
Abstract
Nucleic acid therapeutics were initially considered for treating hereditary diseases, but their potential role in the treatment of acquired diseases such as cancer, neurodegenerative diseases etc., is now widely recognized. The main objective in gene therapy via a systemic pathway is the development of a stable and nontoxic targeted nonviral gene vector that can encapsulate and deliver foreign genetic materials into cells with the transfection efficiency of viral vectors. Targeting of such gene delivery systems to the diseased site can further improve efficacy, minimize toxicity, and also lower the cost of therapy. This chapter discusses various nucleic acid therapeutics, nonviral gene delivery vectors as well as targeting approaches to achieve safe and efficient nucleic acid delivery.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Nucleic acid (NA) therapeutics are deoxyribonucleic or ribonucleic acid (DNA/RNA) based molecules that regulate expression of a particular gene, intracellularly. In the early stages of development, NA based therapy was employed to correct inheritable disorders resulting from a single gene defect such as cystic fibrosis [1] and severe combined immunodeficiency disease (SCID) [2, 3]. Later, this therapy was also useful for complex genetic disorders such as cancer [4, 5], and cardiovascular [6, 7] and neurodegenerative diseases [8, 9]. The first gene therapy clinical trial was in 1990, for treatment of adenosine deaminase (ADA) deficiency [10], a genetic disorder that weakens the immune system to fight against infections. Normal gene encoding for ADA was inserted into the patient’s white blood cells and resulted in normal production of ADA. This outcome together with the successful mapping of the human genome in the year 2003 [11], encouraged many scientists to work in this area and has led to many gene based clinical trials [12] as well as commercial gene products [13, 14].
Despite these promising developments, the full potential of gene therapy remains to be exploited. One of the key challenges to successful NA therapeutics is their inefficient delivery to the desired site. This is a result of their susceptibility to enzymatic degradation, poor cellular uptake and poor endosomal escape. For DNA-based therapeutics (such as plasmid DNA), entry into the nucleus is an additional challenge. Currently, numerous strategies are being pursued to improve delivery of NAs in vitro as well as in vivo. Although high levels of DNA transfection have been achieved by mechanical methods such as microinjection [15, 16] particle bombardment [17] and electroporation [18], these methods may cause local toxicity and are impractical for in vivo purposes. Alternate methods include chemical modification of NA structure in the region of the phosphate backbone, sugars, or bases [19–22]; however, this may compromise NA bioactivity and stability [22, 23]. Accordingly, the use of viral or nonviral delivery vectors is preferred. Viral vectors have very high transfection efficiency due to their innate components that facilitate efficient cellular penetration through the plasma membrane, the endosomal membrane and the nuclear membrane [24, 25]. Nonetheless, viral vectors may be immunogenic as well as toxic [26, 27]. Consequently, utilization of nonviral vectors is favorable in order to facilitate efficient intracellular transport of NAs.
Nonviral vectors such as cationic liposomes and polymers (including dendrimers) interact electrostatically with NAs, thereby protecting these against enzymatic attack, improving their cellular internalization as well as their endosomal escape. However, a major drawback to the use of these cationic substances is their considerable local or systemic toxicity upon administration [28–30]. Accordingly, focus has been diverted towards anionic liposomes/polymers for efficient gene delivery [31–36]. Other nonviral vectors used for gene delivery include PLGA polymeric nanoparticles, which are useful for sustained delivery of NAs. Most recent among the nonviral vectors are “microbubbles” which release NAs upon exposure to ultrasound [37–41].
For safe and efficient delivery, it is imperative to deliver the therapeutic molecule to the desired site while leaving the healthy cells unaffected. This can be attained by using targeted (or site-specific) gene delivery systems. Targeting can be achieved by using polyethylene glycol (PEG) that prolongs particle blood circulation (passive targeting) [42] or by using targeting ligands which bind specifically to receptors that are (over-) expressed in diseased cells (active targeting). Using these approaches tissue-specific [43], cell-specific [35, 44–47] and even organelle-specific targeting [24, 48–54] can be obtained. Additionally other strategies such as ultrasound-triggered microbubble delivery and suicide gene delivery can be employed [37–41] to achieve site-specific delivery.
This chapter describes nucleic acid therapeutics in preclinical and clinical studies, various nonviral vectors employed to deliver these therapeutic molecules, and several strategies adopted to achieve in vitro/in vivo targeted gene delivery.
2 Nucleic Acid Therapeutics
Various NA therapeutics have been employed to treat genetic disorders either by inhibiting expression of harmful proteins (e.g., antisense oligonucleotides, DNAzymes, RNAzymes, aptamers, silencing RNAs and short hairpin RNAs) or by providing a (missing) therapeutic protein (e.g., plasmid DNA). Certain molecules such as microRNA can be used for either of the aforementioned purposes. Figure 8.4 illustrates the site of action of various NA therapeutic molecules. Several of these molecules are currently in clinical trials (Table 8.1). Properties of such NA therapeutics are discussed in the following sections.
2.1 Plasmid DNA (pDNA)
pDNA is a small circular double-stranded DNA that transcribes (to messenger RNA) and translates to produce a “therapeutic” protein. Recombinant pDNA has been utilized to provide “therapeutic” proteins in the body to cure various fatal diseases including cancer. For example Gendicine and Ocorine are pDNA based marketed products (China) that efficiently deliver tumor suppressor gene, p53, for the treatment of head and neck cancer [55]. pDNA can also be employed as vaccines for genetic immunization. These can be prepared by cloning the plasmid with genes encoding for antigen(s) from a specific pathogen. Upon intracellular accumulation in “host” cells, DNA vaccine produces the pathogen’s antigen that activates the “host” immune system [56]. There are currently around 89 clinical trials based on DNA vaccine, mostly for the treatment of cancer or human immunodeficiency virus (HIV) infection [57]. Other than vaccines, pDNA is also used in cancer suicide gene therapy (CSGT). There are two approaches for CSGT: (1) pDNA is cloned with a cancer-specific promoter sequence of a suicidal gene so that it is upregulated (expressed) only in cancer cells, thereby killing them. Examples of cancer specific promoters include carcinoembryonic antigen (gastric and colorectal carcinomas), telomerase (many tumors including ovarian cancer), and prostate specific antigen (prostate cancer) [58]. (2) pDNA is cloned with genes encoding for enzymes that participate in activation of anticancer prodrugs. The prodrug is targeted to the “disease” site and upon enzymatic activation a toxic metabolite is produced which causes tumor cell death. For example pDNA encoding for cytosine deaminase enzyme has been utilized for treating cancer upon activating 5-fluorocytosine [59–61].
2.2 DNAzymes and RNAzymes
DNAzyme (DNA enzyme) is DNA-based enzyme that catalytically cleaves messenger RNA (mRNA), thereby interfering with the translation process. After cleavage, mRNA dissociates with DNAzyme and this prepares DNAzyme to cleave other mRNA molecules [62]. In this manner, DNAzyme can inhibit expression of “harmful” proteins. For example, anti-latent membrane protein-1 (LMP1) DNAzyme has been utilized to effectively knockdown expression of LMP-1 proteins (in mice), which are otherwise responsible for nasopharyngeal carcinoma (NPC) [63]. RNAzyme (RNA enzyme) works in a similar fashion as DNAzyme but is less chemically stable due to the susceptibility of the 2′OH group in the ribose sugar to alkaline hydrolysis [64]. Other examples of DNAzyme/RNAzyme are listed in Table 8.1.
2.3 Aptamers
Nucleic acid aptamers are short single/double stranded NAs that specifically bind (lock and key mechanism) to proteins, other NAs, or even small molecules. Aptamers can be designed to interfere with functioning of “harmful” proteins, either directly (bind to protein) or indirectly (bind to mRNA/DNA). For example, Pegaptanib (Macugen®) is a USFDA approved anti-angiogenic RNA aptamer that prevents blood vessel growth by directly binding to intracellular VEGF165 (isoform of vascular endothelial growth factor (VEGF) that regulates vascular permeability). Pegaptanib is therefore useful in the treatment of wet-age related macular degeneration (wet-AMD) (Table 8.1). ARC1779 is a DNA aptamer in Phase II clinical trials for the treatment of thrombotic microangiopathies and carotid artery disease. ARC1779 specifically interacts with von Willebrand factor (protein that recruits platelets to the damaged arteries) and blocks its binding to platelet membrane glycoprotein receptors, resulting in an antithrombotic effect [65]. Other examples of aptamer-based therapy have been reviewed elsewhere [66, 67].
2.4 Antisense Oligonucleotides
These are short double stranded DNA/RNA molecules that complimentarily bind to a specific mRNA, release RNase H enzymes, thereby causing mRNA hydrolysis and interference in the translation process [68]. Accordingly, antisense oligonucleotides have been successfully employed to inhibit production of “disease causing” proteins and cure conditions such as hypercholesterolemia [57], neuromuscular disorders [69] and inflammatory disorders [70]. For example Alicaforsen is an FDA approved antisense oligonucleotide for the treatment of pouchitis [70]. Pouchitis is an inflammation of the surgically constructed internal pouch created in ulcerative colitis patients who have had their diseased colons removed. Alicaforsen prevents the production of intercellular adhesion molecule 1, or ICAM-1 protein overexpressed in pouchitis. Another antisense drug Mipomersen, in Phase III clinical trials, acts against apo-B protein to treat severe LDL-hypercholesterolemia [57]. Other examples are summarized in Table 8.1.
2.5 shRNA, miRNA, and siRNA
These are the most recent NA therapeutics employed for gene delivery. Short hairpin RNA or shRNA is an expression vector that post-nuclear entry, transcribes to RNA which binds sequence specifically to mRNA and degrades it. For example STMN1-shRNA lipoplexes (Phase-I) have been shown to inhibit production of stathmin 1 (STMN 1) protein. This protein is known to participate in spindle dynamics associated with advanced or metastatic cancer [57]. Micro-RNAs or miRNAs are physiological molecules that play a key role in normal functioning of the human body [71]. Structurally these are small noncoding endogenous oligonucleotides that bind nonspecifically to the 3′UTR (untranslated region) of cytosolic messenger RNA and (up/down) regulates gene expression [72]. However, miRNA levels are usually disrupted in disease conditions. Accordingly miRNAs needs to be exogenously delivered to restore this balance. For example, endogenous levels of microRNA “miR-34,” a tumor suppressor, is usually low in many types of cancers. Accordingly, synthetic miR-34 has been delivered, either directly or with nanoparticles (NOV-340), in various animal models to effectively cure cancer. This could potentially be the first miRNA therapeutic to enter the clinics [73].
Due to nonspecific binding with mRNA, miRNAs could be effective against 1,000s of mRNA molecules with similar sequences [74] but this also increases the probability for off-target effects [75]. Such effects are less common with silencing RNAs (siRNAs) that sequence specifically bind to the mRNA [76]. siRNAs are small 21–25 base nucleotides that function in the cell cytoplasm [77]. Since siRNA is highly specific, compared to miRNA and does not require nuclear delivery (as is the case for shRNA) it is the most preferred in the category of RNA-based therapeutics. Additionally, siRNA is more potent than other therapies such as antisense oligonucleotides since one siRNA molecule can cleave several mRNA molecules (of the same kind) [78]. Many siRNA products are currently in clinical trials for the treatment of a wide range of indications [79] including solid tumors, liver cancer and wet-AMD (Table 8.1).
3 Barriers to Efficient Nucleic Acid Delivery
3.1 Enzymatic/Chemical Instability
Owing to their structure, NAs are prone to attack by exonucleases as well as endonucleases present in the extracellular/intracellular environment. Accordingly, these get degraded much before reaching the desired site of action. Several efforts have been made to improve NA stability. These include chemical modification of the phosphate backbone, sugars or nucleotide bases [19–22]. Phosphodiester bonds in the NA backbone have been substituted with enzymatically resistant bonds such as phosphorothionate [19], boranophosphates [20]. phosphoramidate, and methylphosphonate. Of these, phosphorothionates have achieved the greatest success [80] as evident by the fact that one of the FDA approved NA products, is Vitravene, an antisense phosphorothionate DNA oligonucleotide. Intravitreal injection of Vitravene is used for the treatment of cytomegalovirus retinitis [81, 82]. Whereas such backbone modifications have improved NA stability, high degree of modification may compromise the safety aspect of NAs. For example Amarzguioui et al. reported that 100 % replacement of phosphodiester bonds with phosphorothioate caused significant increase in cytotoxicity although there was remarkable improvement in siRNA serum stability [23].
With respect to sugar modification, the ribose sugar in RNA, has been modified with groups such as 2′NH2, 2′F, 2′OMe, or 2′deoxy [21, 83]. These modifications improved chemical stability of RNA which is otherwise prone to alkaline hydrolysis owing to the presence of 2′OH group [64]. Some researchers have also attempted modification of bases in nucleotides, but this lowered the activity of biomolecules [22].
3.2 Immunogenicity
Nucleic acid therapeutics such as siRNAs could be immunogenic since these trigger activation of inflammatory toll-like receptors (TLR) that further activate interferon (IFN-α) [84], interleukins (IL-6) and other cytokines associated with innate immunity [85], Ribose sugar and multiple uridine bases in siRNA have been observed to be responsible for this triggering mechanism, as these are predominantly identified by TLR7 [86]. Accordingly, modification of the siRNA structure can reduce immunogenicity. For example, when a few 2′ OH groups in ribose sugar were substituted with 2′OMe, siRNA immunogenicity was significantly reduced [87, 88]. The precise mechanism, later discovered by Robbins and co-workers, revealed that 2′OMe acts as an antagonist of TLR7, the primary receptor involved in immune stimulation [89]. These findings were also supported by the work of Cekaite et al. [90]. Besides 2′OMe, other groups such as 2′F [90] and locked NA (LNA) [84] are also useful to inhibit the immune response. In addition to TLR, other cellular receptors such as PKR (dsRNA-binding protein kinase) [91] and RIG-1 (retinoic acid-inducible gene-I) [92] have been implicated in immune-stimulation by siRNA.
3.3 Nuclear Entry
Other than cellular uptake and endosomal release (Sect. 3.4), DNA based therapeutics (pDNA, shRNA) have the additional challenge of entering into the nucleus where transcription occurs (Fig. 8.4). Various strategies have been adopted to improve nuclear entry of these biomolecules and these are discussed in Sect. 5.2.2.1.
3.4 Poor Cellular Uptake and Endosomal Escape
Attributed to their relatively large molecular weight compared to small molecules and the presence of a highly negatively charged backbone, NAs face difficulty in passing through the anionic plasma membrane. Additionally, NAs are unable to escape from endosomes due to the inability of the anionic backbone of NAs to interact with anionic endosomal lipids, leading to their degradation in this low pH environment (Fig. 8.4). The degradation mechanism includes hydrolysis of phosphodiester bonds [93] and/or alterations in base pairing in the acidic pH [94]. To improve cellular uptake as well as the endosomal release capability of NAs, two approaches have been adopted: (a) direct conjugation and (b) delivery vectors. The direct conjugation approach involves chemical conjugation of NAs with moieties (lipids [95], polymers [96], peptides [97]) that can potentiate the cellular uptake and/or endosomal escape of the biomolecule. However, incorrect site of conjugation causing reduced bioactivity [98], charge neutralization of the cationic conjugated moiety by anionic NA [99], and the inability of the conjugated species to protect the NA against enzymatic attack, often limits the potential of this approach. For these reasons, use of delivery vectors is more common.
4 Delivery Vectors
Efficient gene delivery for therapeutic approaches is a demanding task that urges the development of delivery vectors capable of overcoming the aforementioned challenges to efficient NA delivery. Delivery vectors not only protect NAs from enzymatic digestion but also enhance their cellular uptake and endosomal release capability. On the basis of their origin and mechanism of action, delivery vectors can be categorized as viral and nonviral.
4.1 Viral Vectors
Inherent properties of viruses such as effective membrane penetration (plasma, endosome, or nuclear) has been exploited for achieve very high transfection efficiency with viral vectors [24, 25]. However, some viruses (retrovirus) integrate with host genome and excessively produce viral proteins causing a fatal immune response. For example, retrovirus-based delivery of tumor suppressor gene in an X-lined SCID patient caused massive immune response triggered by the viral vector (viral proteins), leading to multiple organ failure and brain death [100]. For safer delivery, researchers have prepared attenuated viruses lacking in virulent components. Currently there are seven attenuated (adeno-associated) viral products in early phase, and two in late phase (III) clinical trials [57]. Despite this success, future prospects of viral vectors are restricted due to the limit size available for cloning therapeutic genes, scale-up as well as manufacturability issues [101]. Consequently, focus is more towards developing safe and efficient nonviral vector based gene delivery systems.
4.2 Nonviral Vectors
Nonviral vectors offer a high level of control with respect to their physicochemical properties and manufacturing on industrial scale. Additionally these are non-immunogenic and relatively safer compared to the viral vectors. However, these vectors have low transfection efficiencies due to lack of innate characteristics of a virus. Tremendous efforts have been made to develop and optimize nonviral carriers which are safe and have transfection efficiency of viral vectors. Various nonviral vectors useful for gene delivery have been discussed in the following sections.
4.2.1 Liposomes
Liposomes are spherical structures with an aqueous core surrounded by a lipid bilayer. These are by far the most commonly used nonviral vectors to facilitate NA delivery. Lipids associate with NAs either via surface complexation or encapsulation of hydrophilic NA molecules within the aqueous core. Biophysical properties of liposomes can be modulated to achieve high NA entrapment, efficient cellular uptake and endosomal escape [32]. Due to this flexibility in designing the liposomal formulations, there are several liposome associated NA products in preclinical [79] and clinical studies (see Table 8.1). Both cationic as well as anionic liposomes have been used for delivery of NAs. Structures of commonly used lipids are shown in Fig. 8.1.

Structures of commonly used nonviral gene delivery vectors. DOTMA 1,2-di-O-octadecenyl-3-trimethylammonium propane, DOTAP 1,2-dioleoyl-3-trimethylammonium propane, DOPG 1,2-dioleoyl-sn-glycero-3-phospho-1′-rac-glycerol, DOPS 1,2-dioleoyl-sn-glycero-3-phospho-l-serine, DOPE 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine, PEI polyethyleneimine, PLGA poly-lactic-co-glycolide, PLA polylactic acid, PPI polypropyleneimine, PAMAM polyamidoamine
4.2.1.1 Cationic Liposomes
Cationic liposomes electrostatically interact with NAs to form the lipid-NA complexes also known as “lipoplexes.” Liposomes help in NA internalization into the cells, as well as its release from endosomes upon interaction with anionic endosomal lipids. The first cationic lipid, DOTMA (Fig. 8.1) was synthesized by Felgner and co-workers [102]. This was followed by other cationic lipids such as DOTAP [103, 104], DOSPA [105], DOSPER [106] and Oligofectamine [44]. Liposomes prepared with these lipids in association with fusogenic lipids (such as di-oleyl phosphatidylethanolamine or DOPE) and/or cholesterol have shown transfection efficiencies in vitro [107–112].
The aforementioned first generation cationic lipids are cytotoxic which restricts their use in vivo [30]. This toxicity could be attributed to their positive charge responsible for nonspecific interactions causing interference with the activity of ion channels, reduction in cellular adhesion and membrane destabilization [113, 114]. Additionally, cationic substances have shown to cause cellular stress in terms of altered actin cytoskeleton and abnormal cell proliferation [115, 116]. Kedmi et al. observed hepatotoxicity and significant weight loss in mice injected with cationic nanoparticles [29]. Ikebe et al. reported triggered release of pro-inflammatory cytokines (such as, IL-2, TNF-α) possibly causing increased risk of angiogenesis and tumor progression [28]. Cationic liposomes also interact with negatively charged cellular components (such as serum proteins, opsonins, and enzymes), consequently leading to serum inactivation [117–119].
To overcome the toxicity and serum inactivation issues, efforts have been made to develop second generation cationic lipids with modified structures. The main components of a cationic lipid which are known to regulate transfection efficiency and cytotoxicity are hydrophobic lipid anchor, the linker, and hydrophilic head group. The lipid anchor is typically either a fatty chain (e.g., derived from oleic or myristic acid) or a cholesterol group, which determines the physical properties of the lipid bilayer, such as flexibility, interaction with membrane lipids, and the rate of lipid exchange [109], and these could indirectly impact NA activity. For example lipid tail unsaturation [120–123], shorter chain length [109, 124–126], and double-tailed lipids [127, 128] have been shown to improve the transfection efficiency. The linker group is an important determinant of the chemical stability, biodegradability, and transfection efficiency of the cationic lipid. Biodegradable lipids are being developed that can be metabolized by various enzymes (e.g., esterases, peptidases) to minimize any toxicity [123, 129]. For example ester linkages are less toxic due to their biodegradability when compared to nonbiodegradable linkers such as ether and carbamate [130–132]. The linker can also provide sites for the introduction of novel side chains to enhance targeting, uptake, and trafficking. The positively charged headgroup on the cationic lipid is responsible for interacting with the negatively charged DNA and is a critical determinant of the transfection and cytotoxicity of liposome formulations. The headgroups differ markedly in structure and may be single- or multiple-charged as primary, secondary, tertiary, and/or quaternary amines. Multivalent headgroups, such as spermine, in a T-shape configuration tend to be more effective than their monovalent counterparts at facilitating gene transfer [109, 133]. Also, the presence of hydroxyalkyl [109], or cyclic groups [134], increases the transfection efficiency. Continued progress toward a comprehensive relationship between lipid structure, its complexation with NA molecules, and the subsequent interaction with the biological system is necessary to facilitate the design of cationic lipids with optimal properties. Such a careful designing of cationic lipids has led to development of commercially available transfection reagents Lipofectamine 2000 (Invitrogen) and RNAiMax (Invitrogen) that are efficient and relatively safer compared to conventional lipid based reagents such as Lipofectin and Lipofectamine [135, 136].
4.2.1.2 Anionic Liposomes
Owing to the cationic charge based toxicity of first generation cationic lipids, significant work has been done to exploit the potential of anionic liposomes for safe and efficient NA delivery [31, 45, 137, 138]. However, entrapment of NAs within these lipids is challenging due to electrostatic repulsion between the two anionic charged species (lipid and NAs). For example, Foged et al. attempted preparing siRNA associated anionic liposomes. These formulations showed only 7–9 % encapsulation efficiency with no activity in HeLa cells [137]. Accordingly, for efficient lipoplex formation, a third moiety is required which can act as a bridging agent between anionic lipids and NAs. For example, Huang’s group prepared anionic lipoplexes using the cationic polymer, poly-l-lysine [35]. Others have prepared anionic liposomal formulations (1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/1,2-dioleoyl-sn-glycero-3-phospho-1′-rac-glycerol (DOPG), to efficiently deliver antisense oligonucleotides into hippocampal neurons [139] or bacterial cells [140]. Our group has developed anionic lipoplexes wherein anionic liposomes (DOPG/DOPE) have been complexed with DNA/siRNA with calcium ion bridges. These systems have shown equivalent transfection efficiency and no cytotoxicity when compared to commercial cationic liposomes, Lipofectamine™ 2000 in CHO-K1 (DNA lipoplexes) [31, 34, 141] and MDA-MB-231 cells (siRNA lipoplexes) [33]. Figure 8.2 represents confocal images that shows efficient (time-dependent) cellular uptake and endosomal release of anionic lipoplexes in MDA-MB-231 cells.

Confocal images of time-dependent (4 h) uptake and endosomal release of siRNA from calcium associated anionic lipoplexes. Cells were stained with Hoescht 33342 (blue) for nucleus and LysoTracker red DND-99 for lysosomes. Overlay images of blue, green, red, and transmitted light channels are shown. Reproduced from ref. [33]
4.2.2 Polymers
Polymers are macromolecules composed of linear or branched chains of repeated units of smaller molecules (monomers). Several cationic polymers have been employed for efficient gene delivery [142] (Fig. 8.1). Cationic polymers often contain high densities of primary, secondary, tertiary or quaternary amines, which are protonatable at neutral pH [143]. Due to the high density of positive charges cationic polymers self-assemble with DNA/RNA to generate condensed structures (polyplexes) that are usually taken up by cells via endocytosis [143]. Polymers facilitate endosomal release of NAs via the “proton sponge” effect [144]. Accordingly, low endosomal pH causes protonation of several amines in the polymer chain resulting in pH buffering. The buffering protons in endosomes attract counter ions (chloride ions) causing osmotic swelling, endosomal rupture, thereby releasing the entrapped NAs.
One of the early polymers used for gene delivery is Poly-l-lysine (PLL). PLL has low efficiency mainly because of its poor endosomal escaping capability [145] and therefore, it is often co-administered with chloroquine or fusogenic peptides [146, 147]. Alternately, endosomal release can be promoted by conjugating PLL with imidazole containing group such as histidine that gets protonated in acidic pH [148, 149]. Another polymer widely used as efficient gene delivery vector is polyethyleneimine (PEI). PEI has a high charge density due to the presence of nitrogen at every third position in the polymer chain [150] and therefore promotes cellular uptake and endosomal release. For example, linear-PEI (Jet-PEI, 22 kDa) has been utilized to efficiently deliver shRNA-pDNA upon intravitreal injection in mice [151]. For other examples see Table 8.2.
Multiple cationic charges on PEI facilitate effective condensation of NAs as well as promote cellular uptake; however, this also increases PEI cytotoxicity. Therefore, low molecular weight PEIs are favored over higher molecular weight PEIs [152, 153]. PEI toxicity is also contributed by the nonbiodegradable nature of these polymers. Recently, biodegradable PEIs with cleavable linkers such as disulfide [154] and amino esters [155, 156] have been prepared that showed low cytotoxicity and high transfection efficiency when compared to the unmodified PEIs. PEIs high charge density results in multi-contact points with the NA, thereby slowing DNA/RNA dissociation from the polymers, after endosomal escape. This issue was combated by employing linear PEIs instead of branched ones. For example Kleemann et al. showed high efficiency and low toxicity with linear PEI/DNA complexes injected into mice trachea, compared to branched PEI/DNA complexes [157].
Due to the nonbiodegradable nature of conventional PEIs, other biocompatible and biodegradable polymers have been exploited for efficient gene delivery. An example includes chitosan which is a linear cationic polymer consisting of glucosamine and N-acetyl d-glucosamine units. Chitosan has shown higher transfection efficiency compared to naked DNA, when injected intratracheally in mice [158]. However, chitosan efficiency was lower than that of PEI (25 kDa) probably due to less effective endosome escaping capability of chitosan. This issue was combated by optimizing degree of deacetylation in chitosan [159]. For example (84 % deacetylated) chitosan/siRNA nanoparticles resulted in efficient knockdown (~44 %) of TNF-alpha gene (inflammation marker) in mouse macrophages and thus were useful in arthritis therapy [160]. Chitosan grafted into PEI has shown to be an efficient (better than PEI alone) and safe gene delivery vector both in vitro as well as in vivo [161].
4.2.3 Dendrimers
Dendrimers are structured polymers that have attained recognition as nano-scale units or carriers for a wide range of applications [162]. These carriers are attractive candidates for gene delivery owing to their small condensed size, ease of preparation and functionalization, and their ability to display multiple copies of surface groups for biological recognition processes. Structurally dendrimers comprise a series of branches around an inner core. This unique core-shell design of dendrimers helps in the incorporation of both hydrophilic and hydrophobic moieties [163]. Dendrimers can be synthesized starting from the central core and working out toward the periphery (divergent synthesis) or in a top-down approach starting from the outermost residues (convergent synthesis) [164].
Two commercially available dendrimers are PAMAM (polyamidoamine) and PPI (polypropyleneimine) (Fig. 8.1) with ethylenediamine and butylenediamine as central core, respectively [163]. Because dendrimers are built from ABn-type monomers, each layer or “generation” of branching units (e.g., G1,G2) doubles or triples the number of peripheral functional groups. Therefore higher generations can accommodate greater payload and there are more sites available for conjugation [165]. For this reason sixth generation PAMAM is known to be an optimal gene transfection reagent (Polyfect™). These dendrimers have been employed to effectively deliver not only plasmid DNA as well as antisense oligonucleotides [166]. Additionally Polyfect™ successfully delayed tumor growth when employed for suicide cancer gene therapy [167, 168]. Other generations of PAMAM have also gained success in gene delivery. For example interleukin-10 gene complexed with G5-PAMAM was effective in improving graft survival in murine cardiac transplant model [169]. PPI dendrimers have shown high efficiency but their application is limited owing to their cytotoxicity at high charge ratios [170]. Cellular uptake of PAMAM and PPI dendrimers with net positive charges, occurs via electrostatic interactions with anionic cell surface followed by release from endosomes by the “proton sponge” effect [171].
Other than PPI and PAMAM, new materials have also been utilized in formulating dendrimers. For example, Ofek et al. prepared novel dendrimers using polyglycerolamine. Dendrimer of this material when complexed with luciferase-siRNA, showed significant gene silencing (85 %) within 24 h (pre-inoculated with U87-Luc human glioblastoma cells) after a single intratumoral injection in mice (dose—20 mg/kg polyglycerolamine, 5 mg/kg luciferase-siRNA). This effect lasted for 3–4 days [172].
4.2.4 Polymeric Nanoparticles
These nanoparticles are polymer based colloidal particles with entrapped moieties such as NAs. Nanoparticles can effectively protect NAs from enzymatic attack and more importantly, facilitate their prolong release thus making these carriers useful for the treatment of chronic ailments such as cancer and inflammatory diseases [173, 174]. Additionally nontoxic nanoparticles can be fabricated by using biocompatible and biodegradable polymers such as PLA (poly-lactic acid) and PLGA (poly-d,l-lactic-co-glycolic acid) (Fig. 8.1). These polymers consist of ester groups that hydrolyze and cause polymer degradation in aqueous environment, thereby releasing the entrapped material. Polymer degradation rate and hence the drug release rate can be controlled based on polymer properties such as molecular weight, co-monomer ratio, glass transition temperature, crystallinity, end groups, etc. [175, 176]. Once the nanoparticles are internalized by cells, endosomal release of entrapped NAs occurs via surface charge reversal of polymer in the nanoparticles. The ionized acidic groups of the polymer gets protonated in the low pH environment of endosomes, and interact with negatively charged endosomal lipids, thereby causing endosome destabilization [173].
PLGA is an FDA approved polymer and therefore is widely used in polymeric nanoparticles. PLGA nanoparticles can be prepared by several techniques including solvent-evaporation, nano-precipitation, solvent displacement [177], emulsion-diffusion [178]. These techniques involve use of organic solvents, vigorous stirring or homogenization. Such harsh conditions may adversely affect stability of NAs potentially lowering their activity. Additionally, limited size of nanoparticles and lack of favorable interactions between anionic polymer groups (in PLGA) and NA may result in low encapsulation efficiency. Several approaches have been adopted to overcome these challenges. As an example, high molecular weight polymers (100 kDa) have been utilized to effectively shield DNA from shear stress during the preparation process. DNA protection, in this case, was probably due to increase in solution’s viscosity attributed to large polymer chains [179, 180]. Increase in nanoparticle encapsulation efficiency has been achieved by: (1) reducing sonication time during nanoparticle formulation process [181]; (2) preparing large size PLGA particles (microspheres) [182]; (3) adding NAs after nanoparticle preparation. The third approach usually results in 80–100 % encapsulation if a bridging agent is employed to associate anionic PLGA nanoparticles with anionic NAs. Several moieties such as chitosan [178], PEI [183] and cetyl-PEI [184] have been utilized as bridging agents.
4.2.5 Microbubbles (Ultrasound-Mediated Delivery)
Microbubbles are micron sized (2–5 μm) gas-filled bubbles formed by purging air or heavy gas (sulphur hexafluoride, perfluoropropane, perfluorobutane) into a solution of proteins, polymers or lipids [38]. Microbubbles burst upon exposure to low frequency ultrasound. Due to this feature, microbubbles can be used for site-specific gene delivery: upon in vivo delivery, microbubbles carry genes until desired site is reached. Ultrasound is then applied to this site causing release of entrapped NAs. This release usually occurs due to ultrasound-triggered cavitation at the bubble-plasma membrane interface [185]. Gene delivery efficiency is dependent upon ultrasound pressure amplitude, pulse frequency as well as duration of exposure [39].
Gene loading in microbubbles can be obtained either by entrapping NAs internally or complexing these onto the bubble surface. Entrapment would protect NAs and therefore is a good technique if naked DNA is being used [37]; however, this method offers lower loading efficiency due to the limited space. To overcome this issue, NAs can be complexed on relatively large surface area of microbubbles but NAs must be pre-complexed with cationic liposomes/polymers for enzymatic stability. For example Lentacker et al. loaded PEGylated DNA-lipoplexes onto microbubble surface via biotin-avidin-biotin linkages. These formulations showed greater transfection efficiency when compared to PEGylated lipoplexes not associated with microbubbles, in human melanoma (BLM) cells [40]. In another study, microbubbles associated lipoplexes were prepared for effective messenger RNA delivery in dendritic cells [41].
Although microbubble technology can provide efficient site-specific delivery, there are some disadvantages associated with it. For example, exposure of microbubbles to ultrasound may increase blood temperature and cause hemolysis. Additionally, ultrasound-mediated microbubble cavitation may exert mechanical pressure and cause cell damage and even cell death [186]. Despite these shortcomings, microbubbles are the only nonviral delivery carriers that can be used for targeted delivery without the use of specific targeting strategies (passive/active).
5 Targeted Delivery
Delivery of a therapeutic molecule directly to the desired site of action not only accelerates therapy benefits but also reduces the chances of drug side effects especially in case of toxic drugs. Additionally, there is improved patient compliance as the dose frequency is lower with targeted systems. Various nonviral delivery vectors (discussed above) have been employed to achieve targeted gene delivery using passive and/or active targeting approaches (Table 8.2).
5.1 Passive Targeting
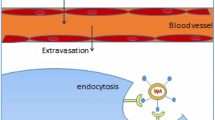
Passive targeting can be achieved by regulating physicochemical parameters of the delivery vector such as size, surface charge or composition, etc. in order to increase blood circulation time usually resulting in improved accumulation in target tissues. Such an approach is specifically useful for cancer therapy since tumor cells have leaky vasculature (200–780 nm fenestrae) [187] and poor lymphatic drainage, the phenomena is known as enhanced permeability and retention (EPR) effect (Fig. 8.3). Therefore, extravasation of tumor cells is achievable with larger sized (100–200 nm) particles that are forbidden in the regions of normal tissues where there are tight junctions. However, the maximum limit for size of particles penetrating tumor tissues depends on the type of tumor. For example PEGylated liposomes of size 126 nm but not 400 nm could penetrate solid tumors, although neither of these particles was detected in healthy cells [188].

Passive targeting approaches utilizing EPR (enhanced permeability and retention) effect in tumor cells: gene delivery systems (large sized/PEGylated) can extravasate tumor cells due to leaky vasculature and stay there for a longer time due to poor lymphatic drainage in tumor vasculature. On the other hand, large/PEGylated delivery systems are unable to penetrate normal tissues due to tight junctions in endothelial cells. However, if some particles pass through normal tissues, these get cleared off rapidly due to the lymphatic drainage
Another approach to prolong particle blood circulation time is by employing polymers such as poly-ethylene glycol (PEG) into the delivery carriers. PEGs are hydrophilic polymers that can easily “fool” macrophages, thereby preventing uptake by reticulo-endothelial system (RES). Accordingly, PEG decorated delivery vectors have delayed clearance and thus there is a greater chance of particles accumulating in the desired site [189]. Additionally, steric repulsion between the PEG chains inhibits close association of delivery vectors. Even if there is an overlap of PEG chains, a region with high osmotic pressure is created in the overlapped region that forces the solvent to enter in, and push the PEGylated nanoparticles away. In this way, PEG chains provide steric stabilization to particles, the efficiency of which depends on the PEG chain length [190]. For these reasons, PEG is useful for long-term gene silencing. For example, mice injected with PEGylated cationic lipoplexes (SNALP) has shown efficient gene silencing of hepatitis B virus for up to 7 days with single dose [191]. Even longer silencing was achieved with single dose i.v. injection of PEGylated lipoplexes wherein tumor suppression was achieved for as long as 3 weeks [192]. Qi et al. showed efficient gene transfection with 6 % PEG-G6-PAMAM dendriplexes injected intramuscularly in neonatal mice [193]. Also William’s lab prepared PEG-PEI conjugates in various molecular weight ratios (PEG:PEI), for effective delivery of antisense oligonucleotides, in vitro as well as in vivo [194]. Although PEG enhances the bio-distribution of gene delivery systems, it interferes with direct interaction of particle surface with cell membrane, thereby reducing the cellular uptake. Thus, PEG chains are usually attached to active targeting ligands that specifically bind to cell surface receptors resulting in efficient cellular internalization [195]. This approach is known as “active targeting” and has been discussed in Sect. 5.2.
5.2 Active Targeting
Unlike passive targeting which is a nonspecific approach, active targeting employs specific targeting moieties that bind efficiently to antigens/receptors (over-)expressed on target cell surfaces (cell-surface targeting). Alternately, ligands with affinity for molecules on the surface of a specific cell organelle, can be employed for organelle-specific targeting. Such approaches remarkably improve gene delivery efficiency and also guarantees minimal side effects. Many researchers are working on cell-surface as well as organelle specific targeting. In general, such active targeting approaches are often coupled with passive targeting approaches (PEGylation) since the combination is known to yield better results [46, 195]. Details on various active targeting approaches are as follows:
5.2.1 Cell-Surface Targeting
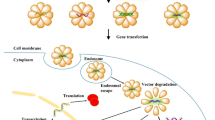
Targeting cell-surface receptors is an attractive concept to achieve specific binding and internalization using the incorporation of cell-binding ligands into delivery carriers. These ligands bind particularly to certain receptors that are selectively expressed or overexpressed on the surface of diseased cells when compared to normal cells. For example tumor cells are fast growing and therefore have greater nutritional requirements compared to normal cells. Consequently, these have overexpressed cell surface receptors required for uptake of nutritional agents such transferrin receptor for iron [196], glycosylated receptor for carbohydrates [197], and folate receptor for folic acid [198]. Accordingly, incorporation of such moieties into nonviral vectors may facilitate rapid and efficient accumulation at the target site. Uptake of cell-surface targeted delivery systems occurs via receptor-mediated endocytosis (RME) as illustrated in Fig. 8.4. Following are the examples of targeting ligands used for cell-surface targeting (see also Table 8.2):

Active targeting approaches for targeted delivery of NAs: Uptake of these particles occurs via receptor-mediated endocytosis (RME). (a) Targeting ligand in a gene delivery system, assist in cell-surface targeting upon specifically binding to cell-surface receptors (transferrin/small molecule/peptide/antibody/aptamer). (b) Early endosomes mature into (c) late endosomes. (d) At this point delivery carrier must assist in endosomal release of NAs which would otherwise (e) degrade in lysosomes. (f) Upon endosomal release, certain NAs (antisense, DNAzyme/RNAzyme, aptamers, miRNA and siRNA) remain in cytoplasm and function here. (g) Others including pDNA/shRNA with/without NLS, penetrate into the nucleus, whereas (h) pDNA with MLS enter mitochondria (“g” and “h” represent organelle-specific targeting). NLS nucleus localization sequence. MLS mitochondrial leader sequence
5.2.1.1 Small Molecules
Folate (folic acid) has a high binding affinity for folate receptors whose expression is upregulated in many tumors [199, 200]. Folate is a preferable ligand due to its small size, non-immunogenicity, temperature and pH stability (over a broad range), and therefore has been widely used in targeted gene delivery systems. Kim et al. showed that folate-PEG-PEI/shRNA complexes efficiently inhibited green fluorescent protein (GFP) expression in KB cells (tumor cells). At N/P ratio of 24/1, folate polyplexes showed 90 % silencing, whereas it was only 60 % in case of non-folate polyplexes [201]. Yoshizawa et al. used PEGylated-folate lipoplexes for efficient knockdown of HER-2 in KB cells [198]. First in vivo “proof of concept” for folate targeting was demonstrated by Hofland et al. wherein folate-PEGylated lipoplexes showed efficient accumulation in tumors when compared to non-folate PEGylated lipoplexes [195].
Asialoglycoprotein receptor (ASGP-R) is expressed predominantly in liver cells and has affinity for a variety of carbohydrates including lactose, galactose, and N-acetylamine. Accordingly, these moieties incorporated into nonviral vectors can be useful for liver [202]. For example DNA lipoplexes prepared with galactosylated liposomes showed significantly higher luciferase expression in HepG2 cells when compared to unmodified liposomes [203]. N-acetylgalactoseamine conjugated -poly(vinyl ether) successfully silenced expression of apolipoprotein and peroxisome proliferator-activated receptor alpha genes, in mouse liver [204]. In a recent study, systemic administration of galactosylated-liposome siRNA nanoparticles markedly helped reduce liver injury in mice infected with hepatitis [205].
Cholesterol and its derivatives have also been used for targeted gene delivery. For example, cholesterol conjugated-apolipoproteinB-siRNA has been utilized to efficiently downregulate apo-B gene expression (57 ± 6 % compared to control), that is otherwise responsible for hypercholesterolemia [206].
Anisamide containing drugs have been shown to have good binding affinity for sigma receptors that are overexpressed in many tumors. Accordingly, anisamide containing delivery systems have been utilized to achieve tumor specific targeting. For example anisamide conjugated PEGylated liposomes have shown efficient luciferase silencing compared to non-anisamide lipoplexes, in lung cancer cells (H-460) [207]. Guo et al. formulated anisamide-PEG-cyclodextrin nanoparticles for siRNA delivery to prostate tumors in mice. After 22 days of injection tumor volume reduced significantly with anisamide-PEG-cyclodextrin siRNA complexes compared to PEG-cyclodextrin siRNA complexes [208].
5.2.1.2 Transferrin
Transferrin, an iron transporting serum glycoprotein, upon binding to the transferrin receptor can be internalized efficiently into the cells (via RME-Fig. 8.4) and through this physiological process, iron is delivered into the cells. Although the transferrin receptor is expressed on almost all cell types, it is overexpression in many rapidly dividing tumors [196]. Accordingly, transferrin bedecked nonviral carriers have been proved to be effective in gene therapy. For example, Kirchies et al. showed tumor targeting with transferrin-PEI/DNA when injected systemically in mice growing neuro2 tumors [209]. Huang et al. demonstrated successful brain targeting of luciferase DNA using transferrin-PEG-PAMAM dendrimers upon intravenous administration in mice [210]. Transferrin-conjugated PEGylated liposomes loaded with anti-BCR-ABL siRNA have been utilized for the treatment of chronic myeloid leukemia in K562 and LAMA-84 cells [211]. A transferrin-containing multi-component siRNA formulation (CALAA-01) is in Phase I clinical trials [212] (Table 8.1).
5.2.1.3 Antibodies or Antibody Fragments
Specific antibodies for certain cell-surface markers can also be used for targeting. For example a monoclonal antibody directed to the CD3 surface marker in human T-cell leukemia enabled efficient gene delivery in vitro [213]. Antibodies have two main fragments: Fc (constant) and Fab (variable). The Fc fragment is responsible for immunogenicity of antibody since it has multiple compliment activation and macrophage binding sites, whereas Fab fragment is non-immunogenic and has antigen binding sites which are responsible of its specific interaction with the antigen. Accordingly, use of antibody fragments has become more common than the whole antibody. For examples, ErbB2, a tumor marker that is highly upregulated in many human breast and prostate cancers, was targeted with a delivery system containing a single-chain antibody [214]. Other examples include the successful use of liposome-polycation-hyaluronic acid modified with single chain antibody fragment (scFv) for dual delivery of siRNA and miRNA when administered systemically in lung-metastasis bearing mice [215]. Cell-type-specific targeting using antibodies was demonstrated by Song et al. In this work, a fusion protein based on protamine coding sequence (F105b) was conjugated at the C terminus to Fab fragment of an HIV-1 envelope antibody. When antibody-F105b siRNA complexes were injected intravenously in mice inoculated with HIV envelope expressing B16 melanoma cells, silencing was specifically observed in HIV-1 expressing cells. When the antibody (HIV-1 envelope) in such a delivery system, was replaced with ErbB2 single-chain antibody (ErbB2-protamine-siRNA), siRNA was observed to accumulate specifically in ErbB2-expressing cancer cells [47]. Pirollo et al. used transferrin antibody targeted lipoplexes. Three intravenous doses showed knockdown of HER-2 gene in mice tumors for 30 days [196]. Santos prepared antagonist G associated targeted PEGylated liposomes for downregulation of Bcl-2 in SCLC SW2 tumor cells [216]. Recent efforts are focusing on antibody-based targeting for treatment/diagnosis of brain cancer. For example Shen et al. prepared PEG-PEI superparamagnetic iron oxide nanoparticles modified with GD2 single chain antibody which binds specifically to neuroblastoma cell-surface receptors. These nanoparticles complexed with Bcl-2 siRNA showed efficient silencing of Bcl-2 gene in human neuroblastoma cells compared to non-targeted nanoparticles (no antibody). Additionally significant apoptosis and suppression of tumor growth was observed with these targeted nanoparticles [217].
5.2.1.4 Peptides
Peptide-based targeting of nonviral delivery vectors has become possible because of our increased understanding of the discrete peptide sequences of proteins involved in cell–cell and effector–cell interactions. Various peptides have been identified as ligands that bind to receptors present specifically in diseased cells [218–222]. Accordingly, delivery systems incorporated with peptide-based ligands can facilitate efficient targeted delivery of genes to the diseased site. For example, Moreira et al. prepared tumor targeted delivery vectors using a hexapeptide analogue, antagonist G, of the neurotransmitter substance P, which binds to receptors on the surface of the small cell lung cancer (SCLC) and blocks the action of multiple neuropeptides, such as vasopressin, gastrin releasing peptide, and bradykinin [223]. Asai et al. isolated peptides specific for tumor angiogenic vasculature by in vivo bio-planning of a phage-displayed peptide library [224]. Phage fused MCF-7 cell specific peptide “DMPGTVLP” incorporated into liposomes, has been utilized for efficient siRNA delivery to breast cancer (MCF-7) cells [218]. In another study, RGD (arginine-glycine-aspartic acid tripeptide) associated PEGylated nanoparticles injected intravenously, have shown significant tumor growth inhibition in nude mice bearing human glioma U87 xenografts, when compared to non-targeted siRNA complexes [221]. Streipe et al. prepared liposomal siRNA delivery systems modified with RGD-peptide for specific recognition by integrin receptors in alveolar rhabdomyosarcoma cells. These targeted systems showed improved silencing over the unmodified ones [220]. Arginine has been used as a targeting ligand by Zhang et al. where a PEGylated octamer of arginine (R8) was decorated on siRNA-loaded cationic liposomes for efficient silencing of HDM2 gene (human double minute gene2) [222]. Kim et al. synthesized poly-l-arginine conjugated PEG (PLR-PEG) and prepared liposomes using the cationic lipid DOTAP, fusogenic lipid DOPE, cholesterol, and PLR-PEG. Such arginine conjugated targeted liposomes were utilized to knockdown GFP in H4II-E and HepG2 cells. Accordingly, it was found that PLG-PEG liposomes were 30 % more efficient than conventional liposomes (non-PEGylated and non-targeted) at N/P ratio of 30:1 [219]. Recently, Siould et al. showed efficient silencing of survivin (an important gene for cancer cell survival) using siRNA conjugated to gastrin-releasing peptide (GRP), which binds specifically to GRP receptors on the surface of breast cancer cells [225].
Other than their application in cell-surface targeting, peptides have also been employed to improve cellular uptake, endosomal escape and/or nucleus localization gene delivery vectors. A majority of peptides show pH-dependent fusogenic and endosomolytic (endosomal escape) activity and are believed to mimic virus like journey into the cell. These peptides also called cell-penetrating peptides (CPPs) are random coil at pH 7.0 but undergo a conformational change into an amphipathic alpha-helix at endosomal pH. This conformational change induces the fusion and lysis of endosomal membrane causing the release of entrapped genes [226]. Certain examples of CPPs include TAT, penetratin, transportan, and INF [227]. Torchillin et al. have shown TAT-liposomes to be more effective and less cytotoxic than liposome control when injected subcutaneously in mice inoculated with Lewis lung carcinoma tumors [228]. Penetratin or transportan have been conjugated to siRNA for knockdown of many target proteins such as luciferase and green fluorescent protein in several cell lines [95]. Since direct conjugation of these peptides may affect NA bioactivity, recent work is focusing on non-covalent association of peptide with DNA/siRNA. For example Simeoni et al. prepared MPG peptide (sequence derived from HIV-1 and NLS) that associates with siRNA via electrostatic and hydrophobic interactions and helps with efficient siRNA delivery in vitro as well as in vivo [44, 229]. Deshayes et al. prepared another peptide (CADY) and formed stable nanostructures with siRNA for potent delivery in primary cells [230]. The nucleus localization potential of peptides has been discussed in details in Sect. 5.2.2.1.
5.2.1.5 Aptamers
Aptamers are NA (Sect. 2.3) or peptide-based molecules that bind specifically to a particular target moiety. As more and more surface molecules are being identified as biomarkers for a particular tumor, aptamer-based cell-surface targeted delivery is gaining importance. For example PSMA (prostrate-specific membrane antigen) molecule occurs predominant on the surface of human prostate cancer cells and therefore incorporation of PSMA in gene delivery systems have been successfully employed for the treatment of prostate cancer [231, 232]. Kurosaki and co-workers developed MUC-1 aptamer containing PEI/pDNA complexes that showed higher transfection efficiency in lung cancer cells (~threefold greater) and tumor-bearing mice (~4-fold greater), compared to non-targeted polyplexes [233]. AS1411 is a DNA aptamer used in the treatment of acute myeloid leukemia. AS1411 targets cancer cell surface protein, nucleolin, gets internalized and inhibits nuclear factor-κB [234]. AS1411 also destabilizes mRNA of the anti-apoptotic protein, B-cell lymphoma protein 2 (BCL-2) [235]. Other examples of aptamer-targeting are included in Table 8.2.
5.2.2 Organelle-Specific Targeting
For certain therapeutic molecules such as pDNA (and shRNA), it is not sufficient to accumulate in cytoplasm. These moieties further need to enter specific cell organelles such as nucleus, mitochondria etc., in order to transcribe and eventually express therapeutic proteins. Penetration of NAs into an organelle is challenging due to their large size and hydrophilicity. However, various (see below) strategies have been adopted to design gene delivery systems for efficient accumulation in a particular cell organelle.
5.2.2.1 Nucleus Targeting
After endosomal release, pDNA/shRNA molecules need to find their way into the nucleus in order to employ therapeutic benefits (Fig. 8.4). Nuclear transfer of pDNA/shRNA usually occurs either via passive entry during cell division and/or by active process via nuclear pores [147, 236]. Besides, non-nuclear pore pathways for DNA nuclear import (in cells that are not undergoing active division) have been reported. For example, it has been speculated that DNA-PEI complexes may enter the nucleus via non-nuclear-pore pathways such as fusion with nuclear membrane phospholipids [237]. Although the entry of these polyplexes into the nucleus may be an effective method of delivery of DNA to the nucleus, the potential for interaction of these polycations with host genes is a significant concern. Accordingly, focus is towards improving nuclear uptake of DNA via nuclear-pore complex (NPC) especially in non-dividing or slowly dividing cells. NPC has a molecular sieve function allowing small molecules (up to 50 KDa) to diffuse through, whereas larger molecules require an active transport mechanism to pass [238]. Active transport of macromolecules (e.g., cytoplasmic proteins) is controlled by short peptides known as nuclear localization sequence (NLS) [239]. NLS has affinity for the proteins of NPC like importin-α. Activated importin-α further activates importin-ß protein which then helps in opening the nuclear-pore complex (see Fig. 8.5). During this active transport the nuclear pore complex opens from ca. 10 to ca. 40 nm and thus can accommodate plasmid DNA, which in its condensed form has a diameter of ca. 25 nm [240, 241]. NLS associated DNA can therefore be utilized to enhance DNA import to nucleus.

Schematic representation of the nuclear localising peptide-conjugated DNA nuclear import mechanism. A well-defined nuclear targeting peptide signal sequence is conjugated to the DNA. Intracellular delivery of the DNA–NLS conjugate is mediated by formation of lipoplexes via the electrostatic interaction of negatively charged DNA molecules with cationic lipid molecules. In the cytoplasm, the importin-α transport receptor binds the DNA–NLS conjugate and together with importin-β mediates interaction with the nuclear pore complex to translocate the import complex into the nucleus. Reproduced from ref. [239]
NLSs are usually characterized by one short peptide sequence containing many lysine and arginine residues [242]. The first NLS sequence (PKKKRKV) was derived from the tumor antigen, simian virus-40 [243]. Various NLSs have been attached with plasmid DNA via covalent or non-covalent association to facilitate nuclear uptake [24, 54, 244, 245]. However, both the approaches have their own challenges. For example, it has been shown that non-covalent association may result in separation of NLS from pDNA in the extracellular environment, if the binding is not strong enough [24, 246]. With respect to direct conjugation approach, NLS-pDNA conjugates have shown reduced transfection efficiency compared to pDNA alone [247]. This is apparently due to the requirement of several NLS molecules/plasmid to cause efficient nuclear uptake since some of the cationic NLSs could neutralize charge on DNA [248, 249]. However, conjugation of multiple NLSs to pDNA may alter its conformation, thereby impeding DNA activity [250]. To combat this issue, spacer moieties between NLS and DNA, such as oligonucleotides [251], psoralen [54] or PEGs [252] have been used. Alternately, NLS has been incorporated into the nonviral vectors, instead of DNA [246, 253]. For example, efficient nuclear uptake of PLGA nanoparticles [254] and HPMA copolymer [54, 255] has been reported when the NLS is attached directly to carrier (nanoparticles/polymer). NLS has also been conjugated to quantum dot (QD) nanoparticles to achieve rapid nuclear uptake [256, 257]. A recent review covers many examples for NLS associated targeted gene delivery systems [258].
5.2.2.2 Mitochondrial Targeting
Mitochondria are membrane-bound organelles whose main function is to produce adenosine triphosphate (ATP), the chemical source of energy. Mitochondria also regulate cell signaling, cell proliferation and apoptosis [259, 260]. Genetic mutation (point mutation, deletions, missense) of mitochondrial DNA (mtDNA) may cause mitochondrial dysfunction leading to metabolic disorders, neurodegenerative diseases and even tumors [48]. Various efforts are being made to cure genetic defects in mtDNA by delivering correct copy of DNA into the mitochondria [50–53].
Researchers have identified certain features which when incorporated into delivery vectors, could facilitate efficient mitochondrial delivery. For example, mitochondria have a very high inner membrane potential (180–200 mV) [49] and therefore attract molecules with high cationic charge density. These molecules need to be amphiphilic so that these can be accumulated onto the mitochondrial surface. For example, vesicles of an amphiphilic cationic surfactant, dequalinium chloride (DQA), were shown to efficiently transport DNA onto the mitochondrial surface [50, 51]. Unlike other cationic lipids (such as DOTAP, DOTMA), DQA release most of the DNA upon encountering cardiolipin-rich anionic membrane of mitochondria when compared to lesser anionic inner-cytosolic membrane. Although with DQAs, DNA could be delivered efficiently onto the surface of mitochondria, DNA internalization was minimal [51]. This issue was resolved with DNA conjugated to mitochondria targeting peptide also known as mitochondrial leader sequence (MLS) (Fig. 8.4) [50]. MLS has also been used with low molecular weight PEI (2,000 Da) to effectively deliver DNA to mitochondria in a cell-free assay [52]. A recent review has included many other examples for effective mitochondrial delivery of NAs using various delivery systems [53].
6 Multifunctional Nano-Carriers
After tremendous research on nonviral delivery carriers, it has been concluded that it is more beneficial to use combinational carriers (more than one type of vector, PEG, targeting ligands) compared to when a single approach is employed. This is evident from various examples in the literature some of which have been enumerated here: PLGA nanoparticles entrapping PEI-siRNA complexes showed greater efficiency in vivo compared to non-PEI containing PLGA nanoparticles [184]. In another strategy, chitosan-siRNA encapsulated in PLGA nanofibers have shown to cause 50 % gene silencing and no significant toxicity (48 h post-transfection) in non-small-cell lung carcinoma cells (H1299) [261]. Xun et al. prepared biodegradable PEI conjugated with small hydrophobic lipid molecules. These conjugates showed high transfection efficiency and low cytotoxicity in human non-small-cell lung carcinoma when compared to PEI (25 kDa) alone [262]. Rui et al, prepared PEI condensed siRNA nanocomplexes and coated these with lipid (egg phosphatidylcholine) and human apolipoprotein A-I, to achieve effective and safe delivery of siRNA in liver cancer (hepatoma) cells [263]. Dextran grafted PEI–DNA complexes incorporated with nuclear localization sequence (Sv7) showed efficient GFP expression in vitro (HepG2, HeLa, 293 T cells) as well as in vivo (mice xenograft model) [264]. A novel triblock copolymer composed of PEG, PLGA, and PLL (mPEG-PLGA-PLL) has shown gene silencing greater than Lipofectamine™ with no cytotoxicity in human lung cancer cells (SPC-A1) [265]. Oishi and coworkers prepared a triblock polymer conjugated to a targeting ligand (lactose), PEG, and siRNA on three different sites for efficient cellular uptake as well as endosomolytic activity [266]. Owing to their great potential and proven success, such multifunctional carriers have become a favorite of researchers working on the development of efficient gene delivery systems.
7 Summary and Future Perspectives
Targeted NA delivery is a challenge that requires the development of delivery vectors that specifically direct NAs to the desired site without adversely affecting healthy cells. As evident from various examples covered in this chapter, several labs have developed safe and efficient targeted gene delivery systems by employing active and/or passive approaches. Despite a remarkable success of these products in preclinical studies, there are only two targeted gene delivery products currently in clinical trials (Table 8.1 and [57]). This could apparently be due to product failure either in the late-stage preclinical (primates) or early-stage clinical trials. Such failures could be the result of wrong selection on animal models (preclinical stage), scale-up issues at industrial scale, or inter-patient variability (pharmacogenomics). Therefore, much work needs to be done to fill this gap between preclinical and clinical stages in order to have numerous targeted nucleic acid products in the clinics and eventually in the market.
References
Yei S et al (1994) Adenovirus-mediated gene transfer for cystic fibrosis: quantitative evaluation of repeated in vivo vector administration to the lung. Gene Ther 1(3):192–200
Aiuti A et al (2002) Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296(5577):2410–2413
Cavazzana-Calvo M et al (2004) Gene therapy for severe combined immunodeficiency. Annu Rev Med 56(1):585–602
Ganly I, Soutar DS, Kaye SB (2000) Current role of gene therapy in head and neck cancer. Eur J Surg Oncol 26(4):338–343
Wahlfors T et al (2006) In vivo enhancement of herpes simplex virus thymidine kinase/ganciclovir cancer gene therapy with polyamine biosynthesis inhibition. Int J Cancer 118(11):2907–2910
Baker AH (2002) Development and use of gene transfer for treatment of cardiovascular disease. J Card Surg 17(6):543–548
Dishart KL et al (2003) Gene therapy for cardiovascular disease. J Biomed Biotechnol 2003(2):138–148
Barkats M et al (1998) Adenovirus in the brain: recent advances of gene therapy for neurodegenerative diseases. Prog Neurobiol 55(4):333–341
Mark HT et al (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11(5):551–555
National Institute of Health (1990) The first gene therapy trial. http://history.nih.gov/exhibits/genetics/sect4.htm#2. Accessed 5 Jul 2013
The Human Genome Management Information System (HGMIS) (2011) About the human genome project: what is the human genome project. http://www.ornl.gov/sci/techresources/Human_Genome/project/about.shtml. Accessed 5 Jul 2013
Gene Therapy Clinical Trials Worldwide 2013. http://www.wiley.com/legacy/wileychi/genmed/clinical/. Accessed 4 Mar 2013
Raty JK et al (2010) Gene therapy: the first approved gene-based medicines, molecular mechanisms and clinical indications. Curr Mol Pharmacol 1(1):13–23
Richards S (2012) Gene therapy arrives in Europe. The Scientist 1(1):2
Masuda T, Akita H, Harashima H (2005) Evaluation of nuclear transfer and transcription of plasmid DNA condensed with protamine by microinjection: the use of a nuclear transfer score. FEBS Lett 579(10):2143–2148
McAllister DV, Allen MG, Prausnitz MR (2000) Microfabricated microneedles for gene and drug delivery. Ann Rev Biomed Eng 2(1):289–313
Yang N-S et al (1990) In vivo and in vitro gene transfer to mammalian somatic cells by particle bombardment. Proc Natl Acad Sci 87(24):9568–9572
Molnar MJ et al (2004) Factors influencing the efficacy, longevity, and safety of electroporation-assisted plasmid-based gene transfer into mouse muscles. Mol Ther 10(3):447–455
Choung S et al (2006) Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun 342(3):919–927
Hall AH et al (2004) RNA interference using boranophosphate siRNAs: structure-activity relationships. Nucleic Acids Res 32(20):5991–6000
Layzer JM et al (2004) In vivo activity of nuclease-resistant siRNAs. RNA 10(5):766–771
Parrish S et al (2000) Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol Cell 6(5):1077–1087
Amarzguioui M et al (2003) Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res 31(2):589–595
Escriou V et al (2003) NLS bioconjugates for targeting therapeutic genes to the nucleus. Adv Drug Deliv Rev 55(2):295–306
Walther W, Stein U (2000) Viral vectors for gene transfer. Drugs 60(2):249–271
Yang Y, Ertl HC, Wilson JM (1994) MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity 1(5):433–442
Yang Y et al (1995) Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol 69(4):2004–2015
Ikebe M et al (2009) Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J Surg Oncol 100(8):725–731
Kedmi R, Ben-Arie N, Peer D (2010) The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 31(26):6867–6875
Lv H et al (2006) Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release 114(1):100–109
Srinivasan C, Burgess DJ (2009) Optimization and characterization of anionic lipoplexes for gene delivery. J Control Release 136(1):62–70
Kapoor M, Burgess DJ (2012) Physicochemical characterization of anionic lipid-based ternary siRNA complexes. Biochim Biophys Acta 1818(7):1603–1612
Kapoor M, Burgess DJ (2012) Efficient and safe delivery of siRNA using anionic lipids: formulation optimization studies. Int J Pharm 432(1–2):80–90
Patil SD, Rhodes DG, Burgess DJ (2004) Anionic liposomal delivery system for DNA transfection. AAPS J 6(4):e29
Lee RJ, Huang L (1996) Folate-targeted, anionic liposome-entrapped polylysine-condensed DNA for tumor cell-specific gene transfer. J Biol Chem 271(14):8481–8487
Kapoor M, Burgess D (2013) Cellular uptake mechanisms of novel anionic siRNA lipoplexes. Pharm Res 30(4):1161–1175
Frenkel PA et al (2002) DNA-loaded albumin microbubbles enhance ultrasound-mediated transfection in vitro. Ultrasound Med Biol 28(6):817–822
Cool SK et al. (2013) Enhancing nucleic acid delivery with ultrasound and microbubbles. In: Nanotechnology for nucleic acid delivery. Springer, New York, p 195–204
Rahim A et al (2006) Physical parameters affecting ultrasound/microbubble-mediated gene delivery efficiency in vitro. Ultrasound Med Biol 32(8):1269–1279
Lentacker I et al (2009) Ultrasound exposure of lipoplex loaded microbubbles facilitates direct cytoplasmic entry of the lipoplexes. Mol Pharm 6(2):457–467
De Temmerman M-L et al (2011) mRNA-Lipoplex loaded microbubble contrast agents for ultrasound-assisted transfection of dendritic cells. Biomaterials 32(34):9128–9135
Ogris M et al (1999) PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther 6(4):595–605
Harris TJ et al (2010) Tissue-specific gene delivery via nanoparticle coating. Biomaterials 31(5):998–1006
Simeoni F et al (2003) Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res 31(11):2717–2724
Pulford B et al (2010) Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrP on neuronal cells and PrP in infected cell cultures. PLoS ONE 5(6):e11085
Arima H et al (2012) Potential use of folate-polyethylene glycol (PEG)-appended dendrimer (G3) conjugate with beta-cyclodextrin as DNA carriers to tumor cells. Cancer Gene Ther 19(5):358–366
Song E et al (2005) Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol 23(6):709–717
Heller A, Brockhoff G, Goepferich A (2012) Targeting drugs to mitochondria. Eur J Pharm Biopharm 82(1):1–18
Vaidya B et al (2009) Targeted nucleic acid delivery to mitochondria. Curr Gene Ther 9(6):475–486
D’Souza GGM, Boddapati SV, Weissig V (2005) Mitochondrial leader sequence-plasmid DNA conjugates delivered into mammalian cells by DQAsomes co-localize with mitochondria. Mitochondrion 5(5):352–358
D’Souza GGM et al (2003) DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J Controlled Release 92(1–2):189–197
Choi JS et al (2006) Low molecular weight polyethylenimine-mitochondrial leader peptide conjugate for DNA delivery to mitochondria. Bull Kor Chem Soc 27(9):1335–1340
Edeas M, Weissig V (2013) Targeting mitochondria: strategies, innovations and challenges: the future of medicine will come through mitochondria. Mitochondrion, 2013. (In Press) Corrected proof: p. http://dx.doi.org/10.1016/j.mito.2013.03.009
Yoo HS, Jeong SY (2007) Nuclear targeting of non-viral gene carriers using psoralen-nuclear localization signal (NLS) conjugates. Eur J Pharm Biopharm 66(1):28–33
Yu H et al (2011) Effects of MDM2 promoter polymorphisms and p53 codon 72 polymorphism on risk and age at onset of squamous cell carcinoma of the head and neck. Mol Carcinog 50(9):697–706
Fioretti D et al (2010) DNA vaccines: developing new strategies against cancer. J Biomed Biotechnol 2010:1–16
Clinical Trials. www.clinicaltrial.gov. Accessed 5 Jul 2013
Malecki M (2012) Frontiers in suicide gene therapy of cancer. J Genet Syndr Gene Ther 2012(3):e114
Kim JH et al (2012) Therapeutic effect of genetically modified human neural stem cells encoding cytosine deaminase on experimental glioma. Biochem Biophys Res Commun 417(1):534–540
Shen L-Z et al (2002) Specific CEA-producing colorectal carcinoma cell killing with recombinant adenoviral vector containing cytosine deaminase gene. World J Gastroenterol 8(2):270–275
Ueda K et al (2001) Carcinoembryonic antigen-specific suicide gene therapy of cytosine deaminase/5-fluorocytosine enhanced by the cre/loxP system in the orthotopic gastric carcinoma model. Cancer Res 61(16):6158–6162
Dass CR (2004) Deoxyribozymes: cleaving a path to clinical trials. Trends Pharmacol Sci 25(8):395–397
Ke X, Yang Y-c, Hong S -l (2011) EBV-LMP1-targeted DNAzyme restrains nasopharyngeal carcinoma growth in a mouse C666-1 xenograft model. Med Oncol 28(1):326–332
Lipkin D, Talbert PT, Cohn M (1954) The mechanism of the alkaline hydrolysis of ribonucleic acids. J Am Chem Soc 76(11):2871–2872
Diener JL et al (2009) Inhibition of von Willebrand factore-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost 7(7):1155–1162
Ni X et al (2011) Nucleic acid aptamers: clinical applications and promising new horizons. Curr Med Chem 18(27):4206
Zhou J, Rossi JJ (2012) Therapeutic potential of aptamer-siRNA conjugates for treatment of HIV-1. BioDrugs 26(6):393–400
Dias N, Stein CA (2002) Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther 1(5):347–355
Aartsma-Rus A, van Ommen G-JB (2009) Progress in therapeutic antisense applications for neuromuscular disorders. Eur J Hum Genet 18(2):146–153
Atlantic Healthcare (2010): Announces it is commencing manufacture and international supply of Alicaforsen for inflammatory bowel disease under named patient supply regulations http://www.atlantichc.com/newsdocs/20100519.pdf.
Wahid F et al (2012) MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta 1803(11):1231–1243
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Bader AG (2012) miR-34a microRNA replacement therapy is headed to the clinic. Front Genet 3(120):1–9
Pillai RS, Bhattacharyya SN, Filipowicz W (2007) Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol 17(3):118–126
Alvarez JP et al (2006) Endogenous and synthetic microRNAs stimulate simultaneous, efficient, and localized regulation of multiple targets in diverse species. Plant Cell 18(5):1134–1151
Ma JB et al (2005) Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature 434(7033):666–670
Elbashir SM et al (2001) Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J 20(23):6877–6888
Haley B, Zamore PD (2004) Kinetic analysis of the RNAi enzyme complex. Nat Struct Mol Biol 11(7):599–606
Kapoor M, Burgess DJ, Patil SD (2012) Physicochemical characterization techniques for lipid based delivery systems for siRNA. Int J Pharm 427(1):35–57
Behlke MA (2008) Chemical modification of siRNAs for in vivo use. Oligonucleotides 18(4):305–319
Grillone LR, Lanz R (2001) Fomivirsen. Drugs Today 37(4):245
Jabs DA, Griffiths PD (2002) Fomivirsen for the treatment of cytomegalovirus retinitis. Am J Ophthalmol 133(4):552–556
Manoharan M (2004) RNA interference and chemically modified small interfering RNAs. Curr Opin Chem Biol 8(6):570–579
Hornung V et al (2005) Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11(3):263–270
Gorden KB et al (2005) Synthetic TLR agonists reveal functional differences between Human TLR7 and TLR8. J Immunol 174(3):1259–1268
Diebold SS et al (2006) Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur J Immunol 36(12):3256–3267
Karikó K et al (2005) Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23(2):165–175
Kim JY et al (2007) Immune activation by siRNA/liposome complexes in mice is sequence- independent: lack of a role for Toll-like receptor 3 signaling. Mol Cells 24(2):247–254
Robbins M et al (2007) 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther 15(9):1663–1669
Cekaite L et al (2007) Gene expression analysis in blood cells in response to unmodified and 2′-modified siRNAs reveals TLR-dependent and independent effects. J Mol Biol 365(1):90–108
Saunders LR, Barber GN (2003) The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J 17(9):961–983
Kato H et al (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23(1):19–28
Oivanen M, Kuusela S, Lennberg H (1998) Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by bronsted acids and bases. Chem Rev 98(3):961–990
Huber R, Fiebig T, Wagenknecht H-A (2003) Pyrene as a fluorescent probe for DNA base radicals. Chem Commun 15:1878–1879
Muratovska A, Eccles MR (2004) Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett 558(1–3):63–68
Oishi M et al (2007) Enhanced growth inhibition of hepatic multicellular tumor spheroids by lactosylated poly(ethylene glycol)-siRNA conjugate formulated in PEGylated polyplexes. ChemMedChem 2(9):1290–1297
Moschos SA et al (2007) Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug Chem 18(5):1450–1459
Chiu Y-L et al (2004) Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol 11(8):1165–1175
Meade BR, Dowdy SF (2007) Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv Drug Deliv Rev 59(2–3):134–140
Thrasher AJ et al (2006) Gene therapy: X-SCID transgene leukaemogenicity. Nature 443(7109):E5–E6, discussion E6–7
Thomas CE, Ehrhardt A, Kay MA (2003) Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 4(5):346–358
Felgner PL et al (1987) Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci 84(21):7413–7417
Kim TW et al (2000) In vivo gene transfer to the mouse nasal cavity mucosa using a stable cationic lipid emulsion. Mol Cells 10(2):142–147
Crook K et al (1998) Inclusion of cholesterol in DOTAP transfection complexes increases the delivery of DNA to cells in vitro in the presence of serum. Gene Ther 5(1):137–143
Felgner PL (1993) LipofectAMINE™ reagent: a new, high efficiency polycationic liposome transfection reagent. Focus 15:73–79
Kott M et al (1998) A new efficient method for transfection of neonatal cardiomyocytes using histone H1 in combination with DOSPER liposomal transfection reagent. Somat Cell Mol Genet 24(4):257–261
Cullis PR, De Kruijff B (1979) Lipid polymorphism and the functional role of lipids in biological membranes. Biochim Biophys Acta 559(4):399–420
Farhood H, Serbina N, Huang L (1995) The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim Biophys Acta 1235(2):289–295
Felgner JH et al (1994) Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. J Biol Chem 269(4):2550–2561
Hafez IM, Cullis PR (2001) Roles of lipid polymorphism in intracellular delivery. Adv Drug Deliv Rev 47(2–3):139–148
Hafez IM, Maurer N, Cullis PR (2001) On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther 8(15):1188–1196
Koltover I et al (1998) An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 281(5373):78–81
Burger HJ et al (1992) Paradoxical transcriptional activation of rat liver cytochrome P-450 3A1 by dexamethasone and the antiglucocorticoid pregnenolone 16 alpha-carbonitrile: analysis by transient transfection into primary monolayer cultures of adult rat hepatocytes. Proc Natl Acad Sci 89(6):2145–2149
Litzinger DC, Huang L (1992) Phosphatidylethanolamine liposomes: drug delivery, gene transfer and immunodiagnostic applications. Biochim Biophys Acta 1113(2):201–227
Park HS, et al. (2004) Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kB. 3589–3593
Soenen SJH et al (2009) The role of nanoparticle concentration-dependent induction of cellular stress in the internalization of non-toxic cationic magnetoliposomes. Biomaterials 30(36):6803–6813
Zelphati O et al (1998) Effect of serum components on the physico-chemical properties of cationic lipid/oligonucleotide complexes and on their interactions with cells. Biochim Biophys Acta 1390(2):119–133
Yang JP, Huang L (1997) Overcoming the inhibitory effect of serum on lipofection by increasing the charge ratio of cationic liposome to DNA. Gene Ther 4(9):950–960
Liu F et al (1997) Factors controlling the efficiency of cationic lipid-mediated transfection in vivo via intravenous administration. Gene Ther 4(6):517–523
Delepine P et al (2000) Cationic phosphonolipids as nonviral vectors: in vitro and in vivo applications. J Pharm Sci 89(5):629–638
Heyes J et al (2005) Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release 107(2):276–287
Loisel S et al (2001) Factors influencing the efficiency of lipoplexes mediated gene transfer in lung after intravenous administration. J Liposome Res 11(2–3):127–138
Kapoor M et al (2011) Novel cationic lipids for safe and efficient siRNA delivery. Control Rel Soc Newslett 28(4):6–9
Balasubramaniam RP et al (1996) Structural and functional analysis of cationic transfection lipids: the hydrophobic domain. Gene Ther 3(2):163–172
Felgner PL et al (1981) Asymmetric incorporation of trisialoganglioside into dipalmitoylphosphatidylcholine vesicles. Biochemistry 20(8):2168–2172
Felgner PL et al (1983) Kinetics of transfer of gangliosides from their micelles to dipalmitoylphosphatidylcholine vesicles. Biochemistry 22(7):1670–1674
Cameron FH et al (1999) A transfection compound series based on a versatile Tris linkage. Biochim Biophys Acta 1417(1):37–50
Pinnaduwage P, Schmitt L, Huang L (1989) Use of a quaternary ammonium detergent in liposome mediated DNA transfection of mouse L-cells. Biochim Biophys Acta 985(1):33–37
Wang J et al (1998) Synthesis and characterization of long chain alkyl acyl carnitine esters. Potentially biodegradable cationic lipids for use in gene delivery. J Med Chem 41(13):2207–2215
Behr JP (1994) Gene transfer with synthetic cationic amphiphiles: prospects for gene therapy. Bioconjug Chem 5(5):382–389
Leventis R, Silvius JR (1990) Interactions of mammalian cells with lipid dispersions containing novel metabolizable cationic amphiphiles. Biochim Biophys Acta 1023(1):124–132
Obika S et al (1997) A symmetrical and biodegradable cationic lipid. Synthesis and application for efficient gene transfection. Bioorg Med Chem Lett 7(14):1817–1820
Lee ER et al (1996) Detailed analysis of structures and formulations of cationic lipids for efficient gene transfer to the lung. Hum Gene Ther 7(14):1701–1717
Majeti BK et al (2004) Enhanced intravenous transgene expression in mouse lung using cyclic-head cationic lipids. Chem Biol 11(4):427–437
Dalby B et al (2004) Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 33(2):95–103
Zhao M et al (2008) Lipofectamine RNAiMAX: an efficient siRNA transfection reagent in human embryonic stem cells. Mol Biotechnol 40(1):19–26
Foged C, Nielsen HM, Frokjaer S (2007) Liposomes for phospholipase A2 triggered siRNA release: preparation and in vitro test. Int J Pharm 331(2):160–166
Halder J et al (2006) Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res 12(16):4916–4924
Lakkaraju A et al (2001) Neurons are protected from excitotoxic death by p53 antisense oligonucleotides delivered in anionic liposomes. J Biol Chem 276(34):32000–32007
Fillion P et al (2001) Encapsulation of DNA in negatively charged liposomes and inhibition of bacterial gene expression with fluid liposome-encapsulated antisense oligonucleotides. Biochim Biophys Acta 1515(1):44–54
Patil SD, Rhodes DG, Burgess DJ (2005) Biophysical characterization of anionic lipoplexes. Biochim Biophys Acta 1711(1):1–11
Pack DW et al (2005) Design and development of polymers for gene delivery. Nat Rev Drug Discov 4(7):581–593
Eliyahu H, Barenholz Y, Domb AJ (2005) Polymers for DNA delivery. Molecules 10(1):34–64
Gao X, Kim K-S, Liu D (2007) Nonviral gene delivery: what we know and what is next. AAPS J 9(1):E92–E104
Zauner W, Ogris M, Wagner E (1998) Polylysine-based transfection systems utilizing receptor-mediated delivery. Adv Drug Deliv Rev 30(1):97–113
Erbacher P et al (1996) Putative role of chloroquine in gene transfer into a human hepatoma cell line by DNA/lactosylated polylysine complexes. Exp Cell Res 225(1):186–194
Wagner E et al (1992) Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle. Proc Natl Acad Sci 89(17):7934–7938
Midoux P, Monsigny M (1999) Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem 10(3):406–411
Pichon C et al (2000) Histidylated oligolysines increase the transmembrane passage and the biological activity of antisense oligonucleotides. Nucleic Acids Res 28(2):504–512
Boussif O et al (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci 92(16):7297–7301
Liao H-W, Yau K-W (2007) In vivo gene delivery in the retina using polyethylenimine. Biotechniques 42(3):285–288
Fischer D et al (1999) A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: effect of molecular weight on transfection efficiency and cytotoxicity. Pharm Res 16(8):1273–1279
Godbey WT, Wu KK, Mikos AG (1999) Size matters: molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J Biomed Mater Res 45(3):268–275
Zhao N, Roesler S, Kissel T (2011) Synthesis of a new potential biodegradable disulfide containing poly (ethylene imine)-poly (ethylene glycol) copolymer cross-linked with click cluster for gene delivery. Int J Pharm 411(1):197–205
Anderson DG et al (2005) Structure/property studies of polymeric gene delivery using a library of poly (I2-amino esters). Mol Ther 11(3):426–434
Y-b L et al (2002) Biodegradable, endosome disruptive, and cationic network-type polymer as a highly efficient and nontoxic gene delivery carrier. Bioconjug Chem 13(5):952–957
Kleemann E et al (2009) Enhanced gene expression and reduced toxicity in mice using polyplexes of low-molecular-weight poly (ethylene imine) for pulmonary gene delivery. J Drug Target 17(8):638–651
Koping-Hoggard M et al (2001) Chitosan as a nonviral gene delivery system. Structure-property relationships and characteristics compared with polyethylenimine in vitro and after lung administration in vivo. Gene Ther 8(14):1108–1121
Lavertu M et al (2006) High efficiency gene transfer using chitosan/DNA nanoparticles with specific combinations of molecular weight and degree of deacetylation. Biomaterials 27(27):4815–4824
Howard KA et al (2008) Chitosan/siRNA nanoparticle-mediated TNF-alpha knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol Ther 17(1):162–168
Wong K et al (2006) PEI-g-chitosan, a novel gene delivery system with transfection efficiency comparable to polyethylenimine in vitro and after liver administration in vivo. Bioconjug Chem 17(1):152–158
Klajnert B, Bryszewska M (2001) Dendrimers: properties and applications. Acta Biochim Pol 48(1):199–208
Dufes C, Uchegbu IF, Schatzlein AG (2005) Dendrimers in gene delivery. Adv Drug Deliv Rev 57(15):2177–2202
Aulenta F, Hayes W, Rannard S (2003) Dendrimers: a new class of nanoscopic containers and delivery devices. Eur Polym J 39(9):1741–1771
Liang P (2002) SAGE Genie: a suite with panoramic view of gene expression. Proc Natl Acad Sci 99(18):11547–11548
Kukowska-Latallo JF et al (1996) Efficient transfer of genetic material into mammalian cells using Starburst polyamidoamine dendrimers. Proc Natl Acad Sci 93(10):4897–4902
Maksimenko AV et al (2003) Optimisation of dendrimers-mediated gene transfer by anionic oligomers. J Gene Med 5(1):61–71
Vincent L et al (2003) Efficacy of dendrimer-mediated angiostatin and TIMP-2 gene delivery on inhibition of tumor growth and angiogenesis: In vitro and in vivo studies. Int J Cancer 105(3):419–429
Wang Y et al (2001) Combination of electroporation and DNA/dendrimer complexes enhances gene transfer into murine cardiac transplants. Am J Transplant 1(4):334–338
Malik N et al (2000) Dendrimers: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Control Release 65(1):133–148
Sonawane ND, Szoka FC, Verkman AS (2003) Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J Biol Chem 278(45):44826–44831
Ofek P et al (2010) In vivo delivery of small interfering RNA to tumors and their vasculature by novel dendritic nanocarriers. FASEB J 24(9):3122–3134
Panyam J, Labhasetwar V (2003) Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev 55(3):329–347
Prabha S, Labhasetwar V (2004) Nanoparticle-mediated wild-type p53 gene delivery results in sustained antiproliferative activity in breast cancer cells. Mol Pharm 1(3):211–219
Park TG (1994) Degradation of poly(d, l-lactic acid) microspheres: effect of molecular weight. J Control Release 30(2):161–173
Park TG, Lu W, Crotts G (1995) Importance of in vitro experimental conditions on protein release kinetics, stability and polymer degradation in protein encapsulated poly (d, l-lactic acid-co-glycolic acid) microspheres. J Control Release 33(2):211–222
Jain RA (2000) The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide) (PLGA) devices. Biomaterials 21(23):2475–2490
Ravi Kumar MNV, Bakowsky U, Lehr CM (2004) Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials 25(10):1771–1777
Wang D et al (1999) Encapsulation of plasmid DNA in biodegradable poly (D, L-lactic-co-glycolic acid) microspheres as a novel approach for immunogene delivery. J Control Release 57(1):9–18
Ando S et al (1999) PLGA microspheres containing plasmid DNA: preservation of supercoiled DNA via cryopreparation and carbohydrate stabilization. J Pharm Sci 88(1):126–130
Koby G et al (2007) Poly (D, L-lactide-co-glycolide acid) nanoparticles for DNA delivery: waiving preparation complexity and increasing efficiency. Biopolymers 85(5–6):379–391
Meuller M et al (2012) Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J Control Release 162(1):159–166
Patil Y, Panyam J (2009) Polymeric nanoparticles for siRNA delivery and gene silencing. Int J Pharm 367(1):195–203
Andersen MA et al (2010) Surface functionalisation of PLGA nanoparticles for gene silencing. Biomaterials 31(21):5671–5677
Koch S et al (2000) Ultrasound enhancement of liposome-mediated cell transfection is caused by cavitation effects. Ultrasound Med Biol 26(5):897–903
Van Wamel A, Bouakaz A, de Jong N (2003) Duration of ultrasound bubbles enhanced cell membrane permeability. In: Ultrasonics. IEEE Symposium 1:917–920
Gaumet M et al (2008) Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm 69(1):1–9
Ishida O et al (1999) Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int J Pharm 190(1):49–56
Woodle MC (1993) Surface-modified liposomes: assessment and characterization for increased stability and prolonged blood circulation. Chem Phys Lipids 64(1–3):249–262
Mosqueira VC et al (2001) Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm Res 18(10):1411–1419
Morrissey DV et al (2005) Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol 23(8):1002–1007
Akinc A et al (2009) Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol Ther 17(5):872–879
Qi R et al (2009) PEG-conjugated PAMAM dendrimers mediate efficient intramuscular gene expression. AAPS J 11(3):395–405
Lutz GJ, Sirsi SR, Williams JH (2008) PEG-PEI copolymers for oligonucleotide delivery to cells and tissues. In: Gene Ther Protocols. Springer. 433:141–150.
Hofland HEJ et al (2002) Folate-targeted gene transfer in vivo. Mol Ther 5(6):739–744
Pirollo KF et al (2007) Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer Res 67(7):2938–2943
Wang Q, Dordick JS, Linhardt RJ (2002) Synthesis and application of carbohydrate-containing polymers. Chem Mater 14(8):3232–3244
Yoshizawa T et al (2008) Folate-linked lipid-based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur J Pharm Biopharm 70(3):718–725
Ross JF, Chaudhuri PK, Ratnam M (1994) Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer 73(9):2432–2443
Weitman SD et al (1992) Cellular localization of the folate receptor: potential role in drug toxicity and folate homeostasis. Cancer Res 52(23):6708–6711
Hwa Kim S et al (2005) Target-specific gene silencing by siRNA plasmid DNA complexed with folate-modified poly (ethylenimine). J Control Release 104(1):223–232
Pathak A, Vyas SP, Gupta KC (2008) Nano-vectors for efficient liver specific gene transfer. Int J Nanomedicine 3(1):31–49
Kawakami S et al (1998) Asialoglycoprotein receptor-mediated gene transfer using novel galactosylated cationic liposomes. Biochem Biophys Res Commun 252(1):78–83
Rozema DB et al (2007) Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci 104(32):12982–12987
Jiang N et al (2012) A novel in vivo sirna delivery system specifically targeting liver cells for protection of ConA-induced fulminant hepatitis. PLoS One 7(9):e44138
Soutschek J et al (2004) Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432(7014):173–178
Li J et al (2010) Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J Control Release 142(3):416–421
Guo J et al (2012) Anisamide-targeted cyclodextrin nanoparticles for siRNA delivery to prostate tumours in mice. Biomaterials 33(31):7775–7784
Kircheis R et al (2001) Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther 8(1):28–40
Huang R-Q et al (2007) Efficient gene delivery targeted to the brain using a transferrin-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. FASEB J 21(4):1117–1125
Mendonca LS et al (2010) Transferrin receptor-targeted liposomes encapsulating anti-BCR-ABL siRNA or asODN for chronic myeloid leukemia treatment. Bioconjug Chem 21(1):157–168
Davis ME (2009) The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm 6(3):659–668
Kircheis R et al (1997) Coupling of cell-binding ligands to polyethylenimine for targeted gene delivery. Gene Ther 4(5):409–418
Li X et al (2001) Single-chain antibody-mediated gene delivery into ErbB2-positive human breast cancer cells. Cancer Gene Ther 8(8):555–565
Chen Y et al (2010) Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther 18(9):1650–1656
Santos AO et al (2010) Design of peptide-targeted liposomes containing nucleic acids. Biochim Biophys Acta 1798(3):433–441
Shen M et al (2012) An MRI-visible non-viral vector for targeted Bcl-2 siRNA delivery to neuroblastoma. Int J Nanomedicine 7:3319–3332
Bedi D et al (2011) Delivery of siRNA into breast cancer cells via phage fusion protein-targeted liposomes. Nanomedicine 7(3):315–323
Kim HK et al (2010) Enhanced siRNA delivery using cationic liposomes with new polyarginine-conjugated PEG-lipid. Int J Pharm 392(1–2):141–147
Streipe S, Rossler J, Suss R (2010) Integrin- and IGF1-receptor-mediated liposomal siRNA delivery to alveolar rhabdomyosarcoma cells. Sci Pharm 78:674
Wang X-L et al (2009) Targeted systemic delivery of a therapeutic siRNA with a multifunctional carrier controls tumor proliferation in mice. Mol Pharm 6(3):738–746
Zhang C et al. (2006) siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J Control Release 112(2): 229–239
Moreira JN et al (2002) Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity. Pharm Res 19(3):265–269
Asai T et al (2002) Anti-neovascular therapy by liposomal DPP-CNDAC targeted to angiogenic vessels. FEBS Lett 520(1–3):167–170
Sioud M, Mobergslien A (2012) Efficient siRNA targeted delivery into cancer cells by gastrin-releasing peptides. Bioconjug Chem 23(5):1040–1049
Plank C et al (1994) The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J Biol Chem 269(17):12918–12924
Martin ME, Rice KG (2007) Peptide-guided gene delivery. AAPS J 9(1):E18–E29
Torchilin VP et al (2003) Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposomes DNA complexes. Proc Natl Acad Sci 100(4):1972–1977
Simeoni F et al. (2005) Peptide-based strategy for siRNA delivery into mammalian cells. In: RNA silencing. Springer. p 251–260
Deshayes S et al (2012) Self-assembling peptide-based nanoparticles for siRNA delivery in primary cell lines. Small 8(14):2184–2188
Dassie JP et al (2009) Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol 27(9):839–846
Liu H et al (1998) Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res 58(18):4055–4060
Kurosaki T et al (2012) Self-assemble gene delivery system for molecular targeting using nucleic acid aptamer. Gene 491(2):205–209
Girvan AC et al (2006) AGRO100 inhibits activation of nuclear factor-kB (NF-kB) by forming a complex with NF-kB essential modulator (NEMO) and nucleolin. Mol Cancer Ther 5(7):1790–1799
Soundararajan S et al (2008) The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res 68(7):2358–2365
Simoes S et al (1999) Successful transfection of lymphocytes by ternary lipoplexes. Biosci Rep 19(6):601–609
Godbey WT, Wu KK, Mikos AG (1999) Tracking the intracellular path of poly (ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci 96(9):5177–5181
Suikkanen S et al (2003) Exploitation of microtubule cytoskeleton and dynein during parvoviral traffic toward the nucleus. J Virol 77(19):10270–10279
Munkonge FM et al (2003) Emerging significance of plasmid DNA nuclear import in gene therapy. Adv Drug Deliv Rev 55(6):749–760
Pante N, Fahrenkrog B, Aebi U (1998) Molecular dissection of nuclear pore complex structure and nucleocytoplasmic transport. Biol Cell 90:275–276
Pante N, Kann M (2002) Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell 13(2):425–434
Adam SA, Geracet L (1991) Cytosolic proteins that specifically bind nuclear location signals are receptors for nuclear import. Cell 66(5):837–847
Kalderon D et al (1984) A short amino acid sequence able to specify nuclear location. Cell 39(3):499–509
Arenal A et al (2004) The SV40 T antigen nuclear localization sequence enhances nuclear import of vector DNA in embryos of a crustacean (Litopenaeus schmitti). Gene 337:71–77
Lim RY (2007) Gate-crashing the nuclear pore complex. Structure 15(8):889–891
Branden LJ, Mohamed AJ, Smith CI (1999) A peptide nucleic acid-nuclear localization signal fusion that mediates nuclear transport of DNA. Nat Biotechnol 17(8):784–787
Tanimoto M et al (2003) No enhancement of nuclear entry by direct conjugation of a nuclear localization signal peptide to linearized DNA. Bioconjug Chem 14(6):1197–1202
Neves C et al (1999) Intracellular fate and nuclear targeting of plasmid DNA. Cell Biol Toxicol 15(3):193–202
Sebestyen MG et al (1998) DNA vector chemistry: the covalent attachment of signal peptides to plasmid DNA. Nat Biotechnol 16(1):80–85
Akita H et al (2006) Evaluation of the nuclear delivery and intra-nuclear transcription of plasmid DNA condensed with μ (mu) and NLS-μ by cytoplasmic and nuclear microinjection: a comparative study with poly-L-lysine. J Gene Med 8(2):198–206
Neves C et al (1999) Coupling of a targeting peptide to plasmid DNA by covalent triple helix formation. FEBS Lett 453:41–45
Nagasaki T et al (2003) Can nuclear localization signals enhance nuclear localization of plasmid DNA? Bioconjug Chem 14(2):282–286
Zanta MA, Belguise-Valladier P, Behr JP (1999) Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci 96(1):91–96
Jeon O et al (2007) Poly(L-lactide-co-glycolide) nanospheres conjugated with a nuclear localization signal for delivery of plasmid DNA. J Drug Target 15(3):190–198
Jensen KD et al (2003) Cytoplasmic delivery and nuclear targeting of synthetic macromolecules. J Control Release 87(1–3):89–105
Burgess DJ, Kapoor M (2010) Quantum dot labeling for assessment of intracellular trafficking of therapeutically active molecules. In: Weissig V, D’Souza GGM (eds) Organelle-specific pharmaceutical nanotechnology. Wiley, New York, NY, pp 535–568
Chen F, Gerion D (2004) Fluorescent CdSe/ZnS nanocrystal-peptide conjugates for long-term, nontoxic imaging and nuclear targeting in living cells. Nano Lett 4(10):1827–1832
Lam AP, Dean DA (2010) Progress and prospects: nuclear import of nonviral vectors. Gene Ther 17(4):439–447
Merkwirth C, Langer T (2009) Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta, Mol Cell Res 1793(1):27–32
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15(22):2922–2933
Chen M et al (2012) Chitosan/siRNA nanoparticles encapsulated in PLGA nanofibers for siRNA delivery. ACS Nano 6(6):4835–4844
Xun M-M et al (2013) Low molecular weight PEI-based biodegradable lipopolymers as gene delivery vectors. Org Biomol Chem 11(7):1242–1250
Rui M et al (2013) Recombinant high density lipoprotein nanoparticles for target-specific delivery of siRNA. Pharm Res 30(5):1203–1214
Y-y L et al (2013) Biocompatible polyethylenimine-graft-dextran catiomer for highly efficient gene delivery assisted with nuclear targeting ligand. Polym Chem 4:2528–2539
Du J et al (2012) Biodegradable nanoparticles of mPEG-PLGA-PLL triblock copolymers as novel non-viral vectors for improving siRNA delivery and gene silencing. Int J Mol Sci 13(1):516–533
Oishi M, Kataoka K, Nagasaki Y (2006) pH-responsive three-layered PEGylated polyplex micelle based on a lactosylated ABC triblock copolymer as a targetable and endosome-disruptive nonviral gene vector. Bioconjug Chem 17(3):677–688
Chu M et al (2013) Biocompatible polyethylenimine-graft-dextran catiomer for highly efficient gene delivery assisted by a nuclear targeting ligand. Polym Chem 4(8):2528–2539
Diez S, Navarro G, de Ilarduya CT (2009) In vivo targeted gene delivery by cationic nanoparticles for treatment of hepatocellular carcinoma. J Gene Med 11(1):38–45
Kim WJ et al (2006) Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther 14(3):343–350
Kircheis R et al (2002) Tumor-targeted gene delivery: an attractive strategy to use highly active effector molecules in cancer treatment. Gene Ther 9(11):731–735
Lee M et al (2007) DNA delivery to the mitochondria sites using mitochondrial leader peptide conjugated polyethylenimine. J Drug Target 15(2):115–122
Matschke J et al (2012) Characterization of Ku702-NLS as bipartite nuclear localization sequence for non-viral gene delivery. PLoS One 7(2):e24615
McNamara JO et al (2006) Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol 24(8):1005–1015
Schiffelers RM et al (2004) Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res 32(19):e149
de Tros Ilarduya C et al (2002) Enhanced gene delivery in vitro and in vivo by improved transferrin-lipoplexes. Biochim Biophys Acta 1561(2):209–221
Weissig V, De’Souza GGM, Torchilin VP (2001) DQAsome/DNA complexes release DNA upon contact with isolated mouse liver mitochondria. J Control Release 75(3):401–408
Wolfrum C et al (2007) Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol 25(10):1149–1157
Wolschek MF et al (2002) Specific systemic nonviral gene delivery to human hepatocellular carcinoma xenografts in SCID mice. Hepatology 36(5):1106–1114
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Controlled Release Society
About this chapter
Cite this chapter
Kapoor, M., Burgess, D.J. (2015). Targeted Delivery of Nucleic Acid Therapeutics via Nonviral Vectors. In: Devarajan, P., Jain, S. (eds) Targeted Drug Delivery : Concepts and Design. Advances in Delivery Science and Technology. Springer, Cham. https://doi.org/10.1007/978-3-319-11355-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-11355-5_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11354-8
Online ISBN: 978-3-319-11355-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)