
Abstract
In this chapter, the development of controlled atmosphere conditions to study gas-solid reactions inside the transmission electron microscope (TEM) will be presented. The two successful approaches to achieve this, namely the use of electron transparent windows and the incorporation of small-bore apertures inside the TEM combined with differential pumping, will be discussed. Finally, we will also describe the state-of-the-art instrumentation available today to study the behavior of nanomaterials in reactive gas environments, which have been largely brought about by the development of aberration correctors, monochromators, specialized TEM holders, as well as faster and more sensitive spectrometers. Examples that highlight the diverse applications in this field will be provided.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Gas-solid reactions
- Windowed cell
- Differential pumping
- Environmental transmission electron microscopy
- Aberration-correction
- Monochromator
- Specialized holders
- Electron energy-loss spectroscopy
- Catalysis
1 Introduction
It was clear to the original pioneers in the 1930s or so that an electron microscope requires a background high vacuum. Indeed, once the transmission electron microscope (TEM) was being applied to examine the microstructure of metals and alloys, specimen contamination, generally from breakdown of residual hydrocarbons in the TEM atmosphere, became a real issue. This was further exacerbated by the introduction of small probe work in the 1970s for microanalysis of chemical composition. The historical trend therefore required increasingly good vacuum conditions especially in the specimen and electron gun chambers.
This evolution clearly contrasts with the desire to study materials in their natural environment, such as atmospheric gaseous conditions or even in liquids. Accordingly, methods needed to be devised whereby the overall microscope vacuum environment could be maintained while viewing the sample in a controlled environment. In this chapter, we will outline the historical progression of these developments alongside the advances in TEM itself.
The main purpose of such experiments is to derive as much information as possible about material interactions with the environment, whether under ambient conditions such as in corrosion or in vivo biological processes, or extreme conditions as might be required for catalytic reactions or non-equilibrium liquid phase processes. This clearly represents a significant broadening to the scope of in situ TEM observations whereby one examines the changes to a material system under a controlled external stimulus such as heating, cooling, electrical bias, and mechanical stress. The behavior of the specimen under high-vacuum conditions already has its own merit as many manufacturing processes are carried out in vacuum, especially for the semiconductor industry. However, the added value of the controlled environment has extensive implications. Indeed, a recent analysis of in situ TEM publications (Sinclair 2013) showed a noticeable increase in recent years attributable mainly to environmental TEM (ETEM) studies, as illustrated in Fig. 1.1, a trend which continues as noted later.

(a) Number of publications identified using the keywords “in situ”, “transmission electron microscopy” and “TEM”, showing a steady increase over the last 20 years and a marked increase in 2012; (b) Breakdown by various subcategories of in situ papers presented at the 2012 Microscopy and Microanalysis Conference (USA) and the 2012 European Microscopy Conference (UK). These subcategories are in situ electrical biasing, electron beam heating, thermal heating, gaseous environment, liquid environment, mechanical loading, and laser stimulation. Reprinted with permission from Sinclair (2013)
Historically, the interest for environmental chambers inside the electron microscope came about as a means to allow wet specimens to be examined in vivo (Marton 1934) while at the same time, minimizing any possible damage and artifacts associated with conventional biological specimen preparation. The presence of an environmental chamber was also thought to be advantageous in reducing specimen contamination produced by the electron beam and residual hydrocarbon gases present in the microscope. In the 1970s, the reason to pursue this expanded to include the study of chemical reactions at elevated temperatures, as described later.
In 1935, Marton (1935) suggested two methods in which gas pressure and composition could be controlled inside the electron microscope. The first involved modifying the specimen holder by placing a pair of electron transparent “windows” above and below the specimen to seal it, and the gas atmosphere, from the column. The second involved modifying the objective pole pieces of the electron microscope, by placing a pair of small apertures above and below the specimen (see also later papers by Ruska 1942, and Abrams and McBain in 1944). Gas leakage into the column is then limited to that which escapes via the apertures. These two approaches have evolved separately, and successfully, alongside the TEM and are referred to as the closed cell (“window”) and “apertures,” respectively, in this chapter.
In the 1970s, the term “controlled atmosphere electron microscopy” (CAEM) was coined by several groups including Feates et al. (1970) and Baker (Baker and Harris 1972; Baker 1979). Despite the sub-nanometer imaging resolution of the TEM (about 0.25 nm or so in the 1970s), its value was found to be limited because materials can only be viewed in a vacuum environment, and the tactic had been to examine a specimen before and after reaction. Of significant interest is the actual appearance of the specimen during reaction. Thus, CAEM is used to describe the technique in TEM in which reactions between gases and solids are studied at very high magnifications, while they are taking place, under realistic conditions of temperature, pressure, and reaction time (Baker 1979). The key design feature of CAEM is the ability to operate at high pressure in the specimen region (3 × 104 Pa, or 225 Torr), while maintaining very low pressure (10−3 Pa, or 10−5 Torr) in the rest of the microscope.
Since the 1970s, there have been a number of comprehensive reviews highlighting the developments in the field of CAEM, or environmental transmission electron microscopy (ETEM) as it is more commonly known today, as well as its diverse applications. Readers can refer to the articles and book chapters by Flower (1973), Allinson (1975), Butler and Hale (1981a), Sharma (2001, 2005), Sharma and Crozier (2005), Gai et al. (2007, 2008), Jinschek and Helveg (2012), publications from the Technical University of Denmark (Hansen and Wagner 2012; Wagner et al. 2012), Jinschek (2014), the Takeda group from Osaka University (Takeda et al. 2015) as well as their references therein. We have elected to present the historical developments in this field in a chronological sequence, with an emphasis toward gas-solid reactions in the TEM. For comprehensive reviews and applications for liquid-solid reactions and interactions, articles and book chapters by Parsons (1974), Parsons et al. (1974), Butler and Hale (1981b) and de Jonge and Ross (2011), and their references therein, are good starting points. A later chapter of this book also discusses liquid phase electron microscopy techniques and applications. We conclude with a number of examples spanning from the 1970s that highlight the diverse applications in this field, and where we think the future in this area lies.
1.1 Window Approach
The first approach to attain environmental conditions inside the transmission electron microscope is by use of a specialized specimen holder combined with a windowed cell. A pair of electron transparent “windows” is placed above and below the specimen to seal it, and the gas atmosphere, from the column. In this method, containment is total—the windows completely seal off the specimen and its surrounding gas environment so that the high vacuum of the microscope remains unchanged. The windowed design has the advantage of working with higher gas pressures (depending on the strength and thickness of the windows). Wet samples can also be imaged with this setup. The reader can refer to a later chapter in this book which describes the developments in the field of liquid cell electron microscopy.
There are several advantages using a specialized holder combined with windowed cells. First of all, the holder can be used in different TEMs without modifications to the microscope column. To date, this approach is the only one that is capable of achieving ambient pressure conditions. However, the specimen geometry and the field of view are usually significantly smaller than in a conventional TEM, and the sample can only be moved or tilted together with the windows since it is sandwiched between them. Additional scattering information from the (usually amorphous) window material is superimposed on the image and diffraction pattern of the sample, leading to compromises in image resolution. The interaction also hinders the acquisition of energy-dispersive X-ray spectroscopy (EDS) signals. The thin films used as windows should have low gas or vapor permeability, and must be able to withstand the pressure difference between the microscope vacuum and the design pressure of the holder device when mounted. There is the possibility of windows rupturing inside the microscope during an experiment, leading to the degradation of the electron column and source vacuum.
The earliest documentation of a closed cell for electron microscopy was by Marton in 1935, who attempted to use two 0.5-μm aluminum foils as windows. In 1944, Abrams and McBain constructed a closed chamber out of two perforated platinum disks covered with plastic film windows less than 100 nm thick, and sealed to vacuum-tightness using wax. It could withstand an atmospheric pressure difference between the inside of the cell and the remainder of the electron microscope. Using this setup, the authors were able to observe the movement of liquid and bubbles inside the electron microscope. However, they found water of one micron thickness to be “practically opaque to electrons.” The lack of fast, continuous recording capability also precluded the possibility of the observations of Brownian movement in colloidal particles. Moreover, the cell was completely sealed and hence capable of operating at only a single (fixed) pressure.
Variable-pressure controlled dynamic experiments were not possible until cells were designed such that a connection existed between the gas space around the specimen and the outside of the microscope. Heide (1962) constructed a specimen holder for a Siemens Elmiskop I electron microscope using two specimen grids with the flat surfaces facing each other. The separation of the two grids was controlled by pieces of thin metal foil. Both specimen grids were covered with a supporting film of low contrast, which could withstand the gas pressure over the central openings, and one of the grids served at the same time as a supporting film for the specimen. The gas inlet was sealed against the microscope column vacuum when the holder was inserted inside the microscope, and gas was injected through a tube placed in the opening of the column along the specimen holder. Air or gas pressures could be varied up to atmospheric pressure with this design.
There were a number of limitations to these earlier basic designs of the closed cells, as highlighted in the review article by Butler and Hale (1981a):
-
1.
The single gas line does not enable experiments to be performed with gas flowing continuously over the specimen.
-
2.
The relatively poor metal-to-metal seal between the apertures and the cartridge results in some leakage into the column.
-
3.
The specimen cannot be heated unless it is supported independently of the windows.
Improvements in these three areas were realized in later designs. Escaig and Sella (1969) incorporated a heater and twin gas lines in their holder so that the temperature of the specimen could be varied while gas continuously circulated over it. Earlier window cells were made out of plastic which were generally not suitable for high temperature work because of damage during heating and were thus employed mainly in the biological field to provide a hydrated environment for in vivo studies. Escaig and Sella (1966, 1968, 1969) made improvements to the windowed cells that consist of triple layers of carbon, nitrocellulose, and silica (facing the specimen), maintaining a gap of approximately 1 mm between window and specimen, which was successfully employed for in situ oxidation of copper, tungsten, and titanium (Escaig and Sella 1972).
Another limitation of the windowed cells is that they tend to bulge under pressure, resulting in a longer electron beam path length at higher pressure. In order to reduce the beam path length and stabilize the films, Fukami and Adachi (1965) developed plastic micro grids consisting of holes of 0.1–10 μm in diameter which were used as the supporting frame for a continuous carbon film. The plastic micro grids were also used as substrates in the production of micro metal grids with thicknesses >30 nm which were stronger and more resistant to heat than their plastic counterparts (Fukami et al. 1966, 1972).
In the late 1960s and 1970s, the development of high-voltage (1 MeV) electron microscopes (HVEM) led to enclosed gas reaction cells with thicker, stronger windows being developed. Dupouy (1968) designed a single gas-line room temperature device with windows constructed out of carbon/colloidion of 60 nm thickness. Twin gas-line top entry designs capable of a range of temperatures were also constructed by Fujita et al. (1976) and Doi et al. (1977). Fujita et al. (1976) built a holder for gas and liquid experiments on a 3MV-class electron microscope, which could be used in the temperature range of −100 to 1000 °C and up to a pressure close to two atmospheres. The window cell consisted of three films whose materials could be vapor-deposited aluminum, SiOx and carbon films, depending on the purpose, and they were supported with metal (Ni) of 300 and 400 mesh in size. The specimen was set directly on the center Ni mesh, or on a vapor-deposited film coated on the Ni mesh. The specimen was directly heated by the application of a current to the Ni mesh. The increased penetration ability of the HVEM led to a significant improvement in contrast at these high operating voltages. Specimen damage was found to depend on the heat conductivity of the supporting film, and decreases when the supporting film has high heat conductivity. The use of crystalline window materials of higher strength also became a consideration for use with HVEM, and single crystal corundum which was ion-beam-thinned to ~180 nm was one possible material option (Allinson 1970). In window-limited holders, the strength and thickness of the window material determines the maximum gas pressure and, in principle, pressures as high as atmospheric pressure can be attained, although the thickness of window material needed to sustain such pressures may not be compatible with instrument performance.
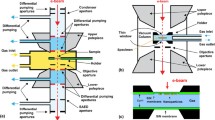
Progress in the development of closed window cell systems was stalled until the 2000s, when holders based on micro-electro-mechanical systems (MEMS) technology for both gas (Creemer et al. 2008; de Jonge et al. 2010; Yokosawa et al. 2012; Yaguchi et al. 2011; Allard et al. 2012; Alsem et al. 2012) and liquid (Williamson et al. 2003; de Jonge et al. 2009; Ring and de Jonge 2010; Klein et al. 2011) TEM applications emerged. This technique uses miniaturized, low mass heating devices and is integrated with a heater and thermal sensor which allows much more rapid thermal response than standard specimen furnace-based heating holders. The specimen drift is also stabilized at a more rapid rate, leading to better control of the reaction process and the imaging experiments. The windows are usually produced using micro-fabrication techniques and consist of a silicon frame with an electron transparent silicon nitride membrane which is tens of nanometers thick. A lot of effort was given to miniaturize the thickness of the gas volume in the windowed cell to confine the gas along the electron beam path into a layer that is as thin as possible (tens of micrometers). Consequently, these MEMS devices allow for correspondingly higher pressures without loss of resolution or damage to the electron source. Atomic resolution with holders like these have been attained, sometimes at local gas pressures of one atmosphere or more (Giorgio et al. 2006; Creemer et al. 2008; de Jonge et al. 2010; Yokosawa et al. 2012; Yaguchi et al. 2011; Allard et al. 2012; Alsem et al. 2012). Improvements are also made to the windowed cell support film. Kawasaki and coworkers (2009) demonstrated that carbon films with thickness <10 nm could withstand pressure differences greater than two atmospheres. A schematic design of the flow system for atmospheric pressure (scanning) TEM ((S)TEM) is illustrated in Fig. 1.2.

Schematic of the flow system for atmospheric pressure (scanning) transmission electron microscopy ((S)TEM). A sample compartment filled with gas at atmospheric pressure is enclosed between two silicon microchips supporting electron-transparent SiN windows. The microchips are separated by a spacer and sealed with epoxy. Gas entry and exit are not shown in this drawing. The flow cell is placed in the vacuum of the electron microscope. Images are obtained by scanning a focused electron beam over nanoparticles attached to the top window and detecting elastically scattered transmitted electrons. The dimensions and angles are not to scale. Reprinted with permission from de Jonge et al. (2010)
1.1.1 Specimen Holders with Gas Injection Systems
An alternative specimen holder design which reduces the number of gas molecules inside the microscope column incorporates a gas injection system was developed by Kamino and coworkers in 2005 (Kamino et al. 2005). This holder is compatible for use with a 300 kV Hitachi high-resolution H-9500 TEM, which has a high-speed turbo molecular pump added to pump the specimen chamber, a 0.3 mm restricting aperture above the specimen chamber to restrict the gas leakage into the upper column, and a safety valve in the gun area to further maintain the safe high vacuum in the gun area. Specimens are supported on a fine tungsten wire which was employed as a heating element of the holder, and gas was injected onto specimens via a nozzle. The maximum pressure near specimens is ~10−2 Pa, while the pressure in the electron gun chamber was kept to ~3 × 10−4 Pa (2 × 10−6 Torr).Footnote 1 The holder requires no self-confined window cell and can heat the sample up to 1000 °C with the injection of gases like H2, O2, air, water vapor, etc. An evaporator could also be added to the design to allow for both synthesis of metal oxide support and deposition of catalyst nanoparticles in situ (Kamino et al. 2006). For pressures up to an atmosphere, a windowed environmental cell holder is necessary. Yaguchi et al. (2011) give details of a windowed environmental cell holder for Hitachi microscopes that builds on the Kamino et al. (2005) design, wherein the tungsten wire passes between two SiN windows of 15 nm thickness with a spacing of about 1 mm, and incorporates a micropressure gauge fabricated using MEMS technology, to measure the pressure inside the windowed cell accurately.
1.2 Aperture Approach
The second approach to controlled atmosphere transmission electron microscopy makes use of small apertures placed above and below the specimen, which restrict the flow of gas from the specimen chamber into the microscope column. The apertures must be large enough to permit the passage of incident, diffracted and scattered electron beams, and yet small enough to avoid too great a gas leak. Leakage into the column vacuum is controlled by the size of the apertures and supplemented by differential pumping. In order to maintain the pressure conditions at the specimen cell while retaining the very low pressure in the rest of the microscope, an equilibrium condition such that the effective pumping speed of the differential pumps exceeds the leak rate of the apertures should be achieved. The first pair of apertures is placed closest to the sample and most of the gas leaking through these apertures is pumped out of the system using a turbo molecular pump. The second pair of apertures is larger than the first pair (because they experience a much smaller pressure drop) and is used to further restrict the leakage of gases into the microscope column.
The main advantage of the aperture type system is that the basic instrument resolution is preserved (i.e., not degraded by the presence of membranes in the windowed cell). Specimen holders that are used in a regular (high-vacuum) TEM are also compatible with this setup. However, the maximum gas pressure is limited to ~103 Pa (tens of Torr) at best. At higher gas pressures, the resolution deteriorates because of the rather thick gas layer (5–10 mm depending on pole-piece gap).
The first aperture environmental cell inside the TEM was reported and constructed by Ito and Hiziya in 1958 to enable direct and continuous observations of chemical reactions in a heated gas environment. The specimen chamber of a JEM electron microscope was modified and diaphragms were introduced to prevent diffusion of the gas from the chamber to the electron source. The gas pressure ranged from about 10−2 to 133 Pa (10−4 to 1 mm Hg), and the specimen could be heated up to 1000 °C using a high temperature furnace which the group had constructed two years earlier (Takahashi et al. 1956).
In the same year, Hashimoto and coworkers (1958) constructed a specimen chamber with a metallic wire with a high melting temperature (e.g., tungsten) as the heating device. Gas pressures of up to ~1.33 Pa (10−2 Torr) could be introduced through a valve of the holder to react with the specimen at elevated temperature. The range of pressure was expanded to about 40 kPa (300 Torr) in a later design which included differential pumping (Hashimoto et al. 1966, 1968). In both versions, the wire was placed between two Pt apertures of diameters 50–100 μm and spaced 0.3–0.5 mm apart. The specimen stage could be taken out of the electron microscope column through an airlock system without breaking the vacuum. These efforts led to the production of a commercially available (JEOLCO JEM AGI attachment) gas reaction stage, and the image resolution achievable was 5–10 nm with an air pressure of 40 kPa (300 Torr).
The Hashimoto cell was adapted by Feates et al. (1970) onto a JEOL 7A microscope with continuous recording facilities. The latter was made possible by incorporating a 20 mm diameter hole in the center of the fluorescent viewing screen to allow the electron beam to strike a secondary screen with a transmission phosphor. The underside of the phosphor was viewed by a 25 mm “Plumbicon” television camera, and the image was displayed on a television screen (300 mm diameter), which increased the effective magnification of the instrument to greater than 2.5 million times. Additional refinements included direct recording of the image on videotape, slow-motion replay facilities, and the provision of permanent records on 16 mm cine film.
In the 1960s and 1970s, a number of electron microscopes were adapted to incorporate gas reaction attachments. Gallegos (1964) converted an existing hot stage for a JEM-6A electron microscope into a gas reactor by attaching a syringe needle to the hot stage. The gas nozzle was shaped into a slit, which facilitated the impingement of gas onto the specimen. The column vacuum was maintained at about 0.1 Pa (1 × 10−3 mm Hg) in this setup. Fryer (1968a, 1968b) investigated the oxidation of graphite catalyzed by palladium by heating the specimen to 500 °C using a heating stage in a Siemens Elmiskop I electron microscope in high vacuum (1.33 × 10−3 Pa, or 10−5 Torr), and then introducing dry air over the specimen by means of a special attachment, and recording the specimen behavior over photographic plates and video tape (by means of closed circuit television). In the same year, Mills and Moodie (1968) designed, constructed, and installed an electron microscope stage on a Hitachi HU-11A microscope which included gas injection capability via a curved capillary. There was no differential pumping mechanism. Rather, the column pumps, working through the same capillary, established a rate adequate for the evacuation of the line between the taps and the valves, and for the adjustment of pressure in the fixed volume.
The need for differential pumping was recognized as a means of increasing the range of allowable pressures in the apertured chamber. Besides the modified Hashimoto (1966, 1968) design, Baker and Harris (1972) incorporated a modified JEOL AGI gas reaction attachment onto a 100 kV JEOL JEM-7A “high-resolution” electron microscope wherein the specimen chamber was evacuated by a three-stage diffusion pump backed by a rotary pump. The attachment consisted of only one aperture, which compromised the maximum allowable pressure up to 30 kPa (or 225 Torr) instead of 40 kPa (300 Torr) in the Hashimoto design, but retained its full diffraction capability. In addition, a glove box was added to the entry port of the microscope to load specimens into the microscope in a high purity argon atmosphere. A high sensitivity Plumbicon camera viewed the underside of the transmission phosphor and its output was monitored and recorded continuously on videotape, with an effective exposure time of 40 ms and a total recording time of 1 h per tape. Ward and Mitchell (1972) from the Cavendish Laboratory in Cambridge also produced an environmental chamber by mounting two 20 μm apertures 0.75 mm apart in the gap of the objective pole piece. The space between the apertures was effectively separated from the rest of the microscope column, and its environment may thus be varied independently. The addition of a diffusion pump with its own backing system to the microscope’s normal pumps enabled the pressure in this space to be increased to one atmosphere (105 Pa) without significantly altering the column pressure.
With the advent of HVEMs in the 1960s and 1970s, Swann and Tighe (Swann and Tighe 1971; Swann et al. 1972; Swann 1972) designed and constructed an environmental cell for the 1 MV AE1-EM7 electron microscope. The environmental specimen chamber was located between the objective pole pieces of the microscope. It consisted of four apertures which were machined concentrically with the upper objective pole piece thus obviating the need for special aperture alignment facilities inside the microscope. The specimen chamber itself was surrounded by a second, differentially pumped chamber which further reduced the leakage rate of the reaction gas into the microscope column. The specimen holder was of the side-entry type and could be translated ±1.5 mm using the normal, side-entry specimen stage. The specimen rod was supported at its tip by a conical socket inserted into the normal side-entry stage. The distance between the specimen apertures was 5.5 mm which was large enough to accommodate a tilting hot stage and yet short enough to obtain satisfactory images of crystalline samples at pressures up to 105 Pa (760 Torr) of He or 20 kPa (150 Torr) of air. The specimen and differential chamber apertures were large enough to transmit complete diffraction rings at 1 MV from crystal planes with spacings larger than 0.05 nm. A movable objective aperture system was provided below the gas reaction cell and was hermetically sealed to prevent gas leaking into the lower part of the microscope column. The superior penetrating power of the high-voltage electron microscope, compared to 100 kV instruments that were commonly available during that time, facilitated the observations of thin foils with micrometer thicknesses (Swann and Tighe 1977). The resolution achievable was about 10 nm in 20 kPa (150 Torr) of air. Increasing gas pressure led to a decrease in image intensity and contrast, with the latter better in dark-field imaging mode at all pressure levels (Fig. 1.3).

Environmental specimen chamber situated between the objective pole pieces of the AEI EM7 one million volt electron microscope, with the side entry, single tilt hot stage shown in position. Reprinted with permission from Swann (1972)
In the 1980s, improvements in the objective lens pole-piece design led to the development of atomic-resolution medium-voltage (200–400 kV) TEMs which allowed in situ heating experiments in vacuum at atomic resolution (e.g., Sinclair et al. 1988). This further led to resurgence in the interest in the environmental cell designs in the 1980s and 1990s, with several groups modifying the objective pole pieces of various TEMs. Differential pumping was incorporated in all the modifications.
Rodriguez and coworkers (1990) modified the specimen chamber region of a JEOL 200CX TEM/STEM to accommodate a specially designed environmental cell and specimen heating stage to continuously observe changes in the appearance of a specimen as it underwent reaction with a beam of atomic oxygen. The environmental cell was differentially pumped making it possible to work at an oxygen pressure of 2 kPa (15 Torr) and at the same time maintain a vacuum of 1.3 × 10−4 Pa (10−6 Torr) in the rest of the microscope column. The microscope setup also included a closed circuit television link in conjunction with a videotape recorder so that dynamic events occurring during the reaction could be captured and replayed later for analyses. The output from the camera was continuously monitored and stored on a Sony ¾″ VO-7600 videocassette recorder. In situ electron diffraction of the reacting specimen was also possible.
Lee and coworkers (1991) designed an objective pole piece for a JEOL 4000 (400 kV) TEM with a gap of 14 mm. Five apertures were used, and additional holes were machined into the pole piece to accommodate the extra gas handling equipment and to provide extra pumping ports. Differential pumping between the primary and secondary apertures, and pumping of the volume surrounding the pole piece and the upper secondary aperture and the condenser stack aperture, was accomplished by four turbo molecular pumps. The point-to-point resolution of the pole piece in the modified state was 0.41 nm. The column vacuum was in the lower 10−5 Pa (10−7 Torr) range with no gas in the cell. At 400 kV, the maximum diffraction angle that could be observed corresponds to 24 mrad (d = 0.08 nm). The cell was pressurized to 9.3 kPa (70 Torr) with H2 gas for several hours with no adverse effects on microscope performance. Images could be recorded on conventional microscope plates or on high-resolution video tape via a Gatan TV camera.
Doole and coworkers (1991) modified a JEOL 4000 EX TEM by introducing differential pumping apertures and achieved a resolution of 0.31 nm at 400 kV under a pressure of 560 Pa (4.2 Torr) of H2 and at temperature of 670 °C. Apart from the gas, the microscope was effectively unaltered (Goringe et al. 1996). Specimen shifts and tilts and heating capabilities were all available, as were the standard imaging apertures. A thin-window for energy-dispersive X-ray spectroscopy (EDS) and a parallel electron energy-loss spectrometer were available for analytical work.
Boyes and Gai (Boyes et al. 1996; Boyes and Gai 1997), then at DuPont, permanently modified the objective lens area of a Philips CM30T TEM/STEM system with a pole-piece gap of 9 mm, by introducing two pairs of apertures above and below the specimen. The apertures were mounted inside the bores of the objective pole pieces rather than between them as in previous designs. With this approach, regular specimen holders compatible with conventional TEMs could be used, as well as objective apertures for diffraction contrast experiments. The controlled environment (ECELL) volume was the regular specimen chamber of the microscope, and was separated from the rest of the column by the apertures in each objective pole piece and by the addition of a gate valve, which was normally closed, in the line to the regular ion-getter pump (IGP) at the rear of the column. A schematic of the basic geometry of their ECELL aperture system is illustrated in Fig. 1.4. A turbo molecular pump as well as an additional IGP was added to the microscope. Differential pumping was introduced at the sample area of the ECELL and at the level of the second condenser (C2) lens and selected area (SA) apertures. (111) lattice fringes of gold islands of 0.24 nm could be resolved at 500 °C in 40 Pa (0.3 Torr) N2 at 200 kV accelerating voltage. The maximum allowable pressure in this design was 5 kPa (50 mbar) (Boyes and Gai 1997). At 200 kV, diffraction patterns extend out to a real space equivalent of <0.07 nm. Regular, small probe scanning TEM (bright-field and annular dark-field) imaging and chemical and crystallographic analyses capabilities were also available.

Schematic of the basic geometry of the ECELL aperture system from Boyes and Gai (1997)
In Arizona State University, Sharma and coworkers (1994) modified a Philips 400-T TEM operated at 120 kV to include a differentially pumped environmental cell, based on the Swann (Swann and Tighe 1971; Swann et al. 1972) design. Then in 1998, a Philips EM 430 (300 kV LaB6 source) high-resolution TEM was modified by fitting a differentially pumped environmental cell (E-cell) in the objective lens pole-piece gap (Sharma and Weiss 1998). It consisted of two sets of apertures placed above and below the sample. Smaller apertures were used for the upper apertures to reduce the leak rate to the electron gun source. The larger lower aperture in the E-cell makes high-angle electron diffraction possible. {422} diffraction rings of Au showing reflections out to 0.083 nm were achieved. The E-cell could handle up to 2.7 kPa (20 Torr) of gas pressure. Regular single- and double-tilt heating holders could be used to heat the samples up to 1300 °C and 850 °C, respectively. This was likely the first E-cell setup to include a post projector energy filter (Gatan Inc.) to filter the inelastic scattering from the gases/thick samples as well as to obtain energy-filtered images (chemical maps). The Gatan Imaging Filter (GIF) could also be used as a parallel electron energy-loss spectrometer to obtain chemical information in the sample composition during the reactions.
In the 2000s, TEM manufacturer Philips Electron Optics (now known as FEI Company) adapted the Boyes and Gai design and further improved the image resolution by incorporating a field emission gun (FEG) and a SuperTwin objective lens which has a narrower pole-piece gap (about 5 mm). An extra pumping stage is introduced to protect the FEG better (Hansen et al. 2006). FEI Company custom built ETEMs for Haldor Topsøe (a chemical company in Denmark) (Hansen et al. 2001), Osaka University in 2005 (Yoshida and Takeda 2005; Yoshida et al. 2007), and Arizona State University (Sharma and Crozier 2005) as part of a collaborative effort.
The latest series of ETEMs from FEI Company is based on the Titan platform, which is a three-condenser lens (scanning) TEM system. Technological improvements in electron microscopy over the past 15 years can be incorporated into these state-of-the-art ETEMs. For example, the spatial resolution is better than 0.1 nm when the microscope is operated at 300 kV with an aberration corrector (Haider et al. 1998a, b) in the image-forming (objective) lens. Spherical aberration correction of the objective lens minimizes image delocalization and leads to directly interpretable TEM images. This is especially crucial for in situ ETEM experiments where reactions and processes are dynamic, leaving little time for the recording of through-focal series images. The addition of a monochromator (Tiemeijer 1999) reduces the energy spread of the incident electron beam to better than 0.2 eV. An energy filter for imaging and electron energy-loss spectroscopy, an energy-dispersive X-ray spectrometer, scanning capabilities for STEM and bright-field and dark-field detectors can be incorporated in the setup. The instrument is also equipped with a residual gas analyzer to monitor the composition of the gases, and a plasma cleaner in the specimen chamber (Fig. 1.5).

Schematic diagram of differentially pumped TEM column (FEG, field emission gun, ion-getter pump (IGP), turbo molecular pump (TMP), residual gas analyzer (RGA), plasma cleaner (PC), first condenser aperture (C1), selected area aperture (SA) in an FEI Titan environmental transmission electron microscope (ETEM). Reprinted with permission from Hansen et al. (2010)
On other fronts, Gai and Boyes (now at the University of York) are developing an ETEM equipped with aberration correctors in both the image- and probe-forming lenses as part of a collaboration with JEOL, which is another major electron microscope company (Gai and Boyes 2009; Boyes et al. 2013; Boyes and Gai 2014a, b). The instrument has both a gas tolerant turbo molecular pumping vacuum system and a wider gap objective lens pole piece in order to accommodate a 1000 °C hot stage, and aims to retain the original full function (S)TEM imaging and chemical analysis capabilities while operating at temperatures of >500 °C in a controlled gas environment. The increase in pole-piece gap is found to have little effect on spatial resolution owing to the aberration correctors. Currently, the system operates in both (E)STEM and (E)TEM modes with up to 5 kPa (50 mbar) in gas pressure.
There is also a possibility of the resurgence of HVEM for environmental studies, as exemplified by researchers at Nagoya University (Tanaka et al. 2013, 2014) who have been developing the reaction science high-voltage electron microscope (RSHVEM), which is a 1 MV high-voltage (S)TEM that can be used to observe nanomaterials under gas, liquids, and illuminating light conditions. Samples of micrometer thicknesses can be observed. The microscope vacuum is supported by a three-stage differential pumping system comprising five turbo molecular pumps. It is also equipped with electron tomography, energy-filtered TEM and electron energy-loss spectroscopy capabilities.
1.3 Development of UHV TEM for Epitaxial Growth Studies
The desire to image atomically clean surfaces and to perform epitaxial growth studies has led to the development of ultrahigh-vacuum (UHV) specimen environments in TEMs. Because of the highly reactive nature of the starting materials, a UHV specimen environment is required so that a clean initial surface can be prepared, usually by annealing at high temperatures. To perform controlled environment studies, McDonald and coworkers (1989) modified a JEOL 200CX high-resolution TEM with point resolution of 0.25 nm, to obtain an UHV environment of ~1.3 × 10−7 Pa (10−9 Torr) (cf. vacuum of 1.3 × 10−5 Pa, or 10−7 Torr in regular TEMs) in the specimen region. This was achieved by isolating the specimen chamber from the manufacturer’s vacuum system, incorporating differential pumping, and using larger pumps, pumping manifolds, improved vacuum seals, and replacing or removing non-vacuum-compatible materials from the specimen chamber. The modified design permitted a variety of different experiments at pressures from <13 Pa (0.1 Torr) to ~1.3 × 10−7 Pa (10−9 Torr). The system was fitted with a calibrated leak valve for the introduction of gases, and in situ heating and evaporation capabilities were also added to the specimen viewing position, where high resolution could be maintained at temperatures exceeding 600 °C. In situ cleaning and thin film deposition capabilities were also added.
Hammar and coworkers from the IBM Research Lab (1996) modified a Hitachi 9000 UHV-TEM operated at 200 kV, with a base pressure in the column of 3 × 10−8 Pa (2 × 10−10 Torr) with the beam on or off. The modifications consist of a gas inlet system for the introduction of oxygen or germane in the column, and a double-tilt heating stage which allows the application of precise dark-field conditions and in which the samples can be heated with direct current up to 1400 °C during observation. This setup has been used extensively to study the growth of nanowires in situ (see Applications section).
2 Applications
Controlled atmosphere transmission electron microscopy has been applied in a variety of materials research problems. In this section, we highlight its major uses in the areas of oxidation and reduction processes, growth of one-dimensional materials as well as catalysis. The reader can also refer to the chapters on growth experiments and catalysis studies in this book for more examples within these topics.
2.1 Oxidation and Reduction Processes
Early applications of CAEM focused on oxidation and reduction processes, often at high temperature, using both aperture- and window-type cells. Using their aperture environmental cell, Ito and Hizaya (1958) observed the oxidation of Al into γ-Alumina at 400 °C in 13.3 Pa (0.1 Torr) of air and the reduction of Cu2O to Cu at 300 °C in 13.3 Pa (0.1 Torr) of H2 by monitoring the changes in the diffraction patterns of the specimens. Hashimoto and coworkers (1958) observed the growth of needle-like tungsten oxide crystals on the surface of a tungsten filament at 850 °C in 400 Pa (3 Torr) of air. Diffraction analyses showed that the needles were composed of γ-tungsten oxide, which transformed to the α-phase (WO3) upon further heating to 1400–1600 °C. WO3 was found to evaporate above 2000 °C. Hashimoto and coworkers (1968, 1966, 1970) performed similar work on ammonium molybdate (NH4)6MO7O24.4H2O at 700 °C in air pressure of 1.3–20 kPa (10–150 Torr), and cuprous iodide (CuI) at 700 °C in hydrogen gas of pressure 6.7–20 kPa (50–150 Torr). Using the gas nozzle apparatus on a JEM microscope, Gallegos (1964) performed oxidation studies on Fe, Cu, and Ni which had been vacuum evaporated onto carbon support grids at high temperatures, as well as the reduction of these oxides.
The environmental cell for the 1 MeV AE1-EM7 high-voltage electron microscope designed by Swann and Tighe (1972) was utilized in a variety of redox experiments, including Ti oxidation at 450–850 °C (Flower and Swann 1974), hematite (Fe2O3) reduction to magnetite (Fe3O4) (Swann 1972; Swann and Tighe 1977), and oxidation of Ni-30Cr and TDNiCr (Flower and Wilcox 1977). It was also used to observe the microstructural changes in Ta during in situ heating in nitrogen (Swann et al. 1973). The high penetrating power of the 1 MeV HVEM permits continuous observation of foils which were mechanically much stronger than those suitable for medium voltage work. It was thus possible to follow the oxidation of a single grain from 100 % metal to 100 % oxide without any problems of foil buckling or fracture. In these experiments, the early stages of nucleation and growth could be followed and the morphology and crystallography of the reactant products could be established, and models to explain the growth kinetics during oxidation (or reduction) could be deduced, albeit at limited resolution (on the order of 10 nm).
Using the JEOL TEM modified with a UHV system (Mcdonald et al. 1989), Ross and Gibson (1992) observed in real time the nature and motion of the silicon (111) surface steps during in situ oxidation inside the microscope, at temperatures from room temperature to 900 °C, by both O2 and H2O at pressures from 1.3 × 10−6 to 0.13 Pa (10−8 to 10−3 Torr), as well as in one atmosphere O2. By using an imaging technique based on the behavior of forbidden reflections in Si, they showed that surface steps of two monolayers in height do not move noticeably during oxidation of several atomic layers of the silicon specimen. In contrast, bilayer step movement was observed in the “etching” of silicon by oxygen at higher temperatures.
With the increasing interest in nanomaterials in recent years, Crozier and coworkers (1998) subjected Pd catalyst particles to oxidation and reduction cycles and studied the changes in particle structure and morphology with in situ electron diffraction and imaging, and found that many of the oxide particles form voids when reduced to Pd metal. The decrease in volume that occurs during reduction is often accompanied by a combination of particle shrinkage and void formation. Koh et al. (2013) reported the first direct study on the oxidation of carbon nanotubes (CNTs) at the resolution of an aberration-corrected environmental transmission electron microscope (ETEM), as the same nanotubes were located and their changes identified as they underwent oxidation at increasing temperatures in situ in the ETEM. Their findings show that only the outside graphene layer is being removed and, on occasion, the interior inner wall is oxidized, presumably due to oxygen infiltrating into the hollow nanotube through an open end or breaks in the tube, contrary to earlier reports based on ex situ observations that CNT oxidation initiates at the cap of the tube and proceeds along its length.
2.2 Catalysis
Another major area which has benefited greatly from the development of environmental TEM is catalysis. The active surface sites of heterogeneous catalysts facilitate conversion of reactants into products and their properties depend on a number of factors including the type and number of surrounding atoms at the surface, the supporting material, and the presence of reactants and products (Molenbroek et al. 2009). A thorough understanding of the nanoparticle structure and its correlation to activity is therefore important to establish rational catalyst design strategies, and electron microscopy is invaluable in this regard owing to its sub-nanometer resolving ability. Although nanoparticles may be synthesized with predefined shapes and surface sites, the morphology of the as-synthesized particles may be influenced by reaction conditions. There is therefore tremendous interest in obtaining atomic level structural and chemical information on the catalysts in situ, in their working conditions. A much more detailed review is given in a later chapter, and we confine our comments to those which illustrate the growth of the field historically.
2.2.1 Catalyst Deactivation Studies
The pioneering experiments of catalytic decomposition of hydrocarbons on metallic surfaces were conducted by Baker and coworkers in the 1970s. Using CAEM (Baker and Harris 1972), Baker and coworkers were able to directly observe the formation of carbon filaments on metal surfaces (Fe, Co, Cr) when reacted in hydrocarbon environments (Baker et al. 1972a, b). The authors noted that the presence of a small particle located at the growing end of each filament appeared to catalyze the growth of the carbonaceous structure. From the experimental data, a mechanism to describe the filament growth process was developed, and activation energies for filament growth were determined (Baker et al. 1973, 1979). The growth of filamentous carbon is a major concern in many industrial processes involving blast furnaces, steam cracking, fuel pins, and boiler tubes of nuclear reactors and various heterogeneous catalyst systems, where the catalyst deactivates due to the spatial limitations imposed on the filamentous carbon growth (coke formation and fouling) by the reactor (Baker 1989, 1990). The motivation for studies relating to the formation of carbon filaments has been the drive to inhibit the growth of this form of carbon deposit.
Besides fouling, catalyst deactivation can also occur from sintering. Gai and Boyes (1997) studied the dynamic effect of Cu-Pd intermetallic alloy particle catalysts supported on carbon at 250 °C in an H2/He and CO/He environments, and found that the particles were more faceted in CO than in H2 and sinter faster in H2. Liu et al. (2004) used in situ ETEM to study the sintering mechanisms on Al2O3-supported Pd catalysts in an oxidizing environment. For fresh catalysts, particle sintering occurred via a traditional ripening and coalescence mechanism and was significant at temperatures from 700 °C. In the case of used catalysts, gasification of carbonaceous material around these particles in an oxidizing environment resulted in movement and coalescence at temperatures as low as 350 °C. Simonsen and coworkers (2010) performed time-resolved imaging of Pt nanoparticles dispersed on amorphous Al2O3 support during exposure to 1 kPa (10 mbar) of synthetic air at 650 °C inside an ETEM, and showed that Pt nanoparticle sintering was mediated by an Ostwald ripening process.
2.2.2 Investigations on Catalysts and Their Supports
The properties of heterogeneous catalysts are often determined by the interactions between the catalyst nanoparticles and their supports (typically metal oxides). Molecules can restructure the surfaces of heterogeneous catalysts under reaction conditions. The nature of the support has important influence on the properties of supported metal catalysts and this has been an interest in the ETEM community over the years. Here, we present a summary of work performed on Au and Cu catalysts, and ceria supports.
-
(a)
Au catalysts
While bulk Au is chemically inert, supported Au nanoparticles (Au NPs) (on selected oxides such as CeO2 and TiO2) catalyze a wide variety of reactions such as CO oxidation (Haruta 1997), water-gas shift reactions (WGSR) (Fu et al. 2003), and selective and total oxidation of hydrocarbons (Hayashi et al. 1998). The catalytic performances of Au markedly depend on particle size, dispersion, supports, and preparation methods. For example, catalytic activity measurements of Au particles of 1–7 nm deposited on rutile TiO2 show that smaller particles have higher activity (Williams et al. 2010). Au nanoparticles supported on TiO2 have been shown to have higher catalytic rates than those supported on Al2O3 in CO oxidation and WSGR (Arrii et al. 2004; Sandoval et al. 2007)
Using an aberration (image)-corrected ETEM, Yoshida, Takeda, and coworkers (Yoshida et al. 2012; Takeda and Yoshida 2013) showed that adsorbed carbon monoxide molecules caused the facets of a Au nanoparticle supported on CeO2 to reconstruct during CO oxidation at room temperature (Fig. 1.6). By operating at a lower accelerating voltage (80 kV), which improved the image contrast of light-element atoms, they also observed bright and dark contrast features extruded from the reconstructed facets of gold nanoparticles. Using image simulations in combination with ab initio electronic calculations, they showed that the electron micrograph allowed deduction of an energetically favorable model for CO adsorption (Fig. 1.7). Uchiyama and coworkers (2011) examined a large number of Au/CeO2 catalysts using ETEM and found that the morphology of Au nanoparticles changes from faceted during CO oxidation in CO/air to becoming more round with decreasing CO partial pressure relative to air. They also examined Au nanoparticles supported on a non-oxide (TiC) crystal and observed no morphological changes in the Au nanoparticles supported on TiC when switching the gases. Using aberration-corrected ETEM, Kuwauchi et al. (2013) show that catalytically active Au NPs move reversibly and stepwise by approximately 0.09 nm on a CeO2 support surface at room temperature and in a reaction environment. The lateral displacements and rotations occur back and forth between equivalent sites, indicating that Au nanoparticles are loosely bound to oxygen-terminated CeO2 and may migrate on the surface with low activation energy.
Fig. 1.6 
Au{100}-hex reconstructed surface under catalytic conditions. GNP supported on CeO2 in (a) a vacuum and (b) a reaction environment (1 vol.% CO in air gas mixture at 45 Pa at room temperature). Two {100} facets are present in the rectangular regions indicated by I and II in (a). The enlarged images of these regions in vacuum and in the CO in air gas mixture are shown at the bottom of (a, b), respectively. In vacuum, the distance between the topmost and the second topmost {100} surface layers of 0.20 nm was the same as the interplanar distance of the {200} planes in crystalline bulk gold. The average distance of the adjoining Au atomic columns on the topmost surface layer of 0.29 nm was the same as the corresponding distance in a {100} plane in crystalline bulk gold. In the reaction environment, both the average distance of the adjoining Au atomic columns and the interplanar distance changed to 0.25 nm. These changes in positions of the Au atomic columns correspond well to those of the Au{100}-hex reconstructed surface structure. To reduce random noise, the images in (a, b) were obtained by averaging four successively acquired images. Figure reprinted with permission from Yoshida et al. (2013)
Fig. 1.7 
Adsorbed CO molecules on a Au{100}-hex reconstructed surface under catalytic conditions. Aberration-corrected ETEM images in (a) vacuum and (b) a reaction environment (1 vol.% CO in air at 100 Pa at room temperature) taken using 80 keV electrons using an under-defocus condition. (c) The observed image in the rectangular region in (b) at higher magnification. (d) A simulated image based on an energetically favorable model. The model is superimposed on the simulated image. The model in (e) plan view along the [001] direction of crystalline gold and (f) cross-sectional view along the [110] direction of crystalline gold to show the undulating topmost Au layer. C atoms, blue colored balls; O atoms, red colored balls; CO adsorbed Au atoms within the surface hexagonal lattice, darker green colored balls; the other Au atoms within the surface hexagonal lattice, brighter green colored balls; the Au atoms within and below the second topmost surface layer, gold colored balls. By fitting the simulated images with the observed images, the viewing direction in (a–c) was determined to be the [1.37, –0.37, 0] direction of the crystalline Au NP. As shown in models (d, e), the [1.37, –0.37, 0] direction is nearly parallel to one side of the surface hexagonal lattice. To reduce random noise, the images in (a–c) were obtained by averaging four successively acquired images. Figure reprinted with permission from Yoshida et al. (2013)
Kuwauchi and coworkers (2012) studied the intrinsic structure of Au/TiO2 catalysts at room temperature under various gas reaction conditions with the ETEM (high vacuum, O2 at 100 Pa, N2 at 100 Pa and 1 % vol CO in air at 100 Pa), and observed that the Au NPs changed their morphology systematically depending on the environment. Based on the ETEM observations, structural evolution diagrams depicting the morphology of the catalysts as a function of electron current density and dose in various gas environments were derived.
-
(b)
Cu catalysts
Another catalyst material that is of interest is Cu. Cu nanoparticles on supports are used as catalysts for methanol synthesis and hydrocarbon conversion processes for fuel cells. Cu alone may work as a methanol synthesis catalyst, but the activity increases significantly for Cu supported by ZnO. Hansen et al. (2002) examined Cu nanoparticles dispersed on ZnO and silica supports and found that for the Cu/ZnO catalysts, the shape of the particles changes with varying gas composition. The addition of water to hydrogen gas transforms the Cu crystals into a more spherical morphology, whereas the removal of H2O from H2 causes the Cu crystals to revert to their original form. For Cu nanocrystals supported on silica, the support was found to have negligible influence on the structure. Vesborg and coworkers (2009) performed time-resolved measurements of the methanol synthesis reaction over a Cu/ZnO-based catalyst using a laminar flow tube reactor and compared the activity measurements with ETEM observations of Cu/ZnO particles during exposure to comparable reaction conditions. Using ETEM, it was revealed that qualitatively, the Cu particles appear to be more flat under more reducing conditions and more spherical when exposed to a more oxidizing environment. The gas-dependent morphology of the Cu nanoparticles is consistent with the dynamic microkinetic model proposed by other research groups. Wagner and coworkers (2003) utilized electron energy-loss spectroscopy (EELS) to investigate metal-support interaction in a Cu/ZnO catalyst by examining the energy-loss near edge structure (ELNES) at the Cu L3 ionization edge as the catalysts are heated in a CO/H2 environment in the ETEM. The intensity of the ELNES reflects modifications in the electronic structure of Cu. They found that in the Cu/SiO2 system, the ELNES intensities were constant, reflecting that no structural changes of the Cu nanocrystals are observed. Changes in the ELNES were observed when the particles were dispersed on a ZnO support, and were interpreted as a tensile strain in the Cu nanoclusters induced by the ZnO support, as well as a Cu-Zn alloy formation induced to a degree depending on the severity of the reduction treatment of the catalyst.
-
(c)
Ceria particles and ceria-based supports
Cerium-based oxides continue to attract considerable attention because of their current and potential use in metal-supported oxide catalyst applications, e.g., in automobile three-way catalytic converters, solid oxide fuel cell anodes, and water-gas shift reactions. The valence state of cerium changes from Ce3+ to Ce4+ during the associated phase transformations \( \left(2{\mathrm{CeO}}_2\leftrightarrow {\mathrm{CeO}}_3+0.5{\mathrm{O}}_2\right) \), and the ability of this material to reversibly accept or contribute oxygen to its surroundings is the basis for its use in catalytic applications where redox processes are important. Parkinson (1989) envisaged feasibility of directly studying atomic rearrangements in catalysts and catalyst supports under reaction conditions by electron microscopy, when he presented HRTEM images of the ceria lattice, taken under flowing nitrogen at 2.7 kPa (20 Torr) which revealed lattice fringes from the {111} planes (separation = 0.312 nm). Such studies show promise of providing a detailed understanding of the mechanisms involved in certain types of heterogeneous catalyst.
Sharma and coworkers (2004) characterized the reduction behavior of high-surface-area (HSA) and low-surface-area (LSA) CeO2 catalysts in hydrogen gas at elevated temperatures using EELS by analyzing the Ce M4,5 white line intensity ratio. The relative intensities of the M5 and M4 peaks in the Ce EEL spectra depend on the oxidation state of Ce. The authors found a direct correlation between the extent of reduction and surface area, with significant Ce reduction occurring by 600 °C in the HSA sample whereas with the LSA sample, significant reduction took place around 700–800 °C.
Wang and coworkers (2008, 2009) followed the dynamic redox process taking place in individual ceria and ceria–zirconia nanoparticles. The samples were progressively heated and cooled while maintaining the same H2-rich atmosphere. Re-oxidation may occur rapidly at temperatures below 600 °C due to residual background oxygen/water vapor in the sample area or in the ETEM column, and the structure and chemistry were monitored using high-resolution TEM imaging, electron diffraction, and electron energy-loss spectroscopy (EELS) (Fig. 1.8). The studies show that for ceria nanoparticles with an average size of 20 nm, oxygen vacancies present in the reduced ceria undergo rapid ordering leading to the immediate formation of Ce2O3. The reduced ceria has a C-type lattice in which the lattice parameter is doubled compared to that of the parent fluorite structure. In the ceria–zirconia nanoparticles, the more active structure has predominantly disordered cations and shows no evidence for oxygen vacancy ordering during reduction.
Fig. 1.8 
Dynamic changes of atomic-level structure characterized by high-resolution imaging (upper row) and electron diffraction (middle row) and chemistry analyzed by the electron energy-loss spectrum (lower row) at (a) 245, (b) 693, and (c) 600 °C in 67 Pa (0.5 Torr) of H2 along the [21-1] zone axis from the same individual ceria nanoparticle during the redox process. (b) After reduction, arrows in the high-resolution image and extra spots along the (220) plane in the electron diffraction pattern show the superstructure formed during reduction. The reversal of Ce M5/M4 intensity indicates the reduction and re-oxidation from Ce4+ to Ce3+ and from Ce3+ to Ce4+ during heating and cooling, respectively. Figure reprinted with permission from Wang et al. (2009)
Cargnello et al. (2013) studied CeO2 which has been found to be an “active support” in terms of increasing the rates of redox reactions. By tailoring the “length” of a ceria-metal interface, through the use of monodisperse Ni, Pd, and Pt nanocrystals, CO oxidation in ceria-based catalysts was found to be greatly enhanced at the ceria-metal interface sites, and the metals deposited on ceria had higher catalytic rates than their alumina-supported counterparts, as evidenced by the much lower temperatures needed to completely oxidize CO.
-
(d)
Catalyst promoters
Besides catalysts themselves, there are also catalyst promoters which cause more than proportional increases in activity or selectivity when added to a catalyst, and are therefore essential to achieve the required activity or selectivity in catalysts. Hansen et al. (2001) performed in situ TEM imaging of Ba-promoted Ru catalysts in their working conditions in an ETEM, and suggested that the increased activity to be related to a two-dimensional barium-oxygen overlayer on the Ru crystals.
-
(e)
Electron energy-loss spectroscopy (EELS) for quantification of gases and catalytic products
With the developments of electron energy-loss spectrometers and their attachments onto modern ETEMs, redox reactions in catalysts and other materials system can be studied by monitoring changes in the EELS spectra of the specimens. EELS thus offers another avenue of TEM characterization in addition to imaging and diffraction capabilities. For example, López-Cartes and coworkers (2003) performed ETEM studies on Rh/Ce0.8Pr0.2O2−x catalysts under H2 conditions at temperatures ranging from 298 to 1223 K, and observed the occurrence of consecutive reduction of Pr4+ and Ce4+ ions, and the formation of an oxygen-deficient Ln16O30 (Ln: Ce, Pr) ordered phase, by analyzing the fine structure in EELS spectra of their M4,5 edges. The group at Arizona State University (Sharma et al. 2004 and Crozier et al. 2008) studied the dynamic changes taking place during redox reactions in ceria and ceria–zirconia nanoparticles in a hydrogen atmosphere by monitoring the white lines in the Ce EELS spectra, and determining the Ce oxidation state by its M5/M4 intensity ratio.
Crozier and Chenna (2011) show that, while modern ETEMs are fitted with residual gas analyzers to monitor the composition of the gases, one can also perform quantitative analysis of gases in an ETEM with in situ EELS with an accuracy of 15 % or better (provided that some precautions are taken during the acquisition to account for the extended gas path lengths associated with the reaction cell). They have since employed this technique to detect and quantify catalytic products directly inside the ETEM (Chenna and Crozier 2012). Ru nanoparticles supported on SiO2 spheres were used as the model catalyst for CO oxidation and CO methanation. CO oxidation reaction was performed by flowing He:CO:CO2 in 50:8:4 ratio in the ETEM. CO has a carbon π* peak of 286.4 eV, and as the temperature of the specimen was increased inside the ETEM, the C π* peak from CO2 (at 289.7 eV) was found to increase in intensity, indicating that the greater percentage of CO had been converted to CO2. There was good agreement between the EELS measurements and the results obtained using an ex situ catalytic reactor. In CO methanation, a CO2 and H2 gas mixture of 4:1 pressure ratio was admitted to the environmental cell and EEL spectra were collected at different temperature. A shoulder peak at about 286.4 eV started to appear at 400 °C, corresponding to CO formation, and became more dominant at increasing temperatures.
A key challenge to successfully applying EELS in catalysis is the peak overlap between reactant and product gases. This can be overcome by the use of electron microscopes equipped with monochromators, which significantly improves the energy resolution from 1 to 0.2 eV or better. Baldi and coworkers (2014) utilized monochromated EELS in an ETEM to observe hydrogen absorption and desorption in individual Pd nanocrystals by directly probing the bulk plasmon resonance mode of the Pd nanoparticle. On hydrogen absorption, individual Pd nanocrystals exhibit large red shifts (exceeding 2 eV) in their bulk plasmon resonance, and these shifts are reversible on desorption. The individual particle spectra are then correlated with particle size and crystallinity using the high spatial resolution of aberration-corrected TEM imaging, and hydrogen loading and unloading isotherms of individual Pd nanocrystals were then deduced.
-
(f)
Photocatalysis
Photocatalysts are also an important class of catalyst materials with applications relating to sustainable energy development. Several groups have recently explored different ways for light irradiation of samples inside the (E)TEM. One approach is the use of specialized lens-based or fiber-based TEM specimen holders that permit specimens to be illuminated with light and electrons at the same time (Shindo et al. 2009; Cavalca et al. 2012). Combining the use of these holders with an ETEM, Cavalca and coworkers (2012, 2013) investigated the photoreduction of cuprous oxide (Cu2O). Water vapor was leaked into the ETEM specimen chamber and the specimen was exposed to light with 405 nm wavelength for various time periods while the electron beam was blanked. Then, the column was evacuated to ~1.3 × 10−4 Pa (10−6 mbar) pressure for 5 h to reduce the water vapor in the column before the electron beam was illuminated on the sample to record changes post-reaction. TEM images show that Cu2O nanocubes were degraded after photoreduction. Diffraction patterns show the transformation of the cubes from Cu2O to Cu, and the change in oxidation state of Cu is also elucidated in the Cu L2,3 edge using EELS. The group at Arizona State University (Miller and Crozier 2013) designed an illuminating system for the ETEM whereby light is introduced through a port on the microscope, and they observed surface amorphization in TiO2 nanocrystals when the latter were irradiated with light while exposed to water vapor inside the ETEM (Zhang et al. 2013).



2.3 Nucleation and Growth of One-Dimensional Nanomaterials
Another application of ETEM is the in situ nucleation and growth of one-dimensional nanomaterials such as carbon nanotubes and nanowires, as described in a later chapter.
Since their discovery in 1991 carbon nanotubes (CNTs) (Iijima 1991) have found an increasing number of applications, most notably as field emission electron sources in displays or in X-ray tubes for medical applications (e.g., Rinzler et al. 1995; de Heer et al. 1995; Wang et al. 1998; Choi et al. 1999; Sugie et al. 2001). Observations of carbon nanotube growth can be made in situ using ETEM and was reported by two groups in 2004. Helveg et al. (2004) formed carbon nanofibers from methane decomposition over Ni nanocrystal catalysts supported on MgAl2O4 (CH4:H2 ratio = 1:1), at temperatures 500–540 °C and total pressure of 200 Pa (1.5 Torr), and observed, in real time, the growth of the nanofibers at the high resolution of the TEM (Fig. 1.9). Carbon nanofibers were found to develop through a reaction-induced reshaping of the Ni nanocrystals, with the nucleation and growth of graphene layers assisted by a dynamic formation and restricting of mono-atomic step edges at the Ni surface. Sharma and Iqbal (2004) used a Ni/SiO2 catalyst and acetylene as the precursor gas. The catalysts–supports were dry loaded on 400 mesh Ni grids, and heated at 400 °C for 30–60 min in vacuum or in 133–266 Pa (1–2 Torr) of H2 using a furnace-based heating holder. When 13.3–200 Pa (100–1500 mTorr) of acetylene (C2H2) was introduced into the ETEM at a temperature ~450 °C, catalytic decomposition of C2H2 on the Ni/SiO2 catalysts led to the growth of carbon nanotubes. Multiwall nanotubes formed at temperatures as low as 450 °C while only single-wall carbon nanotubes (SWNTs) formed at higher temperatures ~700 °C and above. The growth rate, structure, and morphology of the CNTs are found to be dependent upon reaction temperature and pressure. When the growth temperature was maintained at 480 °C, multiwalled CNTs are observed to grow in a zigzag manner, forming waves, spirals or loops, at an average rate of 35–40 nm/s under C2H2 pressures of 2.67–13.3 Pa (20–100 mTorr), whereas SWNTs with diameters ~3.5 nm form at growth rates of 6–9 nm/s observed at C2H2 pressures <1.33 Pa (10 mTorr) (Sharma et al. 2005). When the C2H2 pressure was maintained at 0.13 Pa (1 mTorr) and the growth temperature varied from 450 to 650 °C, both the inner and outer nanotube diameters were found to decrease with increasing temperature (Sharma et al. 2007).

Image sequence of a growing carbon nanofibre. Images (a–h) illustrate the elongation/contraction process. Drawings are included to guide the eye in locating the positions of mono-atomic Ni step edges at the C–Ni interface. The images are acquired in situ with CH4:H2 ratio of 1:1 at a total pressure of 210 Pa (2.1 mbar) with the sample heated to 536 °C. All images are obtained with a rate of 2 frames per sec. Scale bar, 5 nm. Figure reprinted with permission from Helveg et al. (2004)
The catalyst plays an important role in the growth of CNTs via the CVD process. For example, the size of the catalysts dictates the SWNTs diameters grown (Zhang et al. 2002). For surface-bound CVD, the interactions between the catalyst and support play a critical role in controlling catalyst size distribution (Lolli et al. 2006). A fundamental question of interest is the physical and chemical state of the catalyst during the CNT growth process. Yoshida and coworkers (2008) vacuum-evaporated 1 nm-thick Fe on SiO2 and heated the substrate in an ETEM to 600 °C in high vacuum, and subsequently, in a mixture of C2H2:H2 = 1:1 at 10 Pa (0.08 Torr). They concluded, from real-time observations of the nucleation and growth processes of CNTs, that the nanoparticle catalysts are fluctuating crystalline cementite (Fe3C) particles. Hofmann et al. (2009) studied catalyst–support and catalyst–carbon interactions during the CVD growth of SWNTs using a combination of ETEM and in situ, time-resolved X-ray photoelectron spectroscopy, and found the active state of Fe and Ni catalysts to be the crystalline metallic nanoparticles. Pd (silicides) and Au are catalytically less efficient for C2H2 dissociation compared to Fe and Ni.
The growth of ultra-long, vertically oriented SWNT carpets or forests using CVD (Chattopadhyay et al. 2001; Hinds et al. 2004) is also of interest because of their suitability in a number of potential applications. To promote and sustain the growth of SWNT carpets, an approach known as “super growth” was developed by the Iijima group, whereby the introduction of H2O led to an increased activity and lifetime of the catalysts (Hata et al. 2004). Amama and coworkers (2009) showed a posteriori that the addition of H2O promotes super growth of SWNTs by inhibiting Ostwald ripening in the Fe catalyst nanoparticles on Al2O3 support. Kim et al. (2010) demonstrated using both ex situ quantitative measurements of CNT growth rate and in situ ETEM observations that termination of SWNT carpet growth can be linked to the evolution of the catalyst morphology and Fe subsurface migration. Yoshida and coworkers (2009) also demonstrated that the deposition of a proper quantity of Mo on substrates with Fe catalysts leads to the formation of Fe-Mo carbide nanoparticle catalysts of (Fe,Mo)23C6 structures as well as Fe3C. Mo suppresses the nucleation of iron silicates which inhibits catalytic activity in the growth of CNTs, thereby increasing the CNT yield.
The electronic properties of carbon nanotubes are influenced by their chirality (e.g., Odom et al. 1998; Wilder et al. 1998). Existing synthesis methods cannot controllably produce nanotubes with a specific type of conductivity. Harutyunyan and coworkers (2009) show that by varying the noble gas (He or Ar) during thermal annealing of the catalyst in combination with oxidative and reductive species, the yield of nanotubes with metallic conductivity can be significantly increased. TEM characterization reveals a strong difference in the morphological behavior of Fe nanoparticles depending on the gas environment at 500 °C, with all particles in He exhibiting stronger faceting whereas those in Ar were more round shaped.
In situ TEM has also been used to probe the vapor-liquid-solid (VLS) mechanism for the growth of semiconductor nanowires. One of the simplest forms of nanowire growth is by chemical vapor deposition (CVD), whereby a catalytic droplet (commonly a liquid Au-semiconductor eutectic) is formed on a surface and then exposed to a gas supply of the growth material, as illustrated in Fig. 1.10. Ross and coworkers (2010) from IBM studied semiconductor (Si and Ge) nanowires during the growth process in situ using a UHV-TEM which has been modified to enable chemical vapor deposition (CVD) and evaporation processes in situ (Hammar et al. 1996; Ross 2000). In the Si nanowire growth experiments, a Si(111) wafer cleaned with an HF dip and heated above 1200 °C to desorb any surface oxide was evaporated with Au (the catalytic material), and then mounted in the microscope with the Au-covered side vertical. Wire growth was then initiated by heating the specimen (using direct current) and exposing it to disilane gas. When nanowires grow epitaxially, perpendicular to the surface, they are also perpendicular to the electron beam, With this geometry, the nanowire/catalyst interface is parallel to the electron beam so that phenomena at the growth interface can be resolved easily (see Fig. 1.11, and Ross 2010).

(a) Phase diagram for the Au–Si binary system. The typical reaction path for Si nanowire growth is indicated by arrows. (b–f) Schematic diagrams of the process of Si and Ge nanowire growth from Au: (b) Au is deposited on HF-dipped Si. (c) Heating above 370 °C forms AuSi eutectic droplets containing ∼20 % Si. (d) Exposure to a source gas such as disilane which adsorbs preferentially at the droplet surface and supersaturates the droplet with Si. (d, e) Si precipitates beneath the droplet. (f) Heterostructures such as Si/Ge nanowires are grown by changing the source gas. Figure reprinted with permission from Ross (2010)

(a) The pole-piece region of the UHV TEM showing the specimen holder and the capillary tube through which gas is introduced. (b) The specimen, cut from a Si(111) wafer, showing AuSi droplets and the nanowire growth direction. The nanowires are shown as cylinders although their surfaces are actually faceted. (c) Typical TEM image of nanowires during growth, in this case at 550 °C and 1.3 × 10−4 Pa (1 × 10−6 Torr) Si2H6. Figure reprinted with permission from Ross (2010)
In situ growth studies of nanowires have led to new insights into the VLS growth mechanism. Ross and coworkers (2005) found that Si nanowires growing from a Si-Au eutectic show periodic sawtooth facets, which are at an angle to the growth direction. The wire surface is not smooth. Rather, growth occurs at a (111) facet at the end of the wire, and the size and shape of this facet oscillates periodically during growth. At higher temperatures and lower pressures, the presence of small amounts of oxygen was found to change the morphology of Si nanowires grown using the VLS process with Au (Kodambaka et al. 2006). Wires of almost constant diameter and several micrometers in length can be formed in the presence of oxygen, in contrast with wires grown in disilane alone, which show significant changes in diameter over sub-micrometer length. Contrary to the conventional VLS growth model assumptions, Au diffusion during nanowire growth determines the length, shape, and sidewall properties of the nanowires (Hannon et al. 2006). Au can diffuse from one catalyst droplet to another during growth, i.e., Au migrates over the surface as well as up and down the nanowire sidewalls, and Ostwald ripening during growth can lead to the termination of growth due to the disappearance of the Au catalyst. Using the same experimental setup, Kim et al. (2008) obtained quantitative measurements on the nucleation and growth kinetics of Si from a Au-Si eutectic liquid, and by using a simple kinetic model, provided insights into the nanowire growth process.
In some cases, it is also possible to grow nanowires in a high-vacuum TEM without supplying the growth material as a gas precursor. Instead, evaporation from the sample provides the flux required for the growth mechanism to be activated. For example, Stach and coworkers (2003) observed in real-time TEM observations of the growth of GaN nanowires via a self-catalytic VLS mechanism. These nanowires nucleate and grow from Ga droplets formed during thermal decomposition of GaN at elevated temperatures in a vacuum of ~1.3 × 10−5 Pa (1 × 10−7 Torr).
Metals other than Au are also of great interest for VLS growth studies because they allow flexibility in designing electronic properties such as doping type and doping level in as-grown nanowires (Ross 2010). They also provide the opportunity to grow nanowires with different surface structures. Wen et al. (2009) added Al to Au catalysts to raise the eutectic temperature with Si or Ge, and formed compositionally abrupt interfaces in Si-Ge and Si-SiGe heterostructure nanowires using the vapor-solid-solid (VSS) method. Chou et al. (2012) grew Si/Ge nanowires and heterojunctions with compositionally abrupt interfaces using a catalyst based on alloying Ag with Au. Hofmann and coworkers (2008) demonstrated that an environmental TEM with higher base pressure compared to the UHV-TEM can also be used to grow Si nanowires from Pd and Au catalysts.
2.4 Windowed Cell Holder Applications
A key limitation in differentially pumped ETEM systems is that the maximum gas pressure allowed is usually not more than 2 kPa (20 mbar), which is about three orders of magnitude lower than the ambient pressure condition at which many applications are concerned. This “pressure gap” can be bridged by the use of closed window gas cell TEM holders, as described in Sect. 1.1. In this section, some of the applications arising from the use of such closed cell systems in the area of solid-gas reaction studies are described. The reader can also refer to a later chapter about how the windowed cell setup can be used to study specimens in their native liquid medium.
Using the MEMS-based nanoreactor developed by Creemer et al. (2008), Yokosawa and coworkers (2012) investigated the (de)hydrogenation behavior of Pd islands at H2 pressure up to 4.5×105 Pa (4.5 bar) and temperature up to 400 °C, and observed, in electron diffraction mode, Pd crystal lattice expansion and shrinkage owing to hydrogenation and dehydrogenation, respectively. Vendelbo et al. (2014) also used a similar MEMS reactor which has been fitted with quantitative mass spectrometry, to analyze gases exiting the nanoreactor, and reaction calorimetry, to study the oscillatory behavior exhibited in catalytic reactions occurring under fixed conditions (in this case, at 105 Pa, or 1 bar, of CO:O2:He of 3 %:42 %:55 % ratio and temperatures 659–729 K). Pt nanoparticles, which catalyze the oxidation of CO, gradually transformed from a more spherical shape toward a more facetted shape as the CO conversion increased rapidly, and transformed back to the more spherical shape on decrease in CO conversion, and retained that shape until CO conversion increased steeply again. Their studies showed a direct coupling between the oscillatory CO conversion behavior and the Pt nanoparticle shape changes.
With an MEMS-based environmental holder developed by Protochips Inc., Ghassemi and coworkers (2014) studied the mechanism of transformation of two-dimensional Ti3C2, also known as “MXene”, into a TiO2/carbon hybrid material during oxidation at different temperatures and under different heating rates. The specimens which consisted of delaminated Ti3C2Tx (where T stands for the “terminating group” and x is the number of surface groups per Ti3C2 unit cell) flakes were dropcast on holder-compatible chips with electron transparent windows, sealed in ambient air, and flash-oxidized (by increasing the temperature stepwise to 950 °C) or slowly heated to 450 °C at a rate of 0.1 °C/s and maintained at 450 °C for 2 h. With flash oxidation, TiO2 particles were first formed in a planar thin film morphology and subsequently shrank laterally and grew vertically into 3D particles with the anatase phase, whereas slow heating led to the formation of planar nanocrystals of mainly rutile phase.
3 Current Status and Outlook
This chapter review illustrates that numerous researchers have contributed virtually since the original construction of the TEM to improving and applying the ability to observe material reactions in an encompassing environment, at the resolutions uniquely available with the transmission electron microscope, and with its improving analytical capability. Moreover, with the increasing development of specimen holders for both windowed and aperture systems, the breadth of experimentation (as illustrated by the compilation in the present text) is only broadening, and at an impressively rapid rate. An update of the analysis summarized in Fig. 1.1 to include the date of the 2014 International Microscopy Congress is shown in Fig. 1.12. It appears that there has been another significant jump in the numbers of in situ TEM publications, and that the trend noticed in Sinclair (2013) has been firmly established. The environmental (gas and liquid) contributions to this trend remain at over 50 %, as alluded in Fig. 1.12b. It is our conclusion that environmental studies in the TEM will continue to grow, both in numbers and in innovation, and that they will provide ever more insight into material reactions of scientific and technological interest.

(a) Number of publications until 2014 identified using the keywords “in situ”, “transmission electron microscopy” and “TEM”; (b) Breakdown by various subcategories of in situ papers presented at the 2014 Microscopy and Microanalysis Conference (USA) and the 2014 International Microscopy Congress (Czech Republic). These subcategories are in situ electrical biasing, electron beam heating, thermal heating, gaseous environment, liquid environment, mechanical loading, and laser stimulation
Notes
- 1.
We have, throughout this article, used pressures converted to SI units as well as those quoted in the original publications (in parentheses). The conversions are 1 Torr ≡ 1 mm Hg ≡ 1.33 mbar ≡ 133.3 Pa.
References
I.M. Abrams, J.W. McBain, A closed cell for electron microscopy. J. Appl. Phys. 15, 607–609 (1944)
L.F. Allard, S.H. Overbury, W.C. Bigelow, M.B. Katz, D.P. Nackashi, J. Damiano, Novel MEMS-based gas-cell/heating specimen holder provides advanced imaging capabilities for in situ reaction studies. Microsc. Microanal. 18, 656–666 (2012)
D.L. Allinson, Environmental cell for use in a high voltage electron microscope, in Proc. 7th Int. Congr. Electron Microscopy, Grenoble, 1970, pp. 169–170.
D.L. Allinson, Environmental devices in electron microscopy, in Principles and Techniques of Electron Microscopy Biological Applications, ed. by M.A. Hayat, vol. 5 (Van Nostrand Reinhold, New York, 1975)
D.H. Alsem, R.R. Unocic, G.M. Veith, K.L. More, N.J. Salmon, In-situ Liquid and Gas Transmission Electron Microscopy of Nano-Scale Materials. Microsc. Microanal. 18(Suppl 2), 1158–1159 (2012)
P.B. Amama, C.L. Pint, L. McJilton, S.M. Kim, E.A. Stach, P.T. Murray, R.H. Hauge, B. Maruyama, Role of water in super growth of single-walled carbon nanotube carpets. Nano Lett. 9, 44–49 (2009)
S. Arrii, F. Morfin, A.J. Renouprez, J.L. Rousset, Oxidation of CO on gold supported catalysts prepared by laser vaporization: Direct evidence of support contribution. J. Am. Chem. Soc. 126, 1199–1205 (2004)
R.T.K. Baker, P.S. Harris, Controlled atmosphere electron microscopy. J. Physics E: Sci. Instruments 5, 793–797 (1972)
R.T.K. Baker, M.A. Barber, P.S. Harris, F.S. Feates, R.J. Waite, Nucleation and growth of carbon deposits from the nickel catalyzed decomposition of acetylene. J. Catalysis 26, 51–62 (1972a)
R.T.K. Baker, F.S. Feates, P.S. Harris, Continuous electron microscopic observation of carbonaceous deposits formed on graphite and silica surfaces. Carbon 10, 93–96 (1972b)
R.T.K. Baker, P.S. Harris, R.B. Thomas, R.J. Waite, Formation of filamentous carbon from iron, cobalt and chromium catalyzed decomposition of acetylene. J. Catalysis 30, 86–95 (1973)
R.T.K. Baker, In situ electron microscopy studies of catalyst particle behavior. Catal. Rev. Sci. Eng. 19(2), 161–209 (1979)
R.T.K. Baker, Catalytic growth of carbon filaments. Carbon 27, 315–323 (1989)
R.T.K. Baker, Electron microscopy studies of the catalytic growth of carbon filaments, in Carbon Fibers Filaments and Composites, ed. by J.L. Figueiredo et al. (Kluwer Academic, Dordrecht, 1990), pp. 405–439
A. Baldi, T. Narayan, A.L. Koh, J.A. Dionne, In situ detection of hydrogen-induced phase transitions in individual palladium nanocrystals. Nature Mater. 13(12), 1143–1148 (2014)
E.D. Boyes, P.L. Gai, L.G. Hanna, Controlled environment (ECELL) TEM for dynamic in-situ reaction studies with HREM lattice imaging. Mat. Res. Soc. Symp. Proc. 404, 53–60 (1996)
E.D. Boyes, P.L. Gai, Environmental high resolution electron microscopy and applications to chemical science. Ultramicroscopy 67, 219–232 (1997)
E.D. Boyes, M.R. Ward, L. Lari, P.L. Gai, ESTEM imaging of single atoms under controlled temperature and gas environment conditions in catalyst reaction studies. Ann. Phys. (Berlin) 525(6), 423–429 (2013)
E.D. Boyes, P.L. Gai, Aberration corrected environmental STEM (AC ESTEM) for dynamic in-situ gas reaction studies of nanoparticle catalysts. Journal of Physics: Conference Series 522, 012004 (2014a)
E.D. Boyes, P.L. Gai, Visualising reacting single atoms under controlled conditions: Advances in atomic resolution in situ environmental (scanning) transmission electron microscopy (E(S)TEM). C. R. Physique 15, 200–213 (2014b)
E.P. Butler, K.F. Hale, In situ studies of gas-solid reactions, in Dynamic Experiments in the Electron Microscope, ed. by A.M. Glauert. Practical Methods in Electron Microscopy, vol. 9 (North-Holland, Amsterdam, 1981a)
E.P. Butler, K.F. Hale, Wet cell microscopy, in Dynamic Experiments in the Electron Microscope, ed. by A.M. Glauert. Practical Methods in Electron Microscopy, vol. 9 (North-Holland, Amsterdam, 1981b)
M. Cargnello, V.V.T. Doan-Nguyen, T.R. Gordon, R.E. Diaz, E.A. Stach, R.J. Gorte, P. Fornasiero, C.B. Murray, Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 341, 771–773 (2013)
F. Cavalca, A.B. Laursen, B.E. Kardynal, R.E. Dunin-Borkowski, S. Dahl, J.B. Wagner, T.W. Hansen, In situ transmission electron microscopy of light-induced photocatalytic reactions. Nanotechnology 23, 075705 (2012) (6 pp.)
F. Cavalca, A.B. Laursen, J.B. Wagner, C.D. Damsgaard, I. Chorkendorff, T.W. Hansen, Light-induced reduction of cuprous oxide in an environmental transmission electron microscope. ChemCatChem 5, 2667–2672 (2013)
D. Chattopadhyay, I. Galeska, F. Papadimitrakopoulos, Metal-assisted organization of shortened carbon nanotubes in monolayer and multilayer forest assemblies. J. Am. Chem. Soc. 123, 9451–9452 (2001)
S. Chenna, P.A. Crozier, Operando transmission electron microscopy: A technique for detection of catalysis using electron energy-loss spectroscopy in the transmission electron microscope. ACS Catal. 2, 2395–2402 (2012)
W.B. Choi, D.S. Chung, J.H. Kang, H.Y. Kim, Y.W. Jin, I.T. Han, Y.H. Lee, J.E. Jung, N.S. Lee, G.S. Park, J.M. Kim, Fully sealed, high-brightness carbon-nanotube field-emission display. Appl. Phys. Lett. 75, 3129–3131 (1999)
Y.-C. Chou, C.-Y. Wen, M.C. Reuter, D. Su, E.A. Stach, F.M. Ross, Controlling the growth of Si/Ge nanowires and heterojunctions using silver-gold alloy catalysts. ACS Nano 6, 6407–6415 (2012)
J.F. Creemer, S. Helveg, G.H. Hoveling, S. Ullmann, A.M. Molenbroek, P.M. Sarro, H.W. Zandbergen, Atomic-scale electron microscopy at ambient pressure. Ultramicroscopy 108, 993–998 (2008)
P.A. Crozier, R. Sharma, A.K. Datye, Oxidation and reduction of small palladium particles on silica. Microsc. Microanal. 4, 278–285 (1998)
P.A. Crozier, R. Wang, R. Sharma, In situ environmental TEM studies of dynamic changes in cerium-based oxides nanoparticles during redox processes. Ultramicroscopy 108, 1432–1440 (2008)
P.A. Crozier, S. Chenna, In situ analysis of gas composition by electron energy-loss spectroscopy for environmental transmission electron microscopy. Ultramicroscopy 111, 177–185 (2011)
W.A. de Heer, A. Châtelain, D. Ugarte, A carbon nanotube field-emission electron source. Science 270, 1179–1180 (1995)
N. de Jonge, D.B. Peckys, G.J. Kremers, D.W. Piston, Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. U. S. A. 106, 2159–2164 (2009)
N. de Jonge, W.C. Bigelow, G.M. Veith, Atmospheric pressure scanning transmission electron microscopy. Nano Lett. 10, 1028–1031 (2010)
N. de Jonge, F.M. Ross, Electron microscopy of specimens in liquid. Nature Nano 6, 695–704 (2011)
M. Doi, M. Yoshida, M. Nonoyama, S. Arai, T. Imura, A modified window-type environmental cell for use with HVEM, in Proc. 5th Int. Conf. High Voltage Electron Microscopy, Kyoto, 1977, p. 155.
R.C. Doole, G.M. Parkinson, J.M. Stead, High resolution gas reaction cell for the JEM 4000. Institute of Physics Conference Series 119, 157–160 (1991)
G. Dupouy, Electron microscopy at very high voltages, in Advances in optical and electron microscopy, ed. by R. Barer, V.E. Cosslett, vol. 2 (Academic Press, London, 1968), p. 167
J. Escaig, C. Sella, Cellule porte-objet permettent l’observation des specimens chauffes sous atmosphere controlee, in Proc. 6th Int. Cong. Electron Microsc., 1966, vol. 1, p. 177
J. Escaig, C. Sella, Application d’une nouvelle cellule a atmosphere controlee a l’etude de quelques problems d’oxydation et de depot par decomposition termique en phase vapeur, in Proc. 4th Europ. Conf. Electron Microsc., 1968, vol. 1, 241.
J. Escaig, C. Sella, Nouvelle microchambre pour observation en microscopie-electronique, a haute temperature et sous atmosphere controlee. C. r. hebd. Séanc. Acad. Sci., Paris 268, 532 (1969)
J. Escaig, C. Sella, Observation in situ au microscope electronique de l’oxydation desc couches minces metalliques. C. r. hebd. Séanc. Acad. Sci., Paris 274, 27 (1972)
F.S. Feates, H. Morley, P.S. Robinson, Controlled atmosphere electron microscopy, in Proc. 7th Int. Congr. Electron Microscopy, Grenoble, 1970, pp. 295–296
H.M. Flower, High voltage electron microscopy of environmental reactions. J. Micros. 97, 171–190 (1973)
H.M. Flower, P.R. Swann, An in situ study of Titanium oxidation by high voltage electron microscopy. Acta Metall. 22, 1339–1347 (1974)
H.M. Flower, B.A. Wilcox, In situ oxidation of Ni-30 Wt% Cr and TDNiCr in the high voltage electron microscope. Corros. Sci. 17, 253–264 (1977)
J.R. Fryer, Oxidation of graphite catalyzed by palladium. Nature 220, 1121–1122 (1968a)
J.R. Fryer, The direct observation of gas solid reactions inside the electron microscope. Siemens Review 35, 13 (1968b)
Q. Fu, H. Saltsburg, M. Flytzani-Stephanopoulos, Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 301, 935–938 (2003)
H. Fujita, M. Komatsu, I. Ishikawa, A universal environmental cell for a 3MV-class electron microscope and its applications to metallurgical sciences. Jap. J. Appl. Phys. 15(11), 2221–2228 (1976)
A. Fukami, K. Adachi, A new method of preparation of a self-perforated micro plastic grid and its application (I). J. Electron Microscopy 14(2), 112–118 (1965)
A. Fukami, K. Adachi, M. Katoh, On a study of new micro plastic grid and its applications, in Proc. 6th Int. Congr. Electron Microscopy, Kyoto, 1966, pp. 262–264
A. Fukami, K. Adachi, M. Katoh, Micro grid techniques (continued) and their contribution to specimen preparation techniques for high resolution work. J. Electron Microscopy 21(2), 99–108 (1972)
P.L. Gai, E.D. Boyes, Environmental high resolution electron microscopy in materials science, in In-situ microscopy in materials research, ed. by P.L. Gai (Kluwer Academic, Boston, 1997)
P.L. Gai, E.L. Boyes, S. Helveg, P.L. Hansen, S. Giorgio, C.R. Henry, Atomic-resolution environmental transmission electron microscopy for probing gas-solid reactions in heterogeneous catalysis. MRS Bulletin 32, 1044–1050 (2007)
P.L. Gai, R. Sharma, F.M. Ross, Environmental (S)TEM studies of gas-liquid-solid interactions under reaction conditions. MRS Bulletin 33, 107–114 (2008)
P.L. Gai, E.D. Boyes, Advances in atomic resolution in situ environmental transmission electron microscopy and 1Å aberration corrected in situ electron microscopy. Microsc. Res. Tech. 72, 153–164 (2009)
E.J. Gallegos, Gas reactor for hot stage transmission electron microscopy. Rev. Sci. Instrum. 35(9), 1123–1124 (1964)
H. Ghassemi, W. Harlow, O. Mashtalir, M. Beidaghi, M.R. Lukatskaya, Y. Gogotsi, M.L. Taheri, In situ environmental transmission electron microscopy study of oxidation of two-dimensional Ti3C2 and formation of carbon-supported TiO2. J. Mater. Chem. A 2, 14339–14343 (2014)
S. Giorgio, S. Sao Joao, S. Nitsche, D. Chaudanson, G. Sitja, C.R. Henry, Environmental electron microscopy (ETEM) for catalysts with a closed E-cell with carbon windows. Ultramicroscopy 106, 503–507 (2006)
M. Goringe, A. Rawcliffe, A. Burden, J. Hutchison, R. Doole, Observations of solid-gas reactions by means of high-resolution transmission electron microscopy. Faraday Discuss. 105, 85–102 (1996)
M. Haider, S. Uhlemann, E. Schwan, H. Rose, B. Kabius, K. Urban, Electron microscopy image enhanced. Nature 392, 768–769 (1998a)
M. Haider, H. Rose, S. Uhlemann, E. Schwan, B. Kabius, K. Urban, A spherical aberration-corrected 200 kV transmission electron microscope. Ultramicroscopy 75, 53–60 (1998b)
M. Hammar, F. LeGoues, J. Tersoff, M.C. Reuter, R.M. Tromp, In situ ultrahigh vacuum transmission electron microscopy studies of hetero-epitaxial growth. I. Si(009)/Ge. Surf. Sci. 349, 129–144 (1996)
P.L. Hansen, J.B. Wagner, S. Helveg, J.R. Rostrup-Nielsen, B.S. Clausen, H. Topsøe, Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science 295, 2053–2055 (2002)
P.L. Hansen, S. Helveg, A.K. Datye, Atomic-scale imaging of supported metal nanocluster catalysts in the working state. Adv. Catal. 50, 77–95 (2006)
T.W. Hansen, J.B. Wagner, P.L. Hansen, S. Dahl, H. Topsøe, C.J.H. Jacobson, Atomic-resolution in situ transmission electron microscopy of a promoter of a heterogeneous catalyst. Science 294, 1508–1510 (2001)
T.W. Hansen, J.B. Wagner, R.E. Dunin-Borkowski, Aberration-corrected and monochromated environmental transmission electron microscopy: challenges and prospects for materials science. Mater. Sci. Technol. 26(11), 1338–1344 (2010)
T.W. Hansen, J.B. Wagner, Environmental transmission electron microscopy in an aberration-corrected environment. Micros. Microanal. 18, 684–690 (2012)
J.B. Hannon, S. Kodambaka, F.M. Ross, R.M. Tromp, The influence of the surface migration of gold on the growth of silicon nanowires. Nature 440, 69–71 (2006)
M. Haruta, Size- and support-dependency in the catalysis of gold. Catal. Today 36, 153–166 (1997)
A.R. Harutyunyan, G. Chen, T.M. Paronyan, E.M. Pigos, O.A. Kuznetsov, K. Hewaparakrama, S.M. Kim, D. Zakharov, E.A. Stach, G.U. Sumanasekera, Preferential growth of single-walled carbon nanotubes with metallic conductivity. Science 326, 116–120 (2009)
H. Hashimoto, K. Tanaka, E. Yoda, A specimen treating device at high temperature for the electron microscope. J. Electron Microscopy 6, 8–11 (1958)
H. Hashimoto, T. Naiki, T. Eto, K. Fujiwara, M. Watanabe, Y. Nagahama, Specimen chamber for observing the reaction process with gas at high temperature, in Sixth Int. Congress for Electron Microscopy, 1966, pp. 181–182.
H. Hashimoto, T. Naiki, T. Eto, K. Fujiwara, High temperature gas reaction specimen chamber for an electron microscope. Jpn. J. Appl. Phys. 7(8), 946–952 (1968)
S. Hashimoto, S. Urai, H. Yotsumoto, J. Sawamori, Morphology and structure of Mo oxides formed by thermal decomposition of the ammonium salt, in Proc. 7th Int. Congr. Electron Microscopy, Berlin, 1970, p. 87.
K. Hata, D.N. Fubata, K. Mizuno, T. Namai, M. Yumara, S. Iijima, Water-assisted highly efficient synthesis of impurity-free single-walled carbon nanotubes. Science 306, 1362–1364 (2004)
T. Hayashi, K. Tanaka, M. Haruta, Selective vapor-phase epoxidation of propylene over Au/TiO2 catalysts in the presence of oxygen and hydrogen. J. Catal. 178, 566–575 (1998)
H.G. Heide, Electron microscopic observation of specimens under controlled gas pressure. J. Cell. Bio. 13, 147–152 (1962)
S. Helveg, C. Lopez-Cartes, J. Sehested, P.L. Hansen, B.S. Clausen, J.R. Rostrup-Nielsen, F. Abild-Pedersen, J.K. Nørskov, Atomic-scale imaging of carbon nanofibre growth. Nature 427, 426–429 (2004)
B.J. Hinds, N. Chopra, T. Rantell, R. Andrews, V. Gavalas, L.G. Bachas, Aligned multiwalled carbon nanotube membranes. Science 303, 62–65 (2004)
S. Hofmann, S. Sharma, C.T. Wirth, F. Cervantes-Sodi, C. Ducati, T. Kasama, R.E. Dunin-Borkowski, J. Drucker, P. Bennett, J. Robertson, Ledge-flow-controlled catalyst interface dynamics during Si nanowire growth. Nature Mater. 7, 372–375 (2008)
S. Hofmann, R. Blume, C.Y. Wirth, M. Cantoro, R. Sharma, C. Ducati, M. Hävecker, S. Zafeiratos, P. Schnoerch, A. Oestereich, D. Teschner, M. Albrecht, A. Knop-Gericke, R. Schlögl, J. Robertson, State of transition metal catalysts during carbon nanotube growth. J. Phys. Chem. C 113, 1648–1656 (2009)
S. Iijima, Helical microtubules of graphitic carbon. Nature 354, 56–58 (1991)
T. Ito, K. Hiziya, A specimen reaction device for the electron microscope and its applications. J. Electron Microscopy 6, 4–8 (1958)
J.R. Jinschek, S. Helveg, Image resolution and sensitivity in an environmental transmission electron microscope. Micron 43, 1156–1168 (2012)
J.R. Jinschek, Advances in the environmental transmission electron microscope (ETEM) for nanoscale in situ studies of gas–solid interactions. Chem. Commun. 50, 2696–2706 (2014)
T. Kamino, T. Yaguchi, M. Konno, A. Watabe, T. Marukawa, T. Mina, K. Kuroda, H. Saka, S. Arai, H. Makino, Y. Suzuki, K. Kishita, Development of a gas injection/specimen heating holder for use with transmission electron microscope. J. Electron. Microsc. 54, 497–503 (2005)
T. Kamino, T. Yaguchi, M. Konno, A. Watabe, Y. Nagakubo, Development of a specimen heating holder with an evaporator and gas injector and its application for catalyst. J. Electron Microsc. 55, 245–252 (2006)
T. Kawasaki, K. Ueda, M. Ichihashi, T. Tanji, Improvement of windowed type environmental-cell transmission electron microscope for in situ observation of gas-solid interactions. Re. Sci. Instrum. 80, 113701 (2009)
B.J. Kim, J. Tersoff, S. Kodambaka, M.C. Reuter, E.A. Stach, F.M. Ross, Kinetics of individual nucleation events observed in nanoscale vapor–liquid–solid growth. Science 322, 1070–1073 (2008)
S.M. Kim, C.L. Pint, P.B. Amama, D.N. Zakharov, R.H. Hauge, B. Maruyama, E.A. Stach, Evolution in catalyst morphology leads to carbon nanotube growth termination. J. Phys. Chem. Lett. 1, 918–922 (2010)
K.L. Klein, I.M. Anderson, N. de Jonge, Transmission electron microscopy with a liquid flow cell. J. Microscopy 242, 117–123 (2011)
S. Kodambaka, J.B. Hannon, R.M. Tromp, F.M. Ross, Control of Si nanowire growth by oxygen. Nano Lett. 6, 1292–1296 (2006)
A.L. Koh, E. Gidcumb, O. Zhou, R. Sinclair, Observations of carbon nanotube oxidation in an aberration-corrected environmental transmission electron microscope. ACS Nano 7, 2566–2572 (2013)
Y. Kuwauchi, H. Yoshida, T. Akita, M. Haruta, S. Takeda, Intrinsic catalytic structure of gold nanoparticles supported on TiO2. Angew. Chem. Int. Ed. 51, 7729–7733 (2012)
Y. Kuwauchi, S. Takeda, H. Yoshida, K. Sun, M. Haruta, H. Kohno, Stepwise displacement of catalytically active gold nanoparticles on cerium oxide. Nano Lett. 13, 3073–3077 (2013)
T.C. Lee, D.K. Dewald, J.A. Eades, I.M. Robertson, H.K. Birnbaum, An environmental cell transmission electron microscope. Rev. Sci. Instrum. 62, 1438–1444 (1991)
R.-J. Liu, P.A. Crozier, C.M. Smaith, D.A. Hucul, J. Blackson, G. Salaita, In situ electron microscopy studies of the sintering of palladium nanoparticles on alumina during catalyst regeneration processes. Microsc. Microanal. 10, 77–85 (2004)
G. Lolli, L.A. Zhang, L. Balzano, N. Sakulchaicharoen, Y.Q. Tan, D.E. Resasco, Tailoring (n, m) structure of single-walled carbon nanotubes by modifying reaction conditions and the nature of the support of CoMo catalysts. J. Phys. Chem. B 110, 2108–2115 (2006)
C. López-Cartes, S. Bernal, J.J. Calvino, M.A. Cauqui, G. Blanco, J.A. Pérez-Omil, J.M. Pintado, S. Helveg, P.L. Hansen, In situ transmission electron microscopy investigation of Ce(IV) and Pr(IV) reducibility in a Rh (1%)/Ce0.8Pr0.2O2−x catalyst. Chem. Commun. 5, 644–645 (2003)
L. Marton, Electron microscopy of biological objects. Nature 133, 911 (1934)
L. Marton, La microscopie electronique des objets biologiques. Bull. Acad. r. Belg. Cl. Sci. 21, 553 (1935)
M.L. McDonald, J.M. Gibson, F.C. Unterwald, Design of an ultrahigh-vacuum specimen environment for high-resolution transmission electron microscopy. Rev. Sci. Instrum. 60(4), 700–707 (1989)
B.K. Miller, P.A. Crozier, System for in situ UV-visible illumination of environmental transmission electron microscopy samples. Microsc. Microanal. 19, 461–469 (2013)
J.C. Mills, A.F. Moodie, Multipurpose high resolution stage for the electron microscope. Rev. Sci. Instrum. 39, 962–969 (1968)
A.M. Molenbroek, S. Helveg, H. Topsøe, B.S. Clausen, Nano-particles in heterogeneous catalysis. Top Catal 52, 1303–1311 (2009)
T.W. Odom, J.-L. Huang, P. Kim, C.M. Lieber, Atomic structure and electronic properties of single-walled carbon nanotubes. Nature 391, 62–64 (1998)
G.M. Parkinson, High resolution, in-situ controlled atmosphere transmission electron microscopy (CATEM) oh heterogeneous catalysts. Catal. Lett. 2, 303–308 (1989)
D.F. Parsons, Structure of wet specimens in electron microscopy. Science 186, 407–414 (1974)
D.F. Parsons, V.R. Matricardi, R.C. Moretz, J.N. Turner, Electron microscopy and diffraction of wet unstained and unfixed biological objects, in: Advances in Biological and Medical Physics (Volume 15). Edited by J. H. Lawrence, J (W Gofman and T. L. Hayes. Academic Press, New York, 1974)
E.A. Ring, N. de Jonge, Microfluidic System for Transmission Electron Microscopy. Microsc. Microanal. 16, 622–629 (2010)
A.G. Rinzler, J.H. Hafner, P. Nikolaev, L. Lou, S.G. Kim, D. Tomimek, P. Nordlander, D.T. Colbert, R.E. Smalley, Unraveling nanotubes: Field emission from an atomic wire. Science 269, 1550–1553 (1995)
N.M. Rodriguez, S.G. Oh, W.B. Downs, P. Pattabiraman, R.T.K. Baker, An atomic oxygen environmental cell for a transmission electron microscope. Rev. Sci. Instrum. 61(7), 1863–1868 (1990)
F.M. Ross, J.M. Gibson, Dynamic observations of interface propagation during silicon oxidation. Phys. Rev. Lett. 68(11), 1782–1786 (1992)
F.M. Ross, Growth processes and phase transformations studied by in situ transmission electron microscopy. IBM J. Res. Develop. 44, 489–501 (2000)
F.M. Ross, J. Tersoff, M.C. Reuter, Sawtooth faceting in silicon nanowires. Phys. Rev. Lett. 95, 146104 (2005)
F.M. Ross, Controlling nanowire structures through real time growth studies. Rep. Prog. Phys. 73, 114501 (2010) (21 pp.)
F.M. Ross, C.-Y. Wen, S. Kodambaka, B.A. Wacser, M.C. Reuter, E.A. Stach, The growth and characterization of Si and Ge nanowires grown from reactive metal catalysts. Phil. Mag. 90, 4679–4778 (2010)
V.E. Ruska, Beitrag zur ubermikroskopischen Abbildung bei höheren Drucken. Zolloid-Z 100, 212–219 (1942)
A. Sandoval, A. Gomez-Cortes, R. Zanella, G. Diaz, J.M. Saniger, Gold nanoparticles: Support effects for the WGS reaction. J. Mol. Cat. A: Chemical 278, 200–208 (2007)
R. Sharma, K. Weiss, M. McKelvy, W. Glaunsinger, Gas reaction chamber for gas–solid interaction studies by high resolution transmission electron microscopy, in Proceedings of the 52nd Annual Meeting of the Microscopy Society of America, 1994, pp. 494–495
R. Sharma, K. Weiss, Development of a TEM to study in situ structural and chemical changes at an atomic level during gas-solid interactions at elevated temperatures. Micros. Res. Tech. 42, 270–280 (1998)
R. Sharma, Design and applications of environmental cell transmission electron microscope for in situ observations of gas-solid reactions. Microsc. Microanal. 7, 494–506 (2001)
R. Sharma, Z. Iqbal, In situ observations of carbon nanotube formation using environmental transmission electron microscopy. Appl. Phys. Lett. 84, 990–992 (2004)
R. Sharma, P. Crozier, Z.C. Kang, L. Eyring, Observation of dynamic nanostructural and nanochemical changes in ceria-based catalysts during in-situ reduction. Philosophical Magazine 84, 2731–2747 (2004)
R. Sharma, An environmental transmission electron microscope for in situ synthesis and characterization of nanomaterials. J. Mater. Res. 20(7), 1695–1707 (2005)
R. Sharma, P.A. Crozier, Environmental transmission electron microscopy in nanotechnology, in Handbook of Microscopy for Nanotechnology, ed. by N. Yao, Z.L. Wang (Kluwer Academic Publishers, New York, 2005), pp. 531–565
R. Sharma, P. Rez, M.M.J. Treacy, S.J. Stuart, In situ observation of the growth mechanisms of carbon nanotubes under diverse reaction conditions. J. Electron Microsc. 54, 231–237 (2005)
R. Sharma, P. Rez, P. Brown, G. Du, M.M.J. Treacy, Dynamic observations of the effect of pressure and temperature conditions on the selective synthesis of carbon nanotubes. Nanotechnology 18, 125602 (2007) (8pp)
D. Shindo, K. Takahashi, Y. Murakami, K. Yamazaki, S. Deguchi, H. Suga, Y. Kondo, Development of a multifunctional TEM specimen holder equipped with a piezodriving probe and a laser irradiation port. J Electron Microsc 58, 245–249 (2009)
S.B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested, S. Helveg, Direct observations of oxygen-induced platinum nanoparticle ripening studied by in situ TEM. J. Am. Chem. Soc. 132, 7968–7975 (2010)
R. Sinclair, T. Yamashita, M.A. Parker, K.B. Kim, K. Holloway, A.F. Schwartzman, The development of in situ high resolution electron microscopy. Acta Crystallogr. Sec. A 44, 965–975 (1988)
R. Sinclair, In situ high-resolution transmission electron microscopy of material reactions. MRS Bulletin 38, 1065–1071 (2013)
E.A. Stach, P. Pauzauskie, T. Kuykendall, J. Goldberger, P. Yang, Watching GaN nanowires grow. Nano Lett. 3, 867–869 (2003)
H. Sugie, M. Tanemura, V. Filip, K. Iwata, K. Takahashi, F. Okuyama, Carbon nanotubes as electron source in an X-Ray tube. Appl. Phys. Lett. 78, 2578–2580 (2001)
P.R. Swann, N.J. Tighe, High voltage microscopy of gas oxide reactions. Jernkont. Annaler 155, 497–501 (1971)
P.R. Swann, High voltage microscope studies of environmental reactions in electron microscopy and structure of materials, in Proceedings of the Fifth International Materials Symposium, September 13-17, 1971, ed. by G. Thomas, R.M. Fulrath, R.M. Fisher (University of California Press, Berkeley, 1972), pp. 878–904
P.R. Swann, N.J. Tighe, Performance of a differentially pumped environmental cell, in The AEI EM7. Proc. Fifth European Congress on Electron, Microscopy, 1972, pp. 360–361
P.R. Swann, G. Thomas, N.J. Tighe, In situ observations of the nitriding of tantalum. J. Microscopy 97, 24–257 (1973)
P.R. Swann, N.J. Tighe, High voltage microscopy of the reduction of hematite to magnetite. Metallurgical Transactions B 8B, 479–487 (1977)
N. Takahashi, T. Takeyama, K. Ito, T. Ito, K. Mihama, M. Watanabe, High temperature furnace for the electron microscope. J. Electron Microscopy 4, 16–23 (1956)
S. Takeda, H. Yoshida, Atomic-resolution environmental TEM for quantitative in-situ microscopy in materials science. Microscopy 62(1), 193–203 (2013)
S. Takeda, Y. Kuwauchi, H. Yoshida, Environmental transmission electron microscopy for catalyst materials using a spherical aberration corrector. Ultramicroscopy 151, 178–190 (2015)
N. Tanaka, J. Usukura, M. Kusunoki, Y. Saito, K. Sasaki, T. Tanji, S. Muto, S. Arai, Development of an environmental high-voltage electron microscope for reaction science. Microscopy 62(1), 205–215 (2013)
N. Tanaka, J. Usukura, M. Kusunoki, Y. Saito, K. Sasaki, T. Tanji, S. Muto, S. Arai, Development of an environmental high voltage electron microscope and its application to nano and bio-materials. Journal of Physics: Conference Series 522, 012008 (2014)
P.C. Tiemeijer, Measurement of Coulomb interactions in an electron beam monochromator. Ultramicroscopy 78, 53–62 (1999)
T. Uchiyama, H. Yoshida, Y. Kuwauchi, S. Ichikawa, S. Shimada, M. Haruta, S. Takeda, Systematic morphology changes of gold nanoparticles supported on CeO2 during CO oxidation. Angew. Chem. Int. Ed. 50, 10157–10160 (2011)
S.B. Vendelbo, C.F. Elkjær, H. Falsig, I. Puspitasari, P. Dona, L. Mele, B. Morana, B.J. Nelissen, R. van Rijn, J.F. Creemer, P.J. Kooyman, S. Helveg, Visualization of oscillatory behaviour of Pt nanoparticles catalysing CO oxidation. Nat. Mater. 13, 884–890 (2014)
P.C.K. Vesborg, I. Chorkendorff, I. Knudsen, O. Balmes, J. Nerlov, A.M. Molenbroek, B.S. Clausen, S. Helveg, Transient behavior of Cu/ZnO-based methanol synthesis catalysts. J. Catal. 262, 65–72 (2009)
J.B. Wagner, P.L. Hansen, A.M. Molenbroek, H. Topsøe, B.S. Clausen, S. Helveg, In situ electron energy loss spectroscopy studies of gas-dependent metal-support interactions in Cu/ZnO catalysts. J. Phys. Chem. B 107, 7753–7758 (2003)
J.B. Wagner, F. Cavalca, C.D. Damsgaard, L.D.L. Duchstein, T.W. Hansen, Exploring the environmental transmission electron microscope. Micron 43, 1169–1175 (2012)
Q.H. Wang, A.A. Setlur, J.M. Lauerhaas, J.Y. Dai, E.W. Seelig, R.P.H. Chang, A Nanotube-Based Field-Emission Flat Panel Display. Appl. Phys. Lett. 72, 2912–2913 (1998)
R. Wang, P.A. Crozier, R. Sharma, J.B. Adas, Measuring the redox activity of individual catalytic particles in cerium-based oxides. Nano Lett. 8(3), 962–967 (2008)
R. Wang, P.A. Crozier, R. Sharma, Structural transformation in ceria nanoparticles during redox processes. J. Phys. Chem. C 113, 5700–5704 (2009)
P.R. Ward, R.F. Mitchell, A facility for electron microscopy of specimens in controlled environments. Journal of Physics E: Scientific Instruments 5, 160–162 (1972)
C.-Y. Wen, M.C. Reuter, J. Bruley, J. Tersoff, S. Kodambaka, E.A. Stach, F.M. Ross, Formation of compositionally abrupt axial heterojunctions in Si/Ge nanowires. Science 326, 1247–1250 (2009)
J.W.G. Wilder, C. VenemaL, A.G. Rinzler, R.E. Smalley, C. Dekker, Electronic structure of atomically resolved carbon nanotubes. Nature 391, 59–62 (1998)
W.D. Williams, M. Shekhar, W.-S. Lee, V. Kispersky, W.N. Delgass, F.H. Ribeiro, S.M. Kim, E.A. Stach, J.T. Miller, L.F. Allard, Metallic corner atoms in gold clusters supported on rutile are the dominant active site during water-gas shift catalysis. J. Am. Chem. Soc. 132, 14018–14020 (2010)
M.J. Williamson, R.M. Tromp, P.M. Vereecken, F.M. Ross, Dynamic microscopy of nanoscale cluster growth at solid-liquid interface. Nat. Mat. 2, 532–536 (2003)
T. Yaguchi, M. Suzuki, A. Watabe, Y. Nagakubo, K. Ueda, T. Kamino, Development of a high temperature–atmospheric pressure environmental cell for high-resolution TEM. J. Electron Microsc. (Tokyo) 60(3), 217–225 (2011)
T. Yokosawa, T. Alan, G. Pandraud, B. Dam, H. Zandbergen, In-situ TEM on (de)hydrogenation of Pd at 0.5–4.5 bar hydrogen pressure and 20–400°C. Ultramicroscopy 112, 47–52 (2012)
H. Yoshida, S. Takeda, Image formation in a transmission electron microscope equipped with an environmental cell: Single-walled carbon nanotubes in source gases. Phys. Rev. B 72, 195428 (2005)
H. Yoshida, T. Uchiyama, S. Takeda, Environmental Transmission Electron Microscopy Observations of Swinging and Rotational Growth of Carbon Nanotubes. Jap. J. Appl. Phys. 46, L917–L919 (2007)
H. Yoshida, S. Takeda, T. Uchiyama, H. Kohno, Y. Homma, Atomic-scale in-situ observation of carbon nanotube growth from solid state iron carbide nanoparticles. Nano Lett. 8, 2082–2086 (2008)
H. Yoshida, T. Shimizu, T. Uchiyama, H. Kohno, Y. Homma, S. Takeda, Atomic-scale analysis on the role of molybdenum in iron catalyzed carbon nanotube growth. Nano Lett. 9, 3810–3815 (2009)
H. Yoshida, Y. Kuwauchi, J.R. Jinschek, K. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta, S. Takeda, Visualizing gas molecules interacting with supported nanoparticulate catalysts at reaction conditions. Science 335, 317–319 (2012)
L. Zhang, B.K. Miller, P.A. Crozier, Atomic level in situ observation of surface amorphization in anatase nanocrystals during light irradiation in water vapor. Nano Lett. 13, 679–684 (2013)
Y. Zhang, Y. Li, W. Kim, D. Wang, H. Dai, Imaging as-grown single-walled carbon nanotubes originated from isolated catalytic nanoparticles. Appl. Phys. A 74, 325–328 (2002)
Acknowledgements
A.L.K. is grateful for the support from the Stanford Nano Shared Facilities. S.C.L. and R.S. acknowledge funding support from the Center of Nanostructuring for Efficient Energy Conversion (CNEEC) at Stanford University, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award no. DE-SC0001060. R.S. also acknowledges funding from the National Cancer Institute grant CCNE-T U54CA151459-02.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Koh, A.L., Lee, S.C., Sinclair, R. (2016). A Brief History of Controlled Atmosphere Transmission Electron Microscopy. In: Hansen, T., Wagner, J. (eds) Controlled Atmosphere Transmission Electron Microscopy. Springer, Cham. https://doi.org/10.1007/978-3-319-22988-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-22988-1_1
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22987-4
Online ISBN: 978-3-319-22988-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)