
Abstract
One of the most nonnegligible characters of a Transmission Electron Microscope (TEM) is that the interior chamber which contains an electron source, (almost all) lens, apertures, and other functional accessories should maintain a high vacuum or even ultrahigh vacuum during observation. The requirement is determined by the fundamental designed principles, including keeping the high vacuum around the electron source to maintain the capability, and obviating the collisions between the electrons and atmospheric molecules to guarantee the coherence of electron wave. However, the realistic conditions for material operation are usually under gas environments, which contradict the high vacuum environment. Therefore, to compromise the conflict between the TEM apparatus itself and the requirements derived from a large amount of real applications, environmental transmission electron microscope (ETEM) emerged. The word “environmental” in the phrase emphasizes various gaseous surroundings, including different gases and adjustable pressure. The key to solving the problem mainly lies in confining a certain volume of gas only around the specimen to build a gas environment, while maintaining the other chambers under a high vacuum. There are two approaches to achieving the goal nowadays, and both of them are developed well and have unique advantages, which facilitate their broad applications in gas-involved fields, such as catalysis and nanocrystal growth. This chapter will introduce the developing history of gas TEM, and explain two main approaches, then present a summary of applications based on the ETEM and its attachments, and finally the foreseeable prospect and developing tendency.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

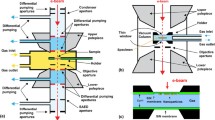

9.1 A History of In Situ Gas TEM
Since the pioneers of transmission electron microscope (TEM) lied the foundation of electron microscopy in 1932 [1, 2], the exploration and development of the TEM apparatus itself and various accessories are being constantly carried out.
In the early stages of development, due to the low magnification and unsatisfying resolution of the TEM, high spatial resolution and sensitivity are the main pursuits of modifications to the microscope. Among the affecting factors, the degree of vacuum is an important and accessible factor to improve the resolution. The scattering of electrons by gas molecules on the electron pathway can influence the coherency of the electron beam (e-beam), which can further affect (degrade) the intensity, resolution, and contrast of imaging. Therefore, the tendency to develop the microscope with high vacuum as much as possible prevailed in the early decades. Special attention was paid to optimizing the specimen chamber, because changing the specimen each time could run the risk of vacuum breaking.
However, the specimen characterized in such a high vacuum environment may not represent the structure or status of it under realistic operating conditions, especially for some gas-sensitive materials or special atomic structures that only existed in a gaseous environment. In addition, the dynamic information of structural and compositional evolution of materials in real environment is missing in these vacuum characterizations, though it is of great significance to understand many physical and chemical processes. This dilemma stimulated the development of environmental TEM techniques, which emphasize simulating realistic gaseous conditions during real-time observation of the specimen.
Actually, the thought of introducing gas into the TEM emerged almost as early as the birth of TEM. The controllable gaseous environment was firstly proposed by Marton in 1935, including two approaches which could accomplish the gas confinement in the close vicinity of the specimen while maintaining the high vacuum in other essential parts of TEM. The first approach is realized by modifying the objective pole pieces, placing pairs of apertures above and below the specimen to confine the gas around the specimen chamber in 1942 [3]; the second approach is creating a sealed (closed) gas cell with electron-transparent windows placed above and below the specimen, which could withstand the pressure difference between the inside and the outside of the cell in 1944 [4].
These two approaches have been subsequently employed in tremendous designs of TEM in the following decades. Both approaches developed well independently and established their special structures and own complete systems, with the amount of applications, respectively. The first method is generally called “opened type” or “aperture”, and another one is named “closed type” or “window”.
At the early stage of development, several researchers carried out TEM observation in reactions by using the residual gases in the microscope column [5, 6]. This method, obviously, suffered from shortcomings including low maximum pressure (~10–3 Torr) and uncontrollable gas composition, so that a variety of chemical reactions may occur. To solve these problems, a means of providing an atmosphere of known composition and stable pressure around the specimen is desired.
In 1958, Ito and Hiziya [7] modified the specimen chamber of an ordinary TEM to directly observe the specimen in a chemical reaction at varied temperatures (room temperature to 1000 °C) in gas atmospheres. The gas distance that electrons passed through in that device was 2 cm, and the gas pressure was limited by suppressing the gas diffusion to the column through doubled diaphragms and a separated pump. Image quality was not worse in the range (10–4 to 1 mm of Hg) of the gas pressure used if the gas of light elements such as air or hydrogen was employed. In 1962, Heide [8, 9] exhibited another solution of the closed type. The specimen chamber was formed by two specimen grids with the flat surfaces facing each other and kept apart at the desired distance by pieces of thin metal foil. Meanwhile, the grids were covered with a supporting film of low contrast, which had to withstand the gas pressure difference from vacuum (10–6 Torr) to 760 Torr.
The available gas pressure in aperture mode was lifted in the following years. In 1968 [10], Hashimoto attained the gas pressure of 300 Torr through modification of TEM, based on the design in which the heating capability up to 1000 °C was achieved [7, 11]. The phrase “controlled atmosphere electron microscopy” (CAEM) was put forward by Baker et al. in the 1970s [12], to describe the technique which enables one to study reactions between gases and solids at very high magnifications, while they were taking place under realistic conditions of temperature, gas, and pressure. The key design idea of CAEM was to create a high-pressure environment in the specimen region while maintaining very low pressure in the rest of the microscope [12]. The pressure in a sealed cell, which was designed by Fujita et al. [13], and exceeded one atmospheric pressure in 1976. The cell consisted of three films that could be filled with gas or liquid on a 3MV-class electron microscope. It could be used for the study of both biological and physical subjects at high pressure (close to two atmospheres).
The importance of differential pumping gradually emerged with the higher desire for controllable pressure in aperture mode. In 1972, Baker and Harris [14] incorporated the gas reaction stage designed by Hashimoto [10, 11] onto a high-resolution TEM. The specimen chamber was evacuated by a three-stage diffusion pump which was backed by a rotary pump. Differential pumping was exploited in Rodriguez’s work [15] in 1990 as well as Lee’s modification [16] in 1991. With five apertures and four turbomolecular pumps employed, 0.41 nm of the point-to-point resolution was achieved in 9.3 kPa of H2 gas for hours. In 1997, Boyes [17] introduced two pairs of apertures above and below the specimen, which were mounted inside the bores of the objective pole pieces rather than between them as in previous designs. The designed custom set of pole pieces for the first stage of differential pumping allowed unrestricted use of regular sample holders in lenses with much lower aberration coefficients. Meanwhile, the apertures restricted the higher angle of diffraction, but useful data can be recorded and provided for convergent beam diffraction pattern analysis with a STEM probe.
In the 2000s, Boyes’ design was adapted by TEM manufacturer Philips Electron Optics (acquired by FEI Company in 1997, and FEI was acquired by ThermoFisher in 2017), with further development in electron source and different objective pole piece gaps.
The next significant progress was the introduction of microelectromechanical systems (MEMS) technology [18,19,20,21] in the window approach. A MEMS-based nanoreactor was invented for the atomic-resolution ETEM study of nanostructured materials under ambient pressures and elevated temperatures. Creemer et al. [18] miniaturized the gas volume and heater into a sealed system, which allows the observation of nanocrystal growth and mobility on a sub-second time scale with a spatial resolution of 0.18 nm, during heating to 500 °C and exposure to 1.2 bar of H2. This technique has a more rapid thermal response and smaller specimen drift compared with the furnace-based heater. Since then, MEMS becomes the main trend in the development of the window approach. Of course, the usage of MEMS already expanded to aperture mode spontaneously with the development and maturity of technology itself.
9.2 In Situ Gas TEM Technologies
Since Marton’s proposal about two approaches which could accomplish the gaseous environment in TEM, great efforts have been devoted to studying and developing both methods in the following decades.
9.2.1 Aperture (Opened) Approach
Aperture approach is modifying the specimen chamber by placing pairs of apertures between or inside [17] the objective pole pieces to confine the gas leakage, and differentially evacuate individual vacuum stages of the microscope column. The position, number, and size of apertures are essential and influential factors which balance the conflict between the passage of electrons and the airtightness of the specimen chamber. The complete system would incorporate the differential pumping system as well, to avoid a gas leak and maintain the high vacuum in the other essential segments of the TEM.
There are no additional membranes, which are made of amorphous material on windows to degrade the resolution completely, meaning that the images are only derived from the specimen itself and gas molecules (disperse uniformly). Meanwhile, the modification of the specimen chamber has little effect on the specimen holder, which means that the normal holders used in a regular TEM are also compatible with the ETEM. In particular, the use of standard TEM specimen holders supports various sample geometries and allows additional (in situ) functionality, such as tomography, optical studies (of photocatalysts), and mechanical testing (of metals). Furthermore, the specimen preparation of aperture mode is also much easier and more convenient than the window mode, given the fact that the completion of a perfectly sealed cell requires several critical processes which should be paid enough attention to. The disadvantages of aperture mode are also obvious. It can’t be used for liquid–solid interactions, and another distinct shortcoming is the maximum gas pressure limited to the size of the apertures, the power of the differential pumping system, and a much thicker gas layer compared with window mode. Actually, the controllable gas pressure in the aperture mode is still lower than the window mode nowadays.
Owing to its robustness and compatibility with specimen types as well as flexibility in experimental conditions [22], the aperture mode was preferred in the early developing period of the gaseous environment.
The aperture environmental cell was constructed by Ito [7]. In the same year, Hashimoto [11, 23] also modified an electron microscope with three lenses to observe the metal-oxide crystal growth on the surface of metal filament (tungsten) under heating and a gaseous environment. The metallic tungsten wire was heated from 700 °C to its melting point (nearly 3000 °C) in a vacuum of 10–2 mm Hg (1.33 Pa). In 1968, Hashimoto [10] improved the design of the specimen stage and elevated the acceptable gas pressure to 300 Torr. The specimen was placed on a film covering the hole in the platinum ribbon and heated by an electric current sent through the ribbon (Fig. 9.1b). The gas was supplied around the specimen through a pipe and overflowed into the vacuum of the microscope column through the two platinum apertures on either side of the ribbon. The overflown gas from the specimen chamber was pumped out separately from the evacuation system of the main column, and the specimen stage can be taken out from the vacuum of electron microscope column through an air lock system without breaking down the vacuum. These efforts led to the production of a commercial gas reaction stage, and the image resolution achievable was 5–10 nm with an air pressure of 40 kPa (300 Torr).

a Cross-section of gas reaction specimen chamber. b Specimen holder. c Cross-section of the electron microscope column in which gas reaction chamber is installed (reproduced with permission from Ref. [10], Copyright 1968, The Physical Society of Japan and The Japan Society of Applied Physics)
In the same year, Fryer [24] investigated the oxidation of graphite catalyzed by palladium by heating the specimen to 500 °C using a Siemens Elmiskop I electron microscope with a heating stage in a vacuum of 10–5 Torr, then let in dry air passing over the specimen by means of special attachment. The behavior was watched and recorded on photographic plates and on videotape by means of closed-circuit television.
Mills and Moodie designed and constructed an electron microscope stage which combined operation at high resolution with various facilities including free movement, tilting, heating and cooling, anticontamination, and gas injection in 1968 [25] as well. A curved silver capillary whose tip engaged in the sleeve of an injector was mounted on the specimen platform and extended inside the capsule to within 5 mm of the specimen cup. The column pumps, working through the capillary, satisfy a rate adequate for the evacuation of the line between the taps and the valves, and for adjustment of the pressure in the fixed volume.
Although the differential pumping system was not employed in Mill’s experiment, the importance and necessity of it displayed gradually, especially catering to the higher gas pressure demand during research.
In 1972, Baker and Harris incorporated a modified gas reaction stage (JEOLCO JEM AGI attachment, commercially available, designed by Hashimoto [10, 11]) onto a JEM 7A “high-resolution” electron microscope. The specimen chamber was evacuated by a three-stage diffusion pump backed by a rotary pump. The attachment omitted the second aperture used in Hashimoto’s design in order to retain the diffraction facility, but with some sacrifice to the maximum attainable gas pressure (up to 30 kPa instead of 40 kPa). Of course, the maximum operating pressure in the cell was dependent upon the area of the aperture, the pumping speed of the auxiliary pump, and the molecular diameter of the gas.
Lee and coworkers [16] employed the aperture-limited, differential pump type in their experiment as well in 1991. A pole piece was designed for a JEOL 4000 that has a gap of 14 mm compared to 4–6 mm in the high-resolution pole pieces. Five apertures were used, with two primary apertures of them located in the body of the cell, two secondary apertures located in the pole piece, and a final aperture located in the lower section of the condenser lens stack. The gap between the principal apertures was reduced to 4.3 mm and a tilt of ±30° can still be obtained from two perpendicular observing directions using this scheme. Another spotlight of the design was the release of restrictions on high-angle diffraction. For an accelerating voltage of 400 keV, the maximum diffraction angle that can be observed corresponds to 1.4°. Four Seiko-Seiki turbomolecular pumps were employed to accomplish the differential pumping (1) between the principal and secondary apertures, (2) in the volume surrounding the pole piece, (3) between the upper secondary aperture and the condenser stack aperture. With this arrangement, the environmental cell can support 20 kPa (150 Torr) of H2 for short periods of time (minutes) or 9.3 kPa (70 Torr) of H2 gas for several hours.
In 1997, Boyes and Gai [17] made a breakthrough in the position of apertures on a Philips CM30T TEM/STEM (scanning TEM) system. They introduced two pairs of apertures above and below the specimen, which were mounted inside the bores of the objective pole pieces rather than between them as in previous designs (Fig. 9.2). This approach allowed unrestricted use of regular sample holders in a relatively narrow gap lens (S = 9 mm) with much lower aberration coefficients (Cs = Cc = 2 mm) than have been possible with previous environmental cell designs. Such design is dedicated to the environmental cell (ECELL, or gas reaction cell), and the controlled environment ECELL volume is the regular sample chamber of the microscope. It was separated from the rest of the column by the apertures in each pole piece and by the addition of a gate valve, which was normally kept closed, in the line to the regular ion getter pump (IGP) at the rear of the column. Pumping ports have been added to the column between the ECELL apertures located in upper objective lens (OL) polepiece and lower OL polepiece, respectively, for the first stage of differential pumping. Then a second stage of pumping between the condenser aperture and upper OL polepiece, and between lower OL polepiece and selected area apertures was accomplished in the modified column liner tubes. The maximum allowable pressure of this apparatus was 50 mbar. In addition, due to the desire for maximum gas pressure attainable, the apertures were normally small enough to restrain gas leakage, which block the high-angle scattered electrons simultaneously, limiting annular dark-field STEM (ADF-STEM) imaging. Here, the relatively large apertures in the cell provided useful angles of diffraction in TEM mode, and for convergent beam diffraction pattern analysis with a STEM probe. The design of the ECELL is outstanding and developed continuously in the following years with the improving capability and expanding research fields [26,27,28,29,30]. And in 2013 [28], they introduced the modification of double aberration-corrected (2AC) JEOL 2200 FS FEG TEM/STEM to provide both ETEM, and for the first time environmental STEM (ESTEM), with full functionalities. These include introducing controlled conditions of the gas environment and high temperature with uncompromised high angle annular dark field (HAADF) Z-contrast ESTEM imaging, low background ESTEM EDS (energy-dispersive X-ray spectroscopy) microanalysis, AC ESTEM EELS (electron energy-loss spectroscopy), and wide-angle electron diffraction analyses of nanoparticle structures and wider crystallography.

Schematic of the basic geometry of the aperture system from Boyes and Gai (reproduced from Ref. [26], copyright 2014, Elsevier Masson SAS)
An inventive gas-providing method with a specialized holder was proposed by Kamino et al. [31] in 2005. The side-entry specimen holder consisting of a heating element and a gas injection nozzle could be attached to a conventional TEM without any modification. A spirally wound fine wire of tungsten with a diameter of 25 mm was used as a heating element (Fig. 9.3a), which was heated by a direct electric current via a power supply unit equipped with batteries. Since a firewire of tungsten was used as the heating element, the application is limited to particles with a diameter of several micrometer or smaller. Definitely, the design provided the capability of high-resolution TEM imaging at very high temperatures. A gas injection nozzle with an inner diameter of 0.5 mm was built near the heating element at a distance of ~1 mm. The experiment was manipulated in a Hitachi H-9500 and a Hitachi-9000NAR, which were ETEM actually (the microscopes have fixed apertures with a diameter of 0.3 mm between the first and second condenser lenses, without any added aperture between the specimen chamber and the electron gun chamber). The pressure of the electron gun chamber was kept in the middle of 10–5 Pa even after the pressure of the specimen chamber reached the middle of 10–2 Pa at the gas-flow rate of ~7 ml · min–1. This pressure in the electron gun chamber was sufficient for working with the LaB6 cathode. In other words, the observation of chemical reactions in a gaseous atmosphere in the range of 10–2 Pa can be carried out in a conventional TEM equipped with a LaB6 cathode without any modifications of the column and/or the pumping systems. In fact, the holder allowed observation of TEM images of the gas-reacted specimen at temperatures of 1300 K in the ~2 × 10–2 Pa gaseous environment at atomic resolution.

Schematic diagram (a) and an external view (b) of the gas injection/specimen heating holder (reprinted with permission from Ref. [31], Copyright 2006, Oxford University Press)
In the 2000s, the design of Boyes and his coworkers was adapted by TEM manufacturer Philips Electron Optics, with further development in electron source and different objective pole piece gaps. Later versions of the in situ ETEM instrument, (which include the CM 200–300 series, and Titan) have been installed in laboratories around the world.
The ETEM installed at the Center for Electron Nanoscopy in the Technical University of Denmark (DTU Cen) [32] is a typical example based on the collaboration of Boys, Gai and Philips. Multilevel turbomolecular and ion getter pumps were equipped successively to sustain high vacuum around the electron source. As shown in Fig. 9.4a, each pumping stage is separated by additional apertures that result in pressure drops, approximately three orders of magnitude lower per aperture, from a pressure of ~1500 Pa in the sample region to below 10–6 Pa in the field emission gun region. The microscope can be operated in two primary modes, including conventional operation (low pressure) which is pumped using the standard ion getter pumps, and ETEM mode whose pumping pathway is through three turbomolecular pump valves. Furthermore, an image CS corrector and a monochromated electron source are both employed to improve the spatial resolution and energy resolution, respectively. When the microscope is operated at an acceleration voltage of 300 kV, the measured energy spread could be better than 200 meV (in high vacuum) with the monochromator, and the point resolution could be brought down to below 0.1 nm with the CS corrector.

a Schematic diagram of differentially pumped TEM column (FEG, field emission gun; IGP, ion getter pump; TMP, turbomolecular pump; RGA, residual gas analyzer; PC, plasma cleaner; C1, first condenser aperture; SA, selected area aperture) (reproduced with permission from Ref. [32], Copyright 2010, Taylor and Francis). b A schematic diagram of differential pumping system suitable for environmental (S)TEM imaging (reproduced with permission from Ref. [33], Copyright 2019, Cambridge University Press). c A schematic diagram of gas injection system suitable for environmental (S)TEM imaging (reproduced with permission from Ref. [34], Copyright 2019, Cambridge University Press)
The ETEM series in FEI developed in the following years, and the up-to-date model is called “Themis ETEM”, which can be combined with optional image Cs corrector, Thermo Scientific™ X-FEG module, and monochromator technology to further extend it to meet the high standards in atomic-resolution STEM imaging and spectroscopy expected from Themis TEM technology. The Themis ETEM features an innovative differentially pumped objective lens, uniquely designed for the ETEM platform. This lens design enables all the same features from a standard Themis S/TEM, such as window-free imaging and compatibility with Themis TEM heating holders for easy sample insertion, while also allowing ample chamber space for a full double-tilt capability to support 3D tomography. Gas inlets allow safely adding of inert and reactive gases to the chamber. Gas pressures in ETEM experiments can be accurately preset from 10–3 Pa up to 2,000 Pa (for N2). Furthermore, The Themis ETEM is equipped with a mass spectrometer to determine the gas composition in either the gas inlet system or the specimen area. A built-in plasma cleaner allows for the cleaning of the specimen area after using a gas.
In 2018, the analytical 200 kV cold field emission TEM (HF5000, Hitachi High-Technologies) equipped with an in house designed probe-forming aberration corrector was introduced and the imaging performance of the environmental real-time STEM was reported by Hitachi Company [33, 34]. Figure 9.4b shows the schematic diagram of the differential pumping system of HF5000 suitable for environmental (S)TEM imaging. An orifice is added between the gun valve and specimen chamber for enhancement of the differential pumping system. The HF5000 can have two gas injection nozzles, one is attached to the specimen chamber and the other is in a filament-heating holder. The gas can be chosen independently (Fig. 9.4c). Gas flow is limited to one nozzle at a time and can be controlled. Maximum allowable flow rate is 3.5 sccm and the calibrated local pressure around the specimen is 10 Pa at the maximum flow. At an accelerating voltage of 200 kV, the TEM resolution (lattice) is 0.102 nm, while the STEM resolution is 78 pm. In addition to retaining key features of previous instruments, such as the automatic correction function and the symmetry-dual Silicon Drift Detector, the HF5000 could simultaneously obtain a secondary electron image with rich high-resolution surface morphology information at the atomic scale besides DF/BF live images. The HF5000 key feature of in situ STEM is a live scanning acquisition (25 frames/s) that allows real-time observation and video recording of dynamic atomic-scale reactions (structure change and elemental diffusion). Meanwhile, the chip-based sample holder is developed using MEMS technology by Norcada and Hitachi High-Technologies Canada. Maximum heating temperature of this holder is 1100 °C and the minimum controllable temperature increment is 1 °C. Heating up speed from room temperature (RT) to 1000 °C is as fast as a few seconds. In other words, HF5000 almost aggregates and fuses core technologies perfected in the development of Hitachi’s HF series of TEMs.
9.2.2 Window (Closed) Approach
The window approach is creating a sealed cell which contains the specimen, with electron-transparent windows which could withstand the atmospheric pressure between the inside and the outside of the cell.
The feature of this mode is the airtightness of the cell, which enjoys conspicuous advantages. Firstly, the whole volume and the specimen itself are confined by the electron-transparent windows, which could sustain higher gas pressure with the development of window material. The length of gas–electron interaction is much shorter compared with the aperture mode (normally several microns), so as to improve the acceptable gas pressure. The atomic resolution can be achieved under one atmosphere or higher (~4.5 bar) [18,19,20, 35,36,37] nowadays. Secondly, a window-friendly cell specimen holder is compatible with different TEMs without any modification to the microscope itself. And the cost of purchasing or modifying a specimen holder is much smaller compared with a differential pumped ETEM. Furthermore, the sealed cell could contain liquid or wet samples, which is the unique advantage of the window mode. Finally, due to the lack of additional apertures which restrict the leakage and diffusion of gases, wide-angle electron diffraction and HAADF imaging could be operated (definitely, HAADF could be implemented in the aperture mode at present).
Unavoidable drawbacks are obvious as well. The unique airtightness is a superiority and a risk simultaneously. The possibility of the fracture of the windows or leakage of the gas during experiments would deteriorate the high vacuum of the column and electron source. Meanwhile, the airtightness results in the complexity and difficulty of the sample loading operation, especially under the demand of maintaining the centering and coupling of top and bottom cell windows. Thirdly, even though the window materials were selected carefully and fabricated finely to sustain the amorphism and homogeneity, the electron-transparent windows still interact with the electrons, resulting in the scattering information superimposed on the image obtained. Furthermore, the scattering effect that results from the amorphous windows is not neglectable, especially since the thickness would influence the quality of imaging immensely. Another easily neglected problem is the contamination from the specimen, which will evaporate/sublimate and condensate on the top window, hindering the observation. Finally, the existence of windows hindered the acquisition of EDS signals as well (it is realizable using specifically designed cells nowadays and is already commercialized, e.g. DENSsolution and Protochips), and local sample loading area and windows confined the observation view, normally much smaller than conventional TEM. Generally, samples could only tilt in one direction with the limitation of the geometrical shape of nanoreactors, which influenced the observation of specific directions.
The materials of the electron-transparent window are essential in the development of the window mode, and they must satisfy the following requirements: (1) be electron-transparent, which is the vital factor; (2) have sufficient strength to resist the pressure difference inside and outside the cell; (3) scatter weakly and non-oriented to preserve the diffraction information of the specimen. The atomic number of the element contained in the window is usually small since a larger atomic mass exacerbates the scattered effect. Initially, the windowed cells were constructed from metal (e.g. Cu/Ni) grids, O-rings, and thin films [8, 38, 39]. Gradually the amorphous material (carbon, polyimide, silicon nitride, silicon dioxide, and alumina) have been used as windows [39].
An enclosed cell to protect the specimen from the high vacuum and electron bombardment was devised by Abrams and McBain in 1944 [4]. The plastic windows scarcely interacted with electrons, and they were liquid-tight and vapor-tight and easily withstand a difference of one atmosphere pressure between the inside of the cell and the remainder of the electron microscope.
In 1962, Heide’s experiment [8, 9] exhibited a successful sealed cell of variable gas pressure. The specimen chamber was formed by two specimen grids with the flat surfaces facing each other and kept apart at the desired distance by pieces of thin metal foil. Both grids were covered with a supporting film of low contrast, which has to withstand the air or gas pressure (760 Torr) over the central openings, meanwhile one of them served as a supporting film for the specimen. The gas was injected through a tube placed in the opening of the column.
So far, the specimen supporting method was placing the specimen on the grid or the supporting film served as the window, which resulted in the difficulty in heating, and the gaseous environment was confined by the single gas line which is incapable of continuous flowing gas.
Escaig and Sella made an important integration of the heater and gas support system in 1969. Twin gas lines in their holder supported the continuous circulation of gas while the temperature of the specimen varied. The windowed cell consisted of triple layers of carbon, nitrocellulose, and silica (facing the specimen), and maintained a gap of approximately 1 mm between window and specimen, which was successfully employed for in situ oxidation of copper, tungsten, and titanium.
In the late 1960s and 1970s, the development of high-voltage (1 meV) electron microscopes with the stronger penetrating power of electrons led to enclosed gas reaction cells with thicker, stronger windows. Fujita [13] designed a new sealed cell with a twin gas line for a 3 MV-class electron microscope. Metal meshes which supported the cell films were chosen from vapor-deposited aluminum, SiOx, and carbon film depending on the purpose. The sealed gas cell can be pressurized up to 195 kPa (1471 Torr) by circulating gas or liquid into the capsule. Meanwhile, the specimen can be heated to 1000 °C or even higher through direct electrization of Ni-mesh or the specimen itself, and cooled with a liquid nitrogen tank.
Images of the crystal lattice of ceria were recorded under flowing nitrogen gas at 20 Torr in 1989 by Parkinson [38]. The key features were that the gas was contained between very thin (ca 5–10 nm), evaporated carbon windows, and the path length of the beam was kept small (10–50 μm). The cell was able to support a pressure difference of circa one atmosphere with a high flow rate (up to 50 ml/min). This was the first time that such high resolution had been achieved under conditions not far from those typical of in situ use (medium voltage, 400 kV). From then on, structural information contains chemical significance became discernible, while the technique began to offer real hope of carrying out fundamental dynamic studies of the activation, reaction, and passivation of gas/solid systems close to the atomic level.
In the following two decades, similar techniques [36, 37, 40,41,42] were developed in the area of film material and simplification of structure. In 2005, Komatsu [40] used a window whose base is a commercially available copper mesh grid with double-layer polyvinyl-formvar/carbon thin films formed by vapor deposition (Fig. 9.5a). Onto this base, additional layers of nylon and amorphous carbon were evaporated to improve both mechanical strength and heat resistance, and the cell pressure could be increased to ~1.3 × 104 Pa. In Giorgio’s experiment [41], the cell was closed by two copper disks where seven holes have been drilled, which were pre-covered by ~10 nm amorphous carbon films, located inside the cell (Fig. 9.5b). The sample (powder) was deposited on the heating wire made of W–Re, which was isolated from the grids by an insulator ceramic. The electrical connections for the heating wire and both tubes for gas circulation were included in the sample holder. The Viton O-rings provided the sealing of the cell toward the vacuum of the TEM column. In 2009, Kawasaki et al. [42] developed a simplified E-cell, thanks to the successful development of remarkably tough thin carbon films as the window material. These films, with a thickness of <10 nm, were found to withstand pressure difference >2 atm. The E-cell at the tip of the specimen holder connected two stainless pipes from the other side of the holder, allowing gases to pass in and out (Fig. 9.5c). In 2010, de Jonge [37] constructed a cell to record the STEM images of gold NPs at atmospheric pressure through a 0.36 mm thick mixture of CO, O2, and He. A sample compartment filled with gas at atmospheric pressure was enclosed between two silicon microchips supported by electron-transparent SiN windows. The microchips were separated by a spacer and sealed with epoxy. Images are obtained by scanning a focused e-beam over NPs attached to the top window and detecting elastically scattered transmitted electrons. In this drawing, gas entry and exit were not shown, and the dimensions and angles were not to scale (Fig. 9.5d). In 2011, Yaguchi [36] developed a new windowed environmental cell holder based on his work 6 years ago, which allows ETEM study of nanomaterials at a maximum temperature of 1500 °C in the gaseous environment at the atmospheric pressure. Figure 9.5e shows the external views of the equipment, and the schematic (Fig. 9.5f) exhibits positions of windows, heating element, gas tubes, and a micropressure gauge. The side-entry type was employed as well with a built-in specimen-heating element of a spiral-shaped fine tungsten wire, which allows heating of specimens up to 1500 °C. Gas pressure inside the environmental cell can be continuously varied from 10–5 Pa to atmospheric pressure while TEM image observation of the specimen is carried out. This design was commercialized by Hitachi in the following years.

a Newly improved multi-layer amorphous film window (reproduced with permission from Ref. [40], Copyright 2005, Oxford University Press). b E-cell heating holder in profile view (reproduced with permission from Ref. [41], Copyright 2006, Elsevier). c Schematic drawing and photograph of the E-cell specimen holder (reproduced with permission from Ref. [42], Copyright 2009, AIP Publishing). d Schematic of the flow system for atmospheric pressure scanning transmission electron microscopy (STEM) (reproduced with permission from Ref. [37], Copyright 2010, American Chemical Society). e External view of the windowed environmental cell holder. f Position of windows, heating element, gas tubes, and a micropressure gauge (1. gas outlet tube; 2. gas inlet tube; 3. micropressure gauge; 4. windows; 5. heating element) (reproduced with permission from Ref. [36], Copyright 2011, Oxford University Press)
Besides the pressure, the heating process was also another challenge impeding the high-quality TEM images. For conventional windowed gas cells based on metal grids, the two metal grids form a metal “furnace”, into which a ceramic encasing a heating filament is placed [43]. The disadvantages, as followings, of this design are obvious, and greatly affected the observation of the specimen: (1) The grids not only sealed the cell, but also served as the heating device for the specimen, and easily expand and retract during the heating and cooling process. It’s one of the major reasons for sample drift, which will typically be several tens of micrometers. Actually, the sample drift during heating/cooling has been a major challenge that disturbed the researchers for a long time; (2) The metal wires, which played the role of heating, always couldn’t sustain the high and stable heating rate, and sometimes it’s still difficult to find and maintain the balance of a stable temperature between the metal wire heating and flowing of the gases.
Until the end of the 2010s, a microelectromechanical system based on closed cells emerged [18,19,20,21, 39, 44,45,46], which utilized the MEMS technology to produce the chips used as cell windows. The MEMS-based nanoreactor employed in the TEM was proposed by Creemer et al. [18]. The nanoreactor (Fig. 9.6a) consists of two facing dies made with thin film technology on a silicon substrate. Each die has a central hole of 1 mm2 that is covered by a 1.2 mm thick membrane of SiNx while the small ovaloids are only carpeted by a 10 nm thin film (Fig. 9.6c). The opposing membranes form the top and bottom of a shallow gas-flow channel. The minimum height of the channel is 4 mm, determined by disk-shaped spacers (SiO2) integrated into one of the membranes. This height corresponds to an atomic density along the beam direction of only 0.2 × 103 atoms/nm2 at 1 bar and RT. The lateral dimensional size (10 μm) and geometric shape (ellipsoid) of ultrathin windows were also carefully selected to satisfy the demanding of pressure difference. In the areas between the windows, the heater is embedded in the form of a spiraled Pt wire (Fig. 9.6c). The MEMS-based technique enabled the integration of the heater into the window membrane, effectively limiting the thermal expansion of the system components and consequently the specimen drift. In the end, the direct observation of samples on a sub-second time scale could reach a spatial resolution of 0.18 nm, during heating to 500 °C and exposure to 1.2 bar of H2.

Illustration of the nanoreactor device. a Schematic cross-section of the nanoreactor. b Optical image of the TEM holder with the integrated nanoreactor and the four electrical probe contacts. c Optical close-up of the nanoreactor membrane. The bright spiral is the Pt heater. The small ovaloids are the electron-transparent windows. The circles are the SiO2 spacers that define the minimum height of the gas channel. d A low-magnification TEM image of a pair of superimposed 10 nm thick windows. Their alignment creates a highly electron-transparent (bright) square through which high-resolution TEM imaging can be performed (reproduced with permission from Ref. [18], Copyright 2008, Elsevier)
In 2009, Allard [44] undertook a project with Protochips Inc. (Raleigh, NC) to develop a new MEMS-based device (Aduro™) for in situ heating experiments that provide sub-Å resolution with rapid specimen heating and cooling. The key component of the MEMS devices is a 150 nm thick, 500 μm square, freestanding membrane made from a conductive ceramic that is suspended on a 3-mm Si chip (Fig. 9.7a). In 2012, Allard [20] incorporated the Aduro heating device into a “closed-cell” configuration, and is capable of exposing specimens to gases at pressures up to 1 atm. The atomic resolution performance of the microscope in high-angle annular dark-field and bright-field imaging modes was demonstrated at elevated temperatures and at 1 atm pressure.

a Top and cross-section view schematic of Protochips heater chip, showing composition of the various elements (reproduced with permission from Ref. [44], Copyright 2009, John Wiley and Sons). b 3D sketch of the all-in-one nanoreactor (reproduced with permission from Ref. [45], Copyright 2011, IEEE). c Schematic diagram of the nanoreactor inserted in the sample holder (reproduced with permission from Ref. [19], Copyright 2012, Elsevier). d Light optical micrograph of the nanoreactor with the gas channel and the reaction zone including the heater spiral and electron-transparent windows (reproduced with permission from Ref. [47], Copyright 2014, Springer Nature)
In 2011, Creemer et al. [45] improved their MEMS nanoreactor to enable atomic-scale imaging of nanostructured materials under high pressures (14 × 105 Pa, 14 bar) and temperatures (660 °C). They integrated the reactor entirely on a single die, with the use of surface micromachining techniques (Fig. 9.7b), which increased the strength and rigidity of pillars holding together the membranes. The employment of a wafer stepper, resulted in a much better alignment of opposite windows (0.1 μm instead of 2 μm), and improved robustness obviously. In the same year, Yokosawa et al. [19] used a modified MEMS-based nanoreactor on the basis of Creemer to observe the (de)hydrogenation of Pd at pressures up to 4.5 bar. Two Si chips with square-shaped 1 μm-thick low stress (silicon-rich) silicon nitride (SiN) membranes, which contain very thin SiN windows (about 20 nm thick), were integrated, and one of the chips (bottom chip) contains a Pt heater and has an inlet and an outlet for the gas. The schematic diagram of the nanoreactor inserted in the sample holder is shown in Fig. 9.7c. Further improvement based on the work of Creemer was exhibited in 2014 [47]. The nanoreactor employed included a unidirectional gas-flow channel (280 μm-wide and 4.5 μm-high), enclosed between two 1 μm-thick SiNx membranes. At the nanoreactor center (Fig. 9.7d), a Mo thin film resistor enabling heating and temperature measurement of a reaction zone at 1 bar pressure, and 18 nm-thick electron-transparent windows facilitated TEM observation. The reactor shown in Fig.9.7b, d also has intrinsic drawbacks: as a special one-piece holder, the sample loading has to be done by flushing the sample into the holder with the risk of plugging.
The development of the window approach promoted the commercialization of integrated MEMS cells. And several companies provide unique holders and chips corresponding to TEMs manufactured by different TEM companies nowadays, such as DENSsolutions, Hummingbird, and Protochips. And the holders used for the window approach with additional functions emerge catering to various requirements depending on the experimental purpose.
The in situ heating and gas reaction cell developed by Protochips is called “atmosphere”. The atomic resolution could reach 1.5 Å with the pressure up to 1 atm, and the temperature ranges from RT to 1000 °C. Protochips developed a proprietary silicon carbide membrane heater that can quickly and accurately reach the highest temperatures without the risk of interacting with the sample or participating in catalyst reactions. The design of the Atmosphere holder enables in situ closed-cell EDS elemental analysis in the TEM. The holder design provides a large line-of-sight solid angle from the sample to the EDS detector, minimizing tilt angle of holder tip and maximizing the count rate.
DENSsolutions also launched a competitive product named as “Climate” system. The atomic resolution could be better than 100 pm (1 Å, depending on microscope configuration) with 2000 mbar (~2 atm) pressure (Climate). The temperature ranges from RT to 1000 °C as well. 4-point probe heating provides the most accurate temperature with 0.01 °C stability even during gas flow, which also allows accurate calorimetry data to be acquired during the experiment. Meanwhile, the Climate G+ system allows direct gas mixing with 3 gas input lines. Using a specially designed and patented mixing valve allows to change the gas composition on the fly and to vary rates with an accuracy of 0.1%. With the defined gas channel and minimal gas volume inside the nanoreactor, the gas environment of the sample can be changed within seconds. The EDS and EELS compatibility is also satisfying. Furthermore, the integration with the optional dedicated DENSsolutions gas analyzer allows full dynamic correlation of the structural and chemical data. Gas analyzation and calorimetry data can also be produced by the Climate system if required, so that it enables to define the optimum experiment conditions before going to the TEM.
No matter which approach (aperture/window) the actual experiment employed, or no matter how complicated the system was designed, the interior process reaction that happened in the CATEM could be clearly simplified as in Fig. 9.8 [48]. On this basis, researchers developed various applications in tremendous fields.

Schematic of the CATEM technique and analytical methods (reproduced with permission from Ref. [48], Copyright 2018, Springer Nature)
9.3 Research Based on In Situ Gas TEM
CATEM has been successfully applied in a variety of research fields. The following application fields will be discussed in this section: reshaping of nanomaterials; redox of nanomaterials; surface reconstruction and segregation; growth of low-dimensional nanomaterials; the catalysts (dynamic observation of catalytic reactions; sintering and dispersion; photocatalysts).
These applications expressed the advances and unique characterization adequately in TEM capabilities embodying imaging and spectroscopy of materials in gaseous environments.
9.3.1 Reshaping of Nanomaterials
Generally, many properties (e.g. optical, magnetic, and electronic) of nanomaterials mainly depend on their shape, size, composition, and surface structure. In particular, the reshaping of the nanomaterial is intuitive and obvious in various gaseous environments, and could significantly influence the properties. Therefore, to in situ observe and characterize the morphology evolution under operational conditions is of great significance to establish the structure–reactivity relationship of nanomaterials.
In 2002, Hansen and his coworkers [49] observed the copper (Cu) nanocrystals underwent reversible shape changes in response to the changes in the gaseous environment. Cu nanocrystals (NCs) with diameters of 3–6 nm were dispersed on ZnO and Si supports, and the interaction relationship between crystals and surfaces was studied. With the environmental change from more oxidizing conditions (mixed gas of H2O and H2) to more reducing conditions (CO and H2), the Cu NCs changed to a more spherical (Fig. 9.9a) and a disklike structure (Fig. 9.9e), respectively. The authors attributed the reason to the different driving forces, respectively, water adsorption on the different Cu facets for the former and the change of Cu/ZnO interface energy (not the relative Cu surface energies) caused by the addition of CO for the latter. Supplementary experiments testified the conclusion: the shape transformations (to more spherical) were found to be reversible with the gas changes (H2–H2/H2O–H2) for both ZnO-supported (Fig. 9.9g–i) and silica-supported Cu NCs, which indicated the influence of H2O, while the transformations (to more disklike, H2–H2/CO) were not observed for silica-supported Cu NCs, and the reason focused on the interface between NCs and the support.

In situ TEM images of a Cu/ZnO catalyst in various gas environments together with the corresponding Wulff constructions of the Cu nanocrystals. a 1.5 mbar of H2: H2O = 3:1 at 220 °C. c 1.5 mbar of H2 at 220 °C. e 5 mbar of H2: CO = 11:1 at 220 °C. g 1.5 mbar of H2 at 220 °C. h 1.5 mbar of H2: H2O = 3:1 at 220 °C. i 1.5 mbar of H2 (reproduced with permission from Ref. [49], Copyright 2002, The American Association for the Advancement of Science)
Reversible shape transformations of faceted Pt NPs during oxidation–reduction cycles were in situ observed by Cabié et al. [50] using ETEM in 2010. The sample holder employed in the present study was closed by two carbon windows, which allows a pressure lower than 10 mbar and a temperature varying between RT and 350 °C. The Pt NPs were square in vacuum observed along the [001] direction, with truncations by (111) facets at the eight corners. Their shape was still close to cubes exposed by (100) surfaces mostly and truncated (111) surfaces at the corners in 3 mbar of O2. Along with the evacuation of O2 and introduction of H2, the (111) surfaces expanded and the (100) surfaces diminished until the shape was close to a cuboctahedron. Finally, when it was reverted to O2, the Pt NPs recovered to cubic shape mainly exposed by (001) facets. The authors believed that the adsorption of gases on different surfaces took effect in the morphological change.
Uchiyama and coworkers systematically investigated the morphology changes of gold NPs (GNP) supported on CeO2 during CO oxidation in 2011 [51]. In CO oxidation environment (1 vol% CO, 21 vol% O2, 78 vol% N2) at 1 mbar pressure, the GNP appeared to be faceted and enclosed by the {111} and {100} facets. Then the GNP became rounded in pure O2 gas and faceted in both inactive N2 gas at 1 mbar and in vacuum, while the GNPs supported on crystalline TiC remained polyhedral in all of the gas environments including pure O2 gas, which indicated that the Au–CeO2 interface played an important role in the catalytic activity. This in situ observation also suggested that CO molecules stabilized the major {111} and {100} facets of GNPs, while oxygen atoms adsorbed on not only the major {111} and {100} facets but also the minority {110} facet of GNPs. A morphology diagram was obtained, which showed the morphology of GNP in different environments.
It should be noted that most prior work was limited to low pressure, which was significantly different from realistic operating conditions. In this regard, the in situ study under conditions that more closer to realistic operating conditions is highly desired. The dynamic morphological evolution of palladium–copper (PdCu) nanocrystal (NC) with atmospheric pressure hydrogen was studied at the atomic scale by Jiang et al. [52] in 2016. The chemically synthesized monodisperse PdCu NCs (Pd/Cu ratio is 1:1) were spherical. Then the PdCu NCs on a SiN chip were loaded into a sealed gas reactor and exposed to 1 bar H2, as Fig. 9.10a shows (along [111] axis). When annealed at 600 K, the spherical PdCu NC started to rotate and showed smoother surfaces, as indicated by the blue dashed line in Fig. 9.10b. Subsequently, accompanied by roll-over and rotation, the NC exhibited four flat surfaces (Fig. 9.10c) and finally became a truncated cube with distinct {001} and {011} facets (Fig. 9.10d). The surface energy of PdCu was calculated by density functional theory (DFT), which indicated the truncated morphology was induced by the order change of the surface energies with hydrogen adsorption. The morphology PdCu was obtained based on Wulff construction (Fig. 9.10k, l) considering hydrogen adsorption, which showed perfect consistency with the experimental images (Fig. 9.10i, j), both from the [100] and [111] zone axes. Furthermore, the PdCu NCs retained the as-synthesized shape (round) when heating in vacuum (10–7 bar) at 600 K in control group, which suggested adsorbed hydrogen-promoting surface diffusion.

a–d Lattice-resolved TEM images and the corresponding FFT patterns showing the morphological evolution of the PdCu NC when exposed to H2 at 1 bar and with heating at 600 K. Scale bar: 10 nm. TEM images and Wulff constructions of the PdCu NCs projected from specific directions: i and k [100]; j and l [111] (reproduced with permission from Ref. [52], Copyright 2016, John Wiley and Sons)

Time-resolved TEM images of the dynamic refacetting of two Pd NPs under 1 bar N2 at 200 °C (a–d). The TEM images showed that the NPs changed from rounded shapes to truncated cuboids (e–h). The atomic-scale images of the indicated parts of (a–d) (reproduced with permission from Ref. [55], Copyright 2018, Royal Society of Chemistry)
Shape evolutions of different metal NPs [53, 54] are commonly believed to happen in reducing or oxidizing gas under high pressure. However, in 2018, Zhang et al. [55] observed an unexpected refacetting process of Pd NP under N2, which was considered as an inert gas, with atmospheric pressure. As shown in Fig. 9.11a, c, the initial morphologies of two Pd NPs after annealing in air at 200 °C were rounded shapes with curved corners. Surprisingly, the NPs underwent obvious refacetting processes after being exposed to 1 bar N2 gas at the same temperature (Fig. 9.11b, d), with flat facets and truncated cuboids appearing. The atomic-scale TEM images (Fig. 9.11e–h) showed that a notable increase in the fraction of Pd {110} surfaces occurred in N2 at atmospheric pressure at 200 °C. The remarkable refacetting of Pd NPs was ascribed to the stabilization of the {110} facets by N2 adsorption by DFT calculations. In comparison, similar experiments under 1 bar Ar pressure were performed, and no refacetting phenomenon was observed at any temperature. The results suggested that N2 was not always an inert gas in high-level pressure, and it should be very careful when using N2 as an inert gas in the pretreatment.
A minireview was published in 2019 by Zhu et al. [56], which combined the in situ environmental transmission electron microscopy experiments and the newly developed multiscale structure reconstruction (MSR) model to study the equilibrium shapes of metal NPs in various reactive environments at atmospheric pressure. The MSR model consists of three parts: the Wulff construction, adsorption isotherms [57], and DFT calculation. The authors corrected the surface tension of a clean surface \({\gamma }_{hkl}\) to be the interface tension \({\gamma }_{hkl}^{int}\) (Eq. (9.1)), where θ(T, P) is the coverage of the gas molecules on the [hkl] surface depending on the temperature and gas pressure, \({E}_{ads}\) refers to the adsorption energy, and \({A}_{at}\) is the surface area per atom of the [hkl] surface:
The equilibrium shape of metal NPs can be constructed at a given temperature and pressure using the derived interface tension. The MSR model achieved great success in reproducing representative experimental results [58, 59], which could be extended to the situations of mixed-gas environments [60]. It was also employed in the works mentioned hereinbefore [52,53,54,55]:
For a supported NP, the contact-surface tension between the metal NP and the support should be considered, according to the Wulff–Kaischew theorem [61]. With gas adsorption, the contact-surface tension can be evaluated by Eq. (9.2), where \({\gamma }_{A}\) is the surface tension of the metal surface in contact with the substrate, \({E}_{adh}\) is the adhesion energy between the metal and the support, \({\theta }^{B}\) is the gas coverage on the support, \({E}_{ads}^{B}\) is the adsorption energy of the gas on the support, and \({A}_{at}^{B}\) is the surface area of the support.
The combination of in situ experiments and theoretical modeling shows that the effect of the environment on the shape of NPs can be predicted quantitatively and efficiently using the MSR model, which facilitated the relationship understanding between the reshaping of the NPs and its catalytic properties in real reaction, further offering possibilities to control the reactivity by real-time regulation of the reaction conditions.
9.3.2 Redox of Nanomaterials
As early as 1958, Ito and Hizaya [7] employed a modified TEM (aperture mode) to investigate the oxidation of Aluminum (Al) thin film and the reduction of oxide copper film/whisker. They obtained the electron microscopic image and diffraction pattern before and after the reaction, which indicated the occurrence of oxidation and reduction. In the following decades, similar in situ redox experiments were performed on other materials such as graphite [24], metals (Fe, Cu, and Ni) [62], and special chemical compound ((NH4)6MO7O24.4H2O) [10].
With the employment of a new window material which increased resolution and contrast in high-voltage electron microscopy, Komatsu and Mori [40] observed the oxidation of copper, reduction of copper oxides, and the growth process of CuO whiskers on a thick (50 μm) Cu film. As shown in Fig. 9.12a–e, a small amount of copper oxide (Cu2O) was formed in the initial stage. Then oxygen gas was introduced into the initially evacuated cell (10–2 Pa) to 1.3 × 103 Pa and the specimen was gradually heated to 470 K. Upon heating in oxygen, Cu2O nucleated on the film surface. As the temperature increased, Cu was oxidized to Cu2O and then completely transformed to fine CuO particles (~10 nm in diameter, in 670 K). And the average grain size increased at 770 K. The reduction of CuO in hydrogen is shown in Fig. 9.12f–h. The same CuO area was reheated after cooling down to RT, and the CuO was completely reduced to Cu at 670 K. The growth of CuO whiskers (Fig. 9.12i–n) in 4 × 103 Pa of oxygen was also recorded using the same apparatus, which is discussed in Sect. 9.3.4.2 Semiconductor/Metal-Oxide 1D Nanomaterials. In general, the research about the oxidation of Cu started to focus on the microscopic domain. However, due to the limitation of the apparatus, the structural evolution at atomic resolution could not be acquired still.

a–e Successive stages of the oxide growth on a 100 nm-thick copper thin film between RT and ~770 K under 1.3 × 103 Pa of oxygen; a′–e′ the corresponding selected area electron diffractions (SAEDs). f–h successive stages of reduction of copper oxide (CuO) between RT and ~670 K under 1.3 × 103 Pa of hydrogen; f′–h′ the corresponding SAEDs. i–n Successive stages of growth of a copper oxide layer (Cu2O) and CuO whiskers under 4 × 103 Pa of oxygen (reproduced with permission from Ref. [40], Copyright 2005, Oxford University Press)
.
In 2016, with ESTEM, Boyes and Gai [30] observed the nucleation of the Cu2O by tracking the oxidation process with real-time HAADF imaging (Fig. 9.13a). The oxidation was proved to be a pressure- and temperature-dependent procedure, since the reaction occurred more quickly at higher pressures (2, 5, and 10 Pa) or temperatures (300–500 °C). The reduction process was also recorded in a similar way (Fig. 9.13c). The Cu formed as a protruding island off the NP at first (with a de-wetting angle, Fig. 9.13b), then the interface length increased and the protrusion lessened with increasing temperature, finally the reduction went to completion and the NP recovered to a single Cu metal NP. The high-resolution dynamic HAADF-ESTEM images indicated that the (111) spacing of Cu and the (111) spacing of Cu2O were related to each other with parallel epitaxy between two phases, and a 7 × 6 or a 6 × 5 lattice matching relationships exsited due to an 18% discrepancy in lattice constant.

a In situ oxidation of Cu carried out at 500 °C in 2 Pa oxygen. The blue arrow indicates the direction in which the interface moved. b A schematic of how the interface angles and wetting angles were defined and measured, and examples of the different cases taken from oxidation on the left and center, and from the reduction on the right (from SI). c In situ reduction of Cu2O in 2 Pa hydrogen over the course of 335 min. In the lower right-hand corner, the temperature profile of the reaction is shown (reproduced with permission from Ref. [30], Copyright 2017, American Chemical Society)

ETEM images showing the growth of an initial NiO crystal on the surface of Ni–Cr alloy in 1 × 10−6 mbar H2O at 350 °C (reproduced with permission from Ref. [69], Copyright 2018, Springer Nature)

Observations at the ending cap of a CNT during oxidation. The inner walls and outer wall of the nanotube at 300 °C a were removed after 1.5 mbar oxidation for 15 min at 300 °C (red arrow and blue triangle). b More etching was observed after the same nanotube was oxidized for 15 min with 1.5 mbar oxygen at 400 °C. c The inset of (c) is shown in (d), where one can see the outermost wall being removed and dangling (black arrow) after oxidation at 400 °C. Scale bars in (a–c) and d represent 5 and 2 nm, respectively (reproduced with permission from Ref. [63], Copyright 2013, American Chemical Society)
Besides typical Cu, other metal NPs were also researched. In 2012, Jeangros et al. [66, 67] studied the redox of nickel particles in ETEM. Images, diffraction patterns, and EELS were acquired to monitor the structural and chemical evolution of the system during the reaction. The structural models that describe NiO reduction and Ni oxidation were also proposed based on the ETEM observation.
The redox usually occurred rapidly on the time scale of second to millisecond, which promoted the development of time-resolved quantitative characterization techniques. In 2018, an approach has been developed that integrated time-resolved in situ electron diffraction and an atmospheric gas cell system, allowing quantitative structural information characterization under ambient pressure with millisecond time resolution. Yu et al. [68] employed this technique in studying the ultrafast oxidation kinetics of Ni NPs at 600 °C with a total pressure of 1000 mbar.
In contrast to the well accepted Wagner and Mott Cabrera models (diffusion-dominated), the oxidation of Ni nanoparticles is linear at the initial stage (<0.5 s), and follows the Avrami Erofeev model (n=1.12) at the following stage, which indicates the oxidation of Ni nanoparticles is a nucleation and growth dominated process.
The presence of water vapor, intentional or unavoidable, is crucial to many materials applications. Phenomenologically, water vapor has been noted to accelerate the oxidation of metals and alloys. However, the atomistic mechanisms behind such oxidation remain elusive. Luo et al. [69] studied the oxidation mechanisms of the single crystalline Ni-10at%Cr alloy in water. As shown in Fig. 9.14, during the growth of NiO on the surface, there were vacancy clusters (white dashed circles) formed by incorporating both Ni and O vacancies, which would migrate, increase in size and annihilate. They revealed that protons derived from water dissociation could occupy interstitial positions in the oxide lattice, consequently lowering vacancy formation energy and decreasing the diffusion barrier of both cations and anions, which led to enhanced oxidation in moist environments at elevated temperatures.
The oxidation of organic materials was also observed in detail. The oxidation of carbon nanotubes (CNTs) at the high resolution of an aberration-corrected environmental TEM (ETEM) was directly studied by Koh and coworkers [63] in 2013. Contrary to the previous thought that CNT oxidation triggered at the end of the tube due to high energy at the cap [64, 65], they observed the outer wall was oxidized and removed first (Fig. 9.15). The blue arrow in Fig. 9.15b started to “peel” away and detached more upon further oxidation at 400 °C, while the wall remained attached to the nanotube cap (black arrow in Fig. 9.15d). Furthermore, on occasion, they observed that the interior wall was oxidized first (red arrow in Fig. 9.15b–c), which was ascribed to oxygen infiltrating into the hollow nanotube through an open end or cracks in the tube.
Besides conventional redox of NPs [70] and alloy, sometimes metal NPs could react with gas which is commonly considered inactive, then format protective layer restraining possible corrosion. Wang and her coworkers [71] turned a native Mg alloy surface into an anti-corrosion coating in excited CO2. As known the native surface film on Mg formed in air mainly consists of Mg(OH)2 and MgO, which is porous and unprotective. By reacting with excited CO2 (activated by e-beam irradiation), researchers fabricated a smooth, compact MgCO3 protective layer (Fig. 9.16) on the Mg alloy surface. A Mg micropillar was fabricated by focused ion beam, which was then exposed to air for 30 min to form a native oxide surface film firstly. After exposure to air for 30 min, a surface oxide layer with a thickness of about 8 nm was formed (Fig. 9.16b). Then it was reacted with the e-beam excited CO2*, producing a compact MgCO3 protective layer on the pillar’s surface without any extra heating. Selected area electron diffraction (SAED) and electron energy loss spectroscopy (EELS) analysis were performed to identify the composition of the final reaction product (Fig. 9.16f, g), which confirmed the presence of both crystalline MgCO3 and MgO. Subsequent immersion tests in an aqueous environment were performed to examine the corrosion resistance of the as-grown MgCO3 films. The results demonstrated that the MgCO3 was effective in preventing the Mg metal from deionized water corrosion.

a Schematic diagram showing how to transform the native surface to MgCO3. b–e Surface evolution of a FIB-fabricated Mg pillar during the exposure in 2 Pa CO2 and e-beam irradiation. TEM images showed transformation process of the native oxide layer to the compact nanocrystalline MgCO3 and MgO layer. f Diffraction pattern of the as-FIBed Mg pillar with an amorphous MgO layer and the EELS spectrum of the surface zone in the yellow circle. g Extra diffraction rings from the newly formed film after treatment in CO2 showed the existence of nanocrystalline MgO and MgCO3. Obvious C-K edge can be seen in the corresponding EELS spectrum (reproduced with permission from Ref. [71], Copyright 2018, Springer Nature)
The observation of nanomaterials with oxidizing, reducing, and inactive atmospheres with the employment of ETEM was an important and extensively studied area. With the development of techniques and accessories, detailed information about structure and composition which is critically needed but hidden deeply could be obtained.
9.3.3 Surface Reconstruction and Segregation
The surface structural change of nanomaterials is another important research field, which attracted tremendous attention in recent years especially with the breakthrough of spatial resolution of ETEM. The surface atoms would occupy a higher proportion for NPs with small sizes, and further influence or even decide the character and property of materials.
Yoshida and coworkers [72] examined a catalyst which is composed of GNPs supported on CeO2 by a 300 keV aberration-corrected ETEM. Observing along Au [110] zone axis at the under-focus condition, a surface reconstruction of the Au/CeO2 powder catalyst was acquired on the {100} surface in reaction environments. As shown in the enlarged images in Fig. 9.17a, the {100} facets remained unreconstructed in vacuum, and the distance of 0.20 nm between the topmost and the second topmost {100} surface layers was the same as the interplanar distance of the {200} planes in crystalline bulk gold. The average distance of the adjoining Au atomic columns on the topmost surface layer was the same as the corresponding distance in a {100} plane in crystalline bulk gold. Under a typical reaction environment (1 volume% CO in air at 45 Pa at RT), as shown in Fig. 9.17b, the Au atomic columns on the topmost and second topmost {100} layers shifted. Both the average distance of the adjoining Au atomic columns and the interplanar distance changed to 0.25 nm. The reconstructed surface Au atomic columns correspond well to those of the Au{100}-hex reconstructed surface. In this reconstructed surface, the Au atoms on the topmost surface layer formed an undulating hexagonal lattice, whereas those on the second layer formed a normal square lattice with slight distortion. The researchers performed the ab initio electronic calculations with regard to CO adsorption on the Au{100}-hex reconstructed surface, and the simulation image based on an energetically favorable model for CO adsorption fit well with the observed image. Therefore, it was deduced that the Au atoms on the topmost layer have unusual bonding configurations with the second surface layer, which sustained the high-density adsorption of CO molecules on the reconstructed surface. The methodology demonstrated in this study has opened an experimental route toward the elucidation of GNP catalytic mechanisms by direct observations of metal atoms and gas species at the particle-support periphery.

Au{100}-hex reconstructed surface under catalytic conditions. GNP supported on CeO2 in a a vacuum and b a reaction environment (1 vol% CO in air gas mixture at 45 Pa at RT). Two {100} facets were present in the rectangular regions indicated by I and II in (a) (reproduced with permission from Ref. [72], Copyright 2012, The American Association for the Advancement of Science)
Besides NPs, reconstruction often occurred on the exposed cleaved surface of the crystal as well, which shows different physical and chemical properties with respect to the bulk-truncated ones. A typical instance is the surface reconstruction of TiO2 (001) surface. Generally speaking, the intrinsic structure of some specific oxides is sensitive to the e-beam and easily destroyed, which precludes direct observations in TEM. Using ETEM, Yuan et al. [73] acquired real-time information on the formation and evolution of the (1 × 4) reconstructed TiO2 (001) surface in oxygen which protects the fragile surface reconstruction. Figure 9.18 displayed the formation process of the reconstruction at 500 °C in an oxygen environment (5 × 10–2 Pa). At the beginning, adsorbed layers (mixture of amorphous organics and TiOx species) covered the (001) surface (Fig. 9.18a, set as t = 0 s). After a few seconds, amorphous layers were gradually removed (Fig. 9.18b, t = 34.9 s) in oxygen with e-beam irradiation, and a crystalline layer appeared on the (001) surface, corresponding to adsorbed Ti or TiOx species. After 56.2 s (Fig. 9.18c), adsorbed species began to exhibit non-uniform contrast; at t = 257.8 s (Fig. 9.18d), adsorbed species finally formed the 4 × periodicity, which remained stable during the following reaction. This evolution was also reflected in the intensity profiles for the outmost two surface layers (Fig. 9.18e–f). These results also confirmed that the (1 × 4) reconstruction could occur on the nanometer-sized TiO2 crystal surface, which had been an open question since it was only observed on the surface of micron-sized single crystals.

Tracking the formation process of the (1 × 4) reconstruction, viewed from [010] direction. a–d Sequential HRTEM images of the anatase TiO2 (001) surface during the reconstruction, acquired at 0, 34.9, 56.2, and 257.8 s. The enlarged images of the dotted rectangles are shown in the lower panels of (a–d), respectively. e, f Intensity profiles along the dashed lines in the lower panels of (a–d) (matching colors). The orange and green lines were acquired from the reconstructed layer and top-surface layer, respectively (reproduced with permission from Ref. [73], Copyright 2016, American Chemical Society)
CeO2 is another widely used catalyst and catalyst support both at the laboratory scale and in industry, due to its ability to release or store oxygen atoms from the environment by altering its valence state (3+/4+) to compensate anionic vacancies in the Ce cationic lattice. However, the temporal evolution of reactive surfaces of CeO2 under a gaseous environment was still ambiguous, which hampered the understanding of its catalytic mechanism. Bugnet et al. [74] visualized and quantified the mobility of Ce atoms at {100} surfaces in high vacuum (HV), O2, and CO2 atmosphere via ETEM. As shown in Fig. 9.19a, the contrast at oxygen atomic positions (indicated by arrows) increased from HV to O2 and CO2 environments, and the intensity variation of the most external Ce atomic layer decreased accordingly. Especially with the introduction of O2 into the TEM chamber, O columns terminated (001) surface as a saturated state, because the oxygen supply compensated the e-beam-induced oxygen loss. This O-terminated surface in turn limited the mobility of the underlying Ce atoms. When CO2 was introduced into the chamber, the surface mobility was completely stopped, and dark dots corresponding to the positions of oxygen atoms (Fig. 9.19a, bottom) had a greater contrast than that in oxygen (Fig. 9.19a, O2), which was attributed to carbonates from adsorbed CO2. Quantitative analysis of atomic intensities at {100} surfaces (Fig. 9.19b) demonstrated a high mobility of Ce atoms under HV. The mobility was lower under the O2 atmosphere and nearly stopped in the CO2 environment.

a Two-second average of 50 frames from recorded video showing the edge-on (001) surface of a nanocube in the [110] orientation under high vacuum at 5 × 10−6 mbar, 5 × 10−2 mbar O2, and 5 × 10−2 mbar CO2 (respectively from top to bottom). b Quantitative analysis of Ce mobility at {100} surfaces. Variation of the standard deviation of atomic column intensities of a ceria nanocube viewed along the [110] direction under high vacuum at 5 × 10−6 mbar, 5 × 10−2 mbar O2, and 2.6 × 10−2 mbar CO2 (respectively from top to bottom). Each dot images a single atomic column position, and its color indicates the variation of intensity over 420 images recorded at 25 fps on a normalized scale (Blue, low variation; red, high variation). Missing columns correspond to positions where columns fully disappear within the time frame spanned by the 420 frames (approximately 17 s) (reproduced with permission from Ref. [74], Copyright 2017, American Chemical Society)
Generally, the reconstruction emerged on the surfaces of metal-oxide more easily, especially when exposed to the reductive atmosphere, the loss of oxygen atoms would influence the surface stoichiometry so as to rearrange the atomic structure and relationship to some extent. Of course, the intrinsic property of oxide, the environmental gas, the temperature, and the e-beam, all take effect on the loss extent of oxygen atoms, which result in different surface structures in the end.
Besides reconstruction, atomic-scale chemical segregation was also observed using ETEM. Dai et al. [75, 76] studied the oxygen-driven element segregation process in a Pt3Co fuel cell–cathode catalyst under atmospheric pressures. Platinum-metal (Pt-M, M = Fe, Co, Ni, etc.) NPs were considered as a promising alternative to traditional Pt NPs as the oxygen reduction reaction (ORR) catalyst in polymer electrolyte membrane fuel cells. However, there were still some unsolved crucial questions, such as the element segregation mechanism during high-temperature annealing in pure oxygen. In situ TEM at atmospheric pressure provided valuable data to derive the corresponding mechanism.
The Pt3Co/C powder sample (a commercial ORR catalyst) [75] was pretreated to create equilibrium-shaped NPs with a random distribution of Pt and Co. Then the sample was heated from RT to 350 °C in 760 Torr of pure oxygen. Figure 9.20a showed a twinned Pt3Co NP after oxygen annealing for more than 10 min. One or two additional atomic layer(s) with a lower contrast formed on the {111} surfaces in the false-colored BF-STEM images. By comparing the marked green and black distance as #1 and #6, it is obvious that the segregated layers have a larger lattice spacing than the inner part. Meanwhile, the concrete data about the length of the side and intersection angle (Fig. 9.20b) are in excellent agreement with the Co sublattice unit cell (Fig. 9.20c) in the structure of CoO. By checking more than 20 Pt3Co NPs, outermost CoO layers are always found on {111}, but not on {100} surfaces, without exception, clearly revealing that facet-dependent oxidation is taking place on the Pt3Co NPs. In comparison, additional atoms were attached on the top {100} surface (yellow arrow in Fig. 9.20f), until a new layer (#14) formed completely at the time t = 32 s (Fig. 9.20g). Because the contrast of the newly formed top layer (#14) is similar to the previous outermost layer #13, it indicated that the atoms newly attached on the {100} surface are closer to pure Pt. Therefore, the authors presumed that Pt atoms possess higher mobility in an oxygen environment, which tend to take place on the {100} Pt3Co surfaces to result in structural fluctuations. In contrast, the CoO layers on the {111} surfaces blocked exposure of underlying Pt to the oxygen environment, and stopped the possible diffusion and reconstruction. The contrast difference of the additional atoms and high spatial resolution of the STEM images played an important role in distinguishing the attached atoms on different surfaces. The outstanding capability in characterizing structure and composition at microscopic domain of ETEM is obviously proved.

In situ observation on the {111} and {100} surfaces of Pt3Co NPs in an oxidizing environment. a–b False-colored BF-STEM images illustrating the periodical unit cell of the oxide layer on {111} Pt3Co surfaces. c Projection of a CoO model along the ⟨110⟩ zone axis. Blue and red spheres represent Co and O atoms, respectively. d–g Sequential STEM images showing the additional Pt layer growth on the {100} surface of the oxidized Pt3Co NP (reproduced with permission from Ref. [75], Copyright 2017, American Chemical Society)

The reversible structural transition of NiAu NPs during the CO2 hydrogenation reaction. a In situ TEM images showing the alloying and dealloying evolution of an individual NiAu particle in the reaction atmosphere (9 ± 0.1 mbar, 25% CO2/75% H2). b, c Surface atom arrangement of a NiAu nanoparticle reconstructed from the full alloy (600 °C) (b) to the Ni@Au core–shell (400 °C) (c); the four parts from left to right in b show the high-resolution TEM (HRTEM) image (with a Thon ring inset), the corresponding enlargement of the surface area, schematic structure, and point analysis of the EELS (with a HAADF inset); the three parts from left to right in c show the HRTEM image (with a Thon ring inset), the corresponding enlargement of the surface area, and phase contrast profile. d Intensity profiles from the integration of diffraction rings of SAED patterns during the reaction. The inset shows 2D profiles stacking along with reaction temperature. e In situ EXAFS of the Au L3 edge and coordination number changes (inset) of the Au–Au, Au–Ni, and Ni–Ni pairs at room temperature, 400 and 550 °C. a.u., arbitrary units (reproduced with permission from Ref. [77], Copyright 2020, Springer Nature)
Recently, Zhang et al. [77] reported reversible segregation and alloy of Ni–Au bimetallic NPs during CO2 hydrogenation. An ETEM was employed for the in situ observation in the reaction atmosphere (9 ± 0.1 mbar, 25% CO2/75% H2). As shown in Fig. 9.21, the core–shell nanoparticle was maintained with a darker Au–rich shell at 450 °C. When the temperature reached 600 °C, the catalysts showed the highest catalytic activity and the Au shell dissolved into the Ni matrix and vanished, forming a mixed NiAu alloy. When the catalysts cooled to 400 °C, the Au shell reappeared, recovering the Ni@Au core–shell structure. The phase evolution was further revealed via in situ electron diffraction seen in Fig. 9.21d that a broadened diffraction peak which corresponded to the NiAu alloy phase emerged between the Au(220) and Ni(220) peaks above 400 °C. If we just investigate the catalysts before and after the catalytic reaction, the Au shell could be thought to be the active site since the NiAu alloy only appeared during the reaction. Combining with other in situ spectroscopies and DFT calculations, the authors thought that the surface Ni atoms offered the active sites for the hydrogenation of CO2 and the surface Au atoms contributed to the selective production of CO. This result gave a universal insight into designing a polynary metal catalyst with reversible structure responding to the environment and reminded us again that the importance of in situ observing the dynamic evolution of catalysts as the ex situ results may lead to a confusing conclusion.
Besides the compositional variations, segregation is usually accompanied by misfit strain and the formation of dislocations in the subsurface region via a surface diffusion and trapping process. Actually, two decades ago, it has been shown that the presence of hydrogen in solution enhanced the dislocation velocity in face-centered cubic, body-centered cubic, and hexagonal close-packed materials. Ferreira [78] proposed that solute hydrogen reduced the elastic interactions between dislocations and elastic centers which act as barriers. Using dynamic, atomic-scale resolution in situ HRTEM and theory modeling, Zhou et al. [79] demonstrated the Au surface segregation in the Cu(Au) solid solution, and focused on the misfit dislocations and its migration and climb facilitated by the segregation. This observed coupling between segregation and dislocation formation has wide relevance, as the partitioning of alloying elements (or impurities) occurs in most multi-component materials under a suitable environmental.
9.3.4 Growth of Low-Dimensional Nanomaterials
The introduction of gas into the TEM facilitated the development of another area which focused on the nucleation and growth of low-dimensional nanomaterials, especially 1D nanowires and nanotubes, which usually enjoy unique and unexpected properties due to quantum confinement effects and the absence of defects. Besides their high surface-to-volume ratios, morphology and crystallography also play important roles in manipulating properties. Therefore, the technique of synthesis of ideal or satisfactory 1D nanomaterials is the basis of further research in extraordinary properties and various applications.
There is no doubt that the traditional ex situ information obtained after the synthesis couldn’t reflect the real-time structural change information during the growing process, which is critical to optimize the synthesis process. ETEM enables the in situ observation during synthesis, and thus direct atomic resolution images in growing processes and kinetic change (morphology, crystallography, defect structure, and chemical composition) could be obtained. It lays the foundation for the understanding of mechanism and improvement of synthesis technology.
For chemical vapor deposition (CVD), there exist three commonly accepted growth mechanisms of low-dimensional nanomaterials as vapor-liquid-solid (VLS), vapor-solid-solid (VSS) and catalyst-free growth [48], and all of them have been observed directly in ETEM [80]. As an ideal cold-wall nanoreactor for CVD, the energy needed to decompose the precursor molecules can be provided either by the e-beam or by thermally heating the substrate.
9.3.4.1 Nanostructure (Island/Dot)
The effect of e-beam on the nucleation and growth of nanostructures is nonnegligible, especially when the temperature was taken into account in synthesis. Electron-beam-induced decomposition (EBID) techniques were extensively used to fabricate periodic arrays of metal/semiconductors. The process involves introducing organometallic source vapor (precursor vapor) into the microscope column and irradiating the target area of the substrate with a focused e-beam.
In 2001, Jiang and coworkers [81] investigated the detailed parameters, such as deposition temperature, source compound, organometallic partial pressure, and electron dose, of EBID to fabricate periodic and aperiodic nanostructures and nanodevices. Metallocene compound, nickelocene (Ni(C2H5)2), was used as an organometallic source and the deposits with edge sharpness under 4 nm had been fabricated, despite the quantity of the deposited material being too small for XPS to detect.
Mitsuishi [82] employed a 200 kV TEM equipped with a gas introduction system satisfying a gaseous flux of 2 × 10–4 Pa L s−1 to fabricate nanometer-sized W dots with a diameter of less than 5 nm in 2003. The precursor employed for the W deposition was tungsten hexacarbonyl [W(CO)6], and the depositions were performed at RT. As shown in Fig. 9.22a, c and b, d, the bright-field TEM and HAADF-STEM images showed the W nanodots deposited on the Si substrate, and the time periods for the large and small dots were about 1.5 and 0.5 s. The depositions were made for 10 s at two regions with different substrate thicknesses. The result showed that the deposition size at the thicker substrate region was only slightly larger, which indicated that the method can also be applied to the bulk substrate used in practical applications.

a TEM bright-field and b HAADF–STEM image of W nanodots deposited on Si. The bright contrast indicates a concentration of the heavy element, W. c The BF and d HAADF–STEM images, in which well-defined nanodots are clearly seen, and are magnifications of the upper right corner of (a) and (b), respectively (reproduced with permission from Ref. [82], Copyright 2003, AIP Publishing). e Z-contrast image of 2D periodic array of GaN dots. Inset shows line profiles through the Z-contrast images, showing a FWHM of 4 nm. f Surface plot of array of dots showing the height of dots derived from the ADF image intensity. Average dot height is 5 nm (reproduced with permission from Ref. [83], Copyright 2004, AIP Publishing)
The deposition of GaN quantum dot arrays with uniform size onto a SiOx substrate was done using a nanolithography technique in 2004 by Crozier [83] as well. The nanolithography technique is based on a combination of sub-nanometer electron-beam-induced chemical vapor deposition and single-source molecular hydride chemistries. A completely inorganic and highly reactive gaseous perdeuterated gallium azide (D2GaN3) was utilized as a hydride source, dissociated exothermically under electron irradiation, resulting in the formation of stochiometric GaN, with byproducts of volatile D2 and N2. The nanolithography and characterization were conducted in a Tecnai ETEM, which is fitted with an environmental cell and essentially operates as a cold-wall CVD reactor. Figure 9.22e, f showed a periodic array of uniform GaN dots grown by exposing the substrate to a precursor pressure of ~30 × 10–5 Torr, evacuating the microscope for 5 min and then depositing a series of dots (the deposition time per dot was 0.5 s). The dots were highly uniform, and displayed an average full width at half-maximum (FWHM) of ~4 nm and a base width of ~9 nm. The diameter of dots was considerably larger than the used e-beam, indicating that the dot size was controlled by the spatial distribution of secondary electrons leaving the substrate surface. In 2005, employing the same device, van Dorp et al. [84] wrote dots with an average FWHM of 1.0 nm, in an array with a spacing of 4.0 nm, further improving the resolution limit of the EBID approach. These experiments demonstrated that EBID is a promising technique for high-resolution resistless lithography, and the ETEM allowed the entire growth process to be observed and controlled in real time as the container and reactor.
9.3.4.2 Semiconductor/Metal-Oxide 1D Nanomaterials
Semiconductor nanowires have important applications in electronic devices, information storage, microelectromechanical systems, and photovoltaics. For many applications, it is essential to control the structure, orientation, and location of the nanowires, and the factors that determine the growth rate, orientation, and epitaxial relationship with the substrate are therefore of key importance [85].
Since the VLS mechanism was proposed by Wagner et al. [86] in 1964, it was extensively studied and verified in the following decades. The characteristic feature of this mechanism is the liquid eutectic mediating the mass transport from the vapor to the solid interface. In 2000, the first real-time observation of semiconductor nanowire growth in an in situ high-temperature TEM unambiguously confirmed the VLS crystal growth mechanism at the nanoscale. Yang and Wu dispersed a small amount of micrometer-sized Ge particles on TEM grids together with solution-made monodispersed Au nanoclusters. In their experiments, they used a conventional TEM without environmental equipment. Therefore, the nucleation and growth were dependent on the Ge evaporation at 900 °C, from a solid Ge source (a thin layer of Ge/C coating). Three well-defined stages have been clearly identified during the process: (I) metal alloying (Ge and Au), (II) crystal nucleation, and (III) axial growth, as shown in Fig. 9.23a [87].

a Schematic illustration of VLS nanowire growth mechanism including three stages (I) alloying, (II) nucleation, and (III) axial growth (reproduced with permission from Ref. [48], Copyright 2017, Springer Nature). b–e Dark-field images in side view during the solid-to-liquid transformation. f Bright-field image showing the droplet after the transformation is complete. g Idealized schematic of the inferred geometry (reproduced with permission from Ref. [88], Copyright 2009, American Physical Society). h–j Bright-field images extracted from a video obtained during nucleation of Si from polycrystalline Au clusters. h Image acquired before opening the leak valve, showing polycrystalline Au particles, as indicated by their faceted shapes and by the interparticle variations in their brightfield contrast. i Image acquired after 65 s, showing the formation of the liquid AuSi alloy as reflected by the rounded shapes in projection and the disappearance of crystalline contrast. j Image acquired after 84 s, showing the appearance of a Si nucleus with lighter contrast at the edge of one AuSi droplet. (k-m) Enlarged images of the boxed regions of (h–j), respectively. Arrows in (m) indicate the interface between liquid AuSi and solid Si (reproduced with permission from Ref. [89], Copyright 2008, The American Association for the Advancement of Science)
Employed a UHV-TEM equipped with facilities for introducing the reactive gas disilane (Si2H6), Kim et al. systematically studied the first and second stages of Si nanowires growth in the Si–Au system, including the size effect [88] during the phase transition and kinetics of individual nucleation [89]. Figure 9.23b–g showed the images of the transition from two-phase Au + AuSi to single-phase AuSi. The first four frames were recorded at an orientation where the crystalline Au provides bright contrast, and the Au particle appeared to consist of a single crystal with multiple twins (visible as dark bands). Figure 9.23f shows the liquid droplet that remained after the crystal had disappeared (bright field), and the idealized schematic of the inferred geometry is illustrated in (g). Similarly, a series of images obtained when introducing the reactive gas disilane to a heated sample illustrated the initial transformation of polycrystalline Au islands (Fig. 9.23h) into eutectic droplets of AuSi (i), followed by the appearance of Si nuclei (j). The Si nucleation occurred at the edge of the droplet, which suggests that this is the energetically favorable location that minimizes the nucleation barrier.
Ross and coworkers further systematically [90,91,92,93] studied steady-state growth of Si and Ge nanowires, considering growth kinetics, sidewall structure, and the phase (liquid or solid) and stability of the catalyst at the nanowire tip. In Si nanowire growth (Fig. 9.24a), the VLS mechanism has a characteristic appearance in the TEM. The droplet, with an amorphous structure and curved surface, rests on the tip of the nanowire. Meanwhile, the droplet/nanowire interface is generally planar, and appears to be composed of Si(111) planes, whatever the growth direction of the nanowire. The dependence of growth rate on pressure, temperature, and particularly nanowire diameter was also observed and recorded in this system, to deduce the growth kinetics of Si nanowires. They suggested that the rate-limiting step was the thermally activated, Au-catalyzed dissociative adsorption of disilane, and that dissociation takes place directly on the catalyst droplet, with no contribution due to adsorption and diffusion from elsewhere. This result was fully consistent with the VLS model, in which the droplet surface was taken to be a preferential site for the adsorption of the growth species [91].

a Sequence of images recorded during the growth of a 26 nm diameter Si nanowire, with the time shown in seconds since the first image. Bright-field imaging conditions were used so that Si appears medium gray and the droplet (AuAuSi) is a dark semicircle (reproduced with permission from Ref. [91], Copyright 2010, IOP Publishing). b Ge nanowire growth from an AuGe liquid droplet. c Ge nanowire growth from solid Au prepared as follows: at constant pressure, the specimen was cooled to solidify the droplets and then returned to the same temperature as (b), where the catalysts remained solid due to hysteresis (reproduced with permission from Ref. [94], Copyright 2007, The American Association for the Advancement of Science). d Si nanowire growth in the VSS modes (AgAu2 at 556 °C and 1 × 10–5 Torr disilane) (reproduced with permission from Ref. [95], Copyright 2012, American Chemical Society)
Besides classical Si and Ge nanowires, the VLS mechanism was also verified in other materials, such as GaN [96] and InAs [97]. Growing III–V GaP nanowires in an ETEM, Chou et al. [98] measured the local kinetics in situ as each atomic plane was added at the catalyst–nanowire growth interface by the VLS process. They found that the growth rate was very sensitive to perturbations using a high V/III ratio, and such sensitivity was absent when growing at a low V/III ratio, creating a highly regular regime that may be optimal for the growth of complex materials. The relationship between local growth kinetics and atomic structure provides insights into understanding the growth mechanism and the requirements for the most precise control over growth.
In the case of Ge nanowires are catalyzed by Au, in situ studies found that nanowires can grow below the eutectic temperature with either liquid or solid catalysts, and the state of the catalyst only depends on the thermal history. The second pathway was called the VSS mechanism. Kodambaka et al. [94] unveiled the phenomenon clearly in 2007. As illustrated in Fig. 9.24b–c, (b) shows a Ge wire at three successive times during VLS growth and the catalyst persisted liquid state (smooth round shape). After cooling the specimen to solidify the catalysts and then reheating to the original temperature, the nanowire growth continued with solid catalyst (Fig. 9.24c, faceted surface). Measurements made on several wires showed that VSS growth was 10–100 times slower than VLS growth even at the same gaseous pressure and temperature, presumably as a result of weaker surface reactivity and/or lower diffusivity through the solid. The principal cause of VLS growth below eutectic temperature excluded the Gibbs–Thomson effect or any direct effect of small size, and finally was identified as the inhibition to nucleation of solid clusters caused by high Ge supersaturation in the droplet. VSS mechanism and similar phenomena were also demonstrated in Si/Ge nanowires growth catalyzed by other catalysts [95, 99,100,101] (Fig. 9.24d).
Besides the common and extensively accepted VLS and VSS growth mechanism, there existed some research [40, 102,103,104,105,106] focused on the catalyst-free pathway, especially for metal-oxide nanowire growth in the presence of oxygen. No external catalysts or solvents were directly used during the growth process, and the nanowires were directly grown on the metal surface through a reaction with oxygen, which was named as vapor–solid (VS) mechanism.
Actually, as early as 1955 [102], Sears’s experiment demonstrated the vapor–solid process in the growth of mercury whiskers. In 2001, Pan et al. [103] deemed that their nanobelts of semiconducting oxides were also governed by VS mechanism because the only source material used was pure oxide powders.
Another possible mechanism explained nanowire growth is accomplished via the incorporation of O from vapor and metal ions through diffusion along the defects, such as twin boundaries, stacking faults, and surfaces, from the substrate to the nanowire tip [40, 48]. As Fig. 9.12i–n shows, the successive stages of growth of copper oxide whiskers in 4 × 103 Pa of oxygen were recorded. At the beginning of the heating (estimated to ~670 K), an oxide film with non-uniform thickness was formed on the copper surface, and consequently the shape of the edge became jagged (Fig. 9.12i–j). After about 40 s, the oxide layer stabilized and the edge became smoother (Fig. 9.12k). Gradually, whiskers started to grow on the oxide layer (Fig. 9.12l–n), and two kinds of whiskers were identified in these micrographs: (i) thick and short and (ii) thin and long. The analysis of grown whiskers suggested that copper atoms easily diffuse along the lattice defects in CuO, and upon reaching the whisker tip, react with the ambient oxygen there. Tokunaga et al. [105] also studied the growth and structure of tungsten oxide nanorods using ETEM. They concluded that it was dominated by the solid diffusion mechanism: cracks occurred in the surface of the natural tungsten oxide layer when the tungsten was heated, after which tungsten diffused through the cracks of natural tungsten oxide layer from the tungsten wire to form a highly crystalline prominence. In 2015, Zhang et al. [104] demonstrated the layer-by-layer growth kinetics statistics in the growth of tungsten oxide nanowires with ETEM.
In 2014, Rackauskas et al. [106] observed the synthesized CuO nanowires in an ETEM by oxidation of pure metallic Cu in the presence of 3.2–7 mbar of O2 in the temperature range of 350–475 °C. They also confirmed the occurrence of the copper diffusion (along the grain boundaries of the oxide layers and to the nanowire tip). Meanwhile, collecting kinetics of the growth, they concluded that nanowire growth should be described as the layer-by-layer formation process, in which nucleation of a new layer is the growth limiting stage, rather than the copper diffusion (along the grain boundaries of the oxide layers and to the nanowire tip).
Based on the growth of semiconductor nanowires, simple nanoscale devices were fabricated and observed over their complete lifecycle from creation to failure with additional electrical measurements. Kallesøe et al. [107] used the VLS method to grow Si nanowires as devices bridged between Si cantilevers. They characterized the formation of the contact between the nanowire and the cantilever, measured the electrical properties and high current failure characteristics of the resulting bridge devices, and related these to the structure. Probably the growth of nanowires in ETEM could be controlled more accurately so as to fabricate more complex structures or special devices in the future.
9.3.4.3 Carbon 1D Nanomaterials
As early as 1971, Baker and Harris [108] had already used the modified controlled atmosphere electron microscope to observe gas reaction, including the investigation of filamentous carbon formation from acetylene, catalyzed by nickel, iron, and cobalt. In spite of the resolution limit, early studies already focused on morphological changes and growing rates, from which kinetic parameters and activation energy could be obtained, so as to help put forward the growth mechanism.
Since carbon nanotubes (CNTs) were discovered in 1991 by Iijima [109], various applications, especially in displays or in X-ray tubes for medical applications, were developed in the following years. These applications require strict control of the diameter, length, and number of walls (such as multi-wall CNTs for field emitters, and single-walled CNTs for transistors) and the chirality. However, it’s difficult to monitor and control the nucleation and growth of CNTs during traditional high-temperature arc-discharge and laser ablation processes. By contrast, CVD is an increasingly important and steady method, especially when ETEM was used as a cold-wall nanoreactor. With the development of accessories and techniques, the ETEM offers an excellent opportunity to satisfy the demand for in situ observation of CNTs growth.
In 2004, there were two groups [110, 111] that completed direct imaging of CNT nucleation and growth process at reaction temperature using ETEM. Helveg et al. [110] presented in situ TEM observations of the formation of carbon nanofibers (including nanotubes) from methane decomposition over supported nickel nanocrystals. The consecutive TEM images of the main equilibrium shape transformed into a highly elongated shape were demonstrated in Fig. 9.25. The formation of more graphene sheets whose basal (002) planes were parallel to the Ni surface was attributed to the elongation of the Ni particle, and it could be concluded that the reshaping of the Ni nanocluster assisted in the alignment of graphene layers into a tubular structure. Specifically, the nucleation and growth of graphene layers were found to be assisted by a dynamic formation and restructuring of mono-atomic step edges at the nickel surface (in Fig. 9.25b–g).

Image sequence of a growing carbon nanofiber. Images a–h illustrated the elongation/contraction process. Drawings were included to guide the eye in locating the positions of mono-atomic Ni step edges at the C–Ni interface. The images were acquired in situ with CH4:H2 = 1:1 at a total pressure of 2.1 mbar with the sample heated to 536 °C (reproduced with permission from Ref. [110], Copyright 2004, Springer Nature)
Sharma and Iqbal [111] reported the in situ observation of carbon filaments and CNTs as well with the employment of ETEM. They used two different catalyst systems (Ni–SiO2 and Co, Mo–MgO) in two flowing precursors, propylene (99.9999%) and welder’s grade acetylene, respectively. Filamentous growth was observed when propylene was leaked over Ni–SiO2 at 400 °C while acetylene, on the other hand, provided the growth of a well-defined nanotube structure. Furthermore, the growth of multi-wall nanotubes (MWNTs) favored at 450 °C, and compared with single- and double-walled CNTs were observed to form predominantly at higher temperatures (700–800 °C).
In the following years, Sharma and coworkers continuously studied the CNTs’ growth under diverse reaction conditions, and they concluded that the growth rate and morphology of the formed nanotubes depended on the synthesis conditions (i.e. temperature and pressure) [80, 101, 112, 113]. For example, catalyzed by Ni–SiO2 with narrow particle size distributions (1.5–3.0 nm), at an average rate of 35–40 nm s−1 under 20–100 mTorr of gas pressures at 480 °C, serpentine-shaped or zigzag multi-wall carbon nanotubes grew, while at an average growth rate of 6–9 nm s−1 at pressure <10 mTorr at the same temperature, straight single-wall carbon nanotubes with nearly uniform diameters (~3.5 nm) formed [112]. Furthermore, the plausible growth mechanism for small-diameter CNTs was also put forward, and shown in Fig. 9.26a–e. The model assumed that nanotubes grow preferentially along <111> direction on certain crystallographic Ni planes (the most active {110}, in (a)), then the particle rotated, or melted and re-crystallized (b), which would stop the growth of the tube momentarily until a new set of active {110} surfaces resume the growth. Epitaxial lattice-matching of the Ni {110} plane to the graphene lattice of the nanotube could induce the Ni particle to re-orient itself so that the Ni {111} aligns with the nanotube axis. The new growth direction would be controlled by the angle between two sets of 110 surfaces (i.e. 110 and 101, 60°). By incorporating 5-ring/7-ring pairs into the graphene lattice, the particle could continue to grow as a fully 3-coordinated seamless tube (c). If the 5-ring/7-ring defect pairs were at low densities, the nanotube segments were straight, punctuated by 60° and 120° bends (d), while they could appear to be continuously curved and of irregular diameter with high densities of defects (e). Figure 9.26f showed a model of such a bend in a SWCNT in detail. The two nanotubes can be merged seamlessly, with fully 3-coordinated carbon atoms, by introducing equal number of 5-rings and 7-rings so that the overall polyhedral curvature of the nanotube does not change. In this model, 3 such 5-ring/7-ring pairs were introduced. The distribution shown here was not unique; many different variants that supported such a bend were possible.

a–e Sketches showing a plausible growth mechanism for small-diameter CNTs. f A hypothetical bend in a SWCNT model induced by three 5-ring/7-ring pairs (reproduced with permission from Ref. [112], Copyright 2005, Oxford University Press)
In another article [113], Sharma and coworkers concluded that straight, single-wall, carbon nanotubes tend to form at high temperatures and low pressures while bent, zigzag, multi-wall carbon nanotubes form at lower temperatures and higher pressures. It’s also perceived that high nucleation and growth rates would reduce the time/or energy required for annealing 5- and 7-ring defects. Furthermore, the overall activity of Ni catalyst particles, i.e. the number of particles active for tube formation, was improved by the addition of a small amount (below 0.2 mol fraction) of Au [114].
Yoshida and Takeda also made intensive and detailed studies on the growth of CNTs. In 2005, they outlined a theory of image information in an ETEM and applied the theory to simulating HRTEM images of SWNT in source gases of ethanol at the actual growth condition [115]. The ETEM observation of the swinging and rotational growth of CNTs in 2007 provided an explanation of the suspended growth mechanism [116]. In 2008, the nucleation and growth process of CNTs catalyzed by NP catalysts of fluctuating crystalline Fe carbide in CVD with C2H2 was acquired with ETEM [117]. 1 nm thick Fe (99.999%) was deposited on the thin SiO2 surface layer of silicon substrates by vacuum evaporation. Then the substrates were set in an FEI Tecnai F20 equipped with an environmental cell, and heated to 600 °C in a vacuum, and subsequently, a mixture of C2H2:H2 = 1:1 was introduced into the ETEM (10 Pa). Figure 9.27 shows the root growth process of a SWNT from an Fe carbide NP catalyst viewed nearly normal to the growth direction. In NP catalyst, various kinds of lattice images appeared and faded out, while all of them were identified as the iron–carbide structure (cementite, Fe3C). The lattice image shown in Fig. 9.27a–d continued for 1–2 s, 1–2 s, 1–2 s, and 7 s, respectively. The phenomenon was accounted for by a change in crystal orientation without physically rotating the whole NP since it remained at nearly the same position and deformed only slightly. In conclusion, the NP catalysts were deemed as fluctuated crystalline cementite, Fe3C.

a–d Growth process of a MWNT from an Fe carbide NP catalyst viewed nearly normal to the growth direction. Enlarged images and Fourier transforms of the dotted square regions in images are shown below, which indicated that phase of catalyst was Fe3C (reproduced with permission from Ref. [117], Copyright 2008, American Chemical Society)
Besides CNT, the formation of different allotropes of carbon on the nanoscale (e.g. graphene layers [118]) was also studied extensively with the employment of ETEM. There is no doubt that the ETEM has the indispensable advantage in observing the dynamics of catalyzed nanostructure growth, which provided the possibilities for understanding the mechanism and promoting the synthesis technology. Furthermore, it’s common that the catalyst would cause some dynamic change during the catalyzation (e.g. in Yoshida’s experiment, the NP catalyst Fe3C already deformed slightly), which would be discussed in the next section.
9.3.5 The Catalyzation
Heterogeneous catalysis plays an essential role in modern industrial manufacturing. Over 85% of commercial chemicals in the industry are catalytically produced, mostly via gas-involved processes [48]. Traditional heterogeneous catalysts consist of metal NPs, which are supported on specific oxide substrates (even specific crystallographic planes). Since the catalytic activity varied with the atomic arrangement on the surface of metal NPs, the interfaces between particles and substrate, the surfacial status of the substrate (oxidation/reduction/doping), it’s of great importance to study the structure and composition of catalyst at the atomic scale. Meanwhile, the catalytic reactions usually occur in an atmospheric environment, which means that the catalysts are likely demonstrating different characteristics and properties in traditional TEM under high vacuum. Therefore, the emergence of ETEM could satisfy the requirement of observation in detail at the atomic scale by maintaining the catalyst under active status in the reaction atmosphere [119].
9.3.5.1 Dynamic Observation of Catalytic Reactions
The catalytic performance of metal catalysts is determined by their structures [120], e.g. size, morphology, composition, crystal structure, metal–support interactions, and so on, while recent studies suggested that the structure of metal catalysts would dynamically change during reaction conditions [121], and their statuses in reaction could be dramatically different from the original/final ones [122, 123]. Therefore, real-time studying the metal catalysts is well expected to give insight into the underlying mechanism and to help further improve the properties of metal catalysts.
The nucleation and growth of CNT is a typical catalytic process, and the catalyst would undergo some dynamic changes in both morphology and composition during the whole growth. For example, the Fe3C catalyst deformed during the reaction (Fig. 9.27). Similarly, in the work of Hofmann et al. [124], the catalyst particle showed a periodical morphology change, from its initial equilibrium shape into a highly elongated shape during carbon nanofibers nucleation. As shown in Fig. 9.28, the Ni particle elongated and then suddenly contracted into a rounder. Typically, the substrate anchorage was overcome during the first contraction and the catalyst crystal lifted off the substrate, resulting in a tip growth mode.

a–d ETEM image sequence showing a growing CNT in 3:1 NH3:C2H2 at 1.3 mbar and 480 °C. The Ni particle elongated and then suddenly contracted into a rounder shape (reproduced with permission from Ref. [124], Copyright 2007, American Chemical Society)
The composition change of the catalyst was also reported. Mazzucco et al. [125] observed and recorded the whole deactivation process of Fe catalyst NP in ETEM, and found that the Fe catalyst transformed into two distinct carbide phases, cementite (Fe3C) and Hägg (Fe5C2) under CNT growth conditions. Moreover, the carbon-rich Hägg carbide was inactive for the competitive process in contrast to the less carbon-rich cementite phase.
As a classical catalyst which is highly active for low-temperature CO oxidation and water–gas shift reaction, Au/CeO2 is studied at the atomic scale to improve and stabilize the catalytic property. Since the interfacial perimeters between metal NPs and oxide substrate probably tip the scale for the catalysis, the structural collapse and reconstruction of the gold–ceria interfacial perimeters under various reaction conditions attracted extensive attention. Using ETEM, Ta et al. [127] directly observed the structural changes of the Au NPs and the interfacial structure (Au/CeO2) under oxidizing and reducing (reactive) atmospheres at 573 K. As shown in Fig. 9.29a, a truncated octahedral Au particle (size: ~3 nm) directly contacted to ceria without any transition layers (Au{111}–CeO2{111}), and the Au–CeO2 interfaces retained their original configuration without appreciable variation in shape and size under an oxidizing gas environment (10 vol%O2/N2). When the same particle was exposed to a reducing and reactive environment (42 vol% CO/6 vol% O2/N2,), the Au–CeO2 interfaces reconstructed with the appearance of disordered CeO2 layers adjacent to the gold particle as shown in Fig. 9.29b, which even increased in thickness over time. The chemical nature of the newly formed ceria layers was identified as CeO1.714, which was partially reduced and probably resulted in the electronic state change of the gold atoms at the perimeter of the gold–ceria interface. Therefore, the authors proposed an anchoring mechanism as Fig. 9.29d shows, the faceted Au NP was constructed of four different domains with different functions.

ETEM images of the Au-673 catalyst (the selected Au NP) under oxidizing and reducing atmospheres at 573 K. a, b A truncated octahedral Au particle of ∼3 nm size under a 10 vol% O2/N2 environment (a) and a 42 vol% CO/6 vol% O2/N2 atmosphere (b). c Schematic depicting a typical gold NP enclosed by {111} and {100} planes. d Atomic scheme of a gold NP anchoring onto a CeO2 nanorod, illustrating the functions of different domains (reproduced with permission from Ref. [127], Copyright 2012, American Chemical Society)
Based on the observation of reversible changes in NPs, the oscillatory CO oxidation behaviors catalyzed by Pt NPs were reported with the employment of a nanoreactor, time-resolved HRTEM, mass spectrometry, and calorimetry in 2014 [47]. The technologies of MEMS and a unidirectional gas-flow channel with a reaction zone satisfied the in situ time-resolved observations of the periodic refacetting of the Pt NPs at 1 bar pressure and elevated temperatures. As shown in Fig. 9.30a, mass spectrometry demonstrated periodic oscillations of O2 and CO pressures in anti-phase, and similar variations are shown in the CO2 pressure. Meanwhile, to keep the nanoreactor temperature constant, the heater power would be reduced (Fig. 9.30b), which was at least 3 orders of magnitude faster than the time scale of reaction oscillations, to compensate the exothermic reaction heat. Finally, the Pt NPs near the reaction zone exit switched between a more spherical and a more facetted morphology (Fig. 9.30c, d). Specifically, as the CO conversion increased rapidly, Pt NPs immediately started a gradual transformation from a more spherical shape toward a more facetted shape (Fig. 9.30d I–III). On the decrease in the CO conversion, the NP transformed back to the more spherical shape (Fig. 9.30d IV) and retained that shape until the CO conversion rose steeply again. Thus, the individual NPs near the exit from the reaction zone could undergo oscillatory and reversible shape changes with a temporal frequency matching the oscillations in reaction power, indicating that the oscillatory CO conversion and the dynamic shape change of the Pt NPs were coupled.

Correlation of oscillatory CO oxidation reaction data with the projected morphology of a Pt NP (Supplementary Movie 1). The gas entering the reaction zone is 1.0 bar of CO:O2:He at 3%:42%:55% and nanoreactor temperature is 659 K. a–c Mass spectrometry of the CO, O2, and CO2 pressures (a), reaction power (b), and shape factor (c) for the Pt NP in d as a function of time. The shape factor corresponds to the relative difference in the area from the best elliptical fit in image I in d (Ref. [4]). The morphology factor is zero for the more spherical shape and deviates for more facetted particles. Part of the reaction oscillation data is highlighted by the red rectangle. d Time-resolved TEM images of a Pt NP at the gas exit of the reaction zone (reproduced with permission from Ref. [47], Copyright 2014, Springer Nature)
Besides the dynamic observation of the catalyst, the oxide substrate was also studied because it usually interacts with the NPs under specific environments. The reconstruction of gold–ceria interfaces in Fig. 9.29b was one manifestation, and another typical and obvious phenomenon was strong metal–support interactions (SMSI) (More details referred to SMSI could be found in [194]). As early as 1978, Tauster [128] had already explored the possibility of strong interactions between group VIII metals and supports containing titanium cations, and proposed the SMSI which was associated with the formation of bonds between the metal and titanium cations or titanium atoms. The definition of SMSI [129, 130] was later extended to include interactions between an active metallic component and any support materials that exhibit similar phenomena to that observed by Tauster. Generally speaking, the phrase was used for catalyst systems consisting of active metal particles on reducible support [131], because the interaction under reducing conditions can induce an electronic effect (charge transfer between support and metal particles), changing the chemisorption capacity of the active phase, and/or preventing the interaction between the gas molecules and the metal surface due to newly formed surface layer.
In 1997, Boyes and Gai [17] had observed the phenomena of catalyst deactivation caused by SMSI in an environmental high-resolution electron microscope (EHREM) at the atomic scale. Owing to the modification of apertures mounted inside the bores of the objective pole pieces rather than between them, the aberration coefficients could low down to 2 mm. Multi-directional resolution of atomic lattice planes with spacings in the range 0.34 (graphitic carbon)-0.23 nm (gold islands with (111) lattice) was routine. Figure 9.31a shows EHREM of finely dispersed Pt/TiO2. In situ dynamic catalyst activation in hydrogen at 300 °C is shown in Fig. 9.31b. Growth of an amorphous Ti–rich overlayer or coating, with a composition close to slightly anion-deficient TiO2 was observed (indicated at C, determined by EDX) on the surface of the same particle at 450 °C in Fig. 9.31c. In addition, there emerged some nm-scale single crystal clusters of Pt metal (arrowed) near the observed particle, and they were not in an epitaxial relationship with the substrate and not encapsulated by titania overlayers during the early development.

a EHREM of finely dispersed Pt/TiO2 prepared by impregnation of TiO2 with Pt solution. b In situ dynamic catalyst activation in hydrogen imaged at 300 °C. The 0.23 nm (111) lattice spacings were clearly resolved in the Pt metal particle (P). c The same particle of Pt (P) imaged at 450 °C, also in H2 (reproduced with permission from Ref. [17], Copyright 1997, Elsevier)
In 2016, Zhang et al. [132] described the atomic migration of a palladium/titania (Pd/TiO2) system, by combining ESTEM and DFT calculations, which visualized the formation of the overlayers at the atomic scale under atmospheric pressure and high temperature. As Fig. 9.32a shows, an amorphous layer started to form on the particles under reducing conditions (H2(5 vol%)/Ar at 1 atm) at 250 °C, arguably through the diffusion of reduced TiOx species from the support to the particle surface, starting from the metal–support boundary (arrow). When the temperature was increased to 500 °C under the same conditions, the amorphous layer crystallized to form a bilayer that was almost epitaxial with the Pd(111) plane (Fig. 9.32b and higher magnification images in C, D). The lattice spacing was ∼2.9 Å for the lower layer and 3.0 Å for the top layer, and the overlayer followed ABCABC stacking of the underneath Pd along [111] direction. The comparison between ABF (Fig. 9.32c) and HAADF (Fig. 9.32d), where the image contrast is the opposite and brightness in the latter case is proportional to atomic number, confirmed that the surface layer was composed of a material with lower atomic number than Pd.

Formation of the TiOx overlayer on Pd nanocrystal in Pd/TiO2. Sequential in situ observations, under reducing conditions (H2(5 vol%)/Ar at 1 atm) of the Pd/TiO2 sample at 250 °C (a), then 500 °C for 10 min (b); c, d are higher magnification ABF and HAADF images, respectively, of a section of part (b) showing the TiOx double layer (reproduced with permission from Ref. [132], Copyright 2016, American Chemical Society)

In situ STEM images of Rh/TiO2 after treatment in 5% H2 and 95% N2 at 550 °C for 10 min (a), which induced the formation of a TiOx SMSI crystalline bilayer, containing exclusively Ti3+, and after treatment in 20CO2:2H2 at 250 °C for 3 h (b), which caused the formation of an amorphous A-SMSI overlayer, containing a mixture of Ti3+ and Ti4+ (reproduced with permission from Ref. [133], Copyright 2017, Springer Nature)
Generally speaking, common SMSI overlayers as mentioned before are crystalline, while there exists special case in which environmental gas participated in the formation of end product, and resulted in amorphous overlayer. Matsubu et al. [133] proposed adsorbate-mediated SMSI (A-SMSI) encapsulation state that formed with the treatment of TiO2-supported Rh NPs in CO2–H2 (CO2-rich) environments at temperatures of 150–300 °C. According to the conditions known to form the traditional SMSI state, a crystalline bilayer of TiOx quickly formed as a conformal coating on large crystalline Rh particles, with Ti exclusively in the Ti3+oxidation state (Fig. 9.33a). In contrast, after 20CO2: 2H2 treatment for three hours at 250 °C, an amorphous overlayer on Rh was observed to form (Fig. 9.33b). In situ electron energy-loss spectroscopy (EELS) measurements with a 1.0–1.5 Å spot size focused at various locations on the overlayer directly proved that Ti existing in a combination of Ti3+ (∼30%) and Ti4+ (∼70%) oxidation states in the amorphous overlayer. Differences in Ti oxidation state for the traditional SMSI and A-SMSI overlayers were hypothesized to result from the presence of HCOx in the A-SMSI overlayer and to be related to the unique reactivity and stability of the A-SMSI overlayer in humid CO2-reduction conditions.
Besides the common surface structure fluctuation and reshaping of nanomaterials in catalyzation, there exist some extremely subtle structural changes during the real-time catalytic process. To reveal the structure–activity relationship, the most direct method is to directly observe the catalytic reaction occurring on the catalyst surface at the atomic level. In 2020, Yuan et al. [134] visualized the gas molecules that adsorb and react dynamically at the specific structure with the employment of spherical aberration-corrected ETEM. Normally, the spatial distribution of active sites on the catalyst surface is disordered, and the molecules adsorbed on the sites do not offer sufficient contrast for TEM identification. To solve this issue, they came up with a strategy to construct a highly ordered active row to enhance the image contrast. They took advantage of the highly ordered four-coordinated Ti(Ti4c) rows on the anatase TiO2 (1 × 4)-(001) surface to facilitate enhanced contrast of adsorbing molecules along the row direction and allow real-time monitoring of H2O species dissociating. As shown in Fig. 9.34a, the reconstructed TiO2 (1 × 4)-(001) surface was obtained after heating in O2, in which the protruding black dots represent the Ti4c rows. Then O2 gas was evacuated and H2O vapor was introduced (Fig. 9.34b). With the raising H2O pressure, two additional small protrusions were observed at the top of the Ti4c rows, and became clearly visible (in Fig. 9.34d). Combined with in situ Fourier transform infrared spectroscopy (FTIR) and density functional theory (DFT), the researchers attributed the twin protrusions to the adsorbed water species, which was composed of two different hydroxyl species. Since TiO2 can catalyze the water gas shift reaction (H2O + CO → H2 + CO2) at elevated temperatures, the researchers introduced CO into the ETEM column after the formation of twin protrusions. They found that the twin-protrusion structure changed dynamically between blur and clearness (Fig. 9.34e, f), which indicated that the adsorbed hydroxyls were reacting with CO molecules, and the Ti4c sites are the reaction sites.

a–d Aberration-corrected in situ ETEM images show the same area of TiO2 (001) surface at 700 °C under oxygen [a 0.001 mbar] and water vapor [b 0.01 mbar; c 1 mbar; d 2.5 mbar] conditions, scale bar, 1 nm; e Sequential ETEM images acquired in the mixed gas environment (1:1 ratio of CO and H2O vapor; gas pressure:5 mbar; temperature: 700 °C), viewed from the [010] direction. Scale bar, 2 nm. f Enlarged ETEM images show the dynamic structural evolution of the Ti row outlined by the dotted rectangle in (e). Scale bar, 0.5 nm (reproduced with permission from Ref. [134], Copyright 2020, The American Association for the Advancement of Science). g and h Aberration-corrected in situ ETEM images of a Au NP supported on a TiO2 (001) surface in g a high-pressure oxygen (6.5 mbar) environment, and h a CO oxidation (total pressure: 4.4 mbar, \({V}_{{O}_{2}}\): \({V}_{CO}\) = 1:2) environment, scale bar, 2 nm; i and j Aberration-corrected in situ ETEM images show the structural evolution of the Au–TiO2 (001) nanocatalyst from an oxygen environment [i 5 mbar] to a reactive environment [j 500 °C, 5 mbar, \({V}_{{O}_{2}}\): \({V}_{CO}\) = 1:2)], scale bar, 2 nm (reproduced with permission from Ref. [135], Copyright 2021, The American Association for the Advancement of Science)
Another challenging and unexpected interface structure change was explored in 2021 by Yuan et al. as well [135]. Supported nanoparticles are widely used as catalysts for heterogeneous reactions, and the active sites in many reactions are located at the perimeter interface (PI), which is generally considered to be rigid. However, by employing the aberration-corrected ETEM (same as the former apparatus), they found that Au nanoparticles could rotate reversibly on the TiO2 (001) surface in different gas environments. Furthermore, by controlling the external oxidative/reductive gaseous environments and temperature, the researchers realized the in situ manipulation of the atomic configuration of the active Au–TiO2 interface. In brief, a perfect epitaxial relationship was found at the interface between the Au NP and the TiO2 (001) interface at high oxygen pressure (Fig. 9.34g, i), while the Au NP rotated by a small angle along the axis perpendicular to the TiO2 (001) surface with the introduction of CO (\({V}_{{O}_{2}}\): \({V}_{CO}\) = 1:2) (Fig. 9.34h, j). The rotation was so subtle that the whole process couldn’t be observed and monitored without the possibility and convenience provided by the ETEM. This result suggests that real-time design of the catalytic interface in operating conditions may be possible.
9.3.5.2 Sintering and Dispersion
Catalyst deactivation was a major concern in many industrial processes, besides SMSI in a specific gaseous environment; sintering was another typical reason which could account for it. The active sites are usually related to the surface area or the specific size, while sintering always result in the decrease of the surface area and changing the size of the catalyst particles. Furthermore, the sintering of supported NP catalysts occurs in various applications, ranging from high-temperature automotive exhaust emission controls to low-temperature fuel cells, made it an urgent demand to stabilize the catalyst distribution, lifting the commercial value. ETEM could provide direct evidence about size/distribution/morphology of NPs, further the dynamic evolution of these information under gaseous environment.
There are two widely accepted sintering mechanisms [130, 136,137,138]: (a) Ostwald ripening (OR) [139], in which the larger particles grow at the expense of small ones, by the diffusion of atoms/clusters due to a difference in chemical potential depending on NP size/surface curvature; (b) Particle migration and coalescence (PMC) [140], in which the independent particles migrated on the substrate surface in a Brownian-like motion and coalesce to other encountered particles. Both mechanisms may co-exist during a real catalytic reaction, and the dominant one is determined by reaction conditions and catalyst characteristics, e.g. loading amount, and weak metal–support interaction (MSI) facilitate the PMC process while strong MSI avails the OR process.
As early as 1974, Baker et al. [141] has observed particle mobility on a graphite surface in a gaseous environment with their modified TEM. In 2004, Liu et al. [142, 143] studied the sintering of palladium NPs on Al2O3 during the catalyst regeneration process. Fresh Pd particle catalyst was located on the Al2O3 surface and sintering occurred in 500 mTorr of steam at 700 °C, via traditional ripening (OR) and coalescence (PMC) mechanisms. As shown in Fig. 9.35a–c, particles marked 1 gradually shrunk in size and finally disappeared, while particles marked 2 coalesced into one. As for the used catalyst, which deactivated apparently due to the coverage of a complex mixture of unsaturated aliphatic hydrocarbons (HC in Fig. 9.35d), Pd particles moved through the hydrocarbon, leaving light contrast tracks in their wake (black arrow in Fig. 9.35e) in 500 mTorr of steam at 350 °C. In this process, the Pd particles catalyzed the gasification of green oil residue, meanwhile the exothermic nature of the reaction raised the temperature and facilitated the coalescence of Pd particles even at much lower temperatures.

a–c Fresh Pd/Al2O3 catalyst a in the as-received condition (RT); after heating in 500 mTorr steam at 700 °C for b 1 h; c 3 h. d–f Used Pd/Al2O3 catalyst heating in 500 mTorr steam at 350 °C for d 0 h, e 1 h, f 2 h (reproduced with permission from Ref. [142], Copyright 2005, Elsevier)
In 2012, Hansen et al. [138] proposed that the sintering process could be divided into three phases via reviewing and analyzing recent in situ observations (Fig. 9.36). Phase I involved rapid loss in catalyst activity (or surface area), phase II was where sintering slows down, and phase III was where the catalyst may reach a stable performance. The experimental data suggested that, in phase I, very little PMC was observed while the disappearance of the smallest particles (OR) was probably responsible for the obvious declining activity. In phase II, sintering slowed down since the small particles already disappeared, and during this stage both OR and PMC could be observed. In phase III, the particles had grown large and been very stable. In this phase, particle migration slowed down, and some other parasitic phenomena, such as support restructuring, turned out to be important or influential, especially at high temperatures. Considering several technical and chemical improvements related to changes in the composition of the NPs or changes in support surface structure, morphology, and chemistry, they endorsed the viewpoint proposed by Moulijn et al. [144], which the movement of atoms holds more weight accounting for the sintering mechanism.

The sintering process could be divided into three phases according to the change of the catalyst activity, and OR could happen during all three phases while PMC mainly occurred during phases II and III (reproduced with permission from Ref. [138], Copyright 2013, American Chemical Society)
As a model system for automotive exhaust abatement, Pt NPs dispersed on a planar, amorphous Al2O3 support were monitored during the exposure to 10 mbar air at 650 °C under in situ TEM by Simonsen et al. in 2010 [145]. Time-resolved image series unequivocally revealed that the sintering of Pt NPs was mediated by an Ostwald ripening process. Benavidez and coworkers [146] performed an ETEM study of Pd/carbon and Pt/SiO2 under high temperatures as well. Anomalous growth patterns were detected wherein some particles grew much larger than others, and they were not caused by random migration and coalescence of particles. The dominant process leading to particle growth was OR, the same as the former research.
Impressive advances had been realized by in situ TEM in several supported catalyst systems, while most of them still focused on determining the dominating sintering mechanism by statistical analysis of particle size evolution from the top view, whose dynamics would respond to environmental and support change. Yuan et al. [147] obtained the atomic-scale information of the interface between metal and support from a side view with the combination of AC-STEM and ETEM. The researchers loaded Au NPs on two typical anatase supports, well-defined Au–TiO2 (101) and Au–TiO2 (001), through impregnation and in situ annealing methods. As shown in Fig. 9.37a, the typical HAADF-STEM image viewed along TiO2 [010] direction exhibited a round-shape Au NP with a small contacting interface, and an incoherent relationship between the NP and support. In contrast, a preferential contacting relationship that Au–TiO2 (001) interface shared the plane with Au (111) was observed in Fig. 9.37b, with a relatively larger interface. Then the samples were heated to 500 °C in an oxygen environment (5 × 10−2 Pa). The Au NPs on the Au–TiO2 (001) surfaces were stable, which was confirmed from both the top view (Fig. 9.37c, d) and the side view (Fig. 9.37e, f) even after several thousand seconds. Though the minimum distance of two particles was less than 0.46 nm, almost all of the particles were firmly attached to the support and kept unchanged. Strong interaction between Au NPs and TiO2 (001) surfaces was obvious. During the sintering of Au–TiO2 (101) catalysts, both PMC and OR processes were observed, as Fig. 9.37g–j shows. The seven particles were labeled from 1 to 7, respectively, then with time going by, initially connected particles 4 and 5 started to coalesce with the diminution of adjacent particle 6. The tendency developed steadily until one large particle was left at the cost of the disappearance of particle 6. It should be noted that the contacting area is still small, indicating a weak interfacial interaction. This work emphasized the facet-dependent sintering behaviors, proposing a feasible method of choosing a suitable support to prevent sintering.

a The HAADF-STEM image shows the typical interface between Au NP and TiO2 (101) surface. b The TEM image shows the typical interface between Au NP and TiO2 (001) surface. c–f The serial TEM images show the sintering behavior of Au NPs on the TiO2 (001) surface, both from top view (c and d) and side view (e and f). The red dashed circles show the initial states of the Au NPs. g–j The serial ETEM images show the sintering behavior of Au NPs on the TiO2 (101) surface. The red and blue dashed circles show the initial and changed states of the Au NPs. The green arrows show the moving direction of the Au NPs (reproduced with permission from Ref. [147], Copyright 2018, John Wiley and Sons)
In 2009, Amama et al. [148] demonstrated that the Ostwald ripening behavior of Fe catalyst films deposited on thin alumina supporting layers was a function of thermal annealing in H2 and H2/H2O. The introduction of H2O to the catalysis of CNT was proposed by Iijima’s group as long ago as 2004 [149], as a prospective agent against amorphous carbon coating, to enhance the activity and lifetime of the catalysts. Amama considered that the addition of H2O inhibited OR through the ability of oxygen and hydroxyl species to reduce diffusion or migration rates of catalyst atoms from one catalyst to another across the sample substrate, on the basis of the ex situ experiment.
Besides the sintering behaviors of tiny clusters (<1 nm) and NPs (<3 nm), the dispersion of a single atom was also recorded using TEM/ETEM/ESTEM. Atomically dispersed catalyst species was reported by Iwasawa et al. [150] as early as 1999 with the usage of EXAFS. In 2011, Zhang et al. [151] proposed the concept, a single-atom catalyst (SAC) that consists of only isolated single atoms anchored to the surface. Normally, the isolated single atoms still possess chemical bonds with surfaces or exist charge transfer. Actually, the SAC inspires an approach to optimize the thermal stability of the catalysts and attracted extensive attention in just a few years [152,153,154,155]. In 2012, Yoshida et al. [156] studied dynamic behaviors of Pt single atoms on amorphous carbon without gaseous introduction. And then they characterized Pt/amorphous carbon (Pt/a-carbon) electrode catalysts in different atmospheres [157], such as hydrogen and air, and a conventional high vacuum of 10–5 Pa. The real-time visualization of atomic behaviors during crystal growth, such as coalescence and surface reconstruction, were acquired with AC-ETEM. Boyes et al. [28] focused on the same area. They demonstrated the dynamic ESTEM imaging of single atoms detected “loose” on the carbon support in 0.02 mbar hydrogen gas at 25, 400, and 500 °C. With the advanced in situ ETEM techniques, the SACs and their dynamics during catalysis could be captured [158, 159].
In most experiments, SACs were stabilized by diverse defects, which limited the fabrication and development of high-metal-loading and thermally stable SACs. In 2019, Lang et al. [160] developed the method that isolated Pt atoms can be stabilized through a strong covalent metal–support interaction, which utilizes the crucial oxide reducibility of support. Firstly, a 1.8 wt% Pt/FeOx catalyst was prepared (denoted as Pt1/FeOx), and subsequently calcined at 800 °C in air for 5 h (denoted as Pt1/FeOx-C800). AC-HAADF-STEM was used to compare the nature of Pt species between two materials. As Fig. 9.38a, b and c, d shows, there only existed isolated Pt atoms dispersed on the support, and there were no Pt nanoclusters or NPs observed even though the sample had experienced 800 °C calcination. Following is the designed in situ ETEM experiment. Colloidal Pt NPs were supported on Fe2O3 at a 1 wt% loading (denoted as 1Pt/Fe2O3-NP). Prior to calcination, about 300 Pt particles with an average particle size of about 3 nm were visible across a randomly picked Fe2O3 support (Fig. 9.38e, f). After heating to 800 °C under a flow of 1 bar O2 for 20 min, the total number of Pt NPs decreased to ~200, since many of the 2–3 nm Pt NPs shrank/or disappeared entirely (Fig. 9.38g, h). Therefore, the researchers proposed that the Fe2O3 support could facilitate the dispersion of Pt NPs and transform into isolated Pt atoms by high-temperature calcination. Three factors which played crucial roles in the dispersion were concluded as high temperature to promote Pt mobility; the presence of molecular O2 to partially oxidize the surface of Pt NPs, thereby enabling vaporization of (mobile) PtO2; and a strong interaction between support and surface Pt atoms.

a, b AC-HAADF-STEM images of Pt1/FeOx and c, d Pt1/FeOx-C800, highlighting atomically dispersed Pt (circled in b). 5 nm scale bar for panel a, 2 nm scale bars for b, d, and 100 nm scale bar for c. e–f HAADF-STEM images of 1Pt/Fe2O3-NP before, and g–h after in situ calcination at 800 °C under 1 bar flowing O2 for 20 min: the yellow squares in panels e and g show the same sample area. A 50 nm scale bars in e and g and 10 nm scale bars in f and h (reproduced with permission from Ref. [160], Copyright 2018, Springer Nature)
The isolated atoms sometimes could coalesce under a specific gaseous environment, or even possess cycles of coalescing to NPs and dissolving back to single atoms. Dai et al. [161] demonstrated the cyclical precipitation–dissolution of Rh NPs in response to redox cycling of the ambient gas. These dynamic evolutions must be studied with the employment of in situ ETEM or gas-cell system to monitor the whole process, or else we could only obtain the ex situ information of the catalyst which easily caused a misunderstanding due to the change of gaseous environment and temperature, and the real catalytic factors or components could not be revealed anymore.
9.3.5.3 Photocatalysis
As an important catalytic process widely used in sustainable energy development, photocatalysis attracted tremendous attention. Considering the outstanding ability of ETEM for real-time observation, introducing light in the ETEM would facilitate the understanding of the photocatalytic mechanism at the atomic scale.
Two mainstream methods allow the introduction of light irradiation into the ETEM nowadays, modifying the specimen holder or the TEM column.
Generally, the modifications of the TEM column have been done using traditional optics [162] (reflective surfaces, lenses, ellipsoidal and/or mirrors, etc.) or optical fibers [163,164,165,166,167]. Actually, as early as 1984, Suzuki et al. [163] had used optical fiber to direct 514.5 nm light from a 4 W Ar-ion laser into the specimen chamber to study dislocation motion in II–VI compounds. In 2004, Yoshida et al. [164] employed the same design to study the photodecomposition process of poly-hydrocarbons on TiO2 catalyst films. The intensity of the light was measured to be 10 mW/cm2 (λ = 360 nm) on the sample plane. This system enabled researchers to change wavelength and intensity by changing the light source easily. With UV light, they observed the decomposition phenomenon of the organic materials, which was considered to be a distinct photocatalytic process catalyzed by TiO2 thin films.
Miller and Crozier [165,166,167] devised a system for in situ illuminating a sample with visible and UV light inside a TEM with the employment of optical fiber as well. The fiber enters the ETEM in a direction perpendicular to the sample rod, and has no physical contact with the sample holder. This configuration allows for movement, tilting, heating, and cooling of the sample with little impact on the optical fiber. They balanced various considerations and showed no notable detrimental effect on the microscope performance while illuminating the sample with over 1 W/cm2 of broadband UV and visible light. Zhang [168] employed the system in the same year to investigate the surface structure of anatase nanocrystals under the conditions of photocatalytic splitting of water (Fig. 9.39a). When the titania was exposed to light and water vapor, the initially crystalline surface converted to an amorphous phase with 1–2 monolayers thick (Fig. 9.39b, c).

a Schematic diagram on in situ TEM showing gases flowing into the sample chamber from an external mixing tank, and exiting through differential pumping apertures in objective lens pole pieces. The temperature is controlled with a heating holder. Light illumination is provided by an optical fiber which is supported by a brass tube perpendicular to the sample rod; the fiber tip is cut at an angle to direct the light onto the sample. b Fresh anatase particles after 40 h in H2O gas at 150 °C, light exposure 12 h; c magnified images of the (101) and (002) surfaces after illumination. The image was taken in 20 s including adjusting the focus (reproduced with permission from Ref. [168], Copyright 2013, American Chemical Society)
The method based on traditional optics evolved slightly slower. In 1995, Ohno and Takeda [162] developed an apparatus of the side-entry type that enabled laser beam introduction using a reflection mirror, optical window, and lens with the modification of a JEOL 2000EX TEM. Thus, the specimen could be illuminated simultaneously by both electron and laser beams.
Picher et al. [169] proposed another approach which used a parabolic mirror to focus the light from an external source and simultaneously collect the sample response in 2014. A free-space, broadband (wavelength λ > 200 nm), high-efficiency (1.5 sr) light delivery and collection system is employed, which is independent of the TEM sample holder. The insertion of a parabolic mirror between the sample holder and the lower objective pole piece of the ESTEM satisfied the delivery and collection of light. Light reaches the parabolic mirror along a free-space beam path through a viewport on a hollow rod into the vacuum system, then be focused on the sample. At the same time, due to the collection of light information of the parabola itself, multiple types of spectroscopy analyses could be integrated into the system, such as Raman spectroscopy. Therefore, it allows the simultaneous collection of microscale spectroscopy data and nanoscale ESTEM images to complement atomic-scale information with a large-scale picture of the structure and kinetics, mitigating the need for two separate in situ measurements. This rod-mirror assembly replaces the objective aperture holder, and both of them are easily interchangeable.
In general, the introduction of light by both optical fiber through a separate port in TEM and employing various mirrors based on traditional optics involves the modification of the microscope, which limited the use of the current system to a single specific microscope while maintaining holder flexibility. Oppositely, the holder-based designs could be used in different microscopes.
Shindo et al. [170] modified double-probe piezo-driving specimen holder by introducing a laser irradiation port in one of two arms. As a result, the new specimen holder consists of a piezo-driving probe and a laser irradiation port, both of which can be 3-dimensionally controlled by using piezoelectric elements and micrometers. While the piezo-driving probe interacts with the specimen set in the holder in several ways, the laser beam causes photo-induced phenomena to occur.
Cavalca [171, 172] followed the same modifying idea of the specimen holder, and developed two novel types of holders that enable in situ illumination: lens-based holder design and fiber-based holder design. The former design comprises a standard TEM specimen holder with a customized tip, whose custom parts can be adapted easily to other holder shafts. The fiber-based design was constructed using a feed-through equipped with single or multiple multi-mode optical fibers, which might be highly desirable for a study of the optical properties of nanomaterials in the future. Cavalca performed the photo-induced degradation of cuprous oxide (Cu2O) experiment using both TEM holder designs, and yielded the same result. Water vapor was leaked into the specimen chamber to a steady-state pressure of ~3 mbar and the sample was exposed to light with λ = 405 nm and a power density of 6 W cm−2 for various time periods. The column was then evacuated to 10−6 mbar for 5 h in order to reduce the water vapor before the specimen was exposed to the e-beam. This procedure excluded the influences induced by e-beam. The morphology of particles evoluted considerably, with change in lattice spacing (electron diffraction patterns) and oxidation state (electron energy loss spectroscopy). A more detailed and complete analysis of the reduction reaction was finished in 2013 [172].
9.4 Conclusions and Outlook
Over the past decades, the introduction of gases in the TEM truly changed the traditional post mortem characterization limited to the vacuum environment. With the developing accessory technologies (such as aberration corrector and microelectromechanical system) and continuously updated design of the TEM, the research boundary expanded gradually and steadily, ranging from structural characterization to atomic-scale activity and property of materials. Accordingly, the various, demanding and stricter restrictions in different exploring fields promoted the improvements in accessory technologies and design.
There is no doubt that there exist universal and concurrent pursuits in almost all TEM research areas, while specific and unique requirements emerged during special experiments. In general, the requirements could be classified as follows.
9.4.1 Spatial and Temporal Resolution
With the incorporation of a monochromator and aberration corrector (probe/image), the spatial and energy resolution in commercially available ETEM already reached the criterion that satisfied many applications. For example, the Themis ETEM (ThermoFisher), which is combined with image Cs corrector, Thermo Scientific X-FEG module, and monochromator technology, could reach the TEM point resolution of 0.12 nm, while the system energy resolution could minimize to 0.25 eV, with the N2 pressure <0.5 mbar. As for the window approach, advancing processing technology for the generation of ultrathin windows could minimize the scattering of electrons, further improving the spatial resolution. Among ultrathin graphene, graphene oxide, and boron nitride membrane as specimen supports for TEM developed in recent years, graphene membrane stands out with an atomic thickness of only one carbon atom used in a liquid cell.
However, the spatial resolution would decline to some extent on account of the scattering of gas molecules doubtless, which is influenced by the pressure and variety of gas as well. Meanwhile, the maximum pressure reported in the literature published is 5 bar (window mode), which is still far below some real reaction conditions. Therefore, the balance between gas pressure and spatial resolution still should be paid enough attention to keep, no matter what kind of methods are employed, e.g. improvement of aperture design.
Another more serious obstacle laid on the temporal resolution due to the demand for the observation across a range of time scale, especially for ultrafast time scale. The employment of ETEM in in situ experiments is usually prepared for monitoring and recording the whole dynamic reaction process, which probably happened in several milliseconds or even nanoseconds. The vital emphasis is the data collection or recording media, which is confined to the electron dose, detection quantum efficiency (DQE) of the camera, and information processability of the computer.
At present, in terms of recording attachment, the temporal and spatial resolution are contradictory performances. Available technologies usually satisfied one of them to sacrifice another one, for example, image acquisition systems allow high spatial resolution (<0.1 nm) images with low temporal resolution (0.033–0.2 s range) or low resolution (a few nanometers in single shot mode) with high temporal resolution [173]. The compromise between them restricts the applications in observing numerous quick chemical reactions.
One of the solutions to the dilemma is the direct electron camera, which is based on complementary metal-oxide semiconductor (CMOS) technology. As the indirect electron detection method, conventional charge-coupled device (CCD) cameras with a capability of 20 frames per second (fps) [174] would have to convert high-energy electrons to photons firstly, which encumbered the response speed. The direct electron camera omits the essential fluorescent coating in CCD, improving the electron collection efficiency a lot. The fps with correspondingly high DQEs of readout rates reached 103 (such as Gatan K3, 1500 frames per second at 5760 × 4092 pixels can be achieved) magnitude. The highly improved temporal resolution provided new possibilities in in situ experiments, while it’s still inadequate in observing transient nucleation events or intermediates during chemical reactions.
Regardless of the camera, another radical method is ultrafast TEM, in the time scale up to femtosecond/nanosecond theoretically. Two principal approaches have emerged in recent decades: the stroboscopic ultrafast electron microscope (4-dimensional ultrafast electron microscope, 4D UEM) [175,176,177] and the nanosecond-time-resolved single-shot instrument (Dynamic Transmission Electron Microscope, DTEM) [178].
4D UEM has expanded the depth and breadth in different application fields, including photon-induced near-field EM [179], diffraction patterns [180], EELS [181], tomography [182], cryo-EM [183], Lorentz imaging [184], imaging in liquid [185], etc. Using this method, temporal, and spatial resolution can be maintained at the optimum levels, but the fact that the specimen must be laser-pumped millions of times means that the process being studied must be perfectly reversible—the sample must recover from the excited state back to the ground state between shots. This limitation allows 4D UEM to study the perfectly reversible chemical and materials processes, but is unavailable to many of the structural phase transformations and the nucleation, growth and mobility of dislocations, voids, and even complex nanostructures [178]. In this regard, 4D UEM may not be appropriate for investigating catalytic reactions.
As for DTEM, since all the information is obtained in a single specimen drive event, this technique is able to measure irreversible and unique material events that could not be studied by the stroboscopic approach. However, the limitation of this method is that space-charge effects in the beam can lead to degradation in resolution, and the essential high current will limit the overall temporal and spatial resolution of the instrument as well [186,187,188,189].
The stroboscopic and single-shot approaches to high-time-resolution in situ TEM are complementary, based on similar physical principles but in different regimes, and occupy specific superiorities, respectively. Limited to the electron–electron interactions and the brightness of the electron source, the spatial and temporal resolutions of the single-shot technique are in the nanometer and nanosecond scale, compared with the sub-picosecond time scale of the stroboscopic approach [189]. The stroboscopic approach is restricted to highly reversible processes, and limited by the emittance which determines the spatial coherence obtainable in a given spot size, which affects contrast and resolution. The detailed interpretation could be consulted in Chap. 10, 4D Ultrafast TEM.
9.4.2 Multiple Stimuli and Characterization Techniques
There is no doubt that researchers always endeavor for observation of the most realistic status of the specimen under various conditions, simulating the actual performance in operating conditions. The ETEM possesses extraordinary prospects in the visualization of dynamic changes in morphology, structure, and chemistry of materials at the atomic scale, especially when it is employed with external stimuli (strain/heating/cooling/electrical biasing/lighting).
Considering the varying stimuli probably induce different manifestations of the specimen, it would naturally take the other common characterization methods into the incorporation of ETEM. A combination of in situ ETEM and other characterization techniques, e.g. gas chromatography, quadrupole mass spectroscopy, in situ spectroscopy (infrared/ultraviolet–visible near-infrared/Raman spectroscopy), in situ XRD, in situ nuclear magnetic resonance (NMR) spectroscopy, and in situ XPS, in situ XAS, would be the fast-growing and fascinating area in the future.
It is clear that spectroscopy was paid a lot of attention, for its outstanding complementary ability to ETEM: (1) the ETEM focused on changes in a small area while the spectroscopy could give a whole overview of the sample to ensure the local effects are representative, or elucidate the specific situation happened actually; (2) the ETEM could observe the morphologic and structural changes, but is slightly weaker in chemical component analysis (EDS and EELS focus on subtle information), which is exactly the superiority of spectroscopy. Definitely, the spatial and temporal resolution of the spectroscopy is another challenge for the synchronous acquisition of information with the TEM images.
As for electron-beam-sensitive materials, the solution which could degrade the electron beam irradiation damage is the pursuit of researchers all the time. Clearly, the diffraction mode is the most common method which satisfies real-time dynamic crystallographic characterization of structure with much lower electron dose interacted with a specimen. Nowadays, no matter whether in ETEM or in a gas-cell holder, the diffraction capability is reserved well and is employed to characterize the beam-sensitive specimen still.
However, since the diffraction methods provided the average results over materials, another technique named as integrated differential phase contrast (iDPC) STEM [190, 191] emerged and developed well in recent years. The iDPC-STEM enabled the linear imaging of the projected electrostatic potential in a lattice, and the resulting contrast is nearly proportional to the atomic number Z instead of its square in the high angle annular dark field (HAADF) STEM, greatly improving the capacity of light-element imaging. Intriguingly, the iDPC technique allowed the researchers [192] to image the adsorbed molecules with a high resolution and signal-to-noise ratio under an ultra-low beam current. The lower damage allows continuous images of the same area so that the temporal changes of structures could be investigated, especially in studying various porous catalysts [193] and inside hydrocarbon pools.
Furthermore, the introduction of gas in TEM sometimes could compensate the electron irradiation damage for specific materials in continuous characterization, e.g. Yuan et al. [73] introduced the protection gas (O2) in the ETEM chamber to compensate the lost oxygen atoms in TiO2, considering the surface structures can be easily damaged by the electron beam. By analogy, the specific gas which could facilitate the repair of the irradiation damage for electron-sensitive materials, especially the oxides, satisfies the requirement of real-time dynamic continuous characterization at the atomic scale in TEM. In other words, the introduction of gas weakened the influence imposed on the specimen by the electron beam to some extent, which provided a possible solution for the characterization of electron-beam-sensitive materials.
9.4.3 Accurate Process Control
It has been a focused problem for a long time since the TEM was invented and would be a continuous problem in the following development: the accurate manipulation of the specimen and external stimuli, including sample position (avoidance of sample drift), uniformity, temperature (enough high/low temperature, and fast real-time rate of temperature change), and gas flux.
The more complex the system designed and the more equipment involved, it would be more difficult to obtain the accurate parameters or control the whole reactor/system accurately, especially when they interacted and influenced themselves. The external stimuli aggravated the situation, especially when the extreme environment is considered to realize in the TEM, e.g. corrosive gaseous environment and multi-stimuli incorporated holder. Definitely, there are some efforts directed to mitigate the problem but a universally available solution is still to be realized.
The electron beam irradiation is also a vital question for TEM characterization. It should be seriously considered, especially for electron-beam-sensitive materials, such as zeolites and metal–organic frameworks. The approach, which would entirely eliminate the influence of the electron beam, or rational model established to deduce and evaluate the influential extent imposed on the specimen by the electron beam, is the pursuit of the researchers.
References
Knoll M, Ruska E (1932) Beitrag zur geometrischen Elektronenoptik. I. Annalen der Physik 404(5):607–640. https://doi.org/10.1002/andp.19324040506
Knoll M, Ruska E (1932) Das Elektronenmikroskop. Z Phys 79(9):699. https://doi.org/10.1007/bf01330526
Ruska E (1942) Beitrag zur übermikroskopischen Abbildung bei höheren Drucken. Kolloid-Zeitschrift 100(2):212–219. https://doi.org/10.1007/bf01519549
Abrams IM, McBain JW (1944) A closed cell for electron microscopy. J Appl Phys 15(8):607–609. https://doi.org/10.1063/1.1707475
Bailey JE (1963) On the oxidation of thin films of zirconium. J Nucl Mater 8(2):259–262. https://doi.org/10.1016/0022-3115(63)90042-4
De Jonghe LC, Thomas G (1971) High voltage electron microscopy studies of phase transformations in cobalt ferrites. Mater Sci Eng 8(5):259–274. https://doi.org/10.1016/0025-5416(71)90092-9
Ito T, Hiziya K (1958) A specimen reaction device for the electron microscope and its applications. J Electron Microsc 6(1):4–8. https://doi.org/10.1093/oxfordjournals.jmicro.a051252
Heide HG (1962) Electron microscopic observation of specimens under controlled gas pressure. J Cell Biol 13(1):147–152. https://doi.org/10.1083/jcb.13.1.147
Heide HG (1960) Elektronenmikroskopie von Objekten unter Atmosphärendruck oder unter Drucken, welche ihre Austrocknung verhindern. Naturwissenschaften 47(14):313–317. https://doi.org/10.1007/bf00628703
Hashimoto H, Naiki T, Eto T, Fujiwara K (1968) High temperature gas reaction specimen chamber for an electron microscope. Jpn J Appl Phys 7(8):946–952. https://doi.org/10.1143/jjap.7.946
Hashimoto H, Tanaka K, Yoda E (1958) A specimen treating device at high temperature for the electron microscope. J Electron Microsc 6(1):8–11. https://doi.org/10.1093/oxfordjournals.jmicro.a051258
Baker RTK (1979) In situ electron microscopy studies of catalyst particle behavior. Catal Rev 19(2):161–209. https://doi.org/10.1080/03602457908068055
Fujita H, Komatsu M, Ishikawa I (1976) A universal environmental cell for a 3MV-class electron microscope and its applications to metallurgical subjects. Jpn J Appl Phys 15(11):2221–2228. https://doi.org/10.1143/jjap.15.2221
Baker RTK, Harris PS (1972) Controlled atmosphere electron microscopy. J Phys E: Sci Instrum 5(8):793–797. https://doi.org/10.1088/0022-3735/5/8/024
Rodriguez NM, Oh SG, Downs WB, Pattabiraman P, Baker RTK (1990) An atomic oxygen environmental cell for a transmission electron microscope. Rev Sci Instrum 61(7):1863–1868. https://doi.org/10.1063/1.1141109
Lee TC, Dewald DK, Eades JA, Robertson IM, Birnbaum HK (1991) An environmental cell transmission electron microscope. Rev Sci Instrum 62(6):1438–1444. https://doi.org/10.1063/1.1142464
Boyes ED, Gai PL (1997) Environmental high resolution electron microscopy and applications to chemical science. Ultramicroscopy 67(1):219–232. https://doi.org/10.1016/S0304-3991(96)00099-X
Creemer JF, Helveg S, Hoveling GH, Ullmann S, Molenbroek AM, Sarro PM, Zandbergen HW (2008) Atomic-scale electron microscopy at ambient pressure. Ultramicroscopy 108(9):993–998. https://doi.org/10.1016/j.ultramic.2008.04.014
Yokosawa T, Alan T, Pandraud G, Dam B, Zandbergen H (2012) In-situ TEM on (de)hydrogenation of Pd at 0.5–4.5 bar hydrogen pressure and 20–400 °C. Ultramicroscopy 112(1):47–52. https://doi.org/10.1016/j.ultramic.2011.10.010
Allard LF, Overbury SH, Bigelow WC, Katz MB, Nackashi DP, Damiano J (2012) Novel MEMS-based gas-cell/heating specimen holder provides advanced imaging capabilities for in situ reaction studies. Microsc Microanal 18(4):656–666. https://doi.org/10.1017/S1431927612001249
Alsem D, Salmon N, Unocic R, Veith G, More K (2012) In-situ liquid and gas transmission electron microscopy of nano-scale materials. Microsc Microanal 18(S2):1158–1159. https://doi.org/10.1017/S1431927612007647
Jinschek JR (2014) Advances in the environmental transmission electron microscope (ETEM) for nanoscale in situ studies of gas–solid interactions. Chem Commun 50(21):2696–2706. https://doi.org/10.1039/c3cc49092k
Hashimoto H, Tanaka K, Yoda E, Araki H (1958) Bundle structure of needle crystal of tungsten oxide. Acta Metall 6(8):557–559. https://doi.org/10.1016/0001-6160(58)90178-0
Fryer JR (1968) Oxidation of graphite catalysed by palladium. Nature 220(5172):1121–1122. https://doi.org/10.1038/2201121b0
Mills JC, Moodie AF (1968) Multipurpose high resolution stage for the electron microscope. Rev Sci Instrum 39(7):962–969. https://doi.org/10.1063/1.1683578
Boyes ED, Gai PL (2014) Visualising reacting single atoms under controlled conditions: advances in atomic resolution in situ environmental (scanning) transmission electron microscopy (E(S)TEM). C R Phys 15(2):200–213. https://doi.org/10.1016/j.crhy.2014.01.002
Gai PL, Boyes ED (2009) Advances in atomic resolution in situ environmental transmission electron microscopy and 1 Å aberration corrected in situ electron microscopy. Microsc Res Tech 72(3):153–164. https://doi.org/10.1002/jemt.20668
Boyes ED, Ward MR, Lari L, Gai PL (2013) ESTEM imaging of single atoms under controlled temperature and gas environment conditions in catalyst reaction studies. Ann Phys 525(6):423–429. https://doi.org/10.1002/andp.201300068
Boyes ED, Gai PL (2014) Aberration corrected environmental STEM (AC ESTEM) for dynamic in-situ gas reaction studies of nanoparticle catalysts. J Phys: Conf Ser 522:012004. https://doi.org/10.1088/1742-6596/522/1/012004
LaGrow AP, Ward MR, Lloyd DC, Gai PL, Boyes ED (2017) Visualizing the Cu/Cu2O interface transition in nanoparticles with environmental scanning transmission electron microscopy. J Am Chem Soc 139(1):179–185. https://doi.org/10.1021/jacs.6b08842
Kamino T, Yaguchi T, Konno M, Watabe A, Marukawa T, Mima T, Kuroda K, Saka H, Arai S, Makino H, Suzuki Y, Kishita K (2006) Development of a gas injection/specimen heating holder for use with transmission electron microscope. J Electron Microsc 54(6):497–503. https://doi.org/10.1093/jmicro/dfi071
Hansen TW, Wagner JB, Dunin-Borkowski RE (2010) Aberration corrected and monochromated environmental transmission electron microscopy: challenges and prospects for materials science. Mater Sci Technol 26(11):1338–1344. https://doi.org/10.1179/026708310x12756557336355
Shirai M, Hanawa A, Kikuchi H, Inada H, Matsumoto H (2019) In-situ observation of catalytic reaction in gas atmosphere using an aberration corrected STEM. Microsc Microanal 25(S2):1526–1527. https://doi.org/10.1017/S1431927619008365
Hanawa A, Kubo Y, Kikuchi H, Nakamura K, Shirai M, Inada H, Matsumoto H, Kawasaki M (2019) Evaluation of high-resolution STEM imaging advancement under gas-environment with open window MEMS holder and gas injection system. Microsc Microanal 25(S2):694–695. https://doi.org/10.1017/S1431927619004203
Hansen TW, Wagner JB (2014) Catalysts under controlled atmospheres in the transmission electron microscope. ACS Catal 4(6):1673–1685. https://doi.org/10.1021/cs401148d
Yaguchi T, Suzuki M, Watabe A, Nagakubo Y, Ueda K, Kamino T (2011) Development of a high temperature–atmospheric pressure environmental cell for high-resolution TEM. J Electron Microsc 60(3):217–225. https://doi.org/10.1093/jmicro/dfr011
de Jonge N, Bigelow WC, Veith GM (2010) Atmospheric pressure scanning transmission electron microscopy. Nano Lett 10(3):1028–1031. https://doi.org/10.1021/nl904254g
Parkinson GM (1989) High resolution, in-situ controlled atmosphere transmission electron microscopy (CATEM) of heterogeneous catalysts. Catal Lett 2(5):303–307. https://doi.org/10.1007/bf00770228
Creemer JF, Helveg S, Kooyman PJ, Molenbroek AM, Zandbergen HW, Sarro PM (2010) A MEMS reactor for atomic-scale microscopy of nanomaterials under industrially relevant conditions. J Microelectromech Syst 19(2):254–264. https://doi.org/10.1109/jmems.2010.2041190
Komatsu M, Mori H (2005) In situ HVEM study on copper oxidation using an improved environmental cell. J Electron Microsc 54(2):99–107. https://doi.org/10.1093/jmicro/dfi032
Giorgio S, Sao Joao S, Nitsche S, Chaudanson D, Sitja G, Henry CR (2006) Environmental electron microscopy (ETEM) for catalysts with a closed E-cell with carbon windows. Ultramicroscopy 106(6):503–507. https://doi.org/10.1016/j.ultramic.2006.01.006
Kawasaki T, Ueda K, Ichihashi M, Tanji T (2009) Improvement of windowed type environmental-cell transmission electron microscope for in situ observation of gas-solid interactions. Rev Sci Instrum 80(11):113701. https://doi.org/10.1063/1.3250862
Wu F, Yao N (2015) Advances in windowed gas cells for in-situ TEM studies. Nano Energy 13:735–756. https://doi.org/10.1016/j.nanoen.2015.03.015
Allard LF, Bigelow WC, Jose-Yacaman M, Nackashi DP, Damiano J, Mick SE (2009) A new MEMS-based system for ultra-high-resolution imaging at elevated temperatures. Microsc Res Tech 72(3):208–215. https://doi.org/10.1002/jemt.20673
Creemer JF, Santagata F, Morana B, Mele L, Alan T, Iervolino E, Pandraud G, Sarro PM (2011) An all-in-one nanoreactor for high-resolution microscopy on nanomaterials at high pressures. In: 2011 IEEE 24th International conference on micro electro mechanical systems, 23–27 Jan 2011, pp 1103–1106. https://doi.org/10.1109/memsys.2011.5734622
Xin HL, Niu K, Alsem DH, Zheng H (2013) In situ TEM study of catalytic nanoparticle reactions in atmospheric pressure gas environment. Microsc Microanal 19(6):1558–1568. https://doi.org/10.1017/s1431927613013433
Vendelbo SB, Elkjær CF, Falsig H, Puspitasari I, Dona P, Mele L, Morana B, Nelissen BJ, van Rijn R, Creemer JF, Kooyman PJ, Helveg S (2014) Visualization of oscillatory behaviour of Pt nanoparticles catalysing CO oxidation. Nat Mater 13(9):884–890. https://doi.org/10.1038/nmat4033
Jiang Y, Zhang Z, Yuan W, Zhang X, Wang Y, Zhang Z (2018) Recent advances in gas-involved in situ studies via transmission electron microscopy. Nano Res 11(1):42–67. https://doi.org/10.1007/s12274-017-1645-9
Hansen PL, Wagner JB, Helveg S, Rostrup-Nielsen JR, Clausen BS, Topsøe H (2002) Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science 295(5562):2053–2055. https://doi.org/10.1126/science.1069325
Cabié M, Giorgio S, Henry CR, Axet MR, Philippot K, Chaudret B (2010) Direct observation of the reversible changes of the morphology of Pt nanoparticles under gas environment. J Phys Chem C 114(5):2160–2163. https://doi.org/10.1021/jp906721g
Uchiyama T, Yoshida H, Kuwauchi Y, Ichikawa S, Shimada S, Haruta M, Takeda S (2011) Systematic morphology changes of gold nanoparticles supported on CeO2 during CO oxidation. Angew Chem Int Ed 50(43):10157–10160. https://doi.org/10.1002/anie.201102487
Jiang Y, Li H, Wu Z, Ye W, Zhang H, Wang Y, Sun C, Zhang Z (2016) In situ observation of hydrogen-induced surface faceting for palladium-copper nanocrystals at atmospheric pressure. Angew Chem Int Ed 55(40):12427–12430. https://doi.org/10.1002/anie.201605956
Zhang X, Meng J, Zhu B, Yu J, Zou S, Zhang Z, Gao Y, Wang Y (2017) In situ TEM studies of the shape evolution of Pd nanocrystals under oxygen and hydrogen environments at atmospheric pressure. Chem Commun 53(99):13213–13216. https://doi.org/10.1039/c7cc07649e
Chmielewski A, Meng J, Zhu B, Gao Y, Guesmi H, Prunier H, Alloyeau D, Wang G, Louis C, Delannoy L, Afanasiev P, Ricolleau C, Nelayah J (2019) Reshaping dynamics of gold nanoparticles under H2 and O2 at atmospheric pressure. ACS Nano 13(2):2024–2033. https://doi.org/10.1021/acsnano.8b08530
Zhang X, Meng J, Zhu B, Yuan W, Yang H, Zhang Z, Gao Y, Wang Y (2018) Unexpected refacetting of palladium nanoparticles under atmospheric N2 conditions. Chem Commun 54(62):8587–8590. https://doi.org/10.1039/c8cc04574g
Zhu B, Meng J, Yuan W, Zhang X, Yang H, Wang Y, Gao Y (2020) Reshaping of metal nanoparticles under reaction conditions. Angew Chem Int Ed 59(6):2171–2180. https://doi.org/10.1002/anie.201906799
Langmuir I (1918) The adsorption of gases on plane surfaces of glass, mica and platinum. J Am Chem Soc 40(9):1361–1403. https://doi.org/10.1021/ja02242a004
Zhu B, Xu Z, Wang C, Gao Y (2016) Shape evolution of metal nanoparticles in water vapor environment. Nano Lett 16(4):2628–2632. https://doi.org/10.1021/acs.nanolett.6b00254
Zhu B, Meng J, Gao Y (2017) Equilibrium shape of metal nanoparticles under reactive gas conditions. J Phys Chem C 121(10):5629–5634. https://doi.org/10.1021/acs.jpcc.6b13021
Meng J, Zhu B, Gao Y (2018) Shape evolution of metal nanoparticles in binary gas environment. J Phys Chem C 122(11):6144–6150. https://doi.org/10.1021/acs.jpcc.8b00052
Henry CR (2005) Morphology of supported nanoparticles. Prog Surf Sci 80(3):92–116. https://doi.org/10.1016/j.progsurf.2005.09.004
Gallegos EJ (1964) Gas reactor for hot stage transmission electron microscopy. Rev Sci Instrum 35(9):1123–1124. https://doi.org/10.1063/1.1718978
Koh AL, Gidcumb E, Zhou O, Sinclair R (2013) Observations of carbon nanotube oxidation in an aberration-corrected environmental transmission electron microscope. ACS Nano 7(3):2566–2572. https://doi.org/10.1021/nn305949h
Ajayan PM, Ebbesen TW, Ichihashi T, Iijima S, Tanigaki K, Hiura H (1993) Opening carbon nanotubes with oxygen and implications for filling. Nature 362(6420):522–525. https://doi.org/10.1038/362522a0
Tsang SC, Harris PJF, Green MLH (1993) Thinning and opening of carbon nanotubes by oxidation using carbon dioxide. Nature 362(6420):520–522. https://doi.org/10.1038/362520a0
Jeangros Q, Hansen TW, Wagner JB, Damsgaard CD, Dunin-Borkowski RE, Hébert C, Van Herle J, Hessler-Wyser A (2013) Reduction of nickel oxide particles by hydrogen studied in an environmental TEM. J Mater Sci 48(7):2893–2907. https://doi.org/10.1007/s10853-012-7001-2
Jeangros Q, Hansen TW, Wagner JB, Dunin-Borkowski RE, Hébert C, Van Herle J, Hessler-Wyser A (2014) Oxidation mechanism of nickel particles studied in an environmental transmission electron microscope. Acta Mater 67:362–372. https://doi.org/10.1016/j.actamat.2013.12.035
Yu J, Yuan W, Yang H, Xu Q, Wang Y, Zhang Z (2018) Fast gas-solid reaction kinetics of nanoparticles unveiled by millisecond in situ electron diffraction at ambient pressure. Angew Chem Int Ed 57(35):11344–11348. https://doi.org/10.1002/anie.201806541
Luo L, Su M, Yan P, Zou L, Schreiber DK, Baer DR, Zhu Z, Zhou G, Wang Y, Bruemmer SM, Xu Z, Wang C (2018) Atomic origins of water-vapour-promoted alloy oxidation. Nat Mater 17(6):514–518. https://doi.org/10.1038/s41563-018-0078-5
Huang X, Jones T, Fedorov A, Farra R, Copéret C, Schlögl R, Willinger M-G (2021) Phase coexistence and structural dynamics of redox metal catalysts revealed by operando TEM. Adv Mater 33(31):2101772. https://doi.org/10.1002/adma.202101772
Wang Y, Liu B, Xa Z, Zhang X, Miao Y, Yang N, Yang B, Zhang L, Kuang W, Li J, Ma E, Shan Z (2018) Turning a native or corroded Mg alloy surface into an anti-corrosion coating in excited CO2. Nat Commun 9(1):4058. https://doi.org/10.1038/s41467-018-06433-5
Yoshida H, Kuwauchi Y, Jinschek Joerg R, Sun K, Tanaka S, Kohyama M, Shimada S, Haruta M, Takeda S (2012) Visualizing gas molecules interacting with supported nanoparticulate catalysts at reaction conditions. Science 335(6066):317–319. https://doi.org/10.1126/science.1213194
Yuan W, Wang Y, Li H, Wu H, Zhang Z, Selloni A, Sun C (2016) Real-time observation of reconstruction dynamics on TiO2(001) surface under oxygen via an environmental transmission electron microscope. Nano Lett 16(1):132–137. https://doi.org/10.1021/acs.nanolett.5b03277
Bugnet M, Overbury SH, Wu ZL, Epicier T (2017) Direct visualization and control of atomic mobility at 100 surfaces of ceria in the environmental transmission electron microscope. Nano Lett 17(12):7652–7658. https://doi.org/10.1021/acs.nanolett.7b03680
Dai S, Hou Y, Onoue M, Zhang S, Gao W, Yan X, Graham GW, Wu R, Pan X (2017) Revealing surface elemental composition and dynamic processes involved in facet-dependent oxidation of Pt3Co nanoparticles via in situ transmission electron microscopy. Nano Lett 17(8):4683–4688. https://doi.org/10.1021/acs.nanolett.7b01325
Dai S, You Y, Zhang S, Cai W, Xu M, Xie L, Wu R, Graham GW, Pan X (2017) In situ atomic-scale observation of oxygen-driven core-shell formation in Pt3Co nanoparticles. Nat Commun 8(1):204. https://doi.org/10.1038/s41467-017-00161-y
Zhang X, Han S, Zhu B, Zhang G, Li X, Gao Y, Wu Z, Yang B, Liu Y, Baaziz W, Ersen O, Gu M, Miller JT, Liu W (2020) Reversible loss of core–shell structure for Ni–Au bimetallic nanoparticles during CO2 hydrogenation. Nat Catal 3(4):411–417. https://doi.org/10.1038/s41929-020-0440-2
Ferreira PJ, Robertson IM, Birnbaum HK (1998) Hydrogen effects on the interaction between dislocations. Acta Mater 46(5):1749–1757. https://doi.org/10.1016/S1359-6454(97)00349-2
Zou L, Yang C, Lei Y, Zakharov D, Wiezorek JMK, Su D, Yin Q, Li J, Liu Z, Stach EA, Yang JC, Qi L, Wang G, Zhou G (2018) Dislocation nucleation facilitated by atomic segregation. Nat Mater 17(1):56–63. https://doi.org/10.1038/nmat5034
Sharma R (2005) An environmental transmission electron microscope for in situ synthesis and characterization of nanomaterials. J Mater Res 20(7):1695–1707. https://doi.org/10.1557/jmr.2005.0241
Jiang H, Borca CN, Xu B, Robertson BW (2001) Fabrication of 2- and 3-dimentional nanostructures. Int J Mod Phys B 15(24–25):3207–3213. https://doi.org/10.1142/s021797920100749x
Mitsuishi K, Shimojo M, Han M, Furuya K (2003) Electron-beam-induced deposition using a subnanometer-sized probe of high-energy electrons. Appl Phys Lett 83(10):2064–2066. https://doi.org/10.1063/1.1611274
Crozier PA, Tolle J, Kouvetakis J, Ritter C (2004) Synthesis of uniform GaN quantum dot arrays via electron nanolithography of D2GaN3. Appl Phys Lett 84(18):3441–3443. https://doi.org/10.1063/1.1736314
van Dorp WF, van Someren B, Hagen CW, Kruit P, Crozier PA (2005) Approaching the resolution limit of nanometer-scale electron beam-induced deposition. Nano Lett 5(7):1303–1307. https://doi.org/10.1021/nl050522i
Gai PL, Sharma R, Ross FM (2008) Environmental (S)TEM studies of gas–liquid–solid interactions under reaction conditions. MRS Bull 33(2):107–114. https://doi.org/10.1557/mrs2008.23
Wagner RS, Ellis WC (1964) Vapor-liquid-solid mechanism of single crystal growth. Appl Phys Lett 4(5):89–90. https://doi.org/10.1063/1.1753975
Wu Y, Yang P (2001) Direct observation of vapor−liquid−solid nanowire growth. J Am Chem Soc 123(13):3165–3166. https://doi.org/10.1021/ja0059084
Kim BJ, Tersoff J, Wen CY, Reuter MC, Stach EA, Ross FM (2009) Determination of size effects during the phase transition of a nanoscale Au-Si eutectic. Phys Rev Lett 103(15):155701. https://doi.org/10.1103/PhysRevLett.103.155701
Kim BJ, Tersoff J, Kodambaka S, Reuter MC, Stach EA, Ross FM (2008) Kinetics of individual nucleation events observed in nanoscale vapor-liquid-solid growth. Science 322(5904):1070–1073. https://doi.org/10.1126/science.1163494
Hannon JB, Kodambaka S, Ross FM, Tromp RM (2006) The influence of the surface migration of gold on the growth of silicon nanowires. Nature 440(7080):69–71. https://doi.org/10.1038/nature04574
Ross FM (2010) Controlling nanowire structures through real time growth studies. Rep Prog Phys 73(11):114501. https://doi.org/10.1088/0034-4885/73/11/114501
Ross FM, Wen CY, Kodambaka S, Wacaser BA, Reuter MC, Stach EA (2010) The growth and characterization of Si and Ge nanowires grown from reactive metal catalysts. Phil Mag 90(35–36):4769–4778. https://doi.org/10.1080/14786435.2010.527713
Ross FM, Tersoff J, Reuter MC (2005) Sawtooth faceting in silicon nanowires. Phys Rev Lett 95(14):146104. https://doi.org/10.1103/PhysRevLett.95.146104
Kodambaka S, Tersoff J, Reuter MC, Ross FM (2007) Germanium nanowire growth below the eutectic temperature. Science 316(5825):729–732. https://doi.org/10.1126/science.1139105
Chou Y-C, Wen C-Y, Reuter MC, Su D, Stach EA, Ross FM (2012) Controlling the growth of Si/Ge nanowires and heterojunctions using silver-gold alloy catalysts. ACS Nano 6(7):6407–6415. https://doi.org/10.1021/nn301978x
Gamalski AD, Tersoff J, Stach EA (2016) Atomic resolution in situ imaging of a double-bilayer multistep growth mode in gallium nitride nanowires. Nano Lett 16(4):2283–2288. https://doi.org/10.1021/acs.nanolett.5b04650
Lenrick F, Ek M, Deppert K, Samuelson L, Reine Wallenberg L (2014) Straight and kinked InAs nanowire growth observed in situ by transmission electron microscopy. Nano Res 7(8):1188–1194. https://doi.org/10.1007/s12274-014-0481-4
Chou YC, Hillerich K, Tersoff J, Reuter MC, Dick KA, Ross FM (2014) Atomic-scale variability and control of III-V nanowire growth kinetics. Science 343(6168):281–284. https://doi.org/10.1126/science.1244623
Wen CY, Reuter MC, Bruley J, Tersoff J, Kodambaka S, Stach EA, Ross FM (2009) Formation of compositionally abrupt axial heterojunctions in silicon-germanium nanowires. Science 326(5957):1247–1250. https://doi.org/10.1126/science.1178606
Wen CY, Reuter MC, Tersoff J, Stach EA, Ross FM (2010) Structure, growth kinetics, and ledge flow during vapor−solid−solid growth of copper-catalyzed silicon nanowires. Nano Lett 10(2):514–519. https://doi.org/10.1021/nl903362y
Hofmann S, Sharma R, Wirth CT, Cervantes-Sodi F, Ducati C, Kasama T, Dunin-Borkowski RE, Drucker J, Bennett P, Robertson J (2008) Ledge-flow-controlled catalyst interface dynamics during Si nanowire growth. Nat Mater 7(5):372–375. https://doi.org/10.1038/nmat2140
Sears GW (1955) A growth mechanism for mercury whiskers. Acta Metall 3(4):361–366. https://doi.org/10.1016/0001-6160(55)90041-9
Pan ZW, Dai ZR, Wang ZL (2001) Nanobelts of semiconducting oxides. Science 291(5510):1947–1949. https://doi.org/10.1126/science.1058120
Zhang Z, Wang Y, Li H, Yuan W, Zhang X, Sun C, Zhang Z (2016) Atomic-scale observation of vapor-solid nanowire growth via oscillatory mass transport. ACS Nano 10(1):763–769. https://doi.org/10.1021/acsnano.5b05851
Tokunaga T, Kawamoto T, Tanaka K, Nakamura N, Hayashi Y, Sasaki K, Kuroda K, Yamamoto T (2012) Growth and structure analysis of tungsten oxide nanorods using environmental TEM. Nanoscale Res Lett 7(1):85. https://doi.org/10.1186/1556-276x-7-85
Rackauskas S, Jiang H, Wagner JB, Shandakov SD, Hansen TW, Kauppinen EI, Nasibulin AG (2014) In situ study of noncatalytic metal oxide nanowire growth. Nano Lett 14(10):5810–5813. https://doi.org/10.1021/nl502687s
Kallesøe C, Wen C-Y, Booth TJ, Hansen O, Bøggild P, Ross FM, Mølhave K (2012) In situ TEM creation and electrical characterization of nanowire devices. Nano Lett 12(6):2965–2970. https://doi.org/10.1021/nl300704u
Baker RTK, Feates FS, Harris PS (1972) Continuous electron microscopic observation of carbonaceous deposits formed on graphite and silica surfaces. Carbon 10(1):93–96. https://doi.org/10.1016/0008-6223(72)90014-0
Iijima S (1991) Helical microtubules of graphitic carbon. Nature 354(6348):56–58. https://doi.org/10.1038/354056a0
Helveg S, López-Cartes C, Sehested J, Hansen PL, Clausen BS, Rostrup-Nielsen JR, Abild-Pedersen F, Nørskov JK (2004) Atomic-scale imaging of carbon nanofibre growth. Nature 427(6973):426–429. https://doi.org/10.1038/nature02278
Sharma R, Iqbal Z (2004) In situ observations of carbon nanotube formation using environmental transmission electron microscopy. Appl Phys Lett 84(6):990–992. https://doi.org/10.1063/1.1646465
Sharma R, Rez P, Treacy MMJ, Stuart SJ (2005) In situ observation of the growth mechanisms of carbon nanotubes under diverse reaction conditions. J Electron Microsc 54(3):231–237. https://doi.org/10.1093/jmicro/dfi037
Sharma R, Rez P, Brown M, Du G, Treacy MMJ (2007) Dynamic observations of the effect of pressure and temperature conditions on the selective synthesis of carbon nanotubes. Nanotechnology 18(12):125602. https://doi.org/10.1088/0957-4484/18/12/125602
Sharma R, Chee S-W, Herzing A, Miranda R, Rez P (2011) Evaluation of the role of Au in improving catalytic activity of Ni nanoparticles for the formation of one-dimensional carbon nanostructures. Nano Lett 11(6):2464–2471. https://doi.org/10.1021/nl2009026
Yoshida H, Takeda S (2005) Image formation in a transmission electron microscope equipped with an environmental cell: single-walled carbon nanotubes in source gases. Phys Rev B 72(19):195428. https://doi.org/10.1103/PhysRevB.72.195428
Yoshida H, Uchiyama T, Takeda S (2007) Environmental transmission electron microscopy observations of swinging and rotational growth of carbon nanotubes. Jpn J Appl Phys 46(37):L917–L919. https://doi.org/10.1143/jjap.46.l917
Yoshida H, Takeda S, Uchiyama T, Kohno H, Homma Y (2008) Atomic-scale in-situ observation of carbon nanotube growth from solid state iron carbide nanoparticles. Nano Lett 8(7):2082–2086. https://doi.org/10.1021/nl080452q
Peng Z, Somodi F, Helveg S, Kisielowski C, Specht P, Bell AT (2012) High-resolution in situ and ex situ TEM studies on graphene formation and growth on Pt nanoparticles. J Catal 286:22–29. https://doi.org/10.1016/j.jcat.2011.10.008
Wu J, Shan H, Chen W, Gu X, Tao P, Song C, Shang W, Deng T (2016) In situ environmental TEM in imaging gas and liquid phase chemical reactions for materials research. Adv Mater 28(44):9686–9712. https://doi.org/10.1002/adma.201602519
Haruta M (1997) Size- and support-dependency in the catalysis of gold. Catal Today 36(1):153–166. https://doi.org/10.1016/S0920-5861(96)00208-8
He Y, Liu J-C, Luo L, Wang Y-G, Zhu J, Du Y, Li J, Mao SX, Wang C (2018) Size-dependent dynamic structures of supported gold nanoparticles in CO oxidation reaction condition. Proc Natl Acad Sci 115(30):7700. https://doi.org/10.1073/pnas.1800262115
Tao F, Crozier PA (2016) Atomic-scale observations of catalyst structures under reaction conditions and during catalysis. Chem Rev 116(6):3487–3539. https://doi.org/10.1021/cr5002657
Zhang S, Nguyen L, Zhu Y, Zhan S, Tsung C-K, Tao F (2013) In-situ studies of nanocatalysis. Acc Chem Res 46(8):1731–1739. https://doi.org/10.1021/ar300245g
Hofmann S, Sharma R, Ducati C, Du G, Mattevi C, Cepek C, Cantoro M, Pisana S, Parvez A, Cervantes-Sodi F, Ferrari AC, Dunin-Borkowski R, Lizzit S, Petaccia L, Goldoni A, Robertson J (2007) In situ observations of catalyst dynamics during surface-bound carbon nanotube nucleation. Nano Lett 7(3):602–608. https://doi.org/10.1021/nl0624824
Mazzucco S, Wang Y, Tanase M, Picher M, Li K, Wu Z, Irle S, Sharma R (2014) Direct evidence of active and inactive phases of Fe catalyst nanoparticles for carbon nanotube formation. J Catal 319:54–60. https://doi.org/10.1016/j.jcat.2014.07.023
Zhou Y, Jin C, Li Y, Shen W (2018) Dynamic behavior of metal nanoparticles for catalysis. Nano Today 20:101–120. https://doi.org/10.1016/j.nantod.2018.04.005
Ta N, Liu J, Chenna S, Crozier PA, Li Y, Chen A, Shen W (2012) Stabilized gold nanoparticles on ceria nanorods by strong interfacial anchoring. J Am Chem Soc 134(51):20585–20588. https://doi.org/10.1021/ja310341j
Tauster SJ, Fung SC, Garten RL (1978) Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J Am Chem Soc 100(1):170–175. https://doi.org/10.1021/ja00469a029
Haller GL, Resasco DE (1989) Metal–support interaction: group VIII metals and reducible oxides. In: Eley DD, Pines H, Weisz PB (eds) Advances in catalysis, vol 36. Academic Press, pp 173–235. https://doi.org/10.1016/S0360-0564(08)60018-8
Liu J (2011) Advanced electron microscopy of metal-support interactions in supported metal catalysts. ChemCatChem 3(6):934–948. https://doi.org/10.1002/cctc.201100090
Su DS, Zhang B, Schlögl R (2015) Electron microscopy of solid catalysts—transforming from a challenge to a toolbox. Chem Rev 115(8):2818–2882. https://doi.org/10.1021/cr500084c
Zhang S, Plessow PN, Willis JJ, Dai S, Xu M, Graham GW, Cargnello M, Abild-Pedersen F, Pan X (2016) Dynamical observation and detailed description of catalysts under strong metal-support interaction. Nano Lett 16(7):4528–4534. https://doi.org/10.1021/acs.nanolett.6b01769
Matsubu JC, Zhang S, DeRita L, Marinkovic NS, Chen JG, Graham GW, Pan X, Christopher P (2017) Adsorbate-mediated strong metal–support interactions in oxide-supported Rh catalysts. Nat Chem 9(2):120–127. https://doi.org/10.1038/nchem.2607
Yuan W, Zhu B, Li X-Y, Hansen Thomas W, Ou Y, Fang K, Yang H, Zhang Z, Wagner Jakob B, Gao Y, Wang Y (2020) Visualizing H2O molecules reacting at TiO2 active sites with transmission electron microscopy. Science 367(6476):428–430. https://doi.org/10.1126/science.aay2474
Yuan W, Zhu B, Fang K, Li X-Y, Hansen Thomas W, Ou Y, Yang H, Wagner Jakob B, Gao Y, Wang Y, Zhang Z (2021) In situ manipulation of the active Au-TiO2 interface with atomic precision during CO oxidation. Science 371(6528):517–521. https://doi.org/10.1126/science.abe3558
Wanke SE, Flynn PC (1975) The sintering of supported metal catalysts. Catal Rev 12(1):93–135. https://doi.org/10.1080/01614947508067523
Granqvist CG, Buhrman RA (1976) Size distributions for supported metal catalysts: coalescence growth versus Ostwald ripening. J Catal 42(3):477–479. https://doi.org/10.1016/0021-9517(76)90125-1
Hansen TW, DeLaRiva AT, Challa SR, Datye AK (2013) Sintering of catalytic nanoparticles: particle migration or Ostwald ripening? Acc Chem Res 46(8):1720–1730. https://doi.org/10.1021/ar3002427
Wynblatt P, Gjostein NA (1976) Particle growth in model supported metal catalysts—I. Theory. Acta Metall 24(12):1165–1174. https://doi.org/10.1016/0001-6160(76)90034-1
Granqvist CG, Buhrman RA (1975) Statistical model for coalescence of islands in discontinuous films. Appl Phys Lett 27(12):693–694. https://doi.org/10.1063/1.88342
Baker RTK, Harris PS, Thomas RB (1974) Direct observation of particle mobility on a surface in a gaseous environment. Surf Sci 46(1):311–316. https://doi.org/10.1016/0039-6028(74)90259-3
Liu RJ, Crozier PA, Smith CM, Hucul DA, Blackson J, Salaita G (2005) Metal sintering mechanisms and regeneration of palladium/alumina hydrogenation catalysts. Appl Catal A 282(1):111–121. https://doi.org/10.1016/j.apcata.2004.12.015
Liu R-J, Crozier PA, Smith CM, Hucul DA, Blackson J, Salaita G (2004) In situ electron microscopy studies of the sintering of palladium nanoparticles on alumina during catalyst regeneration processes. Microsc Microanal 10(1):77–85. https://doi.org/10.1017/s1431927604040188
Moulijn JA, van Diepen AE, Kapteijn F (2001) Catalyst deactivation: is it predictable?: what to do? Appl Catal A 212(1):3–16. https://doi.org/10.1016/S0926-860X(00)00842-5
Simonsen SB, Chorkendorff I, Dahl S, Skoglundh M, Sehested J, Helveg S (2010) Direct observations of oxygen-induced platinum nanoparticle ripening studied by in situ TEM. J Am Chem Soc 132(23):7968–7975. https://doi.org/10.1021/ja910094r
Benavidez AD, Kovarik L, Genc A, Agrawal N, Larsson EM, Hansen TW, Karim AM, Datye AK (2012) Environmental transmission electron microscopy study of the origins of anomalous particle size distributions in supported metal catalysts. ACS Catal 2(11):2349–2356. https://doi.org/10.1021/cs3005117
Yuan W, Zhang D, Ou Y, Fang K, Zhu B, Yang H, Hansen TW, Wagner JB, Zhang Z, Gao Y (2018) Direct in situ TEM visualization and insight into the facet-dependent sintering behaviors of gold on TiO2. Angew Chem 130(51):17069–17073. https://doi.org/10.1002/ange.201811933
Amama PB, Pint CL, McJilton L, Kim SM, Stach EA, Murray PT, Hauge RH, Maruyama B (2009) Role of water in super growth of single-walled carbon nanotube carpets. Nano Lett 9(1):44–49. https://doi.org/10.1021/nl801876h
Hata K, Futaba Don N, Mizuno K, Namai T, Yumura M, Iijima S (2004) Water-assisted highly efficient synthesis of impurity-free single-walled carbon nanotubes. Science 306(5700):1362–1364. https://doi.org/10.1126/science.1104962
Asakura K, Nagahiro H, Ichikuni N, Iwasawa Y (1999) Structure and catalytic combustion activity of atomically dispersed Pt species at MgO surface. Appl Catal A 188(1):313–324. https://doi.org/10.1016/S0926-860X(99)00247-1
Qiao B, Wang A, Yang X, Allard LF, Jiang Z, Cui Y, Liu J, Li J, Zhang T (2011) Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat Chem 3(8):634–641. https://doi.org/10.1038/nchem.1095
Jones J, Xiong H, DeLaRiva AT, Peterson Eric J, Pham H, Challa Sivakumar R, Qi G, Oh S, Wiebenga Michelle H, Pereira Hernández Xavier I, Wang Y, Datye Abhaya K (2016) Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 353(6295):150–154. https://doi.org/10.1126/science.aaf8800
Gänzler AM, Casapu M, Vernoux P, Loridant S, Cadete Santos Aires FJ, Epicier T, Betz B, Hoyer R, Grunwaldt J-D (2017) Tuning the structure of platinum particles on ceria in situ for enhancing the catalytic performance of exhaust gas catalysts. Angew Chem Int Ed 56(42):13078–13082. https://doi.org/10.1002/anie.201707842
Yang X-F, Wang A, Qiao B, Li J, Liu J, Zhang T (2013) Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc Chem Res 46(8):1740–1748. https://doi.org/10.1021/ar300361m
Yao Y, Huang Z, Xie P, Wu L, Ma L, Li T, Pang Z, Jiao M, Liang Z, Gao J, He Y, Kline DJ, Zachariah MR, Wang C, Lu J, Wu T, Li T, Wang C, Shahbazian-Yassar R, Hu L (2019) High temperature shockwave stabilized single atoms. Nat Nanotechnol 14(9):851–857. https://doi.org/10.1038/s41565-019-0518-7
Yoshida K, Bright A, Tanaka N (2012) Direct observation of the initial process of Ostwald ripening using spherical aberration-corrected transmission electron microscopy. J Electron Microsc 61(2):99–103. https://doi.org/10.1093/jmicro/dfr100
Yoshida K, Xudong Z, Bright AN, Saitoh K, Tanaka N (2013) Dynamic environmental transmission electron microscopy observation of platinum electrode catalyst deactivation in a proton-exchange-membrane fuel cell. Nanotechnology 24(6):065705. https://doi.org/10.1088/0957-4484/24/6/065705
DeRita L, Resasco J, Dai S, Boubnov A, Thang HV, Hoffman AS, Ro I, Graham GW, Bare SR, Pacchioni G, Pan X, Christopher P (2019) Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat Mater 18(7):746–751. https://doi.org/10.1038/s41563-019-0349-9
Wei S, Li A, Liu J-C, Li Z, Chen W, Gong Y, Zhang Q, Cheong W-C, Wang Y, Zheng L, Xiao H, Chen C, Wang D, Peng Q, Gu L, Han X, Li J, Li Y (2018) Direct observation of noble metal nanoparticles transforming to thermally stable single atoms. Nat Nanotechnol 13(9):856–861. https://doi.org/10.1038/s41565-018-0197-9
Lang R, Xi W, Liu J-C, Cui Y-T, Li T, Lee AF, Chen F, Chen Y, Li L, Li L, Lin J, Miao S, Liu X, Wang A-Q, Wang X, Luo J, Qiao B, Li J, Zhang T (2019) Non defect-stabilized thermally stable single-atom catalyst. Nat Commun 10(1):234. https://doi.org/10.1038/s41467-018-08136-3
Dai S, Zhang S, Katz MB, Graham GW, Pan X (2017) In situ observation of Rh-CaTiO3 catalysts during reduction and oxidation treatments by transmission electron microscopy. ACS Catal 7(3):1579–1582. https://doi.org/10.1021/acscatal.6b03604
Ohno Y, Takeda S (1995) A new apparatus for in situ photoluminescence spectroscopy in a transmission electron microscope. Rev Sci Instrum 66(10):4866–4869. https://doi.org/10.1063/1.1146166
Suzuki K, Ichihara M, Takeuchi S, Nakagawa K, Maeda K, Iwanaga H (1984) In situ TEM observation of dislocation motion in II–VI compounds. Philos Mag A 49(3):451–461. https://doi.org/10.1080/01418618408233287
Yoshida K, Yamasaki J, Tanaka N (2004) In situ high-resolution transmission electron microscopy observation of photodecomposition process of poly-hydrocarbons on catalytic TiO2 films. Appl Phys Lett 84(14):2542–2544. https://doi.org/10.1063/1.1689747
Miller B, Crozier P (2011) Visible and UV irradiation of ETEM samples for in-situ studies of photocatalysts. Microsc Microanal 17(S2):472–473. https://doi.org/10.1017/s1431927611003230
Miller B, Crozier PA (2012) In situ visible and UV illumination of ETEM samples. Microsc Microanal 18(S2):1074–1075. https://doi.org/10.1017/s1431927612007222
Miller BK, Crozier PA (2013) System for in situ UV-visible illumination of environmental transmission electron microscopy samples. Microsc Microanal 19(2):461–469. https://doi.org/10.1017/s1431927612014122
Zhang L, Miller BK, Crozier PA (2013) atomic level in situ observation of surface amorphization in anatase nanocrystals during light irradiation in water vapor. Nano Lett 13(2):679–684. https://doi.org/10.1021/nl304333h
Picher M, Mazzucco S, Blankenship S, Sharma R (2015) Vibrational and optical spectroscopies integrated with environmental transmission electron microscopy. Ultramicroscopy 150:10–15. https://doi.org/10.1016/j.ultramic.2014.11.023
Shindo D, Takahashi K, Murakami Y, Yamazaki K, Deguchi S, Suga H, Kondo Y (2009) Development of a multifunctional TEM specimen holder equipped with a piezodriving probe and a laser irradiation port. J Electron Microsc 58(4):245–249. https://doi.org/10.1093/jmicro/dfp018
Cavalca F, Laursen AB, Kardynal BE, Dunin-Borkowski RE, Dahl S, Wagner JB, Hansen TW (2012) In situtransmission electron microscopy of light-induced photocatalytic reactions. Nanotechnology 23(7):075705. https://doi.org/10.1088/0957-4484/23/7/075705
Cavalca F, Laursen AB, Wagner JB, Damsgaard CD, Chorkendorff I, Hansen TW (2013) Light-induced reduction of cuprous oxide in an environmental transmission electron microscope. ChemCatChem 5(9):2667–2672. https://doi.org/10.1002/cctc.201200887
Taheri ML, Stach EA, Arslan I, Crozier PA, Kabius BC, LaGrange T, Minor AM, Takeda S, Tanase M, Wagner JB, Sharma R (2016) Current status and future directions for in situ transmission electron microscopy. Ultramicroscopy 170:86–95. https://doi.org/10.1016/j.ultramic.2016.08.007
Faruqi AR, McMullan G (2011) Electronic detectors for electron microscopy. Q Rev Biophys 44(3):357–390. https://doi.org/10.1017/s0033583511000035
Lobastov VA, Srinivasan R, Zewail AH (2005) Four-dimensional ultrafast electron microscopy. Proc Natl Acad Sci USA 102(20):7069. https://doi.org/10.1073/pnas.0502607102
Zewail AH (2006) 4D ultrafast electron diffraction, crystallography, and microscopy. Annu Rev Phys Chem 57(1):65–103. https://doi.org/10.1146/annurev.physchem.57.032905.104748
Zewail AH (2010) Four-dimensional electron microscopy. Science 328(5975):187–193. https://doi.org/10.1126/science.1166135
LaGrange T, Reed BW, Santala MK, McKeown JT, Kulovits A, Wiezorek JMK, Nikolova L, Rosei F, Siwick BJ, Campbell GH (2012) Approaches for ultrafast imaging of transient materials processes in the transmission electron microscope. Micron 43(11):1108–1120. https://doi.org/10.1016/j.micron.2012.04.010
Barwick B, Flannigan DJ, Zewail AH (2009) Photon-induced near-field electron microscopy. Nature 462(7275):902–906. https://doi.org/10.1038/nature08662
Baum P, Yang D-S, Zewail Ahmed H (2007) 4D visualization of transitional structures in phase transformations by electron diffraction. Science 318(5851):788–792. https://doi.org/10.1126/science.1147724
Carbone F, Kwon O-H, Zewail Ahmed H (2009) Dynamics of chemical bonding mapped by energy-resolved 4D electron microscopy. Science 325(5937):181–184. https://doi.org/10.1126/science.1175005
Kwon O-H, Zewail Ahmed H (2010) 4D electron tomography. Science 328(5986):1668–1673. https://doi.org/10.1126/science.1190470
Fitzpatrick AWP, Lorenz UJ, Vanacore GM, Zewail AH (2013) 4D Cryo-electron microscopy of proteins. J Am Chem Soc 135(51):19123–19126. https://doi.org/10.1021/ja4115055
Fu X, Pollard Shawn D, Chen B, Yoo B-K, Yang H, Zhu Y (2018) Optical manipulation of magnetic vortices visualized in situ by Lorentz electron microscopy. Sci Adv 4(7):eaat3077. https://doi.org/10.1126/sciadv.aat3077
Fu X, Chen B, Tang J, Hassan Mohammed T, Zewail Ahmed H (2017) Imaging rotational dynamics of nanoparticles in liquid by 4D electron microscopy. Science 355(6324):494–498. https://doi.org/10.1126/science.aah3582
Armstrong MR, Boyden K, Browning ND, Campbell GH, Colvin JD, DeHope WJ, Frank AM, Gibson DJ, Hartemann F, Kim JS, King WE, LaGrange TB, Pyke BJ, Reed BW, Shuttlesworth RM, Stuart BC, Torralva BR (2007) Practical considerations for high spatial and temporal resolution dynamic transmission electron microscopy. Ultramicroscopy 107(4):356–367. https://doi.org/10.1016/j.ultramic.2006.09.005
King WE, Campbell GH, Frank A, Reed B, Schmerge JF, Siwick BJ, Stuart BC, Weber PM (2005) Ultrafast electron microscopy in materials science, biology, and chemistry. J Appl Phys 97(11):111101. https://doi.org/10.1063/1.1927699
LaGrange T, Armstrong MR, Boyden K, Brown CG, Campbell GH, Colvin JD, DeHope WJ, Frank AM, Gibson DJ, Hartemann FV, Kim JS, King WE, Pyke BJ, Reed BW, Shirk MD, Shuttlesworth RM, Stuart BC, Torralva BR, Browning ND (2006) Single-shot dynamic transmission electron microscopy. Appl Phys Lett 89(4):044105. https://doi.org/10.1063/1.2236263
Reed BW, Armstrong MR, Browning ND, Campbell GH, Evans JE, LaGrange T, Masiel DJ (2009) The evolution of ultrafast electron microscope instrumentation. Microsc Microanal 15(4):272–281. https://doi.org/10.1017/s1431927609090394
Lazić I, Bosch EGT, Lazar S (2016) Phase contrast STEM for thin samples: Integrated differential phase contrast. Ultramicroscopy 160:265–280. https://doi.org/10.1016/j.ultramic.2015.10.011
Yücelen E, Lazić I, Bosch EGT (2018) Phase contrast scanning transmission electron microscopy imaging of light and heavy atoms at the limit of contrast and resolution. Sci Rep 8(1):2676. https://doi.org/10.1038/s41598-018-20377-2
Shen B, Chen X, Cai D, Xiong H, Liu X, Meng C, Han Y, Wei F (2020) Atomic spatial and temporal imaging of local structures and light elements inside zeolite frameworks. Adv Mater 32(4):1906103. https://doi.org/10.1002/adma.201906103
Liu L, Wang N, Zhu C, Liu X, Zhu Y, Guo P, Alfilfil L, Dong X, Zhang D, Han Y (2020) Direct imaging of atomically dispersed molybdenum that enables location of aluminum in the framework of zeolite ZSM-5. Angew Chem Int Ed 59(2):819–825. https://doi.org/10.1002/anie.201909834
The significant role of the atomic surface structure of support in strong metal-support interaction. https://doi.org/10.1002/chem.202104519
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Fang, K., Yuan, W., Wagner, J.B., Zhang, Z., Wang, Y. (2023). In-Situ Gas Transmission Electron Microscopy. In: Sun, L., Xu, T., Zhang, Z. (eds) In-Situ Transmission Electron Microscopy. Springer, Singapore. https://doi.org/10.1007/978-981-19-6845-7_9
Download citation
DOI: https://doi.org/10.1007/978-981-19-6845-7_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-6844-0
Online ISBN: 978-981-19-6845-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)