in Mycobacterium tuberculosis. Science Signal 2:ra12
Ormerod LP (2005) Multidrug-resistant tuberculosis (MDR-TB): epidemiology, prevention and treatment. Br Med Bull 73–74:17–24
Philalay JS, Palermo CO, Hauge KA, Rustad TR, Cangelosi GA (2004) Genes required for intrinsic multidrug resistance in Mycobacterium avium. Antimicrob Agents Chemother 48:3412–3418
Quinting B, Reyrat JM, Monnaie D et al (1997) Contribution of beta-lactamase production to the resistance of mycobacteria to beta-lactam antibiotics. FEBS Lett 406:275–278
Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y (2002) Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 46:3348–3355
Reynolds MG (2000) Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471–1481
Saguy M, Gillet R, Skorski P, Hermann-Le Denmat S, Felden B (2007) Ribosomal protein S1 influences trans-translation in vitro and in vivo. Nucleic Acids Res 35:2368–2376
Sala C, Haouz A, Saul FA, Miras I, Rosenkrands I, Alzari PM, Cole ST (2009) Genome-wide regulon and crystal structure of BlaI (Rv1846c) from Mycobacterium tuberculosis. Mol Microbiol 71:1102–1116
Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL (1999) Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect Immun 67:4531–4538
Scorpio A, Lindholm-Levy P, Heifets L, Gilman R, Siddiqi S, Cynamon M, Zhang Y (1997) Characterization of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrobial Agents Chemother 41:540–543
Scorpio A, Zhang Y (1996) Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med 2:662–667
Senaratne RH, Mobasheri H, Papavinasasundaram KG, Jenner P, Lea EJ, Draper P (1998) Expression of a gene for a porin-like protein of the OmpA family from Mycobacterium tuberculosis H37Rv. J Bacteriol 180:3541–3547
Sergeev R, Colijn C, Murray M, Cohen T (2012) Modeling the dynamic relationship between HIV and the risk of drug-resistant tuberculosis. Sci Transl Med 4:135ra167
Sharp JD, Cruz JW, Raman S, Inouye M, Husson RN, Woychik NA (2012) Growth and translation inhibition through sequence-specific RNA binding by Mycobacterium tuberculosis VapC toxin. J Biol Chem 287:12835–12847
Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE 3rd, Stover CK (1996) Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272:1641–1643
Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML (2005) Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci USA 102:15629–15634
Shi W, Zhang X, Jiang X et al (2011) Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632
Shi W, Zhang Y (2010) PhoY2 but not PhoY1 is the PhoU homologue involved in persisters in Mycobacterium tuberculosis. J Antimicrob Chemother 65:1237–1242
Shiba T, Tsutsumi K, Yano H et al (1997) Inorganic polyphosphate and the induction of rpoS expression. Proc Natl Acad Sci USA 94:11210–11215
Singh A, Gupta R, Vishwakarma RA, Narayanan PR, Paramasivan CN, Ramanathan VD, Tyagi AK (2005) Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J Bacteriol 187:4173–4186
Singh A, Jain S, Gupta S, Das T, Tyagi AK (2003) mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol Lett 227:53–63
Siroy A, Mailaender C, Harder D et al (2008) Rv1698 of Mycobacterium tuberculosis represents a new class of channel-forming outer membrane proteins. J Biol Chem 283:17827–17837
Stephan J, Mailaender C, Etienne G, Daffe M, Niederweis M (2004) Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob Agents Chemother 48:4163–4170
Thayil SM, Morrison N, Schechter N, Rubin H, Karakousis PC (2011) The role of the novel exopolyphosphatase MT0516 in Mycobacterium tuberculosis drug tolerance and persistence. PloS one 6:e28076
Tian J, Bryk R, Itoh M, Suematsu M, Nathan C (2005) Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of alpha-ketoglutarate decarboxylase. Proc Natl Acad Sci USA 102:10670–10675
Tremblay LW, Fan F, Blanchard JS (2010) Biochemical and structural characterization of Mycobacterium tuberculosis beta-lactamase with the carbapenems ertapenem and doripenem. Biochemistry 49:3766–3773
Udwadia ZF (2012) MDR, XDR, TDR tuberculosis: ominous progression. Thorax 67:286–288
Vetting M, Roderick SL, Hegde S, Magnet S, Blanchard JS (2003) What can structure tell us about in vivo function? The case of aminoglycoside-resistance genes. Biochem Soc Trans 31:520–522
Vilcheze C, Av-Gay Y, Attarian R et al (2008) Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Molec Microbiol 69:1316–1329
Viveirosa M, Martins M, Rodrigues L, Machado D, Coutoa I, Ainsad J, Amaral L (2012) Inhibitors of mycobacterial efflux pumps as potential boosters for TB drugs. Expert Rev Anti Infect Ther 10:983–998
Voladri RK, Lakey DL, Hennigan SH, Menzies BE, Edwards KM, Kernodle DS (1998) Recombinant expression and characterization of the major beta-lactamase of Mycobacterium tuberculosis. Antimicrob Agents Chemother 42:1375–1381
Walburger A, Koul A, Ferrari G et al (2004) Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304:1800–1804
Wallis RS, Patil S, Cheon SH et al (1999) Drug tolerance in Mycobacterium tuberculosis. Antimicrobial Agents Chemother 43:2600–2606
Wang F, Cassidy C, Sacchettini JC (2006) Crystal structure and activity studies of the Mycobacterium tuberculosis beta-lactamase reveal its critical role in resistance to beta-lactam antibiotics. Antimicrob Agents Chemother 50:2762–2771
Wang JY, Burger RM, Drlica K (1998) Role of superoxide in catalase-peroxidase-mediated isoniazid action against mycobacteria. Antimicrobial Agents Chemother 42:709–711
Wang X, Mitra N, Secundino I et al (2012) Specific inactivation of two immunomodulatory SIGLEC genes during human evolution. Proc Natl Acad Sci USA 109:9935–9940
Watt B, Edwards JR, Rayner A, Grindey AJ, Harris G (1992) In vitro activity of meropenem and imipenem against mycobacteria: development of a daily antibiotic dosing schedule. Tuber Lung Dis 73:134–136
Wayne LG, Hayes LG (1996) An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64:2062–2069
Wei J, Dahl JL, Moulder JW, Roberts EA, O’Gaora P, Young DB, Friedman RL (2000) Identification of a Mycobacterium tuberculosis gene that enhances mycobacterial survival in macrophages. J Bacteriol 182:377–384
Wolff KA, Nguyen HT, Cartabuke RH, Singh A, Ogwang S, Nguyen L (2009) Protein kinase G is required for intrinsic antibiotic resistance in mycobacteria. Antimicrobial Agents Chemother 53:3515–3519
Wower IK, Zwieb CW, Guven SA, Wower J (2000) Binding and cross-linking of tmRNA to ribosomal protein S1, on and off the Escherichia coli ribosome. EMBO J 19:6612–6621
Xie Z, Siddiqi N, Rubin EJ (2005) Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrobial Agents Chemother 49:4778–4780
Zaunbrecher MA, Sikes RD Jr, Metchock B, Shinnick TM, Posey JE (2009) Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 106:20004–20009
Zhang Y (2012) Drug resistant and persistent tuberculosis: mechanisms and drug development. In: Dougherty TJ, Pucci MJ (ed) Antibiotic discovery and development. Springer Science + Business Media, pp 719–746
Zhang Y, Dhandayuthapani S, Deretic V (1996) Molecular basis for the exquisite sensitivity of Mycobacterium tuberculosis to isoniazid. Proc Natl Acad Sci USA 93:13212–13216
Zhang Y, Heym B, Allen B, Young D, Cole S (1992) The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358:591–593
Zuber B, Chami M, Houssin C, Dubochet J, Griffiths G, Daffe M (2008) Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J Bacteriol 190:5672–5680
Acknowledgments
Work in the Nguyen laboratory is supported by NIH Grant R01 AI087903.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Smith, T., Wolff, K.A., Nguyen, L. (2012). Molecular Biology of Drug Resistance in Mycobacterium tuberculosis . In: Pieters, J., McKinney, J. (eds) Pathogenesis of Mycobacterium tuberculosis and its Interaction with the Host Organism. Current Topics in Microbiology and Immunology, vol 374. Springer, Berlin, Heidelberg. https://doi.org/10.1007/82_2012_279
Download citation
DOI: https://doi.org/10.1007/82_2012_279
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40231-9
Online ISBN: 978-3-642-40232-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)