
Abstract
Psychosocial stress, in particular chronic stress, has emerged as a key factor in the pathophysiology of many chronic diseases, including mental health conditions. Stress is anything that challenges our homeostasis. Several brain regions are involved in stress regulation including the corticolimbic system and the hypothalamic-pituitary-adrenal (HPA) axis, and chronic inflammation has emerged as a potential common pathway underpinning stress-related diseases. The endocannabinoid system (ECS) regulates our responsivity to stress, including the neuroendocrine and behavioral aspects of stress, via the corticolimbic system and the HPA axis, and changes occur within the ECS under conditions of acute and chronic stress. The gut microbiome is also involved in our stress regulation and reactions, and it is linked to the ECS also. An understanding of the impact of stress is important since stress increases the risk of mental health problems including anxiety, depression, sleep problems, and others.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
The term “stress” and its meaning has evolved over time since Selye first popularized it in a paper in Nature in 1936 where he defined stress as “non-specific responses of the body to any demand upon it” [1, 2]. The definition of stress has evolved from one primarily related to an effect to include the concept of stress as a cause of illness [2], including stress in the psychosocial sense.
Hill et al. [3] define stress as anything that presents as a challenge to homeostasis and is typically something that threatens our well-being. It may be a real threat, or a perceived one, but people exposed to the same stress are not necessarily affected in the same manner [4]. Emotions cannot be divorced from stress since they drive the creation and interpretation of factors or events that are causes of stress, and the physiological responses that occur while experiencing stress become encoded as part of the interpreted emotion [2].
Stress is one of the most important and often overlooked etiological and pathological factors in many illnesses, particularly those of chronic nature. While stress is difficult to quantify and sometimes to even describe, it has become a feature of modern, westernized life, and as humans, we are becoming sicker. The element of stress, as a cause, an effect, an exacerbating factor, and/or an enabling factor, is likely to be involved in many illnesses, including mental health conditions. In particular, stress in the sense of a psychosocial stressor appears a feature of modern life.
While some level of stress in our lives may be useful, and indeed serves an important function in survival, it is when stress becomes chronic, unrelenting, prolonged, overwhelming, or extreme that it can start to become unhealthy and affect our mental and physical health. Inadequate control of stress responses contributes to the risk of mental health disorders and increases mortality [5, 6].
Stress can increase the risk of many illnesses including cancer, hypertension, cardiovascular disease, stroke, gastrointestinal problems (including inflammatory bowel disease), a weakened immune system, and others [7, 8]. Long-term stress increases the risk of mental health problems including anxiety and depression, sleep problems, substance abuse, and pain [7]. Psychosocial stress plays a clear role in the development and continuance of symptoms of major mental health illnesses including depression and schizophrenia [9]. Stress also alters our responses to emotional stimuli [10].
Chronic stress is a risk factor for development and exacerbation of mental illnesses including depression, anxiety, and post-traumatic stress disorder (PTSD) [11,12,13]. Thus, by understanding the biochemical adaptations that occur with chronic stress, it might be possible to discover targets and treatments for many different mental health conditions [14].
There is evidence that the endocannabinoid system (ECS) is involved in our stress response (both acute and chronic) and in stress regulation. It is altered when stress is chronic and when there is early life stress. The gut microbiome also has a role to play in stress regulation, and the two (ECS and gut microbiome) form a close relationship.
This Chapter
In this chapter, we will examine the brain regions involved in stress regulation, what happens to the autonomic nervous system stress response and the neuroendocrine stress response, how inflammation is involved in stress, and how stress impacts on the ECS as well as how the ECS is involved in stress regulation. We will also look at the impact of early life stress on mental health and the ECS functioning, the gut microbiome’s involvement in stress, and how the “endocannabinoidome ,” a term referring to the extended ECS, is involved in mental health.
Key Brain Structures Involved in the Regulation of Stress
There are several key brain regions involved in the processing and regulation of stress. Sensory information is processed by the thalamus and primary sensory cortical centers and then transmitted to the amygdala via corticothalamic afferent nerves. Two important structures associated with the regulation of stress and emotions are the corticolimbic system (prefrontal cortex, amygdala, hippocampus) and the hypothalamic-pituitary-adrenal (HPA) axis [15].
Corticolimbic System
The corticolimbic system is made up of:
-
Prefrontal cortex (PFC)
-
Amygdala (especially the basolateral amygdala, BLA)
-
Hippocampus
The corticolimbic system processes a wide range of cognitive and behavioral responses including emotional regulation, decision-making, mnemonic function, and motor programming and control [16]. The corticolimbic system integrates emotion with cognition and also is involved in pain modulation [17]. As clinicians know, pain is known to have a strong emotional component. Chronic pain is a disorder with anxio-depressive symptoms as well as cognitive deficits, and the corticolimbic system is implicated in pathophysiology of maladaptive pain [17, 18].
The brain areas that make up the corticolimbic system are all involved in the stress response, as well as in processing and regulation of emotions, fear, reward, emotional memory, anxiety-related behaviors, and drug abuse, and the ECS is widely distributed throughout these regions [3, 19, 20]. These brain regions regulate normal emotional and cognitive development and behavior [21].
The structures of the corticolimbic system are all connected to each other with both feedforward and feedback processes occurring, where stimulation of the ventral hippocampus and mPFC activates principal cells and interneurons in the basal amygdala and innervated principal cells project back from the basal amygdala to the mPFC [22]. There are also connections between the corticolimbic system and other parts of the brain, and some of these will be explored throughout various chapters. For example, the medial PFC (mPFC) projects to several brain regions involved in control of motivated behavior, anxiety, and depression [22].
Roles of the Components of the Corticolimbic System
The roles of the basolateral amygdala (BLA) are believed to include modulating learning after stress, memory consolidation, and hippocampus activity during learning and stress [23]. Activation of the BLA involved several aspects of the stress response including pain and anxiety, HPA axis activation, and alterations in cognitive processes through connections with other parts of the amygdala and hypothalamus [24]. It is understood that stress-induced release of corticotropin-releasing hormone in the amygdala contributes to emotional responses to exposure to stress (e.g., anxiety) [6]. Reference to past experience and retrieval of spatial memory are mediated via cross-talk between the mPFC and the hippocampus, and the amygdala is also involved in retrieval of fear associations, something clearly relevant in post-traumatic stress disorder [22]. There is also thalamic input to the mPFC that projects information in relation to affective-motivational behaviors [22].
The amygdaloid complex regulates anxiety and fear, providing valence to memories via its connections to the hippocampus which is where memory consolidation occurs. The hippocampus is involved in feedback regulation of the HPA axis [6].
The prefrontal cortex regulates activity of the amygdala and hippocampus, controlling emotional stress responses and facilitating active coping, and contributes to resilience to stress [6], meanwhile excitatory outputs from the medial PFC and the hippocampus control activity and output of the amygdala [22].
Hypothalamic-Pituitary-Adrenal (HPA) Axis
The hypothalamic-pituitary-adrenal (HPA) axis, believed to be responsible for our main endocrine response to stress, is made up of:
-
Hypothalamus (including the paraventricular nucleus, PVN)
-
Pituitary
-
Adrenal glands
Anxiety and mood disorders have been found to be associated with a dysfunctional stress response by the HPA axis [25]. Physiological stressors (which disturb internal homeostasis) utilize a bottom-up circuit to activate the HPA axis, via direct recruitment of the HPA axis by brainstem nuclei, while in contrast, psychological stressors utilize top-down processes to activate the HPA axis and produce a neuroendocrine response [26,27,28]. The hypothalamus, involved in the HPA axis stress response, integrates input from limbic and sensory brain areas to regulate HPA axis activation [6].
The paraventricular nucleus (PVN) of the hypothalamus is a key structure in the physiological stress response and integrates pre-autonomic and neuroendocrine control of energy homeostasis, fluid balance, and our stress response [29].
Other Areas
There are other brain regions that connect with these regions that are involved in emotional processing, regulation, and responses. For example, here are some of them:
Nucleus Accumbens
It is located in the rostral and ventral forebrain; receives input from the amygdala, hippocampus, and from cells of the stria terminalis; is believed to be the primary site mediating reward behavior and and to play a role in addictive behaviors associated with drug use [15].
Anterior Cingulate Cortex (ACC)
It has extensive connections to brain areas involved in emotional processing (including the amygdala), autonomic functions (e.g., lateral hypothalamus, brainstem centers), memory (e.g., hippocampus), and reward (e.g., orbitofrontal cortex, ventral striatum) [30]; integrates neuronal circuitry for “affect regulation” (ability to manage uncomfortable emotions) via connections to both the “emotional” limbic system and the “cognitive” prefrontal cortex [30]; and is involved in assessing emotions, emotional learning, and autonomic regulation, with the posterior portion involved in emotional regulation, autonomic regulation, and pain-related affect [30].
Medial Cingulate Cortex
It is “cognitive” in nature and is involved in several functions such as emotional appraisal, monitoring of conflict, selection and execution of responses, and others [30].
The Stress Response
Several stress response systems come into play following exposure to stress including the autonomic nervous system (in particular the sympathetic nervous system), the HPA axis, increased negative emotions and behavioral responses such as increased vigilance that help us avoid a threat [6].
Our central nervous system (CNS) is in communication with all other vital organ systems in our body in two ways: direct transmission of neural signals through the autonomic nervous system and via the neuroendocrine system through hormone release into the circulation affecting downstream organs [31]. The stress response thus has two components: an autonomic response and a neuroendocrine response [24].
The stress response to acute stressors or agents is considered adaptive; it mobilizes necessary resources in response to a threat, real or perceived. However, chronic activation of the stress pathways can be etiological factors in illness and/or exacerbate existing pathologies through alterations in metabolic processes, immune dysregulation, and prolonged systemic drive [2].
The Autonomic Stress Response
In the autonomic stress response , there is stimulation of sympathetic motor and hormonal outputs via descending neural circuits originating in hypothalamic pre-autonomic control centers. This leads to release of catecholamines in the brain and in the circulation [24].
The central nervous system (CNS) involves the brain and spinal cord, and the peripheral nervous system involves the autonomic nervous system and somatic nervous systems. There are two divisions of the autonomic nervous system (ANS):
-
The sympathetic nervous system (SNS)
-
The parasympathetic nervous system (PNS)
In terms of evolution, the SNS has evolved to keep us safe from predators in the past. In more recent times, our threats are not wild animals (unless you work in a game reserve possibly) but have become other immediate environmental threats. Think about what happens if you step out onto a busy road and suddenly there’s a car you didn’t see bearing down on you. It’s an immediate threat, and you want your SNS response to become activated fast—it is an appropriate response to an immediate threat to existence. However, we also know that there are other forms of existential threat, perhaps not as immediate but more insidious, psychosocial in nature, that we might experience under more chronic conditions. It is when our physiological stress responses become disproportionate and chronic that is dangerous to our well-being.
Under a threat, perceived or real, the SNS response (also called the “fight or flight” response) mobilizes the body’s energy resources such as glucose and oxygen and reallocates these quickly to those systems which must be activated (fast) to help us fight or get away (e.g., our skeletal muscles, heart, and vasculature). Our heart rate, blood pressure, and respiration rate and skin conductance all increase in response to a threat, and meanwhile, energy is diverted away from those areas of the body that are not important at that point in time for our survival (e.g., digestive system, reproductive system) [31]. The SNS is paired with the PNS, much like an accelerator (SNS) and a brake (PNS) on the car [32]: once the threat has passed, the PNS reduces the activation of the SNS and energy and blood flow once more resumes being supplied to areas requiring it for normal physiological functioning [31].
Sympathetic Nervous System Mechanisms of Action
The SNS is linked neuroanatomically to the limbic system and brainstem and reaches various areas of the body peripherally through the sympathetic ganglia (located bilateral to the spinal cord, extending from the neck to coccyx). There are two neural networks that constitute the SNS and use norepinephrine as a neurotransmitter:
-
Alpha-adrenergic fibers: in the cardiovascular system, these constrict blood vessels.
-
Beta-adrenergic fibers: in the cardiovascular system, these dilate blood vessels, and importantly, these fibers alone innervate the heart and cause increased heart rate and contractility [31]
Parasympathetic Nervous System Neuroanatomical Connections
In contrast, the PNS connects directly to peripheral organs via several of the cranial nerves, including the vagal nerve (ninth cranial nerve) and the pelvic splanchnic nerves that provide afferent as well as efferent channels. The PNS uses acetylcholine in particular as its neurotransmitter. The vagal nerve transmits signals to the heart which leads to reduced heart rate and reduced cardiac contractility, but it does not have any direct influence on arterial vasoconstriction or vasodilation (unlike the SNS). Thus, in terms of heart functioning, both heart rate and blood pressure are influenced by the SNS and PNS (since both innervate the heart) [31].
Measurement of ANS Activity
Popular outcome variables for measuring ANS functioning include heart rate, blood pressure, and heart rate variability (HRV) [31].
Neuroendocrine Stress Response
In contrast to the fast action of the ANS which utilizes nerve impulses, the neuroendocrine response to stress utilizes hormones traveling via the bloodstream, and for this reason, the neuroendocrine response is slower than the ANS responses to stressors [31].
The adrenal glands release two key stress hormones which impact on cardiovascular and other systems’ functions: cortisol from the adrenal cortex and adrenalin (epinephrine) from the adrenal medulla . The CNS constantly monitors the levels of these stress-responsive hormones, and there are negative feedback loops in operation to adjust their levels [31].
Both cortisol and epinephrine influence the SNS:
-
Cortisol: influences the sensitivity of the SNS beta-adrenergic receptors; enhances the sensitivity of SNS alpha-adrenergic receptors, catecholamine synthesis, and glucose production; release is mediated by the HPA axis.
-
Epinephrine: influences the sensitivity of the SNS beta-adrenergic receptors; release is mediated by the sympathoadrenomedullary axis (epinephrine is released from the adrenal medulla into the circulation following direct SNS transmission from the brainstem) [2, 31].
Herman et al. [2] argue in that the case of the two “stress systems,” the sympathoadrenomedullary axis and the HPA axis, their relationship with stressors is only one of their functions and in fact, both systems are mainly concerned with metabolism on a day-to-day basis, in that both mobilize energy in the liver to provide resources needed to respond to a challenge (glucocorticoids involved in gluconeogenesis, proteolysis, and lipolysis and epinephrine involved in glycogenolysis and glycolysis).
The HPA Axis in Stress
Continuing from above, Herman et al. [2] make an important point that the HPA axis is not a “stress system” per se, but rather a normal metabolic system that gets co-opted or recruited by stressors (causal agents of stress). There are several areas of the brain which are involved in its regulation and which play a role when there is a perceived or real threat. The functioning of the HPA axis in stress is described well by Herman et al. [2], and the following summary draws heavily from them.
The HPA Axis as a Metabolic System
The control of the functioning of the HPA axis is complex. The HPA axis is under circadian control, and peak activity corresponds to when we wake up, logical as we need to mobilize energy necessary for the activities of our day. The processes which result in the release of glucocorticoids are simply part of the normal metabolic role of the HPA axis which includes facilitation of glyconeogenesis, lipolysis, and proteolysis [2]. It is not just on waking that our glucocorticoid levels increase—levels of glucocorticoids also increase when we anticipate a meal. However, as Herman et al. [2] explain, under a condition of stress, this process essentially gets diverted to produce the glucocorticoids needed to deal with a stressor, some kind of threat to homeostasis (and of course, this could be psychosocial in nature). They make an important point relevant to research and the clinic: in attempting to quantify HPA axis activation in the context of investigating the body’s response to stress and/or in illness (i.e., by measuring changes in baseline glucocorticoid levels), one needs to take into account both stressor-elicited secretion and endogenous release patterns (e.g., circadian rhythms) [2].
Regulation of the HPA Axis
There are many factors that can activate the HPA axis which might be consistent with the need to provide energy quickly, and this includes disruptions to metabolism, thermoregulation , and fluid balance which are under the homeostatic control of the hypothalamus [2].
The paraventricular nucleus (PVN) of the hypothalamus is a central player in the HPA axis response. The classic understanding of the chain of events occurring in HPA axis activation is that PVN releases corticotropin-releasing hormone (CRH) into the portal vessels. The CRH travels to the anterior pituitary gland and binds with CRH receptors, and this stimulates the anterior pituitary to produce adrenocorticotropic hormone (ACTH) which is released into the systemic circulation. ACTH stimulates the release of glucocorticoid hormones (in humans this is mainly cortisol) from the adrenal cortex [3]. The glucocorticoids mobilize energy stores and produce several different effects on the cardiovascular, immune, metabolic, and neural systems [3]. There is then feedback mechanisms which serve to shut off the HPA axis response and restore homeostasis (discussed later).
As with anything in the body, there is a great deal more complexity involved. A key role of the PVN is integration of pre-autonomic and neuroendocrine control of energy homeostasis, fluid balance, and our stress response [29]. If a stressor that could pose a threat to homeostasis is encountered, information from the stressor is integrated across various different brain circuits prior to reaching the PVN. For example, the PVN is stimulated by afferents from the brainstem and hypothalamus which transmit information. The bed nucleus of the stria terminalis (BST) may be involved in the relay of contextual information [2]. Meanwhile, the subnuclei of the amygdala indirectly drive the HPA axis response via disinhibition mediated by GABAergic connections to PVN-projecting neurons in the hypothalamus and BST [2].
Very importantly, emotions must be considered within the context of stressors and HPA axis activation, since emotions can drive generation and interpretation of stressors [2]. When we look at post-traumatic stress disorder (PTSD) in a later chapter, we can also see how memory (in particular fear memories or memories of a traumatic event) can feed into the HPA axis and how the HPA axis “stress response” becomes dysfunctional [33].
Herman et al. [2] point out that there is a three-step amplification process that occurs as we move from the CRH to ACTH to glucocorticoids, i.e., fg/ml for CRH, pg/ml for ACTH, and then ng/ml for glucocorticoids, and this amplification process can be altered by the anterior pituitary and adrenals via various mechanisms. For example, sympathetic activation can increase glucocorticoid production by the adrenals, and negative glucocorticoid feedback can block release of ACTH from the pituitary [2].
Glucocorticoid signaling affects many systems. Glucocorticoids bind with glucocorticoid receptors and the mineralocorticoid receptors, and these act via many different mechanisms including acting as membrane receptors or transcription factors to effect changes in genes, and their effects on their targets can be fast (minutes) or can last weeks [2]. Glucocorticoid receptors are widely distributed in the CNS, and mineralocorticoid receptors are also present in several brain regions, including some areas such as the hippocampus where they co-localize with glucocorticoid receptors [2, 34].
Turning Off the HPA Axis
Under normal circumstances, the body tries to restore homeostasis since elevated glucocorticoids, if sustained, cause detrimental effects on health. There are complex and distributed negative feedback processes in the brain and periphery that serve to inhibit the PVN neurons and thereby turn off the HPA axis response [2, 3]. Glucocorticoid receptors in various brain regions including the hippocampus and PFC inhibit the HPA axis through glucocorticoid-mediated negative feedback [2, 3]. The corticolimbic system (i.e., PFC, amygdala, hippocampus) which communicates with the PVN and is involved in positive regulation of the HPA axis is also involved in its inhibition [35]. For example, neurons in the PFC and hippocampus inhibit the HPA axis via hypothalamic and BST GABAergic relays to the PVN [2]. There are also peripheral mechanisms of feedback, for example, from adipocytes [2].
Under normal circumstances, all works well, and following a stressful situation and activation of the HPA axis , negative feedback processes occur, and homeostasis is restored. However, under chronic stress conditions, HPA axis dysregulation can occur, and at the neurological level, this is probably due to inappropriate balance between the excitatory and inhibitory inputs impacting on the output of the PVN [2]. In early stages, this HPA axis dysfunction may be characterized by oversecretion of cortisol, but as time goes by, this can turn into what has been termed “adrenal exhaustion” where not enough cortisol is produced (with consequent low energy levels experienced by the patient) [36].
As you can see, the regulation of the stress response is quite complex. To take a deep dive, please read Herman et al.’s [2] paper.
Measurement of Neuroendocrine Activity
Commonly, measurements of neuroendocrine activity involve analysis of urine, blood, or saliva samples. Cortisol measurements need to take into account its normal diurnal fluctuations in secretion when investigating neuroendocrine responses to stress [31].
Sex Differences in the Stress Response System Along the HPA Axis
Substantial research has shown that there are sex differences in the stress response, in numerous components of the HPA axis, and these might explain at least partly why certain diseases are more prevalent in males and females [37]. This includes sex differences in basal cortisol levels, stress responsivity, and glucocorticoid receptors [37].
There are some differences in what has been found with animal data in comparison to human data. Animal data has demonstrated that the glucocorticoid levels generated are higher in females than males after HPA axis stimulation, but human research data has not been quite as clear-cut. Some studies have found no difference for HPA axis response to physical activity, and others have found either no difference in cortisol or higher cortisol in young men compared with young women under various psychological stress situations (real-life stress or laboratory stress tests) [37].
In general, it seems that adult men generate greater increases in cortisol in response to psychological stress than women [37]. This lower cortisol response found in women may be related to hypoactivity of the HPA axis, and it possibly might explain the greater prevalence of autoimmune conditions in women, while the greater stress reactivity and higher cortisol levels might possibly explain the higher prevalence of cardiovascular disease in men [37]. It is not clear yet if such differences between male and female humans in adrenocortical responses to stress are present at birth—there are some studies that suggest there is no difference and at least one study that suggests there may be [37, 38].
When the Stress Response Becomes Pathological
Allostasis is the body’s attempt to stabilize physiological functioning by modifying energy distribution to our vital organs, and the size and duration of the response will vary depending on the threat and the health status of the individual [31]. Allostatic load was defined as “the cost of chronic exposure to fluctuating or heightened neural or neuroendocrine response resulting from repeated or chronic environmental challenge” [39]. Another way of thinking about allostatic load is that it represents the “wear and tear” on the brain and body due to persistent and sustained activations of the stress response systems and the loss of the systems that normally buffer stress responses [6].
The ANS and neuroendocrine stress responses function to help us survive, as we have seen, but the activation of these systems is not always proportional to the nature of the threat. Stress responses can become dysfunctional and consequently negatively impact on health. Larkin et al. [31] explain that failure to maintain allostasis (dynamic homeostasis) in response to an acute environmental stress can lead to patterns of dysfunction of the ANS and/or neuroendocrine responses during chronic stress, and these can be quite different to what is seen in acute stress (e.g., hyperarousal in acute stress may lead to hypo-arousal in chronic stress).
As explained by Larkin et al. [31], dysfunctional ANS and neuroendocrine stress responses include (1) exaggerated reactivity to stress (characterized by increased heart rate, blood pressure, and levels of stress hormones such as cortisol), (2) blunted reactivity to stress, and (3) prolonged recovery from stress (e.g., prolonged recovery of blood pressure and heart rate associated with mental stress), which represent three patterns of “allostatic load,” and all of which can place a cumulative burden on body functioning that leads to increased disease risk (including CVD) [31, 40, 41]. Meanwhile, low stress reactivity (blunted reactivity) has been found to be associated with lower immune function, obesity, depression, and addictive behaviors including smoking and alcoholism (but not CVD) [31, 42].
There has been some interesting research investigating stress reactivity and cardiovascular disease (described in Larkin et al. [31]). Exaggerated cardiovascular reactivity to laboratory challenge can predict future cardiovascular morbidity and mortality [41]. Increased ANS reactivity, demonstrated, for example, by magnitude of heart rate and blood pressure reactivity, has been found to be associated with various stressful life situations such as being employed in stressful occupations or being a caregiver for a dementia sufferer [31, 43, 44]. In one study, chronic and momentary work stress (significantly) independently predicted greater heart rate reactivity after adjusting for heart rate, age, smoking, caffeine, and momentary physical activity levels [43].
In another study, caregivers with high levels of social support showed a typical age-related decrease in heart rate reactivity, but those with low levels of social support showed an age-related increase in heart rate reactivity [44] which underscores not only the stressful nature of being a caregiver but also the power of social support. Human beings are social beings, and research shows clearly that social connections and social therapy can have tremendous benefits on reducing mortality and morbidity, including in relation to serious illnesses such as cancer [8].
Stress and Inflammation
The relationship between stress and inflammation is well-known: stress can be proinflammatory [45]. There is increasing evidence that excessive inflammation plays a key role in the pathophysiology of stress-related diseases and that chronic, mild inflammation provides a common pathway of stress-related diseases [46]. In fact, it has been estimated that 75–90% of diseases are related to activation of the stress system. Common stress-related diseases include cardiovascular disease , metabolic disease (e.g., diabetes), mental health conditions (depression , Alzheimer’s disease ), neurodegenerative diseases, and cancer [46].
Stressful events trigger multiple neurochemical, neurotransmitter, and hormonal changes mainly through activation of the sympathetic nervous system (SNS) and the HPA axis. While traditionally, the link between stress and disease focused on the SNS and HPA axis, inflammation has more recently emerged as a promising biological mechanism linking stress and disease [46]. Chronic stress can lead to inflammation through production of proinflammatory cytokines such as IFNγ and TNFα [47,49,52,50,51]. Increased levels of inflammatory cytokines have been found in people with major depression [50, 52] , and we will look at that in more detail in a later chapter.
While inflammation can induce changes in the brain and body functioning which can contribute to a range of psychiatric conditions [45], it has been argued by several authors that even though for an individual patient, inflammation may be a or even the cause of depression, depression and other psychiatric conditions are not inflammatory conditions per se [45, 53]. The argument is that not every patient with depression has increased inflammation [53] and that inflammation may be one of several causes [45]. Psychological stress and inflammation can also underpin other conditions in the body such as coronary heart disease (CHD) [54] and cancer [8].
How Does Stress Activate Inflammatory Changes?
Stress can activate an inflammatory response both centrally and peripherally. When the HPA axis is activated, glucocorticoids are released, and these are immunosuppressive and anti-inflammatory, and indeed research indicates that glucocorticoids can reduce many types of proinflammatory cytokines including IL-6 and TNF-α as well as increasingly anti-inflammatory cytokines (e.g., IL-10, TNF-β). However, despite this, glucocorticoids have also been found to have a proinflammatory effect on the immune system. Pro- and anti- inflammatory mechanisms depend on both the type and intensity of the stressor involved. For example, acute stressors tend to enhance immune function, chronic stressors suppress immune function , and intense stressors overactivate the immune system, resulting in an imbalance between pro- and anti-inflammation effects [46]. To bring things back to the basics, there is a lack of balance and illness. We see this departure from balance in many different ways in the body when there is illness, something that ancient medical systems such as Chinese medicine recognized and sought then and seek now to identify and address as part of its fundamental approach to both diagnosis and treatment.
Centrally, neuroinflammation can occur under conditions of stress, with increased microglia activation , increased proinflammatory cytokines, and increased peripherally derived monocytes and macrophages occurring. It is thought that a triad of an overactivated immune system, increased activity via sympathetic nervous system pathways, and reduced glucocorticoid responsiveness may jointly activate the inflammatory responses associated with stress and that glucocorticoids, catecholamines, cytokines, and other chemicals released as a result of stress, in the main, mediate the proinflammatory effect induced by stress [46].
As we will see in later chapters, the theme of inflammation appears again and again in the pathophysiology of various mental health conditions, and while it is not the only pathological factor involved in such conditions, it appears to be a relatively consistent one. When we discuss the therapeutic attributes of medicinal cannabis, one of the key ones is its effect on inflammation.
Endocannabinoid System and Stress Regulation
The endocannabinoid system (ECS) regulates our responsivity to stress and emotional behavior, including the neuroendocrine and behavioral aspects of stress, seeking to inhibit stress and reduce its negative impacts [21, 24]. The ECS is also involved in the regulation of mood, anxiety, reward, and extinction of fear learning (which becomes dysregulated in post-traumatic stress disorder ) [20]. When there is disruption of endocannabinoid signaling, it recapitulates many of the effects of stress such as HPA axis activation [24].
Animal research indicates that endocannabinoid signaling is altered by stress as well as mediating or modulating stress responses [55], with mobilization of 2-AG occurring downstream of glucocorticoid receptor activation [56] and AEA increases occurring in response to corticotropin-releasing hormone receptor activation, secondary to FAAH inhibition [57, 58]. The endocannabinoid/CB1 receptor signaling can inhibit the activation of the HPA axis by stress and also enhance recovery following termination of the stress response [58].
Several Lines of Evidence that the ECS Is a Regulator of the Stress Response
Several lines of evidence provide support for contention that the ECS is an important regulator of the stress response, including the following:
-
Cannabis consumption in humans is associated with reduced anxiety and perceptions of stress and greater relaxation [24].
-
Animal studies indicate that disruption of endocannabinoid signaling produces a neurobehavioral phenotype which displays characteristics of the stress response including increased anxiety, activation of the HPA axis, hypervigilance, and impaired cognitive flexibility [24].
-
Components of the ECS (AEA, 2-AG, their degradative enzymes, and CB1 receptors) are structures found in the corticolimbic system and HPA axis [24].
-
Evidence of presence of the CB1 receptors in sympathetic neuron terminals and cells of the adrenal medulla [6].
Research suggests that both the sympathetic nervous system stress response and the neuroendocrine stress response (mediated by the HPA axis) are regulated by the ECS [6, 58].
Bidirectional Relationship Between Stress and the ECS
However, the relationship between the ECS and stress is not unidirectional. In addition to mediating and modulating the effects of stress on the brain, the ECS is altered by exposure to stress, and there is much animal research evidencing the roles of the endocannabinoids and CB1 receptor in brain adaptations as a result of exposure to repeated stress. Chronic stress exposure can increase an individual’s vulnerability to mental illness, and because of the association of the ECS to our stress systems, it may be an important therapeutic target for prevention and treatment of mental illnesses associated with stress [6].
ECS and the Autonomic Nervous System Stress Response
There is evidence that the ECS is involved in modulation of the sympathetic nervous system (SNS) stress response , as CB1 receptors have been found on sympathetic neuron terminals and cells of the adrenal medulla and CB1 receptor agonists have been shown to inhibit the release of the norepinephrine and epinephrine from these, as well as inhibit vasoconstriction and inflammation, both mediated by the SNS [6]. As yet though, the source and trigger for endocannabinoids innervating CB1 receptors on SNS terminals are not yet known [6]. Circulating levels of AEA and 2-AG in the peripheral circulation are increased with stress exposure [58]. As occurs in the CNS, the ECS in the periphery is activated by exposure to a stressor and serves to buffer the response through inhibition of release of catecholamines epinephrine and norepinephrine [6].
ECS, the Corticolimbic System, and the HPA Axis
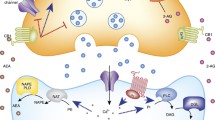
The ECS is involved in the homeostasis of the corticolimbic system, and signaling in corticolimbic structures is critical for regulating emotional behaviors like anxiety and stress in adults [19]. ECS involvement is evidenced by the fact that CB1 receptors are widely expressed and the two key endocannabinoids, anandamide (AEA) and 2-arachidonylglycerol (2-AG) , are synthesized in particular in the BLA (and also in the central nucleus), as well as in the hippocampus, medial PFC, and nucleus accumbens [24, 59, 60].
The basolateral amygdala (BLA) is believed to be an important site for ECS regulation of the HPA axis, acting as a “gatekeeper,” though there may be other sites also involved [24]. When experiments injecting CB1 receptor antagonists into the PVN or the mPFC were performed, no effect on basal corticosterone levels was found, but when injected into the BLA, an increase in HPA axis activity was demonstrated, supporting the BLA’s important role in HPA axis regulation [24].
The ECS does play an important role within the PFC, maintaining top-down regulatory mechanisms that serve to dampen stress and thereby cultivate resilience to effects associated with exposure to stress [6]. The ECS is also a negative regulator of amygdalar reactivity through CB1 receptors located in the amygdala [6].
We also find that CB1 receptors are expressed and AEA and 2-AG are synthesized in the hypothalamus, including in the PVN portion which, as we saw earlier, plays a central role in the neuroendocrine stress response via the HPA axis [29]. On a related note, research in rats also suggests that CB1 receptors in the ventromedial hypothalamus play a role in modulating the unconditioned fear-related defensive behavioral reactions [61].
ECS and Regulation of the HPA Axis
The ECS regulates the HPA axis in the maintenance of basal and stress-induced responses [25]. The ECS regulates basal and circadian HPA axis activation: for example, AEA levels in the hypothalamus are highest between 7.00 am and 11.00 am and low between 15.00 and 3.00 am [62].
There is a bidirectional relationship between the ECS and the HPA axis. Endocannabinoid signaling contributes to the regulation of the HPA axis, and conversely, the HPA axis helps regulate endocannabinoid signaling [3]. AEA is believed to represent the “tonic” signal, while 2-AG represents the “phasic” signal of the ECS [24]. Endocannabinoid tone, specifically AEA tone in the BLA in particular, provides a steady-state inhibition of the HPA axis, mediated via glutaminergic neurons, contributing to maintenance of low basal glucocorticoid levels during basal conditions and limiting HPA axis activity, and if this is disrupted, the HPA axis is activated [3, 25, 63].
The postulated mechanism by which AEA regulates the HPA axis is that a tonic level of AEA/CB1 receptor signaling in the BLA “gates” glutaminergic inputs to key neurons in the BLA, suppressing activation of the HPA axis [3]. Disruption of this AEA tone by stress activates the HPA axis with the resultant secretion of glucocorticoids [24]. Once the HPA axis is activated, the negative feedback mechanism comes into play to reduce the amounts of circulating glucocorticoids and reduce the HPA axis activity, thereby bringing the body back into balance.
The potential mechanism for glucocorticoid-mediated “fast feedback” inhibition of the HPA axis (this can occur in minutes) involves glucocorticoids inducing endocannabinoid signaling through a rapid process in CRH neurons of the PVN [3]. Glucocorticoids induce G-protein-dependent release of endocannabinoids and nitric oxide which inhibit glutaminergic inputs and enhance GABAergic inputs to CRH neurons in the PVN [2, 64]. In this way, they decrease the excitatory drive to the HPA axis [3].
Interestingly, there may be hormonal cross-talk occurring in PVN cells (in the hypothalamus) between glucocorticoids (which stimulate endocannabinoid synthesis and release) and leptin (which blocks glucocorticoid-induced endocannabinoid synthesis and suppression of excitation in the PVN) which may modulate synaptic excitation via endocannabinoid release in the hypothalamus. This could represent a mechanism for integration of the neuroendocrine regulation of energy homeostasis, fluid balance, and the stress response [29].
Let’s now look in more detail at what happens to the components of the ECS under conditions of acute and chronic stress.
How Is the ECS Involved in Acute and Chronic Stress?
In general, stress reduces levels of AEA and increases levels of 2-AG in most corticolimbic brain regions that have been studied. Current evidence suggests that AEA contributes to a tonic-like mechanism, regulating the basal synaptic transmission, while 2-AG acts as a phasic signal, activating CB1 receptors in a burst-like manner [24]. The purpose of the increase in 2-AG is to buffer and limit the detrimental effects of stress on the brain, and it plays a role in terminating the stress-induced HPA axis activation. It also contributes to habituation to stress [24].
However, as we saw in Chap. 2, the concept of the ECS has been extended to include the extended ECS, termed the “endocannabinoidome ” which refers to bioactive derivatives, enzymes, transporters, and a plethora of receptor targets modulated by ECS components [65, 66]. We also find evidence that several components of theendocannabinoidome come into play in stress. We will now look at how the key components of the ECS are involved in acute and chronic stress.
Acute Stress
Stress regulates both AEA and 2-AG levels. Glucocorticoids rapidly increase synaptic concentrations of 2-AG via membrane glucocorticoid receptors that are coupled to 2-AG synthesis, and in this way, activation of the HPA axis is directly linked to increased 2-AG levels [6].
Acute stress triggers a delayed increase in 2-AG levels: a moderate increase in 2-AG content in the medial PFC and a greater increase in 2-AG content in the hypothalamus [24]. However, some animal experiments using restraint stress have found there is no change in 2-AG levels in the amygdala, at least immediately after a certain time period of such stress [24, 67]. Differences in findings of such studies may reflect differences in stress models used. Elevated levels of corticosterone levels (corticosterone in animals is the correlate of cortisol in humans) are understood to mediate the increase in 2-AG in response to stress, but how it elevates AEA is still not understood [24].
In contrast, stress decreases AEA levels via its catabolic enzyme FAAH [6]. In response to acute stress, CRH is released, and this binds with the G-protein-coupled receptor CRH receptor 1, resulting in a fast increase in fatty acid amide hydrolase (FAAH, the enzyme responsible for AEA degradation) activity following activation of receptors on glutaminergic neurons in the BLA [6, 57]. Since FAAH degrades AEA, levels of AEA drop, leading to disinhibition of BLA neurons and increased output from neurons in the amygdala, which then impacts downstream, activating the HPA axis [3, 25]. Since AEA synthesis is constitutive and probably exerts tonic activation of CB1 receptors (i.e., maintaining low levels of neurotransmitter release at synapses that are regulated in this way), the removal of the AEA-induced tone (i.e., the decrease in AEA signaling) leads to reduced CB1 receptor signaling and increased neurotransmitter release (remember that the principal way in which endocannabinoids work is to inhibit the release of neurotransmitters), which then facilitates activation of the HPA axis and increases emotional and anxiety-like behavior, impairment of fear extinction, and suppression of cell proliferation in the neurogenic region of the hippocampus [6, 24, 25].
Animal studies indicate that acute stress reduces AEA content in the BLA and the hippocampus; however, its effect on the medial PFC is more complex and evidence is less consistent in the literature, with some experimentally induced stress experiences reducing AEA content and others not altering it or in the case of a mechanical stress like foot-shock (often used in mice experiments), elevating it [24].
There have been few investigations of the effect of acute stress on CB1 receptors in those regions of the brain associated with stress regulation. One study found that there was no effect of acute stress on density of CB1 receptors in the amygdala [27].
Chronic Stress
Chronic stress can downregulate the ECS, with resultant excessive stress responses and poor adaptation [6]. The sequela of chronic stress, in particular prolonged exposure to stress hormones (e.g., cortisol), is often illness, including depression, cardiovascular disease, and cancer [8, 68].
Various animal experiments suggest that under conditions of chronic stress, the endocannabinoid signaling in the paraventricular nucleus (PVN, of the hypothalamus) is impaired, resulting in impaired “fast-feedback” inhibition of the HPA axis, something that may help explain the elevated glucocorticoid levels in chronic stress [3, 69].
Under chronic stress, the following changes in the ECS occur:
-
CB1 receptors are downregulated: Generally, a downregulation of CB1 receptor expression in most parts of the brain involved in the stress response occurs, with the exception of the medial prefrontal cortex (where research indicates homotypic and heterotypic stressors increase CB1 receptor expression at the level of mRNA, protein, and receptor binding).
-
AEA is decreased: Sustained exposure to glucocorticoids upregulates CRH signaling which consequently maintains increased FAAH and therefore decreased AEA levels. This contributes to the activation of the HPA axis, manifestation of anxiety, impairment of fear extinction, suppression of cell proliferation in the neurogenic region of the hippocampus, and development of anhedonia and hyperalgesia.
-
2-AG is increased: Increases in 2-AG levels (found in acute stress) are amplified under conditions of chronic (repeated) stress, possibly mediated by reductions in MAGL (which degrades 2-AG) [13], leading to habituation of corticosterone responses (in animals, the equivalent of cortisol in humans). Increased 2-AG levels help terminate the stress-induced activation of the HPA axis as well as contribute to habituation to stress [24].
In animal experiments which are where much of our knowledge of mechanisms of action are gleaned, the effects of chronic stress on AEA, 2-AG, and CB1 receptors depend on the nature of the stress experienced , including whether it is homotypic (repeated exposure to the same stressor, e.g., restraint stress, social defeat stress) or heterotypic stress (exposure to different types of stress) [24]. Table 3.1 sets out the different effects of chronic homotypic stress and heterotypic stress on the endocannabinoids and CB1 receptors.
Mice experiments have demonstrated that stress-induced development of anxiety-like behavior is accompanied by the transient appearance of 2-AG-mediated long-term depression at GABAergic synapses in the basolateral amygdala (BLA, involved in motivation, affective regulation, and emotional learning) which is mediated partly by downregulation of MAGL. It appears that endocannabinoid synaptic plasticity at inhibitory synapses within the amygdala and increases in 2-AG levels prevent the behavioral and synaptic adaptations to chronic stress and suggests the possibility that pharmacological enhancement of 2-AG levels might be a useful approach to treating stress-induced mental health disorders [13].
The Endocannabinoidome (Extended ECS) and Stress
As mentioned previously, the extended ECS has been termed the “endocannabinoidome” (be careful of the spelling; it is not –“diome” as in gut microbiome). The endocannabinoidome refers to bioactive derivatives, enzymes, transporters, and a plethora of receptor targets modulated by ECS components [65, 66]. Members of the extended ECS are also involved in responses to stress. For example, TRPV1 plays an important role in regulating CNS function in response to stress. There is evidence that TRPV1 can modulate glial and neuronal activity, mediating several pathways including glial reactivity, cytokine release, synaptic transmission and plasticity [73]. Another example is that peroxisome proliferator-activated receptor γ (PPARγ) is expressed in brain regions involved in regulation of psychological stress and aging such as the hippocampus, and may play a role in preventing the effects of aging and stress on the brain. It is also known that PPARγ agonists are able to reduce the physiological stress response [74]. These are only two examples to simply indicate that the ECS involvement in stress regulation extends beyond the classic cannabinoid receptors.
Genes, Stress, and the Endocannabinoid System
Polymorphisms in genes coding for components of the ECS may influence an individual’s stress responses. Single nucleotide polymorphisms (SNPs) in CNR1, the gene that codes for CB1 receptors, have been found to be associated with stress-induced activation of the ventromedial PFC (the brain region which plays an important role in regulation of emotions) as measured using magnetic resonance imaging, while under non-stress conditions, this genotype was associated with enhanced cross-talk with the vmPFC and amygdala. Research suggests that there may be a protective effect of this particular genotype of the CB1 receptor polymorphism (CNR1; rs1049353) against stress-related psychopathologies [75].
Social Isolation
Rat experiments in which rats were socially isolated from weaning to adulthood found that the expression levels of CB1 receptors, DAGL-α, DAGL-β, MAGL, and NAPE-PLD mRNA, were significantly higher in several brain areas particularly the prefrontal area, cortical layers, and several thalamic regions. DAGL-β mRNA levels were significantly higher in the substantia nigra and ventral tegmental area, while FAAH mRNA expression was significantly lower in several prefrontal areas, cortical layers, and the caudate putamen [76].
Environmental Enrichment
Animal research (in mice) indicates that exposure to environmental enrichment during early life stages was associated with differential regulation of genes encoding for various components of the ECS. An experiment compared expression of CB1, receptors, FAAH, and MAGL in various brain regions in mice reared under conditions of environmental enrichment compared to standard environments, from weaning to adulthood. They found that environmental enrichment increased CB1 receptor mRNA levels in the hypothalamus and BLA but decreased them in the basomedial amygdala. They also found that FAAH mRNA levels were higher in the hypothalamus and BLA in mice reared under environmental enrichment conditions (no change in the basomedial amygdala), but MAGL mRNA levels were not affected in any of the brain regions investigated. The researchers surmised that these regional changes induced by environmental enrichment could indicate that early exposure to environmental enrichment can induce changes in the ECS that may result in reduced responses to stress, something they confirmed in findings that environmentally enriched mice had reduced stress responses (as measured in a novelty-induced suppression of feeding test) [77].
Early Life Stress and the Endocannabinoid System
The endocannabinoid receptors in the corticolimbic structures are critical to normal development, and if these receptors are altered by early life stress, brain development can be altered resulting in emotional and cognitive dysfunction . Animal and human research indicates that environmental stress during periods of high neural plasticity (e.g., during pregnancy, childhood, and adolescence) can cause emotional disturbances [19, 21, 78,79,80,81]. The effects of early life stress may emerge later in life, during adolescence or adulthood [21, 79]. For example, early life stress is associated with post-traumatic stress disorder (PTSD) and major depression in adulthood [78].
Early Life Stress, Corticolimbic System, and HPA Axis Development
Early life stress influences the development of the corticolimbic system and the HPA axis [21, 79] and increases vulnerability to adult psychopathology [79]. The corticolimbic system plays a key role in mediating the effects of early life stress on brain dysfunction later in life, and early life stress is a long-term risk factor for dysfunctional development of those limbic structures involved in stress and emotion regulation [79].
Severe early stress produces a chain of events involving stress-induced programming of glucocorticoid, noradrenergic, and vasopressin-oxytocin stress systems which augment stress responses. These then produce effects on neurogenesis, synaptic over production and pruning, and nerve myelination during sensitive developmental periods, resulting in reductions in size of areas of the brain (mid-portions of the corpus callosum); attenuated development of areas including the left neocortex, hippocampus, and amygdala; and abnormal frontotemporal electrical activity and decreased functional activity of the cerebellar vermis. These changes may be the mechanism through which early stress increases the risk of developing mental health conditions including depression, PTSD, ADHD, and others [82].
Stress Responsivity
The ECS plays a crucial role in regulating stress responsivity and emotional behavior during the child’s development and is sensitive to early life stress in a sex- and region-dependent way. The effect of early life stress on the ECS is bidirectional, that is, the ECS can reduce stress through its anxiolytic activity and environmental stress can alter the ECS anxiolytic capacity [21].
Findings from Animal Models
Animal experiments have demonstrated that maternal deprivation and social isolation in early life affect several parts of the ECS in neonate, adolescent, and adult rat brains, including altering CB1 mRNA expression and producing changes of cannabinoid receptors in corticolimbic and striatal brain areas [19, 21, 81]. In a rat experiment, using a maternal deprivation as a model for early life stress, researchers measured the expression of genes coding for CB1 and CB2 receptors, TRPV1 and GPR55 receptors, FAAH, MAGL, and enzymes involved in endocannabinoid synthesis (N-acyl phosphatidyl-ethanolamine phospholipase D and diacylglycerol lipase) in various brain regions: frontal cortex, ventral and dorsal striatum, dorsal hippocampus, and amygdala. They found that maternal deprivation increased the expression of all genes measured in the frontal cortex in adolescent male rats, but in females this increase was in the hippocampus [81].
Since ECS signaling in the amygdala plays an important role in production of anxiety and excitation of the HPA axis , it is possible that impairment of CB1 receptor signaling may sensitize the person to stress [21, 79].
Early Life Stress and Genetic Patterns in Humans
Early life stress in humans has been associated with specific genetic patterns including CACNA1C which encodes for an essential calcium channel that is part of ECS signaling. This channel relates to neuronal excitability, plasticity, and neurogenesis and was associated with childhood trauma leading to vulnerability to anxiety and depression [83]. Single nucleotide polymorphisms (SNPs) in CNR1 (gene coding for CB1 receptors) and the gene encoding for FAAH (specifically the genetic variant rs324420) are associated with bipolar disorder and major depression [84]. And even variants in CNR2 (codes for CB2 receptors) and the FAAH gene (functional polymorphism C385A) interact with childhood trauma and in anxious and depressive phenotypes [85]. Furthermore, Juhasz et al. [86] found that variants in the CNR1 gene are associated with high neuroticism and low agreeableness and interact with recent negative life events to predict current depressive symptoms. It would therefore appear evident that genes play a significant potential role in defining susceptibility to life stresses, personality types, and behavioral responses. More research is needed to define the exact influence of the various endocannabinoidome permutations on personality and psychopathology.
Sex Differences in How Early Life Stress Affects the ECS
In a similar way that there are sex differences between the stress response along the HPA axis, evidence suggests that there are sex differences in the ECS. Greater CB1R G-protein has been found in the hippocampus in males than females, but females showed greater CB1R G-protein activation, implying a more sensitive response of CB1Rs. It is possible that sex differences in the ECS could explain different responses to stressors [87].
Research indicates that early life stress decreases CB1 receptors in adulthood in males in the striatum, PFC, and amygdala (findings in females are not as clear) and increases gene expression for enzymes involved in endocannabinoid degradation in various brain regions (frontal cortex, striatum, hippocampus, and amygdala). Males also showed increased dorsal hippocampus prostaglandin products of arachidonic branch of the ECM.
In a rat experiment, early life stress demonstrated regional and sex-specific effects including:
-
Impairing peripheral basal corticosterone (adult males only)
-
Decreasing 2-AG and AEA in the cerebellar interpositus nucleus (males only)
-
Decreasing 2-AG in the cerebellar Crus (females only)
-
Increasing dorsal hippocampus prostaglandins (males only)
-
Impairing social preference (females only) [87]
The Gut and Stress
The gut is understood to play a critical role in the functioning of the immune system, influencing inflammation and the nervous system [88]. The gut “microbiome” refers to the microorganisms within the gut which form part of a multidirectional communication network with the brain, the microbiome-gut-brain axis [89]. The ECS is an important translational mediator for this communication.
The gut microbiota play a key role in the regulation of the gut-brain axis [90]. Vagal and spinal afferent pathways provide the means of communication between the gut microbes and the central nervous system [90]. The gut microbiota also modulate immune signaling from the gut to the brain via the induction of various cytokines [90].
The Gut Microbiome and Stress
The gut microbiome is involved in the stress response. There is increasing evidence that the gut microbiome and brain and immune and endocrine systems are all in constant communication, and the gut microbiome is a key player in the stress response [8]. The microbiome-gut-brain axis is believed to mediate the interaction between stress, the HPA axis, and the immune system [91] and to regulate moods and emotions. Stress can modulate the microbiota, and the microbiota can alter the set point for stress sensitivity [89].
The gut microbiota regulates the production of many neurotransmitters and their precursors including GABA, dopamine, acetylcholine, tryptophan, serotonin, and norepinephrine, and neurotransmitter imbalances or deficiencies are associated with mental health problems including depression and anxiety [89, 90]. Changes in GABA receptor expression are implicated in the pathogenesis of anxiety and depression, both of which are associated with functional bowel problems [92]. The gut microbiota can also secrete and upregulate essential proteins and metabolites involved in neuropeptide and gut hormone release, such as short-chain fatty acids and brain-derived neurotrophic factor [90].
Microbiota Changes and Stress
Several preclinical studies have demonstrated clear links between stress and gut microbiota changes [8]. For example:
-
Germ-free mice have an exaggerated HPA axis response to restraint stress which was reversed by mono-colonization with Bifidobacterium infantis [93].
-
Lack of normal gut microbiota in mice is associated with decreased expression of BDNF in the hippocampus, a key protein involved in neuronal plasticity and cognition [94].
-
Absence of the gut microbiota in rats has been found to exacerbate behavioral responses to acute stress and was associated with an altered neurotransmitter turnover rate in areas of the brain known to regulate reactivity to stress and anxiety-like behavior [95].
-
Treatment of mice with Lactobacillus rhamnosus (JB-1) induced changes in GABA mRNA expression in areas of the brain and reduced stress-related corticosterone and anxiety and depression-related behavior, with the vagus nerve implicated as the major modulatory pathway between the gut and the brain [92].
Research also indicates that depression is associated with dysregulated gut microbiota composition [89].
The Gut Regulates the HPA Axis
Evidence indicates that gut microbes may be involved in the development and functioning of the HPA axis [90, 93, 96]. As we have seen, dysregulation of the HPA axis occurs in stress, as well as in anxiety and depression. The gut microbiota can contribute to increased cortisol and inflammation, and inflammation can also feed into microbiota alterations through its negative effects on the gastrointestinal system [90, 97].
Leaky Gut and Mental Health
The gut has an important role in regulating intestinal permeability and maintaining the intestinal barrier. Deficits in intestinal permeability may be an etiological factor in chronic low-grade inflammation that can occur in some individuals suffering depression [98]. Gastrointestinal disorders , including exacerbations of their symptoms/signs, are often associated with stress.
Normally the intestine maintains tight junctions between the cells lining it. If these are compromised, the intestine lining becomes permeable or “leaky” [88], the so-called “leaky gut.” When there are high levels of cortisol and inflammatory mediators, these can increase the intestinal permeability, and Gram-negative bacteria (which have an additional lipopolysaccharide [LPS] exterior membrane) can move into the bloodstream [90]. This can then induce chronic inflammation in the CNS [90, 99]. Inflammatory gastrointestinal conditions such as irritable bowel syndrome, understood to involve compromised intestinal permeability, often co-exist with high rates of mental health conditions such as depression [100], lending further credence to the role of the gut in regulation of moods and emotion via the gut-brain axis [90].
A US study found that the prevalence and median levels of serum IgM and IgA against LPS of enterobacteria were significantly higher in patients with major depression than in controls [52]. Their results suggest that increased gastrointestinal permeability (leaky gut) with increased translocation of Gram-negative bacteria is involved in the pathophysiology of depression [52]. Systemic LPS and administration of proinflammatory cytokines can cause chronic central neuroinflammation, and central neuroinflammation is known to induce a sickness behavior complex that is similar to the symptoms of major depression (e.g., anorexia, malaise, loss of interest, etc.) [52].
The Endocannabinoidome , the Gut, and Links to Mental Health
There is a growing body of data that suggests that the gut, the brain, and the ECS are all interlinked when it comes to, at least some, mental health conditions. It is clear that the ECS plays a critical role in the functioning of the gut, given its wide distribution in the gut where it regulates most gut functions:
-
Gastrointestinal motility
-
Gastrointestinal inflammation and gut permeability
-
Nausea and vomiting
-
Visceral sensation
-
Immune homeostasis in the gut
-
Energy balance
ECS dysfunction is associated with pathology in the gut including inflammatory bowel disease (IBD) , irritable bowel syndrome, and obesity [102], and of course these conditions are inflammatory in nature. For example, CB1 receptor activation in the intestinal epithelium contributes to development of obesity and metabolic disease [104], and polymorphisms in the CNR1 gene (encodes for CB1 receptors) have been found to be associated with forms of irritable bowel syndrome [105]. Ulcerative colitis, one of the types of IBD, has been found to induce changes in the expression of the ECS in human colonic tissue [106].
Patients with visceral (abdominal) pain often find this is exacerbated by stress, and there is now evidence that the ECS modulates chronic stress-associated visceral hyperalgesia [103, 107]. CB1 receptors in sensory ganglia (that innervate the gut) control visceral sensation , and transcription of CNR1 (the gene encoding for CB1 receptors) is modified through epigenetic processes under conditions of chronic stress. There appears a potential mechanism to explain the link between stress and abdominal pain: chronic stress induces visceral hyperalgesia through region-specific changes in endocannabinoid and endovanilloid (endogenous ligand for TRPV1) pathways in sensory neurons that innervate the pelvic viscera [103].
Inflammation, the Gut, and the ECS
It appears that the gut, the ECS, and at least some mental health conditions (e.g., depression) may be linked, and this may be through inflammation. We have already seen that the gut microbiome plays a critical role in the functioning of the immune system, thereby influencing inflammation and the nervous system [88], and we also know that the gut is involved in the stress response. We also find that depression is associated with dysregulation of the gut microbiota composition, and in a later chapter, we will see that the ECS is altered in depression. There is also evidence that inflammation is involved in the pathogenesis of several other mental health conditions, including anxiety, autism spectrum disorder, and Alzheimer’s disease (discussed in later chapters). If inflammation is indeed involved in the pathophysiology of mental health conditions such as depression [52], then since one of the roles of the ECS is to control inflammation, it stands to reason that the ECS may play a role in those mental health conditions [50], and where “leaky gut” is part of the pathogenesis of a mental health condition such as depression, it would not be unreasonable to posit that the ECS within the gut may be important in the body’s attempts to bring the gut back into balance.
Understanding the potential or indeed likely link between the gut microbiome, the ECS, stress, and mental health reminds us that when treating mental health conditions such as depression, we should address any deficiency or dysfunction of the gut microbiota as part of a holistic treatment approach. This generally includes examining a patient’s diet and bacterial and possibly parasite exposures carefully. Of course, this is an area where more research is needed.
The role of the ECS in the gut and how it links with regulation of stress, emotions, and mental health conditions are an area of research that will no doubt expand in the future. In Chinese medicine, the link between emotions and dysfunction of organ systems, including (but not confined to) the digestive system, forms an important part of its theoretical model of physiological functioning of the body [108]. Biomedical research is in the process of elucidating the mechanisms of how emotions and the body are interrelated, albeit through the lens of its own (biomedical) model.
Cannabis Exposure and Stress Responsivity
Research indicates that the use of cannabis (i.e., typically containing high levels of tetrahydrocannabinol, the exogenous cannabinoid that is associated with dose-dependent intoxication) during adolescence, a time of maturation of the stress response system, can cause long-term alterations in stress responsivity, and cannabis use in adolescence has been associated with mental health conditions including depression and substance abuse disorder in adulthood in some preclinical and human studies [109, 110, 111] (discussed more in other chapters). For this reason, the recreational use of cannabis in adolescents should be avoided—this is a time of much neural plasticity and development of the brain and the ECS; therefore, exposure to any environmental toxins that can negatively impact on the synaptic pruning and refining occurring during this sensitive developmental phase should be prevented.
Conclusion
Stress is an increasing feature of our westernized lifestyles, and there is evidence linking it to many diseases including increased risk of cardiovascular disease, cancer, and mental health problems including anxiety and depression, sleep problems, and substance abuse. Stress impacts on the sympathetic nervous system and the HPA axis, and increasingly evidence implicates inflammation which may provide a common underlying pathway linking stress and stress-related illnesses. The ECS is involved in the regulation of stress and emotions, and when there is chronic stress, we also see changes in the ECS. The gut has gained increased importance within the scientific community, and it is clear that the ECS is critical in its homeostatic regulation. It is therefore not surprising that there is increasing evidence linking the gut microbiome, the ECS, and the stress response. Inflammation appears to be one underlying factor linking some mental health illnesses, stress, the gut microbiome, and the ECS. In treating mental health conditions, it therefore is prudent to remember to address any dysfunction in the gut.
Summary of Changes Associated with Acute and Chronic Stress
-
Acute and chronic stress: AEA is reduced and 2-AG increased.
-
Reduction of AEA occurs relatively quickly in response to stress and is mediated by CRH activating CRHR1 receptors to increase hydrolysis of AEA by increased FAAH.
-
Under chronic stress: ongoing exposure to glucocorticoids upregulates CRH signaling, and the FAAH increases and AEA decreases are maintained.
-
In acute stress, decreased AEA signaling contributes to anxiety, activation of the HPA axis, impairment of fear extinction, and suppression of cell proliferation in the hippocampus, and additionally, under chronic stress conditions, the more sustained reduction in AEA also contributes to anhedonia and hyperalgesia.
-
Increase in 2-AG is delayed and likely to be mediated by increased corticosterone. Amplification of 2-AG responses under conditions of repeated stress, which also results in habituation of corticosterone responses, may be mediated by reduced MAGL [24, 25]
References
Selye H. A syndrome produced by diverse nocuous agents. Nature. 1936;138(3479):32.
Herman JP, Nawreen N, Smail MA, Cotella EM. Brain mechanisms of HPA axis regulation: neurocircuitry and feedback in context Richard Kvetnansky lecture. Stress. 2020;23(6):617–32.
Hill MN, Patel S, Campolongo P, et al. Functional interactions between stress and the endocannabinoid system: from synaptic signalling to behavioral output. J Neurosci. 2010;30(45):14980–6.
Pearlin L. Stress and mental health: a conceptual overview. In: Horwitz AV, Scheid TL, editors. A handbook for the study of mental health: social contexts, theories and systems. Cambridge University Press; 1999. p. 161–75.
Chiang JJ, Turiano NA, Mroczek DK, Miller GE. Affective reactivity to daily stress and 20-year mortality risk in adults with chronic illness: findings from the National Study of Daily Experiences. Health Psychol. 2018;37:170–8.
deRoon-Cassini T, Stollenwerk RM, Beatka M, Hillard CJ. Meet your stress management professionals: the endocannabinoids. Trends Mol Med. 2020;26(10):953–68.
CAMH. Stress. CAMH, 2021. Available at: www.camh.ca. Accessed 5 Jan 2021.
O’Brien K, Sali A. A Clinician’s guide to integrative oncology: what you should be talking about with Cancer patients and why? US: Springer; 2017.
Dinan TG. Stress: the shared common component in major mental illnesses. Eur Psychiatry. 2005;20(S3):S326–8.
Weymar M, Schwabe L, Löw A, Hamm AO. Stress sensitizes the brain: increased processing of unpleasant pictures after exposure to acute stress. J Cognitive Neuroscience. 2012;24(7):111–1518.
Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167(6):648–62.
McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci. 2004;1032:1–7.
Sumislawski JJ, Ramikie TS, Patel S. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36:2750–61.
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873–904.
Willis MA, Haines DE. The Limbic System. In: Haines DE, Mihailoff GA, editors. Fundamental neuroscience for basic and clinical applications. 5th ed. Philadelphia: Elsevier; 2018.
Prager E. The corticolimbic system in health: implications for learning, memory and disease. Advanced Science News. Jan 27, 2017. Available at: https://www.advancedsciencenews.com/corticolimbic-system-health-disease/. Accessed 18 Jan 2021.
Rusbridge C. Neurobehavioral disorders: the corticolimbic system in health and disease. Vet Clin North Am Small Anim Pract. 2020;50(5):1157–81.
Thompson JM, Neugbauer V. Cortico-limbic pain mechanisms. Neurosci Lett. 2019;702:15–23.
Dow-Edwards D, Silva L. Endocannabinoids in brain plasticity: cortical maturation, HPA axis function and behaviour. Brain Res. 2017;1654:157–64.
Korem N, Zer-Aviv TM, Ganon-Elazar E, et al. Targeting the endocannabinoid system to treat anxiety-related disorders. J Basic Clin Physiol Pharmacol. 2016;27(3):193–202.
Goldstein Ferber S, Trezza V, Weller A. Early life stress and development of the endocannabinoid system: a bidirectional process in programming future coping. Dev Psychobiol. 2019;00:1–10.
Hubner C, Bosch D, Gall A, et al. Ex vivo dissection of optogenetically activated mPFC and hippocampal inputs to neurons in the basolateral amygdala: implications for fear and emotional memory. Front Behav Neurosci. 2014;8:64.
Waddell J, Bangasser DA, Shors TJ. The basolateral nucleus of the amygdala is necessary to induce the opposing effects of stressful experience on learning in males and females. J Neurosci 2008;28:5290–4.
Morena M, Patel S, Bains JS, Hill MN. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology. 2016;41:80–102.
Parker LA. Cannabinoids & emotional regulation. In: Cannabinoids and the brain. Massachusetts: MIT Press; 2017.
Herman JP, Figueiredo HF, Mueller NK, et al. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamic-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–80.
Hill MN, McLaughlin RJ, Morrish AC, et al. Suppression of amygdalar endocannabinoid signalling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34:2733–45.
Pecoraro N, Dallman MF, Warne JP, et al. From Malthus to motive: how the HPA axis engineers the phenotype, yoking needs to wants. Prog Neurobiol. 2006;79:247–340.
Malcher-Lopez R, Di S, Marcheselli VS, et al. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–50.
Stevens FL, Robin A, Hurley RA, Taber KH. Anterior cingulate cortex: unique role in cognition and emotion. J Neuropsychiatry Clin Neurosci. 2011;23:2.
Larkin KT, Brown LA, Tiani AG. Chapter 5. The autonomic and neuroendocrine response to stress. In: Chantler PD, Larkin KT, editors. Cardiovascular implications of stress and depression. Academic Press; 2020. p. 87–110.
Andreassi JL. Psychophysiology: human behavior & physiological response, 5th ed. In: Larkin KT, Brown LA, Tiani AG, editors. Chapter 5. The autonomic and neuroendocrine response to stress. Academic Press; 2007. p. 87–110.
Ney LJ, Matthews A, Bruno R, Felmingham KL. Cannabinoid interventions for PTSD: where to next? Prog Neuropsychopharmacol Biol Psychiatry. 2019;93:124–40.
Herman JP. Regulation of adrenocorticosteroid receptor mRNA expression in the central nervous system. Cell Mol Neurobiol. 1993;13(4):349–72.
Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409.
Patel R. HPA axis dysfunction explained: the facts you need to know. WebFMD. Nov 3, 2019. Available at: https://webfmd.com/hpa-axis-dysfunction/. Accessed 11 Jan 2021.
Goel N, Workman JL, Lee TT, et al. Sex differences in the HPA axis. Compr Physiol. 2014;4:1121–55.
Davis M, Emory E. Sex differences in neonatal stress reactivity. Child Dev. 1995;66:14–27.
McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153(18):2093–101.
Chida Y, Stepto A. Greater cardiovascular responses to laboratory mental stress are associated with poor subsequent cardiovascular risk status: a meta-analysis of prospective evidence. Hypertension. 2010;55(4):1026–32.
Panaite V, Salomon K, Jin A, Rottenberg J. Cardiovascular recovery from psychological and physiological challenge and risk for adverse cardiovascular outcomes and all-cause mortality. Psychosom Med. 2015;77(3):215–26.
Lovallo WR. Do low levels of stress reactivity signal poor states of health? Biol Psychol. 2011;86(2):121–8.
Lumley MA, Chi W, Wiholm C, et al. The relationship of chronic and momentary work stress to cardiac reactivity in female managers: feasibility of a smart phone-assisted assessment system. Psychosom Med. 2014;76:512–8.
Uchino BN, Kiecolt-Glaser JK, Cacioppo JT. Age-related changes in cardiovascular response as a function of a chronic stressor and social support. J Pers Soc Psychol. 1992;63(5):839–46.
Raison C. Introduction: the inflammation connection. Psychiatric Times. Apr 2018. Available at: https://www.psychiatrictimes.com/special-reports/introduction-inflammation-connection. Accessed 8 Jan 2020.
Liu Y-Z, Wang Y-X, Jiang C-L. Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci. 2017;11:316. https://doi.org/10.3389/fnhum.2017.00316.
Lutgendorf SK, Sood AK. Biobehavioral factors ad cancer progression: physiological pathways and mechanisms. Psychosom Med. 2011;73:724–30.
Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1- like response in stress-induced anxiety. Cytokine. 1998;10(4):313–8.
Maes M, Lin AH, Delmeire L, et al. Elevated serum interleukin-6 (IL-6) and IL-6 receptor concentrations in posttraumatic stress disorder following accidental man-made traumatic events. Biol Psychiatry. 1999;45(7):833–9.
Reiche EM, Nunes SO, Morimoto HK. Stress, depression, the immune system and cancer. Lancet Oncol. 2004;5(10):617–25.
Song C, Kenis G, van Gastel A, et al. Influence of psychological stress on immune-inflammatory variables in normal humans. Part II. Altered serum concentrations of natural anti-inflammatory agents and soluble membrane antigens of monocytes and T lymphocytes. Psychiatry Res. 1999;85(3):293–303.
Maes M, Kubera M, Leunis J-C. The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuroendocrinol Lett. 2008;29(1):117–24.
Miller MH. Five things to know about inflammation and depression. Psychiatric Times. Apr 30, 2018. Available at: https://www.psychiatrictimes.com/special-reports/five-things-know-about-inflammation-and-depression. Accessed 8 Jan 2020.
Wirtz PH, von Känel R. Psychological stress, inflammation, and coronary heart disease. Curr Cardiol Rep. 2017;19:111.
Hillard CJ. Stress regulates endocannabinoid-CB1 receptor signalling. Semin Immunol. 2014;26:380–8.
Di S, Malcher-Lopes R, Marcheselli VL, et al. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and GABA inputs to hypothalamic magnocellular neurons. Endocrinology. 2005;146:4292–301.
Gray JM, Vecchiarelli HA, Morena M, et al. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–92.
Hillard CJ. Circulating endocannabinoids: from whence do they come and where are they going? Neuropharmacology. 2018;43:155–72.
Hill MN, Carrier EJ, McLaughlin RJ, et al. Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem. 2008;106(6):2322–36.
Ramikie TS, Nyilas R, Bluette RJ, et al. Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutaminergic synapses. Neuron. 2014;81:1111–25.
Khan AU, Falconi-Sobrinho LL, Dos Anjos-Garcia T, et al. Cannabidiol-induced panicolytic-like effects and fear-induced antinociception impairment: the role of the CB 1 receptor in the ventromedial hypothalamus. Psychopharmacology (Berl) 2020;37(4):1063–79.
Liedhegner ES, Sasman A, Hillard CJ. Brain region-specific changes in N-acylethanolamine contents with time of day. J Neurochem. 2014;128:491–506.
Steiner MA, Marsicano G, Nestler EJ, et al. Antidepressant-like behavioural effects of impaired cannabinoid receptor type 1 signalling coincide with exaggerated corticosterone secretion in mice. Psychoneuroendocrinology. 2008;33:54–67.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23(12):4850–7.
Di Marzo V, Wang J, editors. The endocannabinoidome. The world of endocannabinoids and related mediators. US: Elsevier Inc; 2014.
Piscitelli F, Carta G, Bisogno T, et al. Effect of dietary krill oil supplementation on the endocannabinoidome of metabolically relevant tissues from high-fat-fed mice. Nutr Metab (Lond). 2011;8:51.
Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005;21:1057–69.
Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5(7):374–81.
Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–96.
Hill MN, Patel S, Carrier EJ, Rademacher DJ, et al. Downregulation of endocannabinoid signalling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30:508–15.
Lee TT, Hill MN. Age of stress exposure modulates the immediate and sustained effects of repeated stress on corticolimbic cannabinoid CB(1) receptor binding in male rats. Neuroscience. 2013;249:106–14.
Patel S, Kingsley PJ, Mackie K, et al. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signalling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–709.
Ho KW, Ward NJ, Calkins DJ. TRPV1: a stress response protein in the central nervous system. Am J Neurodegener Dis 2012;1(1):1–14.
Ulrich-Lai YM, Ryan KK. PPARγ and stress: implications for aging. Exp Gerontol 2013;48(7):671–6.
Wirz L, Reuter M, Felten A, Schwabe L. An endocannabinoid receptor polymorphism modulates affective processing under stress. Soc Cogn Affect Neurosci. 2018;13(11):1177–89.
Robinson SA, Loiacono R, Christopoulos A et al. The effect of social isolation on rat brain expression of genes associated with endocannabinoid signalling. Brain Research 2010;1343:153–67.
El-Rawas R, Thiret N, Nader J, et al. Early exposure to environmental enrichment alters the expression of genes of the endocannabinoid system. Brain Res. 2011;1390:80–9.
Davidson RJ, McEwan BS. Social influences on neuroplasticity: stress and interventions to promote well-being. Nat Neurosci. 2012;15(5):689–95.
Hill MN, Eiland L, Lee TT, et al. Early life stress alters the developmental trajectory of corticolimbic endocannabinoid signalling in male rats. Neuropharmacology. 2019;146:154–62.
Marco EM, Viveros MP. The critical role of the endocannabinoid system in emotional homeostasis: avoiding excess and deficiencies. Mini Rev Med Chem. 2009;9:1407–15.
Marco EM, Echeverry-Alzate V, Lopez-Moreno JA, et al. Consequences of early life stress on the expression of endocannabinoid-related genes in the rat brain. Behav Pharmacol. 2014;25:547–56.
Teicher MH, Andersen SL, Polcari A, et al. Developmental neurobiology of childhood stress and trauma. Psychiatr Clin North Am. 2002;25(2):397–426.
Lazary J, Eszlari N, Kriko E, et al. Genetic analyses of the endocannabinoid pathway in association with affective phenotypic variants. Neurosci Lett. 2021;744:135600.
Monteleone P, Bifulco M, Maina G, et al. Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression. Pharmacol Res. 2010;61:400–4.
Lazary J, Eszlari N, Juhasz G, Bagdy G. A functional variant of CB2 receptor gene interacts with childhood trauma and FAAH gene on anxious and depressive phenotypes. J Affect Disord. 2019;257:716–22.
Juhasz G, Chase D, Pegg E, et al. CNR1 gene is associated with high neuroticism and low agreeableness and interacts with recent negative life events to predict current depressive symptoms. Neuropsychopharmacology. 2009;34:2019–27.
Moussa-Tooks, et al. Long-term aberrations to cerebellar endocannabinoids induced by early-life stress. Sci Rep. 2020;10:7236.
Perlmutter D. Brain maker. Great Britain: Yellow Kite; 2015.
Sherwin E, Rea K, Dinan TG, Cryan JF. A gut (microbiome) feeling about the brain. Curr Opin Gastroentero. 2016;32(2):96–102.
Simpson CA, Diaz-Arteche C, Eliby D, et al. The gut microbiota in anxiety and depression – a systematic review. Clin Psychol Rev. 2021;83:101943.
De Palma G, Collins SM, Bercik P, Verdu EF. The microbiota-gut-brain axis in gastrointestinal disorders: stressed bugs, stressed brain or both? J Physiol. 2014;592(Part 14):2989–97.
Bravo JA, Forsythe P, Chew MV, et al. Ingestion of Lactobacillus strain regulates emotional behaviour and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A. 2011;108(38):10650–5.
Sudo N, Chida Y, Aiba Y, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol. 2004;558(Pt1):263–75.
Mayer EA, Knight R, Mazmanian SK, et al. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34(46):15490–6.
Crumeyrolle-Arias M, Jaglin M, Bruneau A, et al. Absence of the gut microbiota enhances anxiety-like behavior and neuroendocrine response to acute stress in rats. Psychoneuroendocrinology. 2014;42:207–17.
Foster JA, Rinaman L, Cryan JF. Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol Stress 2017;7:124–36.
Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–35.
Kelly JR, Kennedy PJ, Cryan JF, et al. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front Cell Neurosci. 2015;9:932.
Huang TT, Lai JB, Du YL, et al. Current understanding of gut microbiota in mood disorders: an update of human studies. Front Genet. 2019;10:98. https://doi.org/10.3389/fgene.2019.00098.
Simpson CA, Schwartz OS, Simmons JG. The human gut microbiota and depression: widely reviewed, yet poorly understood. J Affect Dis. 2020;274:73–5.
Acharya N, Penukonda S, Shcheglova T, et al. Endocannabinoid system acts as a regulator of immune homeostasis in the gut. Proc Natl Acad Sci U S A. 2017;114(19):5005–10.
DiPatrizio NV. Endocannabinoids in the gut. Cannabis Cannabinoid Res. 2016;1:1.
Sharkey KA, Wiley JW. The role of the endocannabinoid system in the brain-gut axis. Gastroenterology. 2016;151(2):252–66.
Cani PD. Crosstalk between the gut microbiota and the endocannabinoid system: impact on the gut barrier function and the adipose tissue. Clin Microbiol Infect. 2012;18(4):50–3.
Camilleri M, Kolar GJ, Vazquez-Roque MI, et al. Cannabinoid receptor 1 gene and irritable bowel syndrome: phenotype and quantitative traits. Am J Phys. 2013;304:G553–60.
Marquez L, Suarez J, Iglesias M, et al. Ulcerative colitis induces changes on the expression of the endocannabinoid system in the human colonic tissue. PLoS One. 2009;4:e6893.
Storr MA, Sharkey KA. The endocannabinoid system and gut-brain signalling. Curr Opin Pharmacol. 2007;7:575–82.
O’Brien KA, Xue CC. The theoretical framework of Chinese medicine. In: Leung P-C, Xue CC, Cheng Y-C, editors. A comprehensive guide to Chinese medicine. 2nd ed. New Jersey: World Scientific Press; 2016.
Andersen SL, Teicher MH. Desperately driven and no brakes: development stress exposure and subsequent risk for substance abuse. Neurosci Behav Rev. 2009;33(4):516–24.
McCormick CM, Green MR. From the stressed adolescent to the anxious and depressed adult: investigations in rodent models. Neuroscience. 2013;249:242–57.
Lee TTY, Gorzalka BB. Timing is everything: evidence for a role of corticolimbic endocannabinoids in modulating hypothalamic–pituitary–adrenal axis activity across developmental periods. Neuroscience. 2012;204:17–30.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
O’Brien, K., Blair, P. (2021). The Endocannabinoid System, Stress, and Mental Health. In: Medicinal Cannabis and CBD in Mental Healthcare. Springer, Cham. https://doi.org/10.1007/978-3-030-78559-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-78559-8_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78558-1
Online ISBN: 978-3-030-78559-8
eBook Packages: MedicineMedicine (R0)