
Abstract
Cultivated foxtail millet (Setaria italica) and its wild progenitor (S. viridis) have collectively been considered as tractable model species for studying C4 photosynthesis, stress biology, and biofuel traits. Being cultivated in arid and semiarid tropics of the world, these species are well adapted to harsh environments such as drought, heat, and salinity. This adaptation or acclimation potential of Setaria spp. has drawn research interest, and attempts have been made to dissect the molecular mechanisms of stress tolerance. Compared to other stresses, drought response has been studied extensively in S. italica and many drought-responsive genes encoding for transcription factors, signaling molecules, and enzymes have been identified and characterized. Several genome-wide studies have reported on identification of stress-responsive gene family members, and speculated on the potential for expansion and neofunctionalization of paralogs in these gene families. In this context, this chapter discusses the key genetic determinants identified for stress tolerance in S. italica and demonstrates their use in improving drought tolerance. In addition, strategies for identification of genes underlying stress tolerance are also described. Little effort has so far been made towards understanding the stress-tolerance characteristics of Setaria as compared to studies reported in other crops. Comprehensive functional studies along with the use of integrated -omics approaches are required to elucidate the genetics and genomics of stress tolerance in Setaria, as it is important to develop climate change resilient crops to meet the growing demand for food and feed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Foxtail millet (Setaria italica)
- Green foxtail (Setaria viridis)
- Drought stress
- Gene expression
- Transcription factors
- Small RNAs
- Next-generation sequencing
1 Introduction
Anthropogenic emissions of greenhouse gases have significantly contributed to global warming , resulting in increased atmospheric temperatures and unpredictable rainfall, which has serious impacts on agricultural productivit y (IPCC 2014). Drought is one immediate outcome of global climate change and poses severe threats to agriculture, with the degree of its effect depending on onset time, duration, and intensity. Global temperature has been increased by 1.2 °C over the past century, and it is projected to rise by an average of 3 °C by 2010 (IPCC 2014), which would markedly affect the survival and yield of food crops. Occurrence of drought stress at the reproductive stage of field crops causes an average yield loss of more than 50 % (Venuprasad et al. 2007). In Australia, wheat production was halved after a ±2 °C temperature variation (Asseng et al. 2011). The adverse effects of climate change and decrease in arable land as well as a growing world population that is expected to reach nine billion by 2050 demands immediate action for doubling crop yields to meet the challenge of food and nutrition security (Karp and Richter 2011). Among cultivated crops, C3 staple cereals such as wheat and rice are the worst affected by stresses imposed by climate change, particularly drought (Lal 2010). However, the productivity of underutilized grasses such as millets are affected less by drought as they are C4 crops with better water use efficiency and are tolerant to a broad spectrum of biotic and abiotic stresses (Sadras et al. 2011). Furthermore, millets are cultivated in the arid and semiarid tropics of the world, where there is limited availability of rainfall and irrigation.
Most millets belong to the subfamily Panicoideae of the Poaceae and generally have a short life cycle, produce characteristic small grains, and can withstand dry and elevated temperature conditions. Most importantly, millets can survive on nutritionally poor soils with little compromise on yield. Among millets, Setaria italica (foxtail millet) and S. viridis (green foxtail), members of the tribe Paniceae, are considered as a model for studying C4 photosynthesis and stress biolog y (Doust et al. 2009; Brutnell et al. 2010, 2015; Li and Brutnell 2011; Wang et al. 2011; Lata et al. 2013; Muthamilarasan and Prasad 2015). Reports have suggested the origin of cultivated S. italica from wild S. viridis ~11,000 years ago in Northern China (Yang et al. 2012). Presently, S. italica is being widely cultivated in tropical and subtropical regions of China, India, sub-Saharan Africa, and America for food and feed (Dwivedi et al. 2011). The prominent attributes of both S. italica and S. viridis (collectively, Setaria) include small diploid genomes (~490 Mb), short growing cycle (~90 days), small stature, C4 traits, potential stress tolerance, and significant genetic colinearity with biofuel grasses and major cereals (Doust et al. 2009; Brutnell et al. 2010, 2015; Li and Brutnell 2011; Lata et al. 2013; Diao et al. 2014; Muthamilarasan and Prasad 2015). Therefore, considering Setaria as a model, the genomes of S. italica “Yugu1” and S. viridis “A10” were sequenced by the Joint Genome Institute—U.S. Department of Energy (Bennetzen et al. 2012) and the genome of S. italica “Zhang gu” and “A2” was sequenced by the Beijing Genomics Institute, China (Zhang et al. 2012; Lata and Prasad 2013a). In addition, the transcriptome of S. italica tissues such as root, leaf, stem, and spica (tassel) from young seedlings has also been sequenced (Zhang et al. 2012).
The ability of these plants to tolerate or avoid stress by acclimatization and adaptation suggests that a repository of genetic diversity essential for enhancing yield stability exists in the germplasm of both S. italica and its wild progenitor S. viridis. Therefore, identification of key genetic determinants of drought tolerance in Setaria using QTL mapping, association mapping, and screening by recurrent selection is imperative, as this would enable the transfer of genes into other crops using genomics-assisted breeding. The first comparative transcriptome of S. italica cultivar “Mar51” (drought tolerant; Zhang et al. 2005) in response to drought stress using subtracted cDNA libraries reported the up-regulation of 95 and 57 ESTs in roots and shoots, respectively (Zhang et al. 2007). These expressed sequence tags (ESTs) showed tissue-specific expression patterns, and it was deduced that activation of glycolysis metabolism in roots is the first response to drought stress (Zhang et al. 2007). Similar subtractive hybridization analyses were performed by Lata et al. (2011) and Puranik et al. (2011a) in stress-tolerant S. italica cv. “Prasad” during drought and salinity stress. These studies reported 327 and 159 differentially expressed transcripts in drought and salt stressed libraries, respectively. Comparative analysis of these differentially expressed transcripts from both libraries revealed that only 10 % of them are similar (Puranik et al. 2011a). This demonstrated the existence of gene sets which are distinct for drought and salt stress, suggesting the presence of unique tolerance mechanisms to circumvent each stress.
The release of the S. italica genome sequence has expedited investigations on stress-related studies, and many reports are now available on the identification of stress-responsive genes that might confer durable tolerance (Lata et al. 2011, 2014; Mishra et al. 2012a, b, 2013; Puranik et al. 2013; Muthamilarasan et al. 2014a, b; Wang et al. 2014a, b; Zhu et al. 2014; Yadav et al. 2015a; Kumar et al. 2015). Further characterization of these genes through transgene-based approaches to understand their role in molecular, cellular, and physiological processes of drought tolerance would enable the transfer of this knowledge to other related crop species. Though studies on deciphering the mechanism of drought tolerance in S. italica commenced a decade ago, similar investigations in S. viridis have yet not been reported (Muthamilarasan and Prasad 2015). In this context, this chapter presents an overview of research efforts made towards identifying and characterizing the genetic determinants of drought tolerance in S. italica and the strategies to transfer these genes/QTLs into modern crop germplasm using genomic approaches.
2 Drought-Responsive Transcription Factors
The response of plants to drought stress is a complex process involving multiple dynamic responses at physiological, biochemical, and molecular levels. The stress signal perceived is communicated via sophisticated signal transduction networks to initiate the activity of stress-responsive transcription factors (TFs). Comparative transcriptome analysis of drought-tolerant S. italica cv. “Prasad” using a subtractive hybridization technique has identified two important stress-responsive transcription factors belonging to the DREB (dehydration-responsive element-binding proteins) and NAC (NAM, ATAF, and CUC) families (Lata et al. 2011). Transcript profiling of SiDREB2 and SiNAC2 genes using qRT-PCR in two S. italica cultivars with contrasting tolerance to dehydration stress (tolerant cv. “Prasad,” susceptible cv. “Lepakshi”) revealed significant up-regulation of these genes in the tolerant cultivar (Lata et al. 2011), suggesting putative involvement of these genes in stress-responsive mechanisms.
DREB is a subfamily of AP2/ERF (APETALA2/ethylene-responsive element-binding factor) TFs and participates in regulation of stress-responsive gene expression through ABA-independent pathways. This subfamily comprises two main subgroups, DREB1 and DREB2, which are involved in responses to chilling and drought, respectively (Lata and Prasad 2011). Cloning and characterization of SiDREB2 revealed that it is a nuclear localized 234 amino acid protein (25.7 kDa) encoded by 1119 bp cDNA (Lata et al. 2011). The SiDREB2 protein comprises a 58 amino acid AP2/ERF DNA-binding domain along with two functional amino acids, valine and glutamic acid, at the 14th and 19th residues, respectively. These two amino acids were identified in the DBA-binding domain and deduced to be important for binding with their respective cis-elements. Sequence alignment showed that the AP2/ERF DNA-binding domain of SiDREB2 was highly conserved among AP2/ERF TFs of other Poaceae members (Lata et al. 2011). Expression profiling of SiDREB2 in four S. italica cultivars (drought tolerant cv. “Prasad” and “IC-403579”; susceptible cv. “Lepakshi” and “IC-480117”) during different time-points of drought, salinity, and cold stress showed the up-regulation of this gene (up to 12-fold) in tolerant cultivars in response to drought and salinity. The strong responsiveness of SiDREB2 to drought and salinity in “Prasad” and “IC-403579” may be positively correlated to the tolerance behavior of these cultivars (Lata et al. 2011).
Sequence analysis of the SiDREB2 gene in 43 contrasting S. italica cultivars identified a synonymous SNP associated with dehydration tolerance at the 558th base pair (an A/G transition) (Lata et al. 2011). An allele-specific marker (ASM) was developed from this SNP and validated in a core set of 170 S. italica accessions (Lata et al. 2011). The regression of lipid peroxidation (LP) and relative water content (RWC) on this ASM demonstrated that the SiDREB2-associated trait contributes to ~27 % and ~20 % of the total variation in LP and RWC, respectively (Lata and Prasad 2012, 2013b).
A genome-wide survey was conducted using in silico approaches based on the role of DREB TFs in stress response (Lata et al. 2014). The study revealed 171 AP2/ERF-encoding genes in the S. italica genome (Fig. 16.1), of which 48 were DREB TFs identified by phylogenetic and domain architecture analysis. Transcript profiling of candidate genes was performed in drought-tolerant foxtail millet cultivar “IC-403579” exposed to 20 % polyethylene glycol (PEG 6000) and 250 mM sodium chloride, with transcript abundance analyzed at 1 h (early) and 24 h (late) post-stress treatments. The DREB gene SiAP2/ERF-002 was highly expressed in both phases of drought stress, and SiAP2/ERF-084 as well as SiAP2/ERF-090 was up-regulated in the late phase of drought and salinity stress. Hormonal treatment studies reported higher expression of SiAP2/ERF-084 and SiAP2/ERF-090 during the early phase of ethephone (converted into ethylene by the plant) and salicylic acid treatments, respectively. SiAP2/ERF-002 was up-regulated in both early and late phases of ethephone and salicylic acid treatments (Lata et al. 2014). The study identified SiAP2/ERF-002 as a potential candidate gene for further functional validation and overexpression studies with a view towards its utilization in crop improvement programs for stress tolerance.

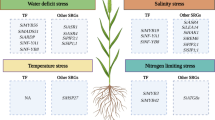
Chromosomal location of S. italica stress-responsive gene family members validated in response to drought stress. Vertical bars represent chromosomes and numbers in the left indicate the physical position (in Mbp). The respective gene names are provided in the right and the color codes correspond to each gene family. Genes which could be potential candidates for further functional characterization for drought stress tolerance are indicated by a star symbol (Refer Table 16.1)
NAC TFs are well known for their regulatory role in biotic as well as abiotic stress in many crop plants (Puranik et al. 2012). A subtractive hybridization study in S. italica also identified a significant up-regulation of SiNAC2 in both drought (Lata et al. 2011) and salinity stress libraries (Puranik et al. 2011a). Molecular characterization of SiNAC2 showed that the full length cDNA is 2051 bp with an open reading frame of 1386 bp encoding a protein of 462 amino acids (51.12 kDa). The full length SiNAC protein has a conserved NAC domain at its N-terminal (156 amino acids) along with a hypervariable C-terminal region. Using an electrophoretic mobility shift assay (EMSA), the DNA-binding site in the SiNAC2 protein has also been identified (Puranik et al. 2011b). Subcellular localization studies suggested that the SiNAC2 protein is membrane localized, and nuclear localization is also observed after deletion of the C-terminus. Expression profiling of SiNAC in S. italica cv. “Prasad” (drought tolerant) and “Lepakshi” (drought susceptible) in response to salinity and drought stress showed relatively higher levels of SiNAC transcripts in the tolerant cultivar, suggesting a positive role of SiNAC in stress response (Puranik et al. 2011b).
Similar reports are also available in other crop plants such as rice (SNAC1, Hu et al. 2006; OsNAC045, Zheng et al. 2009), soybean (GmNAC2, GmNAC3, GmNAC4, Pinheiro et al. 2009), and wheat (TaNAC4, Xia et al. 2010; TaNAC2a, Tang et al. 2012), substantiating the role of NAC TFs in diverse stress responses including drought tolerance. In view of this, a genome-wide analysis for identification and characterization of NAC TFs in S. italica was performed by Puranik et al. (2013). The study identified 147 SiNAC genes (Fig. 16.1), classified into 11 subfamilies. Of the 147 SiNAC genes, 50 candidate genes were chosen for quantitative expression analysis in response to various abiotic stresses. During drought stress, SiNAC062, SiNAC064, SiNAC070, and SiNAC128 were observed to be up-regulated during the early phase, whereas SiNAC024, SiNAC093, SiNAC100, SiNAC101, and SiNAC128 were up-regulated during the late phase of stress. The study identified SiNAC128 as a potential candidate for further in-depth characterization (Puranik et al. 2013).
Availability of the S. italica draft genome sequence in the public domain has facilitated the identification and characterization of a few important stress-responsive TFs namely, MYB and C2H2-type zinc fingers. MYB and C2H2 proteins represent the largest TF families in plants, playing crucial roles in various developmental and stress-responsive processes (Ambawat et al. 2013). Considering their significance, comprehensive genome-wide surveys were conducted, which led to the identification of 209 and 124 gene family members of MYB and C2H2, respectively (Fig. 16.1). Phylogenetic analysis categorized SiMYB proteins into ten groups (I–X) and SiC2H2 proteins into five groups (I–V). Comparative analysis of SiMYB and SiC2H2 protein sequences with their orthologs in sorghum, maize, and rice showed a remarkable conservation in overall protein structure (Muthamilarasan et al. 2014a, b).
Expression analysis of SiMYB and SiC 2 H 2 candidate genes in response to abiotic stresses and hormone treatments using qRT-PCR revealed specific and/or overlapping expression patterns of these genes. In the case of SiMYB genes, 11 candidate genes were chosen for expression profiling and three genes (SiMYB124, SiMYB126, and SiMYB150) showed significant up-regulation during drought stress (Muthamilarasan et al. 2014a). Among the nine SiC 2 H 2 genes selected for expression studies, SiC2H2_031 showed a gradual up-regulation with maximum expression at 48th hour (h) of drought stress, whereas SiC2H2_78, SiC2H2_85, and SiC2H2_94 showed higher expression during the early phase of drought stress. Altogether, these studies have identified potential candidate genes for further functional validation and utilization in crop improvement programs for stress tolerance (Table 16.1).
In view of the role of TFs in modulating stress-responsive gene regulatory networks, TF-encoding genes in S. italica genome have been identified and the TFs in silico characterized (Bonthala et al. 2014). The study identified 2297 putative TFs and categorized them in 55 families. This information is available in the Foxtail millet Transcription Factor Database (http://59.163.192.91/FmTFDb/) in which complete details of the TFs are compiled, including their sequences, physical positions, tissue-specific gene expression data, gene ontologies, and phylogeny (Bonthala et al. 2014). This database will be useful in pinpointing candidate TFs for stress-related studies and for performing large-scale investigations.
Though TFs are reported to be effectual in enhancing stress tolerance of transgenic plants by regulating the expression of broad-spectrum stress-related genes, the lack of an efficient transformation system for expressing/overexpressing the candidate TFs is a bottleneck in Setaria genomics (Diao et al. 2014) (but see Chaps. 20 and 21). As a consequence, the detailed molecular, cellular, and physiological mechanisms responsible for variation in drought tolerance among foxtail millet lines have not yet been elucidated.
3 Stress-Responsive Proteins in Drought Tolerance
Other than transcription factors, various stress-responsive proteins have been reported to play roles in conferring tolerance to drought stress (Hasanuzzaman et al. 2013). One such protein is WD40, which largely functions as a platform for protein–protein interactions and is involved in several biological process, such as signal transduction, transcriptional regulation, protein modification, cytoskeleton assembly, vesicular trafficking, DNA damage and repair, cell death, and cell cycle progression (Mishra et al. 2012a; Zhang and Zhang 2015). Reports have shown the association of these proteins with abiotic stress tolerance in crop plants (Zhu et al. 2008; Lee et al. 2010; Mishra et al. 2012a; Kong et al. 2015). In S. italica, ESTs encoding for putative WD-domain containing proteins and 14-3-3 like proteins were identified from a salinity and dehydration stress subtractive cDNA library (Lata et al. 2011; Puranik et al. 2011a). The full length cDNA of SiWD40 was deduced to be 1795 bp long with an ORF of 1314 bp, which encodes for a 437 amino acid protein (43.9 kDa). Protein modeling and analysis of SiWD40 revealed an eight blade β-propeller architecture at the C-terminus, with each blade comprising a four-stranded antiparallel β sheet (Mishra et al. 2012b). Transcript profiling of the SiWD40 gene in S. italica stress tolerant cv. “IC-403579” at different time-points of drought stress revealed a steady-state transcript accumulation from early to late phase with maximum expression at the 48th h of drought stress. Subcellular localization studies have shown the localization of the SiWD40 protein in the nucleus, and EMSA along with transactivation assays have revealed the regulation of SiWD40 gene expression by dehydration-responsive elements (DRE) (Mishra et al. 2012b).
Global analyses of WD40 protein-encoding genes in the S. italica genome showed the presence of 225 SiWD40 genes, classified into five subfamilies (Mishra et al. 2013) (Fig. 16.1). Expression analysis of 13 candidate WD40 genes in response to drought, salinity, and cold stresses has been performed. Among the candidate SiWD40 genes, SiWD40-028, SiWD40-037, SiWD40-063, SiWD40-106, SiWD40-156, and SiWD40-203 showed a gradual rise in expression levels and an average higher expression at 12–24 h (Mishra et al. 2013). This study suggests that SiWD40 proteins might play a prominent role in dynamically integrating multiple regulatory pathways mediating tolerance to abiotic stresses.
14-3-3 proteins are reported to regulate plant growth and development, and stress responses through protein–protein interactions, by binding with phosphoserine/phosphothreonine residues in the target proteins (Li and Dhaubhadel 2011; Denison et al. 2011). Bioinformatic prediction of 14-3-3 gene family members revealed the presence of 8 genes in the S. italica genome (Kumar et al. 2015) (Fig. 16.1). Further characterization revealed large variation in their structure, chromosomal localization, and protein properties, and in silico expression profiling indicated their higher expression in all the four investigated tissues of S. italica namely, roots, stems, leaves, and spikes. Comparative mapping to identify the orthologous genes in other grasses showed a high degree of conservation throughout the family. Subcellular localization studies showed differential localization of Si14-3-3_a, Si14-3-3_d, Si14-3-3_f, and Si14-3-3_h proteins within the cell. Si14-3-3_f was localized in cytoplasm and nuclear membrane, whereas the other three members were ubiquitously distributed (Kumar et al. 2015).
Transcript profiling of Si14-3-3 genes in response to drought and salinity stress as well as ABA, SA, and MeJA treatments indicated that these genes have varied expression patterns. During drought stress, a relatively high expression of Si14-3-3_a, Si14-3-3_c, Si14-3-3_d, Si14-3-3_f, and Si14-3-3_g was reported at the early phase. Further downstream characterization indicated the interaction of Si14-3-3 with a nucleocytoplasmic shuttling phosphoprotein (SiRSZ21A) in a phosphorylation-dependent manner, demonstrating that Si14-3-3 might regulate the splicing events by binding with phosphorylated SiRSZ21A (Kumar et al. 2015). The demonstration of an interaction between Si14-3-3 and SiRSZ21A provides novel clues on the involvement of 14-3-3 proteins in splicing events. In this context, it would be interesting to investigate the protein–protein interaction behavior of 14-3-3 proteins during environmental stresses.
Cytokinins are reported to participate in various aspects of plant growth, development, and stress adaptations (Havlova et al. 2008), and the level of cytokinins is fine-tuned by cytokinin oxidase/dehydrogenases (CKXs) (Gajdosová et al. 2011). In maize and soybean, CKX genes have responded to drought and salinity stress (Vyroubalova et al. 2009; Le et al. 2012). Considering this, Wang et al. (2011) conducted a comprehensive genome-wide survey and identified 11 SiCKX genes in the S. italica genome. Phylogenetic analysis of SiCKX proteins with rice and Arabidopsis orthologs classified them in two groups. The relative transcript levels of SiCKX genes in germinating embryos under drought stress showed higher expression of all SiCKX genes except SiCKX2 and SiCKX11 (Wang et al. 2011), substantiating the role of SiCKX genes in response to drought stress.
Aldehyde dehydrogenases (ALDHs) are a conserved gene family encoding NAD (P)+-dependent enzymes, which catalyze the irreversible oxidation of broad-spectrum endogenous and exogenous aromatic and aliphatic aldehydes into corresponding carboxylic acids (Yoshida et al. 1998). Studies have reported the involvement of these ALDHs in guarding plants from various biotic and abiotic stresses by indirectly detoxifying cellular reactive oxygen species (ROS) and/or reducing lipid peroxidation (Singh et al. 2013). In view of this, Zhu et al. (2014) performed a genome-wide analysis and identified 20 ALDH genes in S. italica. The study categorized these SiALDH genes into ten gene families and examined their duplication and divergence, chromosomal distribution, gene structure, and orthologous relationships with rice. Further, the SiALDH genes were subjected to quantitative expression analysis in response to drought, salt, high and low temperature, and hydrogen peroxide stress treatments. In the event of drought stress, all the SiALDH2 genes were up-regulated except SiALDH2C1, SiALDH3H2, and SiALDH11A1. Of note, SiALDH2C4 showed maximum expression at the 6th hour of drought stress (Zhu et al. 2014) and is a potential candidate for modifying drought stress response.
Late embryogenesis abundant (LEA) proteins are accumulated in seeds during the later stage of development before the desiccation phase, and these proteins are also reported to function in protecting the plants from environmental stresses (He et al. 2012; Liu et al. 2013). Reports have shown the accumulation of LEA proteins during drought stress, and their overexpression confers stable tolerance to water deficit in transgenic plants (Xu et al. 1996; Colmenero-Flores et al. 1999; Goyal et al. 2005; Tolleter et al. 2010). In S. italica, a novel member of the atypical subgroup 5C LEA gene named SiLEA14 was functionally characterized (Wang et al. 2014a, b). The full length sequence of SiLEA14 has 821 bp, encoding a 170 amino acid LEA protein (18.77 kDa), which is cytosol localized. Expression levels of SiLEA14 under drought stress showed an immediate induction within 0.5 h of stress initiation and maximum expression was reported at 12 h. Overexpression of SiLEA14 in Arabidopsis and S. italica enhanced the tolerance of transgenic plants to drought stress, with a higher accumulation of proline and sugar (Fig. 16.1; Wang et al. 2014a, b). The findings suggested that overexpression of SiLEA14 gene in crop plants might improve tolerance to drought stress, but further experimental validation is required to support this conclusion.
Studies have been performed to identify the members of the RNA silencing machinery, such as Dicer-like (DCL), Argonaute (AGO), and RNA-dependent RNA polymerase (RDR) genes, and understand how they regulate gene expression during abiotic stress in Arabidopsis (Henderson et al. 2006), rice (Kapoor et al. 2008), maize (Qian et al. 2011), tomato (Bai et al. 2012), and poplar (Zhao et al. 2015). Similar efforts have identified 8 DCL, 19 AGO, and 11 RDR genes in S. italica (Yadav et al. 2015a) (Fig. 16.1). These genes have been characterized using in silico approaches, and expression profiling was performed for candidate genes (4 DCL, 5 AGO, and 3 RDR) in response to salinity and drought stress in two S. italica cultivars (cv. “IC04,” stress tolerant; cv. “IC41,” stress susceptible). The results found a differential expression pattern of candidate genes at two different time-points, stresses and cultivars, thus suggesting the participation of these genes in a complex molecular network of stress response (Yadav et al. 2015a). Significantly higher expression levels of SiDCL01, SiDCL06, SiAGO08, SiAGO018, and SiRDR07 during drought stress suggests that these genes should be further functionally analyzed. Altogether, the identification, characterization, and expression profiling of stress-responsive protein-encoding genes in S. italica has established the putative role of respective proteins in complex networks of pathways to perform diverse physiological, molecular, and cellular functions in response to drought and other stresses. Though the expression profiling studies provide a clue to the role of these proteins in imparting stress tolerance (Table 16.1), comprehensive functional characterization is required to confirm their functionality and durability in conferring tolerance.
4 Small RNAs in Drought-Regulated Gene Expression
Small RNAs (sRNAs) are a part of noncoding RNAs, and they comprise two major classes namely, microRNAs (miRNAs) and endogenous small interfering RNAs (siRNAs). Both miRNAs and siRNAs are identified as modulators of gene expression at the post-transcriptional level and have emerged as key players in stress responses (Sunkar et al. 2007). miRNAs regulate the expression of the target transcript by binding to reverse complementary sequences, causing cleavage of the target RNA, whereas siRNAs bind to the target sequence in a similar manner and direct DNA methylation (Khraiwesh et al. 2012). Involvement of miRNAs in various biotic and abiotic stresses including drought (Zhao et al. 2007; Liu et al. 2008; Zhou et al. 2010), cold (Zhou et al. 2008), salinity (Liu et al. 2008; Sunkar et al. 2008), bacterial infection (Navarro et al. 2006), UV-B radiation (Zhou et al. 2007), and mechanical stress (Lu et al. 2005) have been well documented. Advances in high-throughput sequencing and small RNA profiling have facilitated the sequencing of small RNA libraries of drought stress samples for identification of drought-related miRNAs (Ding et al. 2013; Rajwanshi et al. 2014).
The genomic and CDS sequences of S. italica were analyzed for plant miRNA sequences, and 355 mature miRNAs (Sit-miR) were identified and classified into 53 families (Khan et al. 2014). Secondary structures and putative targets of Sit-miRs were then identified, followed by chromosomal localization, comparative mapping, and tissue-specific expression profiling. Northern blot analysis and stem-loop RT-qPCR of candidate Sit-miRs in response to different abiotic stresses in two S. italica cultivars (“IC-403579,” stress tolerant; “IC-480117,” stress susceptible) were performed. The analysis showed up- and down-regulation of candidate Sit-miRs during various stresses. In the drought-tolerant cultivar, Sit-miR156c, Sit-miR397a, Sit-miR393, Sit-miR160d, and Sit-miR6248a were down-regulated. Up-regulation of Sit-miR162a, Sit-miR167b, and Sit-miR171b was also observed in the tolerant cultivar when compared to the expression levels of respective Sit-miRs in susceptible cultivars (Khan et al. 2014). The complete data of identified sit-miRs including chromosomal location, length, MFE, AMFE, sequences of pre-miRNA and mature miRNA, secondary structure, and target gene information have been made available to the global research community through an open access web resource, Foxtail millet miRNA Database (http://59.163.192.91/FmMiRNADb/index.html; Khan et al. 2014).
The use of two S. italica cultivars “IC-403579” (stress tolerant) and “IC-480117” (stress susceptible) in all the functional genomics studies discussed above encouraged us to construct four small RNA libraries from control and drought-stressed seedlings of these cultivars, which were then sequenced using the Illumina HiSeq 2000 platform (Yadav et al. 2016). A total of 55 known miRNAs (representing 23 miRNA families) and 136 novel miRNAs (representing 47 miRNA families) were identified in this study. Other downstream analyses such as chromosomal positioning, structure and target prediction, target annotation and validation, and expression profiling were performed, and a few candidate novel dehydration-responsive Sit-miRs were validated by stem-loop quantitative real-time PCR. Further functional characterization of these Sit-miRs are in progress (Yadav et al. 2016). Of note, this study showed differential expression pattern of Sit-miRs in response to drought, which may play an important role in providing the contrasting tolerance characteristics of these cultivars.
A similar bioinformatic approach was followed by Khan et al. (2014), with some modifications in the filtering criteria used by Han et al. (2014), and 271 miRNAs belonging to 44 families were predicted. The study identified 23 pairs of sense/antisense miRNAs and 18 miRNA clusters as well as 432 targets for 38 miRNA families. Of these, 43 miRNAs were chosen for tissue-specific expression profiling in S. italica leaves, roots, stems, and spikes, and five predicted targets of four miRNAs were experimentally validated using 5′-RLM-RACE (Han et al. 2014). In another study, two small RNA libraries constructed from shoot tissue of S. italica inbred line “Yugu1” were sequenced using the Illumina HighSeq 2000 platform, and 43 known miRNAs, 172 novel miRNAs, and 2 miRNA precursor candidates were identified (Yi et al. 2013). Targets of these miRNAs were predicted and annotated, followed by validation of candidate miRNAs by stem-loop RT-PCR in four tissues (Yi et al. 2013). Though these studies are insightful in understanding the miRNAome of S. italica, the role of identified miRNAs in response to drought and other stresses remains elusive.
Investigation of genome-wide transcriptome reconfiguration in S. italica challenged by drought stress was performed by Qi et al. (2013). RNA and sRNA libraries were constructed from drought stressed and unstressed (control) whole seedlings of S. italica “Yugu1” and sequenced. Among the sRNAs, 24-nt (nucleotide) sRNAs were found to be predominant followed by 21, 22, and 23-nt sRNA. The study inferred that decreased levels of 24-nt siRNA around genic regions have a negative role in influencing gene expression in response to drought stress. Particularly, the differential expression analysis identified the maximum levels of 19 long noncoding RNAs during drought stress and, among these, two natural antisense transcripts (NATs of Si003758m and Si038715m) showed drought-regulated expression patterns (Qi et al. 2013). The generated raw reads from the studies of Yi et al. (2013) and Qi et al. (2013) are available in the NCBI SRA database under accession numbers SRA062640 and SRA062827.
Taken together, it is understood that identification and characterization of target genes is important for delineating the role of sRNAs. Prediction of sRNAs and their targets followed by their functional analysis will assist in understanding the complex miRNA- and siRNA-mediated regulatory networks controlling stress-responsive machinery in Setaria.
5 Strategies for Identifying Genetic Determinants of Drought Tolerance
Comprehensive molecular investigations have elucidated the role of a few known genes and gene families in stress responsiveness of S. italica, but so far no novel gene has been reported for stress tolerance. The genome annotation data of S. italica “Zhang gu” and “A2” has identified 1517 foxtail millet-specific gene families, of which 586 genes were annotated as “response to water” [GO:0009415, defined as any process that results in a change in state or activity of a cell or an organism (in terms of movement, secretion, enzyme production, gene expression, etc.) as a result of a stimulus reflecting the presence, absence, or concentration of water] (Zhang et al. 2012). This demonstrates the existence of unexplored genetic determinants which could be responsible for stress-tolerance behavior and adaptation of Setaria to arid and semiarid environments. Furthermore, advances in next-generation sequencing (NGS) technologies and high-throughput analysis platforms provide an excellent opportunity to explore the genome and transcriptome of Setaria for pinpointing genes/alleles/QTLs responsible for drought tolerance.
Reports have indicated that drought tolerance is a complex trait dynamically controlled by numerous genes, and it is therefore imperative to identify the candidate genes to decipher the mechanism of drought response in Setaria, which would expedite genetic improvement either by molecular breeding or transgene-based approaches. Until now, genetic studies using approaches like subtractive cDNA hybridization, qRT-PCR, and cDNA microarray have been performed in Setaria (Diao et al. 2014; Muthamilarasan and Prasad 2015), but mapping of QTL at the gene level through map-based cloning has not been reported. In this context, NGS platforms can provide a comprehensive insight into sequence variations in the genome, and whole genome resequencing (WGR) could assist in detecting polymorphisms, develop high-throughput markers, understand epigenetic modifications, identify splice variants, and perform expression profiling and DNA footprinting (Fig. 16.2) (Delseny et al. 2010). NGS is also used for identification of candidate genes and variants underlying important traits by linkage mapping, genome-wide association mapping, and genotyping-by-sequencing (Fig. 16.2) (Varshney et al. 2014).

An integrated strategy of classical and reverse genetics for dissecting the drought-tolerance mechanisms and enhancing tolerance is illustrated. DT drought tolerance, DS drought susceptibility, GWAS genome-wide association study, QTLs quantitative trait loci, NIL near-isogenic line, IL isogenic line, MABC marker-assisted backcrossing, MARS marker-assisted recurrent selection, MAS marker-assisted selection, GWS genome-wide selection
Identifying and utilizing the sequence variation present in the genome is imperative for crop genetics and breeding. Availability of the S. italica genome sequence in public databases (Phytozome: http://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Sitalica; Foxtail millet Database: http://foxtailmillet.genomics.org.cn/page/species/index.jsp; PlantGDB: http://www.plantgdb.org/SiGDB/; Setaria italica Functional Genomics Database: http://structuralbiology.cau.edu.cn/SIFGD/) has enabled the development of high-throughput molecular markers such as microsatellites (Pandey et al. 2013; Kumari et al. 2013; Zhang et al. 2014), intron-length polymorphisms (Muthamilarasan et al. 2013), miRNA-based (Yadav et al. 2014) and transposable elements-based markers (Yadav et al. 2015b), and development of a marker database (http://www.nipgr.res.in/foxtail.html; Suresh et al. 2013). The genome sequence information has also served as a reference in whole genome resequencing (WGR) of 916 S. italica accessions collected from different eco-geographical zones of the world and the construction of a high density haplotype map using 85 million single nucleotide polymorphisms, which revealed genomic variations among these accessions (Jia et al. 2013). Similarly, the S. italica genome sequence data facilitates WGR of cultivated and wild varieties of Setaria with contrasting phenotypes to identify novel genes/alleles/QTLs underlying drought response and to execute NGS-based genomics-assisted breeding for drought tolerance. Though the S. viridis genome has also been sequenced (Bennetzen et al. 2012), the lack of publically available sequence information has for a long period significantly impeded the development of genetic and genomic resources in this important model species. However, recent developments have made this data available in the web portal “Setariabase” (http://www.sviridis.org; Brutnell et al. 2015).
In addition to WGR, transcriptome sequencing could be useful for elucidating the transcriptional and post-transcriptional regulation of genes in response to drought stress and to understand the global expression pattern of the Setaria genome (Deyholos 2010). Transcriptomes of four S. italica tissues namely root, stem, leaf, and spike, as well as RNA-seq data of whole seedlings under drought conditions is already available in the NCBI SRA database (Zhang et al. 2012; Qi et al. 2013). In the case of S. viridis, RNA-seq data of leaf, stem, node, crown, root, spikelet, floret, and seed tissues at three developmental stages including seed germination, vegetative growth, and reproduction is available in NCBI SRA (Xu et al. 2013). Using these data as a reference, transcriptomes of Setaria accessions with distinctive phenotypes could be sequenced and compared to identify novel transcripts, which could be responsible for the trait-of-interest. Besides transcriptome sequencing, a few small RNA libraries have also been sequenced in control and stressed conditions, and miRNAs modulating drought stress-associated processes and gene networks identified (Yi et al. 2013; Qi et al. 2013; Yadav et al. 2016). Moreover, examining the DNA methylation profiles and small RNA profiles of drought-stressed libraries will facilitate the identification of genes and/or regions that are regulated by miRNA/siRNA-mediated DNA methylation, which could contribute to epigenetic inheritance of drought stress tolerance.
Proteomics offers versatile approaches for identifying drought-responsive proteins and corresponding genes (Kamal et al. 2010). More importantly, proteomics techniques are potent tools to delineate stress-responsive proteins and their corresponding genes even when genome sequence information is not available. A suggested pathway for such an analysis could include the resolution of total protein of Setaria cultivars subjected to drought stress and control conditions on two-dimensional gel electrophoresis, followed by in-gel digestion and MALDI-TOF mass spectrometry analysis . Subsequent in silico protein and nucleotide BLAST searches (e.g., using the MASCOT program; Brosch et al. 2009) would reveal differentially expressed proteins and their corresponding genes. Thus, proteomics could bridge the gap between transcriptome and metabolome and complement genomics approaches.
Recently, research has shifted from understanding the physiological and molecular responses of crop plants exposed to individual stress to those exposed to a combination of stresses, as numerous reports have demonstrated that the response of plants to concurrent stresses is unique and not directly extrapolated from the individual stress responses (Mittler 2006; Atkinson and Urwin 2012; Rasmussen et al. 2013; Suzuki et al. 2014; Ramegowda and Senthil-Kumar 2015). All the stress-related investigations conducted in Setaria were in response to different stresses applied individually and so far studies on the impact of combined/concurrent stress have not been performed. However, we can speculate that combinations of stresses could activate complex pathways controlled by different signaling events, which might be unique to Setaria. Therefore, research programs should focus on understanding the tolerance mechanism of Setaria to a combination of biotic and abiotic stress conditions, especially the stresses which mimic field conditions.
In the “-omics” era, identifying the genetic determinants for drought tolerance might not be a difficult task, but understanding the exact role of those genetic determinants in improving drought tolerance is challenging due to the lack of efficient Setaria transformation systems (Diao et al. 2014; Muthamilarasan and Prasad 2015; Brutnell et al. 2015). The availability of high-throughput genetic transformation systems has accelerated the maturation of rice and Arabidopsis genomics, but limited availability of such efficient protocols in Setaria has impeded further molecular studies in this model crop (Diao et al. 2014; Muthamilarasan and Prasad 2015). Further advances in transformation are reported in this volume (Chaps. 20 and 21). Irrespective of this, many candidate genes have been identified in S. italica which could be utilized in improving abiotic stress adaptation of other cereal crops.
6 Conclusions
Global climate change has caused irreversible damage to the earth’s environment, which includes rise in atmospheric temperature, melting of glaciers, increase in sea levels, and changes in rainfall patterns. Emission of greenhouse gases due to extreme anthropogenic activities and extensive deforestation regimes accelerate the effects of climate change. These adverse conditions have severe impact on yields of crop plants, especially cereals (IPCC 2014) while, on the other hand, agricultural productivity needs to increase globally by an estimated 60 % by 2050 to meet the food and feed demands of a growing population (FAO 2015). Development of climate change resilient crops is the principal solution for this aggravating problem, but genes that enable growth and reproduction in adverse environments might have been lost in all the presently cultivated crops during domestication and improvement. Reports have shown that genetic diversity for stress tolerance, which enhances yield stability could be present in traditional landraces, wild relatives, and genetically close crop plants which are well adapted to adverse environments (Khoury et al. 2013; Atwell et al. 2014; Brutnell et al. 2015).
Being cereal crops, Setaria sp. and major cereals show high levels of synteny at the genome level, and, as discussed in the above sections, S. italica and S. viridis are tractable models for understanding stress biology and C4 photosynthesis. Both species are tolerant to drought stress, which is the major impact of climate change, and in view of this, attempts have been made to identify the genetic determinants of drought stress using various molecular genetic and genomic approaches. At present, functional characterization is required to confirm the drought responsiveness of the identified genes and once confirmed, these could be introgressed into major cereals for generating elite cultivars with durable stress tolerance.
References
Ambawat S, Sharma P, Yadav NR, Yadav RC. MYB transcription factor genes as regulators for plant responses: an overview. Physiol Mol Biol Plants. 2013;19:307–21.
Asseng S, Foster I, Turner NC. The impact of temperature variability on wheat yields. Global Change Biol. 2011;17:997–1012.
Atkinson NJ, Urwin PE. The interaction of plant biotic and abiotic stresses: from genes to the field. J Exp Bot. 2012;63:3523–43.
Atwell BJ, Wang H, Scafaro AP. Could abiotic stress tolerance in wild relatives of rice be used to improve Oryza sativa? Plant Sci. 2014;215–216:48–58.
Bai M, Yang GS, Chen WT, Mao ZC, Kang HX, Chen GH, Yang YH, Xie BY. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene. 2012;501:52–62.
Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, Estep M, Feng L, Vaughn JN, Grimwood J, Jenkins J, Barry K, Lindquist E, Hellsten U, Deshpande S, Wang X, Wu X, Mitros T, Triplett J, Yang X, Ye CY, Mauro-Herrera M, Wang L, Li P, Sharma M, Sharma R, Ronald PC, Panaud O, Kellogg EA, Brutnell TP, Doust AN, Tuskan GA, Rokhsar D, Devos KM. Reference genome sequence of the model plant Setaria. Nat Biotechnol. 2012;30:555–61.
Bonthala VS, Muthamilarasan M, Roy R, Prasad M. FmTFDb: a foxtail millet transcription factors database for expediting functional genomics in millets. Mol Biol Rep. 2014;41:6343–8.
Brosch M, Yu L, Hubbard T, Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res. 2009;8:3176–81.
Brutnell TP, Wang L, Swartwood K, Goldschmidt A, Jackson D, Zhu XG, Kellogg E, Van Eck J. Setaria viridis: a model for C4 photosynthesis. Plant Cell. 2010;22:2537–44.
Brutnell TP, Bennetzen JL, Vogel JP. Brachypodium distachyon and Setaria viridis: model genetic systems for the grasses. Annu Rev Plant Biol. 2015;65:715–41.
Colmenero-Flores JM, Moreno LP, Smith CE, Covarrubias AA. Pvlea-18, a member of a new late-embryogenesis-abundant protein family that accumulates during water stress and in the growing regions of well-irrigated bean seedlings. Plant Physiol. 1999;120:93–104.
Delseny M, Han B, Hsing YI. High throughput DNA sequencing: the new sequencing revolution. Plant Sci. 2010;179:407–22.
Denison FC, Paul AL, Zupanska AK, Ferl RJ. 14-3-3 proteins in plant physiology. Semin Cell Dev Biol. 2011;22:720–7.
Deyholos MK. Making the most of drought and salinity transcriptomics. Plant Cell Environ. 2010;33:648–54.
Diao X, Schnable J, Bennetzen JL, Li J. Initiation of Setaria as a model plant. Front Agri Sci Eng. 2014;1:16–20.
Ding Y, Tao Y, Zhu C. Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot. 2013;64:3077–86.
Doust AN, Kellogg EA, Devos KM, Bennetzen JL. Foxtail millet: a sequence-driven grass model system. Plant Physiol. 2009;149:137–41.
Dwivedi S, Upadhyaya H, Senthilvel S, Hash C, Fukunaga K, Diao X, Santra D, Baltensperger D, Prasad M. Millets: genetic and genomic resources. Plant Breed Rev. 2011;35:247–375.
FAO. The post-2015 development agenda and the millennium development goals. 2015. http://www.fao.org/post-2015-mdg/14-themes/climate-change/en/. Accessed 10 Apr 2015.
Gajdosová S, Spíchal L, Kamínek M, Hoyerová K, Novák O, Dobrev PI, Galuszka P, Klíma P, Gaudinová A, Zizková E, Hanus J, Dancák M, Trávnícek B, Pesek B, Krupicka M, Vanková R, Strnad M, Motyka V. Distribution, biological activities, metabolism, and the conceivable function of cis-zeatin-type cytokinins in plants. J Exp Bot. 2011;62:2827–40.
Goyal K, Walton LJ, Tunnacliffe A. LEA proteins prevent protein aggregation due to water stress. Biochem J. 2005;388:151–7.
Han J, Xie H, Sun Q, Wang J, Lu M, Wang W, Guo E, Pan J. Bioinformatic identification and experimental validation of miRNAs from foxtail millet (Setaria italica). Gene. 2014;546:367–77.
Hasanuzzaman M, Nahar K, Alam MM, Roychowdhury R, Fujita M. Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int J Mol Sci. 2013;14:9643–84.
Havlova M, Dobrev PI, Motyka V, Storchova H, Libus J, Dobra J, Malbeck J, Gaudinova A, Vankova R. The role of cytokinins in responses to water deficit in tobacco plants over-expressing trans-zeatin O-glucosyltransferase gene under 35S or SAG12 promoters. Plant Cell Environ. 2008;31:341–53.
He S, Tan L, Hu Z, Chen G, Wang G, Hu T. Molecular characterization and functional analysis by heterologous expression in E. coli under diverse abiotic stresses for OsLEA5, the atypical hydrophobic LEA protein from Oryza sativa L. Mol Genet Genomics. 2012;287:39–54.
Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, Jacobsen SE. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet. 2006;38:721–5.
Hu H, Dai M, Yao J, Xiao B, Xianghua L, Zhang Q, Xiong L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc Natl Acad Sci U S A. 2006;A103:12987–92.
IPCC. The Intergovernmental Panel on Climate Change: synthesis report. 2014. http://www.ipcc.ch/report/ar5/syr/. Accessed 15 Apr 2015.
Jia G, Huang X, Zhi H, Zhao Y, Zhao Q, Li W, Chai Y, Yang L, Liu K, Lu H, Zhu C, Lu Y, Zhou C, Fan D, Weng Q, Guo Y, Huang T, Zhang L, Lu T, Feng Q, Hao H, Liu H, Lu P, Zhang N, Li Y, Guo E, Wang S, Wang S, Liu J, Zhang W, Chen G, Zhang B, Li W, Wang Y, Li H, Zhao B, Li J, Diao X, Han B. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat Genet. 2013;45:957-96.
Kamal AHM, Kim KH, Shin KH, Choi JS, Baik BK, Tsujimoto H, Heo HY, Park CS, Woo SH. Abiotic stress responsive proteins of wheat grain determined using proteomics technique. Aust J Crop Sci. 2010;4:196–208.
Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, Kapoor S. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics. 2008;9:451.
Karp A, Richter GM. Meeting the challenge of food and energy security. J Exp Bot. 2011;62:3263–71.
Khan Y, Yadav A, Suresh BV, Muthamilarasan M, Yadav CB, Prasad M. Comprehensive genome-wide identification and expression profiling of foxtail millet [Setaria italica (L.)] miRNAs in response to abiotic stress and development of miRNA database. Plant Cell Tiss Organ Cult. 2014;118:279–92.
Khoury CK, Greene S, Wiersema J, Maxted N, Jarvis A, Struik PC. An inventory of crop wild relatives of the United States. Crop Sci. 2013;53:1496–508.
Khraiwesh B, Zhu JK, Zhu J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta. 2012;1819:137–48.
Kong D, Li M, Dong Z, Ji H, Li X. Identification of TaWD40D, a wheat WD40 repeat-containing protein that is associated with plant tolerance to abiotic stresses. Plant Cell Rep. 2015;34:395–410.
Kumar K, Muthamilarasan M, Bonthala VS, Roy R, Prasad M. Unraveling 14-3-3 proteins in C4 panicoids with emphasis on model plant Setaria italica reveals phosphorylation-dependent subcellular localization of RS splicing factor. PLoS One. 2015;10:e0123236.
Kumari K, Muthamilarasan M, Misra G, Gupta S, Subramanian A, Parida SK, Chattopadhyay D, Prasad M. Development of eSSR-markers in Setaria italica and their applicability in studying genetic diversity, cross-transferability and comparative mapping in millet and non-millet species. PLoS One. 2013;8:e67742.
Lal R. Managing soils for a warming earth in a food-insecure and energy-starved world. J Plant Nutr Soil Sci. 2010;173:4–15.
Lata C, Prasad M. Role of DREBs in regulation of abiotic stress responses in plants. J Exp Bot. 2011;14:4731–48.
Lata C, Prasad M. Validation of an allele-specific marker associated with dehydration stress tolerance in a core set of foxtail millet accessions. Plant Breed. 2012;132:496–9.
Lata C, Prasad M. Setaria genome sequencing: an overview. J Plant Biochem Biotechnol. 2013a;22:257–60.
Lata C, Prasad M. Association of an allele-specific marker with dehydration stress tolerance in foxtail millet suggests SiDREB2 to be an important QTL. J Plant Biochem Biotechnol. 2013b;23:119–22.
Lata C, Bhutty S, Bahadur RP, Majee M, Prasad M. Association of an SNP in a novel DREB2-like gene SiDREB2 with stress tolerance in foxtail millet [Setaria italica (L.)]. J Exp Bot. 2011;62:3387–401.
Lata C, Gupta S, Prasad M. Foxtail millet: a model crop for genetic and genomic studies in bioenergy grasses. Crit Rev Biotechnol. 2013;33:328–43.
Lata C, Mishra AK, Muthamilarasan M, Bonthala VS, Khan Y, Prasad M. Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS One. 2014;9:e113092.
Le DT, Nishiyama R, Watanabe Y, Vankova R, Tanaka M, Seki M, Ham LH, Yamaguchi-Shinozaki K, Shinozaki K, Tran LSP. Identification and expression analysis of cytokinin metabolic genes in soybean under normal and drought conditions in relation to cytokinin levels. PLoS One. 2012;7:e42411.
Lee S, Lee J, Paek K-H, Kwon S-Y, Cho H, Kim S, Park J. A novel WD40 protein, BnSWD1, is involved in salt stress in Brassica napus. Plant Biotechnol Rep. 2010;4:165–72.
Li P, Brutnell TP. Setaria viridis and Setaria italica, model genetic systems for the panicoid grasses. J Exp Bot. 2011;62:3031–7.
Li X, Dhaubhadel S. Soybean 14-3-3 gene family: identification and molecular characterization. Planta. 2011;233:569–82.
Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14:836–43.
Liu Y, Wang L, Xing X, Sun L, Pan J, Kong X, Zhang M, Li D. ZmLEA3, a multifunctional group 3 LEA protein from maize (Zea mays L.), is involved in biotic and abiotic stresses. Plant Cell Physiol. 2013;54(6):944–59.
Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL. Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell. 2005;17:2186–203.
Mishra AK, Puranik S, Bahadur RP, Prasad M. The DNA-binding activity of an AP2 protein is involved in transcriptional regulation of a stress-responsive gene, SiWD40, in foxtail millet. Genomics. 2012a;100:252–63.
Mishra AK, Puranik S, Prasad M. Structure and regulatory networks of WD40 protein in plants. J Plant Biochem Biotechnol. 2012b;21:32–9.
Mishra AK, Muthamilarasan M, Khan Y, Parida SK, Prasad M. Genome-wide investigation and expression analyses of WD40 protein family in the model plant foxtail millet (Setaria italica L.). PLoS One. 2013;9:e86852.
Mittler R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006;11:15–9.
Muthamilarasan M, Prasad M. Advances in Setaria genomics for genetic improvement of cereals and bioenergy grasses. Theor Appl Genet. 2015;128:1–14.
Muthamilarasan M, Venkata Suresh B, Pandey G, Kumari K, Parida SK, Prasad M. Development of 5123 intron-length polymorphic markers for large-scale genotyping applications in foxtail millet. DNA Res. 2013;21:41–52.
Muthamilarasan M, Khandelwal R, Yadav CB, Bonthala VS, Khan Y, Prasad M. Identification and molecular characterization of MYB transcription factor superfamily in C4 model plant foxtail millet (Setaria italica L.). PLoS One. 2014a;9:e109920.
Muthamilarasan M, Bonthala VS, Mishra AK, Khandelwal R, Khan Y, Roy R, Prasad M. C2H2-type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct Integr Genomics. 2014b;14:531–43.
Muthamilarasan M, Bonthala VS, Khandelwal R, Jaishankar J, Shweta S, Nawaz K, Prasad M. Global analysis of WRKY transcription factor superfamily in Setaria identifies potential candidates involved in abiotic stress signaling. Front Plant Sci. 2015a;6:910.
Muthamilarasan M, Khan Y, Jaishankar J, Shweta S, Lata C, Prasad M. Integrative analysis and expression profiling of secondary cell wall genes in C4 biofuel model Setaria italica reveals targets for lignocellulose bioengineering. Front Plant Sci. 2015b;6:965.
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JD. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006;312:436–9.
Pandey G, Misra G, Kumari K, Gupta S, Parida SK, Chattopadhyay D, Prasad M. Genome-wide development and use of microsatellite markers for large-scale genotyping applications in foxtail millet [Setaria italica (L.)]. DNA Res. 2013;20:197–207.
Pinheiro GL, Marques CS, Costa MDBL, Reis PAB, Alves MS, Carvalho CM, Fietto LG, Fontes EPB. Complete inventory of soybean NAC transcription factors: sequence conservation and expression analysis uncover their distinct roles in stress response. Gene. 2009;444:10–23.
Puranik S, Jha S, Srivastava PS, Sreenivasulu N, Prasad M. Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J Plant Physiol. 2011a;168:280–7.
Puranik S, Kumar K, Srivastava PS, Prasad M. Electrophoretic mobility shift assay reveals a novel recognition sequence for Setaria italica NAC protein. Plant Signal Behav. 2011b;6:1588–90.
Puranik S, Sahu PP, Srivastava PS, Prasad M. NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 2012;17:369–81.
Puranik S, Sahu PP, Mandal SN, Venkata Suresh B, Parida SK, Prasad M. Comprehensive genome-wide survey, genomic constitution and expression profiling of the NAC transcription factor family in foxtail millet (Setaria italica L.). PLoS One. 2013;8:e64594.
Qi X, Xie S, Liu Y, Yi F, Yu J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol Biol. 2013;83:459–73.
Qian Y, Cheng Y, Cheng X, Jiang H, Zhu S, Cheng B. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 2011;30:1347–63.
Rajwanshi R, Chakraborty S, Jayanandi K, Deb B, Lightfoot DA. Orthologous plant microRNAs: microregulators with great potential for improving stress tolerance in plants. Theor Appl Genet. 2014;127:2525–43.
Ramegowda V, Senthil-Kumar M. The interactive effects of simultaneous biotic and abiotic stresses on plants: mechanistic understanding from drought and pathogen combination. J Plant Physiol. 2015;176:47–54.
Rasmussen S, Barah P, Suarez-Rodriguez MC, Bressendorff S, Friis P, Costantino P, Bones AM, Nielsen HB, Mundy J. Transcriptome responses to combinations of stresses in Arabidopsis. Plant Physiol. 2013;161:1783–94.
Sadras V, Grassini P, Steduto P. Status of water use efficiency of main crops. In: The state of world’s land and water resources for food and agriculture (SOLAW). Rome: FAO, London: Earthscan. 2011. http://www.fao.org/fileadmin/templates/solaw/files/thematic_reports/TR_07_web.pdf. Accessed 2 Feb 2015.
Singh S, Brocker C, Koppaka V, Chen Y, Chen Y, Jackson BC, Matsumoto A, Thompson DC, Vasiliou V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med. 2013;56:89–101.
Sunkar R, Chinnusamy V, Zhu J, Zhu JK. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007;12:301–9.
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8:25.
Suresh BV, Muthamilarasan M, Misra G, Prasad M. FmMDb: a versatile database of foxtail millet markers for millets and bioenergy grasses research. PLoS One. 2013;8:e71418.
Suzuki N, Rivero RM, Shulaev V, Blumwald E, Mittler R. Abiotic and biotic stress combinations. New Phytol. 2014;203:32–43.
Tang Y, Liu M, Gao S, Zhang Z, Zhao X, Zhao C, Zhang F, Chen X. Molecular characterization of novel TaNAC genes in wheat and overexpression of TaNAC2a confers drought tolerance in tobacco. Physiol Plant. 2012;144:210–24.
Tolleter D, Hincha DK, Macherel D. A mitochondrial late embryogenesis abundant protein stabilizes model membranes in the dry state. Biochim Biophys Acta. 2010;1798:1926–33.
Varshney RK, Terauchi R, McCouch SR. Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biol. 2014;12:e1001883.
Venuprasad R, Lafitte HR, Atlin GN. Response to direct selection for grain yield under drought stress in rice. Crop Sci. 2007;47:285–93.
Vyroubalova S, Vaclavikova K, Tureckova V, Novak O, Smehilova M, Hluska T, Ohnoutkova L, Frebort I, Galuszka P. Characterization of new maize genes putatively involved in cytokinin metabolism and their expression during osmotic stress in relation to cytokinin levels. Plant Physiol. 2009;151:433–47.
Wang L, Peterson RB, Brutnell TP. Regulatory mechanisms underlying C4 photosynthesis. New Phytol. 2011;190:9–20.
Wang M, Li P, Li C, Pan Y, Jiang X, Zhu D, Zhao Q, Yu J. SiLEA14, a novel atypical LEA protein, confers abiotic stress resistance in foxtail millet. BMC Plant Biol. 2014a;14:290.
Wang Y, Liu H, Xin Q. Genome-wide analysis and identification of cytokinin oxidase/dehydrogenase (CKX) gene family in foxtail millet (Setaria italica). Crop J. 2014b;2:244–54.
Xia N, Zhang G, Sun YF, Zhu L, Xu LS, Chen XM, Liu B, Yu YT, Wang XJ, Huang LL, Kang ZS. TaNAC8, a novel NAC transcription factor gene in wheat, responds to stripe rust pathogen infection and abiotic stresses. Physiol Mol Plant Pathol. 2010;74:394–402.
Xu D, Duan X, Wang B, Hong B, Ho T, Wu R. Expression of a late embryogenesis abundant protein gene, HVA1, from barley confers tolerance to water deficit and salt stress in transgenic rice. Plant Physiol. 1996;110:249–57.
Xu J, Li Y, Ma X, Ding J, Wang K, Wang S, Tian Y, Zhang H, Zhu XG. Whole transcriptome analysis using next-generation sequencing of model species Setaria viridis to support C4 photosynthesis research. Plant Mol Biol. 2013;83:77–87.
Yadav CB, Muthamilarasan M, Pandey G, Khan Y, Prasad M. Development of novel microRNA-based genetic markers in foxtail millet for genotyping applications in related grass species. Mol Breed. 2014;34:2219–24.
Yadav CB, Muthamilarasan M, Pandey G, Prasad M. Identification, characterization and expression profiling of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in foxtail millet. Plant Mol Biol Rep. 2015a;33:43–55.
Yadav CB, Bonthala VS, Muthamilarasan M, Pandey G, Khan Y, Prasad M. Genome-wide development of transposable elements-based markers in foxtail millet and construction of an integrated database. DNA Res. 2015b;22:79–90.
Yadav A, Khan Y, Prasad M. Dehydration-responsive miRNAs in foxtail millet: genome-wide identification, characterization and expression profiling. Planta. 2016;243:749–66.
Yang X, Wan Z, Perry L, Lu H, Wang Q, Zhao C, Li J, Xie F, Yu J, Cui T, Wang T, Li M, Ge Q. Early millet use in northern China. Proc Natl Acad Sci U S A. 2012;109:3726–30.
Yi F, Xie S, Liu Y, Qi X, Yu J. Genome-wide characterization of microRNA in foxtail millet (Setaria italica). BMC Plant Biol. 2013;13:212.
Yoshida A, Rzhetsky A, Hsu LC, Chang C. Human aldehyde dehydrogenase gene family. Eur J Biochem. 1998;251:549–57.
Zhang C, Zhang F. The multifunctions of WD40 proteins in genome integrity and cell cycle progression. J Genomics. 2015;3:40–50.
Zhang JP, Wang MY, Bai YF, Jia JP, Wang GY. Rapid evaluation on the drought tolerance of foxtail millet at seedling stage. J Plant Genet Resour. 2005;6:59–62.
Zhang J, Liu T, Fu J, Zhu Y, Jia J, Zheng J, Zhao Y, Zhang Y, Wang G. Construction and application of EST library from Setaria italica in response to dehydration stress. Genomics. 2007;90:121–31.
Zhang G, Liu X, Quan Z, Cheng S, Xu X, Pan S, Xie M, Zeng P, Yue Z, Wang W, Tao Y, Bian C, Han C, Xia Q, Peng X, Cao R, Yang X, Zhan D, Hu J, Zhang Y, Li H, Li H, Li N, Wang J, Wang C, Wang R, Guo T, Cai Y, Liu C, Xiang H, Shi Q, Huang P, Chen Q, Li Y, Wang J, Zhao Z, Wang J. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotechnol. 2012;30:549–54.
Zhang S, Tang C, Zhao Q, Li J, Yang L, Qie L, Fan X, Li L, Zhang N, Zhao M, Liu X, Chai Y, Zhang X, Wang H, Li Y, Li W, Zhi H, Jia G, Diao X. Development of highly polymorphic simple sequence repeat markers using genome-wide microsatellite variant analysis in Foxtail millet [Setaria italica (L.) P. Beauv]. BMC Genomics. 2014;2:15.
Zhao B, Liang R, Ge L, Li W, Xiao H, Lin H, Ruan K, Jin Y. Identification of drought-induced microRNAs in rice. Biochem Biophys Res Commun. 2007;354:585–90.
Zhao K, Zhao H, Chen Z, Feng L, Ren J, Cai R, Xiang Y. The Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in Populus trichocarpa: gene structure, gene expression, phylogenetic analysis and evolution. J Genet. 2015;94(2):317–21.
Zheng X, Chen B, Lu G, Han B. Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem Biophys Res Commun. 2009;379:985–9.
Zhou X, Wang G, Zhang W. UV-B responsive microRNA genes in Arabidopsis thaliana. Mol Syst Biol. 2007;3:103.
Zhou X, Wang G, Sutoh K, Zhu JK, Zhang W. Identification of cold-inducible microRNAs in plants by transcriptome. Biochim Biophys Acta. 2008;1779:780–8.
Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot. 2010;61:4157–68.
Zhu J, Jeong JC, Zhu Y, Sokolchik I, Miyazaki S, Zhu JK, Hasegawa PM, Bohnert HJ, Shi H, Yun DJ, Bressan RA. Involvement of Arabidopsis HOS15 in histone deacetylation and cold tolerance. Proc Natl Acad Sci U S A. 2008;105:4945–50.
Zhu C, Ming C, Zhao-shi X, Lian-cheng L, Xue-ping C, You-zhi M. Characteristics and expression patterns of the aldehyde dehydrogenase (ALDH) gene superfamily of foxtail millet (Setaria italica L.). PLoS One. 2014;9:e101136.
Acknowledgements
Studies on millet genomics in Dr. Manoj Prasad’s laboratory are supported by the Department of Biotechnology, Govt. of India and Core Grant of National Institute of Plant Genome Research (NIPGR), New Delhi, India. Mr. Mehanthan Muthamilarasan acknowledges the award of a Research Fellowship from the University Grants Commission, New Delhi, India. Authors also thank Ms. Jananee Jaishankar, NIPGR for critically reading this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Muthamilarasan, M., Prasad, M. (2017). Genetic Determinants of Drought Stress Tolerance in Setaria . In: Doust, A., Diao, X. (eds) Genetics and Genomics of Setaria. Plant Genetics and Genomics: Crops and Models, vol 19. Springer, Cham. https://doi.org/10.1007/978-3-319-45105-3_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-45105-3_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45103-9
Online ISBN: 978-3-319-45105-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)