
Abstract
The complementary actions of the innate and adaptive immune systems often provide effective host defense against microbial pathogens and harmful environmental agents. Germline-encoded pattern recognition receptors (PRRs) endow the innate immune system with the ability to detect and mount a rapid response against a given threat. Members of several intracellular PRR families, including the nucleotide-binding domain and leucine-rich repeat containing receptors (NLRs), the AIM2-like receptors (ALRs), and the tripartite motif-containing (TRIM) protein Pyrin/TRIM20, nucleate the formation of inflammasomes. These cytosolic scaffolds serve to recruit and oligomerize the cysteine protease caspase-1 in filaments that promote its proximity-induced autoactivation. This oligomerization occurs either directly or indirectly through intervention of the bipartite adaptor protein ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD), which is needed for the domain interaction. Caspase-1 cleaves the precursors of the inflammatory cytokines interleukin (IL)-1β and IL-18 and triggers their release into the extracellular space, where they act on effector cells to promote both local and systemic immune responses. Additionally, inflammasome activation gives rise to a lytic mode of cell death, named pyroptosis, which is thought to contribute to initial host defense against infection by eliminating replication niches of intracellular pathogens and exposing them to the immune system. Inflammasome-induced host defense responses are the subject of intense investigation, and understanding their physiological roles during infection and the regulatory circuits that are involved is becoming increasingly detailed. Here, we discuss current understanding of the activation mechanisms and biological outcomes of inflammasome activation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: PRRs, PAMPs, and DAMPs
The complementary qualities of the innate and adaptive immune systems allow vertebrates to mount a highly tailored and efficacious host defense against intruding pathogens, while avoiding immunopathology. The efficacy of such combined immune response for defense against pathogens and harmful environmental agents is illustrated by the presence of innate and adaptive immune subsystems in early vertebrates such as the lamprey and mammals alike (Boehm 2012). The innate immune system detects and mounts the initial response to the threat. In mammals, the innate immune system encompasses a diversity of physical and chemical barriers. This includes mucosal membranes that interface the extracellular environment, a complement system that tags invading agents for removal, and professional phagocytes that clear the infectious agent. Phagocytes such as macrophages and neutrophils also release inflammatory mediators to recruit additional immune cells into the affected area (Palm and Medzhitov 2009a). Adaptive immunity is directed by dendritic cells and other antigen-presenting cells (APCs) that relay information about the harmful agent to lymphocytes. Pathogen-derived peptide antigens are presented to T-lymphocytes in association with major histocompatibility complex (MHC) proteins, whereas multiple mechanisms may govern how B-lymphocytes encounter antigens (Cyster 2010; Palm and Medzhitov 2009b). The highly specific adaptive immune system produces B- and T-lymphocytes with diverse antigen receptors, and clonal expansion of the cells recognizing foreign material culminates in its targeted removal. In addition, the adaptive immune system is capable of immune memory that provides protection from reinfection with the same pathogen (Koch and Radtke 2011).
While antigen receptor gene rearrangements in lymphocytes enable the adaptive immune system to recognize seemingly any antigen, innate immune cells rely on only a fixed set of germline-encoded ‘pattern recognition receptors’ (PRRs) to detect pathogens (Takeuchi and Akira 2010). PRRs are expressed on many cell types that may come in contact with microbes, including hematopoietic cells, fibroblasts, endothelial cells, and epithelial cells that line mucosal membranes. Given the limited repertoire of PRRs that is available to the host, it may be unsurprising that—rather than signaling out a particular microbe—PRRs guard conserved microbial signatures termed ‘pathogen-associated molecular patterns’ (PAMPs) that may signal infection by a certain class of pathogens. Microbial nucleic acids, bacterial secretion systems, and components of the microbial cell wall that are not produced by eukaryotes are the examples of such conserved microbial factors that are sensed by PRRs. Nevertheless, damaged host cells as well may trigger PRR activation by releasing danger-associated molecular patterns (DAMPs) such as uric acid crystals, ATP, high-mobility group box 1 (HMGB1), and the heat-shock proteins Hsp70 and Hsp90 (Takeuchi and Akira 2010). Detection of DAMPs by PRRs is thought to primarily promote tissue repair, although excessive release might elicit a severe inflammatory response exacerbating tissue damage in several infectious and autoinflammatory and autoimmune diseases (Lotze et al. 2007).
PRR families may be subdivided into genuine transmembrane receptors that survey the extracellular environment and endosomes for DAMPs and PAMPs, and those that reside in cytosolic compartments. Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) are prime examples of the first class, while the RIG-I-like receptor (RLR), the AIM2-like receptor (ALR), and the nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins all respond to pathogens at intracellular compartments (Takeuchi and Akira 2010). PRRs are also frequently classified according to the PAMPs and DAMPs they sense, or the immune signaling pathways they control. For instance, members of several PRR families may detect microbial DNA or RNA molecules and engage signaling cascades that culminate in activation of members of the nuclear factor-kappa B (NF-κB), activator protein 1 (AP1), and interferon regulatory factor (IRF) transcription factor families (Battistini 2009; Kim and Choi 2010; Vallabhapurapu and Karin 2009). The concerted activities of these key inflammatory transcription factors lead to the production of type I interferons (IFN-I), inflammatory cytokines, and other pro-inflammatory or microbicidal proteins (Takeuchi and Akira 2010). In addition to these comprehensive transcriptional reprograming events, innate immune cells are equipped with PRRs that may assemble cytosolic multi-protein complexes called ‘inflammasomes.’ Inflammasomes are regarded as key elements in the innate immune response of mammalian hosts in providing protection against invading micro-organisms. By definition, inflammasomes are scaffolds for activation of the inflammatory cysteine-dependent aspartate-specific protease caspase-1. This protease is chiefly known for its key role in maturation and secretion of the inflammatory cytokines interleukin IL-1β and IL-18 (Lamkanfi and Dixit 2014). Additionally, caspase-1 activation may result in a programmed, lytic cell death of myeloid cells that has been named ‘pyroptosis’ (Cookson and Brennan 2001). Based on a wealth of experimental evidence gathered in the past 2 decades, these two inflammasome-dependent biological responses (cytokine production and pyroptosis) contribute importantly to the immune system’s ability to resolve the threat and restore homeostasis. Here, we will review and discuss current understanding of inflammasome biology with an emphasis on recent developments in control of microbial infections. A brief introduction of the different inflammasomes characterized to date will be followed by a discussion of the roles of particular inflammasome complexes in microbial infections. Finally, mechanisms regulating inflammasome activation will be discussed along with specific examples illustrating the importance of tight regulation of inflammasome activation.
2 PRRs as Inflammasome Scaffolds
Selected members of the ‘Hematopoietic interferon-inducible nuclear protein family’ (Hin200) and the ‘Tripartite Motif Family’ (TRIM) PRR families assemble inflammasomes in their own right (Fig. 1), but the majority of inflammasomes relies on NLR proteins for pathogen sensing and scaffold assembly. NLRs are defined by the combined presence of a nucleotide-binding and oligomerization domain (NACHT/NBD) and leucine-rich repeat (LRR) motifs, typically located in the central and most carboxy-terminal regions of the proteins (Kanneganti et al. 2007). Most NLRs in addition contain an amino-terminal protein interaction domain of the baculovirus inhibitor repeat (BIR), Pyrin (PYD), and caspase recruitment domain (CARD) types, thus allowing their subclassification based on domain architecture (Fig. 1). The NLR family consists of 22 human and 34 murine members that play diverse roles in the mammalian immune and reproductive systems (Kanneganti et al. 2007; Van Gorp et al. 2014). NLRs are considered an evolutionary ancient PRR family as supported by the identification of over 200 NLR genes in the genome of the sea urchin Strongylocentrotus purpuratus (Rast et al. 2006), and the presence of NLRs in zebrafish (Laing et al. 2008; Stein et al. 2007) and tetrapods (Hansen et al. 2011). However, NLR genes appear to have been lost during speciation of particular animal species. For instance, NLRs are absent from the genome of the model nematode species Caenorhabditis elegans and the insects Drosophila melanogaster (fruit fly) and Apis mellifera (honey bee). However, they are present in other insects such as Culex quinquefasciatus (southern house mosquito) and Aedes aegypti (yellow fever mosquito) (Lange et al. 2011). Although genuine NLR genes have so far only been identified in animals, homologs with functionally related domains and motifs have been described in Hydra, fungi, and plants as well (Lange et al. 2011). The comparable domain architecture of the pathogen-resistant (R-) proteins of higher plants in which carboxy-terminal LRR motifs combine with a centrally located NB-ARC domain that is structurally related to the NACHT ATPase of NLRs represents an nice example of convergent evolution (Chisholm et al. 2006). Moreover, also R-proteins are central mediators of antimicrobial resistance mechanisms that are collective referred to as the ‘hypersensitive response’ in plants (Hulbert et al. 2001).

Inflammasome components and their domain architecture. Members of the nucleotide-binding domain and leucine-rich repeat containing receptors (NLR) family contain a central ‘neuronal apoptosis inhibitor protein (NAIP), CIITA (MHC class II transcription activator), HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein)’ (NACHT) ATPase domain, and leucine-rich repeat (LRR) motifs. NLR family members may be further subdivided into NLRP, containing a Pyrin domain (PYD); NLRC, containing a caspase recruitment domain (CARD); and NLRB, containing a Baculovirus inhibitor repeat (BIR) domain. The latter subset is frequently referred to as the NAIP proteins. The AIM2-like receptor (ALR) family is composed of a PYD and a dsDNA-binding ‘hematopoietic interferon-inducible nuclear protein with a 200 amino acid repeat’ (Hin200) domain. Finally, the inflammasome scaffold protein Pyrin consists of PYD, B-Box-type zinc finger (BB), and coiled-coil (CC) domains that are followed by a carboxy-terminal B30.2 domain in the human, but not its murine ortholog. Inflammasome assembly involves homotypic CARD and PYD interactions between its components
The physiological roles of most mammalian NLRs are largely obscure, but some members have well-defined roles in the regulation of inflammatory gene transcription. Among the best characterized examples are the intracellular peptidoglycan receptors NOD1 and NOD2 (nucleotide-binding oligomerization domain containing proteins 1/2) that promote RIPK2- and CARD9-dependent transcription of NF-κB- and AP1-target genes (Wilmanski et al. 2008). Additionally, transcription of major histocompatibility class II (MHC II) genes in antigen-presenting cells requires the NLR protein class II trans-activator (CIITA) (Wilmanski et al. 2008). Another subset of NLRs—namely NLRP1, NLRP3, NLRC4, and NAIP—promotes immune responses at the post-translational level by initiating inflammasome signaling (Lamkanfi and Dixit 2014).
Signal-induced PRR clustering is thought to recruit and promote the nucleation of procaspase-1 filaments in which the protease zymogens undergo proximity-induced autoactivation. Caspase-1 may be recruited in these inflammasome scaffolds through direct homotypic interactions involving the CARD motifs of procaspase-1 and that of the nucleating PRR (NLRC4, NLRP1b), or indirectly through homotypic interactions with the bipartite PYD/CARD inflammasome adaptor protein ASC in the case of PYD-containing NLRP3, AIM2, and Pyrin receptors. Notably, inflammasome-induced activation of caspase-1 by each of these inflammasomes is associated with ASC-dependent autoproteolytic cleavage of caspase-1, but automaturation does not appear essential for pyroptosis induction in the context of the NLRP1b and NLRC4 inflammasomes (Broz et al. 2010b; Van Opdenbosch et al. 2014). Nevertheless, ASC does contribute to efficient secretion of bioactive IL-1β and IL-18 in response to triggers of these respective inflammasomes.
IL-1β is responsible for generation of fever, lymphocyte activation, and guiding the transmigration of leukocytes into the stress location (Dinarello 2009). As such, IL-1β induces both a systemic and a local response to infection and injury. IL-18 does not have this pyrogenic activity, but it orchestrates IFN gamma production, leading to control over the Th1 population. Depending on the cytokine environment, IL-18 can as well orchestrate the Th2 population. Furthermore, IL-18 regulates ROS production, expression of cell adhesion molecules and expression of other chemokines/cytokines (Dinarello 2009). Considering the broad impact of these proinflammatory cytokines on the hosts’ immune responses, a strict control of their activity is needed. One mechanism by which this is accomplished involves transcriptional control of their expression levels. ProIL-1β is virtually absent in naïve myeloid cells, but its mRNA levels are highly responsive to NF-κB-dependent transcriptional upregulation. By contrast, proIL-18 is constitutively present in the cytosol of naïve macrophages and dendritic cells. The differential regulation of proIL-1β and proIL-18 at the transcriptional level is illustrated by the observation that the NLRP1b and NLRC4 inflammasomes can be induced to secrete mature IL-18—but not IL-1β—in the absence of prior TLR engagement (Nystrom et al. 2013; Van Opdenbosch et al. 2014). In this context, the requirement for cytokine maturation may be regarded as another safeguard against accidental release of IL-1β and IL-18. Indeed, unlike most other cytokines that are secreted through the classical secretory pathway, proIL-1β and proIL-18 are produced as biologically inactive cytosolic precursors that await caspase-1-dependent cleavage for release of their bioactive forms (Gu et al. 1997; Lamkanfi 2011). Several mechanisms have been proposed by which the latter cytokines may be released, the most recent being another prominent outcome of inflammasome activation, namely pyroptosis. Pyroptosis is a lytic form of cell death characterized by cytoplasmic swelling and early rupture of the plasma membrane that requires the protease activities of either caspase-1 or caspase-11 (Cookson and Brennan 2001; Kayagaki et al. 2011). Pyroptosis is thought to constitute a defensive innate immune strategy of the host that eliminates the replicative niche of intracellularly replicating pathogens and exposes them to other immune cells (Aachoui et al. 2013; Casson et al. 2013; Miao et al. 2010a). Additionally, pyroptosis-associated cell lysis further releases DAMPs such as IL-1α and HMGB1 into the extracellular environment (de Gassart and Martinon 2015; Lamkanfi et al. 2010).
3 Inflammasome Activation Mechanisms
3.1 The NLRP3 Inflammasome
The NLRP3 inflammasome is by far responding to the largest set of activating agents (Lamkanfi and Dixit 2014). It is also rather unique among inflammasomes in that it requires an NF-κB-mediated signal prior to its activation. This so-called ‘signal 1’ or ‘priming’ step involves transcriptional upregulation of NLRP3 together with proIL-1β (Bauernfeind et al. 2009) and is defective in mice lacking the NF-κB regulator A20/TNFAIP3 (Vande Walle et al. 2014). Non-transcriptional mechanisms that involve its de-ubiquitination and/or IL-1 receptor-associated kinase (IRAK-1) kinase activity have also been proposed to additionally control NLRP3 inflammasome activation (Juliana et al. 2012; Lin et al. 2014; Lopez-Castejon et al. 2013; Py et al. 2013). Together, these priming mechanisms establish effective checkpoints that prevent accidental NLRP3 inflammasome activation.
In the presence of such priming signals, NLRP3 inflammasome assembly and activation is induced when the host cell is exposed to a ‘signal 2’. NLRP3 inflammasome activation may be induced by the components of bacterial [Staphylococcus aureus, Streptococcus pneumoniae, Listeria monocytogenes (Mariathasan et al. 2006; McNeela et al. 2010; Wu et al. 2010)]; viral [Influenza A virus (IAV); Encephalomyocarditis virus (EMCV); Vesicular stomatitis virus (VSV)] (Allen et al. 2009; Kanneganti et al. 2006; Rajan et al. 2011); as well as fungal (Candida albicans, Aspergillus fumigatus, Cryptococcus neoformans) (Gross et al. 2009; Guo et al. 2014; Hise et al. 2009; Said-Sadier et al. 2010) origin. In addition, DAMPs such as millimolar concentrations extracellular ATP, calcium pyrophosphate dehydrate and monosodium urate (Mariathasan et al. 2006; Martinon et al. 2006, 2009), environmental crystals (alum, silica, asbestos) (Dostert et al. 2008; Eisenbarth et al. 2008; Hornung et al. 2008; Martinon et al. 2006), β-fibrils and aggregates (β-amyloid, β-glucans) (Halle et al. 2008; Kumar et al. 2009), and ionophores (nigericin, maitotoxin) (Mariathasan et al. 2006; Perregaux and Gabel 1994) all engage the NLRP3 inflammasome. The NLRP3 inflammasome may also be engaged by intracellular LPS. Unlike for the ‘canonical’ stimuli above, caspase-11 and its human orthologs caspases 4 and 5 are required for cytosolic LPS-induced NLRP3 inflammasome activation (Baker et al. 2015; Hagar et al. 2013; Kayagaki et al. 2011; Kayagaki et al. 2013; Schmid-Burgk et al. 2015; Shi et al. 2014). In this ‘non-canonical’ signaling pathway, caspase-11 induces pyroptosis independently of the NLRP3 inflammasome, while cleavage of proIL-1β and proIL-18 is relayed through the NLRP3 inflammasome (Kayagaki et al. 2011). The importance of non-canonical NLRP3 inflammasome signaling in Gram-negative infections is highlighted by the resistance of caspase-11-deficient mice to LPS-induced lethality (Kayagaki et al. 2011; Wang et al. 1998), and their increased susceptibility to enteric pathogens (Broz et al. 2012; Gurung et al. 2012; Knodler et al. 2014). Recent work revealed cleavage of the gasdermin D as a central commonality of pyroptosis induction by caspases 1 and 11 (Kayagaki et al. 2015; Shi et al. 2016, 2015). Gasdermin D cleavage also links LPS-induced caspase-11 activation with engagement of the NLRP3 inflammasome, but how it integrates with mechanisms of canonical inflammasome activation is not fully clarified.
Since the diverse canonical NLRP3 inflammasome-activating agents listed above are structurally and chemically unrelated, a direct ligand sensing model for the NLRP3 inflammasome is highly unlikely. Instead, NLRP3 activation is thought to involve a defined cellular event or secondary messenger that is commonly and selectively triggered by these NLRP3-activating agents. In this respect, K+ is among the most frequently cited ions in regulation of NLRP3 activation. A drop in intracellular K+ levels is regarded as a prerequisite for activation of NLRP3 because it accompanies NLRP3 inflammasome activation by a diversity of agents, and because preventing K+ efflux by cultivating cells in media containing high extracellular K+ concentrations prevents caspase-1 activation by the NLRP3 inflammasome (Franchi et al. 2007; Munoz-Planillo et al. 2013; Perregaux and Gabel 1994; Petrilli et al. 2007). However, also activation of the Nlrp1b inflammasome was reported to be sensitive to high K+ concentrations (Fink et al. 2008; Wickliffe et al. 2008). Na+, Ca2+, and Cl− are other candidates for ion flux-mediated NLRP3 activation. For instance, exchanging Na+ for Li+, choline or K+ in iso-osmotic media of LPS-primed macrophages was shown to prevent ATP-induced caspase-1 activation (Perregaux and Gabel 1998). The specific role of intracellular Ca2+ is a matter of debate, with reports arguing against its requirement but rather correlating its involvement with simultaneous K+ efflux induction (Katsnelson et al. 2015; Munoz-Planillo et al. 2013); or implicating Ca2+ fluxing as a critical step in NLRP3 inflammasome activation (Lee et al. 2012; Yaron et al. 2015).
Mitochondrial dysfunction—sometimes coupled with ionic flux deregulation—has also been proposed to control NLRP3 activation, with potential involvement of cardiolipin release, oxidized mitochondrial DNA, and loss of the mitochondrial membrane potential as secondary messengers for NLRP3 activation (Iyer et al. 2013; Nakahira et al. 2011; Shimada et al. 2012). Despite the ambiguities surrounding the molecular events regulating NLRP3 activation, Nek7 was recently established as a key NLRP3-binding partner that selectively controls activation of the NLRP3 inflammasome (He et al. 2016; Schmid-Burgk et al. 2016; Shi et al. 2016, 2015). This finding may provide a novel grasping point for further dissection of NLRP3 activation mechanisms.
The need for tight regulation of NLRP3 inflammasome activation is best highlighted by the existence of autoinflammatory diseases caused by gain-of-function mutations in NLRP3. These cryopyrin-associated periodic syndromes (CAPS) cover three autoinflammatory diseases. Familial cold autoinflammatory syndrome (FCAS) is associated with cold-induced fevers, rash, and constitutional symptoms. Muckle–Wells syndrome (MWS) is not triggered by exposure to cold and additionally features the possible occurrence of hearing loss (Hoffman et al. 2001). Fever, chronic meningitis, eye inflammation, hearing loss, skin rash, and a deforming arthropathy all are clinical aspects of neonatal-onset multisystem inflammatory disease (NOMID) (Aksentijevich et al. 2002). Over 80 disease-associated NLRP3 mutations have been reported, most of them being situated within or in the close vicinity of the central NACHT domain. The effectiveness of anti-IL-1 therapies in CAPS patients illustrates the pathogenic role of IL-1β in these diseases (Yu and Leslie 2011). The identification of small-molecule inhibitors that prevent NLRP3 inflammasome activation might one day enable therapeutic strategies that selectively prevent cytokine secretion by this inflammasome only (Coll et al. 2015; Lamkanfi et al. 2009).
3.2 The NLRP1 Inflammasome
NLRP1 was one of the first NLRs reported to assemble an inflammasome (Martinon et al. 2002). NLRP1 undergoes autocleavage in a unique ‘function to find’ domain (FIIND) that lays between its carboxy-terminal CARD and LRR motifs (Chavarria-Smith and Vance 2013; Finger et al. 2012; Frew et al. 2012). Whereas humans encode a single NLRP1 gene, mice may express up to three different NLRP1 isoforms, known as Nlrp1a, Nlrp1b, and Nlrp1c (Boyden and Dietrich 2006). Moreover, human NLRP1 is equipped with both an amino-terminal PYD and a carboxy-terminal CARD, the former being absent in its murine homologs. Nevertheless, the murine paralogs appear to have non-redundant roles in immune signaling. Nlrp1b is critical for inflammasome assembly in macrophages upon exposure to lethal toxin (LeTx) of Bacillus anthracis, the causative agent of anthrax (Fig. 2). LeTx is a bipartite toxin consisting of a protective antigen (PA) and a lethal factor (LF) subunit, in which PA functions as a pore-forming unit enabling cytosolic delivery of the zinc metalloprotease LF subunit (Bann 2012). LF protease activity is required for Nlrp1b inflammasome activation because catalytically inactive mutants of LF fail to activate the Nlrp1b inflammasome (Levinsohn et al. 2012). This suggests that instead of directly sensing the presence of LF, Nlrp1b monitors LF protease activity in the cytosol. Notably, the inflammasome adaptor ASC is critical for LeTx-induced caspase-1 autocleavage, but the induction of pyroptosis proceeded unhampered in the absence of ASC specks (Guey et al. 2014; Van Opdenbosch et al. 2014). ASC was as well dispensable for Nlrp1b-dependent IL-1β and IL-18 secretion, although it could enhance this output. These observations suggest that direct recruitment of procaspase-1 to Nlrp1b platforms induces conformational changes in caspase-1 that suffice for its activation, while ASC-dependent caspase-1 autocleavage may serve to lock the protease in its active conformation. Next to its role in sensing LeTx activity, Nlrp1b was recently identified as a type 1 diabetes-susceptibility gene in the NOD mouse model (Motta et al. 2015), and implicated in protection from dextran sodium sulfate-induced colitis in mice (Williams et al. 2015). Also the Nlrp1a isoform was shown to induce inflammasome responses in vivo (Masters et al. 2012). Hematopoietic progenitor cells expressing gain-of-function mutations in Nlrp1a had ASC-independent, but caspase-1-mediated induction of pyroptosis, which prevented their proliferation and differentiation. These mice consequently suffered from leukopenia and anemia during hematopoietic stress (Masters et al. 2012). Identified single-nucleotide polymorphisms (SNPs) in the human NLRP1 locus were shown to confer an increased risk for the development of autoimmune diseases as well, with genetic variants being linked to Addison’s disease, generalized vitiligo and type I diabetes (Jin et al. 2007; Spritz 2007; Zurawek et al. 2010). Other studies further linked NLRP1 variants to systemic lupus erythematosus and rheumatoid arthritis (Levandowski et al. 2013; Magitta et al. 2009).

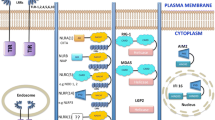
Simplified scheme of inflammasome triggers and signaling events. AIM2, Pyrin, and inflammasome-assembling NLR family members detect—directly or through a secondary messenger—the presence of a pathogen and/or cellular damage. This results in their oligomerization and inflammasome assembly in which inactive procaspase-1 precursors are recruited directly or through the bipartite adaptor protein ASC. Oligomerization of procaspase-1 leads to its autoactivation, and active caspase-1 catalyzes the maturation and secretion of the inflammatory cytokines interleukin IL-1β and IL-18. Additionally, inflammasome activation may result in a lytic mode of programmed cell death termed pyroptosis. A non-canonical mode of inflammasome signaling is engaged upon detection of bacterial LPS in the cytosolic compartment by murine caspase-11 and its human orthologs caspases 4 and 5. Caspase-11 directly induces pyroptosis through cleavage of gasdermin D and promotes secretion of IL-1β and IL-18 through the NLRP3 inflammasome
3.3 The NLRC4 Inflammasome
The upstream mechanisms promoting assembly and activation of the NLRC4 inflammasome probably are the best understood among inflammasome scaffolds (Fig. 2). NLRC4 has an amino-terminal CARD motif that allows direct recruitment of procaspase-1 in the absence of ASC (Broz et al. 2010b; Van Opdenbosch et al. 2014). Also the molecular constituents by which bacterial pathogens such as Salmonella enterica serovar Typhimurium (Lara-Tejero et al. 2006; Mariathasan et al. 2004; Miao et al. 2006), Legionella pneumophila (Amer et al. 2006; Zamboni et al. 2006), Pseudomonas auruginosa (Miao et al. 2008; Sutterwala et al. 2007), Shigella flexneri (Suzuki et al. 2007, 2014), and Listeria monocytogenes (Wu et al. 2010) trigger NLRC4 inflammasome activation have been mapped. The virulence of these bacteria strongly depends on flagellin-mediated motility and the delivery of pathogenic effector molecules in the host cell cytosol through bacterial type III and IV secretion systems (T3SS and T4SS, respectively). Intracellular detection of flagellin and/or T3SS components is what engages the NLRC4 inflammasome. Notably, NLRC4 does not detect the presence of these virulence factors directly, but their unwarranted presence in the cytosol is signaled by NLR family apoptosis inhibiting protein (NAIP) isoforms that act upstream of NLRC4 in the pathway. A full-length human NAIP isoform as well as mouse Naip5 and Naip6 serve as cytosolic flagellin receptors (Kofoed and Vance 2011; Kortmann et al. 2015; Miao et al. 2006). A shorter isoform of human NAIP and its murine Naip1 ortholog recognizes T3SS needle proteins (Miao et al. 2010b; Zhao et al. 2011), whereas Naip2 binds T3SS rod proteins (Kofoed and Vance 2011; Suzuki et al. 2014; Zhao et al. 2011). Notably, biochemical studies with chimeric Naip fusions pointed to the region surrounding the central NACHT as the bacterial ligand-binding domain of Naip proteins (Tenthorey et al. 2014). In addition to NAIP-mediated detection of flagellin and the bacterial T3SS, phosphorylation of NLRC4 at Ser533 was shown to be required for inducing NLRC4 inflammasome activation upon S. Typhimurium infection (Qu et al. 2012). Subsequent studies in Naip5-deficient macrophages showed that flagellin-dependent NLRC4 phosphorylation proceeds independently of Naip5, and that flagellin mutants which were unable to induce NLRC4 inflammasome activation retained their ability to induce potent NLRC4 Ser533 phosphorylation (Matusiak et al. 2015). Together, these findings suggest a two-step Nlrc4 activation mechanism in which Ser533 phosphorylation primes NLRC4 for subsequent Naip5-dependent activation of the NLRC4 inflammasome.
Notably, the NLRC4 inflammasome is not only linked to in vivo host defense against bacterial pathogens (Broz et al. 2010a; Miao et al. 2010a), but also may lead to the development of autoinflammatory disease in patients. Recently, three de novo SNPs in the NACHT domain of the NLRC4 gene were described to associate with autoinflammatory disease: Nlrc4-MAS (NLRC4T337S) (Canna et al. 2014), SCAN4 (NLRC4V341A) (Romberg et al. 2014), and FCAS-like syndrome (NLRC4H443P) (Kitamura et al. 2014). Although the three identified missense mutations induce somewhat distinct clinical syndromes, they have spontaneous activation of the NLRC4 inflammasome as the unifying underlying etiology. Nlrc4-MAS and SCAN4 patients further produce extremely high levels of circulating IL-18, and increased inflammasome-induced macrophage cell death was observed in SCAN4 patients (Canna et al. 2014; Romberg et al. 2014).
3.4 The AIM2 Inflammasome
Absent in melanoma 2 (AIM2) is a member of the HIN200 family/AIM-2-like receptor (ALR) family of PRRs that are characterized by an N-terminal PYD and the presence of one or two DNA-binding hematopoietic IFN inducible nuclear protein with 200 amino acid (Hin200) domains (Hornung et al. 2009). AIM2 assembles an inflammasome scaffold when dsDNA of bacterial or viral origin is bound to its Hin200 domain (Choubey 2012; Fernandes-Alnemri et al. 2009; Jin et al. 2012; Rathinam et al. 2010; Sauer et al. 2010). As such, AIM2 endows myeloid cells with the ability to produce IL-1β and IL-18 and induce pyroptosis upon recognition of bacterial and viral nucleic acids in the cytosol of infected macrophages (Rathinam et al. 2010; Sagulenko et al. 2013). The AIM2 inflammasome was shown to be critically involved in controlling infection by Francisella tularensis, the causative agent of tularemia (Fernandes-Alnemri et al. 2010; Jones et al. 2010; Rathinam et al. 2010), and further mediates host defense against Listeria monocytogenes (Tsuchiya et al. 2010; Wu et al. 2010) and the viral pathogens mouse cytomegalovirus (MCMV) (Alnemri 2010; Kanneganti 2010; Rathinam et al. 2010) and vaccinia virus (VV) (Fernandes-Alnemri et al. 2010; Rathinam et al. 2010).
3.5 The Pyrin Inflammasome
The human MEFV gene encodes Pyrin/TRIM20, a member of the TRIM family that is composed of an amino-terminal PYD motif that is followed by a centrally located coiled-coil and B-box region that mediates its oligomerization (Vajjhala et al. 2014; Yu et al. 2007). Unlike in rodents, this architecture is further extended to the carboxy-terminus with a B30.2 domain in human Pyrin (Fig. 1). The expression of Pyrin is largely restricted to myeloid cells with studies showing that monocyte differentiation toward macrophages significantly reduces the expression of Pyrin (Seshadri et al. 2007). Recently, Pyrin was shown to form an inflammasome that activates caspase-1 and mediates secretion of IL-1β in response to RhoA-inactivating bacterial toxins (Xu et al. 2014). As a result, the Pyrin inflammasome is engaged in macrophages and monocytes that have been infected with Clostridium difficile or Burkholderia cenocepacia (Gavrilin et al. 2012; Xu et al. 2014). The former is a Gram-positive obligate anaerobic pathogen that is the primary cause of nosocomial diarrhea in hospitalized patients and elderly people. B. cenocepacia is an opportunistic pathogen that causes progressive respiratory inflammation in immunocompromised patients. The above studies suggest that Pyrin may indirectly respond to the enzymatic activity of RhoA-modifying toxins, but more studies are needed to clarify the molecular chain of events leading to Pyrin inflammasome activation.
Notably, more than 280 mutations have been identified in MEFV that cause familial Mediterranean fever (FMF), the most common monogenic autoinflammatory disease worldwide. FMF has a largely autosomal recessive inheritance, although patients with apparent dominant inheritance have also been documented (Balow et al. 1997; Consortium 1997). Notably, the majority of FMF-associated mutations is clustered in the carboxy-terminal B30.2 domain of human Pyrin that is absent in its mouse ortholog (Touitou et al. 2004). As the name suggests, FMF is particularly common in Southern Europe, the Mediterranean Basin, the Middle East, and the Caucasus, frequently affecting Jewish, Turkish, Armenian, Arab, and Italian populations. In these regions, the prevalence of FMF is between 1 in 500 and 1 in 1000, and MEFV mutations are very common, with the carrier rate reaching up to 1:5 in these endemic regions. This restricted geographical distribution suggests the existence of an as yet unknown selective advantage for heterozygous carriers in the Mediterranean Basin. Although the disease is less prevalent in Northern Europe (with estimated frequencies of 1:75,000), the disease has spread over the world with migrations of South European, North African, and Middle Eastern populations over the past centuries and in more recent times (Ozen and Bilginer 2014). FMF usually has a childhood onset and is characterized by recurrent attacks of fever associated with serositis. Its main long-term complication is amyloid A (AA) amyloidosis, a severe manifestation with poor prognosis. The microtubule polymerization inhibitor colchicine remains the therapeutic choice to prevent both FMF attacks and complications in most patients, and IL-1-targeting biologicals such as anakinra appear effective in patients with colchicine-refractory disease (Chae et al. 2006; Masters et al. 2009; Ng et al. 2010).
4 Concluding Remarks
Understanding of inflammasome biology is increasing at an impressive pace, with work during the past few years revealing the ligands and detailing the signaling mechanisms of the NLRC4, Pyrin, AIM2, and other inflammasomes. Activation of the different inflammasomes is increasingly recognized to be regulated by a variety of post-translational mechanisms, with each scaffold having its private peculiarities. In addition, disease-associated mutations in Pyrin, NLRP3, NLRC4, and NLRP1 have been shown to cause autoinflammatory diseases with variable clinical presentations. This realization has also led to studies demonstrating the remarkable efficacy of anti-IL-1 therapies in these rare genetic syndromes. Despite the tremendous progress made on these and other fronts, quite some gaps remain in our understanding of inflammasome biology. Further progress is needed in understanding the molecular mechanisms promoting caspase-1 activation by the different inflammasome platforms. The mechanisms by which gasdermin D cleavage promotes pyroptosis warrant further investigation, and the physiological role of pyroptosis in release of cytokines and DAMPs in the context of infections, autoinflammation, and autoimmune diseases is an exciting area of continued study. Newly gained insight in these and other aspects of inflammasome biology likely is to offer novel opportunities for targeted treatment of inflammatory diseases.
References
Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA (2013) Caspase-11 protects against bacteria that escape the vacuole. Science 339(6122):975–978
Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R, Goldsmith D, Dent P, Rosenberg HF, Austin F, Remmers EF, Balow JE Jr, Rosenzweig S, Komarow H, Shoham NG, Wood G, Jones J, Mangra N, Carrero H, Adams BS, Moore TL, Schikler K, Hoffman H, Lovell DJ, Lipnick R, Barron K, O’Shea JJ, Kastner DL, Goldbach-Mansky R (2002) De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 46(12):3340–3348
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30(4):556–565
Alnemri ES (2010) Sensing cytoplasmic danger signals by the inflammasome. J Clin Immunol 30(4):512–519
Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Nunez G (2006) Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem 281(46):35217–35223
Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, Tanzer MC, Monteleone M, Robertson AA, Cooper MA, Alvarez-Diaz S, Herold MJ, Bedoui S, Schroder K, Masters SL (2015) NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 45(10):2918–2926
Balow JE Jr, Shelton DA, Orsborn A, Mangelsdorf M, Aksentijevich I, Blake T, Sood R, Gardner D, Liu R, Pras E, Levy EN, Centola M, Deng Z, Zaks N, Wood G, Chen X, Richards N, Shohat M, Livneh A, Pras M, Doggett NA, Collins FS, Liu PP, Rotter JI, Fischel-Ghodsian N, Gumucio D, Richards RI, Kastner DL (1997) A high-resolution genetic map of the familial Mediterranean fever candidate region allows identification of haplotype-sharing among ethnic groups. Genomics 44(3):280–291
Bann JG (2012) Anthrax toxin protective antigen–insights into molecular switching from prepore to pore. Protein Sci 21(1):1–12
Battistini A (2009) Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J Interferon Cytokine Res 29(12):765–780
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183(2):787–791
Boehm T (2012) Evolution of vertebrate immunity. Curr Biol 22(17):R722–R732
Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38(2):240–244
Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM (2010a) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207(8):1745–1755
Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010b) Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8(6):471–483
Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490(7419):288–291
Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, Chapelle D, Plass N, Huang Y, Villarino AV, Biancotto A, Fleisher TA, Duncan JA, O’Shea JJ, Benseler S, Grom A, Deng Z, Laxer RM, Goldbach-Mansky R (2014) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46(10):1140–1146
Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S (2013) Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 9(6):e1003400
Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL (2006) The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA 103(26):9982–9987
Chavarria-Smith J, Vance RE (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9(6):e1003452
Chisholm ST, Coaker G, Day B, Staskawicz BJ (2006) Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124(4):803–814
Choubey D (2012) DNA-responsive inflammasomes and their regulators in autoimmunity. Clin Immunol 142(3):223–231
Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21(3):248–255
Consortium FF (1997) A candidate gene for familial Mediterranean fever. Nat Genet 17(1):25–31
Cookson BT, Brennan MA (2001) Pro-inflammatory programmed cell death. Trends Microbiol 9(3):113–114
Cyster JG (2010) B cell follicles and antigen encounters of the third kind. Nat Immunol 11(11):989–996
de Gassart A, Martinon F (2015) Pyroptosis: caspase-11 unlocks the gates of death. Immunity 43(5):835–837
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320(5876):674–7
Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453(7198):1122–1126
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458(7237):509–513
Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11(5):385–393
Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin J, Gough PJ (2012) Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem 287(30):25030–25037
Fink SL, Bergsbaken T, Cookson BT (2008) Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA 105(11):4312–4317
Franchi L, Kanneganti TD, Dubyak GR, Nunez G (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282(26):18810–18818
Frew BC, Joag VR, Mogridge J (2012) Proteolytic processing of Nlrp1b Is required for inflammasome activity. PLoS Pathog 8(4):e1002659
Gavrilin MA, Abdelaziz DH, Mostafa M, Abdulrahman BA, Grandhi J, Akhter A, Abu Khweek A, Aubert DF, Valvano MA, Wewers MD, Amer AO (2012) Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol 188(7):3469–3477
Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J (2009) Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459(7245):433–436
Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, Kurimoto M, Tanimoto T, Flavell RA, Sato V, Harding MW, Livingston DJ, Su MS (1997) Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 275(5297):206–209
Guey B, Bodnar M, Manié SN, Tardivel A, Petrilli V (2014) Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci USA 111(48):17254–17259
Guo C, Chen M, Fa Z, Lu A, Fang W, Sun B, Chen C, Liao W, Meng G (2014) Acapsular Cryptococcus neoformans activates the NLRP3 inflammasome. Microbes Infect 16(10):845–854
Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, Vogel P, Lamkanfi M, Kanneganti TD (2012) Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287(41):34474–34483
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341(6151):1250–1253
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9(8):857–865
Hansen JD, Vojtech LN, Laing KJ (2011) Sensing disease and danger: a survey of vertebrate PRRs and their origins. Dev Comp Immunol 35(9):886–897
He Y, Zeng MY, Yang D, Motro B, Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530(7590):354–7
Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA (2009) An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5(5):487–497
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet 29(3):301–305
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9(8):847–856
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458(7237):514–518
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39(1):285–312
Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39(2):311–323
Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, Spritz RA (2007) NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med 356(12):1216–1225
Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, Hornung V, Latz E, Bowie AG, Fitzgerald KA, Xiao TS (2012) Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36(4):561–571
Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A 107(21):9771–9776
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287(43):36617–36622
Kanneganti TD (2010) Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol 10(10):688–698
Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281(48):36560–36568
Kanneganti TD, Lamkanfi M, Nunez G (2007) Intracellular NOD-like receptors in host defense and disease. Immunity 27(4):549–559
Katsnelson MA, Rucker LG, Russo HM, Dubyak GR (2015) K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194(8):3937–3952
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479(7371):117–121
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341(6151):1246–1249
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526(7575):666–671
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta (BBA) Mol Basis Dis 1802(4):396–405
Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K (2014) An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med 211(12):2385–2396
Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA (2014) Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16(2):249–256
Koch U, Radtke F (2011) Mechanisms of T cell development and transformation. Annu Rev Cell Dev Biol 27:539–562
Kofoed EM, Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477(7366):592–595
Kortmann J, Brubaker SW, Monack DM (2015) Cutting edge: inflammasome activation in primary human macrophages is dependent on flagellin. J Immunol 195(3):815–819
Kumar H, Kumagai Y, Tsuchida T, Koenig PA, Satoh T, Guo Z, Jang MH, Saitoh T, Akira S, Kawai T (2009) Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J Immunol 183(12):8061–8067
Laing KJ, Purcell MK, Winton JR, Hansen JD (2008) A genomic view of the NOD-like receptor family in teleost fish: identification of a novel NLR subfamily in zebrafish. BMC Evol Biol 8:42
Lamkanfi M (2011) Emerging inflammasome effector mechanisms. Nat Rev Immunol 11(3):213–220
Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157(5):1013–1022
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM (2009) Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187(1):61–70
Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM (2010) Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185(7):4385–4392
Lange C, Hemmrich G, Klostermeier UC, Lopez-Quintero JA, Miller DJ, Rahn T, Weiss Y, Bosch TC, Rosenstiel P (2011) Defining the origins of the NOD-like receptor system at the base of animal evolution. Mol Biol Evol 28(5):1687–1702
Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, Flavell RA, Galan JE (2006) Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med 203(6):1407–1412
Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ (2012) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492(7427):123–127
Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, McFann KK, Holland PJ, Fain PR, Dinarello CA, Spritz RA (2013) NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome. Proc Natl Acad Sci USA 110(8):2952–2956
Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8(3):e1002638
Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, Nanda S, Cohen P, Thomas JA, Pasare C (2014) IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci USA 111(2):775–780
Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, Kirov A, Prudovsky I, Swanton E, Brough D (2013) Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J Biol Chem 288(4):2721–2733
Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, DeVera ME, Liang X, Tör M, Billiar T (2007) The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev 220(1):60–81
Magitta NF, Boe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, Undlien DE, Joner G, Njolstad PR, Kvien TK, Forre O, Knappskog PM, Husebye ES (2009) A coding polymorphism in NALP1 confers risk for autoimmune Addison’s disease and type 1 diabetes. Genes Immun 10(2):120–124
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430(6996):213–218
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440(7081):228–232
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10(2):417–426
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440(7081):237–241
Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265
Masters SL, Simon A, Aksentijevich I, Kastner DL (2009) Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol 27:621–668
Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ, Cengia LH, Henley KJ, Collinge JE, Kastner DL, Feigenbaum L, Hilton DJ, Alexander WS, Kile BT, Croker BA (2012) NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37(6):1009–1023
Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard JC, Kanneganti TD, Lamkanfi M (2015) Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci USA 112(5):1541–1546
McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Petrilli V, Andrew PW, Kadioglu A, Lavelle EC (2010) Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 6(11):e1001191
Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A (2006) Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7(6):569–575
Miao EA, Ernst RK, Dors M, Mao DP, Aderem A (2008) Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci USA 105(7):2562–2567
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010a) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11(12):1136–1142
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010b) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107(7):3076–3080
Motta VN, Markle JG, Gulban O, Mortin-Toth S, Liao KC, Mogridge J, Steward CA, Danska JS (2015) Identification of the inflammasome Nlrp1b as the candidate gene conferring diabetes risk at the Idd4.1 locus in the nonobese diabetic mouse. J Immunol 194(12):5663–5673
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38(6):1142–1153
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3):222–230
Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL (2010) Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139(2):542–552, 552 e1-3
Nystrom S, Antoine DJ, Lundback P, Lock JG, Nita AF, Hogstrand K, Grandien A, Erlandsson-Harris H, Andersson U, Applequist SE (2013) TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J 32(1):86–99
Ozen S, Bilginer Y (2014) A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol 10(3):135–147
Palm NW, Medzhitov R (2009a) Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227(1):221–233
Palm NW, Medzhitov R (2009b) Immunostimulatory activity of haptenated proteins. Proc Natl Acad Sci USA 106(12):4782–4787
Perregaux D, Gabel CA (1994) Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269(21):15195–15203
Perregaux DG, Gabel CA (1998) Human monocyte stimulus-coupled IL-1β posttranslational processing: modulation via monovalent cations. Am J Physiol Cell Physiol 275(6):C1538–C1547
Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14(9):1583–1589
Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49(2):331–338
Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Komuves L, Cupp JE, Arnott D, Monack D, Dixit VM (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490(7421):539–542
Rajan JV, Rodriguez D, Miao EA, Aderem A (2011) The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol 85(9):4167–4172
Rast JP, Smith LC, Loza-Coll M, Hibino T, Litman GW (2006) Genomic insights into the immune system of the sea urchin. Science 314(5801):952–956
Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11(5):395–402
Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK, Huttner AJ, West B, Podoltsev NA, Boggon TJ, Kazmierczak BI, Lifton RP (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 46(10):1135–1139
Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ (2013) AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20(9):1149–1160
Said-Sadier N, Padilla E, Langsley G, Ojcius DM (2010) Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS ONE 5(4):e10008
Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA (2010) Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7(5):412–419
Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V (2015) Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45(10):2911–2917
Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V (2016) A Genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 291(1):103–109
Seshadri S, Duncan MD, Hart JM, Gavrilin MA, Wewers MD (2007) Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1 processing and release. J Immunol 179(2):1274–1281
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514(7521):187–192
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575):660–665
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, Lyon S, Scott L, Quan J, Sun Q, Russell J, Arnett S, Jurek P, Chen D, Kravchenko VV, Mathison JC, Moresco EM, Monson NL, Ulevitch RJ, Beutler B (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17(3):250–8
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36(3):401–414
Spritz RA (2007) The genetics of generalized vitiligo and associated autoimmune diseases. Pigment Cell Res 20(4):271–278
Stein C, Caccamo M, Laird G, Leptin M (2007) Conservation and divergence of gene families encoding components of innate immune response systems in zebrafish. Genome Biol 8(11):R251
Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA (2007) Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204(13):3235–3245
Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3(8):e111
Suzuki S, Franchi L, He Y, Munoz-Planillo R, Mimuro H, Suzuki T, Sasakawa C, Nunez G (2014) Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcdelta. PLoS Pathog 10(2):e1003926
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820
Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE (2014) Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell 54(1):17–29
Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, Shoham N, Aganna E, Hugot JP, Wise C, Waterham H, Pugnere D, Demaille J, Sarrauste de Menthiere C (2004) Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat 24(3):194–198
Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, Dewamitta SR, Shen Y, Fang R, Mitsuyama M (2010) Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J Immunol 185(2):1186–1195
Vajjhala PR, Kaiser S, Smith SJ, Ong QR, Soh SL, Stacey KJ, Hill JM (2014) Identification of multifaceted binding modes for pyrin and ASC pyrin domains gives insights into pyrin inflammasome assembly. J Biol Chem 289(34):23504–23519
Vallabhapurapu S, Karin M (2009) Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27:693–733
Van Gorp H, Kuchmiy A, Van Hauwermeiren F, Lamkanfi M (2014) NOD-like receptors interfacing the immune and reproductive systems. FEBS J 281(20):4568–4582
Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M (2014) Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun 5:3209
Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D, Kanneganti TD, van Loo G, Lamkanfi M (2014) Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512(7512):69–73
Wang S, Miura M, Jung Y-K, Zhu H, Li E, Yuan J (1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92(4):501–509
Wickliffe KE, Leppla SH, Moayeri M (2008) Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol 10(2):332–343
Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, Heid B, Cecere TE, Allen IC (2015) The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol 194(7):3369–3380
Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS (2008) NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol 83(1):13–30
Wu J, Fernandes-Alnemri T, Alnemri ES (2010) Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol 30(5):693–702
Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, Wang F, Shao F (2014) Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513(7517):237–241
Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, Tian Y, Glenn HL, Meldrum DR (2015) K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis 6:e1954
Yu JR, Leslie KS (2011) Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep 11(1):12–20
Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, McCormick M, Zhang Z, Alnemri ES (2007) Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell 28(2):214–227
Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, Kuida K, Mariathasan S, Dixit VM, Flavell RA, Dietrich WF, Roy CR (2006) The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol 7(3):318–325
Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477(7366):596–600
Zurawek M, Fichna M, Januszkiewicz-Lewandowska D, Gryczynska M, Fichna P, Nowak J (2010) A coding variant in NLRP1 is associated with autoimmune Addison’s disease. Hum Immunol 71(5):530–534
Acknowledgments
We apologize to colleagues whose work is not cited due to space constraints. A.W. is supported in part by a fellowship from the Fund for Scientific Research-Flanders. Work in ML’s laboratory is supported by grants from VIB, Ghent University (BOF 01N02313, BOF 01J11113, BOF14/GOA/013), the Odysseus Foundation (grant Nr. G0C4913N to A.W. and M.L.), the Fund for Scientific Research-Flanders (grant G011315N), and the European Research Council (grant 281600).
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dubois, H., Wullaert, A., Lamkanfi, M. (2016). General Strategies in Inflammasome Biology. In: Backert, S. (eds) Inflammasome Signaling and Bacterial Infections. Current Topics in Microbiology and Immunology, vol 397. Springer, Cham. https://doi.org/10.1007/978-3-319-41171-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-41171-2_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41170-5
Online ISBN: 978-3-319-41171-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)