
Abstract
The endosymbiosis of mitochondria and the resulting increase in energy supply thus conferred upon the eukaryotic cell enabled the evolution of multicellular organisms and complex organs, such as the brain. As a result, the brain and other organs with high energy demands, depend heavily upon mitochondrial metabolism for normal function, and as a consequence, defects in mitochondrial function lead to neurodegenerative disorders. However, the mechanisms linking mitochondrial defects to hallmarks of neurodegeneration, such as the protein aggregates are also extracellular epigenetic alterations, abnormal gene expression, systemic inflammation, and protein aggregates, remain unclear.
Emerging evidence demonstrates that mitochondria are not only power-generating organelles, but also engage in signaling at multiple levels. During the bioenergetic decline associated with aging, dysfunctional mitochondria generate signals of stress (SOS) that can trigger and/or amplify neurodegenerative processes. In this chapter, we describe emerging mechanisms for mitochondrial signaling at four different levels. Mitochondria communicate (1) with each other via fusion and specialized inter-mitochondrial junctions, (2) with cytoplasmic components via posttranslational modifications that shift signaling pathways and promote protein aggregation, (3) with the nucleus to regulate epigenetic modifications and gene expression, and (4) with the systemic environment where they alter neuroendocrine and inflammatory processes that impact neuronal function. The relevance of mitochondrial signaling to neurodegeneration is discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mitochondrial signaling
- Mitochondrial signals of stress (SOS)
- Epigenetics
- Transcriptional regulation
- Retrograde signaling
- Mitochondrial damage-associated molecular patterns (mtDAMPs)
- Inflammation
- Protein aggregates
- Microglia
- Neuroendocrine regulation
1 Introduction
Mitochondria evolved from an α-proteobacterium engulfed by the proto-eukaryotic cell over 1.5 billion years ago [1]. The endosymbiosis of this aerobic bacterium was a turning point in the evolution of complex life, with the larger amount of energy afforded by mitochondrial oxidative phosphorylation (OXPHOS), enabling the regulation of a complex genome comprised of >25,000 genes [2]. This biologically unprecedented marriage of OXPHOS with complex genetic material culminated in the development of multicellular organisms, tissues, and interdependent organ systems [3]. The brain, with its unparalleled structural and functional complexity, exemplifies the product of multicellular evolution enabled by mitochondria. Likely as a result of this evolutionary interdependence, the structure and function of the brain and of its constituent neurons are closely linked to energy metabolism in general, and to mitochondrial function in particular.
Unlike the traditional “powerhouse of the cell” concept would suggest, the role of mitochondria is not limited to energy production. As beneficial symbionts, mitochondria perform several other “non-energetic” (i.e., other than ATP synthesis) functions that impact cell function and fate. These crucial functions include, but are not limited to, calcium (Ca2+) uptake and release that regulates energy production itself [4], intracellular signaling and vesicular endocytosis [5], and cell death [6]; reactive oxygen species (ROS) production to alter gene expression and protein function [7, 8]; macromolecule biosynthesis including heme, hormones, and purines/pyrimidines to enable cell growth and replication [9]; and the balance of specific cell death effectors [10].
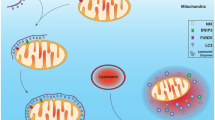
Not unlike their bacterial ancestors that communicate and behave as colonies through regulated processes of quorum sensing [11–14], mitochondria participate in signaling at a number of levels. Signaling is defined as the exchange of information between two compartments, involving transmission and/or reception of a chemical or molecular signal. In this chapter, we consider mitochondrial signaling at four levels (Fig. 5.1).

Overview of mitochondrial signaling at four different levels. (1) Mitochondria exchange information with each other via inter-organelle fusion, the release of soluble mediators, and inter-mitochondrial junctions (IMJs) (see [38] for details). (2) Mitochondrial redox signaling shifts signal transduction pathways in the cytoplasm by modifying redox-sensitive amino acids and altering the activity of kinases, phosphatases, and proteases. (3) Mitochondrial metabolic intermediates and ROS travel to the nucleus where they impact epigenetic and transcriptional processes that alter gene expression profiles. (4) Mitochondria release signals that travel to the systemic circulation, influencing the behavior of surrounding cells and tissues, and the physiological function of neuroendocrine, cardiovascular, and nervous systems that indirectly feedback onto the brain to impact neuronal structure and function, and contribute to neurodegeneration
First, we describe evidence of chemical and physical communication within the mitochondrial network. Second, we discuss how mitochondrial signals of stress (SOS) control the intracellular environment of the aging brain and alter protein function and folding in the cytoplasm. Third, mitochondrial signals that shift the epigenetic landscape in the nucleus and influence gene expression are discussed. Finally, we present emerging evidence that mitochondria exert systemic effects, in part via modulation of the immune and neuroendocrine networks relevant to the pathophysiology of neurodegenerative diseases. Overall, the emerging picture is one where mitochondria behave as signaling hubs, dynamically integrating endogenous and environmental factors to dictate neurological health by communicating with each other, within other cellular compartments, and systemically.
2 Mitochondria: Inter-mitochondrial Signaling
Within the cell cytoplasm, mitochondria interact with each other and engage in mitochondria-mitochondria signaling via a number of mechanisms. The field of research around inter-organellar signaling being relatively recent, some caveats must be pointed out as we discuss it in the context of neuronal dysfunction and the brain. Some of the mechanisms described in this section such as the inter-mitochondrial junctions (IMJs) have been reported in neurons, yet their functional relevance to neuronal pathophysiology is still a matter of speculation. In contrast, mitochondrial fusion as a means of exchanging molecular information has been extensively investigated in neurons and its involvement in neurodegeneration reviewed elsewhere (see Chap. 7). In different cell types, mitochondrial signaling involves the release of diffusible signals such as ROS, Ca2+, and apoptotic inducers, which propagate in a wavelike fashion through the mitochondrial network [15–17]. These mechanisms have been identified and investigated mostly in certain cell types (e.g., cardiomyocytes), but may also occur to some extent in neurons. Thus, these mechanisms will not be discussed here.
This section focuses on two major mechanisms for inter-mitochondrial communication: (i) molecular exchange through mitochondrial fusion and (ii) communication through inter-mitochondrial junctions (IMJs) that connect adjacent mitochondria.
2.1 Mitochondria-Mitochondria Signaling Through Fusion and Molecular Exchanges
Mitochondria continuously change their size, shape, and cristae architecture [18], which allows them to adapt to the fluctuating demands of their cellular environment and react with the appropriate signal [19]. Whereas mitochondrial fusion appears important to preserve OXPHOS and prevent cell death during starvation [20], cristae remodeling may impact bioenergetic efficiency by enhancing respiratory supercomplex formation [21]. Mitochondrial cristae remodeling also regulates the release of mitochondrial SOS, including proapoptotic signals (reviewed in [18]). Furthermore, mitochondrial fusion and its reciprocal process of fission are necessary to maintain mitochondrial DNA (mtDNA) integrity and renew distal mitochondria for proper synaptic function [22, 23].
In addition to impacting intrinsic functional properties of mitochondria, the dynamic process of mitochondrial fusion is critical for the exchange of proteins and mtDNA among the mitochondrial network. Mitochondria contain >1,000 proteins that are normally distributed throughout the mitochondrial network within the cytoplasm. Ablation of the profusion factors mitofusin 1 and 2 and (Mfn1/Mfn2) leads to substantial protein heterogeneity among the mitochondrial network, with some mitochondria preferentially accumulating certain proteins and lacking others [24]. Protein heterogeneity is a distinguishing feature in neurons, with the proteome of synaptic and non-synaptic mitochondria differing by about 20 % [25]. However, the significance of selective fusion and fission in mitochondrial protein distribution for proper neuronal function, and the relevance to neurodegeneration, remains unknown.
Mitochondrial dynamics also contributes to mitochondrial axonal trafficking, the process by which mitochondria commute toward and away from synaptic terminals [26]. Because mitochondria constantly generate signals, the act of transporting mitochondria from one cellular compartment (e.g., cell body) to another (e.g., synaptic terminal) should be regarded as a form of intracellular signaling.
Importantly, mitochondrial fusion also results in the exchange of genetic material from neighboring mitochondria. In the context of mtDNA heteroplasmy, i.e., when some mitochondria contain mtDNA mutations and others have normal mtDNA, the mixing of mitochondrial contents through fusion enables functional complementation of gene products, thus maintaining mitochondrial homeostasis despite the presence of mitochondrial defects [27]. Mitochondrial functional complementation in heteroplasmic cells does not occur with “kiss-and-run” contact, but requires fusion via both Mfn2 (outer mitochondrial membrane fusion) and optic atrophy 1 (OPA1) (fusion of the inner membrane) and exchange of DNA nucleoids, although mtDNA transcription/translation appears dispensable [28]. Mitochondrial fusion also prevents severe heterogeneity in membrane potential among cells with mtDNA heteroplasmy [28]. Fusion-dependent mtDNA exchange can be regarded as a “stable” form of inter-mitochondrial signaling, where the normal mtDNA transmitted through fusion acts as the signal that promotes normal respiratory chain function among a mutant mtDNA bearing mitochondrion. Beyond permitting mtDNA exchange, mitochondrial fusion also appears essential to preserve mtDNA integrity, since mtDNA mutations accumulate in the absence of mitochondrial fusion [24].
As evidence that mitochondria-mitochondria signaling via membrane fusion is important in the context of neurodegeneration, ablation of mitofusins in the mouse promotes cell loss and cerebellar neurodegeneration [22]. In humans, a growing list of mutations in genes encoding the machinery necessary for mitochondrial fusion and cristae remodeling cause neurological disease affecting both the central and peripheral nervous systems [29].
2.2 Mitochondria-Mitochondria Signaling via Inter-mitochondrial Junctions
Even in the absence of fusion, electrochemical information can be passed from one mitochondrion to its neighbor(s) [30]. This form of signaling was originally described in cardiomyocytes [31] and more recently skeletal muscle [32], constituting the basis for the “cable” theory. Elongated “mitochondrial cables” extending up to >30 um have also been reported in dendrites of hippocampal neurons [33, 34]. This theory of cables stipulates that mitochondria behave as electrically coupled conduits that carry information about their electrical charge (membrane potential) throughout the mitochondrial network of a cell [35]. In various cell types such as cardiomyocytes, where mitochondrial membrane potential may spontaneously oscillate over time, clusters of physically juxtaposed mitochondria exhibit a substantial degree of coordination [36, 37], revealing the existence of inter-mitochondrial signaling without mitochondrial fusion.
Studies in the 1980s suggested the existence of physical structures connecting electrically coupled mitochondria in cardiomyocytes, termed inter-mitochondrial junctions (IMJs) [31]. Closer quantitative examination of IMJs by electron tomography, which provides substantial spatial resolution and three dimensionality, revealed the presence of structures characterized by enhanced electron density and physical tethering of juxtaposed mitochondria through a <10-nm gap [38]. These gap junction-like IMJs are evolutionary conserved from mollusk to mammal and inducible by the physical rapprochement of adjacent mitochondria through a synthetic linker molecular system [38]. More interestingly in the context of signaling is that cristae organization is altered at IMJs. When two mitochondria are joined by an IMJ, cristae from both mitochondria become coordinated in space, often exhibiting substantial deformation and bending to achieve near-perfect alignment [38].
Similar to synapses in the brain (which regulate the exchange of information from one cell to another), IMJs appear dynamically strengthened or weakened based on their activity. For example, presynaptic terminals of more metabolically demanding, tonic synapses contain more IMJs than phasic synapses that exhibit more temporally spaced firings in the brain [39]. Evidence from a different tissue, skeletal muscle, also indicates that low energy requirements during inactivity tend to reduce the number of IMJs [40], while exercise increases their numbers [41], indicating a certain level of IMJ plasticity. The transmitochondrial coordination of cristae represents the first physical demonstration of inter-mitochondrial signaling, but the functional significance for the brain, neurons, or mitochondria themselves remains unclear.
3 Mitochondria: Cellular Signaling
Mitochondria generate diffusible signals that are transmitted – or communicated – to both the cytoplasm and the cell nucleus. Many of these diffusible signals are propagated by redox-based posttranslational reactions that alter signal transduction pathways and protein processing in the cytoplasm, and activate transcription factors that translocate to the nucleus. In addition, mitochondrial signals, such as metabolic intermediates, are the epigenetic language that directs chromatin accessibility. This section discusses mitochondrial signaling to both cytoplasmic and nuclear compartments and the relevance of mitochondrial signals of stress (SOS) in neurodegenerative pathogenesis (Fig. 5.2).

Mitochondrial signals of stress: from mitochondrial dysfunction to neurodegenerative disease. Common stressors such as aging, psychological and metabolic stress, and exposure to toxins cause mitochondrial dysfunction and aberrant morphology, including donut-shaped mitochondria ( ). Mitochondria respond by sending signals of stress (SOS), which induce defined molecular changes within the cytoplasm and nucleus that directly contribute to the established hallmarks of neurodegenerative disease, including protein aggregates and transcriptional dysregulation. OXPHOS oxidative phosphorylation, Cyt c cytochrome c, PTMs posttranslational modifications, NFTs neurofibrillary tangles, LBs Lewy bodies
). Mitochondria respond by sending signals of stress (SOS), which induce defined molecular changes within the cytoplasm and nucleus that directly contribute to the established hallmarks of neurodegenerative disease, including protein aggregates and transcriptional dysregulation. OXPHOS oxidative phosphorylation, Cyt c cytochrome c, PTMs posttranslational modifications, NFTs neurofibrillary tangles, LBs Lewy bodies
3.1 Mitochondrial Bioenergetics and Redox Signaling
Mitochondrial redox chemistry is not only central to the production of ATP by OXPHOS, but also a critical means by which mitochondria communicate to the nucleus, regulate enzymatic activity, control cell death, and stimulate the immune system. During OXPHOS, up to 2 % of the electrons leak to molecular oxygen and produce the superoxide radical (O2 ∙−) [8]. The mitochondrial matrix antioxidant, manganese superoxide dismutase (MnSOD), converts O2 ∙− to hydrogen peroxide (H2O2), which may act as a signaling molecule in the cytosol/nucleus or generate more toxic ROS, such as the hydroxyl radical (∙OH). The majority of mitochondrial ROS (mtROS) are produced by the OXPHOS complexes I and III, but other mitochondrial enzymes such as NADPH oxidase-4 (Nox4), monoamine oxidases (MAOs), and some TCA cycle enzymes are also important in neural redox regulation [7, 42].
While mtROS are often viewed as indiscriminate molecular villains, they are in fact mitochondrial signaling molecules that produce a wide range of physiological effects dependent on the cellular context in which they are produced. Low levels of mtROS can induce gene expression changes to preserve mitochondrial function during stress, such as those that extend the lifespan of C. elegans [43]. Physiologically appropriate fluctuations in mtROS modulate the redox status of the mitochondria and the cell via several key redox couples: NADPH/NADP+, NADH/NAD+, thioredoxins 1 and 2 (Trx 1 and Trx2) (SH)2/SS, glutathione (GSH/GSSG), and cysteine/cysteine (CyS/CySS) [44]. These redox nodes are interconnected within and between cellular compartments. For instance, the mitochondrial and cytoplasmic NADH/NAD+ redox states are linked via the activity of the malate/aspartate NAD(H) and α-glycerophosphate shuttles, which may be particularly important in neurons and less so in astrocytes [45]. Transient opening of the mitochondrial permeability transition pore (PTP) can also rapidly release NAD+ and NADH into the cytoplasm [46], although the functional significance of this mechanism in the brain remains unclear.
The intramitochondrial redox state is intimately coupled to mitochondrial energetics by nicotinamide nucleotide transhydrogenase (NNT) [47]. NNT relies on the proton motive force generated during OXPHOS to regenerate NADPH from NADP+, in order to power NADPH-dependent reductases that recycle the other redox couples in the mitochondrial matrix. Thus, by linking mitochondrial proton motive force to interconnected redox control nodes within and outside mitochondria, mitochondria may communicate the energetic state with the rest of the cell and organism. When mitochondrial bioenergetics is compromised, the redox nodes act as “barometers” that induce mitophagy or apoptosis, depending on the magnitude of the insult [48–51]. The importance of these redox nodes to cellular homeostasis is evidenced by the embryonic lethality that occurs when components are genetically deleted [52–55] and neuronal cell death that ensues when the system is overwhelmed by excess mtROS [56].
The downstream signaling pathways initiated by mtROS are determined by the reactivity, charge, and location of the particular species. In the aging brain, impairments in OXPHOS produce O2 ·− primarily within the mitochondrial matrix and inner membrane. In order for O2 ·− to react with mitochondrial components, the target must outcompete micromolar concentrations of MnSOD in the matrix [57]. One signaling molecule that meets these stringent requirements is nitric oxide (NO·). In contrast to O2 ·−, NO· possesses a relatively long half-life for a “reactive species,” which allows it to readily diffuse into the mitochondria from cytoplasmic sources where it can induce neuroprotective pathways. In mitochondria, the close proximity of NO· and O2 ·− generates the highly toxic species peroxynitrite (ONOO−) by a spontaneous reaction that occurs seven times faster than that of O2 ·− and MnSOD [58]. This reaction occurs predominantly in neuroinflammatory conditions when NO· is present at higher concentrations [59, 60]. In most contexts, ONOO- causes permanent protein damage, although it may protect the brain from ischemic episodes associated with stroke [61, 62].
The signaling potential of superoxide is increased in many neurodegenerative diseases due to nitration and permanent inactivation of MnSOD [63, 64]. O2 ·− is more selective than most mtROS, but readily reacts with iron-sulfur (Fe-S) centers of nonheme proteins, such as respiratory complexes I–III and TCA cycle enzymes. This reaction releases free ferrous iron (Fe2+), which generates the highly reactive hydroxyl radical (·OH) via Fenton chemistry, thereby initiating a chain of lipid peroxidation, protein modifications, and DNA damage within or near the inner mitochondrial membrane (IMM) [57].
Cardiolipin, the major phospholipid of the IMM, is particularly susceptible to peroxidation by ROS due to its high degree of unsaturation. During mitochondrial stress, mtROS or cytochrome c (cyt c) may oxidize cardiolipin, which dissolves its association with cyt c [65, 66]. Oxidized cardiolipin then translocates to the outer membrane, recruits mitophagic or apoptotic machinery, and releases cyt c into the cytosol [67]. If the cytosolic redox balance is also shifted to a prooxidant state, the released cyt c will remain oxidized and able to initiate activation of the apoptosome [68, 69]. Therefore, mtROS initiate cell death pathways by direct modification of lipids and proteins, but the amplification of these signals necessary to effectuate intrinsic apoptosis requires modulation of redox nodes in multiple compartments. In addition, mtROS may also induce neuronal death via caspase-independent mechanisms involving IMM compromise and Ca2+ dysregulation [56].
Due to their transient nature, it is currently difficult to directly measure ROS in vivo, and therefore analysis of ROS levels is largely dependent on the molecular footprints left in their wake of destruction [42]. Recent efforts to develop mitochondria-targeted mass spectrometry probes are under development and could circumvent this technical limitation in preclinical studies [70]. Among all organs of the body, the brain is perhaps most vulnerable to oxidative damage due to its high utilization of oxygen, increased levels of polyunsaturated fatty acids and redox transition metals, and relatively low levels of antioxidants. Elevation of virtually every established marker of oxidative damage has been documented in brain tissue from the most prevalent neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Lewy body disease (LBD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and Friedreich’s ataxia (FA) [71, 72].
Interestingly, the evidence suggests that bioenergetic defects precede the onset of diagnostic symptoms [73]. Furthermore, neurological symptoms often emerge without or before the accumulation of proteinaceous lesions [74] that are generally considered the key determinants of neurodegenerative disease pathogenesis [72, 75, 76]. Therefore, the chronological disassociation between the emergence of diagnostic symptoms and proteinaceous lesions [77] suggests that the pathological processing of amyloid, tau, and alpha-synuclein could be a consequence of more proximal pathological stimuli, such as mitochondrial SOS produced by progressive mitochondrial dysfunction in aging [78].
3.2 Mitochondrial Distress Signals and Pathological Protein Processing
Several mechanisms link mitochondrial signaling with protein misfolding and aggregation. The causative nature of mitochondrial dysfunction in this association is supported by animal models and patients that develop aggregates and neurodegeneration after exposure to mitochondrial toxins [79, 80], as well as patient-derived transmitochondrial cybrids [81], in which the mtDNA from AD, PD, and ALS patients is combined with an otherwise healthy cell and induces neurodegenerative disease-associated proteinopathy and cell death [82]. The next three sections focus on recent insights linking signals from dysfunctional mitochondria to the most common protein lesions found in neurodegenerative disease patient brains: amyloid plaques, tau tangles, and α-synuclein Lewy bodies.
3.2.1 Amyloid Precursor Protein to Plaques
Since the discovery of amyloid plaques by Alois Alzheimer in the 1920s, the amyloid hypothesis has enjoyed the etiological spotlight. The main protein component of plaques is the amyloid-β (Aβ) peptide, a molecule of 40–42 amino acids with unclear function, derived from the proteolytic cleavage of the integral membrane amyloid precursor protein (APP). Interestingly, the key amyloidogenic proteases are influenced by fluctuations in mitochondria function and oxidative stress [83–88]. While mitochondrial SOS alter Aβ production and clearance in cellular and animal models, the pathophysiological relationship between mitochondrial distress and amyloid production remains ambiguous. As discussed below, since mitochondrial SOS occur early in AD and may act in concert to generate Aβ, which then localizes to mitochondria, one possibility is that the accumulation of “abnormal” Aβ represents a hormetic response to mitochondrial stress in neurons.
Indeed, APP cleavage products can act as antioxidants and Aβ monomers shift neuronal glucose metabolism away from mitochondria to glycolysis [89–91]. This metabolic shift would confer neuroprotection by decreasing mtROS produced by OXPHOS while boosting endogenous antioxidant defenses by doubling NADPH levels and reducing the redox control nodes. In other studies, when the mtROS-responsive, Aβ-generating secretases are inhibited, neuronal death ensues, unless Aβ fragments are exogenously added [92]. Therefore, the induction of β- and γ-secretases by mitochondrial dysfunction may initially serve as a survival signal during oxidative stress by generating neuroprotective Aβ species. Aβ monomers preserve neuronal survival in classic models of mitochondria-dependent cell death that are mediated by mtROS [78, 80], but Aβ oligomers have the opposite effect [93–95], such that this pathophysiological role of Aβ depends on the particular species and the aggregation state [96, 97].
Interestingly, immunotherapeutic clearance of amyloid from transgenic mice and AD patients has adverse consequences [98–100]. Recent studies suggest that these effects involve the ability of amyloid to influence Ca2+ flux [101] associated with AD mitochondrial impairments, which points to a second potential role of amyloid in mediating mitochondrial SOS. Taken together, the available evidence suggests that dysfunctional mitochondria in the aging brain may initially signal to induce Aβ production via mtROS in order to prevent oxidative damage and Ca2+ dysregulation and preserve neuronal health.
3.2.2 Tau to Neurofibrillary Tangles
Mitochondrial redox signaling modulates aberrant processing of another key protein related to age-dependent dementia and AD: the microtubule-stabilizing protein tau [102]. Tau normally functions to promote microtubule assembly and stabilization for neurite trafficking. In neurodegeneration, tau disengages from the microtubule and becomes hyperphosphorylated into neurofibrillary tangles (NFTs). Similar to amyloid processing, mtROS may initiate the process in AD by inducing caspases 3 and 7 [88], which cleave tau into neurotoxic fragments [103]. Mitochondrial redox signaling activates the key kinases currently implicated in tau hyperphosphorylation, including the stress-activated kinases JNK and p38 [104], as well as glycogen synthase kinase-3β (GSK-3ß) [105, 106]. mtROS may also inhibit peptidyl-prolyl isomerase (Pin1), which is necessary for tau dephosphorylation and microtubule binding [107–109]. Oxidative impairment of Pin1 appears crucial in the clinical presentation of tau pathology, as it is increased in presymptomatic individuals and inversely correlates with NFT levels and neurodegeneration in the AD brain [110]. Therefore, early mitochondrial SOS may culminate in the pathological processing of tau, which then destabilizes microtubules and further impairs mitochondrial signaling by inhibiting mitochondrial dynamics and trafficking between the synapses and cell body [26, 111].
3.2.3 α-Synuclein to Lewy Bodies
Lewy bodies are composed primarily of aggregated α-synuclein (α-syn), along with ubiquitin and tau, and represent the key histological hallmark shared by PD and LBD. α-Syn is a ubiquitously expressed protein of unknown function that localizes to various cellular compartments, depending on the conformational changes induced by inherited mutations, or environmental conditions, such as increased oxidative stress [112–115].
In normal conditions, α-syn is found in the cytosol where it regulates the dopamine synthesis pathway [116] and within mitochondria-associated ER membranes (MAMs) where it may participate in a number of key signaling pathways, including the regulation of mitochondrial Ca2+ transients and mitochondrial dynamics [117]. PD-associated mitochondrial dysfunction induces α-syn [71, 118, 119], which may represent a retrograde signal designed to dampen the bioenergetic burden of synaptic transmission by blocking vesicle exocytosis and neurotransmitter release [120], to preserve neuronal Ca2+ homeostasis at MAMs [117], or to decrease oxidative stress associated with dopamine autoxidation [116] within the PD brain [119].
However, in the event that mitochondrial dysfunction persists, excess mtROS may cause α-syn nitration in dopaminergic neurons, thereby impairing its ability to bind membranes [121]. In this regard, nitration of α-syn in “sporadic” PD patients may produce the same effect as familial PD mutations in the α-syn gene that decrease its lipid-binding ability and redirect it from MAMs to the mitochondria [122]. Mitochondria-localized α-syn may impair complex I function directly or increase its sensitivity to other modifiers [123–126]. Complex I inhibition by α-syn could then serve a second signaling role by amplifying mtROS formation and changing α-syn physical conformation to promote aggregation and LB formation [127].
This section has thus far focused on the effect of mitochondrial signaling in the cytoplasmic compartment, where mitochondrial SOS lead to the accumulation and aggregation of proteins associated with neurodegenerative diseases. The following section discusses mitochondrial signaling to the nucleus, which affects gene expression.
3.3 Metabolic Signaling and the Epigenetic Language
Contrary to popular conception, mitochondria do not float randomly amid the cytoplasm. Their cellular distribution is highly regulated by energy needs and other intracellular cues [128]. While most attention is given to the synaptic mitochondria required to supply the estimated 4.7 billion ATP molecules per second of a single neuron’s activity [129], mitochondria within the cell body are also critical for neuronal function via retrograde signaling. In the soma, mitochondria are positioned in the perinuclear region, in close proximity to the chromatin [130]. The chromatin is the DNA-protein structure that modulates gene accessibility by either “closing” portions of the genome to repress transcription or “opening” them to promote their expression. Mitochondrial regulation of gene expression is facilitated by two major factors. The first is “topological,” consisting in the perinuclear positioning of mitochondria only a few hundred nanometers away from the nuclear pores through which molecular signals travel [130]. The second and more studied factor that enables mitochondria-nuclear signaling is the nature of biochemical signals that induce epigenetic modifications, most of which are produced by mitochondria. The latter aspect constitutes the focus of this section.
As part of their normal metabolism, mitochondria produce most of the biochemical substrates and cofactors required for epigenetic (i.e., “on top of” genes) modifications that remodel chromatin structure. These substrates and cofactors include, but are not limited to, acetyl-coenzyme A (Ac-CoA), succinyl-CoA, SAM these (S-adenosyl methionine), ATP, α-ketoglutarate, NAD+ (nicotinamide adenine dinucleotide), and FAD (flavin adenine dinucleotide) [131, 132]. Posttranslational modifications require mitochondrial energy input and metabolism and consist of acetylation, succinylation, methylation, phosphorylation, and other labile alterations of the core chromatin component, the histones. The DNA itself is also modified via cytosine methylation, hydroxymethylation, and other modifications. This partially heritable collection of epigenetic modifications constitutes the relatively malleable “epigenetic landscape” that orchestrates gene expression, enabling a single genome to support multiple, drastically different cellular phenotypes (i.e., neurons vs. fibroblasts [133]). In neurodegenerative diseases, the epigenetic landscape of the brain is altered [134], including early changes in DNA methylation in Alzheimer’s disease [135]. The link between mitochondrial intermediate metabolism and epigenetic modifications suggests a role for mitochondrial dysfunction and resulting signals in the transcriptional reprogramming that may underlie neurodegeneration.
In a study of human cells with varying levels of mitochondrial dysfunction, it was recently discovered that the majority (>67 %) of the human genome is under mitochondrial regulation [136]. Indeed, whereas low levels of mitochondrial dysfunction in the presence of 20–30 % of mtDNA mutation load induced epigenetic modifiers and repressed cell growth, higher levels (60–90 %) of the same mtDNA mutation caused opposing transcriptional changes, inducing anaerobic glycolytic metabolism and transcriptional programs associated with cell senescence [136]. A single nucleotide change in the mtDNA can thus exert a wide range of transcriptional changes in the cell nucleus.
In addition to metabolites, mtROS also travel to the nucleus where they promote an oxidized nuclear environment favorable to the expression of redox-responsive genes [137]. Beyond regulating gene expression under normal circumstances, mtROS signals may also accelerate telomere shortening and aging [138, 139]. MtROS further activate oxidative stress-sensitive, pro-inflammatory pathways such as nuclear factor-kappa B (Nf-kB) [140], involved in general inflammatory response to chronic stress in human monocytes and microglia [141].
Mitochondrial bioenergetic signals are also transduced to the nucleus via different transcription factors and co-activators including but not restricted to Nf-kB [142]. Upon activation, transcription factors located in the cytoplasm translocate to the nucleus through nuclear pores, where they interact with chromatin components and the DNA itself. Canonical mito-nuclear signaling mechanisms include the energy sensors AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α), nuclear factor erythroid 2-related factor (NRF2) and p53 induced by oxidative stress, and Ca2+-sensitive calcineurin pathways [143, 144]. These and others have been shown to be altered in neurodegenerative conditions [145]. Therefore, multiple pathways exist whereby progressive mitochondrial dysfunction is communicated to the nucleus where it modifies gene expression via the epigenome.
Genetic and biochemical mitochondrial defects alter cellular bioenergetics and can lead to energy deficiency due to ATP depletion, which is generally believed to contribute to neurodegeneration. In addition, this section has outlined alternative pathways whereby dysfunctional mitochondria produce biochemical SOS, to which the cell and its plastic (epi)genome have evolved molecular sensitivity. These mitochondrial signals – oxidative stress and metabolic intermediates – travel to the nucleus to induce epigenetic modifications, many of which are post-translational modifications of regulatory proteins, that impact gene expression. From an evolutionary perspective, the coupling of genome remodeling epigenetic processes with mitochondrial signals in the brain was likely an important step for the evolution of species in an environment where energy supply and demand vary widely over time [146]. Multiple mechanisms thus exist to link mitochondrial metabolism to fundamental aspects of neuronal function.
3.4 Mitochondria as Synaptic Neuromodulators
Evidence for mitochondrial signaling at the synaptic level has recently expanded our conception of mitochondria not only as powerhouses but also as neuromodulators. In addition to mitochondrial ATP that influences synaptic function [147], mitochondrial Ca2+ uptake via the mitochondrial calcium uniporter (MCU) also regulates presynaptic neurotransmitter release [5]. On the other hand, abnormal mitochondrial morphology (i.e., donut shaped) in presynaptic terminals has been associated with abnormal synapse structure (smaller active zone, fewer docked neurotransmitter vesicles) and impaired short-term memory, indicating a link between mitochondrial morphology and higher cognitive function [148], although the mechanism by which this occurs remains unclear.
In mice, mitochondrial DNA heteroplasmy (i.e., a mixture of two mtDNA types differing by 91 nucleotides) impairs memory retention and alters animal behavior [149], demonstrating a link between mitochondrial genetics and cognitive function. Live cell imaging studies of synaptic neurotransmitter release further showed how the movement of mitochondria into a presynaptic terminal dynamically potentiates presynaptic vesicle release and reduces fatigability, whereas the absence of a mitochondrion in a presynaptic terminal leads to fatigability in response to consecutive stimulation [150]. Collectively, this positions mitochondria as neuromodulators within the brain [151, 152], illustrating a mechanism other than cell death and neurodegenerative changes by which mitochondrial signaling may affect cognitive function.
4 Mitochondria: Systemic Signaling
The previous section discussed how the state of mitochondria directs processes within the cell cytoplasm and nucleus and their role at the synapse. In addition, possibly as a result of the evolutionary intertwine between mitochondrial energetics and the development of multicellular organisms, organ systems such as the cardiovascular and endocrine axes are also functionally regulated by the bioenergetic state of mitochondria. In this scenario, mitochondrial signals act in a cell non-autonomous manner to influence systemic neuroendocrine processes linked to neuronal function.
Early evidence for mitochondrial signaling at the systemic level came from exercise physiology studies in patients with inherited mitochondrial myopathy. There is normally a tight coupling between whole body energy expenditure and cardiorespiratory responses during exercise, where a one-unit increase in oxygen consumption leads to a five-unit increase in cardiac output [153]. In contrast, patients with mitochondrial disease (m.3243A > G, single deletion) show exaggerated cardiac output and ventilatory responses that are disproportionate to energy demand [153], suggesting that dysfunctional mitochondria signal to the central nervous system, via an currently unknown mechanism, to exaggeratedly mobilize oxygen delivery systems (heart rate, and ventilation). A similar response was also observed at the level of skeletal muscle, where muscle fibers with respiratory chain-deficient mitochondria are selectively surrounded by a greater number of capillaries than adjacent fibers with normal mitochondria [154], indicating that defective mitochondria communicate their dysfunctional state via SOS to neighboring endothelial cells, stimulating the growth of capillaries (i.e., angiogenesis). Collectively, the evidence suggests that dysfunctional mitochondria engage in signaling not only with each other and the cell nucleus but also with surrounding cells and across the organism where they influence broad physiological functions.
The remaining section discusses recent lines of research that aim to understand the effect of mitochondrial function on neuroendocrine, metabolic, and inflammatory processes. Because neuroendocrine, metabolic, and inflammatory mediators impact neuronal health [155], mitochondrial systemic signaling represents an emerging pathway by which mitochondrial dysfunction may influence the etiology and progression of neurodegenerative diseases.
4.1 Mitochondrial Functions Regulate Neuroendocrine Systems
Two major neuroendocrine axes that release neuroactive hormones in the systemic circulation have been linked to mitochondrial function: the hypothalamic-pituitary-adrenal (HPA) axis, which leads to the secretion of cortisol (corticosterone in rodents), and the sympathetic-adrenal-medullary (SAM) axis, a division of the autonomic nervous system (ANS), which leads to the secretion of the catecholamines noradrenaline and adrenaline. Both cortisol and the catecholamines can influence brain structure and function by acting on the glucocorticoid receptor and adrenergic receptors on afferent sensory nerves, respectively [156, 157].
Recent evidence in animal models suggests that mtDNA variants can alter stress-reactive corticosterone (CORT) production in mice [158, 159] and the CORT-generating cells of the adrenal cortex are exquisitely sensitive to intramitochondrial oxidative stress [160]. Likewise, mutations or deletions of the redox-regulating enzyme NNT lead to hypocortisolemia in humans [160] and mice [161], respectively. In patients with mutations in the mitochondrial adenine nucleotide translocator (ANT), which impairs ATP/ADP exchange across the IMM, resting circulating catecholamine levels are also almost double that of healthy controls [162], indicating that both HPA and SAM axes’ activities are modulated by mitochondrial dysfunction.
Mitochondrial respiratory chain dysfunction may also be associated with ANS dysfunction, involving increased sympathetic vagal nerve drive as evidenced by increased resting heart rate, and increased high frequency power RR interval variability (RRV) in patients with the m.3243A>G mutation [163]. Mitochondrial disorders are also associated with excessive epinephrine and norepinephrine secretion by the SAM axis during exercise [164].
A recent study in mice with either mtDNA point mutations affecting OXPHOS capacity or deletions of nuclear-encoded NNT or ANT1 genes demonstrated that in addition to gene expression in the hippocampus, the major neuroendocrine systems are under substantial mitochondrial regulation [161]. In response to psychological stress, mitochondrial defects may in some cases double corticosterone output while blunting catecholamine release [161]. Mitochondrial dysfunction may also promote stress-induced hypercortisolemia and hyperglycemia, which are associated with brain atrophy and cognitive decline [165]. These mitochondria-regulated neuroendocrine perturbations may cause neuronal insult directly by further damaging mitochondrial function and leading to the accumulation of mtDNA defects [165, 166] and indirectly by dysregulating the metabolic and immune systems [167], curtailing brain plasticity and adaptation to stress [156].
4.2 Mitochondrial Immunogenic Signals
Another common feature of late-onset neurodegenerative diseases that may be modulated by mitochondrial SOS is inflammation (Fig. 5.3). As we age, our metabolic and immune systems undergo a process of senescence characterized by a progressive decline in mitochondrial and immune function, which correlates with an increased frequency of infection and neurodegenerative disease [78, 168]. Inflammation in neurodegenerative diseases occurs without evidence of externally acquired pathogens, suggesting that endogenous factors may instead initiate inflammation (i.e., sterile inflammation) [169]. Interestingly, a growing body of literature indicates that mitochondrial SOS may trigger the immune systems and promote chronic low-grade inflammation implicated in neurodegenerative diseases.

Mitochondrial signals of stress and neuroinflammation. In neurodegenerative diseases, microglia become “primed” and contribute to inflammatory changes within the brain. Here, we propose that neuronal mitochondrial dysfunction initiates microglial activation by releasing immunogenic signals of stress (SOS). Mitochondrial SOS include reactive oxygen species ( ROS) and damage-associated molecular patterns (DAMPs;
ROS) and damage-associated molecular patterns (DAMPs;  mtDNA, RNA, and N-formyl peptides) that activate toll-like receptors 4 and 9 (TLR4/TLR9), inducing the innate immune response from microglia and systemic macrophages. In the aging brain, chronic stimulation by mitochondrial SOS shifts the response of “primed” microglia from primarily phagocytic and anti-inflammatory (M2) to pro-inflammatory (M1) and induces their proliferation. TLR4/TLR9 stimulation activates M1 microglia by MyD88 or MAPK, which sends NF-kB from the cytosol to the nucleus to induce proIL-1β and NLRP3 transcription. When activated by oxidized mtDNA, the NLRP3-inflammasome cleaves pro-caspase-1 and induces the release of cytokines (IL-4, IL-8, IL-1B, and TNF-α) that promote neuroinflammation, gliosis, and neuronal damage. Secondary insult from protein aggregates (
mtDNA, RNA, and N-formyl peptides) that activate toll-like receptors 4 and 9 (TLR4/TLR9), inducing the innate immune response from microglia and systemic macrophages. In the aging brain, chronic stimulation by mitochondrial SOS shifts the response of “primed” microglia from primarily phagocytic and anti-inflammatory (M2) to pro-inflammatory (M1) and induces their proliferation. TLR4/TLR9 stimulation activates M1 microglia by MyD88 or MAPK, which sends NF-kB from the cytosol to the nucleus to induce proIL-1β and NLRP3 transcription. When activated by oxidized mtDNA, the NLRP3-inflammasome cleaves pro-caspase-1 and induces the release of cytokines (IL-4, IL-8, IL-1B, and TNF-α) that promote neuroinflammation, gliosis, and neuronal damage. Secondary insult from protein aggregates ( ) or glucocorticoids may exaggerate this response in sensitive brain regions, contributing to the mood disorders, cognitive decline, and cell death associated with neurodegenerative disease. Microglia may also contribute to elimination of dysfunctional mitochondria within the axon terminal, in a process whereby neurons outsource mitophagy [223]
) or glucocorticoids may exaggerate this response in sensitive brain regions, contributing to the mood disorders, cognitive decline, and cell death associated with neurodegenerative disease. Microglia may also contribute to elimination of dysfunctional mitochondria within the axon terminal, in a process whereby neurons outsource mitophagy [223]
The most common risk factors for neurodegenerative diseases include aging, metabolic and immune disorders, chemical toxin exposure, and psychological trauma [170]. These factors impact mitochondrial function by either promoting the accumulation of mtDNA mutations/deletions, decreased OXPHOS capacity, or excessive mtROS production, which can trigger the release of mitochondria-derived damage-associated molecular patterns (mtDAMPs), such as mtDNA fragments and some mitochondrial proteins [169]. Progressive impairment of mitochondrial function and integrity and the associated release of immunogenic molecules may thus steadily stimulate the innate immune system and promote inflammation both systemically and within the brain [171]. Here, we discuss the interplay between mitochondrial SOS and the immune system, which can be divided into two main categories.
4.2.1 Mitochondrial SOS as Inflammatory Triggers
The circular mtDNA of bacterial origin and resultant N-formyl peptides are recognized as foreign molecules by the immune system [169]. N-formyl peptides and mtDNA itself constitute mtDAMPs that are released following mitochondrial stress, particularly oxidative stress [172]. The release of mtDAMPs engages the innate immune system through the intracellular DNA-sensing system cGAS [173] and toll-like receptors (TLRs) [174]. TLRs are present in dendritic cells, macrophages, and microglia in the CNS, as well as nonimmune cells such as neurons and epithelial cells, where they activate the inflammasome and pro-inflammatory gene expression [175]. In microglia and neuronal cultures, mtDAMPs cause dopaminergic cell death [176] and increased amyloidosis with AD-associated inflammation [177]. In animal models, mtDAMP inflammasome activation is associated with cognitive impairment [178] and neurodegenerative disease [176, 179, 180].
Other mitochondrial components have also been reported in human plasma and suggested to play signaling roles that may influence the brain (Table 5.1). They include cardiolipin, which may act as a mtDAMP when oxidized [181]; mitochondrial proteins encoded in the nuclear genome such as cyt c [182], heat shock protein 60 (Hsp60) [183], and prohibitins [184]; and mtDNA-encoded proteins derived from alternative open reading frames (altORFs) such as humanin [185] and MOTS-c [186] (see Table 5.1). Circulating Hsp60 levels correlate with neurodegenerative disease risk factors, such as psychological stress and circulating cholesterol levels [183], indicating a potential immunogenic link between psychosocial factors, mitochondrial stress, and neuronal demise [187]. Mitochondria-derived molecules are thus emerging as a source of signals that may chronically instigate systemic inflammation in neurodegenerative disease.
Just as mtDAMPs correlate with known mediators of neurodegeneration, new evidence suggests that established neurodegenerative disease therapeutics prevent mtDAMP release. The anti-inflammatory signal acetylcholine (ACh) prevents stress-induced release of mtDNA via binding a putative mitochondrial nicotinic ACh receptor [172]. Therefore, it is plausible that the therapeutic benefit of nicotine and acetylcholinesterase (AChE) inhibitors for NDs [188] may not only be related to mitochondrial signaling in cell death [189], but also to mitochondria-related inflammatory signaling.
4.2.2 Mitochondria Regulate Intracellular Immunogenic Responses
Infected immune cells require energized mitochondria to recruit the mitochondrial antiviral signaling (MAVS) protein, which aggregates on the mitochondrial outer membrane and initiates antiviral signaling [190]. This signal stimulates the cellular antiviral response by activating nuclear translocation of NF-kB and interferon regulatory factors (IRFs) where they stimulate transcription of type I interferons and pro-inflammatory cytokine genes associated with “primed” microglia in aging and neurodegenerative diseases [191]. Ablating mitochondrial membrane potential inhibits this response [192], whereas mitochondrial ROS and mtDAMPs potentiate it [193, 194]. Interestingly, the mtDNA variants that govern mtROS and membrane potential also correlate with risk for neurological disorders and alter the metabolic-immune axis in a manner that mirrors the circulating cytokine profiles in patients with mood disorders and neurodegenerative disease [195–199].
Taken together, this relationship suggests that genetic or environmental insults to mitochondria may induce the release of SOS (mtDAMPs and mtROS) that drive neuroinflammation by activating the innate immune system and priming microglia (see Fig. 5.3). In turn, primed microglia in the aged brain inappropriately respond to immune challenges presented by mitochondrial dysfunction, as evident by their decreased phagocytic activity and sustained inflammatory signals in neurodegenerative disease [200]. The result of this faulty mitochondrial-immune network may amplify protein aggregates, neuronal death, and neurodegenerative disease progression.
4.3 Mitochondria as Mediators of Common Risk Factors in Neurodegenerative Disease
The risk of developing late-onset neurodegenerative disease is intimately tied to lifestyle factors such as physical activity [201] and psychosocial stress [202]. As discussed below, physical activity and chronic psychosocial stress induce biological patterns that correlate with downstream changes in biomarkers of increased [203] or decreased neurodegenerative disease risk [204, 205], respectively. Mitochondria may thus contribute to translate the lifestyle and environmental risk factors into the biological changes that cause neurodegenerative disease.
Physical inactivity, or sedentary behavior, promotes disease risk by producing a diabetes-like physiological state of metabolic oversupply that damages mitochondria [206]. Metabolic stress arises from excess energetic substrates (glucose, lipids), impairs mitochondrial dynamics, increases mtROS production, and leads to accumulation of mtDNA damage [206]. Diabetes exemplifies this state of metabolic dysregulation, where circulating glucose levels are continuously elevated. As a result, diabetes is associated with brain atrophy and cognitive decline in aging [165] and AD patients [207]. Hyperglycemia increases Aβ levels in the hippocampus [208]. On the other hand, exercise counteracts metabolic oversupply systemically [206] and within the brain by inducing mitochondrial biogenesis and neurogenesis and may stimulate clearance of Aβ [209–211]. The metabolic state, regulated by the combined effect of physical activity and diet, imparts substantial effects on neuronal health and the risk of neurodegenerative diseases [201].
Likewise, psychological stress in the social and work environment, which arises from real or perceived threat to the self, increases the risk of neurodegenerative disease. Psychosocial stress at work (i.e., low job control and high demand) is associated with increased risk for dementia and AD later in life [212]. Likewise, longitudinal studies demonstrated that chronic life stress during middle age is associated with brain atrophy and white matter lesions later in life [213] and increased risk for depression, dementia, and neurodegenerative diseases [202, 214], underscoring the negative impact of various stressors on neuronal health.
In seeking to resolve the biological connections responsible for these associations, it is relevant to note that hormones secreted during stress, including cortisol which becomes dysregulated during chronic stress and neurodegenerative disease, impact mitochondria both directly via the mitochondrial glucocorticoid receptor [166, 215] and indirectly by promoting metabolic stress systemically [167]. Excess stress-associated cortisol and metabolic stress can both impair neuronal mitochondria function and lead to neuroinflammation and dopaminergic death associated with PD [216]. On the other hand, physical activity/exercise confers protection against age-related hippocampal atrophy and memory decline [217] and reduces AD risk [218] and PD progression [219]. Interestingly, physical activity may also buffer against stress-associated cellular changes such as telomere shortening [220]. The exact mechanisms for the beneficial effects of physical activity and the damaging effect of chronic stress on brain structure and function remain unclear [156]. However, evidence that stress damages mitochondria, whereas physical activity promotes mitochondrial biogenesis [221], and that mitochondrial signaling is essential to exercise-induced neurogenesis (i.e., the formation of new neurons) in the adult brain [222], suggests that mitochondria constitute a hub where the action of stressors and resiliency factors intersect to influence brain structure and function.
5 Conclusion
The road from mitochondrial dysfunction to neurodegeneration takes many turns, and the trajectory from normal to abnormal brain function can be influenced by multiple mitochondrial signals. In addition to their central role in energy production, mitochondria perform a number of functions essential to neuronal activity and synaptic transmission. In this chapter, we have considered emerging facets of mitochondrial biology related to signal transduction from mitochondria to mitochondria, mitochondria to cytoplasm, mitochondria to nucleus, and mitochondria to the systemic circulation.
Mitochondrial dysfunction may result from primary inherited genetic defects or from acquired structural and functional changes with aging and chronic stressors (i.e., mitochondrial allostatic load) [167]. In turn, dysfunctional mitochondria produce abnormal signals of stress (i.e., SOS) that propagate to other cellular compartments and influence systemic regulatory processes.
This chapter integrated experimental, clinical, and epidemiological evidence that posits mitochondria as a proximal mediator of established pathogenic processes that define neurodegenerative disease. Why would mitochondrial signals influence such a broad number of physiological and molecular processes? A partial justification for this “mitocentric” perspective may in part rest in biological events that occurred as part of the evolution of complex life. To reiterate, mitochondria played a central and necessary role in the evolution of multicellular organ systems, of which the brain is arguably the most intricate product. The cellular machinery involved in neurological functioning, including synaptic transmission, and those of its basic cellular behaviors including replication, differentiation, and growth, as well as the underlying process of transcriptional regulation, have thus relied and become dependent upon mitochondrial biochemical outputs (ATP, mtROS, and intermediate metabolites).
As a result, multiple evolutionary-carved paths exist to translate signals of mitochondrial dysfunction into maladaptive cellular processes contributing to neurodegeneration. Future work will be needed to elucidate the molecular and physiological basis for mitochondrial signaling within and beyond the cell. A fuller understanding of the role of mitochondria in the process of neurodegeneration should provide new opportunities to design interventions to preserve optimal brain function throughout the lifespan.
References
Margulis L, Bermudes D. Symbiosis as a mechanism of evolution: status of cell symbiosis theory. Symbiosis. 1985;1:101–24.
Lane N, Martin W. The energetics of genome complexity. Nature. 2010;467(7318):929–34.
Wallace DC. Bioenergetics, the origins of complexity, and the ascent of man. Proc Natl Acad Sci U S A. 2010;107 Suppl 2:8947–53.
Glancy B, Balaban RS. Role of mitochondrial ca(2+) in the regulation of cellular energetics. Biochemistry. 2012;51(14):2959–73.
Marland JR, Hasel P, Bonnycastle K, Cousin MA. Mitochondrial calcium uptake modulates synaptic vesicle endocytosis in central nerve terminals. J Biol Chem. 2016;291(5):2080–6.
Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13(9):566–78.
Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–81.
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95.
Papadopoulos V, Miller WL. Role of mitochondria in steroidogenesis. Best Pract Res Clin Endocrinol Metab. 2012;26(6):771–90.
Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24(12):761–70.
Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–46.
Nadell CD, Bucci V, Drescher K, Levin SA, Bassler BL, Xavier JB. Cutting through the complexity of cell collectives. Proc Biol Sci R Soc. 2013;280(1755):20122770.
Braschi E, McBride HM. Mitochondria and the culture of the Borg: understanding the integration of mitochondrial function within the reticulum, the cell, and the organism. Bioessays. 2010;32(11):958–66.
Picard M, Burelle Y. Mitochondria: starving to reach quorum?: insight into the physiological purpose of mitochondrial fusion. Bioessays. 2012;34(4):272–4.
Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 2010;6(1):e1000657.
Pacher P, Hajnoczky G. Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 2001;20(15):4107–21.
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–14.
Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2015;78:505–31.
Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. 2013;304(6):R393–406.
Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13(5):589–98.
Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155(1):160–71.
Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–62.
Oettinghaus B, Schulz JM, Restelli LM, Licci M, Savoia C, Schmidt A, et al. Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 2016;23(1):18–28.
Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–9.
Stauch KL, Purnell PR, Fox HS. Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J Proteome Res. 2014;13(5):2620–36.
Sheng ZH. Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol. 2014;204(7):1087–98.
Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28(3):272–5.
Yang L, Long Q, Liu J, Tang H, Li Y, Bao F, et al. Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell Mol Life Sci. 2015;72(13):2585–98.
Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369(23):2236–51.
Santo-Domingo J, Giacomello M, Poburko D, Scorrano L, Demaurex N. OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J. 2013;32(13):1927–40.
Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J Cell Biol. 1988;107(2):481–95.
Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523(7562):617–20.
Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–87.
Popov V, Medvedev NI, Davies HA, Stewart MG. Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J Comp Neurol. 2005;492(1):50–65.
Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26(1):23–9.
Kurz FT, Aon MA, O’Rourke B, Armoundas AA. Spatio-temporal oscillations of individual mitochondria in cardiac myocytes reveal modulation of synchronized mitochondrial clusters. Proc Natl Acad Sci U S A. 2010;107(32):14315–20.
Kurz FT, Aon MA, O’Rourke B, Armoundas AA. Cardiac mitochondria exhibit dynamic functional clustering. Front Physiol. 2014;5:329.
Picard M, McManus MJ, Csordas G, Varnai P, Dorn 2nd GW, Williams D, et al. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat Commun. 2015;6:6259.
Brodin L, Bakeeva L, Shupliakov O. Presynaptic mitochondria and the temporal pattern of neurotransmitter release. Philos Trans R Soc Lond Ser B Biol Sci. 1999;354(1381):365–72.
Picard M, Azuelos I, Jung B, Giordano C, Matecki S, Hussain S, et al. Mechanical ventilation triggers abnormal mitochondrial dynamics and morphology in the diaphragm. J Appl Physiol. 2015;118(9):1161–71.
Picard M, Gentil BJ, McManus MJ, White K, St Louis K, Gartside SE, et al. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J Appl Physiol. 2013;115(10):1562–71.
Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA. Mitochondrial ROS metabolism: 10 years later. Biochem Biokhim. 2015;80(5):517–31.
Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157(4):897–909.
Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348.
McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol. 2006;71(4):399–407.
Dumas JF, Argaud L, Cottet-Rousselle C, Vial G, Gonzalez C, Detaille D, et al. Effect of transient and permanent permeability transition pore opening on NAD(P)H localization in intact cells. J Biol Chem. 2009;284(22):15117–25.
Gameiro PA, Laviolette LA, Kelleher JK, Iliopoulos O, Stephanopoulos G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J Biol Chem. 2013;288(18):12967–77.
Ma S, Zhang X, Zheng L, Li Z, Zhao X, Lai W, et al. Peroxiredoxin 6 is a crucial factor in the initial step of mitochondrial clearance and is upstream of the PINK1-Parkin pathway. Antioxid Redox Signal. 2015;24:486–501.
Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener. 2011;6:34.
Sabens EA, Distler AM, Mieyal JJ. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson’s disease. Biochemistry. 2010;49(12):2715–24.
Angeles DC, Gan BH, Onstead L, Zhao Y, Lim KL, Dachsel J, et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum Mutat. 2011;32(12):1390–7.
Shi ZZ, Osei-Frimpong J, Kala G, Kala SV, Barrios RJ, Habib GM, et al. Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc Natl Acad Sci U S A. 2000;97(10):5101–6.
Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279(2):324–9.
Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178(1):179–85.
Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25(5):1980–8.
McManus MJ, Murphy MP, Franklin JL. Mitochondria-derived reactive oxygen species mediate caspase-dependent and -independent neuronal deaths. Mol Cell Neurosci. 2014;63:13–23.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18(4):195–9.
Khan M, Dhammu TS, Matsuda F, Annamalai B, Dhindsa TS, Singh I, et al. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2015;1630:159–70.
Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis JAD. 2010;20 Suppl 2:S369–79.
Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–14.
Lacza Z, Snipes JA, Kis B, Szabó C, Grover G, Busija DW. Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Res. 2003;994(1):27–36.
Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, et al. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am J Pathol. 2006;168(5):1608–18.
Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y, Shimizu K, Umegae N, et al. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann Neurol. 2000;47(4):524–7.
Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem. 2011;286(30):26334–43.
Orrenius S, Zhivotovsky B. Cardiolipin oxidation sets cytochrome c free. Nat Chem Biol. 2005;1(4):188–9.
Birk AV, Chao WM, Liu S, Soong Y, Szeto HH. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim Biophys Acta. 2015;1847(10):1075–84.
Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10(12):1477–83.
Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci Off J Soc Neurosci. 2002;22(15):6480–90.
Logan A, Shabalina IG, Prime TA, Rogatti S, Kalinovich AV, Hartley RC, et al. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell. 2014;13(4):765–8.
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95.
Murray J, Tsui WH, Li Y, McHugh P, Williams S, Cummings M, et al. FDG and amyloid PET in cognitively normal individuals at risk for late-onset alzheimer’s disease. Adv Mol Imaging. 2014;4(2):15–26.
Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180–95.
Majd S, Power JH, Grantham HJ. Neuronal response in Alzheimer’s and Parkinson’s disease: the effect of toxic proteins on intracellular pathways. BMC Neurosci. 2015;16:69.
Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16(2):109–20.
Su B, Wang X, Nunomura A, Moreira PI, Lee H, Perry G, et al. Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res. 2008;5(6):525–32.
Wallace DC. Mitochondrial DNA, mutations in disease and aging. Environ Mol Mutagen. 2010;51(5):440–50.
Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog Brain Res. 2010;184:17–33.
Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–80.
Swerdlow RH, Parks JK, Cassarino DS, Maguire DJ, Maguire RS, Bennett Jr JP, et al. Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurology. 1997;49(4):918–25.
Swerdlow RH. Role and treatment of mitochondrial DNA-related mitochondrial dysfunction in sporadic neurodegenerative diseases. Curr Pharm Des. 2011;17(31):3356–73.
Tan JL, Li QX, Ciccotosto GD, Crouch PJ, Culvenor JG, White AR, et al. Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. PLoS One. 2013;8(4):e61246.
Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10(3):279–88.
Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104(3):683–95.
Quiroz-Baez R, Rojas E, Arias C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem Int. 2009;55(7):662–70.
Bredesen DE, John V, Galvan V. Importance of the caspase cleavage site in amyloid-β protein precursor. J Alzheimers Dis JAD. 2010;22(1):57–63.
McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31(44):15703–15.
Soucek T, Cumming R, Dargusch R, Maher P, Schubert D. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron. 2003;39(1):43–56.
Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci Off J Soc Neurosci. 2002;22(12):4833–41.
Kontush A, Berndt C, Weber W, Akopyan V, Arlt S, Schippling S, et al. Amyloid-beta is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic Biol Med. 2001;30(1):119–28.
Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci Off J Soc Neurosci. 2003;23(13):5531–5.
Devi L, Ohno M. Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis. 2012;45(1):417–24.
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670–5.
Ronnback A, Pavlov PF, Mansory M, Gonze P, Marliere N, Winblad B, et al. Mitochondrial dysfunction in a transgenic mouse model expressing human amyloid precursor protein (APP) with the Arctic mutation. J Neurochem. 2015;136(3):497–502.
Kontush A, Atwood CS. Amyloid-beta: phylogenesis of a chameleon. Brain Res Brain Res Rev. 2004;46(1):118–20.
Giuffrida ML, Caraci F, De Bona P, Pappalardo G, Nicoletti F, Rizzarelli E, et al. The monomer state of beta-amyloid: where the Alzheimer’s disease protein meets physiology. Rev Neurosci. 2010;21(2):83–93.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–33.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–21.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet (London, England). 2008;372(9634):216–23.
Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, Staufenbiel M, et al. Decreased amyloid-beta and increased neuronal hyperactivity by immunotherapy in Alzheimer’s models. Nat Neurosci. 2015;18(12):1725–7.
Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2015;12:15–27.
Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. 2005;64(2):104–12.
Gao F, Chen D, Hu Q, Wang G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS One. 2013;8(8):e72046.
Chiara F, Gambalunga A, Sciacovelli M, Nicolli A, Ronconi L, Fregona D, et al. Chemotherapeutic induction of mitochondrial oxidative stress activates GSK-3alpha/beta and Bax, leading to permeability transition pore opening and tumor cell death. Cell Death Dis. 2012;3:e444.
Alavi Naini SM, Soussi-Yanicostas N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxidative Med Cell Longev. 2015;2015:151979.
Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol Dis. 2015;76:13–23.
Innes BT, Sowole MA, Gyenis L, Dubinsky M, Konermann L, Litchfield DW, et al. Peroxide-mediated oxidation and inhibition of the peptidyl-prolyl isomerase Pin1. Biochim Biophys Acta. 2015;1852(5):905–12.
Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, et al. Pin1 in Alzheimer’s disease. J Neurochem. 2006;98(6):1697–706.
Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, et al. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: a redox proteomics analysis. Neurobiol Aging. 2006;27(7):918–25.
DuBoff B, Gotz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75(4):618–32.
Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, et al. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport. 1999;10(4):717–21.
Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. 2001;276(45):41958–62.
Nam MK, Han JH, Jang JY, Yun SE, Kim GY, Kang S, et al. A novel link between the conformations, exposure of specific epitopes, and subcellular localization of alpha-synuclein. Biochim Biophys Acta. 2015;1850(12):2497–505.
Zhou W, Zhu M, Wilson MA, Petsko GA, Fink AL. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J Mol Biol. 2006;356(4):1036–48.
Lou H, Montoya SE, Alerte TNM, Wang J, Wu J, Peng X, et al. Serine 129 phosphorylation reduces the ability of α-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem. 2010;285(23):17648–61.
Cali T, Ottolini D, Negro A, Brini M. Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287(22):17914–29.
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis. 2009;34(2):279–90.
McLean JR, Hallett PJ, Cooper O, Stanley M, Isacson O. Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol Cell Neurosci. 2012;49(2):230–9.
Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48.
Hodara R, Norris EH, Giasson BI, Mishizen-Eberz AJ, Lynch DR, Lee VM, et al. Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem. 2004;279(46):47746–53.
Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, et al. Alpha-synuclein is localized to mitochondria-associated ER membranes. J Neurosci Off J Soc Neurosci. 2014;34(1):249–59.
Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, et al. Alpha-synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett. 2009;454(3):187–92.
Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem. 2002;277(7):5411–7.
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283(14):9089–100.
Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179(1):9–16.
Barrett PJ, Timothy Greenamyre J. Post-translational modification of alpha-synuclein in Parkinson’s disease. Brain Res. 2015;1628(Pt B):247–53.
Lackner LL. Shaping the dynamic mitochondrial network. BMC Biol. 2014;12:35.
Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, et al. Quantitative imaging of energy expenditure in human brain. NeuroImage. 2012;60(4):2107–17.
Picard M. Mitochondrial synapses: intracellular communication and signal integration. Trends Neurosci. 2015;38(8):468–74.
Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502(7472):489–98.
Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal. 2014;26(7):1598–603.
Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–8.
Salminen A, Haapasalo A, Kauppinen A, Kaarniranta K, Soininen H, Hiltunen M. Impaired mitochondrial energy metabolism in Alzheimer’s disease: impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog Neurobiol. 2015;131:1–20.
De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17(9):1156–63.
Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci U S A. 2014;111(38):E4033–42.
Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, et al. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5(231):ra47.
Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5(5):e110.
Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging. 2012;4(1):3–12.
Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, et al. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci Off J Soc Neurosci. 2008;28(34):8624–34.
Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64(4):266–72.
Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. 2014;122(12):1271–8.
Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion. 2013;13(6):577–91.
Arany Z. PGC-1 coactivators and skeletal muscle adaptations in health and disease. Curr Opin Genet Dev. 2008;18(5):426–34.
Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ. Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol. 2012;8(9):518–30.
McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43(1):2–15.
Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156(4):825–35.
Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci U S A. 2014;111(1):486–91.
Sharpley MS, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K, et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151(2):333–43.
Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 2013;4(3):413–9.
Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;2:139.
Picard M, McEwen BS. Mitochondria impact brain function and cognition. Proc Natl Acad Sci U S A. 2014;111(1):7–8.
Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. 2003;126(Pt 2):413–23.
Taivassalo T, Ayyad K, Haller RG. Increased capillaries in mitochondrial myopathy: implications for the regulation of oxygen delivery. Brain. 2012;135(Pt 1):53–61.
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873–904.
McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, et al. Mechanisms of stress in the brain. Nat Neurosci. 2015;18(10):1353–63.
Tank AW, Lee WD. Peripheral and central effects of circulating catecholamines. Compr Physiol. 2015;5(1):1–15.
Gimsa U, Kanitz E, Otten W, Ibrahim SM. Behavior and stress reactivity in mouse strains with mitochondrial DNA variations. Ann N Y Acad Sci. 2009;1153:131–8.
Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, et al. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci U S A. 2015;112(48):E6614–23.
Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet. 2012;44(7):740–2.
Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, et al. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci. 2015;112(48):E6614–23.
Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci U S A. 2013;110(9):3453–8.
Bates MG, Newman JH, Jakovljevic DG, Hollingsworth KG, Alston CL, Zalewski P, et al. Defining cardiac adaptations and safety of endurance training in patients with m.3243A>G-related mitochondrial disease. Int J Cardiol. 2013;168(4):3599–608.
Jeppesen TD, Orngreen MC, Van Hall G, Vissing J. Lactate metabolism during exercise in patients with mitochondrial myopathy. Neuromuscul Dis NMD. 2013;23(8):629–36.
van Elderen SG, de Roos A, de Craen AJ, Westendorp RG, Blauw GJ, Jukema JW, et al. Progression of brain atrophy and cognitive decline in diabetes mellitus: a 3-year follow-up. Neurology. 2010;75(11):997–1002.
Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U S A. 2009;106(9):3543–8.
Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nat Rev Endocrinol. 2014;10(5):303–10.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol Ser A Biol Med Sci. 2014;69 Suppl 1:S4–9.
Masters SL, Walsh PT. Release of the mitochondrial endosymbiont helps explain sterile inflammation. Proc Natl Acad Sci U S A. 2010;107(10):E32.
Brown RC, Lockwood AH, Sonawane BR. Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect. 2005;113(9):1250–6.
Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur J Immunol. 2014;44(5):1552–62.
Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, et al. Alpha 7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014;20:350–8.
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–7.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–14.
Gao X, Hu X, Qian L, Yang S, Zhang W, Zhang D, et al. Formyl-methionyl-leucyl-phenylalanine-induced dopaminergic neurotoxicity via microglial activation: a mediator between peripheral infection and neurodegeneration? Environ Health Perspect. 2008;116(5):593–8.
Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, et al. Mitochondrial lysates induce inflammation and Alzheimer’s disease-relevant changes in microglial and neuronal cells. J Alzheimers Dis JAD. 2015;45(1):305–18.
Chen L, Na R, Boldt E, Ran Q. NLRP3 inflammasome activation by mitochondrial reactive oxygen species plays a key role in long-term cognitive impairment induced by paraquat exposure. Neurobiol Aging. 2015;36(9):2533–43.
Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, et al. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis JAD. 2012;30(3):617–27.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8.
Wan M, Hua X, Su J, Thiagarajan D, Frostegard AG, Haeggstrom JZ, et al. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis. 2014;235(2):592–8.
Pullerits R, Bokarewa M, Jonsson IM, Verdrengh M, Tarkowski A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatol (Oxford, England). 2005;44(1):32–9.
Shamaei-Tousi A, Steptoe A, O’Donnell K, Palmen J, Stephens JW, Hurel SJ, et al. Plasma heat shock protein 60 and cardiovascular disease risk: the role of psychosocial, genetic, and biological factors. Cell Stress Chaperones. 2007;12(4):384–92.
Mengwasser J, Piau A, Schlag P, Sleeman JP. Differential immunization identifies PHB1/PHB2 as blood-borne tumor antigens. Oncogene. 2004;23(44):7430–5.
Lee C, Yen K, Cohen P. Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab TEM. 2013;24(5):222–8.
Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443–54.
Lehnardt S, Schott E, Trimbuch T, Laubisch D, Krueger C, Wulczyn G, et al. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J Neurosci Off J Soc Neurosci. 2008;28(10):2320–31.
Perez XA. Preclinical evidence for a role of the nicotinic cholinergic system in Parkinson’s disease. Neuropsychol Rev. 2015;25(4):371–83.
Akaike A, Takada-Takatori Y, Kume T, Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci MN. 2010;40(1–2):211–6.
West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11(6):389–402.
Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90.
Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4(158):ra7.
West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–80.
Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun. 2015; doi: 10.1016/j.bbi.2015.10.009 (in press).
Gomez-Duran A, Pacheu-Grau D, Lopez-Gallardo E, Diez-Sanchez C, Montoya J, Lopez-Perez MJ, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet. 2010;19(17):3343–53.
Karabatsiakis A, Bock C, Salinas-Manrique J, Kolassa S, Calzia E, Dietrich DE, et al. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl Psychiatr. 2014;4:e397.
Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, Derbeneva O, Khusainova R, Khidiyatova I, et al. A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson’s disease. Ann N Y Acad Sci. 2008;1147:1–20.
Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, LaFerla F, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820(5):553–64.
Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial-nuclear interactions. Hum Mol Genet. 2014;23(13):3537–51.
Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217–24.
Mattson MP. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 2012;16(6):706–22.
Johansson L, Guo X, Waern M, Ostling S, Gustafson D, Bengtsson C, et al. Midlife psychological stress and risk of dementia: a 35-year longitudinal population study. Brain. 2010;133(Pt 8):2217–24.
Herrera AJ, Espinosa-Oliva AM, Carrillo-Jimenez A, Oliva-Martin MJ, Garcia-Revilla J, Garcia-Quintanilla A, et al. Relevance of chronic stress and the two faces of microglia in Parkinson’s disease. Front Cell Neurosci. 2015;9:312.
Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2015;23(1):128–42.
Fiuza-Luces C, Garatachea N, Berger NA, Lucia A. Exercise is the real polypill. Physiol (Bethesda, Md). 2013;28(5):330–58.
Picard M, Turnbull DM. Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes. 2013;62(3):672–8.
Morris JK, Vidoni ED, Honea RA, Burns JM. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol Aging. 2014;35(3):585–9.
Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichle ME, Perez R, et al. Hyperglycemia modulates extracellular amyloid-beta concentrations and neuronal activity in vivo. J Clin Invest. 2015;125(6):2463–7.
Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci Off J Soc Neurosci. 2005;25(17):4217–21.
Moore KM, Girens RE, Larson SK, Jones MR, Restivo JL, Holtzman DM, et al. A spectrum of exercise training reduces soluble Abeta in a dose-dependent manner in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2015;85:218–24.
Leal MC, Magnani N, Villordo S, Buslje CM, Evelson P, Castano EM, et al. Transcriptional regulation of insulin-degrading enzyme modulates mitochondrial amyloid beta (Abeta) peptide catabolism and functionality. J Biol Chem. 2013;288(18):12920–31.
Wang HX, Wahlberg M, Karp A, Winblad B, Fratiglioni L. Psychosocial stress at work is associated with increased dementia risk in late life. Alzheimers Dement. 2012;8(2):114–20.
Johansson L, Skoog I, Gustafson DR, Olesen PJ, Waern M, Bengtsson C, et al. Midlife psychological distress associated with late-life brain atrophy and white matter lesions: a 32-year population study of women. Psychosom Med. 2012;74(2):120–5.
Djamshidian A, Lees AJ. Can stress trigger Parkinson’s disease? J Neurol Neurosurg Psychiatry. 2014;85(8):878–81.
Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta. 2011;1813(10):1814–21.
de Pablos RM, Herrera AJ, Espinosa-Oliva AM, Sarmiento M, Munoz MF, Machado A, et al. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J Neuroinflammation. 2014;11:34.
Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108(7):3017–22.
Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788–94.
Goldman JG, Weintraub D. Advances in the treatment of cognitive impairment in Parkinson’s disease. Mov Disord. 2015;30(11):1471–89.
Puterman E, Lin J, Blackburn E, O’Donovan A, Adler N, Epel E. The power of exercise: buffering the effect of chronic stress on telomere length. PLoS One. 2010;5(5):e10837.
Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM. Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol. 2011;111(4):1066–71.
Steib K, Schaffner I, Jagasia R, Ebert B, Lie DC. Mitochondria modify exercise-induced development of stem cell-derived neurons in the adult brain. J Neurosci Off J Soc Neurosci. 2014;34(19):6624–33.
Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111(26):9633–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing
About this chapter
Cite this chapter
Picard, M., McManus, M.J. (2016). Mitochondrial Signaling and Neurodegeneration. In: Reeve, A., Simcox, E., Duchen, M., Turnbull, D. (eds) Mitochondrial Dysfunction in Neurodegenerative Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-28637-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-28637-2_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28635-8
Online ISBN: 978-3-319-28637-2
eBook Packages: MedicineMedicine (R0)