
Abstract
Inflammasomes are cytosolic innate immune surveillance systems that recognize a variety of danger signals, including those from pathogens. Listeria monocytogenes is a Gram-positive intracellular bacterium evolved to live within the harsh environment of the host cytosol. Further, L. monocytogenes can activate a robust cell-mediated immune response that is being harnessed as an immunotherapeutic platform. Access to the cytosol is critical for both causing disease and inducing a protective immune response, and it is hypothesized that the cytosolic innate immune system, including the inflammasome, is critical for both host protection and induction of long-term immunity. L. monocytogenes can activate a variety of inflammasomes via its pore-forming toxin listeriolysin-O, flagellin, or DNA released through bacteriolysis; however, inflammasome activation attenuates L. monocytogenes, and as such, L. monocytogenes has evolved a variety of ways to limit inflammasome activation. Surprisingly, inflammasome activation also impairs the host cell-mediated immune response. Thus, understanding how L. monocytogenes activates or avoids detection by the inflammasome is critical to understand the pathogenesis of L. monocytogenes and improve the cell-mediated immune response generated to L. monocytogenes for more effective immunotherapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
1.1 Overview
Listeria monocytogenes is a Gram-positive facultative intracellular pathogen that has evolved to survive in a variety of severe environments and infect a wide range of hosts. These features combine to make L. monocytogenes an important foodborne pathogen most frequently associated with unpasteurized dairy products, deli meats, and more recently, fresh produce (Ferreira et al. 2014). Infection with L. monocytogenes can result in a noninvasive gastroenteritis that is likely severely underreported (Swaminathan and Gerner-Smidt 2007). More importantly, systemic listeriosis poses a severe risk to the immunocompromised, including the very young and old, as well as pregnant women. Complications of listeriosis include septicemia, meningitis, encephalitis, abortion, and still-borne births, resulting in a strikingly high mortality rate of 20–30 % even with proper antibiotic therapy (Swaminathan and Gerner-Smidt 2007).
While L. monocytogenes remains an important foodborne pathogen, there is an increasing interest in harnessing the robust cell-mediated immune response induced upon L. monocytogenes infection for use as a potential immunotherapy. Due to its almost exclusive intracellular lifecycle, described more in detail below, L. monocytogenes stimulates a robust CD8+ T-cell response. Additionally, L. monocytogenes is genetically tractable and is able to break self-tolerance, key factors in making L. monocytogenes an extremely promising cancer immunotherapeutic platform (Le et al. 2012). As access to the cytosol is an essential prerequisite for both the pathogenesis and the induction of immunity following L. monocytogenes infection, understanding how bacteria are sensed by the innate immune system in this environment has been a focal point of L. monocytogenes research for the past decade. Particularly, activation and avoidance of the inflammasome, a critical innate immune signaling pathway that all cytosolic pathogens must deal with, has become a recent focus.
1.2 Life Cycle
Listeria monocytogenes can infect a variety of cell types either through phagocytosis by myeloid cells or through active invasion of epithelial cells or hepatocytes with its virulence factors, internalin A or internalin B, respectively (Mengaud et al. 1996; Shen et al. 2000). Upon entry into the host cell, L. monocytogenes is initially contained within a phagocytic vacuole. Using its cholesterol-dependent pore-forming toxin listeriolysin-O (LLO, encoded by the gene hly) and a pair of phospholipases (PlcA and PlcB), L. monocytogenes escapes from the phagosome and enters into the cytosol (Portnoy et al. 1988; Mengaud et al. 1991; Camilli et al. 1991; Vazquez-Boland et al. 1992; Hamon et al. 2012).
Unlike most bacterial pathogens, L. monocytogenes is able to not only survive but also flourish and replicate within the host cytosol. This is a property unique to L. monocytogenes and other cytosol-adapted pathogens as intracellular pathogens that mislocalize to the cytosol or non-intracellular pathogens that are placed within the cytosol are unable to survive or replicate (Beuzón et al. 2000; Goetz et al. 2001; Slaghuis et al. 2004; Creasey and Isberg 2012). These studies suggest that L. monocytogenes has evolved specific adaptations to deal with cytosolic stresses, cell autonomous defense mechanisms, and innate immune detection in the cytosol, including the inflammasome. For example, L. monocytogenes modifies its peptidoglycan through N-deacetylation and O-acetylation to avoid killing by lysozyme and detection by the innate immune system (Boneca et al. 2007; Rae et al. 2011). L. monocytogenes can also modulate the production of the innate signaling cytokine IL-6 through its virulence factor InlH (Personnic et al. 2010); misregulation of these factors results in the attenuation of L. monocytogenes, demonstrating the importance of L. monocytogenes carefully controlling its detection by the host (Boneca et al. 2007; Personnic et al. 2010; Rae et al. 2011).
Once in the cytosol, L. monocytogenes avoid exposure to the extracellular milieu and the host defenses found there by utilizing the virulence factor ActA to hijack host actin and propel itself through membrane protrusions into neighboring cells (Kocks et al. 1992). Both Δhly and ΔactA L. monocytogenes mutants are attenuated; however, of these, only ΔactA L. monocytogenes are able to mount a protective cell-mediated immune response (Portnoy et al. 1988; Goossens and Milon 1992; Bahjat et al. 2009), further highlighting the importance of the cytosol and cytosolic innate immune recognition in stimulating robust CD8+ T cell-mediated immunity.
1.3 Innate Immune Response
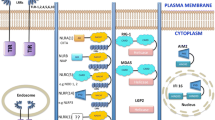
Once L. monocytogenes accesses the cytosol, it is rarely exposed to extracellular host defenses; as such, detection by cytosolic innate immune sensors is likely critical for combating infection. Likewise, as L. monocytogenes must access the cytosol to cause disease, avoidance or direct inhibition of the cytosolic innate immune system is likely critical for L. monocytogenes’ virulence. Indeed, multiple cytosolic innate immune pathways have been demonstrated to recognize L. monocytogenes infection (reviewed in Witte et al. 2012). Briefly, peptidoglycan fragments of L. monocytogenes can activate nucleotide binding oligomerization domain (NOD) proteins leading to the phosphorylation of p38 mitogen-activated protein kinase (MAPK) and transcription factor NF-κB activation, further leading to inflammatory cytokine production (O’Riordan et al. 2002; Opitz et al. 2006). Further, NOD signaling can synergize with interferon (IFN) signaling to enhance the host innate response (Leber et al. 2008), although it should be noted that type I IFN induction has been shown to inversely correlate with host protection (O’Connell et al. 2004; Auerbuch et al. 2004; Carrero et al. 2006). More recently, cyclic dinucleotides secreted by L. monocytogenes have been shown to be recognized by the STING/IRF3 pathway resulting in the production of type I IFNs (Woodward et al. 2010; Sauer et al. 2011b). Additionally, as will be highlighted in this chapter, L. monocytogenes has been reported to activate the inflammasome through a variety of receptors, ultimately resulting in the activation of caspase-1 (Fig. 1). Caspase-1 activation leads to an inflammatory cell death, pyroptosis, release of the pro-inflammatory cytokines IL-1β and IL-18, and the production and release of lipid mediators known as eicosanoids (Fig. 2). The engagement of these innate pathways resulting in the production of pro-inflammatory cytokines and the recruitment of innate immune cells is hypothesized to be critical in the development of adaptive immunity.

In vitro mechanisms of activation and avoidance of the inflammasome by L. monocytogenes. L. monocytogenes lipoproteins and flagellin can signal through toll-like receptors (TLRs) via MyD88 and IRAK1 to activate the transcription factor NF-κB and prime cells for inflammasome activation by up-regulating IL-1β and NLRP3 transcripts. L. monocytogenes can then activate the NLRP3 inflammasome via a K+ efflux secondary to extracellular or intracellular LLO-induced pores. Vacuole rupture by LLO can result in cathepsin B release that along with p60 can also activate the NLRP3 inflammasome. Flagellin can activate the NLRC4 inflammasome, while bacteriolysis and the subsequent DNA release can activate the AIM2 inflammasome. L. monocytogenes limits the activation of any of these inflammasomes via the tight control of LLO or flagellin, or by unknown mechanisms of combating cytosolic stress and limiting bacteriolysis

Activation of the inflammasome attenuates L. monocytogenes through macrophage recruitment. In wild-type L. monocytogenes infection, L. monocytogenes minimally activates caspase-1 and maintains its intracellular niche, ultimately promoting a pathogenic infection. In contrast, inflammasome-activating L. monocytogenes results in robust caspase-1 activation and pyroptosis, release of IL-1β and IL-18, and eicosanoids. These effects result in expulsion of L. monocytogenes, increased activated macrophage recruitment, and potentially an influx of neutrophils. Increased macrophage recruitment restricts L. monocytogenes infection
In this chapter, we will focus on how L. monocytogenes activates different inflammasomes. We will also discuss the downstream consequences of inflammasome activation on the pathogenesis of L. monocytogenes. Finally, we will discuss how engagement of the inflammasome influences the innate and adaptive immune responses to L. monocytogenes, with a focus on its impact on the development of L. monocytogenes as an immunotherapeutic platform.
2 Activation of Different Inflammasomes
2.1 NLRP3 Activation
NLRP3 belongs to the Nod-like receptor family of pattern recognition receptors and recognizes a variety of danger signals including ATP, uric acid crystals, and toxins (Mariathasan et al. 2006). L. monocytogenes was first observed to activate the NLRP3 inflammasome in the seminal paper by Mariathasan et al. (2006) who demonstrated that cytosolic L. monocytogenes can activate the NLRP3 inflammasome, while vacuole-contained L. monocytogenes fail to activate the inflammasome, suggesting that the cytosolic recognition is critical. Concurrently, Kannengati et al. (2006) observed that L. monocytogenes total RNA could stimulate the NLRP3 inflammasome, further suggesting that L. monocytogenes could activate the NLRP3 inflammasome through a variety of mechanisms. More recently, activation of the Nrlp3 inflammasome specifically in unprimed bone marrow-derived macrophages infected with L. monocytogenes has been reported (Wu et al. 2010; Fernandes-Alnemri et al. 2013; Lin et al. 2014). However, the importance of NLRP3 in L. monocytogenes infection has also been questioned, as Franchi et al. (2007) failed to find any difference in caspase-1 processing and IL-1β release in Nlrp3−/− cells following L. monocytogenes infection. Similarly, Sauer et al. (2010) failed to find any significant role for the NLRP3 inflammasome during L. monocytogenes infection.
These differing observations beg the questions: “Which components of L. monocytogenes stimulate the NLRP3 inflammasome and what role, if any, does it play under physiologic conditions?” Pore-forming toxins have been demonstrated to activate the NLRP3 inflammasome, and Mariathasan and colleagues hypothesized that LLO could act like many other NLRP3 agonists and result in intracellular K+ disruptions to activate the NLRP3 inflammasome (Mariathasan et al. 2006). While their original interpretation was that the importance of LLO was to facilitate the access of L. monocytogenes to the cytosol, it is also possible that pore formation, and not cytosolic access, was the critical function of LLO in NLRP3 activation during their studies, particularly as these studies used a high MOI of 50. Additionally, Meixenberger et al. (2010) suggest that cathepsin B release from LLO-damaged phagosomes can stimulate the NLRP3 inflammasome. While some data have directly suggested that cytosolic access of intact bacteria is required for L. monocytogenes NLRP3 activation, more data are consistent with a role of extracellular LLO in NLRP3 activation (Meixenberger et al. 2010; Hamon and Cossart 2011; Sakhon et al. 2013). Extracellular LLO can induce caspase-1 processing and IL-1β release depending on NLRP3 (Meixenberger et al. 2010) by stimulating a K+ efflux (Hamon and Cossart 2011). Blocking bacterial uptake with cytochalasin D still results in the activation of caspase-1, suggesting that low amounts of LLO from extracellular L. monocytogenes can activate the inflammasome (Hamon and Cossart 2011). Further, purified LLO that maintains pore-forming ability, but not hemolytically inactive LLO (LLOW492A), can activate caspase-1 (Sakhon et al. 2013), and these results are independent of the cholesterol-binding activity (Hara et al. 2008; Hamon and Cossart 2011). Taken together, these results suggest that pore formation from extracellular LLO can trigger K+ efflux resulting in NLRP3 inflammasome activation. However, LLO expression, activity, and stability are tightly regulated during L. monocytogenes infection (Glomski et al. 2002; Schnupf et al. 2006a, b), thus limiting the likelihood of high extracellular concentrations of LLO during infection in vivo and potentially suggesting that NLRP3 activation by LLO is an in vitro artifact.
While LLO is believed to be the primary molecule triggering the NLRP3 inflammasome, both bacterial RNA (Kanneganti et al. 2006) and the secreted virulence factor, p60 encoded by the gene invasion-associated secreted endopeptidase (iap), from L. monocytogenes have been shown to activate the NLRP3 inflammasome (Schmidt and Lenz 2012). The N-terminal LysM and SH3 domain region (L1S) of p60 stimulates IL-1β and IL-18 release, independent of the act of pyroptotic cell death, in bone marrow-derived dendritic cells (Schmidt and Lenz 2012). Given the recent reports about the importance of gasdermin D-dependent host cell death in cytokine secretion (Shi et al. 2015; Kayagaki et al. 2015), it is unclear how IL-1β and IL-18 are being secreted in this scenario; however, Schmidt and Lenz found that ROS inhibition with the inhibitor DPI impaired IL-1β secretion, while IL-18 secretion remained intact. Further, in BMDCs from mice on a 129S6 background that lack caspase-11, IL-1β secretion was impaired, while IL-18 was unaffected (Schmidt and Lenz 2012). These observations suggest different licensing mechanisms for IL-1β and IL-18, potentially allowing either the host cell or the invading pathogen to fine-tune the downstream consequences of inflammasome activation.
Most work examining L. monocytogenes and the NLRP3 inflammasome has been done with murine cells. However, human peripheral blood mononuclear cells (PBMCs) infected with L. monocytogenes can undergo LLO-induced NLRP3-dependent inflammasome activation that depends on phagosomal acidification and cathepsin B release (Meixenberger et al. 2010). These results suggest that the observed differences for the role of the NLRP3 inflammasome may also stem from the cell types used. More importantly, the state of the cells used in experiments matters. Multiple groups were able to find a role for the NLRP3 inflammasome in the first hour of L. monocytogenes infection when using unprimed cells (Wu et al. 2010; Fernandes-Alnemri et al. 2013; Lin et al. 2014). Interestingly, for L. monocytogenes to induce a rapid NLRP3-dependent inflammasome activation, intact toll-like receptor (TLR) signaling must be present as MyD88−/− cells fail to activate the inflammasome (Fernandes-Alnemri et al. 2013; Lin et al. 2014). Further, this response is dependent on the MyD88 downstream signaling molecule IL-1 receptor-associated kinase (IRAK1) as IRAK1−/− cells also fail to activate the inflammasome and secrete IL-1β and IL-18 (Thomas et al. 1999; Fernandes-Alnemri et al. 2013; Lin et al. 2014). In the presence of priming, however, IRAK1 is dispensable for inflammasome activation (Lin et al. 2014); additionally, a prolonged infection results in NLRP3-independent inflammasome activation (Lin et al. 2014). These results suggest that L. monocytogenes can rapidly activate the NLRP3 inflammasome early in infection, as long as an intact TLR pathway is present. Many groups do not use unprimed cells, as priming is generally required for inflammasome activation in vitro (Latz et al. 2013), potentially explaining the observed discrepancies in the importance of NLRP3.
2.2 NLRC4 Activation
The NLRC4 inflammasome recognizes flagellin and type III secretion system components (Miao et al. 2010b). As L. monocytogenes is a Gram-positive pathogen and lacks type III secretion components, its flagellin is likely the key activator of the NLRC4 inflammasome. In line with this, L. monocytogenes engineered to hyperexpress flagellin can activate the NLRC4 inflammasome (Sauer et al. 2011a; Warren et al. 2011). Additionally, ΔflgK L. monocytogenes lack the adaptor molecule between the flagellar hook and flagellin monomers, and infection with this strain results in excess flagellin secretion and hyperactivation of the NLRC4 inflammasome (Warren et al. 2008). However, L. monocytogenes likely avoids the activation of the NLRC4 inflammasome by shutting off flagellin expression in mammalian hosts (Peel et al. 1988; Shen and Higgins 2006). As such, L. monocytogenes inflammasome activation was shown to be independent of NLRC4 (Franchi et al. 2007; Sauer et al. 2010). Similarly, infection with L. monocytogenes deficient in flagellin (ΔflaA) resulted in similar caspase-1 processing and IL-1β release (Warren et al. 2008) as infection with wild-type L. monocytogenes. Taken together, these results suggest that although L. monocytogenes flagellin can activate the NLRC4 inflammasome, likely in a manner similar to other bacterial flagellin being dependent on the adaptor molecule Naip5 (Kofoed and Vance 2011; Zhao et al. 2011), L. monocytogenes largely avoids the activation of this inflammasome through the control of its flagellin in vivo.
2.3 AIM2 Activation
As opposed to the NLR family members described above, AIM2 (absent in melanoma) is a member of the interferon-inducible class of genes (DeYoung et al. 1997). The AIM2 inflammasome through its HIN200 domain recognizes double-stranded DNA, rich in adenine and thymine that is mislocalized to the cytosol (Fernandes-Alnemri et al. 2009; Hornung et al. 2009). Initial evidence for the involvement of AIM2 in L. monocytogenes infection came from observations that ΔflaA mutant L. monocytogenes could still induce caspase-1 activation in the absence of NLRP3 (Warren et al. 2008; Wu et al. 2010). However, caspase-1 activation was abrogated in Aim2−/− cells, suggesting that the AIM2 inflammasome is responsible for the remaining caspase-1 activation (Wu et al. 2010; Kim et al. 2010). Additionally, Warren et al. (2010) observed that a component of lysed L. monocytogenes could activate the AIM2 inflammasome. Similarly, transfected L. monocytogenes’ genomic DNA could also activate the AIM2 inflammasome (Warren et al. 2010). Using Hoechst-labeled L. monocytogenes, Wu et al. (2010) were able to observe colocalization of L. monocytogenes with GFP-labeled AIM2, further suggesting that L. monocytogenes DNA is able to activate the AIM2 inflammasome.
Finally, Sauer et al. (2010) demonstrated that bacteriolysis within the cytosol resulted in the activation of the AIM2 inflammasome, identifying a likely mechanism for how bacterial DNA accesses the cytosol to be recognized by AIM2. These experiments demonstrated that wild-type L. monocytogenes lyses at low levels throughout the course of infection resulting in the recognition of cytosolic DNA by AIM2. Treatment of cytosolically replicating L. monocytogenes with the bactericidal β-lactam antibiotic, ampicillin, but not the bacteriostatic antibiotic chloramphenicol, triggered the increased bacteriolysis and subsequently increased inflammasome activation (Sauer et al. 2010). Additionally, a L. monocytogenes mutant was identified that was defective for cytosolic survival leading to increased cytosolic bacteriolysis and ultimately increased AIM2-dependent inflammasome activation. Finally, L. monocytogenes engineered to lyse specifically within the host cytosol through the expression of the bacteriophage proteins holin and lysin resulted in a significant bacteriolysis and subsequent cell death (Sauer et al. 2010). Importantly, these data implicated cytosolic survival and avoidance of cell autonomous defenses as important mechanisms of avoiding detection by the AIM2 inflammasome. Further, a similar mechanism of cytosolic bacteriolysis was subsequently found to be the mechanism of AIM2 activation following Francisella tularensis infection using a similar reporter system (Peng et al. 2011).
2.4 Other NLR Engagement
The inflammasomes mentioned above are the traditionally described inflammasomes as well as the primary inflammasomes that L. monocytogenes has been demonstrated to engage. Beyond the receptors described above, other NLRs including NLRC5 and NLRP6 have been studied in regard to L. monocytogenes infection. The NLRC5 inflammasome largely regulates MHC class I-associated gene expression (Meissner et al. 2010). Nlrc5−/− mice infected with L. monocytogenes have impaired CD8+ T-cell responses, both in the number and in the ability to produce the effector cytokine, IFNγ. CD4+ T-cell responses are not impaired in these mice, consistent with the role of NLRC5 in regulating MHC I-associated gene expression (Yao et al. 2012; Biswas et al. 2012). NLRC5 has also been shown to associate with NLRP3 and contribute to NLRP3 inflammasome activation (Davis et al. 2011; Yao et al. 2012). As discussed above, L. monocytogenes has been shown, in some cases, to activate the NLRP3 inflammasome, although the specific contribution of NLRC5 to this activation will require additional studies. That said, infection of Nlrc5−/− mice with L. monocytogenes resulted in partial impairments in caspase-1 processing and IL-1β secretion, suggesting a role for this protein in a more traditional inflammasome-dependent response (Yao et al. 2012). Finally, Nlrc5−/− mice infected with L. monocytogenes have higher bacterial burdens (Yao et al. 2012; Biswas et al. 2012), although it is unclear whether this is due to deficient CD8+ T-cell responses or to a more canonical inflammasome-dependent infection control mechanism.
NLRP6 has been shown to have roles in altering the gut microbiota to promote gut inflammation and ultimately tumorigenesis (Elinav et al. 2011; Normand et al. 2011; Chen et al. 2011). Interestingly, Nlrp6−/− mice infected orally with L. monocytogenes are largely protected from lethal L. monocytogenes infection, with 75 % surviving lethal challenge. Further, Nlrp6−/− mice harbor lower bacterial burdens in their spleens and livers and fail to lose weight following infection (Anand et al. 2012). These effects appear to be independent of the gut microbiota composition, but instead depend on increases in circulating monocytes and neutrophils in Nlrp6−/− mice after L. monocytogenes infection both systemically in the blood and locally in the peritoneal cavity (Anand et al. 2012). Though NLRP6 seems to have important roles in L. monocytogenes infection, this appears to occur independent of canonical inflammasome activation as caspase-1 activation and IL-1β processing are not impaired following L. monocytogenes infection in Nlrp6−/− macrophages (Anand et al. 2012). Similarly, in human PBMCs, NLRP6 is dispensable for canonical inflammasome activation by L. monocytogenes (Meixenberger et al. 2010). Taken together, these results suggest that though NLRP6 has a role in L. monocytogenes pathogenesis, it is likely independent of the effects from inflammasome signaling.
Finally, among the remaining NLRs, a few have been tested for L. monocytogenes-dependent phenotypes. HEK293 cells reconstituted with NLRP2 or NLRP12 result in increases in IL-1β production when infected with L. monocytogenes, suggesting that L. monocytogenes can also engage these inflammasomes, although which components of L. monocytogenes are recognized by these receptors remains unknown (Tsuchiya et al. 2010). Conversely, in human PBMCs, NLRP1 and NLRP12 are dispensable for inflammasome activation by L. monocytogenes (Meixenberger et al. 2010). These results suggest that L. monocytogenes can potentially engage other NLRs; however, the effects of this engagement are unclear and the mechanisms by which they are activated require more in-depth future studies.
In conclusion, although under in vitro conditions L. monocytogenes has been found to activate the NLRP3, NLRC4, and AIM2 inflammasomes (Fig. 1), the in vivo relevance of each of inflammasomes is unclear. As discussed in more detail below and as shown in Fig. 1, molecules that engage the NLRP3 and NLRC4 inflammasomes are under the tight control in physiologic settings, thus limiting their ability to be sensed by their respective inflammasomes. Although AIM2 is the most likely relevant inflammasome in vivo, L. monocytogenes also has yet undefined ways of avoiding cytosolic bacteriolysis, suggesting that overall, L. monocytogenes limits inflammasome activation.
3 Avoidance of Inflammasome Activation
Avoidance of cell death is critical for the virulence of L. monocytogenes as mutants that misregulate LLO activity in the cytosol cause necrosis and are highly attenuated in vivo (Glomski et al. 2003). Similarly, L. monocytogenes that hyperactivate the inflammasome or induce apoptosis are also attenuated (Sauer et al. 2011a; Warren et al. 2011; Theisen and Sauer, unpublished results). Thus, although L. monocytogenes can activate multiple inflammasomes during infection in vitro, it is likely that it largely avoids doing so during infection in vivo to protect its intracellular niche. As a pathogen that utilizes a replication niche rich in inflammasome receptors, it is critical that L. monocytogenes has evolved mechanisms to avoid detection or actively inhibit the inflammasome.
3.1 Avoidance of NLRP3
As described above, LLO, through its pore-forming activity, is capable of activating the NLRP3 inflammasome, likely when found in high concentrations extracellularly. However, expression, stability, and activity of LLO are tightly regulated to prevent host cell toxicity, either directly through pore formation and necrosis or potentially through pore-induced NLRP3 activation. LLO expression is both transcriptionally and post-transcriptionally regulated through the activity of the PrfA transcription factor and the 5′ UTR, respectively (Shen and Higgins 2005; Schnupf et al. 2006a). Additionally, LLO in the host cytosol is ubiquitinated and targeted for proteosomal degradation (Schnupf et al. 2006b). Finally, and perhaps most importantly, LLO toxicity is limited at the level of pore-forming activity such that LLO is maximally active only at an acidic pH, a characteristic unique to LLO among the cholesterol-dependent cytolysins (Glomski et al. 2002). Each of these levels of regulation contributes to minimizing the toxicity of LLO to maximize virulence as misregulated LLO can lead to cytotoxicity and result in severe attenuation of virulence (Glomski et al. 2003). Taken together, these studies demonstrate that L. monocytogenes carefully regulates the expression of its pore-forming toxin to limit host cell damage and maintain its virulence, in the process likely avoiding a robust activation of the NLRP3 inflammasome under physiologic conditions.
3.2 Avoidance of NLRC4
Similar to the tight regulation of LLO, L. monocytogenes has developed exquisite control over expression of its flagellin, regulating the expression through temperature sensing as well as yet to be identified signals found in the host cell cytosol. At the physiologic temperature of 37 °C, L. monocytogenes limits the expression of its flagellin through the transcriptional repressor, MogR. However, at temperatures below 37 °C, such as those found in the external environment, L. monocytogenes must express flagellin (Peel et al. 1988), and MogR repression must be removed.
Repression by MogR is relieved by the anti-repressor GmaR (Shen et al. 2006). Thus, the balance between these two factors is important in the expression of flagellin. As an added measure to ensure that flagellin is not expressed at 37 °C, there is post-transcriptional regulation of GmaR so that MogR is the dominant component regulating flagellin expression (Kamp and Higgins 2009). Therefore, similar to what is observed for LLO, although flagellin can activate the NLRC4 inflammasome, L. monocytogenes tightly limits its expression in the presence of inflammasome components. Whether this level of regulation evolved to facilitate the avoidance of TLR5 detection or inflammasome activation is unclear; however given that the majority of L. monocytogenes’ lifecycle in the host is in the cytosol, it is enticing to imagine a strong selective pressure exerted by inflammasome detection leading to the tight control of flagellin expression. Indeed, recent evidence suggests that, contrary to previous reports, primary human macrophages utilize a specific splice variant of NLR family, apoptosis inhibiting protein (NAIP) to facilitate the recognition of flagellin through the NLRC4 inflammasome (Kortmann et al. 2015).
3.3 Avoidance of AIM2
In addition to regulating LLO and flagellin to avoid detection by the inflammasome, as a pathogen that makes its life in the cytosol, fidelity during replication and evasion of host defense mechanisms is essential for L. monocytogenes to avoid detection by the AIM2 inflammasome. Work by Werner Goebel’s laboratory suggests that cytosolic pathogens have developed unique defenses to survive the harsh environment, that is, the host cytosol. Non-cytosolic pathogens that are placed into the cytosol by microinjection fail to replicate and in some cases are killed, whereas cytosol-adapted pathogens survive and thrive (Goetz et al. 2001; Slaghuis et al. 2004). Further, additional studies demonstrated that even intracellular pathogens that mislocalize to the cytosol, such as ΔsifA mutant Salmonella enterica subsp. Typhimurium, are unable to survive (Beuzón et al. 2000). Finally, Legionella pneumophila ΔsdhA mutants mislocalize to the cytosol, subsequently lyse, and activate the inflammasome (Creasey and Isberg 2012; Ge et al. 2012). Taken together, these data suggest that bacteria such as L. monocytogenes that are able to survive and replicate within the harsh environment of the cytosol must have evolved mechanisms to deal with these stresses and/or active host defenses. Recent evidence suggests that the loss of these survival adaptations ultimately results in the lysis of L. monocytogenes in the cytosol, leading to AIM2 activation and virulence attenuation, further highlighting the importance of inflammasome avoidance in promoting L. monocytogenes virulence (Sauer et al. 2010, Daniel Pensinger, Grischa Chen and JD Sauer unpublished data).
The specific adaptations that allow L. monocytogenes to survive within the cytosol of cells and avoid AIM2 inflammasome activation are largely unknown. L monocytogenes Δyvck mutants lyse in the macrophage cytosol, resulting in hyperactivation of the AIM2 inflammasome and virulence attenuation (Sauer et al. 2010). The function of YvcK, a conserved protein of unknown function, in maintaining cell wall integrity specifically in the cytosol remains unknown, though the loss of yvck sensitizes L. monocytogenes to lysozyme and numerous cell wall-acting antibiotics in vitro, suggesting that yvck plays an integral role in dealing with cell wall stress (Burke et al. 2014, Daniel Pensinger and JD Sauer unpublished data). Similarly, L. monocytogenes deficient in either the N-deacetylation (Δpgd) or O-acetylation (Δoat) of peptidoglycan are sensitive to lysozyme and lyse within the host cytosol (Rae et al. 2011). Finally, Witte et al. (2012) reported a number of L. monocytogenes mutants that hyperinduce host cell death and undergo bacteriolysis in the host cell cytosol, though these mutants were not fully characterized as activating the AIM2 inflammasome. However, it is likely given their lysis phenotypes correlating with increased host cell death that these genes are required for the avoidance of host cytosolic defense mechanisms to promote the avoidance of the AIM2 inflammasome. Importantly, maintaining bacterial integrity in the cytosol as a mechanism of avoiding AIM2 detection is a conserved virulence mechanism as Peng et al. (2011) recently demonstrated that a series of Francisella tularensis mutants that hyperactivate the inflammasome do so due to cytosolic bacteriolysis. Taken together, these data suggest that maintenance of the cell wall is a critical determinant of L. monocytogenes virulence, in part due to the necessity of avoiding AIM2 activation.
3.4 Active Inhibition of the Inflammasome
Although pathogens such as Yersinia spp. and Pseudomonas aeruginosa express molecules that actively inhibit inflammasome activation (Sutterwala et al. 2007; Brodsky et al. 2010), there is little evidence of L. monocytogenes actively inhibiting the inflammasome. Instead, it is presumed that the lack of robust inflammasome activation observed during L. monocytogenes infection is due to avoiding detection. These data are supported by the studies demonstrating that L. monocytogenes engineered to activate the inflammasome through ectopic expression of flagellin are able to potently activate the inflammasome (Sauer et al. 2011a; Warren et al. 2011). Likewise, L. monocytogenes-infected macrophages are still capable of robustly responding to transfected DNA, suggesting that L. monocytogenes is not actively inhibiting AIM2 inflammasome activation (JD Sauer unpublished observations). While it is possible, and maybe even likely given its importance in host defense, that L. monocytogenes actively inhibits pyroptosis under certain conditions or for the activation of specific inflammasomes, studies to date have not identified such a mechanism. The lone potential example is during in vivo infection where L. monocytogenes can activate cholesterol 25-hydroxylase (Ch25h) downstream of liver X receptors (LXRs) through type I INF production (Zou et al. 2011). Ch25h results in the expression of Cd5l, a pro-survival gene that limits the activation of both caspase-3 and caspase-1, ultimately limiting host cell inflammasome activation and pyroptosis. Further, this response seems to be conserved among other downstream targets of LXRs (Zou et al. 2011), suggesting that L. monocytogenes may activate this pathway to limit caspase-1 activation and preserve its niche.
4 Role in Pathogenesis
4.1 Role of Caspase-1/11
As described above, L. monocytogenes can interact with many different inflammasomes in vitro, culminating in the activation of caspase-1. Initial studies of murine caspase-1 were actually studies of both caspase-1 and caspase-11 double deletions (Kayagaki et al. 2011). Caspase-11 has largely been implicated in Gram-negative infections recognizing the LPS moiety of many of these pathogens (Wang et al. 1998; Kayagaki et al. 2011). L. monocytogenes has been found to induce caspase-11 expression upon infection; however, genetic deletion of caspase-11 still allows caspase-1 to function normally (Akhter et al. 2012), suggesting that caspase-11 is dispensable in L. monocytogenes infection.
Initial reports suggested that mice deficient in caspase-1/11 had impaired clearance of L. monocytogenes, resulting in an increased death rate, particularly at late time points in infection (Tsuji et al. 2004). Mice were rescued with exogenous IL-18 or IFNγ, suggesting that the production of these critically important cytokines is impaired in caspase-1/11−/− mice (Tsuji et al. 2004). While these initial experiments suggest a critical role for caspase-1/11 in controlling L. monocytogenes infection, further experiments have failed to find a similar role. Both Sauer et al. (2011a) and Tsuchiya et al. (2014) observed no increased susceptibility to L. monocytogenes in caspase-1/11−/− mice at 2 days post infection. Similarly, there was no increased susceptibility to L. monocytogenes in zebrafish lacking caspase-1 (Vincent et al. 2015). These studies are consistent with the tight levels of regulation observed with potential inflammasome agonists as well as the critical nature of host cell survival that has previously been reported to promote L. monocytogenes virulence. The differences between the initial characterization of caspase-1 and more recent studies likely stem from the time points examined, the background of mice used in the initial studies (C57Bl/6 J × 129SV/J), as well as the differences in the strains of L. monocytogenes used for each of the studies. Taken together, the more recent studies suggest that caspase-1 is dispensable for the control of L. monocytogenes early during infection, likely due to L. monocytogenes avoiding the activation of the inflammasome and maintaining its intracellular niche. However, it is possible that later in infection when bacterial burdens are high, the inflammasome plays a role in host protection. Importantly, as briefly discussed above and in more detail below, under conditions in which L. monocytogenes does activate the inflammasome, due to ectopic flagellin expression or mutations that result in detection by AIM2, caspase-1 activation and the subsequent pyroptosis severely attenuate virulence, further highlighting the importance of inflammasome evasion for L. monocytogenes pathogenesis.
4.2 Role of ASC
ASC is an adaptor molecule required for cytokine secretion and pyroptosis downstream of both the NLRP3 and AIM2 inflammasomes (Broz et al. 2010). In the case of CARD-containing inflammasomes, such as the NLRC4 inflammasome, ASC is only required for cytokine secretion and is dispensable for pyroptosis (Broz et al. 2010). Thus, as discussed above, the majority of inflammasome activation by L. monocytogenes requires signaling through ASC for both pyroptosis and cytokine secretion (Fig. 1). As such, ASC−/− peritoneal macrophages fail to activate caspase-1 or secrete IL-1β and IL-18 (Ozören et al. 2006). Consistent with observations that the loss of caspase-1 has no effect on L. monocytogenes virulence in vivo (Sauer et al. 2011a; Tsuchiya et al. 2014), the loss of ASC results in similar susceptibility to L. monocytogenes (Sauer et al. 2011a). Interestingly, recent work by Tsuchiya suggests that ASC deficiency may in some cases result in enhanced host protection during L. monocytogenes infection (Tsuchiya et al. 2014). ASC−/− mice are able to withstand a bacterial load of 1 × 106 colony forming units (CFU) of L. monocytogenes, a log higher than the LD50 for wild-type C57Bl/6 mice, and harbor lower bacteria burdens in their livers. The investigators report that ASC promotes the production of the anti-inflammatory cytokine IL-10 and that protection in ASC-deficient mice is likely due to reduced IL-10 production (Tsuchiya et al. 2014). Taken together, these results suggest that similar to the loss of caspase-1, the loss of ASC does not promote the virulence of L. monocytogenes, and in fact may result in enhanced protection, although this phenotype may be independent of ASC’s role in caspase-1 activation (Tsuchiya et al. 2014).
4.3 Role of IL-1β and IL-18
IL-1β and IL-18 have been examined in the context of L. monocytogenes infection well before these cytokines were known to be downstream of the inflammasome. Initial reports of IL-1 receptor-deficient (IL-1R1−/−) mice that lack signaling of IL-1α and IL-1β suggested that these mice are more susceptible to L. monocytogenes (Labow et al. 1997); however, this was done in a mixed background (B6/129) and is likely confounded by the fact that 129 mice are more susceptible to L. monocytogenes at baseline (Gahan and Collins 1995). Further examination on a clean C57Bl/6 background suggests that there is no alteration in acute virulence in the absence of IL-1 signaling (Glaccum et al. 1997). Similarly, mice deficient just in IL-1β show no differences in susceptibility (Zheng et al. 1995), suggesting that IL-1 signaling is dispensable during initial infection.
Priming of cells via signal one, largely through NF-κB signaling, is critical in up-regulating IL-1β transcripts (Latz et al. 2013). In TLR2−/− macrophages, IL-1β secretion is impaired following L. monocytogenes infection likely through transcriptional impairment (Ozören et al. 2006). Further, the loss of tumor progression locus 2 (TPL2), a factor critical in orchestrating cytokine responses downstream of TLR signaling, impairs IL-1β secretion following L. monocytogenes infection through impairments of IL-1β transcript up-regulation (Mielke et al. 2009). Interestingly, the loss of TPL2 increases the susceptibility of mice to L. monocytogenes, though the regulation of other key innate cytokines such as TNF-α and IFN-γ is similar regardless of whether TPL2 is present or not (Mielke et al. 2009). Even though TPL2 influences the levels of IL-1β, the increased susceptibility of TPL2−/− mice does not recapitulate the phenotype of IL-1R1−/− mice; thus, the increased susceptibility of TPL2−/− mice is not likely due to the changes in IL-1β but instead to some other undescribed effect of loss of TPL2.
Characterization of the role of IL-18, particularly in acute infection, has also resulted in conflicting reports. Initial studies suggested that IL-18 is critical in acute infection, and the lack of IL-18 leads to an increased susceptibility (Neighbors et al. 2001). IL-18 is critical in stimulating IFN-γ, a crucial cytokine for controlling L. monocytogenes infection, and thus, decreases in IL-18 production result in less IFN-γ production, leading mice to succumb to infection. While these observations were made in initial reports, the authors also observed decreases in TNF-α production, another critical cytokine in innate susceptibility, and suggest that the corresponding lack of TNF-α from IL-18-deprived mice is largely what is responsible for susceptibility to infection. More recent studies in both IL-18−/− and IL-18Rα−/− contradict these earlier results and suggest that IL-18−/− mice are actually more resistant to L. monocytogenes, particularly at high doses (Lochner et al. 2008; Tsuchiya et al. 2014). These results are in line with reports that suggest that ASC−/− mice, which also lack IL-18 secretion, are less susceptible to L. monocytogenes (Tsuchiya et al. 2014). The reason for this decreased susceptibility could stem from an increased influx of leukocytes in the initial 72 h postinfection that are resistant to apoptosis (Lochner et al. 2008).
While most IL-1β and IL-18 signaling is believed to occur downstream of the inflammasome, there have been caspase-1-independent observations of IL-1β and IL-18 secretion (Uchiyama et al. 2013; Tsuchiya et al. 2014). One such mechanism relies on Fas (CD95/Apo-1) signaling. Peritoneal exudate cells (PECs) deficient in Fas secreted less IL-1β and IL-18 after L. monocytogenes infection (Uchiyama et al. 2013), suggesting that Fas is required for cytokine secretion. Interestingly, this mechanism is caspase-1, caspase-11, NLRP3, and NLRC4 independent, while it is dependent on ASC (Uchiyama et al. 2013). In line with the reports that suggest IL-1β maturation can occur through caspase-8 activation (Gringhuis et al. 2012; Bossaller et al. 2012), L. monocytogenes can activate caspase-8 in a Fas-dependent manner, suggesting that in this instance, IL-1β and IL-18 could be processed through caspase-8 and not caspase-1. Additionally, IL-18 can be processed to its mature form through neutrophil serine proteases (Sugawara et al. 2001). In caspase-1/11−/− mice infected with lethal doses of L. monocytogenes, elevated neutrophil serine protease activity was observed that correlated with the increased IL-18 production (Tsuchiya et al. 2014). Taken together, while not all studies are consistent, and differences in mouse backgrounds, L. monocytogenes strains, doses, and infection kinetics could account for discrepancies, the preponderance of data suggests that the inflammatory cytokines downstream of inflammasome activation are dispensable for host defense during L. monocytogenes infection.
4.4 Innate Immune Cell Infiltrate
During murine L. monocytogenes infection, there is a robust, initial infiltrate of myelomonocytic cells (MMCs) consisting of neutrophils and Ly6Chi monocytes into the splenic white pulp (Waite et al. 2011; Williams et al. 2013). Because L. monocytogenes largely avoids activating the inflammasome, much of the work examining the host response to pyroptosis has used an engineered strain of L. monocytogenes that ectopically expresses flagellin resulting in hyperactivation of the inflammasome and attenuation (Sauer et al. 2011a; Warren et al. 2011; Williams et al. 2013; Vincent et al. 2015). Infection with inflammasome-activating L. monocytogenes resulted in an earlier MMC infiltrate that penetrates into the deep T-cell zones of the spleen; however, this infiltrate is less robust and clears earlier than with wild-type infection, corresponding to a drop in burden (Williams et al. 2013). Similar early innate immune cell infiltrates consisting of predominantly macrophages were observed in zebrafish infected with inflammasome-activating L. monocytogenes ultimately resulting in attenuation (Fig. 2) (Vincent et al. 2015). Previous work with Salmonella enterica subsp. Typhimurium similarly engineered to robustly activate the inflammasome suggests that pyroptosis expels bacteria for uptake by bactericidal neutrophils ultimately resulting in phagocyte oxidase-dependent attenuation (Miao et al. 2010a). However, neutrophil depletion does not specifically rescue virulence defects following the activation of pyroptosis by L. monocytogenes (Sauer et al. 2011a; Vincent et al. 2015). Instead, depletion of macrophages rescues the virulence defect of inflammasome-activating L. monocytogenes (Vincent et al. 2015). Why the attenuation of L. monocytogenes is independent of neutrophils while S. typhimurium is dependent on neutrophils is unclear, although it is important to note that the depletion of neutrophils was not directly assessed in the Salmonella studies; rather, the loss of phagocyte oxidase implicated the role for neutrophils (Miao et al. 2010a). In wild-type L. monocytogenes infection, neutrophils are dispensable for defense, while inflammatory monocytes are critical (Shi et al. 2011), suggesting that attenuation following inflammasome activation likely depends on the cell type critical in clearing the pathogen normally.
5 Adaptive Immune Response to L. monocytogenes
The specific mechanisms by which L. monocytogenes infection triggers robust CD8+ T cell-dependent cell-mediated immune responses are largely unknown. Generally, the initial innate response is posited to inform the type and robustness of the adaptive immune response (Janeway 1989). It was hypothesized that activation of the inflammasome and its resulting robust innate inflammatory response would contribute to mounting a better adaptive immune response. As discussed below, surprising results suggest that activation of the inflammasome is detrimental for the activation of L. monocytogenes-triggered adaptive immunity (Sauer et al. 2011a). Importantly, understanding how CD8+ T-cell responses form following L. monocytogenes infection and how we can improve them is critical in the development of L. monocytogenes as a cancer immunotherapeutic platform.
5.1 Protective Immunity
To understand how the inflammasome impacts the generation of adaptive immunity, multiple groups independently generated strains of L. monocytogenes that hyperactivate the inflammasome (Sauer et al. 2011a; Warren et al. 2011). Both groups utilized expression systems whereby flagellin expression was activated exclusively in the cytosol of infected macrophages, limiting flagellin detection to the NAIP5/NLRC4 inflammasome while limiting the potential impact of TLR5-mediated flagellin recognition. Warren et al. (2011) observed that mice immunized with flagellin-expressing L. monocytogenes could survive a subsequent lethal challenge, suggesting that the activation of the inflammasome allows a protective immune response to form. While all mice immunized with a high dose of inflammasome-activating bacteria survived the subsequent lethal dose challenge, no wild-type control mice were analyzed as a comparison, and neither T-cell responses nor bacterial burdens after lethal challenge were examined to determine whether the response was better or worse than that activated by non-inflammasome-activating L. monocytogenes.
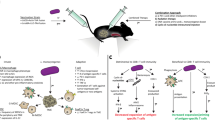
In contrast, similar immunization studies with inflammasome-activating L. monocytogenes found that adaptive immune responses were impaired. While immunization of mice with high doses of attenuated inflammasome-activating or nonactivating L. monocytogenes both conferred some degree of protection from the subsequent lethal challenge, mice were significantly less protected when immunized with strains that activated the inflammasome. More strikingly, when immunized at low doses, inflammasome-activating strains were unable to confer any protection above mock-immunized mice, whereas non-inflammasome-activating immunizations led to a significant protection from the subsequent lethal challenge (Sauer et al. 2011a). Similarly, immunization with inflammasome-activating L. monocytogenes resulted in an impaired antigen-specific primary CD8+ T-cell response, with about half as many antigen-specific CD8+ T cells forming compared to immunization with non-inflammasome-activating strains (Sauer et al. 2011a). Further, these CD8+ T-cell deficits persist into the memory stage (Sauer et al. 2011a), and levels of the recall CD8+ T cells at 2 days after challenge are also impaired (Theisen and Sauer, unpublished results). Interestingly, the recall response examined at 5 days post challenge results in similar levels of expanded antigen-specific CD8+ T cells (Sauer et al. 2011a), suggesting that the rate of expansion upon recall is slower and likely contributes to the differences seen in bacterial burdens upon challenge. Finally, caspase-1 mice immunized with L. monocytogenes mount more robust protective immune responses compared to the wild-type (Sauer et al. 2011a, JD Sauer unpublished data). Taken together, these data suggest that at high immunizing doses, some protection can be conferred from L. monocytogenes strains that activate the inflammasome, but that this response is impaired compared to strains that avoid inflammasome activation, and at lower immunizing doses, T-cell activation is insufficient to confer the protection from a subsequent lethal challenge (Fig. 3).

Inflammasome-associated inflammation impairs host cell-mediated immune responses to L. monocytogenes. In wild-type L. monocytogenes infection, L. monocytogenes minimally activates caspase-1 and, through a not fully understood mechanism, induces a robust multifunctional CD8+ T-cell response. In contrast, activation of the inflammasome by L. monocytogenes results in robust caspase-1 activation and impairment of multifunctional CD8+ T-cell generation through the inflammasome-associated inflammatory milieu, independent of IL-1β and IL-18. Inflammasome activation also results in a robust, early recruitment of myelomonocytic cells that while attenuating L. monocytogenes do not impair CD8+ T-cell generation
How inflammasome activation inhibits L. monocytogenes-stimulated immunity remains unclear. Inflammasome-activating strains of L. monocytogenes are attenuated in vivo (Sauer et al. 2011a; Warren et al. 2011; Vincent et al. 2015); however, some of the adaptive immunity studies have been done with Δact L. monocytogenes to normalize the differences in bacterial burden (Sauer et al. 2011a; Williams et al. 2013). Further, the depletion of MMCs, which partially rescues inflammasome-activating L. monocytogenes virulence defects, does not affect the defects in the development of CD8+ T cells or protective immunity (Fig. 3) (Williams et al. 2013), suggesting that deficits in adaptive immunity are independent of virulence defects and are consistent with evidence that immunity is independent of bacterial burden (Mercado et al. 2000). Interestingly, inflammasome activation is required for T-cell responses to a variety of other pathogens, including Candida albicans, Schistosoma mansoni, and Bordetella pertussis (Dunne et al. 2010; Ritter et al. 2010; van de Veerdonk et al. 2011). However, these responses largely require CD4+ T cell-dependent Th1 or Th17 responses, not the CD8+ T-cell response that L. monocytogenes induces, suggesting that inflammasome activation may have different roles in regulating the adaptive immune response pending the type of response required for pathogen control.
5.2 Influence of Cytokines
Preliminary data suggest that inflammasome-dependent inhibition of immunity is largely mediated by the inflammatory milieu and not deficits in antigen presentation or costimulation. However, the specific inflammatory mediators that inhibit the generation of robust cell-mediated immunity remain unknown (Erin Theisen and JD Sauer, unpublished data). We and others have examined how deficiency of either IL-1β or IL-18 influences the adaptive immune response. In our hands, the loss of IL-1β, IL-18, or both still results in impairments in the protective immune response following the immunization with inflammasome-activating L. monocytogenes, suggesting that these cytokines do not negatively influence the development of adaptive immunity. Further, IL-18−/− or IL-18Rα−/− mice do not have impairments in generating antigen-specific CD8+ T cells that have effector functions (Lochner et al. 2008; Haring and Harty 2009). Similarly, the loss of IL-18Rα does not impact the rate of contraction or formation or maintenance of memory T cells (Haring and Harty 2009). Interestingly, it seems that the loss of IL-18 does result in fewer CD4+ T cells; however, this ultimately does not impact the generation of protective immunity as IL-18−/− or IL-18Rα−/− mice harbor similar burdens of L. monocytogenes following the lethal challenge (Lochner et al. 2008; Haring and Harty 2009).
Taken together, these results suggest that inflammasome activation is detrimental to the influence of adaptive immunity; however, the reason for this inhibition remains largely unknown. We hypothesize that an undescribed inflammatory component is responsible for these defects.
6 Concluding Remarks
In this chapter, we have discussed the mechanisms by which L. monocytogenes can activate the inflammasome. However, in vivo data suggest that under physiologic conditions, L. monocytogenes largely avoids activating the inflammasome as evidenced by multiple studies that demonstrate no change in virulence in caspase-1-deficient mice and attenuation when L. monocytogenes is engineered to robustly activate the inflammasome (Sauer et al. 2011a; Warren et al. 2011; Vincent et al. 2015).
Attenuated L. monocytogenes is currently being developed as a cancer immunotherapeutic platform, and early returns in multiple phase I and II clinical trials have been exceptionally positive (Le et al. 2015). How L. monocytogenes is able to mount a CD8+ T-cell response that is able to break self-tolerance remains unknown. It was initially hypothesized that inflammasome activation and its associated inflammation would positively influence cell-mediated immunity; however, inflammasome activation ultimately impairs the adaptive immune response generated to L. monocytogenes (Sauer et al. 2011a; Williams et al. 2013). It is possible that although the inflammasome is detrimental in the context of strong non-self-antigens, it could be beneficial in the context of less robust or self-antigens. Studies to assess this possibility are currently underway.
While L. monocytogenes largely avoids inflammasome activation, immunization of caspase-1/11−/− mice results in better protective immune responses than wild-type mice, suggesting that even low-level inflammasome activation is detrimental to the development of adaptive immunity (Sauer et al. 2011a). Additionally, current platforms of L. monocytogenes immunotherapy are limited in the complexity of antigens they can produce, and for the production of more complex antigens, the host must be able to transcribe and translate the antigens—similar to the idea of immunization with plasmid DNA. Platforms of L. monocytogenes designed to release plasmid DNA encoding complex tumor antigens through engineered bacteriolysis or antibiotic-mediated lysis (van Pijkeren et al. 2010; Sauer et al. 2010) are severely inhibited due to inflammasome activation. Thus, understanding and modulating inflammasome activation by L. monocytogenes is critical for its success as an immunotherapeutic platform.
References
Akhter A, Caution K, Abu Khweek A et al (2012) Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37:35–47. doi:10.1016/j.immuni.2012.05.001
Anand PK, Malireddi RKS, Lukens JR et al (2012) NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488:389–393. doi:10.1038/nature11250
Auerbuch V, Brockstedt DG, Meyer-Morse N et al (2004) Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 200:527–533. doi:10.1084/jem.20040976
Bahjat KS, Meyer-Morse N, Lemmens EE et al (2009) Suppression of cell-mediated immunity following recognition of phagosome-confined bacteria. PLoS Pathog. doi:10.1371/journal.ppat.1000568
Beuzón CR, Méresse S, Unsworth KE et al (2000) Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19:3235–3249. doi:10.1093/emboj/19.13.3235
Biswas A, Meissner TB, Kawai T, Kobayashi KS (2012) Cutting edge: impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J Immunol 189:516–520. doi:10.4049/jimmunol.1200064
Boneca IG, Dussurget O, Cabanes D et al (2007) A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci USA 104:997–1002. doi:10.1073/pnas.0609672104
Bossaller L, Chiang P-I, Schmidt-Lauber C et al (2012) Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol 189:5508–5512. doi:10.4049/jimmunol.1202121
Brodsky IE, Palm NW, Sadanand S et al (2010) A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7:376–387. doi:10.1016/j.chom.2010.04.009
Broz P, von Moltke J, Jones JW et al (2010) Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8:471–483. doi:10.1016/j.chom.2010.11.007
Burke TP, Loukitcheva A, Zemansky J, et al (2014) Listeria monocytogenes is resistant to lysozyme through the regulation, not the acquisition, of cell wall-modifying enzymes. J Bacteriol 196:3756–3767. doi: 10.1128/JB.02053-14
Camilli A, Goldfine H, Portnoy DA (1991) Listeria monocytogenes mutants lacking phosphatidylinositol-specific phospholipase C are avirulent. J Exp Med 173:751–754
Carrero JA, Calderon B, Unanue ER (2006) Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med 203:933–940. doi:10.1084/jem.20060045
Chen GY, Liu M, Wang F et al (2011) A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186:7187–7194. doi:10.4049/jimmunol.1100412
Creasey EA, Isberg RR (2012) The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc Natl Acad Sci USA 109:3481–3486. doi:10.1073/pnas.1121286109
Davis BK, Roberts RA, Huang MT et al (2011) Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol 186:1333–1337. doi:10.4049/jimmunol.1003111
DeYoung KL, Ray ME, Su YA et al (1997) Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 15:453–457. doi:10.1038/sj.onc.1201206
Dunne A, Ross PJ, Pospisilova E et al (2010) Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol 185:1711–1719. doi:10.4049/jimmunol.1000105
Elinav E, Strowig T, Kau AL et al (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745–757. doi:10.1016/j.cell.2011.04.022
Fernandes-Alnemri T, Yu J-W, Datta P et al (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513. doi:10.1038/nature07710
Fernandes-Alnemri T, Kang S, Anderson C et al (2013) Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol 191:3995–3999. doi:10.4049/jimmunol.1301681
Ferreira V, Wiedmann M, Teixeira P, Stasiewicz MJ (2014) Listeria monocytogenes persistence in food-associated environments: epidemiology, strain characteristics, and implications for public health. J Food Prot 77:150–170. doi:10.4315/0362-028X.JFP-13-150
Franchi L, Kanneganti T-D, Dubyak GR, Núñez G (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282:18810–18818. doi:10.1074/jbc.M610762200
Gahan CG, Collins JK (1995) Non-dystrophic 129 REJ mice are susceptible to i.p. infection with Listeria monocytogenes despite an ability to recruit inflammatory neutrophils to the peritoneal cavity. Microb Pathog 18:355–364. doi:10.1006/mpat.1995.0032
Ge J, Gong Y-N, Xu Y, Shao F (2012) Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc Natl Acad Sci USA 109:6193–6198. doi:10.1073/pnas.1117490109
Glaccum MB, Stocking KL, Charrier K et al (1997) Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol 159:3364–3371
Glomski IJ, Gedde MM, Tsang AW et al (2002) The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol 156:1029–1038. doi:10.1083/jcb.200201081
Glomski IJ, Decatur AL, Portnoy DA (2003) Listeria monocytogenes mutants that fail to compartmentalize Listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun 71:6754–6765. doi:10.1128/IAI.71.12.6754-6765.2003
Goetz M, Bubert A, Wang G et al (2001) Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci USA 98:12221–12226. doi:10.1073/pnas.211106398
Goossens PL, Milon G (1992) Induction of protective CD8+ T lymphocytes by an attenuated Listeria monocytogenes actA mutant. Int Immunol 4:1413–1418
Gringhuis SI, Kaptein TM, Wevers BA et al (2012) Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol 13:246–254. doi:10.1038/ni.2222
Hamon MA, Cossart P (2011) K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore-forming toxins. Infect Immun 79:2839–2846. doi:10.1128/IAI.01243-10
Hamon MA, Ribet D, Stavru F, Cossart P (2012) Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol 20:360–368. doi:10.1016/j.tim.2012.04.006
Hara H, Tsuchiya K, Nomura T et al (2008) Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J Immunol 180:7859–7868
Haring JS, Harty JT (2009) Interleukin-18-related genes are induced during the contraction phase but do not play major roles in regulating the dynamics or function of the T-cell response to Listeria monocytogenes infection. Infect Immun 77:1894–1903. doi:10.1128/IAI.01315-08
Hornung V, Ablasser A, Charrel-Dennis M et al (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. doi:10.1038/nature07725
Janeway CA (1989) Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54(Pt 1):1–13
Kamp HD, Higgins DE (2009) Transcriptional and post-transcriptional regulation of the GmaR antirepressor governs temperature-dependent control of flagellar motility in Listeria monocytogenes. Mol Microbiol 74:421–435. doi:10.1111/j.1365-2958.2009.06874.x
Kanneganti T-D, Ozören N, Body-Malapel M et al (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440:233–236. doi:10.1038/nature04517
Kayagaki N, Warming S, Lamkanfi M et al (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121. doi:10.1038/nature10558
Kayagaki N, Stowe IB, Lee BL et al (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671. doi:10.1038/nature15541
Kim S, Bauernfeind F, Ablasser A et al (2010) Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol 40:1545–1551. doi:10.1002/eji.201040425
Kocks C, Gouin E, Tabouret M et al (1992) L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531
Kofoed EM, Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595. doi:10.1038/nature10394
Kortmann J, Brubaker SW, Monack DM (2015) Cutting edge: inflammasome activation in primary human macrophages is dependent on flagellin. J Immunol 195:815–819. doi:10.4049/jimmunol.1403100
Labow M, Shuster D, Zetterstrom M et al (1997) Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J Immunol 159:2452–2461
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. doi:10.1038/nri3452
Le DT, Dubensky TW, Brockstedt DG (2012) Clinical development of Listeria monocytogenes-based immunotherapies. Semin Oncol 39:311–322. doi:10.1053/j.seminoncol.2012.02.008
Le DT, Wang-Gillam A, Picozzi V et al (2015) Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 33:1325–1333. doi:10.1200/JCO.2014.57.4244
Leber JH, Crimmins GT, Raghavan S et al (2008) Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog 4:e6. doi:10.1371/journal.ppat.0040006
Lin K-M, Hu W, Troutman TD et al (2014) IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci USA 111:775–780. doi:10.1073/pnas.1320294111
Lochner M, Kastenmüller K, Neuenhahn M et al (2008) Decreased susceptibility of mice to infection with Listeria monocytogenes in the absence of interleukin-18. Infect Immun 76:3881–3890. doi:10.1128/IAI.01651-07
Mariathasan S, Weiss DS, Newton K et al (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232. doi:10.1038/nature04515
Meissner TB, Li A, Biswas A et al (2010) NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA 107:13794–13799. doi:10.1073/pnas.1008684107
Meixenberger K, Pache F, Eitel J et al (2010) Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol 184:922–930. doi:10.4049/jimmunol.0901346
Mengaud J, Braun-Breton C, Cossart P (1991) Identification of phosphatidylinositol-specific phospholipase C activity in Listeria monocytogenes: a novel type of virulence factor? Mol Microbiol 5:367–372
Mengaud J, Ohayon H, Gounon P et al (1996) E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84:923–932
Mercado R, Vijh S, Allen SE et al (2000) Early programming of T cell populations responding to bacterial infection. J Immunol 165:6833–6839
Miao EA, Leaf IA, Treuting PM et al (2010a) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11:1136–1142. doi:10.1038/ni.1960
Miao EA, Mao DP, Yudkovsky N et al (2010b) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107:3076–3080. doi:10.1073/pnas.0913087107
Mielke LA, Elkins KL, Wei L et al (2009) Tumor progression locus 2 (Map3k8) is critical for host defense against Listeria monocytogenes and IL-1 beta production. J Immunol 183:7984–7993. doi:10.4049/jimmunol.0901336
Neighbors M, Xu X, Barrat FJ et al (2001) A critical role for interleukin 18 in primary and memory effector responses to Listeria monocytogenes that extends beyond its effects on interferon production. J Exp Med 194:343–354
Normand S, Delanoye-Crespin A, Bressenot A et al (2011) Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA 108:9601–9606. doi:10.1073/pnas.1100981108
O’Connell RM, Saha SK, Vaidya SA et al (2004) Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 200:437–445. doi:10.1084/jem.20040712
O’Riordan M, Yi CH, Gonzales R et al (2002) Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci 99:13861–13866. doi:10.1073/pnas.202476699
Opitz B, Püschel A, Beermann W et al (2006) Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J Immunol 176:484–490
Ozören N, Masumoto J, Franchi L et al (2006) Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J Immunol 176:4337–4342
Peel M, Donachie W, Shaw A (1988) Temperature-dependent expression of flagella of Listeria monocytogenes studied by electron microscopy, SDS-PAGE and western blotting. J Gen Microbiol 134:2171–2178. doi:10.1099/00221287-134-8-2171
Peng K, Broz P, Jones J et al (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell Microbiol 13:1586–1600. doi:10.1111/j.1462-5822.2011.01643.x
Personnic N, Bruck S, Nahori M-A et al (2010) The stress-induced virulence protein InlH controls interleukin-6 production during murine listeriosis. Infect Immun 78:1979–1989. doi:10.1128/IAI.01096-09
Portnoy DA, Jacks PS, Hinrichs DJ (1988) Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med 167:1459–1471
Rae CS, Geissler A, Adamson PC, Portnoy DA (2011) Mutations of the Listeria monocytogenes peptidoglycan N-deacetylase and O-acetylase result in enhanced lysozyme sensitivity, bacteriolysis, and hyperinduction of innate immune pathways. Infect Immun 79:3596–3606. doi:10.1128/IAI.00077-11
Ritter M, Gross O, Kays S et al (2010) Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci USA 107:20459–20464. doi:10.1073/pnas.1010337107
Sakhon OS, Victor KA, Choy A et al (2013) NSD1 mitigates caspase-1 activation by listeriolysin O in macrophages. PLoS ONE 8:e75911. doi:10.1371/journal.pone.0075911
Sauer J-D, Witte CE, Zemansky J et al (2010) Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7:412–419. doi:10.1016/j.chom.2010.04.004
Sauer J-D, Pereyre S, Archer KA et al (2011a) Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci USA 108:12419–12424. doi:10.1073/pnas.1019041108
Sauer J-D, Sotelo-Troha K, von Moltke J et al (2011b) The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79:688–694. doi:10.1128/IAI.00999-10
Schmidt RL, Lenz LL (2012) Distinct licensing of IL-18 and IL-1β secretion in response to NLRP3 inflammasome activation. PLoS ONE 7:e45186. doi:10.1371/journal.pone.0045186
Schnupf P, Hofmann J, Norseen J et al (2006a) Regulated translation of listeriolysin O controls virulence of Listeria monocytogenes. Mol Microbiol 61:999–1012. doi:10.1111/j.1365-2958.2006.05286.x
Schnupf P, Portnoy DA, Decatur AL (2006b) Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell Microbiol 8:353–364. doi:10.1111/j.1462-5822.2005.00631.x
Shen A, Higgins DE (2005) The 5’ untranslated region-mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol 57:1460–1473. doi:10.1111/j.1365-2958.2005.04780.x
Shen A, Higgins DE (2006) The MogR transcriptional repressor regulates nonhierarchal expression of flagellar motility genes and virulence in Listeria monocytogenes. PLoS Pathog 2:e30. doi:10.1371/journal.ppat.0020030
Shen Y, Naujokas M, Park M, Ireton K (2000) InlB-dependent internalization of Listeria is mediated by the met receptor tyrosine kinase. Cell 103:501–510. doi:10.1016/S0092-8674(00)00141-0
Shen A, Kamp HD, Grundling A, Higgins DE (2006) A bifunctional O-GlcNAc transferase governs flagellar motility through anti-repression. Genes Dev 20:3283–3295. doi:10.1101/gad.1492606
Shi C, Hohl TM, Leiner I et al (2011) Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J Immunol 187:5293–5298. doi:10.4049/jimmunol.1101721
Shi J, Zhao Y, Wang K et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. doi:10.1038/nature15514
Slaghuis J, Goetz M, Engelbrecht F, Goebel W (2004) Inefficient replication of Listeria innocua in the cytosol of mammalian cells. J Infect Dis 189:393–401. doi:10.1086/381206
Sugawara S, Uehara A, Nochi T et al (2001) Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol 167:6568–6575
Sutterwala FS, Mijares LA, Li L et al (2007) Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204:3235–3245. doi:10.1084/jem.20071239
Swaminathan B, Gerner-Smidt P (2007) The epidemiology of human listeriosis. Microbes Infect 9:1236–1243. doi:10.1016/j.micinf.2007.05.011
Thomas JA, Allen JL, Tsen M et al (1999) Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol 163:978–984
Tsuchiya K, Hara H, Kawamura I et al (2010) Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J Immunol 185:1186–1195. doi:10.4049/jimmunol.1001058
Tsuchiya K, Hara H, Fang R et al (2014) The adaptor ASC exacerbates lethal Listeria monocytogenes infection by mediating IL-18 production in an inflammasome-dependent and -independent manner. Eur J Immunol 44:3696–3707. doi:10.1002/eji.201444673
Tsuji NM, Tsutsui H, Seki E et al (2004) Roles of caspase-1 in Listeria infection in mice. Int Immunol 16:335–343
Uchiyama R, Yonehara S, Tsutsui H (2013) Fas-mediated inflammatory response in Listeria monocytogenes infection. J Immunol (Baltimore, Md 1950) 190:4245–4254. doi:10.4049/jimmunol.1203059
van de Veerdonk FL, Joosten LAB, Shaw PJ et al (2011) The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur J Immunol 41:2260–2268. doi:10.1002/eji.201041226
van Pijkeren JP, Morrissey D, Monk IR et al (2010) A novel Listeria monocytogenes-based DNA delivery system for cancer gene therapy. Hum Gene Ther 21:405–416. doi:10.1089/hum.2009.022
Vazquez-Boland JA, Kocks C, Dramsi S et al (1992) Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect Immun 60:219–230
Vincent WJB, Freisinger CM, Lam P-Y et al (2015) Macrophages are required for inflammasome-dependent host defense in vivo. Cell Microbiol. doi:10.1111/cmi.12536
Waite JC, Leiner I, Lauer P et al (2011) Dynamic imaging of the effector immune response to listeria infection in vivo. PLoS Pathog 7:e1001326. doi:10.1371/journal.ppat.1001326
Wang S, Miura M, Jung YK et al (1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92:501–509
Warren SE, Mao DP, Rodriguez AE et al (2008) Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol 180:7558–7564
Warren SE, Armstrong A, Hamilton MK et al (2010) Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol 185:818–821. doi:10.4049/jimmunol.1000724
Warren SE, Duong H, Mao DP et al (2011) Generation of a Listeria vaccine strain by enhanced caspase-1 activation. Eur J Immunol 41:1934–1940. doi:10.1002/eji.201041214
Williams CR, Dustin ML, Sauer J-D (2013) Inflammasome-mediated inhibition of Listeria monocytogenes-stimulated immunity is independent of myelomonocytic function. PLoS ONE 8:e83191. doi:10.1371/journal.pone.0083191
Witte CE, Archer KA, Rae CS et al (2012) Innate immune pathways triggered by Listeria monocytogenes and their role in the induction of cell-mediated immunity. Adv Immunol 113:135–156. doi:10.1016/B978-0-12-394590-7.00002-6
Woodward JJ, Iavarone AT, Portnoy DA (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 80(328):1703–1705
Wu J, Fernandes-Alnemri T, Alnemri ES (2010) Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in Caspase-1 activation by Listeria monocytogenes. J Clin Immunol 30:693–702. doi:10.1007/s10875-010-9425-2
Yao Y, Wang Y, Chen F et al (2012) NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res 22:836–847. doi:10.1038/cr.2012.56
Zhao Y, Yang J, Shi J et al (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi:10.1038/nature10510
Zheng H, Fletcher D, Kozak W et al (1995) Resistance to fever induction and impaired acute-phase response in interleukin-1β-deficient mice. Immunity 3:9–19. doi:10.1016/1074-7613(95)90154-X
Zou T, Garifulin O, Berland R, Boyartchuk VL (2011) Listeria monocytogenes infection induces prosurvival metabolic signaling in macrophages. Infect Immun 79:1526–1535. doi:10.1128/IAI.01195-10
Acknowledgments
The authors would like to thank Dr. Laurie Ristow and Grischa Chen for critical reading of this manuscript. The authors would also like to thank other members of the Sauer laboratory for commentary on the figures. This work is funded by the NIH (R01 CA188034) to J-D.S., and an AAI Careers in Immunology Fellowship to E.T.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Theisen, E., Sauer, JD. (2016). Listeria monocytogenes and the Inflammasome: From Cytosolic Bacteriolysis to Tumor Immunotherapy. In: Backert, S. (eds) Inflammasome Signaling and Bacterial Infections. Current Topics in Microbiology and Immunology, vol 397. Springer, Cham. https://doi.org/10.1007/978-3-319-41171-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-41171-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41170-5
Online ISBN: 978-3-319-41171-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)