
Abstract
Stress was characterized by Hans Selye, in 1936, as the “stereotyped biological response to any demand.” Corticotrophin-releasing factor (CRF) and its related peptides urocortins (Ucns 1,2,3) are the mediators of stress. They are present both in the central nervous system and in the gastrointestinal (GI) tract where they exert their biological actions on target cells through activation of two receptors, CRF1 and CRF2. Stress is able to modulate numerous functions of the GI tract such as motility, secretion, permeability, sensitivity, and microbiota. Classically, stress delays gastric emptying while stimulating colonic transit and secretion, increases intestinal permeability and visceral sensitivity, and modifies intestinal microbiota. Through these various effects at the level of the GI tract, stress is involved in the pathophysiology of irritable bowel syndrome (IBS), a functional digestive disorder, as well as inflammatory bowel disease (IBD), Crohn’s disease, and ulcerative colitis. Targeting these CRF1/CRF2 signaling pathways by selective antagonists/agonists should be of clinical interest in the domain of IBS and IBD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Autonomic nervous system
- Corticotrophin-releasing factor
- Gastrointestinal
- Hypothalamus
- Inflammatory bowel disease
- Irritable bowel syndrome
- Stress
- Sympathetic nervous system
- Urocortins
- Vagus nerve
Stress-Definition
In 1936, Hans Selye defined the concept of stress as the “stereotyped biological response to any demand” [168] and elaborated the concept of the general adaptation syndrome. Later, the hypothalamic factor named corticotrophin-releasing factor (CRF) which stimulates ACTH release by the rat pituitary was discovered [77], positioning the hypothalamic–pituitary adrenal (HPA) axis as a key element in this concept. In 1981, Vale and his group [196] identified the 41-aa peptide CRF characterized from ovine hypothalami and later the cloning of CRF receptors and the development of specific CRF receptor antagonists [157]. In 2003, McEwen and Wingfield [121] defined the concept of allostasis as a process through which organisms actively adjust to both predictable and unpredictable events with allostatic overload being the cause of serious pathophysiology.
History of Stress Influence on Gut Functions and Diseases
In 1833, William Beaumont [15], a US Army surgeon, was the first to describe the influence of fear and anger on gastric acid secretion of his patient Alexis St. Martin, a Canadian trapper with a permanent gastric fistula caused by a musket shot. In 1902, Walter Cannon [42] observed gastrointestinal (GI) motility disturbances in the cat faced with an aggressive dog. Numerous other stress models have confirmed the effect of stress on GI motility in rodents [185]. Nowadays, the effect of psychological, physical, and immunological stressors on GI secretion, motility, epithelial permeability, visceral sensitivity, microbiota, and inflammation is clearly demonstrated, and stress has a major influence in irritable bowel syndrome (IBS) and most likely plays a role in inflammatory bowel disease (IBD). The circuitries and biochemical coding, particularly CRF and its receptors, involved in the stress response have been characterized through (i) the use of the immediate early gene c-fos, a marker of neuronal activation, and its relation to the autonomic regulation of gut functions [33] and (ii) its combination with the identification of neuromediators activated by stress models and (iii) the use of CRF-overexpressing as well as knockout animals.
The present review will describe first the state of knowledge on the distribution of the CRFergic system including CRF and its related peptides urocortins (Ucns), and their CRF receptors, in the brain and in the GI tract and the effects and mechanisms of stress on gastrointestinal motility, secretion, permeability, visceral sensitivity, and microbiota. Lastly, the implication of stress in the pathophysiology of IBS and IBD [Crohn’s disease (CD) and ulcerative colitis (UC)] will be highlighted.
Mechanisms of Stress
Stress Peptides and Their Receptors
CRF and CRF-Related Peptides (Fig. 7.1)

The CRF peptide family and its receptors and receptor antagonists (With permission from Taché and Bonaz [185]). CRF corticotrophin-releasing factor
CRF is a member of a family of mammalian CRF-related peptides including Ucn 1, Ucn 2 (known as stresscopin-related peptide), and Ucn 3 (known as stresscopin) [12] with 40, 38, and 38 aa in length, respectively. During stress, CRF is released from the hypothalamus and is vehiculated through a portal venous network to the anterior pituitary where it triggers the release of ACTH which stimulates the release of cortisol, the stress steroid, by the adrenal glands, i.e., the HPA axis (Fig. 7.2). CRF also acts directly in the central nervous system (CNS) as a neurotransmitter and neuromodulator through the projections of CRF-containing neurons to areas of the central autonomic network that controls the central response to stress. The intracerebral injection of CRF mimics behavioral (anxiety/depression, alterations of feeding), autonomic (sympathetic and sacral parasympathetic activation, vagal inhibition), immune, metabolic, and visceral responses induced by various systemic or cognitive stressors [12, 188]. The hypothalamic peptide arginine–vasopressin (AVP) acts in synergy with CRF to stimulate ACTH secretion in acute stress conditions, while a habituation of the hypothalamic CRF is observed in chronic stress conditions where AVP takes over from CRF [31].

The hypothalamic–pituitary–adrenal axis and the autonomic nervous system (With permission from Bonaz and Bernstein [27])). ACTH adrenocorticotropic hormone, AP area postrema, CRF corticotrophin-releasing factor, DMN dorsal motor nucleus of the vagus nerve, EN epinephrine, GC glucocorticoids, HPA hypothalamic–pituitary adrenal axis, LC locus coeruleus, LPS lipopolysaccharides, NE norepinephrine, PVN paraventricular nucleus of the hypothalamus, RVM rostral ventrolateral medulla
CRF Receptors
CRF and Ucns exert their biological actions on target cells through activation of two receptors, CRF1 (415 aa) and CRF2 (411 aa), that belong to the B1 class subfamily of 7-transmembrane domain G protein-coupled receptors and are encoded by two distinct genes [83].
CRF1a, the main functional variant of CRF1, is greatly expressed in the brain and some peripheral tissues in mammals [83]. Alternative splicing of the primary transcript encoding CRF1 can lead to other variants (n = 8), named CRF1b–i, all of which display impaired signaling [83]. The functional significance of these other transcripts is still poorly characterized. Selective peptide CRF1 agonists (cortagine and stressin1-A) have been developed [155]. Three functional CRF2 variants (2a,b,c) have been identified in humans [83], whereas only 2a and 2b are expressed in other mammals.
CRF1 and CRF2 have distinct affinities for the CRF family of peptides [12, 83]. CRF has a higher (10- to 40-fold) affinity for CRF1 than for CRF2. Ucn 1 binds CRF2 with a greater (100-fold) affinity than CRF and CRF1 with greater (sixfold) affinity than CRF. Ucns 2 and 3 exhibit high selectivity only for CRF2, with a slightly higher affinity for CRF2b versus CRF2a. CRF2a, CRF2b, and CRF2c are almost identical with high affinity for Ucn 1, Ucn 2, and Ucn 3 and lower affinity for rat/human CRF. In non-primate mammals, CRF2a is expressed only by neurons and CRF2b mainly in the periphery and by nonneuronal cells of the brain, whereas CRF2c is found only in the amygdala of the human brain. The binding of CRF family peptides to their receptors has been characterized [83]. The long N-terminal extracellular domain of CRF receptors primarily interacts with the C-terminal residues of CRF, and the N-terminal residues of CRF interact with the transmembrane region of the receptor, resulting in conformational changes that enable G protein activation. Stimulation of CRF1a, CRF2a, and CRF2b activates adenylyl cyclase/cAMP–protein kinase A signaling pathways through coupling and activation of Gαs proteins. CRF receptors modulate various kinases, including phosphokinases A, B, and C, and can phosphorylate and activate mitogen-activated protein kinase (MAPK), particularly the ERK1/2 and p38/MAPK pathways. CRF receptors also stimulate transient calcium mobilization in certain cell types by phospholipase C activation in vitro and PKC activation in vivo.
CRF receptor antagonists have been developed to characterize the functions of CRF receptors in the behavioral (anxiety, decreased feeding, and drug seeking), cognitive (arousal and anxiety), neuroendocrine (ACTH release), autonomic (activation of the sympathetic nervous system), immunological, and visceral (hypertension and alterations in gut motor function) responses to stress [50]. α-helical CRF9–41 was the first CRF receptor antagonist [157] followed by subsequent CRF antagonists such as d-Phe12CRF12–41 and astressin. Astressin-B was developed later, as the most efficacious and still effective 24 h after a single peripheral injection [158]. Peripheral administration of these peptide antagonists has a poor penetrance into the brain and could thus be used to selectively block physiological processes of the GI tract. Selective CRF2 peptide antagonists, binding equally to CRF2a, b, and c with little or no affinity for CRF1 receptors, have been developed [156] such as antisauvagine-30 and the long-acting analog astressin2-B. Non-peptide selective CRF1 antagonists that cross the blood–brain barrier depending on their lipophilicity have been developed, e.g., CP-154,526, antalarmin, NBI 30775, and NBI 35965 [50]. The activation of the brain CRF/CRF1 signaling pathway plays a major role in the coordination of many physiological responses to adaptive stress through the activation of the HPA axis, autonomic nervous system (ANS), and changes in cardiovascular, GI, and immune functions in rodents and primates [80, 115]. The abnormal increase of central CRF1 signaling could contribute to the pathogenesis of anxiety and depression as well as to IBS [11, 80, 161]. CRF2 receptors in the brain dampen and/or facilitate the proper recovery of the CRF1-initiated behavioral, endocrine, and visceral responses to stress [154] and have a primary role in the anorexic response to exogenous administration of CRF and Ucns [177]. CRF-binding protein (332-aa) isolated in different species functions as an endogenous antagonist by sequestrating CRF ligands, thus modulating the access of CRF and related peptides to CRF receptors [16].
Expression of the CRFergic System in the Central Nervous System and the Gastrointestinal Tract
In the brain, these peptides are expressed in distinct regions. CRF is essentially expressed by the neurons of the parvocellular part of the paraventricular nucleus of the hypothalamus (PVH) involved in the activation of the HPA axis, in the prefrontal and cingulate cortices which mediate behavioral and cognitive components of stress, as well as in the central nucleus of the amygdala (CeA) [182], a limbic structure involved in the processing of emotion [29]. CRF is also present in the Barrington’s nucleus, which is located adjacent to the locus coeruleus (LC) and regulates the sacral parasympathetic nucleus and thus the autonomic control of the rectum, left colon, and bladder [199]. CRF is also found in the hippocampus, bed nucleus of the stria terminalis, nucleus accumbens, several thalamic areas, substantia nigra, raphe, periaqueductal gray, cerebellum, and spinal cord.
Ucn 1 neurons are mainly localized in the Edinger–Westphal nucleus (EWN) and lateral superior olive of the pons in rodents, humans, and nonhuman primates and at a lesser extent in the olfactory bulb, supraoptic nucleus (SON), ventromedial and magnocellular regions of the PVH, lateral hypothalamic area, several cranial nerve motor nuclei, and in neurons of the ventral horn of the spinal cord in experimental animals and humans [25, 97]. In the rat, the highest density of Ucn 1 immunoreactive fibers is found in the lateral septum, with more moderate levels in the SON, PVH, periaqueductal gray, EWN, dorsal raphe, nucleus of the solitary tract (NTS), parabrachial nucleus, substantia nigra, and interpeduncular nucleus, as well as throughout the length of the spinal cord. The only Ucn 1 projections are those from the EWN to the spinal cord and lateral septum. There is little neuroanatomical overlap between the brain distribution of CRF and Ucn 1.
Ucn 2 mRNA is expressed in the PVH, SON, and arcuate nucleus of the hypothalamus and in the LC as well as in several cranial nerve motor nuclei and the ventral horn of the spinal cord [113, 152]. The neuroanatomical distribution of Ucn 2-containing fibers has not been characterized.
The distribution of Ucn 3 in the brain differs from that of CRF and Ucn 1 and Ucn 2. Ucn 3 is essentially located in the median preoptic nucleus, perifornical area between the fornix and the PVH, dorsal division of the medial amygdaloid nucleus, and superior paraolivary nucleus [107, 113]. Major Ucn 3 terminal fields are found in the forebrain, including the amygdala, the lateral septum, and the ventromedial hypothalamus.
CRF and Ucn 1 and Ucn 2 are localized in several stress-related nuclei involved in the modulation of GI functions and the integration of afferent signals from visceral origin, such as the PVH, SON, Barrington nucleus, and LC.
CRF ligands and receptors are also expressed in the GI tract in animals and humans where they can act directly on the gut in a paracrine or autocrine manner, thus supporting a role for peripheral CRF and/or Ucns in the modulation of GI functions [38, 47, 48, 143, 144, 178].
CRF is present in guinea pig cecal smooth muscle cells and in epithelial and submucosal immune cells of the GI tract as well as in epithelial cells of the colonic mucosa including endocrine (enterochromaffin) cells, in the myenteric and submucosal plexuses along the GI tract. Numerous CRF-containing nerve terminals reached the circular smooth muscle layer and submucosal arterioles. Ucn 1 has been described in the rat duodenum and in human gastric tissue, in parietal cells and others, as well as in the human colonic mucosa and in the submucosal and myenteric plexuses along the small intestine and colon of the rat. Ucn 2 is mainly distributed in the stomach, while low levels are observed in the small intestine and colon. In contrast, Ucn 3 is highly expressed in the gut, mainly in the stomach, small intestine, and colon, but not in the ileocecal region, cecum, and esophagus.
CRF receptors: CRF1 is present in the rat colon in goblet and stem cells of the crypts as well as on surface epithelial cells, lamina propria, and preferentially in the myenteric nervous plexus. CRF1 and CRF2 are expressed in the lamina propria of the human colon [133], particularly at the level of the colonic myenteric plexus [125]. CRF2a mRNA is expressed in human colonic epithelial HT-29 cells, with little expression of the b and c splice variants [95]. CRF2 receptors are highly expressed in the rat upper gut, including the esophagus and stomach.
Neuroanatomy of the Brain–Gut Axis (Figs. 7.2 and 7.3)

Regions where the activation of CRF receptors influences gastric and colonic motor functions through neural pathways innervating the gut (With permission from Taché and Bonaz [185]). AP area postrema, DMN dorsal motor nucleus of the vagus nerve, DVC dorsal vagal complex, ENS enteric nervous system, NTS nucleus tractus solitaries, PVN paraventricular nucleus of the hypothalamus (m magnocellular, p parvocellular), SPN sacral parasympathetic nucleus
The CNS and the gut communicate bidirectionally through the ANS (i.e., the brain–gut axis) and the circumventricular organs. The gut contains a “little brain,” i.e., the enteric nervous system (ENS), which contains as many as neurons as in the spinal cord (400–600 million) and can function independently of the CNS for the programming of motility and secretion [66]. Some neuropeptides and receptors are present in both the ENS and the CNS. The function of the GI tract is modulated by both the ENS and the ANS which is composed of the sympathetic (i.e., the splanchnic nerves) and parasympathetic nervous system, i.e., the vagus nerves (VN), the largest visceral sensory nerve in the body, and the sacral parasympathetic nucleus represented by the pelvic nerves, which are mixed systems. The VN contains 80–90 % of afferent fibers carrying information from the abdominal organs to the brain [4] with the exception of pelvic viscera for which information is transmitted to the sacral (S2–S4) spinal cord by the pelvic nerves with central projections similar to other spinal visceral afferents. The VN carries mainly mechanoreceptor and chemosensory information of the gut. Vagal afferents do not encode painful stimuli but are able to modulate nociceptive processing in the spinal cord and the brain [149]. The VN is described as the sixth sense based on how sensory vagal information is processed in the CNS [210]. The sympathetic nerves contain 50 % afferent fibers. Visceral afferents that enter via spinal nerves (i.e., splanchnic and pelvic nerves), at thoracic five–lumbar two segments of the spinal cord, carry information concerning temperature as well as nociceptive visceral inputs related to mechanical, chemical, or thermal stimulation through C and Aδ afferents, which will reach conscious perception. The afferent information of the ANS reaches the CNS at both the spinal cord, for the splanchnic nerves, and the NTS in the dorsal medulla, and sacral parasympathetic (S2–S4) for the VN and pelvic nerves, respectively. At the level of the spinal cord, sympathetic afferents are integrated at the level of laminae I, II outer, V, VII (indirectly), and X, and the information is then projected to the upper level through the spinothalamic and spinoreticular tracts, the dorsal column with projection to the thalamus (ventral posterolateral nucleus, intralaminar nucleus) and the cerebral cortex (insular, anterior cingulate, dorsolateral prefrontal cortex, etc.). At the level of the NTS, vagal afferent information is integrated according to a visceral somatotopy in subnuclei (e.g., medial, commissural, gelatinosus) [3] then projecting to the parabrachial nucleus, in the pons, according to a viscerotopic organization, which in turn sends projections to numerous structures in the brainstem, hypothalamus, basal forebrain, thalamus, and cerebral cortex [65]. Among the cerebral cortex, the insular cortex acts as a visceral (e.g., GI) cortex through an NTS–parabrachial–thalamocortical pathway according to a viscerotopic map. The insular cortex has connections with the limbic system (bed nucleus of the stria terminalis and CeA) and with the lateral frontal cortical system [165]. The NTS also sends projections to the ventrolateral medulla, the parvocellular part of the PVH, and the amygdala/bed nucleus of the stria terminalis contributing to visceral perception. The NTS receives convergent afferents from both the spinal cord (i.e., laminae I, V, VII, and X) and the VN; some of these afferents can be at the origin of autonomic reflex responses. This convergence is also observed at the level of the parabrachial nucleus and ventrolateral medulla [164], thus arguing for a relationship of pain with visceral sensations. The dorsal vagal complex (DVC), located in the brainstem, in the floor of the fourth ventricle, and the PVH are the best candidates for the action of stress and CRF/Ucns in the brain. The DVC is composed of the NTS, the dorsal motor nucleus of the VN (DMNV), which contains the perikarya of efferent vagal preganglionic neurons, and area postrema (AP), which is a circumventricular organ lying dorsally in the middle third of the rostrocaudal extent of the DVC. The PVH and DVC, as well as limbic structures, contain neurons bearing CRF2a [24] and are known to influence autonomic nervous outflow. The messages coming from the gut are integrated in the central autonomic nervous system which contains brain regions involved in the autonomic, endocrine, motor, and behavioral responses [164]. This brain network can be divided into executive structures, mainly hypothalamic, and coordinating structures, mainly included in the limbic system, and high level control structures, mainly the frontal cortex. The LC and the Barrington nucleus are also important parts of this network. The LC, the largest group of noradrenergic neurons located in the pons, mediates emotional arousal, autonomic, and behavioral responses to stress and attention-related processes through its dense projections to most areas of the cerebral cortex [60]. The Barrington nucleus is ventrolaterally located to the LC and projects to the sacral parasympathetic nucleus to increase the motility of the distal recto-colon through an LC-Barrington nucleus interaction [198]. The central autonomic nervous system, in turn, adapts the response of the digestive tract through the efferent ANS (the VNs, the sacral parasympathetic pelvic nerves, and the splanchnic nerves) through reflex loops which are essentially unconscious or become conscious in pathological conditions. Descending pathways that control somatic as well as visceral pain by modulating at the spinal cord-level visceral informations have been described. These pathways are both inhibitory, thus producing analgesia as represented by projections from the periaqueductal gray to the rostroventral medulla and LC descending fibers to the spinal cord, and facilitatory producing hyperalgesia (rostroventral medulla and OFF and ON cells) [195]. Many of the regions of the CNS that we have described above contain the CRFergic system. Among them, the PVH, Barrington’s nucleus/LC, and amygdala are well positioned to participate in the reciprocal brain–gut interactions as it pertains to sensory information from the colon and reflex behavioral and autonomic responses to the viscera.
The circumventricular organs are highly vascularized structures with fenestrated capillaries located around the third and fourth ventricles and characterized by the lack of a blood–brain barrier; they represent points of communication between the blood, the brain, and the cerebrospinal fluid [17]. They are represented by the subfornical organ, median eminence, pineal gland, area postrema, and organum vasculosum of the lamina terminalis. They are involved in sodium and water balance, cardiovascular regulation, metabolic and energetic balance, immune function, regulation of body temperature, vomiting, and reproduction.
Stress Effect on Gastrointestinal Functions
Motility and Secretion (Fig. 7.3)
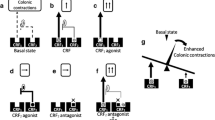
Acute stress is currently known to delay gastric emptying and small bowel transit while increasing colonic motility in experimental animals as well as in healthy humans [185, 188]. The effects of stress on gastric emptying are reproduced by central (intracerebroventricular or intracisternal) injections of CRF or Ucns, mainly performed in rodents and dogs, and are prevented by central (intracerebroventricular or intracisternal) injection of either nonselective CRF1/CRF2 antagonists (α-helical CRF9 − 41, d-Phe12CRF12 − 41, astressin, astressin-B) or selective CRF2 antagonist (astressin2-B), while selective CRF1 antagonists (CP-154,526, antalarmin, NBI 27914) have no effect, thus arguing that CRF and Ucns act primarily through interaction with CRF2 receptors [185]. In contrast to gastric motor function, few data are available about the effects of stress on small intestinal motility, the role of brain CRF/urocortins and their receptors being often studied in parallel with those observed in the stomach. Central injection of CRF and Ucn 1 induces inhibition of duodenal, small intestinal transit, and propulsive motility in rats and dogs [39, 93]. Central injection of CRF1 preferential agonists such as ovine CRF, rat/human CRF, and Ucn 1 induces increase in colonic transit and defecation [33]. Central injection of Ucn 2 and 3, selective CRF2 agonists, is ineffective in mice at a dose similar to that of CRF [116], while this effect is blocked by central administration of either nonselective CRF1/CRF2 receptor antagonists or selective CRF1 antagonists [33, 115]. In addition, central or peripheral injection of a selective CRF1 antagonist reduces stress-induced increase of colonic transit time. CRF1 invalidated mice have significantly less defecation in an open field test than their wild-type littermates [13, 115, 188]. Central injection of a selective CRF2 antagonist (astressin2-B), at a dose which inhibits the CRF2-induced delay in gastric emptying, is not able to reverse the increase in colonic transit following central injection of CRF in rodents [188], thus confirming that CRF1 is the receptor involved in stress-induced colonic motility disturbances.
Numerous data have established the involvement of peripheral CRF signaling in the modulation of secretory function under stress conditions via activation of both CRF1 and CRF2 receptors and activation of cholinergic enteric neurons, mast cells, and possibly serotonergic pathways [101].
The HPA axis is not involved in stress-induced inhibition of gastric emptying. These effects are mediated by the ANS. The effect of CRF and Ucn 1 is mediated by the VN and abolished by vagotomy [93, 103, 130]. The effect of Ucn 2 requires the integrity of the sympathetic nervous system and peripheral α-adrenergic receptors while not altered by vagotomy [54]. The effect of stress-induced CRF1-dependent activation of colonic motility is mediated by the parasympathetic nervous system, i.e., the celiac branch of the VN, which innervate the proximal colon, and the sacral parasympathetic fibers, which innervate the distal part of the colon and the rectum [128, 130]. The intrinsic nervous system of the colon, as represented by myenteric cholinergic and nitrergic neurons, is involved in the effector mechanism of parasympathetic activation, as well as the action of serotonin (5-HT), from enterochromaffin cells and/or enteric neuron origin, on 5-HT3 and 5-HT4 receptors. The PVN and LC/Barrington’s complex are involved in central CRF1 signaling-induced increase in colonic motor function. Changes in c-fos and CRF gene expression in the Barrington nucleus have been reported under acute and chronic stress [86], and CRF-synthesizing neurons in the Barrington nucleus project to the noradrenergic LC as well as to the intermediolateral column of the sacral spinal cord, which contains the sacral parasympathetic nucleus innervating the descending colon [197]. There are also CRF efferent projections directly from the PVH to the LC, thus modulating the activity of LC neurons and integrating autonomic responses in brain by influencing LC neurons [151]. Water avoidance stress activates the PVN and LC/Barrington’s nuclei and CRF gene transcription in the PVH, whereas icv injection of the nonselective CRF1/CRF2 antagonist α-helical CRF9 − 41 reduces Fos expression in these nuclei in correlation with defecation score [33]. In agreement with a role for CRF/CRF1 signaling in the PVN in stress-induced increase in colonic motility, α-helical CRF9 − 41 injected directly into the PVN blocks partial restraint- and water avoidance-induced stimulation of colonic transit and defecation, and various neurogenic and systemic stressors activate the transcription of the CRF1 receptor gene in the PVN ([31]; 130). CRF increases the firing rate of noradrenergic neurons in the LC and releases noradrenalin into the brain prefrontal cortex, an input of the LC, which results in arousal and anxiogenic behavior [198]. Consequently, CRF/CRF1 signaling pathways in the PVN and LC/BN complex may physiologically regulate the behavioral and autonomic responses to stress that influence colonic function as part of the brain–gut axis [129, 198]. These CRF/CRF1 signaling pathways may play a role in diarrhea-predominant IBS patients with psychic comorbidities such as anxiety and depression [115].
Intestinal Permeability
The digestive tract is characterized by an epithelial monolayer essentially composed of enterocytes and colonocytes in the small bowel and colon, respectively, and to a lesser extent mucin-secreting goblet cells, enteroendocrine cells, and others such as intraepithelial lymphocytes. Its integrity is preserved by both cell polarity and the interactions between the cell adhesion complexes, represented by tight junctions and adherens junctions, with the actin cytoskeleton [79]. In physiological conditions, a limited quantity of antigens goes through the epithelial barrier, playing an important role in the induction of normal immune tolerance toward foreign food antigens and the resident microbiota [88]. The gut is the largest immune organ with the majority of immune cells present in the mucosa. Consequently, a mucosal barrier function is necessary to separate the luminal compartment from the body proper and control antigen-induced inflammation. Any anatomical or functional damage to the intestinal epithelium may allow access of luminal contents to the mucosa then stimulating an inflammatory response in a susceptible host, as observed in IBD, IBS, and celiac disease. This mucosal barrier function is regulated by neuroendocrine and immune factors; thus, it is not surprising that stress, and the CRFergic system, can have a major impact. In contrast to the effects of stress on GI motility, fewer studies have evaluated its effect on intestinal permeability. However, abnormal intestinal permeability has been described in IBS [40] and IBD [175], two GI disorders where stress has a role in their pathogeny [27, 132]. The influence of stress on mucosal function has been recently recognized in short acute stress, early life trauma, and chronic ongoing psychological stress in rats [174]. Indeed, restraint stress or water avoidance stress performed in Wistar–Kyoto rats, a stress-sensitive strain, induces ion secretion, a decrease in barrier function, and an enhancement of small (e.g., proinflammatory bacterial tripeptides) and large molecules (e.g., horseradish peroxidase, HRP) across the epithelium through the transcellular and paracellular pathways [94, 167]. These effects are reproduced by ip injection of CRF 1h before stress [163] and blunted by ip injection of the specific nonselective CRF receptor antagonist, alpha-helical CRF9–41 as well as by pretreatment of the animals with adrenergic and cholinergic antagonists and a mast cell stabilizer. Acute immobilization stress or iv injection of CRF increases colonic mucin release through cholinergic-, adrenergic-, and mast cell-dependent mechanisms [43]. Immobilization stress induces increase in mast cell protease II and mucine release from colonic explants; these effects are prevented by peripheral administration of alpha-helical CRF9-41 [43]. The effects of acute stress on permeability returned to normal within 24 h. These data argue for a complex mechanism of action involving peripheral CRF pathways, mast cell activation, mucine secretion, and increase of intestinal permeability. Mast cells regulate barrier physiology in normal as well as inflamed intestine in rats and humans [19]. Despite convincing evidence of intestinal expression of CRF-like peptides and receptors and its production by local immunocytes and enteroendocrine cells [47, 185], only recently a role for subepithelial CRF–mast cell loops in the regulation of colonic permeability in human biopsies has been reported [204]. Santos et al. [162] have recently shown that the stress-like peptides CRF and sauvagine stimulated ion secretion and macromolecular permeability in the distal colon of WKY rats in vitro. Epithelial responses were inhibited by both the nonselective CRF receptor antagonist astressin and the mast cell stabilizer doxantrazole and significantly reduced in tissues from Ws/Ws rats. These data argue for the involvement of mucosal/submucosal CRF receptors through mast cell-dependent pathways. Neonatal Sprague–Dawley rats, a strain not normally sensitive to stress in contrast to Wistar–Kyoto rats, separated from their mothers from days 2 to 21 for 4 h per day then housed normally until day 60 and then submitted at day 90 to either water avoidance stress or sham stress for 30 min, present an increase of permeability to macromolecules but not in controls [173]. More recently, CRF2 signals through Src/ERK pathway induce the alteration of cell–cell junctions and the shuttle of p120ctn and Kaiso in the nucleus. In HT-29 cells, this signaling pathway also leads to the remodeling of cell adhesion by the phosphorylation of focal adhesion kinase and a modification of actin cytoskeleton and focal adhesion complexes [58].
Gastrointestinal Inflammation
Numerous data argue for a direct autocrine/paracrine effect of CRF on inflammation. CRF reaches the site of inflammation either through nerve terminals or by local release of the peptide by epithelial, endothelial, or resident immune cells. CRF, Ucns, and their receptors are present in mast cells, mouse splenocytes, lymphocytes, monocytes–macrophages, and TH cells [73]. CRF has an opposite effect at the central and peripheral level. Indeed, central CRF has an anti-inflammatory role through the activation of the HPA axis and the release of glucocorticoids, while peripheral CRF acts as a proinflammatory mediator [147, 205]. CRF signals play a role in augmenting LPS-induced proinflammatory cytokine production by macrophages through CRF1 since these effects are inhibited by antalarmin [1]. Peripheral mechanisms by which CRF and Ucn are involved in the development and progression of colitis are not completely clear. Several factors seem involved: the type of inflammatory stimulus, intestinal cell phenotype, type of CRF receptor, and phase of colitis (acute versus chronic). CRF2 and its specific ligand, Ucn 2, appear to mediate proinflammatory responses in the gut. Indeed, CRF-deficient mice develop substantially reduced local inflammatory responses [68] and have dramatically reduced ileal fluid secretion, epithelial cell damage, and neutrophil transmigration 4 h after intraluminal C. difficile toxin A [5]. This effect is counterbalanced by administration of the selective CRF2 antagonist astressin 2B. The CRF system is also involved in the inflammatory response associated with IBD [74]. Colonic biopsies from patients with active UC show significantly increased CRF immunoreactive lamina propria mononuclear cells and macrophages [92]. In UC patients without glucocorticoid treatment, Ucn 1-positive cells and plasma cells increased in proportion to the severity of inflammation but are significantly lower in number in glucocorticoid-treated patients. Ucn 1 mRNA was expressed in lamina propria plasma cells, and both CRF1 and CRF2a mRNAs were also partially coexpressed in these cells and macrophages. Ucn 1 therefore may act as a possible local immune-inflammatory mediator in UC [166]. Ucn 2 mRNA is expressed in normal conditions in the small intestine and colon, while in a rat model of chemically induced colitis, Ucn 2 levels are increased, whereas expression levels of its only identified receptor, CRF2, are decreased. This suggests that Ucn 2 exerts its effects only in part via CRF2 [44]. The endogenous upregulation of Ucn 2 following the local inflammatory process accounts for the downregulation of the respective receptor, being either a causing or a resulting effect. Exposure of human colonic epithelial cells HT-29 to C. difficile toxin A or TNF-alpha induces an increase expression of CRF2 mRNA and protein and stimulation of NCM460 colonocytes overexpressing CRF2a with Ucn 2 results in a time- and concentration-dependent increase in IL8 production as well as to an activation of NF-kappaB and MAP kinase in these cells. In addition, expression of Ucn 2 and CRF2 mRNA was increased in mucosal samples of patients with IBD (CD and UC) and after exposure of human intestinal xenografts to C. difficile toxin A. These data suggest that Ucn 2 has proinflammatory effects in human intestinal cells via CRF2a and may be involved in the pathophysiology of colitis in humans [131]. CRF2-deficient mice develop substantially reduced intestinal inflammation and had lower intestinal mRNA expression of the potent chemoattractant keratinocyte chemokine and MCP 1 when exposed to intraluminal C. difficile toxin A. This effect is mimicked by the selective CRF2 antagonist astressin 2B before toxin A exposure. Only Ucn 2, but not other Ucn, was significantly upregulated by ileal administration of toxin A at 4 h compared with buffer exposure [95]. These data argue for CRF2-mediated intestinal inflammation via the release of proinflammatory mediators at the colonocyte level. La Fleur et al. [99] have shown that, in an experimental model of toxin-induced intestinal inflammation, inhibition of CRF ablated the inflammatory response, while Ucn 2 dsRNA treatment did not modify the inflammatory response to toxin. Gonzales-Rey et al. [71] investigated the potential therapeutic effect of Ucn 1 in a murine model of TNBS-induced colitis and showed that Ucn 1 significantly improved the clinical and histopathological severity of colitis and increased the survival rate of mice with colitis. Importantly, Ucn 1 treatment was effective in established colitis and avoided recurrence of the disease. This work identifies Ucn 1 as a potent anti-inflammatory factor. Few data are available regarding the role of the CRFergic system as a pro- or anti-inflammatory system at the level of the stomach. In the upper GI, CRF2 appears to be the most prominent. Indeed, CRF1- and CRF2-positive cells are present in the oxyntic gland and the submucosal blood vessels. No specific CRF1 is observed in the antrum. CRF2 is present in the gastric glands along with immunoreactive Ucn. Thus, both CRF receptor subtypes are expressed in the upper GI tissues with a distinct pattern and regional differences suggesting a differential function. A paracrine CRF-like circuit is present in human stomach composed of Ucn and its CRF2 receptor compared to the CRF/CRF1 circuit in the human colon. Chatzaki et al. [49] examined the presence of CRF and Ucn transcripts and peptides in human gastric mucosa and the association between CRF and Ucn and H. pylori gastritis. They observed the presence of the Ucn transcript in biopsies obtained by gastroscopy from normal and inflamed gastric mucosa, while the CRF transcript was not detectable. Immunoreactive Ucn was localized in gastric epithelial cells and in inflammatory elements of the surrounding negative for Ucn gastric stroma. The level of immunoreactive Ucn was higher in gastric biopsies from patients with active H. pylori gastritis than in controls. Eradication of H. pylori was followed by a dramatic increase of ir-Ucn levels, while nonresponders to the eradication treatment did not show any significant change in ir-Ucn levels. These data suggest that in human gastric epithelium, Ucn is present and plays an important physiological role, while CRF is absent. In addition, and in contrast to what has been observed for CRF in ulcerative colitis [92], a highly significant but negative correlation has been found between Ucn levels and gastric inflammation, suggesting that Ucn may exert an anti-inflammatory effect in the gastric mucosa. The Ucn/CRF2 system in the upper gastrointestinal tract plays a completely different role in gastric inflammatory response compared with that of the CRF/CRF1 system in the inflammatory response in the colon. These differences between upper and lower GI tracts, vis-à-vis their CRF paracrine circuits, may partially explain the lack of severe chronic inflammatory diseases in the stomach compared with the colon in IBD.
Microbiota
There are 10–100 trillion bacterial cells in our gut, ten times more than the number of somatic and germ cells in our body. The number of species is estimated between 500 and 800, and eighty percent of these species are unculturable [59]. New molecular techniques allow inventory of the gut’s resident bacterial species without having to culture them. Bacterial genes outnumber our body’s genes by as much as 100–1 [7]. The human microbiota is necessary to the healthy functioning of the GI tract and contributes to the intestinal development, metabolic transformations, and protections against enteric infections [75]. Bacteria in the gut have an important role in the immune response, including inflammation [106]. There is a new concept on the bidirectional communication between the nervous system and commensal, pathogenic, and probiotic organisms, i.e., the microbiota–brain–gut axis where gut microorganism can activate the VN; this activation plays a critical role in mediating effects on the brain and behavior. The VN appears to differentiate between nonpathogenic and potentially pathogenic bacteria even in the absence of overt inflammation and mediates signals that can induce both anxiogenic and anxiolytic effects, depending on the nature of the stimulus [62]. Mice treated orally with Campylobacter jejuni showed vagally mediated activation in the NTS in the absence of intestinal inflammation [70]. This axis is vital for maintaining homeostasis and may be involved in the etiology of several metabolic and mental dysfunctions/disorders. Commensal microbiota can affect the postnatal development of brain systems involved in the endocrine response to stress. Indeed, an exaggerated response of the HPA axis to stress (higher plasma ACTH and corticosterone elevation) was observed in germ-free mice that was reversed by reconstitution of the microbiota; germ-free mice also exhibited reduced brain-derived neurotrophic factor (BDNF) expression levels in the cortex and hippocampus [181]. The exaggerated HPA stress response by germ-free mice was reversed by reconstitution with Bifidobacterium infantis. Thus, commensal microbiota can affect the postnatal development of the HPA stress response in mice. Prevention of intestinal barrier impairment by a probiotic attenuates HPA response to an acute psychological stress in rats [2]. In addition, germ-free mice show reduced anxiety-like behavior in comparison to specific pathogen-free mice, a phenotype accompanied by changes in plasticity-related genes in the hippocampus and amygdala [136], two key structures in the adaptation to the stress response. The intestinal microbiota influences central (i.e., hippocampal) levels of BDNF, which regulates dendritic architecture and spines, and behavior independently of the ANS, gastrointestinal-specific neurotransmitters, or inflammation [18]. Consumption of a fermented milk product with probiotic for 4 weeks by healthy women altered brain intrinsic connectivity or responses to emotional attention tasks [192]; thus, neuroimaging seems to be an interesting tool to study the microbiome–gut–brain axis. In regular functioning conditions, the intestinal barrier is able to prevent most environmental and external antigens to interact openly with the numerous and versatile elements that compose the mucosal-associated immune system. Stress is well known to induce an increase of intestinal permeability that allows bacteria to cross the epithelial barrier to activate mucosal immune response (Killiaan et al. 1998) and to translocate to secondary lymphoid organs [10], thus stimulating the innate immune system. Stress is able to modify the intestinal microbiota [9]. Indeed, exposure of mice to a social stressor affects the structure of the intestinal microbiota and increases the circulating level of cytokines; this effect is reversed by antibiotics [8]. Changes in the intestinal microbiota reduce resistance to infectious challenge with intestinal pathogens [9]. These data provide evidence for the interplay between stress, the intestinal microbiota, and the immune response. This can in turn have significant impact on the host and affect behavior, visceral sensitivity, and inflammatory susceptibility [52]. The sympathetic nervous system, through the release of catecholamines, e.g., norepinephrine, a stress mediator, stimulates growth of bacteria [111]. Stress-mediated changes may shift the microbial colonization patterns on the mucosal surface and alter the susceptibility of the host to infection; these changes in host–microbe interactions may also influence neural activity in stress-responsive brain areas [110].
Visceral Sensitivity
Stress increases visceral perception and emotional response to visceral events by a perturbation of the brain–gut axis at its different levels, central (brain and spinal cord), peripheral (gut), and the ANS [100]. Genetic models of depression and anxiety, such as the high-anxiety Wistar–Kyoto rats or Flinders Sensitive Line, rats [139] or deleting CRF1, exhibit a decrease of colonic sensitivity to colonic distension [194] while models overexpressing CRF1 exhibit enhanced response [127]. These data argue for the filiation stress–anxiety–inflammation and visceral hypersensitivity. CRF signaling, both at the central and peripheral level, is a key element involved in this effect. Data argue for an equally important contribution of the peripheral CRF/CRF1 signaling locally expressed in the gut to the GI stress response [101]. Indeed, mast cell degranulation in the colon under stress and peripheral administration of CRF induces visceral hypersensitivity through the release of their mediators (histamine, tryptase, prostaglandin E2, nerve growth factor, CRF, TNF) that can stimulate or sensitize sensory afferents [201].
Among the brain structures involved in stress-induced visceral hypersensitivity, the amygdala, a major extrahypothalamic source of CRF, is a key element. Indeed, an activation of the amygdala is observed in experimental models of somatovisceral and visceral pain [171, 172] as well as experimental models of stress-induced GI disturbances [31–33] and colitis [145] as well as in a model of visceral pain in healthy volunteers [6] and IBS patients [30]. Activation of corticosteroid receptor in the CeA is involved in the induction of anxiety and visceral hypersensitivity [135]. Implants of corticosterone micropellets in the CeA increase anxiety-like behavior as well as visceral hypersensitivity to colonic distension and increased responsiveness of viscera-sensitive lumbosacral spinal neurons that mediate visceromotor reflexes to colorectal distension [134]. Water avoidance stress performed for 7 consecutive days induced visceral hypersensitivity that is abolished by glucocorticoid receptor and mineralocorticoid receptor antagonists in the amygdala. Wistar–Kyoto rats express a greater amount of CRF and CRF1 mRNA in the CeA and PVH [36] and depict colonic hypersensitivity to luminal distension which is reversed by peripheral administration of a CRF1 antagonist as well as into the CeA, thus strengthening the role of CRF1 receptor in the amygdala in visceral hypersensitivity mechanism [90]. CRF neurons in the CeA project directly to the LC and increase the firing rate of LC neurons, thus increasing noradrenaline release in the vast terminal fields of this ascending noradrenergic system. The expression of CRF in the LC is increased in WKY rats, and a selective CRF1 receptor antagonist dampened the activation of LC neurons by colorectal distension and intracisternal CRF in rats [96]. CRF1 and CRF2 in the amygdala mediate opposing effects on nociceptive processing, i.e., pro- and antinociceptive effects of CRF, respectively [89]. Low concentrations of CRF facilitate nociceptive processing in the CeA neurons through CRF1, while higher concentrations of CRF have inhibitory effects through CRF2 receptors, in agreement with the concept that CRF2 receptors serve to dampen or reverse CRF1-initiated responses [185]. These results clarify to some extent the controversial role of CRF in pain modulation. Administration of CRF into the CeA significantly increased the number of abdominal muscle contractions in response to colorectal distension in male Wistar rats; this effect was dampened by injection of the CRF1 antagonist, CP-154526. Colorectal distension increased noradrenaline in the CeA which was further increased by CRF and inhibited by CRF1 antagonist. These data suggest that CRF in the CeA sensitizes visceral nociception via CRF1 with release of noradrenaline [180]. The insular cortex is also a key region of the pain matrix, more particularly involved in visceral pain (i.e., colorectal distension) [6, 30]. Bilateral insular cortex lesions in rats markedly inhibit visceral hypersensitivity induced by chronic stress, thus strengthening its role in the pathophysiology of stress-related visceral hypersensitivity [208]. Chronic stress increases DNA methylation and histone acetylation of genes that regulate visceral pain sensation in the peripheral nervous system of rats. These results have potential therapeutic implications when blocking epigenetic regulatory pathways in specific regions of the spinal cord [84].
A temporary disruption of the gut microbiota in early life induces specific and long-lasting changes in visceral sensitivity in male rats, a hallmark of stress-related functional disorders of the brain–gut axis such as IBS [138]. Early life adversity is known to induce visceral hypersensitivity through ovarian hormones, specifically estradiol, and signaling within the HPA axis, either through reduced negative feedback or increased facilitation, with specific changes in amygdala-mediated mechanisms. Stress-induced visceral hypersensitivity following maternal separation is transferred across generations, this transfer depending on maternal care [200].
Implication in Functional Digestive Disorders and Inflammatory Bowel Diseases
Functional Digestive Disorders: Irritable Bowel Syndrome (Fig. 7.4)

The relationship between early life, psychological factors, physiology, subjective experience of symptoms, behavior, and outcome in irritable bowel syndrome (With permission from Mulak and Bonaz [132])). CNS central nervous system, ENS enteric nervous system, IBS irritable bowel syndrome, MD medical doctor
IBS is the most common functional digestive disorders, with an estimated prevalence rate in the general population of 10–15 % in industrialized countries. IBS is characterized by abdominal pain, bloating, and altered bowel habits without any organic cause [132]. Women have a higher prevalence of symptoms than men. IBS accounts for up to 12 % of visits to primary care doctors and 28 % of visits to gastroenterologists [41]. IBS is associated with a significant impairment in quality of life, a high rate of absence from work, and a significant increase in healthcare costs. Extraintestinal manifestations are frequently associated to digestive symptoms such as headache, arthralgia, urinary manifestations, insomnia, and fatigue. Fibromyalgia is often observed in IBS and conversely [46]. Psychiatric comorbidity, mainly major depression, anxiety, and somatoform disorders, is observed in 20–50 % of IBS patients [67]; stress is involved in such disorders and psychiatric disorders precede the onset of the GI symptoms [184]. Numerous data argue for a role of stress in the pathophysiology of IBS. Patients with IBS report more stressful life events than medical comparison groups or healthy subjects. Stress is strongly associated with symptom onset and symptom severity in IBS patients. Illness experience, healthcare-seeking behavior, and treatment outcome are adversely affected by stressful life events, chronic social stress, anxiety disorders, and maladaptive coping style. A history of emotional, sexual, or physical abuse is found in 30–50 % of IBS patients [35]. A majority of patients with IBS have a visceral hypersensitivity as represented by lower pain thresholds to intestinal distension compared to healthy controls [153]. Among the peripheral mechanisms of this visceral hypersensitivity, low-grade inflammation in the GI tract could favor modifications of neuronal plasticity [37, 119] and mast cells could also be involved by sensitizing visceral afferent terminals [76]. A postinfectious IBS has been observed in 4–30 % following bacterial gastroenteritis [78]; perceived stress, anxiety, somatization, and negative illness beliefs at the time of infection in favor of a cognitive–behavioral model of IBS were predictors of postinfectious IBS [176]. Modifications in central sensory processing are described in IBS [30]. A spinal hypersensitivity has been evoked in IBS patients as well as a supraspinal cause where stress is of primary importance [132]. A defect of descending pain inhibition pathways is also evoked [206], as described in fibromyalgia patients [102]. Globally, IBS is assimilated to a central sensitization syndrome as observed for chronic fatigue syndrome, fibromyalgia, posttraumatic stress disorders, headaches, restless legs syndrome, and others [209].
Early life traumas, known to increase the risk of developing IBS later in life, play a major role in the development of mood and anxiety disorders and increased CRF signaling [81]. Neonatal maternal separation (MS), as an early life trauma, is a model of IBS in rodents, leading to chronic dysregulation in the limbic–HPA axis [150]. This has led to the biopsychosocial model of IBS [189] and the concept that IBS is due to a brain–gut axis dysfunction consistent with an upregulation in neural processing between the gut and the brain. Globally, there is a hypervigilance state that explains the visceral hypersensitivity observed in these patients most likely through a central (and peripheral) hyper-CRFergic state. In addition, pain is a stress per se that could amplify such disturbances, contributing to its chronicity. Since the ANS is the link between the gut and the brain, it is not surprising in this context of an abnormal brain–gut axis to observe an important dysautonomia, with a high sympathetic and a low parasympathetic tone, whatever the positive or negative affective adjustment [142].
Functional brain imaging studies have allowed a better understanding of the pathophysiology of IBS. Indeed, an abnormal brain processing to visceral pain has been described in IBS patients [30, 193] particularly in brain loci of the pain matrix such as the somatosensory, insular, prefrontal, and cingular cortices as well as subcortical loci such as the thalamus, the amygdala, and the periaqueductal gray. In addition there is a major influence of cognitive–affective processes, including arousal, attention, conditioning and negative affect, and coping strategies, on GI sensations, and its central correlates in health and IBS [118, 142, 202]. The role of the central and/or peripheral CRF system is gaining clinical recognition as part of the neurobiological common denominator of IBS symptoms susceptible to stress and anxiety/depression [63, 186]. Elevated concentrations of CRF in the CSF are observed in patients with anxiety and vulnerability to stress as well as in those suffering from obsessive–compulsive disorders, posttraumatic stress disorders, or childhood trauma, and CRF in the CSF is a predictor of perceived aversive early life experiences [105]. In patients suffering from fibromyalgia, known to have comorbidity with IBS, CSF levels of CRF are associated with both pain symptoms and autonomic dysfunction [122]. An overactivity of the HPA axis and enhanced plasma CRF response to mental stress has been described in IBS patients [146]. IBS patients have previously been proposed to have an exaggerated brain–gut response to CRF [64]. Basal levels of noradrenaline are higher in IBS patients than in controls [82] indicating enhanced activity of the sympathetic nervous system, known to selectively increase visceral sensitivity [87]. An alteration in central noradrenergic signaling is observed in IBS; early life trauma may be one mediator of these abnormalities [20]. In healthy controls, CRF iv decreases rectal pain threshold to distension and mimics an IBS-specific visceral response [137]. Peripheral injection of α-helical CRF9 − 41 prevents rectal electrical stimulation-induced enhanced sigmoid colonic motility, visceral perception, and anxiety in IBS patients compared to controls without altering the HPA axis [161] and improves decreased alpha power spectra and increases beta power spectra of electroencephalogram in IBS patients [190]. The induction of IBS-like symptoms in healthy subjects and heightened sensitivity in IBS patients are alleviated by a peptide CRF antagonist targeting CRF1 receptor [63] that may provide a new therapeutic avenue in the treatment of IBS [115]. However, in women with diarrhea-predominant IBS, a CRF1 antagonist did not significantly alter colonic or other regional transit or bowel function, thus requiring further study [183].
The medical treatment of IBS is disappointing, often targeting symptoms. In the meantime, because of the complexity of the pathophysiology of IBS, one might wonder that such an ideal treatment would not exist. Nonmedical treatments, as represented by cognitive–behavioral therapy or hypnosis, are of interest [61, 104].
Inflammatory Bowel Diseases (Fig. 7.5)

Actors and pathways through which stress may play a role in the pathophysiology of inflammatory bowel disease (With permission from Bonaz and Bernstein [27]). HRV heart rate variability, VN vagus nerves, SN sympathetic nerves
IBDs are organic diseases classically divided in CD and UC involving the digestive tract, and particularly the small bowel and colon, starting early in life (between 15 and 30 years), and evolving by flares alternating with periods of remissions of variable duration. Symptoms are characterized by abdominal pain, diarrhea, fever, weight loss, and extraintestinal manifestations. The rising incidence of IBD in Western countries supports the hypothesis that “Westernization” of our lifestyle has led to the increased incidence of IBD. Today, there is no medical treatment susceptible to cure definitively IBD; the treatment is only suspensive. The pathophysiology of IBD is multifactorial involving immunological, genetic, infectious, and environmental factors [85]. Among the latter, the role of stress is evoked based on experimental and clinical data [27]. IBDs are models of “brain–gut” interactions; they represent an interoceptive (immunogenic) and exteroceptive (psychological) stress involving the neuroendocrine–immune axis. The ANS has a key role in the relation between stress and digestive inflammation. A dysautonomia is reported in IBD, as represented by a sympathetic dysfunction in CD [109] and a vagal dysfunction in UC [108]. This dysautonomia could explain the differential effect of tobacco, nicotine being a parasympathetic activator, which is protective in UC and deleterious in CD. Stress may play a deleterious role in IBD through different pathways close to the ones described for IBS [27] (Fig. 7.5): (1) Activation of mast cells in the intestinal mucosa, in close contact with sympathetic and VN terminals, induces the release of their mediators (see above) that increase intestinal permeability and activate the mucosal immune function [23, 187]; (2) Catecholamines, acting through α- and β-adrenergic receptors, mediate stress-induced increases in peripheral and central inflammatory cytokines and activation of the inflammatory nuclear factor-kB signaling pathway [91]. Classically, the SNS has a proinflammatory role [120], (3) The VN has a dual anti-inflammatory effect both through its afferents, activating the anti-inflammatory HPA axis, and efferents through the cholinergic anti-inflammatory pathway [28]. Indeed, acetylcholine (ACh) released at the distal end of VN efferents decreases the production of proinflammatory cytokines such as TNF by human macrophages through alpha7 nicotinic ACh receptor (a7nAChR) expressed by macrophages [140]. VN stimulation (VNS) has been shown to reduce the systemic inflammatory response to endotoxin [34] and intestinal inflammation [57, 124] and could be a nonpharmacological treatment of IBD [28, 51]. The VN also modulates the immune activity of the spleen either directly through connections with the splenic sympathetic nerve [159] or indirectly [114]. Stress has a proinflammatory effect based on its activation of the SNS and adrenomedullary activity while inhibiting the VN [185, 207]. (4) The activity of the sympathovagal balance and the HPA axis, monitored by heart rate variability and cortisol, is linked to the activity of the prefrontal cortex (PFC) and amygdala, respectively [191]. The hypoactivity of the PFC and the enhancement of amygdala activity are strongly influenced by stress [53]. A dysregulation of the amygdala–PFC equilibrium induces an imbalance between the HPA axis and the ANS, as observed in IBD [179], and thus a proinflammatory condition. There is an imbalance between the HPA axis and the vagal tone in CD patients with an inverse association between vagal tone and TNF-alpha level [141], (5) Habituation of the hypothalamic CRFergic system is observed in chronic stress conditions [31]. Chronic colitis suppresses CRF gene activation in the hypothalamus and plasma corticosterone level and dampens the counter-regulatory anti-inflammatory mechanisms during water avoidance stress, thus contributing to the stress-related worsening of colitis [98]. A predisposition to a hyporeactive HPA axis and an inhibition of the central response to a chronic interoceptive stress may favor inflammation in IBD, (6) As described above, the CRFergic system is present in the GI tract and may play an anti- or proinflammatory role. The peripheral CRFergic system forms an interacting and balanced system, and the differences observed among studies depend on the model of inflammation and the receptors activated and the ligands; an imbalance of this system could favor GI inflammation, (7) There is a microbial basis of IBD [55]. Stress-induced increased intestinal permeability allows bacteria to cross the epithelial barrier to activate mucosal immune response [94] and to translocate to secondary lymphoid organs [10] to stimulate the innate immune system. The sympathetic nervous system, through the release of catecholamines (e.g., norepinephrine), stimulates growth of bacteria [111]. The intestinal microbiota may act as a mediator in the communication between the gut and the brain (i.e., the microbiota brain–gut axis), (8) Modification of the stress axis early in development, as observed in early life traumas, could induce a maladaptive control of neuroendocrine immune axis. Indeed, the HPA axis is programmed by early life events, and neonatal inflammatory stimuli exert long-term changes in HPA activity and immune regulation in adult animals [169]. Experimental colitis induces a significantly higher inflammatory reaction in MS animals [14]. Deficient maternal care in rats increases glucocorticoid receptor promoter methylation leading to decreased expression in the hippocampus, a recognized target for glucocorticoid feedback [123]. There is a link between early life stress and depression that may predispose to increased inflammation both under baseline conditions and following stress [56]. MS mice develop a behavioral pattern reminiscent of depression and are more susceptible to inflammation; this vulnerability is reversed by tricyclic antidepressants [203]. Experimental depression in mice is followed by impaired parasympathetic function and increased susceptibility to an experimental colitis that was reduced by desmethylimipramine, through a vagally dependent enhanced parasympathetic function [69]. Most of the data presented above are described in experimental conditions. However, there are now increasing data arguing for a role of stress in IBD patients. There is consistent evidence that psychological factors play a role both in the pathophysiology and course of IBD and in how patients deal with IBD. In a population-based cohort of IBD patients, significantly more people in the persistently inactive disease group indicated they had experienced no stressful events compared to those in the persistently active disease group. Only the psychological factors, including occurrence of a major life event, high perceived stress, and high negative mood during a previous 3-month period, were significantly associated with the occurrence of a flare [21]. On multivariate logistic regression analyses of these variables, only high perceived stress was associated with increased risk of flare. Perception of stress is a key factor, which incorporates the individual’s appraisal of the demands created by stress in general and resources to cope with stress. The interaction between perceived stress and avoidance coping was predictive of earlier relapse in quiescent CD [26]. Chronic stress, including caregiving and marital discord, and perceived stress are associated with increases in CRP and other inflammatory mediators [126]. Stress increased LPS-stimulated cytokines, leukocyte and NK cell counts, platelet activation, and reactive oxygen metabolites production and reduced rectal mucosal blood flow in a study of rectal mucosa of UC patients compared to healthy controls [117]. Sympathetic nerve fibers and neurotransmitters are lost in inflamed areas of the colon in both CD but not in noninflamed ileum [112]; thus, stress may generate symptoms from the uninflamed areas where sympathetic nerve fibers are intact. Perhaps, then, stress may contribute to spreading of the inflammatory lesion. Recent reviews have concluded that stress has an impact on the course of disease, but the jury is still out as to whether cognitive therapies or psychotropic medications can positively influence the course of IBD [72, 148].
At least 33–57 % of IBD patients are able, during the course of their disease, to switch from IBD to IBS [170] most likely through modifications of neuroplasticity induced by chronic inflammation [37]. Proinflammatory cytokines (IL1, IL6, and TNF-α) could favor visceral hyperalgesia via an activation of spinal immune-like glial cells inducing release of proinflammatory substances (IL1, IL6, TNF-α, prostaglandins, nitric oxide) triggering the amplification of pain by modulating the excitability of spinal neurons. However, if visceral hypersensitivity is classically described in IBS, we [160] and others [22, 45] found a visceral hyposensitivity in IBD patients in remission.
Conclusions
Major advances have been made in the domain of stress and CRF signaling pathways in the brain and, more recently, in the gut. The CRFergic system mediates the effects of psychological, physical, and immunological stressors on hormonal responses, anxiety, mood, feeding behavior, and GI functions. Conclusive experimental data show that activation of brain and colonic CRF1 pathways mimics the features of stress-induced stimulation of colonic motility, defecation/watery diarrhea, intestinal permeability, and visceral hypersensitivity described in pathological conditions such as IBS. In contrast, in the upper gut, the brain and gastric CRF2 signaling systems are more prominently involved in CRF ligands- and stress-related suppression of gastric motor function. CRF2 signaling has proinflammatory properties in the lower GI tract but anti-inflammatory properties in the upper GI tract. Targeting these CRF1/CRF2 signaling pathways by selective antagonists/agonists should be of clinical interest in the domain of IBS and IBD.
References
Agelaki S, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun. 2002;70(11):6068–74.
Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, Houdeau E, Fioramonti J, Bueno L, Theodorou V. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology. 2012;37(11):1885–95.
Altschuler SM, Escardo J, Lynn RB, Miselis RR. The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology. 1993;104(2):502–9.
Altschuler SM, Bao XM, Bieger D, Hopkins DA, Miselis RR. Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. J Comp Neurol. 1989;283(2):248–68.
Anton PM, Gay J, Mykoniatis A, Pan A, O’Brien M, Brown D, Karalis K, Pothoulakis C. Corticotropin-releasing hormone (CRH) requirement in Clostridium difficile toxin A-mediated intestinal inflammation. Proc Natl Acad Sci U S A. 2004;101(22):8503–8.
Baciu MV, Bonaz BL, Papillon E, Bost RA, Le Bas JF, Fournet J, Segebarth CM. Central processing of rectal pain: a functional MR imaging study. AJNR Am J Neuroradiol. 1999;20(10):1920–4.
Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307(5717):1915–20.
Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25(3):397–407.
Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect Immun. 2010;78(4):1509–19.
Bailey MT, Engler H, Sheridan JF. Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J Neuroimmunol. 2006;171(1–2):29–37.
Bale TL. Sensitivity to stress: dysregulation of CRF pathways and disease development. Horm Behav. 2005;48(1):1–10.
Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–57.
Bale TL, Lee KF, Vale WW. The role of corticotropin-releasing factor receptors in stress and anxiety. Integr Comp Biol. 2002;42(3):552–5.
Barreau F, Ferrier L, Fioramonti J, Bueno L. Neonatal maternal deprivation triggers long term alterations in colonic epithelial barrier and mucosal immunity in rats. Gut. 2004;53(4):501–6.
Beaumont W. Nutrition classics. Experiments and observations on the gastric juice and the physiology of digestion. By William Beaumont. Plattsburgh. Printed by F. P. Allen. 1833. Nutr Rev. 1977;35(6):144–5.
Behan DP, De Souza EB, Lowry PJ, Potter E, Sawchenko P, Vale WW. Corticotropin releasing factor (CRF) binding protein: a novel regulator of CRF and related peptides. Front Neuroendocrinol. 1995;16(4):362–82.
Benarroch EE. Circumventricular organs: receptive and homeostatic functions and clinical implications. Neurology. 2011;77(12):1198–204. doi:10.1212/WNL.0b013e31822f04a0.
Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J, Deng Y, Blennerhassett P, Macri J, McCoy KD, Verdu EF, Collins SM. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology. 2011;141(2):599–609, 609.e1-3.
Berin MC, Kiliaan AJ, Yang PC, Groot JA, Kitamura Y, Perdue MH. The influence of mast cells on pathways of transepithelial antigen transport in rat intestine. J Immunol. 1998;161(5):2561–6.
Berman S, Suyenobu B, Naliboff BD, Bueller J, Stains J, Wong H, Mandelkern M, Fitzgerald L, Ohning G, Gupta A, Labus JS, Tillisch K, Mayer EA. Evidence for alterations in central noradrenergic signaling in irritable bowel syndrome. Neuroimage. 2012;63(4):1854–63. doi:10.1016/j.neuroimage.2012.08.028. Epub 2012 Aug 21.
Bernstein CN, Singh S, Graff LA, Walker JR, Miller N, Cheang M. A prospective population-based study of triggers of symptomatic flares in IBD. Am J Gastroenterol. 2010;105(9):1994–2002.
Bernstein CN, Niazi N, Robert M, Mertz H, Kodner A, Munakata J, Naliboff B, Mayer EA. Rectal afferent function in patients with inflammatory and functional intestinal disorders. Pain. 1996;66(2–3):151–61.
Bischoff SC. Physiological and pathophysiological functions of intestinal mast cells. Semin Immunopathol. 2009;31(2):185–205.
Bittencourt JC, Sawchenko PE. Do centrally administered neuropeptides access cognate receptors?: an analysis in the central corticotropin-releasing factor system. J Neurosci. 2000;20(3):1142–56.
Bittencourt JC, Vaughan J, Arias C, Rissman RA, Vale WW, Sawchenko PE. Urocortin expression in rat brain: evidence against a pervasive relationship of urocortin-containing projections with targets bearing type 2 CRF receptors. J Comp Neurol. 1999;415(3):285–312.
Bitton A, Dobkin PL, Edwardes MD, Sewitch MJ, Meddings JB, Rawal S, Cohen A, Vermeire S, Dufresne L, Franchimont D, Wild GE. Predicting relapse in Crohn’s disease: a biopsychosocial model. Gut. 2008;57(10):1386–92.
Bonaz BL, Bernstein CN. Brain-gut interactions in inflammatory bowel disease. Gastroenterology. 2013;144(1):36–49.
Bonaz B, Picq C, Sinniger V, Mayol JF, Clarençon D. Vagus nerve stimulation: from epilepsy to the cholinergic anti-inflammatory pathway. Neurogastroenterol Motil. 2013;25(3):208–21.
Bonaz B, Pellissier S, Sinniger V, Clarençon D, Peinnequin A, Canini F. The irritable bowel syndrome: how stress can affect the amygdala activity and the brain-gut axis. In: Ferry DB, editor. The amygdala – a discrete multitasking manager. Rijeka, Croatie: InTech; 2012. p. 339–74.
Bonaz B, Baciu M, Papillon E, Bost R, Gueddah N, Le Bas JF, Fournet J, Segebarth C. Central processing of rectal pain in patients with irritable bowel syndrome: an fMRI study. Am J Gastroenterol. 2002;97(3):654–61.
Bonaz B, Rivest S. Effect of a chronic stress on CRF neuronal activity and expression of its type 1 receptor in the rat brain. Am J Physiol. 1998;275(5 Pt 2):R1438–49.
Bonaz B, Taché Y. Corticotropin-releasing factor and systemic capsaicin-sensitive afferents are involved in abdominal surgery-induced Fos expression in the paraventricular nucleus of the hypothalamus. Brain Res. 1997;748(1–2):12–20.
Bonaz B, Taché Y. Water-avoidance stress-induced c-fos expression in the rat brain and stimulation of fecal output: role of corticotropin-releasing factor. Brain Res. 1994;641(1):21–8.
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–62.
Bradford K, Shih W, Videlock EJ, Presson AP, Naliboff BD, Mayer EA, Chang L. Association between early adverse life events and irritable bowel syndrome. Clin Gastroenterol Hepatol. 2012;10(4):385–90.e1-3.
Bravo JA, Dinan TG, Cryan JF. Alterations in the central CRF system of two different rat models of comorbid depression and functional gastrointestinal disorders. Int J Neuropsychopharmacol. 2011;14(5):666–83.
Brierley SM, Linden DR. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat Rev Gastroenterol Hepatol. 2014;11(10):611–27.
Buckinx R, Adriaensen D, Nassauw LV, Timmermans JP. Corticotrophin-releasing factor, related peptides, and receptors in the normal and inflamed gastrointestinal tract. Front Neurosci. 2011;5:54.
Bueno L, Fioramonti J. Effects of corticotropin-releasing factor, corticotropin and cortisol on gastrointestinal motility in dogs. Peptides. 1986;7(1):73–7.
Camilleri M, Gorman H. Intestinal permeability and irritable bowel syndrome. Neurogastroenterol Motil. 2007;19(7):545–52.
Camilleri M. Pathophysiology in irritable bowel syndrome. Drug News Perspect. 2001;14(5):268–78.
Cannon WB. The movements of the intestines studied by means of the Röntgen Rays. J Med Res. 1902;7(1):72–5.
Castagliuolo I, Lamont JT, Qiu B, Fleming SM, Bhaskar KR, Nikulasson ST, Kornetsky C, Pothoulakis C. Acute stress causes mucin release from rat colon: role of corticotropin releasing factor and mast cells. Am J Physiol. 1996;271(5 Pt 1):G884–92.
Chang J, Hoy JJ, Idumalla PS, Clifton MS, Pecoraro NC, Bhargava A. Urocortin 2 expression in the rat gastrointestinal tract under basal conditions and in chemical colitis. Peptides. 2007;28(7):1453–60.
Chang L, Munakata J, Mayer EA, Schmulson MJ, Johnson TD, Bernstein CN, Saba L, Naliboff B, Anton PA, Matin K. Perceptual responses in patients with inflammatory and functional bowel disease. Gut. 2000;47(4):497–505.
Chang L. The association of functional gastrointestinal disorders and fibromyalgia. Eur J Surg Suppl. 1998;583:32–6.
Chatzaki E, Murphy BJ, Wang L, Million M, Ohning GV, Crowe PD, Petroski R, Taché Y, Grigoriadis DE. Differential profile of CRF receptor distribution in the rat stomach and duodenum assessed by newly developed CRF receptor antibodies. J Neurochem. 2004;88(1):1–11.
Chatzaki E, Crowe PD, Wang L, Million M, Taché Y, Grigoriadis DE. CRF receptor type 1 and 2 expression and anatomical distribution in the rat colon. J Neurochem. 2004;90(2):309–16.
Chatzaki E, Charalampopoulos I, Leontidis C, Mouzas IA, Tzardi M, Tsatsanis C, Margioris AN, Gravanis A. Urocortin in human gastric mucosa: relationship to inflammatory activity. J Clin Endocrinol Metab. 2003;88(1):478–83.
Chen C. Recent advances in small molecule antagonists of the corticotropin-releasing factor type-1 receptor-focus on pharmacology and pharmacokinetics. Curr Med Chem. 2006;13(11):1261–82.
Clarençon D, Pellissier S, Sinniger V, Kibleur A, Hoffman D, Vercueil L, David O, Bonaz B. Long term effects of low frequency (10 hz) vagus nerve stimulation on EEG and heart rate variability in Crohn’s disease: a case report. Brain Stimul. 2014;7(6):914–6.
Collins SM, Bercik P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology. 2009;136(6):2003–14.
Czéh B, Perez-Cruz C, Fuchs E, Flügge G. Chronic stress-induced cellular changes in the medial prefrontal cortex and their potential clinical implications: does hemisphere location matter? Behav Brain Res. 2008;190(1):1–13.
Czimmer J, Million M, Taché Y. Urocortin 2 acts centrally to delay gastric emptying through sympathetic pathways while CRF and urocortin 1 inhibitory actions are vagal dependent in rats. Am J Physiol Gastrointest Liver Physiol. 2006;290(3):G511–8. Epub 2005 Oct 13.
Dalal SR, Chang EB. The microbial basis of inflammatory bowel diseases. J Clin Invest. 2014;124(10):4190–6.
Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A. 2007;104(4):1319–24.
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–51.
Ducarouge B, Pelissier-Rota M, Lainé M, Cristina N, Vachez Y, Scoazec JY, Bonaz B, Jacquier-Sarlin M. CRF2 signaling is a novel regulator of cellular adhesion and migration in colorectal cancer cells. PLoS One. 2013;8(11):e79335.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–8.
Foote SL, Bloom FE, Aston-Jones G. Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63(3):844–914.
Ford AC, Quigley EM, Lacy BE, Lembo AJ, Saito YA, Schiller LR, Soffer EE, Spiegel BM, Moayyedi P. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol. 2014;109(9):1350–65.
Forsythe P, Bienenstock J, Kunze WA. Vagal pathways for microbiome-brain-gut axis communication. Adv Exp Med Biol. 2014;817:115–33.
Fukudo S. Role of corticotropin-releasing hormone in irritable bowel syndrome and intestinal inflammation. J Gastroenterol. 2007;42 Suppl 17:48–51.
Fukudo S, Nomura T, Hongo M. Impact of corticotropin-releasing hormone on gastrointestinal motility and adrenocorticotropic hormone in normal controls and patients with irritable bowel syndrome. Gut. 1998;42(6):845–9.
Fulwiler CE, Saper CB. Subnuclear organization of the efferent connections of the parabrachial nucleus in the rat. Brain Res. 1984;319(3):229–59.
Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9(5):286–94.
Garakani A, Win T, Virk S, Gupta S, Kaplan D, Masand PS. Comorbidity of irritable bowel syndrome in psychiatric patients: a review. Am J Ther. 2003;10(1):61–7.
Gay J, Kokkotou E, O’Brien M, Pothoulakis C, Karalis KP. Corticotropin-releasing hormone deficiency is associated with reduced local inflammation in a mouse model of experimental colitis. Endocrinology. 2008;149(7):3403–9. doi:10.1210/en.2007-1703.
Ghia JE, Blennerhassett P, Collins SM. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J Clin Invest. 2008;118(6):2209–18.
Goehler LE, Gaykema RP, Opitz N, Reddaway R, Badr N, Lyte M. Activation in vagal afferents and central autonomic pathways: early responses to intestinal infection with Campylobacter jejuni. Brain Behav Immun. 2005;19(4):334–44.
Gonzalez-Rey E, Fernandez-Martin A, Chorny A, Delgado M. Therapeutic effect of urocortin and adrenomedullin in a murine model of Crohn’s disease. Gut. 2006;55(6):824–32.
Graff LA, Walker JR, Bernstein CN. Depression and anxiety in inflammatory bowel disease: a review of comorbidity and management. Inflamm Bowel Dis. 2009;15(7):1105–18.
Gravanis A, Margioris AN. The corticotropin-releasing factor (CRF) family of neuropeptides in inflammation: potential therapeutic applications. Curr Med Chem. 2005;12(13):1503–12.
Gross KJ, Pothoulakis C. Role of neuropeptides in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(7):918–32.
Guarner F. Enteric flora in health and disease. Digestion. 2006;73 Suppl 1:5–12.
Gué M, Del Rio-Lacheze C, Eutamene H, Théodorou V, Fioramonti J, Buéno L. Stress-induced visceral hypersensitivity to rectal distension in rats: role of CRF and mast cells. Neurogastroenterol Motil. 1997;9(4):271–9.
Guillemin R, Rosenberg B. Humoral hypothalamic control of anterior pituitary: a study with combined tissue cultures. Endocrinology. 1955;57(5):599–607.
Gwee KA, Graham JC, McKendrick MW, Collins SM, Marshall JS, Walters SJ, Read NW. Psychometric scores and persistence of irritable bowel after infectious diarrhoea. Lancet. 1996;347(8995):150–3.
Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660–9.
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5(4):453–79.
Heim C, Newport DJ, Miller AH, Nemeroff CB. Long-term neuroendocrine effects of childhood maltreatment. JAMA. 2000;284(18):2321.
Heitkemper M, Jarrett M, Cain K, Shaver J, Bond E, Woods NF, Walker E. Increased urine catecholamines and cortisol in women with irritable bowel syndrome. Am J Gastroenterol. 1996;91(5):906–13.
Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27(3):260–86.
Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology. 2015;148(1):148–157.e7.
Hugot JP, Zouali H, Lesage S, Thomas G. Etiology of the inflammatory bowel diseases. Int J Colorectal Dis. 1999;14(1):2–9.
Imaki T, Shibasaki T, Wang XQ, Demura H. Intracerebroventricular administration of corticotropin-releasing factor antagonist attenuates c-fos mRNA expression in the paraventricular nucleus after stress. Neuroendocrinology. 1995;61(4):445–52.
Iovino P, Azpiroz F, Domingo E, Malagelada JR. The sympathetic nervous system modulates perception and reflex responses to gut distention in humans. Gastroenterology. 1995;108(3):680–6.
Iweala OI, Nagler CR. Immune privilege in the gut: the establishment and maintenance of non-responsiveness to dietary antigens and commensal flora. Immunol Rev. 2006;213:82–100.
Ji G, Neugebauer V. Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol. 2007;97(6):3893–904.
Johnson AC, Tran L, Schulkin J, Greenwood-Van Meerveld B. Importance of stress receptor-mediated mechanisms in the amygdala on visceral pain perception in an intrinsically anxious rat. Neurogastroenterol Motil. 2012;24(5):479–86, e219.
Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005;135(4):1295–307.
Kawahito Y, Sano H, Mukai S, Asai K, Kimura S, Yamamura Y, Kato H, Chrousos GP, Wilder RL, Kondo M. Corticotropin releasing hormone in colonic mucosa in patients with ulcerative colitis. Gut. 1995;37(4):544–51.
Kihara N, Fujimura M, Yamamoto I, Itoh E, Inui A, Fujimiya M. Effects of central and peripheral urocortin on fed and fasted gastroduodenal motor activity in conscious rats. Am J Physiol Gastrointest Liver Physiol. 2001;280(3):G406–19.
Kiliaan AJ, Saunders PR, Bijlsma PB, Berin MC, Taminiau JA, Groot JA, Perdue MH. Stress stimulates transepithelial macromolecular uptake in rat jejunum. Am J Physiol. 1998;275(5 Pt 1):G1037–44.
Kokkotou E, Torres D, Moss AC, O’Brien M, Grigoriadis DE, Karalis K, Pothoulakis C. Corticotropin-releasing hormone receptor 2-deficient mice have reduced intestinal inflammatory responses. J Immunol. 2006;177(5):3355–61.
Kosoyan HP, Grigoriadis DE, Taché Y. The CRF(1) receptor antagonist, NBI-35965, abolished the activation of locus coeruleus neurons induced by colorectal distension and intracisternal CRF in rats. Brain Res. 2005;1056(1):85–96.
Kozicz T, Yanaihara H, Arimura A. Distribution of urocortin-like immunoreactivity in the central nervous system of the rat. J Comp Neurol. 1998;391(1):1–10.
Kresse AE, Million M, Saperas E, Taché Y. Colitis induces CRF expression in hypothalamic magnocellular neurons and blunts CRF gene response to stress in rats. Am J Physiol Gastrointest Liver Physiol. 2001;281(5):G1203–13.
la Fleur SE, Wick EC, Idumalla PS, Grady EF, Bhargava A. Role of peripheral corticotropin-releasing factor and urocortin II in intestinal inflammation and motility in terminal ileum. Proc Natl Acad Sci U S A. 2005;102(21):7647–52.
Larauche M, Mulak A, Taché Y. Stress and visceral pain: from animal models to clinical therapies. Exp Neurol. 2012;233(1):49–67.
Larauche M, Kiank C, Tache Y. Corticotropin releasing factor signaling in colon and ileum: regulation by stress and pathophysiological implications. J Physiol Pharmacol. 2009;60 Suppl 7:33–46.
Lautenbacher S, Rollman GB. Possible deficiencies of pain modulation in fibromyalgia. Clin J Pain. 1997;13(3):189–96.
Lee C, Sarna SK. Central regulation of gastric emptying of solid nutrient meals by corticotropin releasing factor. Neurogastroenterol Motil. 1997;9(4):221–9.
Lee HH, Choi YY, Choi MG. The efficacy of hypnotherapy in the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Neurogastroenterol Motil. 2014;20(2):152–62.
Lee R, Geracioti Jr TD, Kasckow JW, Coccaro EF. Childhood trauma and personality disorder: positive correlation with adult CSF corticotropin-releasing factor concentrations. Am J Psychiatry. 2005;162(5):995–7.
Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330(6012):1768–73.
Li C, Vaughan J, Sawchenko PE, Vale WW. Urocortin III-immunoreactive projections in rat brain: partial overlap with sites of type 2 corticotrophin-releasing factor receptor expression. J Neurosci. 2002;22(3):991–1001.
Lindgren S, Stewenius J, Sjölund K, Lilja B, Sundkvist G. Autonomic vagal nerve dysfunction in patients with ulcerative colitis. Scand J Gastroenterol. 1993;28(7):638–42.
Lindgren S, Lilja B, Rosén I, Sundkvist G. Disturbed autonomic nerve function in patients with Crohn’s disease. Scand J Gastroenterol. 1991;26(4):361–6.
Lyte M, Vulchanova L, Brown DR. Stress at the intestinal surface: catecholamines and mucosa-bacteria interactions. Cell Tissue Res. 2011;343(1):23–32.
Lyte M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: microbial endocrinology in the design and use of probiotics. Bioessays. 2011;33(8):574–81.
Magro F, Vieira-Coelho MA, Fraga S, Serrão MP, Veloso FT, Ribeiro T, Soares-da-Silva P. Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Dig Dis Sci. 2002;47(1):216–24.
Mano-Otagiri A, Shibasaki T. Distribution of urocortin 2 and urocortin 3 in rat brain. J Nippon Med Sch. 2004;71(6):358–9.
Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014;182:65–9.
Martinez V, Taché Y. CRF1 receptors as a therapeutic target for irritable bowel syndrome. Curr Pharm Des. 2006;12(31):4071–88.
Martinez V, Wang L, Million M, Rivier J, Taché Y. Urocortins and the regulation of gastrointestinal motor function and visceral pain. Peptides. 2004;25(10):1733–44.
Mawdsley JE, Macey MG, Feakins RM, Langmead L, Rampton DS. The effect of acute psychologic stress on systemic and rectal mucosal measures of inflammation in ulcerative colitis. Gastroenterology. 2006;131(2):410–9.
Mayer EA, Naliboff BD, Chang L. Basic pathophysiologic mechanisms in irritable bowel syndrome. Dig Dis. 2001;19(3):212–8.
Mayer EA, Gebhart GF. Basic and clinical aspects of visceral hyperalgesia. Gastroenterology. 1994;107(1):271–93.
McCafferty DM, Wallace JL, Sharkey KA. Effects of chemical sympathectomy and sensory nerve ablation on experimental colitis in the rat. Am J Physiol. 1997;272(2 Pt 1):G272–80.
McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43(1):2–15.
McLean SA, Williams DA, Stein PK, Harris RE, Lyden AK, Whalen G, Park KM, Liberzon I, Sen A, Gracely RH, Baraniuk JN, Clauw DJ. Cerebrospinal fluid corticotropin-releasing factor concentration is associated with pain but not fatigue symptoms in patients with fibromyalgia. Neuropsychopharmacology. 2006;31(12):2776–82.
Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13(7):269–77.
Meregnani J, Clarençon D, Vivier M, Peinnequin A, Mouret C, Sinniger V, Picq C, Job A, Canini F, Jacquier-Sarlin M, Bonaz B. Anti-inflammatory effect of vagus nerve stimulation in a rat model of inflammatory bowel disease. Auton Neurosci. 2011;160(1–2):82–9.
Miampamba M, Maillot C, Million M, Taché Y. Peripheral CRF activates myenteric neurons in the proximal colon through CRF(1) receptor in conscious rats. Am J Physiol Gastrointest Liver Physiol. 2002;282(5):G857–65.
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732–41.
Million M, Wang L, Stenzel-Poore MP, Coste SC, Yuan PQ, Lamy C, Rivier J, Buffington T, Taché Y. Enhanced pelvic responses to stressors in female CRF-overexpressing mice. Am J Physiol Regul Integr Comp Physiol. 2007;292(4):R1429–38.
Million M, Wang L, Martinez V, Taché Y. Differential Fos expression in the paraventricular nucleus of the hypothalamus, sacral parasympathetic nucleus and colonic motor response to water avoidance stress in Fischer and Lewis rats. Brain Res. 2000;877(2):345–53.
Mönnikes H, Schmidt BG, Tebbe J, Bauer C, Taché Y. Microinfusion of corticotropin releasing factor into the locus coeruleus/subcoeruleus nuclei stimulates colonic motor function in rats. Brain Res. 1994;644(1):101–8.
Mönnikes H, Schmidt BG, Raybould HE, Taché Y. CRF in the paraventricular nucleus mediates gastric and colonic motor response to restraint stress. Am J Physiol. 1992;262(1 Pt 1):G137–43.
Moss AC, Anton P, Savidge T, Newman P, Cheifetz AS, Gay J, Paraschos S, Winter MW, Moyer MP, Karalis K, Kokkotou E, Pothoulakis C. Urocortin II mediates pro-inflammatory effects in human colonocytes via corticotropin-releasing hormone receptor 2alpha. Gut. 2007;56(9):1210–7.
Mulak A, Bonaz B. Irritable bowel syndrome: a model of the brain-gut interactions. Med Sci Monit. 2004;10(4):RA55–62.
Muramatsu Y, Fukushima K, Iino K, Totsune K, Takahashi K, Suzuki T, Hirasawa G, Takeyama J, Ito M, Nose M, Tashiro A, Hongo M, Oki Y, Nagura H, Sasano H. Urocortin and corticotropin-releasing factor receptor expression in the human colonic mucosa. Peptides. 2000;21(12):1799–809.
Myers B, Dittmeyer K, Greenwood-Van Meerveld B. Involvement of amygdaloid corticosterone in altered visceral and somatic sensation. Behav Brain Res. 2007;181(1):163–7.
Myers B, Greenwood-Van Meerveld B. Corticosteroid receptor-mediated mechanisms in the amygdala regulate anxiety and colonic sensitivity. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1622–9.
Neufeld KM, Kang N, Bienenstock J, Foster JA. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol Motil. 2011;23(3):255–64, e119.
Nozu T, Kudaira M. Corticotropin-releasing factor induces rectal hypersensitivity after repetitive painful rectal distention in healthy humans. J Gastroenterol. 2006;41(8):740–4.
O’Mahony SM, Felice VD, Nally K, Savignac HM, Claesson MJ, Scully P, Woznicki J, Hyland NP, Shanahan F, Quigley EM, Marchesi JR, O’Toole PW, Dinan TG, Cryan JF. Disturbance of the gut microbiota in early-life selectively affects visceral pain in adulthood without impacting cognitive or anxiety-related behaviors in male rats. Neuroscience. 2014;277:885–901.
Overstreet DH, Djuric V. A genetic rat model of cholinergic hypersensitivity: implications for chemical intolerance, chronic fatigue, and asthma. Ann N Y Acad Sci. 2001;933:92–102.
Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med. 2003;9(5–8):125–34.
Pellissier S, Dantzer C, Mondillon L, Trocme C, Gauchez AS, Ducros V, Mathieu N, Toussaint B, Fournier A, Canini F, Bonaz B. Relationship between vagal tone, cortisol, TNF-alpha, epinephrine and negative affects in Crohn’s disease and irritable bowel syndrome. PLoS One. 2014;9(9):e105328.
Pellissier S, Dantzer C, Canini F, Mathieu N, Bonaz B. Psychological adjustment and autonomic disturbances in inflammatory bowel diseases and irritable bowel syndrome. Psychoneuroendocrinology. 2010;35(5):653–62.
Porcher C, Peinnequin A, Pellissier S, Meregnani J, Sinniger V, Canini F, Bonaz B. Endogenous expression and in vitro study of CRF-related peptides and CRF receptors in the rat gastric antrum. Peptides. 2006;27(6):1464–75.
Porcher C, Juhem A, Peinnequin A, Sinniger V, Bonaz B. Expression and effects of metabotropic CRF1 and CRF2 receptors in rat small intestine. Am J Physiol Gastrointest Liver Physiol. 2005;288(5):G1091–103.
Porcher C, Sinniger V, Juhem A, Mouchet P, Bonaz B. Neuronal activity and CRF receptor gene transcription in the brains of rats with colitis. Am J Physiol Gastrointest Liver Physiol. 2004;287(4):G803–14.
Posserud I, Agerforz P, Ekman R, Björnsson ES, Abrahamsson H, Simrén M. Altered visceral perceptual and neuroendocrine response in patients with irritable bowel syndrome during mental stress. Gut. 2004;53(8):1102–8.
Radulovic M, Spiess J. Immunomodulatory role of the corticotropin-releasing factor. Arch Immunol Ther Exp (Warsz). 2001;49(1):33–8.
Rampton DS. The influence of stress on the development and severity of immune-mediated diseases. J Rheumatol Suppl. 2011;88:43–7.
Randich A, Gebhart GF. Vagal afferent modulation of nociception. Brain Res Brain Res Rev. 1992;17(2):77–99.
Renard GM, Rivarola MA, Suárez MM. Sexual dimorphism in rats: effects of early maternal separation and variable chronic stress on pituitary-adrenal axis and behavior. Int J Dev Neurosci. 2007;25(6):373–9.
Reyes BA, Valentino RJ, Xu G, Van Bockstaele EJ. Hypothalamic projections to locus coeruleus neurons in rat brain. Eur J Neurosci. 2005;22(1):93–106.
Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB, Gulyas J, Rivier J, Vale WW, Sawchenko PE. Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci U S A. 2001;98(5):2843–8.
Ritchie J. Pain from distension of the pelvic colon by inflating a balloon in the irritable colon syndrome. Gut. 1973;14(2):125–32.
Rivier CL, Grigoriadis DE, Rivier JE. Role of corticotropin-releasing factor receptors type 1 and 2 in modulating the rat adrenocorticotropin response to stressors. Endocrinology. 2003;144(6):2396–403.
Rivier J, Gulyas J, Kunitake K, DiGruccio M, Cantle JP, Perrin MH, Donaldson C, Vaughan J, Million M, Gourcerol G, Adelson DW, Rivier C, Taché Y, Vale W. Stressin1-A, a potent corticotropin releasing factor receptor 1 (CRF1)-selective peptide agonist. J Med Chem. 2007;50(7):1668–74.
Rivier J, Gulyas J, Kirby D, Low W, Perrin MH, Kunitake K, DiGruccio M, Vaughan J, Reubi JC, Waser B, Koerber SC, Martinez V, Wang L, Taché Y, Vale W. Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists. J Med Chem. 2002;45(21):4737–47.
Rivier J, Rivier C, Vale W. Synthetic competitive antagonists of corticotropin-releasing factor: effect on ACTH secretion in the rat. Science. 1984;224(4651):889–91.
Rivier JE, Kirby DA, Lahrichi SL, Corrigan A, Vale WW, Rivier CL. Constrained corticotropin releasing factor antagonists (astressin analogues) with long duration of action in the rat. J Med Chem. 1999;42(16):3175–82.
Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105(31):11008–13.
Rubio A, Pellissier S, Picot A, Dantzer C, Bonaz B. The link between negative affect, vagal tone, and visceral sensitivity in quiescent Crohn’s disease. Neurogastroenterol Motil. 2014;26(8):1200–3.
Sagami Y, Shimada Y, Tayama J, Nomura T, Satake M, Endo Y, Shoji T, Karahashi K, Hongo M, Fukudo S. Effect of a corticotropin releasing hormone receptor antagonist on colonic sensory and motor function in patients with irritable bowel syndrome. Gut. 2004;53(7):958–64.
Santos J, Yates D, Guilarte M, Vicario M, Alonso C, Perdue MH. Stress neuropeptides evoke epithelial responses via mast cell activation in the rat colon. Psychoneuroendocrinology. 2008;33(9):1248–56.
Santos J, Saunders PR, Hanssen NP, Yang PC, Yates D, Groot JA, Perdue MH. Corticotropin-releasing hormone mimics stress-induced colonic epithelial pathophysiology in the rat. Am J Physiol. 1999;277(2 Pt 1):G391–9.
Saper CB. The central autonomic nervous system: conscious visceral perception and autonomic pattern generation. Annu Rev Neurosci. 2002;25:433–69.
Saper CB. Convergence of autonomic and limbic connections in the insular cortex of the rat. J Comp Neurol. 1982;210(2):163–73.
Saruta M, Takahashi K, Suzuki T, Torii A, Kawakami M, Sasano H. Urocortin 1 in colonic mucosa in patients with ulcerative colitis. J Clin Endocrinol Metab. 2004;89(11):5352–61.
Saunders PR, Santos J, Hanssen NP, Yates D, Groot JA, Perdue MH. Physical and psychological stress in rats enhances colonic epithelial permeability via peripheral CRH. Dig Dis Sci. 2002;47(1):208–15.
Selye H. A syndrome produced by diverse nocuous agents. 1936. J Neuropsychiatry Clin Neurosci. 1998;10(2):230–1.
Shanks N, Windle RJ, Perks PA, Harbuz MS, Jessop DS, Ingram CD, Lightman SL. Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proc Natl Acad Sci U S A. 2000;97(10):5645–50.
Simrén M, Axelsson J, Gillberg R, Abrahamsson H, Svedlund J, Björnsson ES. Quality of life in inflammatory bowel disease in remission: the impact of IBS-like symptoms and associated psychological factors. Am J Gastroenterol. 2002;97(2):389–96.
Sinniger V, Mouchet P, Bonaz B. Effect of nor-trimebutine on neuronal activation induced by a noxious stimulus or an acute colonic inflammation in the rat. Life Sci. 2005;77(23):2927–41.
Sinniger V, Porcher C, Mouchet P, Juhem A, Bonaz B. c-fos and CRF receptor gene transcription in the brain of acetic acid-induced somato-visceral pain in rats. Pain. 2004;110(3):738–50.
Söderholm JD, Yates DA, Gareau MG, Yang PC, MacQueen G, Perdue MH. Neonatal maternal separation predisposes adult rats to colonic barrier dysfunction in response to mild stress. Am J Physiol Gastrointest Liver Physiol. 2002;283(6):G1257–63.
Söderholm JD, Perdue MH. Stress and gastrointestinal tract. II. Stress and intestinal barrier function. Am J Physiol Gastrointest Liver Physiol. 2001;280(1):G7–13.
Söderholm JD, Peterson KH, Olaison G, Franzén LE, Weström B, Magnusson KE, Sjödahl R. Epithelial permeability to proteins in the noninflamed ileum of Crohn’s disease? Gastroenterology. 1999;117(1):65–72.
Spence MJ, Moss-Morris R. The cognitive behavioural model of irritable bowel syndrome: a prospective investigation of patients with gastroenteritis. Gut. 2007;56(8):1066–71.
Stengel A, Taché Y. CRF and urocortin peptides as modulators of energy balance and feeding behavior during stress. Front Neurosci. 2014;8:52.
Stengel A, Taché Y. Corticotropin-releasing factor signaling and visceral response to stress. Exp Biol Med (Maywood). 2010;235(10):1168–78.
Straub RH, Herfarth H, Falk W, Andus T, Schölmerich J. Uncoupling of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis in inflammatory bowel disease? J Neuroimmunol. 2002;126(1–2):116–25.
Su J, Tanaka Y, Muratsubaki T, Kano M, Kanazawa M, Fukudo S. Injection of corticotropin-releasing hormone into the amygdala aggravates visceral nociception and induces noradrenaline release in rats. Neurogastroenterol Motil. 2015;27(1):30–9.
Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, Kubo C, Koga Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol. 2004;558(Pt 1):263–75.
Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36(3):165–86.
Sweetser S, Camilleri M, Linker Nord SJ, Burton DD, Castenada L, Croop R, Tong G, Dockens R, Zinsmeister AR. Do corticotropin releasing factor-1 receptors influence colonic transit and bowel function in women with irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2009;296(6):G1299–306.
Sykes MA, Blanchard EB, Lackner J, Keefer L, Krasner S. Psychopathology in irritable bowel syndrome: support for a psychophysiological model. J Behav Med. 2003;26(4):361–72.
Taché Y, Bonaz B. Corticotropin-releasing factor receptors and stress-related alterations of gut motor function. J Clin Invest. 2007;117(1):33–40.
Taché Y, Martinez V, Wang L, Million M. CRF1 receptor signaling pathways are involved in stress-related alterations of colonic function and viscerosensitivity: implications for irritable bowel syndrome. Br J Pharmacol. 2004;141(8):1321–30.
Taché Y, Perdue MH. Role of peripheral CRF signalling pathways in stress-related alterations of gut motility and mucosal function. Neurogastroenterol Motil. 2004;16 Suppl 1:137–42.
Taché Y, Martinez V, Million M, Wang L. Stress and the gastrointestinal tract III. Stress-related alterations of gut motor function: role of brain corticotropin-releasing factor receptors. Am J Physiol Gastrointest Liver Physiol. 2001;280(2):G173–7.
Tanaka Y, Kanazawa M, Fukudo S, Drossman DA. Biopsychosocial model of irritable bowel syndrome. J Neurogastroenterol Motil. 2011;17(2):131–9.
Tayama J, Sagami Y, Shimada Y, Hongo M, Fukudo S. Effect of alpha-helical CRH on quantitative electroencephalogram in patients with irritable bowel syndrome. Neurogastroenterol Motil. 2007;19(6):471–83.
Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361–72.
Tillisch K, Labus J, Kilpatrick L, Jiang Z, Stains J, Ebrat B, Guyonnet D, Legrain-Raspaud S, Trotin B, Naliboff B, Mayer EA. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology. 2013;144(7):1394–401. 1401.e1–4.
Tillisch K, Mayer EA, Labus JS. Quantitative meta-analysis identifies brain regions activated during rectal distension in irritable bowel syndrome. Gastroenterology. 2011;140(1):91–100.
Trimble N, Johnson AC, Foster A, Greenwood-van Meerveld B. Corticotropin-releasing factor receptor 1-deficient mice show decreased anxiety and colonic sensitivity. Neurogastroenterol Motil. 2007;19(9):754–60.
Tsuruoka M, Wang D, Tamaki J, Inoue T. Descending influence from the nucleus locus coeruleus/subcoeruleus on visceral nociceptive transmission in the rat spinal cord. Neuroscience. 2010;165(4):1019–24.
Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213(4514):1394–7.
Valentino RJ, Kosboth M, Colflesh M, Miselis RR. Transneuronal labeling from the rat distal colon: anatomic evidence for regulation of distal colon function by a pontine corticotropin-releasing factor system. J Comp Neurol. 2000;417(4):399–414.
Valentino RJ, Miselis RR, Pavcovich LA. Pontine regulation of pelvic viscera: pharmacological target for pelvic visceral dysfunctions. Trends Pharmacol Sci. 1999;20(6):253–60.
Valentino RJ, Page ME, Luppi PH, Zhu Y, Van Bockstaele E, Aston-Jones G. Evidence for widespread afferents to Barrington’s nucleus, a brainstem region rich in corticotropin-releasing hormone neurons. Neuroscience. 1994;62(1):125–43.
van den Wijngaard RM, Stanisor OI, van Diest SA, Welting O, Wouters MM, Cailotto C, de Jonge WJ, Boeckxstaens GE. Susceptibility to stress induced visceral hypersensitivity in maternally separated rats is transferred across generations. Neurogastroenterol Motil. 2013;25(12):e780–90.
van den Wijngaard RM, Klooker TK, de Jonge WJ, Boeckxstaens GE. Peripheral relays in stress-induced activation of visceral afferents in the gut. Auton Neurosci. 2010;153(1–2):99–105.
Van Oudenhove L, Coen SJ, Aziz Q. Functional brain imaging of gastrointestinal sensation in health and disease. World J Gastroenterol. 2007;13(25):3438–45.
Varghese AK, Verdú EF, Bercik P, Khan WI, Blennerhassett PA, Szechtman H, Collins SM. Antidepressants attenuate increased susceptibility to colitis in a murine model of depression. Gastroenterology. 2006;130(6):1743–53.
Wallon C, Yang PC, Keita AV, Ericson AC, McKay DM, Sherman PM, Perdue MH, Söderholm JD. Corticotropin-releasing hormone (CRH) regulates macromolecular permeability via mast cells in normal human colonic biopsies in vitro. Gut. 2008;57(1):50–8.
Webster EL, Torpy DJ, Elenkov IJ, Chrousos GP. Corticotropin-releasing hormone and inflammation. Ann N Y Acad Sci. 1998;840:21–32.
Willer JC, Bouhassira D, Le Bars D. Neurophysiological bases of the counterirritation phenomenon: diffuse control inhibitors induced by nociceptive stimulation. Neurophysiol Clin. 1999;29(5):379–400.
Wood SK, Woods JH. Corticotropin-releasing factor receptor-1: a therapeutic target for cardiac autonomic disturbances. Expert Opin Ther Targets. 2007;11(11):1401–13.
Yi L, Sun H, Ge C, Chen Y, Peng H, Jiang Y, Wu P, Tang Y, Meng Q, Xu S. Role of insular cortex in visceral hypersensitivity model in rats subjected to chronic stress. Psychiatry Res. 2014;220(3):1138–43.
Yunus MB. Role of central sensitization in symptoms beyond muscle pain, and the evaluation of a patient with widespread pain. Best Pract Res Clin Rheumatol. 2007;21(3):481–97.
Zagon A. Does the vagus nerve mediate the sixth sense? Trends Neurosci. 2001;24(11):671–3.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bonaz, B. (2016). Stress and the Gastrointestinal System. In: Constantinescu, C., Arsenescu, R., Arsenescu, V. (eds) Neuro-Immuno-Gastroenterology. Springer, Cham. https://doi.org/10.1007/978-3-319-28609-9_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-28609-9_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28607-5
Online ISBN: 978-3-319-28609-9
eBook Packages: MedicineMedicine (R0)