
Abstract
Radiation therapy (RT) has historically been the most common approach used to achieve local tumor control in cancer patients. However, emerging evidences over the last decades suggest an important role for RT in modulating or amplifying the antitumor immune response upon induction of cancer cell death through its direct cytocidal effect. RT alters multiple components of the tumor microenvironment which affect both the immune cell phenotype and function as well as the interactions between tumor and the immune system. Despite the documented immunostimulatory effects, RT alone rarely induces effective antitumor immunity resulting in systemic tumor rejection. RT can also reinforce immunosuppressive mechanisms within the tumor microenvironment, which negatively impacts on the tumor response to RT. Preclinical and clinical data show that combination RT and immunotherapy can elicit powerful antitumor efficacy through either strengthening the immune activation or counteracting immune suppression. In this review, we summarize the immunological changes in the tumor microenvironment upon exposure to radiation. We also highlight radiation triggered molecular and cellular pathways that may contribute to immune evasion and tumor recurrence. Rational and optimized combination of RT and immunotherapy to achieve synergistic antitumor activities for systemic eradication of cancer cells and development of durable antitumor immunity will also be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Introduction
Radiation therapy (RT) is a well-established conventional cancer treatment modality that is administrated up to 50 % of the cancer patients [1]. Tumorical effect of RT lies in its ability to cause DNA damage in irradiated cancer cells. RT is frequently used to achieve local or regional control of cancers either alone or in combination with other treatments, e.g., surgery or chemotherapy. Although RT has been well recognized for its direct cytotoxic and cytostatic effect on neoplastic cells, it is increasingly clear that the immune system also has a major role in the long-term control of tumor growth by RT [2, 3]. This may provide an immunological basis for the abscopal effect of RT [4]. The mechanisms underlying RT-induced antitumor immune responses are complex and involve an active interaction between irradiated cancer cells and the tumor stroma. Historically the tumor cell itself has been the focus to improve the outcomes of RT, while the interplay between the tumor cells and tumor microenvironment (TME) were largely ignored.
The immune compartment within the tumor stroma is primarily constituted of resident or recruited leukocytes with both lymphoid and myeloid origins. Depending on their phenotype and activation state, these immune cells can either promote or suppress tumor progression as well as modulate the therapeutic response to anticancer treatments, including RT [5–7]. Although RT transiently depletes resident leukocytes via direct cytocidal activity, the rebound effects of immune cells following RT are known to impact on tumor response. In this review, we describe immunological changes in the TME following RT and discuss how this immune profile alteration may promote radio-resistance and tumor recurrence. We will reveal the capability of RT to provoke or modulate an immune response and the immune system’s role in regulating the local or regional effects of RT implicate the potential rationale of combination RT with immunotherapy. Lastly, we will discuss how RT may exploited to counteract tumor-mediated immune evasion and how immunotherapy can be integrated into RT regimen to achieve improved treatment outcome by promoting immune activation and/or overcoming tumor-associated immune suppression.
7.2 Immune Stimulatory Effects of RT
7.2.1 Induction of Immune Stimulatory Factors by RT
RT can directly stimulate production of immunostimulatory cytokines and chemokines from both tumor cells and tumor stroma. Tumor necrosis factor-α (TNF-α), which can enhance the radiation lethality of tumor cells upon treatment with X-rays in human sarcoma cells [8]. Irradiation of B16 mouse melanoma tumors induces production of interferon-γ (IFN-γ), which can act directly on cancer cells to induce upregulation of surface the major histocompatibility complex class I (MHC-I), an antigen-presenting molecule critical for T cell mediated recognition and elimination of cancer cells [9, 10]. Clinically, serum IFN-γ levels were found to increase in a dose-dependent fashion in the 56 of 63 patients with esophageal squamous cell carcinoma who were treated with RT alone. The remaining 7 patients whose IFN-γ levels remained unchanged in response to RT developed local recurrence despite radiation [11]. Type I interferons (IFN-α and IFN-β) not only play important roles in the immune responses to viral infection, but are also actively involved in anti-tumor immunity [12]. RT can induce production of Type I interferons through activation of intracellular DNA sensors, such as the STING-dependent pathway [13, 14]. Induction of type I interferon within the TME is required for generation of type I interferon-dependent innate and adaptive antitumor immunity by potentiating the cross-priming capacity of tumor-infiltrating dendritic cells (DCs) as well as recruitment and effector function of CD8+ T effector cells [15, 16]. It is demonstrated that RT substantially enhances the secretion of the chemokine (C-X-C motif) ligand 16 (CXCL16) by mouse and human breast cancer cells. Upon binding to its receptor CXCR6 on T helper cells (Th1 cells) or activated CD8+ effector T cells, CXCL16 plays an important role in recruiting antitumor immune effector cells [17]. Therefore, it is suggested that in addition to the direct cytotoxic effects, RT can exert its immune stimulating effects through triggering the production of immune activating cytokines or chemokines, which might be additive to radiation lethality through autocrine and paracrine mechanisms.
7.2.2 RT-Increased Antigen Presentation Within the TME
Activation of naïve T cells requires both the recognition of antigen-MHC complexes by the T cell antigen receptor and additional costimulatory signals, including B7 molecules (CD80 and CD86) on the antigen-presenting cells (APCs) [18, 19]. Killing of cancer cells and the subsequent inflammatory responses can make the tumors visible to the immune system if released tumor antigens are taken up by DCs and presented to T cells along with effective co-stimulation signals [20]. RT can induce extensive immunogenic alterations of dying and surviving tumor cells within the TME. The resulting stress and death of tumor cells could activate tumor-specific immune responses through the liberation of damage-associated molecular patterns (DAMPs) upon binding to their corresponding pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) on APCs [21]. It was recently reported that RT triggered immunogenic cell death (ICD), which is defined by translocation of stress protein calreticulin from the endoplasmic reticulum (ER) to the tumor cell-surface or extracellular release of the nuclear high-mobility group protein-1 (HMGB1) and adenosine triphosphate (ATP) [22–25]. Although TLRs in the mammalian immune system was first described as innate receptors recognizing pathogen-associated molecules, there is growing evidence that the TLRs also sense and respond to DAMPs, endogenous molecules or signals associated with cellular stress and tissue injury [26]. In breast cancer patients with loss-of-function alleles in TLR4, which mediates a signaling response to the HMGB1 stimulation, relapse occurs more quickly after chemoradiation compared with patients with wild-type alleles, indicating that the mode of host response to cancer cell death can affect clinical outcomes of cancer therapy [27].
As opposed to normal self-antigens, immune responses can be biased against the tumor-specific antigens that the immune system has been tolerated without resulting in side effects associated with standard cytotoxic therapies [24, 27]. Studies in mouse models revealed that the antigenic repertoire of tumor cells was substantially altered following RT. Radiation-induced exposure of antigenic peptides have been identified as a mechanism underlying RT-elicited antitumor immune response [28]. The ‘danger’ signals, including immunostimulatory cytokines, generated by radiation within the TME can activate DCs phenotypically and functionally for effective cross-presentation of tumor antigens [29]. Intratumoral injection of DCs alone does not show evident antitumor effects in mice with squamous cell carcinoma; however, significant tumor regression were observed when combined with chemoradiation, suggesting that the immune environment conditioned by the RT favors DC activation and fosters generation of antitumor immunity [30]. In both mice and humans, activation of tumor antigen-specific T cell immunity following RT requires TLR4 on DCs. Efficient processing and cross-presentation of antigens from dying tumor cells by DCs during RT are dependent on signaling through TLR4 and its adaptor MyD88 [27]. Similarly, local high-dose irradiation of B16 tumors results in activation of tumor-associated DCs as well as the consequent mobilization of tumor-reactive CD8+ T cells [31]. Ablative RT can dramatically improve the cross-priming capacity of tumor-infiltrating DCs. The autocrine effect of type I IFNs is required for the enhanced cross-priming ability of DCs after their infiltration into the irradiated tumor tissues [15]. A recent study demonstrated that adaptor protein STING in DCs and downstream type I IFN signaling are essential for RT-induced adaptive immune responses [13]. In addition, the cytokine secretory profile and its relevance to DC function upon direct radiation exposure have been noted. DCs show enhanced expression of IL-2, IL-12 and IFN-γ after exposure to low dose irradiation, which is positively correlated with their enhanced capability to prime T cells compared with non-irradiated DCs [32].
7.2.3 Activation and Recruitment of T Cells by RT
In addition to direct damage to the tumor cell DNA, accumulating data supports the notion that T cell recruitment and activation represent important mechanisms mediating the antitumor effect of RT. Stone et al. provided the first evidence supporting T cell repertoire dependent tumor response to RT by comparing the efficacy of RT in immunocompetent and T cell deficient mice [33]. RT can also remodel the abnormal tumor vessels and facilitate efficient tumor infiltration of anti-tumor T cells in a transgenic mouse model of insulinoma with multiple carcinogenesis. The remodeling of the tumor vasculature directly affects lymphocyte extravasation and effector function [34]. Up-regulation of vascular cell adhesion molecule (VCAM)-1 after RT promotes T cell infiltration into mouse B16 melanomas [10, 35]. Recruitment of CD8+ cytotoxic T lymphocytes (CTLs) into 4 T1 mammary tumors was found to depend on RT-induced CXCL16 release from tumor cells [17]. Indeed, the chemokine CXCL16 has been identified as a prognostic factor that correlates with improved survival and increased numbers of tumor-infiltrating lymphocytes in colorectal cancer and renal cell carcinoma [36, 37]. Prostate cancer patients developed detectable tumor-specific CD4+ and CD8+ T cells responses following RT that were undetectable prior to the treatment [38]. RT has been reported to dramatically increase the T-cell priming in lymphoid tissues or tumor tissue. The efficacy of RT can be abolished upon depletion of CD8+ T cells through administration of anti-CD8 monoclonal antibodies [39, 40]. Combination of RT with Th1 cell therapy augments the generation of tumor-infiltrating CTLs, resulting in complete regression of mouse EG7 lymphomas, which suggests that CD4+ T cells are also critically involved in RT-induced CTL response and tumor eradication [40].
7.2.4 Other Immune Cells Activated by RT
Natural killer (NK) cells are considered to be innate lineage cells based on their characteristic that there are no specific antigen receptors on their surface unlike T and B cells. NK cells play an important role in antitumor immunity by directly targeting tumor cells through cytolysis or the secretion of soluble immune mediators [41]. Ionizing radiation can increase the expression of natural-killer group 2 member D (NKG2D) ligands in human cancer cell lines, including KM12, NCI-H23, HeLa and A375. This makes the irradiated cancer cells more susceptible to NK cell-mediated cytotoxicity via the activating receptors NKG2D [42, 43].
7.3 Immune Suppressive Mechanisms Engaged by RT
7.3.1 Immunosuppressive Factors Induced by RT
Although RT can induce immune responses to tumor antigens, it is not sufficient to prime T cells specific for endogenous antigens that can efficiently reject the poorly immunogenic tumors. This may be attributed to the preexisting immune suppression in the TME and the immunosuppressive factors induced by RT. Activation of TGF-β is an early as well as a persistent event in tumors exposed to RT [44, 45]. Serum levels of TGF-β during the course of RT was evaluated in patients with non-small-cell lung cancer to adaptively deliver higher doses of radiation [46]. TGF-β plays a dual role both limiting tumor growth and stimulating tumor cells progression. Although TGF-β seems to be an antitumor factor at the early stages of cancer progression, it eventually becomes protumorigenic [47, 48]. In the mouse mammary tumor virus-polyoma virus middle T antigen (MMTV-PyVmT) transgenic model of metastatic breast cancer, RT significantly increases the circulating TGF-β and lung metastases, which can be suppressed by the deficiency of type II TGF-β receptor. This implicates RT induced TGF-β as a pro-metastatic signal for tumor cells [49]. TGF-β neutralization in mice bearing 4 T1 mammary tumors enhances radiation sensitivity and significantly delays tumor growth [50]. A recent study reported that TGF-β activity is a major obstacle that hinders the ability of RT to induce antigen-specific anti-tumor immunity. Neutralization of TGF-β by antibody injection during RT effectively rescues a CD8+ T-cell response against poorly immunogenic mouse carcinomas [51].
Hypoxia-inducible factors (HIFs) are the main molecular transcriptional factors in the hypoxia response [52, 53]. HIF-1 is highly induced in the irradiated tumors and high HIF-1 activity is often used as an independent predictor of poor prognosis after RT [54–56]. Expression of HIF-1 and HIF-2 is strongly associated with RT failure in patients with head and neck squamous cell carcinoma [57]. HIF-1 can stimulate the production of stromal-derived factor-1 (SDF-1), a chemokine that recruit tumor-promoting and immunosuppressive myeloid cells through the chemokine receptor CXCR4 [58, 59].
7.3.2 Tumor-Associated Macrophages
Tumor-associated macrophages (TAMs), derived from circulating monocytes, make up a critical component of immune cells in solid tumors [60, 61]. Although a few studies showed that RT enhances the anti-tumor properties of TAMs, including enhanced cytolytic activity and increased secretion of IL-12 and IL-18 [62, 63], there exists extensive literature indicating that TAMs enhance resistance to RT. CD11b+ myeloid cells, including TAMs, are believed to be the major source of pro-tumor growth factors that support angiogenic programs during tumor progression, e.g., vascular endothelial growth factor (VEGF) and matrix metalloproteinase-9 (MMP-9) [58]. Murine tumors are more sensitive to RT when are transplanted in CD18 hypomorphic or CD11b knockout mice. Resistance of tumors to RT is partially restored by rescue of CD18 hypomorphism with the reconstitution of wild-type bone marrow [64].
Depletion of TAMs by injection of liposomal clodronate prior to RT enhances tumor control, emphasizing an important role of TAM for modulating tumor response to RT. Radiation exposure upregulates VEGF expression in macrophages and VEGF-neutralization subcutaneously improves the antitumor potency of RT [65]. Recently, it was revealed that CD11b+ monocytes/macrophages restored the damaged vasculature by promoting vasculogenesis and growth of surviving cancer cells following RT in a human glioblastoma xenograft model. Blocking the influx of CD11b+ monocytes/macrophages by pharmacologic inhibition of HIF-1 or SDF-1-CXCR4 pathway can prevent tumor recurrence [66]. Similar observation was made using human breast and lung carcinoma xenografts, further supporting a critical role of myeloid cells, primarily macrophages, in promoting tumor regrowth after RT. It is proposed that TAMs facilitate tumor recurrence by promoting the survival of endothelial cells (ECs) and tumor revascularization [67]. Studies of three murine tumors (TRAMP-C1 prostate adenocarcinoma, ALTS1C1 astrocytoma, and GL261 glioma) demonstrate that CD11blow/F4/80+ macrophages locate at the junctions between central necrotic and surrounding hypoxic regions in the irradiated tumors. Hypoxia-aggregated TAMs are more polarized toward an immunosuppressive and pro-angiogenic M2 phenotype, indicated by the higher expression of arginase I [68]. Thus, despite a potential stimulatory effect of radiation on cytolytic activity of macrophages, the recruitment and alternative activation of macrophages in the TME shifts the balance toward immunosuppression and pro-angiogenesis that benefits tumor recurrence.
7.3.3 Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of myeloid cells, comprised of myeloid progenitor cells and immature myeloid cells [69, 70]. MDSCs are often expanded in tumor-bearing hosts and have been well documented to act as a suppressor of antitumor immunity [71–73]. MDSCs are believed to be one of the mechanisms by which cancers escape from immune surveillance or resist immunotherapy [71–73]. MDSCs are characterized as CD11b+Gr-1+ cells in mice [69, 70] and CD11b+CD14–CD33+ in human [74]. Two distinct subsets of MDSCs have been identified in mice, i.e., monocytic MDSCs (M-MDSCs; CD11b+Ly6G−Ly6Chigh) and granulocytic MDSCs (G-MDSCs; CD11b+Ly6G+Ly6Cint), both characterized by the expression of Gr-1 on the cell surface [75]. CSF signaling has been documented to expand and recruit myeloid cells or MDSCs to the tumor sites [76, 77]. Use of selective inhibitor of colony-stimulating factor 1 receptor (CSF1R) can suppress tumor growth more effectively when combined with RT, highlighting the significance of CSF1/CSF1R signaling in the recruitment of myeloid cells (e.g., MDSCs) that limit the efficacy of RT [78]. The role of MDSCs in limiting the efficacy of RT has also been demonstrated in RT in combination with Sunitinib, an angiogenesis inhibitor [79, 80]. A recent study found that Sunitinib treatment decreased M-MDSC levels and enhanced T-cell proliferative activity in cancer patients with oligometastases [81]. Moreover, the synergetic effect of Sunitinib and stereotactic body radiotherapy (SBRT) was only seen in the responders whose CD11b+CD33+ myeloid cell populations were reduced by Sunitinib [81].
7.3.4 Regulatory T Cells
FoxP3+ regulatory T (Treg) cells are suppressive immune cells that promote tumor progression through suppressing anti-tumor immune responses [82–84]. Treg cell ablation in a polyoma middle-T antigen-driven tumor model significantly reduces tumor burden and improves overall survival following RT. Combining Treg cell ablation with RT could provide beneficial effects for the poorly immunogenic malignancies [85]. Epidermal mononuclear phagocytes Langerhans cells (LCs) are resistant to the depletion by high dose irradiation. Upon exposure to RT, LCs upregulates MHCII molecule and induces the expansion of Treg cells that can dampen anti-tumor immunity [86, 87].
7.4 Combining RT with Immunotherapy to Improve Therapeutic Index
RT alone is often insufficient to achieve a permanent cure in many clinical scenarios. This is primarily a result of insufficient radiation doses to control tumor without resulting in unacceptable toxicity related to normal tissues. This also suggests that despite the numerous pro-immunogenic or immunostimulating effects, RT as a sole modality fails in shifting the immunosuppressive TME. Systemic antitumor responses following local RT or abscopal responses are also extremely rare in clinical practice. However, RT-induced systemic abscopal response through development of effective and durable antitumor immunity can be promoted by additional immune manipulation. Therefore, it provides a scientific rationale for integrating RT with immunotherapy to amplify the systemic antitumor immunity and to improve overall therapeutic outcomes.
Irradiated tumor cells have been shown to be a source of tumor-associated antigens which can elicit anti-tumor T cell responses after capture and presentation by DCs [88]. Combination of RT and concurrent administration of DCs may result in in situ vaccination against tumors. Injection of unpulsed autologous DCs directly into irradiated D5 melanoma or MCA 205 sarcoma tumors was shown to activate tumor-specific reactive T cells and generate a potent systemic antitumor response causing regression of established tumors [89]. In a recent phase I clinical trial of combining external beam RT and intraprostatic DC injection, patients with high-risk prostate cancer showed increased tumor-infiltrating CD8+ T-cells as well as prostate specific CD8+ T-cells in the peripheral blood [90]. In patients with high-risk soft tissue sarcoma that received this combinational therapy, 9 of 17 patients developed tumor-specific immune responses and 12 patients remained free of progression 1 year after treatment [91]. Another recent trial was conducted in 40 patients with recurrent, metastatic, or locally advanced tumors [92]. Patients were treated with conformal RT and autologous DCs pulsed with autologous tumor cell lysates or tumor-specific peptides. Of 9 patients with evaluable tumor response outside the RT target site, 22 % had a partial response and 33 % had stable disease, indicating that the combination of RT and DC-based vaccination induces measurable clinical responses [92].
An alternative approach that combined local RT and concomitant expansion of DCs in vivo through systemic administration of fms-like tyrosine kinase-3 ligand (Flt3L) was also shown to improve survival of animals bearing Lewis lung carcinoma by generating a long-term tumor-specific immune response [93]. The use of Flt3L was also shown to facilitate abscopal effect of RT, indicated by inhibition of both the irradiated breast tumor and the contralateral untreated tumor [94].
Manipulation of the TLR signaling can increase the functional activation of APCs and provide co-stimulation signals to T cells, thereby facilitating an effective adaptive antitumor immunity after RT [95, 96]. A phase II trial conducted in patients with recurrent anaplastic glioma showed that combined RT and intramuscular injection of poly-ICLC, a TLR3 agonist, improved 1-year overall survival compared to RT alone [97]. The TLR7 agonist imiquimod has been approved for the treatment of basal cell skin carcinomas and melanomas. Topical imiquimod can synergize with RT to inhibit tumor growth in a mouse model of skin-involving breast cancer, which is associated with increased number of tumor-infiltrating CD11c+, CD4+ and CD8+ cells [98]. Based on a recent clinical study demonstrating imiquimod-induced immune rejection of skin metastases in breast cancer patients [99], a trial is ongoing to test combination of imiquimod and RT for improving therapeutic outcomes in brain cancer (ClinicalTrials.gov: #NCT01400672).
Our studies have recently identified scavenger receptor A (SRA) or CD204, a pattern recognition innate receptor, as an immunosuppressive molecule expressed on DCs that dampens DC function and T cell activation against several cancers by suppressing DC-intrinsic TLR signaling [100–102]. Our subsequent work revealed that absence of SRA/CD204 significantly increased the immunogenicity of ionizing radiation–treated mouse prostate cancer cells [103], which provides a scientific rationale for combining RT with in situ vaccination using SRA/CD204-downregulated DCs. We showed that intratumoral administration of SRA/CD204-silenced DCs, not DC counterparts without genetic modification, profoundly enhanced the control of RT-treated mouse prostate tumor as well as its metastases, which was mainly mediated by IFN-γ-producing CTLs [104]. These preclinical evidence supports the further development of TLR or SRA-targeting strategies for combinational use with RT to convert the tumor into an effective individualized vaccine.
Adoptive cell therapy (ACT) is a passive cancer immunotherapy by transferring of tumor-specific T cells that have been expanded ex vivo to cancer patients [105, 106]. Local irradiation of mouse MC38 colon tumors causes up-regulation of Fas on tumor cells and potentiates tumor eradication by adoptively transferred antigen-specific CTLs [107]. Local tumor irradiation combined with intratumoral DC vaccination regimens significantly enhances the therapeutic efficacy of ACT in a mouse liver cancer model, evidenced by reduced local tumor size, decreased metastasis, and prolonged survival. The enhanced antitumor activity is correlated with the activation of endogenous CD4+ T cells [108]. Therapeutic vaccination represents an active immunotherapy that aims to stimulate T cell response against specific tumor antigens. The synergistic antitumor effect of RT and therapeutic immunization is supported by a preclinical study in colon tumor-bearing mice that received RT plus recombinant vaccinia encoding carcinoembryonic antigen [109]. In a randomized phase II trial patients received RT alone or RT plus a viral vaccine targeting tumor antigen PSA and co-stimulatory B7.1. The results showed an elevated PSA-specific T cell response in the combination group compared to the RT alone arm [110].
Immune modulators that target immunosuppressive signaling in T cells to overcome immune suppression and restore and/or sustain antitumor function of T cells for tumor eradication have shown promise in cancer patients [111]. Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death 1 (PD-1) are two primary immune checkpoint molecules that inhibits T cell activation upon binding to their ligands, B-7 molecules and PD-L1, respectively [20, 111, 112]. As T cell activation relies on the engagement of both antigen receptor and the costimulatory molecule CD28 [113], it is conceivable that using RT to increases antigen availability and APC activation in conjunction with anti-CTLA-4 or anti-PD-L1 therapy will further augment antitumor immune responses.
Anti-CTLA-4 antibody, ipilimumab, has been approved by FDA in the treatment of patients with metastatic melanoma [114]. Combination of local RT of 4T1 mouse mammary carcinoma with the anti-CTLA-4 monoclonal antibodies 9H10 significantly elicited an anti-tumor immunity that inhibited metastases [115]. In another preclinical study administration of 9H10 was shown to optimize tumor response to fractionated RT by inducing an abscopal effect involving activation of CD8+ T cells [116]. The abscopal effect was also reported in a patient with metastatic melanoma following treatment with RT plus ipilimumab. Clinical observation obtained several months after last dose of RT revealed that tumor masses in the spleen and hilar lymph nodes eventually reached the point of stable minimal disease [4]. Complete response in both the primary tumor and the metastatic lesions was also achieved in another patient with asymptomatic melanoma treated with ipilimumab and concurrent RT [117]. A recent case report described that a patient with non-small-cell lung carcinoma also showed abscopal response upon the combination therapy [118]. A phase III trial that evaluated RT combined with ipilimumab therapy in metastatic castration-resistant prostate cancer (mCRPC) was recently completed [119]. The trial did not meet its primary endpoint, however, there was an improvement in overall survival of patients treated with RT plus and ipilimumab compared to RT plus placebo arm (11.2 months vs. 10 months; p = 0.053) [119]. Currently, more than ten phase I/II clinical trials that are testing the combination of RT and ipilimumab for treatment of multiple cancer types including melanoma, head and neck cancer, and cervical cancer are ongoing.
PD-1 receptor is another important immune checkpoint molecule that downregulates T cell-mediated immune responses. Expression of PD-1 on T cells in the TME is an indicator of their exhaustion that is often associated with an impaired T cell response. Overexpression of the PD-1 ligand (PD-L1), also known as B7 homolog ligand 1 (B7-HL1), in a variety of malignant cancers such as renal, lung, ovarian, breast, head and neck cancers, represents one of the mechanisms responsible for tumor immune evasion [20, 120]. Low doses of fractionated RT increases the tumor expression of PD-L1 in a number of syngeneic mouse cancer models, which is attributed to IFN-γ produced by CD8+ T cells [121]. Upregulation of the PD-L1 on tumor cells were shown to contribute to radio-resistance of cancer and suppress the antitumor function of tumor-infiltrating T cells [121–123]. Recently, antibodies targeting PD-L1 (BMS-936559, MEDI4736, MPDL3280A) and its receptor PD-1 (Nivolumab, Pidilizumab, Lambrolizumab) have been developed to overcome PD-1/PD-L1 signaling-mediated immune suppression. Clinical studies showed that anti-PD-1/PD-L1 antibodies have achieved significantly increased objective response (~20–30 %) in the treatment of several types of cancers including non-small cell lung cancer, melanoma, and renal-cell cancer [124–126]. Studies using pre-clinical models demonstrated the synergistic effects of RT combined with PD-1 checkpoint inhibitors. Adding anti-PD-1 antibody to combination therapy of RT and anti-CD137 therapy resulted in cure of the primary mammary tumors [127]. Treatment with RT in conjunction with anti-PD-1 antibody resulted in synergistic inhibition of mouse glioma, TUBO mammary carcinoma, and MC38 colon adenocarcinoma, probably through increasing the infiltration of IFN-γ- or TNF-α-expressing CTLs while decreasing the accumulation of Treg and MDSCs within the TME that normally suppress T cell function [122, 128]. Despite the impressive clinical responses resulted from immune checkpoint inhibitors, optimization is required to overcome multiple non-redundant mechanism of immune resistance. A recent phase I trial reported that melanoma patients with high expression of PD-L1 did not respond to RT plus anti-CTLA4 therapy [129]. Mouse studies found that this resistance was due to upregulation of PD-L1 on melanoma cells during RT and consequent T-cell exhaustion, which allows tumors to escape anti-CTLA4 therapy. Thus, triple combination of RT, anti-CTLA4 and anti-PD-L1 treatments, which enhances the diversity of the T-cell receptor repertoire of intratumoral T cells, inhibits Treg cells, thereby increasing the ratio of CTL to Treg, and reverses T-cell exhaustion, can achieve maximum antitumor response by engaging distinct mechanisms [129].
In addition to targeting immune checkpoint molecules to rescue and sustain T cell functions in the TME, other approaches directed to promote co-stimulation can also enhance T cell priming and effector function. OX40 signaling is one of the co-stimulatory mechanisms involved in T cell activation [130–132]. Administration of OX40 agonistic antibodies in combination with RT significantly extends the mouse survival in a model of primary sarcoma by augmenting the activity of tumor antigen-specific CTLs following RT [133]. Clinical trials of combining RT and OX40 agonist for treatment of metastatic prostate cancer (ClinicalTrials.gov: #NCT01303705) and breast cancer (ClinicalTrials.gov: #NCT01642290) are ongoing.
7.5 Challenges and Opportunities
Encouraging preclinical results of combining RT and immunotherapy for cancer treatment have stimulated clinical translation of this combinatorial therapeutic modality. However, clinical data from trials that combined RT with other immune modulating agents, e.g., vaccines, immune checkpoint blockade, have only shown a modest promise. While these outcomes provide the rationale for current clinical trials of both treatments, further investigation is required to realize the full potential of the combination.
The immunogenic alterations in the TME induced RT at molecular and cellular levels are just beginning to be elucidated. The immunoregulatory effects of dose and fractionation schedules as well as delivery during RT remain to be further defined. The timing of RT relative to immune manipulation should also be carefully determined in preclinical and clinical studies. Extrapolation of data derived from mouse models may have its own limitation because currently most animal studies involve radiation to tumor xenografts. Use of spontaneous transgenic mouse model instead of transplantation mouse model to address these questions may better guide the optimal design of clinical RT in combination with immunotherapy. Mechanistic understanding of immunological and biological changes in the TME has an important impact on capitalization of tumor destruction capacity of radiation and immune augmentation.
The immunosuppressive effect of the TME remains a major hurdle for clinical success of combined RT and immunotherapy. The promising results observed in clinical trials with immune checkpoint blockade therapy highlights the therapeutic potential by modification of the TME to overcome immunosuppressive pathways. Exploring and identifying additional and non-redundant immunosuppressive molecules or mechanisms that operate in the TME will provide new therapeutic targets to synergize with RT to mount an effective and durable antitumor immunity.
Personalized medicine has been an important part of therapeutic endeavors in the field of cancer research and treatment. Emerging data suggests that each patient’s own immune system have the potential to develop a tailored immune response to the unique clonal populations within the tumors. Thus, immunotherapy is at the forefront of personalize anticancer therapy and precision oncology. With the advantage of its very focused and localized nature, RT is ideal for combination with proper modulations of the immune system and has a great potential to convert the tumor into an individualized vaccine. Ongoing and future randomized trials with RT and immune-based combinations will help determine if such regimens can change the RT paradigm and, more importantly, revolutionize cancer treatment (Table 7.1).
7.6 Concluding Remarks
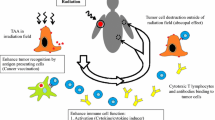
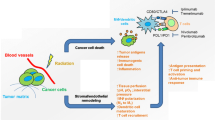
The effort in improving the therapeutic efficacy of RT have focused on the capability of ionizing radiation to kill neoplastic cells while sparing normal healthy tissues. However, accumulating evidence supports the immune modulating effects of RT (Fig. 7.1a). RT-mediated destruction of cancer cells releases tumor antigens along with ‘danger’ signals or PAMPs that defines the immunogenicity of tumor or ICD. These immunostimulatory factors result in recruitment and activation of APCs (e.g., DCs), which facilitates subsequent T cell priming and antitumor immune response. However, it is recognized that multiple mechanisms in the TME, which involve induction of immunosuppressive factors (TGF-β, CTLA-4 and PD-1) and recruitment of immunosuppressive cells (TAM, MDSC, Treg cell), dampen or impair immune effector function and promote immune tolerance. The substantial expansion and recruitment of myeloid cells following RT is known to facilitate tumor revascularization and possibly immune suppression as well. Therefore, it is unlikely RT alone is capable of generating an effective immune response that can eradicate tumors locally and abscopally. Nevertheless, interaction of RT and the immune system offers new opportunity to strengthen and improve antitumor response by strategically combining RT and immune interventions or immunotherapy, e.g., in situ DC vaccination, TLR activation, immune checkpoint blockade, immune co-stimulation (Fig. 7.1b). Cancer immunotherapy has emerged as a viable therapeutic option and with multiple agents in clinical development the immunotherapeutic portfolio is expected to expand significantly in the near future. While more research is needed to precisely understand and rationally optimize the protocol of this combinatorial treatment, clinical studies have started to show the promise in improved treatment outcome, which we believe may lead to ultimate elimination of cancers and metastases by amplifying immune-mediated abscopal effects after standard RT.

Exploiting the interplay between radiation therapy (RT) and the immune system to improve cancer therapeutic index. (a) RT can induce immunogenic cell death associated with release of tumor-associated antigens (TAA) with damage-associated molecular patterns (DAMPs), which recruit and stimulate dendritic cells (DCs) via toll-like receptor (TLR) for antigen cross-presentation. Subsequent priming of CD8+ T cells though engaging surface T cell receptor (TCR) contributes to cancer cell killing during RT. However, concurrent increase in the number of pro-angiogenic and immunosuppressive cell types, such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Treg), as well as induction of immunosuppressive factors, such as cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1), compromise immune effector function within the tumor microenvironment (TME) and promote cancer escape and recurrence. (b) While RT-induced immune response is not adequate to achieve systemic eradication of tumors, integrating RT with immunotherapy could amplify the systemic antitumor immunity and improve overall treatment outcomes. Selectively stimulation of TLRs or strategic administration of ex vivo expanded DCs can enhance DC function and antigen cross-presentation by providing additional co-stimulatory signals. Adoptive transfer of tumor-reactive T cells, either ex vivo expanded T cells or genetically modified T cells, can further strengthen the effector arm of antitumor mechanisms since radiation exposure sensitize cancer cells to the killing of T cells. Additionally, immune checkpoint inhibitors can be used to rescue the effector function of T cells by counteracting the immunosuppressive signaling mediated by CTLA-4 or PD-1/PD-L1. Other approaches directed to block the recruitment and/or to overcome the immunosuppressive activity of myeloid cells (e.g., TAMs, MDSCs) can also boost antitumor immune response. The increased magnitude and duration of systemic activation tumor-specific T cells combined with direct tumoricidal effect of RT could lead to synergetic and optimized destruction of local/regional tumors as well as metastases
References
Harrington KJ, Billingham LJ, Brunner TB, Burnet NG, Chan CS, Hoskin P, Mackay RI, Maughan TS, Macdougall J, McKenna WG, Nutting CM, Oliver A, Plummer R, Stratford IJ, Illidge T. Guidelines for preclinical and early phase clinical assessment of novel radiosensitisers. Br J Cancer. 2011;105:628–39.
Barker HE, Paget JTE, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15:409–25.
Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, Sedrak C, Jungbluth AA, Chua R, Yang AS, Roman RA, Rosner S, Benson B, Allison JP, Lesokhin AM, Gnjatic S, Wolchok JD. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925–31.
Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–91.
Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25:198–213.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci U S A. 1989;86:10104–7.
Kageshita T, Nakamura T, Yamada M, Kuriya N, Arao T, Ferrone S. Differential expression of melanoma associated antigens in acral lentiginous melanoma and in nodular melanoma lesions. Cancer Res. 1991;51:1726–32.
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180:3132–9.
Ma JL, Jin L, Li YD, He CC, Guo XJ, Liu R, Yang YY, Han SX. The intensity of radiotherapy-elicited immune response is associated with esophageal cancer clearance. J Immunol Res. 2014;2014:794249.
Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu YX, Weichselbaum RR. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52.
Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, Komatsu K, Akira S, Kawai T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A. 2013;110:2969–74.
Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–96.
Lim JY, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol Immunother. 2014;63:259–71.
Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, Demaria S. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–107.
Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48.
Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259:368–70.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61.
Schaue D, McBride WH. Links between innate immunity and normal tissue radiobiology. Radiat Res. 2010;173:406–17.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848–50.
Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–63.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, Griekspoor A, Mesman E, Verreck FA, Spits H, Schlom J, van Veelen P, Neefjes JJ. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75.
Moyer JS, Li J, Wei S, Teitz-Tennenbaum S, Chang AE. Intratumoral dendritic cells and chemoradiation for the treatment of murine squamous cell carcinoma. J Immunother. 2008;31:885–95.
Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, Schwendener R, Pruschy M, Knuth A, van den Broek M. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. 2012;189:558–66.
Shigematsu A, Adachi Y, Koike-Kiriyama N, Suzuki Y, Iwasaki M, Koike Y, Nakano K, Mukaide H, Imamura M, Ikehara S. Effects of low-dose irradiation on enhancement of immunity by dendritic cells. J Radiat Res. 2007;48:51–5.
Stone HB, Peters LJ, Milas L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J Natl Cancer Inst. 1979;63:1229–35.
Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462–70.
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–23.
Gutwein P, Schramme A, Sinke N, Abdel-Bakky MS, Voss B, Obermuller N, Doberstein K, Koziolek M, Fritzsche F, Johannsen M, Jung K, Schaider H, Altevogt P, Ludwig A, Pfeilschifter J, Kristiansen G. Tumoural CXCL16 expression is a novel prognostic marker of longer survival times in renal cell cancer patients. Eur J Cancer. 2009;45:478–89.
Hojo S, Koizumi K, Tsuneyama K, Arita Y, Cui Z, Shinohara K, Minami T, Hashimoto I, Nakayama T, Sakurai H, Takano Y, Yoshie O, Tsukada K, Saiki I. High-level expression of chemokine CXCL16 by tumor cells correlates with a good prognosis and increased tumor-infiltrating lymphocytes in colorectal cancer. Cancer Res. 2007;67:4725–31.
Tabi Z, Spary LK, Coleman S, Clayton A, Mason MD, Staffurth J. Resistance of CD45RA- T cells to apoptosis and functional impairment, and activation of tumor-antigen specific T cells during radiation therapy of prostate cancer. J Immunol. 2010;185:1330–9.
Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, Weichselbaum RR, Fu YX. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–95.
Takeshima T, Chamoto K, Wakita D, Ohkuri T, Togashi Y, Shirato H, Kitamura H, Nishimura T. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res. 2010;70:2697–706.
Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142:847–56.
Kim JY, Son YO, Park SW, Bae JH, Chung JS, Kim HH, Chung BS, Kim SH, Kang CD. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp Mol Med. 2006;38:474–84.
Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3–12.
Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA. Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest. 1994;93:892–9.
Wang J, Zheng H, Sung CC, Richter KK, Hauer-Jensen M. Cellular sources of transforming growth factor-beta isoforms in early and chronic radiation enteropathy. Am J Pathol. 1998;153:1531–40.
Anscher MS, Marks LB, Shafman TD, Clough R, Huang H, Tisch A, Munley M, Herndon 2nd JE, Garst J, Crawford J, Jirtle RL. Using plasma transforming growth factor beta-1 during radiotherapy to select patients for dose escalation. J Clin Oncol. 2001;19:3758–65.
Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment—tumorigenesis and therapy. Nat Rev Cancer. 2005;5:867–75.
Desai S, Kumar A, Laskar S, Pandey BN. Cytokine profile of conditioned medium from human tumor cell lines after acute and fractionated doses of gamma radiation and its effect on survival of bystander tumor cells. Cytokine. 2013;61:54–62.
Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, Freeman ML, Arteaga CL. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–13.
Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, Babb JS, Lonning SM, DeWyngaert JK, Formenti SC, Barcellos-Hoff MH. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17:6754–65.
Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75:2232–42.
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309.
Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res. 2012;18:1207–13.
Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, Semenza GL. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001;61:2911–6.
Koukourakis MI, Giatromanolaki A, Sivridis E, Simopoulos C, Turley H, Talks K, Gatter KC, Harris AL. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2002;53:1192–202.
Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–41.
Koukourakis MI, Bentzen SM, Giatromanolaki A, Wilson GD, Daley FM, Saunders MI, Dische S, Sivridis E, Harris AL. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol. 2006;24:727–35.
Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205.
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64.
Guo C, Buranych A, Sarkar D, Fisher PB, Wang XY. The role of tumor-associated macrophages in tumor vascularization. Vasc Cell. 2013;5:20.
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–7.
Lambert LE, Paulnock DM. Modulation of macrophage function by gamma-irradiation. Acquisition of the primed cell intermediate stage of the macrophage tumoricidal activation pathway. J Immunol. 1987;139:2834–41.
Shan YX, Jin SZ, Liu XD, Liu Y, Liu SZ. Ionizing radiation stimulates secretion of pro-inflammatory cytokines: dose-response relationship, mechanisms and implications. Radiat Environ Biophys. 2007;46:21–9.
Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8.
Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, Weichselbaum RR. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70:1534–43.
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705.
Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70:5679–85.
Chiang CS, Fu SY, Wang SC, Yu CF, Chen FH, Lin CM, Hong JH. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol. 2012;2:89.
Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–44.
Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol. 2010;199:273–81.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79.
Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–600.
Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, Wu YC, Chu Y, Chung FT, Kuo CH, Lee KY, Lin SM, Lin HC, Wang CH, Yu CT, Kuo HP. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2009;136:35–45.
Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31.
Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–94.
Kleibeuker EA, Griffioen AW, Verheul HM, Slotman BJ, Thijssen VL. Combining angiogenesis inhibition and radiotherapy: a double-edged sword. Drug Resist Updat. 2012;15:173–82.
Roskoski Jr R. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356:323–8.
Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M, Sung M, Ferris R, Kao J, Wang LH, Pan PY, Ko EC, Chen SH. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent Sunitinib and stereotactic body radiotherapy. Clin Cancer Res. 2015;21:4073–85.
Byrne WL, Mills KH, Lederer JA, O’Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. 2011;71:6915–20.
Liu Z, Kim JH, Falo Jr LD, You Z. Tumor regulatory T cells potently abrogate antitumor immunity. J Immunol. 2009;182:6160–7.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307.
Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210:2435–66.
Liauw SL, Connell PP, Weichselbaum RR. New paradigms and future challenges in radiation oncology: an update of biological targets and technology. Sci Transl Med. 2013;5:173sr172.
Price JG, Idoyaga J, Salmon H, Hogstad B, Bigarella CL, Ghaffari S, Leboeuf M, Merad M. CDKN1A regulates Langerhans cell survival and promotes Treg cell generation upon exposure to ionizing irradiation. Nat Immunol. 2015;16:1060–8.
Strome SE, Voss S, Wilcox R, Wakefield TL, Tamada K, Flies D, Chapoval A, Lu J, Kasperbauer JL, Padley D, Vile R, Gastineau D, Wettstein P, Chen L. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res. 2002;62:1884–9.
Teitz-Tennenbaum S, Li Q, Rynkiewicz S, Ito F, Davis MA, McGinn CJ, Chang AE. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res. 2003;63:8466–75.
Finkelstein SE, Rodriguez F, Dunn M, Farmello MJ, Smilee R, Janssen W, Kang L, Chuang T, Seigne J, Pow-Sang J, Torres-Roca JF, Heysek R, Biagoli M, Shankar R, Scott J, Antonia S, Gabrilovich D, Fishman M. Serial assessment of lymphocytes and apoptosis in the prostate during coordinated intraprostatic dendritic cell injection and radiotherapy. Immunotherapy. 2012;4:373–82.
Finkelstein SE, Iclozan C, Bui MM, Cotter MJ, Ramakrishnan R, Ahmed J, Noyes DR, Cheong D, Gonzalez RJ, Heysek RV, Berman C, Lenox BC, Janssen W, Zager JS, Sondak VK, Letson GD, Antonia SJ, Gabrilovich DI. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys. 2012;82:924–32.
Shibamoto Y, Okamoto M, Kobayashi M, Ayakawa S, Iwata H, Sugie C, Mitsuishi Y, Takahashi H. Immune-maximizing (IMAX) therapy for cancer: combination of dendritic cell vaccine and intensity-modulated radiation. Mol Clin Oncol. 2013;1:649–54.
Chakravarty PK, Guha C, Alfieri A, Beri V, Niazova Z, Deb NJ, Fan Z, Thomas EK, Vikram B. Flt3L therapy following localized tumor irradiation generates long-term protective immune response in metastatic lung cancer: its implication in designing a vaccination strategy. Oncology. 2006;70:245–54.
Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, Formenti SC. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58:862–70.
Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–67.
Roses RE, Datta J, Czerniecki BJ. Radiation as immunomodulator: implications for dendritic cell-based immunotherapy. Radiat Res. 2014;182:211–8.
Butowski N, Lamborn KR, Lee BL, Prados MD, Cloughesy T, DeAngelis LM, Abrey L, Fink K, Lieberman F, Mehta M, Ian Robins H, Junck L, Salazar AM, Chang SM. A North American brain tumor consortium phase II study of poly-ICLC for adult patients with recurrent anaplastic gliomas. J Neurooncol. 2009;91:183–9.
Dewan MZ, Vanpouille-Box C, Kawashima N, DiNapoli S, Babb JS, Formenti SC, Adams S, Demaria S. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin Cancer Res. 2012;18:6668–78.
Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, Hochman T, Shuman N, Axelrod D, Speyer J, Novik Y, Tiersten A, Goldberg JD, Formenti SC, Bhardwaj N, Unutmaz D, Demaria S. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748–57.
Wang XY, Facciponte J, Chen X, Subjeck JR, Repasky EA. Scavenger receptor-A negatively regulates antitumor immunity. Cancer Res. 2007;67:4996–5002.
Yi H, Guo C, Yu X, Gao P, Qian J, Zuo D, Manjili MH, Fisher PB, Subjeck JR, Wang XY. Targeting the immunoregulator SRA/CD204 potentiates specific dendritic cell vaccine-induced T-cell response and antitumor immunity. Cancer Res. 2011;71:6611–20.
Yu X, Yi H, Guo C, Zuo D, Wang Y, Kim HL, Subjeck JR, Wang XY. Pattern pecognition scavenger receptor CD204 attenuates toll-like receptor 4-induced NF-{kappa}B activation by directly inhibiting ubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J Biol Chem. 2011;286:18795–806.
Guo C, Yi H, Yu X, Hu F, Zuo D, Subjeck JR, Wang XY. Absence of scavenger receptor A promotes dendritic cell-mediated cross-presentation of cell-associated antigen and antitumor immune response. Immunol Cell Biol. 2012;90:101–8.
Guo C, Yi H, Yu X, Zuo D, Qian J, Yang G, Foster BA, Subjeck JR, Sun X, Mikkelsen RB, Fisher PB, Wang XY. In situ vaccination with CD204 gene-silenced dendritic cell, not unmodified dendritic cell, enhances radiation therapy of prostate cancer. Mol Cancer Ther. 2012;11:2331–41.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–8.
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308.
Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman CN, Hodge JW. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol. 2003;170:6338–47.
Teitz-Tennenbaum S, Li Q, Davis MA, Wilder-Romans K, Hoff J, Li M, Chang AE. Radiotherapy combined with intratumoral dendritic cell vaccination enhances the therapeutic efficacy of adoptive T-cell transfer. J Immunother. 2009;32:602–12.
Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J, Hodge JW. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004;64:4328–37.
Gulley JL, Arlen PM, Bastian A, Morin S, Marte J, Beetham P, Tsang KY, Yokokawa J, Hodge JW, Menard C, Camphausen K, Coleman CN, Sullivan F, Steinberg SM, Schlom J, Dahut W. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin Cancer Res. 2005;11:3353–62.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106.
Lu P, Wang YL, Linsley PS. Regulation of self-tolerance by CD80/CD86 interactions. Curr Opin Immunol. 1997;9:858–62.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11:728–34.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88.
Hiniker SM, Chen DS, Reddy S, Chang DT, Jones JC, Mollick JA, Swetter SM, Knox SJ. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol. 2012;5:404–7.
Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res. 2013;1:365–72.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, Ng S, Maio M, Franke FA, Sundar S, Agarwal N, Bergman AM, Ciuleanu TE, Korbenfeld E, Sengelov L, Hansen S, Logothetis C, Beer TM, McHenry MB, Gagnier P, Liu D, Gerritsen WR, Investigators CA. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12.
Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–5.
Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, Stratford IJ, Poon E, Morrow M, Stewart R, Jones H, Wilkinson RW, Honeychurch J, Illidge TM. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74:5458–68.
Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–95.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33.
Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, Duret H, Yagita H, Johnstone RW, Smyth MJ, Haynes NM. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 2012;72:3163–74.
Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, Hammers HJ, Mathios D, Tyler B, Brem H, Tran PT, Pardoll D, Drake CG, Lim M. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9.
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, Minn AJ. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7.
Humphreys IR, Loewendorf A, de Trez C, Schneider K, Benedict CA, Munks MW, Ware CF, Croft M. OX40 costimulation promotes persistence of cytomegalovirus-specific CD8 T Cells: A CD4-dependent mechanism. J Immunol. 2007;179:2195–202.
Ohshima Y, Yang LP, Uchiyama T, Tanaka Y, Baum P, Sergerie M, Hermann P, Delespesse G. OX40 costimulation enhances interleukin-4 (IL-4) expression at priming and promotes the differentiation of naive human CD4(+) T cells into high IL-4-producing effectors. Blood. 1998;92:3338–45.
Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–10.
Gough MJ, Crittenden MR, Sarff M, Pang P, Seung SK, Vetto JT, Hu HM, Redmond WL, Holland J, Weinberg AD. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33:798–809.
Acknowledgements
The present study was supported in part by National Institutes of Health Grants CA175033, CA154708 (X-Y.W), Department of Defense W81XWH-11-/0481, W81XWH-13-/0455. X-Y.W. is the Mary Anderson Harrison Distinguished Professor in Cancer Research in the VCU Massey Cancer Center.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Guo, C., Harris, T., Wang, XY. (2016). Aiming the Immune System to Improve the Antitumor Efficacy of Radiation Therapy. In: Anscher, M., Valerie, K. (eds) Strategies to Enhance the Therapeutic Ratio of Radiation as a Cancer Treatment. Springer, Cham. https://doi.org/10.1007/978-3-319-45594-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-45594-5_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45592-1
Online ISBN: 978-3-319-45594-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)