
Abstract
The hypothalamic-pituitary-adrenal (HPA) axis plays a fundamental role in the maintenance of basal and stress-related homeostasis. This neuroendocrine axis consists of three distinct components located in the hypothalamus, the pituitary gland and the adrenal cortex. Glucocorticoids, the end-products of the HPA axis, exert their diverse actions in virtually all tissues through their ubiquitously expressed glucocorticoid receptor. A broad spectrum of pathologic conditions affecting any of the three anatomical parts results in hypo- or hyper-activation of the HPA axis and subsequent clinical manifestations of glucocorticoid deficiency or excess, respectively. Moreover, our ever-increasing understanding of glucocorticoid receptor signaling has allowed us to approach diagnostically and therapeutically these endocrine diseases in a more integrated way. However, the pathophysiology, differential diagnosis and therapy of some of these disorders still remain challenging. In this chapter, we describe the physiologic and endocrine aspects of the HPA axis, and we present the clinical manifestations and therapeutic management of its most common disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
All living organisms maintain a complex dynamic equilibrium, called homeostasis, which may be threatened, or perceived as threatened, by a wide variety of extrinsic or intrinsic adverse challenges or forces, the stressors [1, 2]. To cope with these stressful stimuli, living organisms have developed a highly complex regulatory system, the stress system, which, upon activation, leads to a programmed repertoire of physiologic and behavioral central nervous system (CNS) and peripheral adaptive responses [1–3]. If the stress response is either inadequate or excessive or prolonged, it may adversely influence personality development and behavior, and may impair fundamental physiologic functions, such as growth, development, metabolism, reproduction, circulation and the immune response [3]. The hypothalamic-pituitary-adrenal (HPA) axis is one of the two major arms of the stress system, which acts synergistically with the locus caeruleus/norepinephrine-autonomic nervous systems to adjust homeostasis. This neuroendocrine axis consists of three components: the paraventricular nuclei (PVN) located in the hypothalamus, the corticotrope cells located in the pituitary gland and the zona fasciculata residing in the adrenal cortices. Glucocorticoids, the end-products of the HPA axis, are cholesterol-derived molecules that exert their pleiotropic actions through the glucocorticoid receptor (GR), which acts as an intracellular ligand-induced transcription factor, influencing the transcription rate of many thousands of genes in a positive or negative way [4]. Consequently, the dysfunction of the HPA axis at any of the above levels may represent the main pathogenetic mechanism underlying several pathologic conditions, which may strongly influence human health and quality of life. In this chapter, we describe the functional components of the HPA axis, we provide an overview of the endocrine effects of glucocorticoids through their cognate receptor, and we finally present the most common diseases associated with dysfunction of this axis.
1.1 Historical Development of the Stress and the HPA Axis
…he who knows things from their beginning and origins understands them better… (Aristotle, 4th Century BCE).
The concepts of stress go back to Greek antiquity [1, 2]. Pythagoras was the first to use the term harmony to describe the equilibrium of the human organism or the balance of the entire universe (The harmony of the Cosmos). This harmony is constantly challenged by disturbing forces, while other counteracting forces help harmony to be reestablished. Shortly afterwards, Alcmaeon of Croton used the term isonomia to describe the balance of opposing forces. Empedocles of Argigentum proposed that all matter consisted of four basic elements (= rhizomata or roots), the earth, the water, the air and the fire, which were in a dynamic opposition to one another, and that balance was a necessary prerequisite for the health of living organisms [2, 5]. The Empedoclean doctrine seems to be the precursor of the Hippocratic humoral system. Hippocrates, the father of medicine, equated health to a harmonious equilibrium of the four humors: blood, phlegm, black and yellow bile, corresponding to the heart, the brain, the liver and the spleen, respectively. The balance of the humors is termed eucrasia, whereas their imbalance or dyscrasia causes disease [2]. Hippocrates also suggested that the disturbing forces that produced dyscrasia and the counteracting forces that reestablish eucrasia both derived from nature. He, therefore, introduced the concept that “Nature is the healer of disease” [2]. Many years later, Epicurus suggested that the mind could be one of these healing forces and wrote that ataraxia, or “imperturbability of mind” and aponia (no pain) represented most desirable states [2].
In the years of Renaissance, Thomas Sydenham proposed that an individual’s adaptive response to the disturbing forces that lead to systematic disharmony could result in pathologic changes. Two hundred years later, Claude Bernard, one of the world’s greatest physiologists, pointed out that cells are surrounded by an internal medium, the milieu intérieur, which provides a steady state. He wrote that “it is the fixity of the milieu intérieur, which is the condition of free and independent life” [6]. Fifty years later, Walter Bradford Cannon coined the term homeostasis (from the Greek homoios, or similar, and stasis, or position) for all the physiologic processes that maintain the steady state of the organism [2, 6]. He published the concept of homeostasis in his book entitled “Wisdom of the Body” in 1932. Cannon also described an animal’s response to threat. The concept of “fight or flight reaction” suggests that the adaptive response to stress is associated with catecholamine secretion due to activation of the sympathetic nervous system, priming the animal for fight against the threat or fleeing to save its life [2, 6]. This response was considered to be the first stage (acute stress response) of the general adaptation syndrome (GAS) suggested later by Hans Selye.
Hans Hugo Bruno Selye, the father of the concept of stress, observed that patients with different diseases manifested many of the same ‘nonspecific’ symptoms as a common response to stressors. Selye, and before him Cannon, borrowed the term “stress” from physics and defined it as the mutual actions of forces that happen across any section of body [2, 6]. He postulated that the presence of stereotypic psychological and physiological manifestations occurring in seriously ill patients represented the consequences of severe long-lasting adaptational responses. He named this condition the “General Adaptation or Stress Syndrome” and redefined Sydenham’s concept in the “diseases of adaptation”. In addition to clearly defining stress, Selye coined the term heterostasis (from the Greek heteros, or other), later named as allostasis, initially meaning “stability through change” [2, 6].
In parallel with the evolution of stress concepts, numerous advances in neuroendocrinology uncovered the physiologic biochemical effectors of the stress response and the central loci responsible for the regulation of their production and release. The locus caeruleus/autonomic nervous system and the HPA axis represent the major arms of the stress system. Coined by the physiologist John Newport Langley in 1898, the autonomic nervous system was shown to play an important role in the fight or flight reaction and the maintenance of homeostasis, especially in the cardiovascular system [6]. On the other hand, our understanding of the HPA axis has improved significantly in the last century. The pituitary gland had long been considered to be an autonomous regulator of many endocrine glands, such as the thyroid, the gonads and the adrenal cortex. However, experiments by Geoffrey Harris and William Rowan and others led to an acceptance of the novel concept that the pituitary gland functions under the control of the CNS. Evidence supporting the neurohumoral hypothesis of anterior pituitary control came from the pituitary stalk section and pituitary grafting experiments of Geoffrey Harris and Dora Jacobsohn, from the characterization of some of the neurohormonal molecules by Andrew Schally and Roger Guillemin, and from the proof that these neurohormones were directly secreted into hypophyseal portal veins [6]. Corticoctropin-releasing factor (CRF), a 41-amino acid peptide that controls the production and secretion of adrenocorticotropic hormone (ACTH), was isolated and sequenced by Wylie Vale and associates in 1981 [6]. Finally, glucocorticoids, the effector molecules of the HPA axis, were isolated and synthesized by Edward Kendall and Tadeus Reichstein [6]. Philip Hench was the first who tested cortisone, a synthetic pre-glucocorticoid, in patients with rheumatoid arthritis, and showed the anti-inflammatory effect of this compound [6]. Kendall, Hench, and Reichstein were jointly awarded the Nobel Prize for physiology and medicine in 1950. Despite the isolation and synthesis of glucocorticoids in the middle of the twentieth century, their mechanisms of pleiotropic actions were – and still remain – an enigma. In 1985, the human glucocorticoid receptor (hGR) cDNA was isolated by expression cloning, predicting two protein forms, of 777 (alpha) and 742 (beta) amino acids, which differ at their carboxyl termini [7]. Since then, the GR-mediated anti-inflammatory and immunosuppressive effects have been elucidated and accumulating evidence from in vitro and in vivo studies supports the stochastic (randomly determined) nature of glucocorticoid signaling [8].
1.2 The HPA Axis
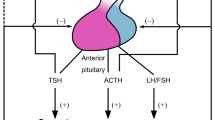
The HPA axis consists of the paraventricular nuclei (PVN) located in the hypothalamus, the pituitary gland and the adrenal cortices. A broad spectrum of internal and external signals from various organs of the human body are detected and subsequently integrated by the CNS, which transduces this information to the PVN and other brain loci, such as the hypothalamic suprachiasmatic nucleus (SCN), the amygdala and the raphe nuclei of the brain stem, and controls the activity of the PVN [9]. The SCN functions also as a central circadian rhythm center (master CLOCK), under the control of light/dark changes during the 24 h day, and generates the circadian rhythmicity of the HPA axis through synaptic connections with the PVN, leading to the diurnal fluctuation of circulating concentrations of glucocorticoids [10]. Neurons of the PVN produce and release corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) into the hypophyseal portal system [9, 11]. Subsequently, CRH and, to a lesser degree, AVP trigger the synthesis and secretion of ACTH by the corticotrope cells of the anterior pituitary gland [12, 13] (Fig. 6.1). CRH is the strongest and probably the most important stimulator of ACTH secretion. AVP, although a potent synergistic factor of CRH, has little ACTH secretagogue activity on its own. ACTH is synthesized as part of a large precursor molecule of 214 amino acids, pro-opiomelanocortin (POMC). Depending on the cleavage enzymes, which are expressed in a tissue-specific fashion, ACTH and several other peptides, e.g. NH2-terminal peptide, joining peptide, β- or γ-lipotropins, β-endorphin, α-melanocyte-stimulating hormone (MSH) and corticotropin-like intermediate peptide (CLIP), are produced [14]. ACTH is secreted in regular pulses of variable amplitude over 24 h, with peak concentrations attained in the early morning hours (04:00–08:00 h), thus forming the basis of the circadian pattern of ACTH and cortisol secretion. Upon binding to its cognate transmembrane G-protein-coupled receptor of the adrenocortical cells of the zona fasciculata of the adrenal cortex, ACTH induces the activity of proteins and the expression of genes implicated in the biosynthetic pathway of glucocorticoids (cortisol in humans, corticosterone in rodents) [15] Glucocorticoids are cholesterol-derived molecules, which are released into the systemic circulation in a pulsatile, circadian and stress-related fashion, and exert their numerous actions in almost all tissues and organs. However, these steroid hormones also have negative feedback effects on both PVN CRH and AVP synthesis and secretion, and inhibit pituitary synthesis of POMC, as well as ACTH secretion, forming a closed, tightly regulated, negative feedback loop [9] (Fig. 6.1).

The HPA axis and systemic actions of glucocorticoids. CRH corticotropin-releasing hormone, ACTH adrenocorticotropic hormone (Figure 6.1 was prepared using image vectors from Servier Medical Art (www.ervier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/))
1.3 Glucocorticoids and Glucocorticoid Receptor
Glucocorticoids, the end-products of the HPA axis, play a fundamental role in the maintenance of basal and stress-related homeostasis, and regulate a broad spectrum of physiologic functions, such as the cardiovascular tone, the intermediary metabolism primarily through catabolic actions, the quantity and the quality of the inflammatory and immune response and many other functions, by altering the transcription rate of a significant percentage of genes in a positive or negative way [3, 4, 8] (Fig. 6.1). Glucocorticoids exert their diverse actions through an intracellular, ubiquitously expressed protein, the human glucocorticoid receptor (hGR), which belongs to the nuclear receptor superfamily of transcription factors [16].
The hGR gene is located on the short arm of chromosome 5 and consists of 10 exons [16]. The alternative use of two distinct exons, 9α and 9β, generates two main isoforms, the hGRα and the hGRβ [17]. The hGRα is a 777-amino acid protein and represents the classic isoform, which binds endogenous and synthetic glucocorticoids, and mediates all genomic effects of these hormones in almost every cell type in the human organism [17]. On the other hand, the hGRβ contains 742 amino acids, does not bind glucocorticoids, and acts as a dominant negative regulator on the hGRα-mediated transcriptional activity through several mechanisms, such as heterodimerization and competition with hGRα for binding to glucocorticoid response elements (GREs) in the promoter regions of target genes, or for interacting with nuclear receptor coregulators (coactivators, corepressors) [17–21]. Furthermore, the hGRα mRNA was demonstrated to be translated from eight alternative initiation sites into functionally distinct hGRα protein isoforms by ribosomal leaky scanning or ribosomal shunting [22]. Given that hGRβ mRNA translation is initiated by the same 5′ termini, it is possible that the above translation mechanisms could give rise to eight different hGRβ proteins as well [8]. Thus, the 16 amino terminal hGRα and hGRβ protein isoforms may form 256 homo- or hetero-dimers to transduce the glucocorticoid signal.
At the cellular level, hGRα is primarily localized in the cytoplasm of the target cell in the absence of a ligand, as part of a multiprotein complex consisting of heat shock proteins and immunophilins [16] (Fig. 6.2). Upon ligand-binding, hGRα undergoes conformational change, dissociates from the rest of proteins, and translocates into the nucleus through the nuclear pore, via an active ATP-dependent process mediated by its nuclear localization signal (NL)-1 and −2 [19]. Within the nucleus, the ligand-activated GR directly interacts as a homo- or hetero-dimer with specific DNA sequences, the glucocorticoid response elements (GREs), in the promoter regions of target genes [19]. The GR contains two transactivation domains, activation function (AF)-1 and −2, located at its NTD and LBD, respectively, through which it interacts with protein complexes, such as the nuclear receptor coactivators p160, p300/CREB-binding protein (CBP) and p300/CBP-associated factor (p/CAF) complexes and the SWI/SNF and vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein (DRIP/TRAP) chromatin-remodeling complexes; it influences the activity of the RNA polymerase II and its ancillary factors, thereby modulating the transcription rates of glucocorticoid-responsive genes [16] (Fig. 6.2).

Glucocorticoid signaling pathway. GR glucocorticoid receptor, HSP heat shock proteins, FKBP immunophilins, p160 nuclear receptor coactivators p160, SWI/SNF switching/sucrose non-fermenting complex, DRIP/TRAP vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein complex (Figure 6.2 was prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/))
Alternatively, hGRα may modulate gene expression independently of its DNA binding, by interacting with and altering the functions of other known transcription factors, such as NF-κB, STAT-5, and AP-1, possibly as monomer [16]. These interacting factors, in turn, may alter the actions of the glucocorticoid receptor acting on positive or negative GREs.
1.4 Disorders of the HPA Axis
The disorders of the HPA axis can be categorized according to the anatomical component(s) that is/are primarily affected: (1) the hypothalamus, (2) the pituitary gland, (3) the adrenal glands and (4) other tissues and organs, including alterations of glucocorticoid availability or action at target tissues (Fig. 6.3). These disorders may result in either elevated or reduced concentrations or effects of circulating glucocorticoids; thus, symptoms and signs are associated with increased or decreased actions of these hormones in the responsive tissues, (Table 6.1).

The HPA axis and associated disorders. The disorders of the HPA axis can be categorized according to the anatomical component(s) that is/are primarily affected: (1) the hypothalamus, (2) the pituitary gland, (3) the adrenal glands and (4) the glucocorticoid-responsive cell. Mutations or polymorphisms of the hGR gene may impair normal glucocorticoid signal transduction, thus altering tissue sensitivity to glucocorticoids. CRH corticotropin-releasing hormone, ACTH adrenocorticotropic hormone, GR glucocorticoid receptor (Figure was prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/))
Indeed, chronic glucocorticoid excess results in central obesity, buffalo hump, purple striae of the skin, hypertension, osteoporosis, glucose intolerance/overt diabetes mellitus, dyslipidemia and leukocytosis, all of which are referred to as the “Cushing phenotype” [23]. Patients with pathologically increased concentrations of cortisol are at great risk of severe cardiovascular complications, such as ischemic heart/cerebral disease [23]. Moreover, they are more susceptible to infections secondary to any pathogens, since cortisol suppresses strongly the activity of the immune system.
On the other hand, patients with isolated glucocorticoid deficiency or adrenal insufficiency including aldosterone deficiency may present with fatigue, vomiting, nausea, hypotension, hypoglycemia, hyponatremia, hyperkalemia in the absence of aldosterone secretion and eosinophilia [23]. The decreased concentrations of cortisol lead to compensatory elevations in plasma ACTH concentrations, which result in hyperpigmentation of the skin and buccal mucosa in these patients.
1.4.1 Hypothalamic Disorders Associated with Alterations in the HPA Axis
Many space-occupying lesions of the hypothalamus, including craniopharyngioma, Rathke’s pouch cysts, head trauma, inflammatory or infiltrative diseases (encephalitis, meningitis, sarcoidosis, histiocytosis X) and brain malignant tumors (primary or metastatic) lead to destruction of the PVN neurons producing CRH or to dissection of the pituitary stalk that transfers CRH from the hypothalamus to the pituitary gland, thus causing decreased function of the HPA axis [24]. These conditions are often associated with dysfunction of other endocrine axes, such as the hypothalamic-pituitary-gonadal or thyroid axes and the growth hormone/insulin-like growth factor-I axis.
Prader-Willi syndrome is a rare genetic disorder, which is characterized by symptoms associated with hypothalamic dysfunction, such as hyperphagia, obesity, sleep disorders, hypogonadism, hypocortisolism, learning disabilities/borderline intellectual functioning and hypotonia [25].
Patients with the melancholic type of major depression show hyperactivity of the HPA axis and have increased cerebrospinal fluid CRH and norepinephrine, plasma ACTH and serum cortisol concentrations [26, 27]. This sustained hyperfunction of the HPA axis leads to central obesity, hypertension, impaired glucose tolerance, decreased libido and osteoporosis, all of which result in impaired quality of life.
One of the major eating disorders, anorexia nervosa, is also characterized by elevated cortisol concentrations via activation of the HPA axis caused by strict and prolonged energy restriction, as well as by mental stress [28]. Patients with anorexia nervosa demonstrate increased concentrations of CRH in their cerebrospinal fluid [28]. However, in contradistinction to depressed patients, they do not show any clinical manifestations suggestive of glucocorticoid excess. Indeed, they present with symptoms and signs associated with reduced action of glucocorticoids in target tissues, possibly due to lack of food substrate and changes of proteins that may influence the actions of glucocorticoids through the hGR, such as the AMP-activated protein kinase (AMPK) [29].
Any conditions associated with uncoupling of the cortisol circadian rhythm that is under the influence of the SCN CLOCK system, and the circadian peripheral tissue sensitivity to glucocorticoids, under the control of the peripheral CLOCK, may cause the development of components of the metabolic syndrome and its detrimental cardiovascular complications [10, 30]. Night-shift workers and people frequently exposed to jet lag are at an increased risk for developing symptoms and signs of chronic hypercortisolemia, such as central obesity, insulin resistance, dyslipidemia and hypertension, which represent the cardinal clinical manifestation of metabolic syndrome. These pathologic conditions were recently explained at the molecular and cellular level, when it was convincingly shown that the transcription factor “circadian locomotor output cycle kaput” (CLOCK) physically interacts with GR, and catalyzes enzymatically the acetylation of a lysine cluster, located in the hinge region of the receptor and leading to decreased sensitivity to glucocorticoids [10, 30, 31]. This effect of CLOCK on GR can lead to “functional” tissue hypersensitivity to glucocorticoids via uncoupling of circadian cortisol concentrations and peripheral tissue responsiveness to these hormones [10, 30, 31].
2 Pituitary Disorders Associated with Alterations in the HPA Axis
2.1 Cushing Disease (and Syndrome)
Cushing disease is part of Cushing syndrome, which is characterized by elevated concentrations of circulating cortisol and symptoms and signs of glucocorticoid excess. This disease is caused by semi-autonomous neoplasia or hyperplasia of corticotropes that result in over-secretion of ACTH from the pituitary gland. In contrast, Cushing syndrome indicates a disorder that derives from the adrenal glands, caused by either an adrenocortical adenoma or carcinoma, or from an ectopic site secreting CRH or ACTH (~15 % of ACTH-dependent forms of Cushing disease). Ectopic secretion of ACTH from non-pituitary tissues includes small cell carcinoma of the lung and lung carcinoids, pancreatic or adrenomedullary tumors, as well as other neoplasias [32].
The first manifestation of Cushing disease may be the loss of the diurnal rhythm in ACTH and cortisol secretion. Patients with either Cushing disease or syndrome present with “Cushingoid features”, including moon facies, “buffalo hump”, central obesity, purple skin striae, hypertension, insulin resistance/overt diabetes mellitus, mood changes, menstrual irregularity and osteoporosis [33]. The diagnosis should be made after careful medical history and physical examination, and confirmed by endocrinologic evaluation and imaging studies. Cushing disease is mostly caused by pituitary adenomas. They are usually microadenomas with a diameter less than 1 cm, and are sometimes quite difficult to detect even in the MRI scan with contrast material [34]. Bilateral inferior petrosal sinus sampling with CRH stimulation is helpful in some cases to localize pituitary adenomas in the pituitary including the site of the tumor inside the pituitary gland. The therapeutic approach of patients with Cushing disease starts with resection of the ACTH-secreting pituitary adenoma by transsphenoidal surgery [35]. However, 10–15 % of patients with remission may recur in the future. Bilateral adrenalectomy may be performed in difficult cases in which tumors cannot be successfully resected [35]. Radiation may also be applied to the pituitary gland in combination with medical therapy, usually with mitotane or ketoconazol.
2.2 Pituitary Disorders That Result in Hypofunction of the HPA Axis
Any space-occupying lesions in the pituitary gland may lead to hypofunction of the HPA axis and secondary adrenal insufficiency [24]. Pituitary adenomas, cysts, craniopharyngiomas, ependymomas, meningiomas and rarely carcinomas may interfere with ACTH secretion. In addition to these lesions, infections or infiltrative processes, including lymphocytic hypophysitis, haemochromatosis, tuberculosis, meningitis, sarcoidosis, actinomycosis, histiocytosis X and Wegener’s granulomatosis can result in low ACTH release [24]. Sheehan’s syndrome is caused by pituitary apoplexy after labor and consequent pan-hypopituitarism, and used to be a major cause of secondary adrenal insufficiency. However, its incidence has been dramatically reduced because of the improved maternal care in developed countries.
Mutations in genes encoding crucial transcription factors and molecules essential for corticotrope survival and function can cause developmental defects and/or dysfunction in these cells leading to hypofunction of the HPA axis. So far, pathologic mutations have been identified in the genes expressing the transcription factors HESX homolog 1 (HESX1), orthodenticle homeobox 2 (OTX2), LIM homeobox 4 (LHX4), SRY (sex-determining region Y)-box 2 and 3 (SOX2 and SOX3), PROP paired-like homeobox 1 (PROP1) and T-box 19 (TBX19) [36]. Patients harboring these mutations may present with short stature, metabolic disorders, craniofacial abnormalities and learning difficulties/mental retardation associated with abnormalities in the CNS. The onset of adrenal insufficiency occurs generally early in life, although cases owing to PROP1 mutations usually manifest in adolescence.
Congenital pro-opiomelanocortin deficiency is another cause of secondary adrenal insufficiency. The molecular basis of this genetic condition has been ascribed to inactivating mutations in the pro-opiomelanocortin (POMC) gene. Patients develop a characteristic syndrome of hypocortisolism, early onset morbid obesity, and alterations in skin and hair pigmentation [37].
Achalasia-Adrenal Insufficiency-Alacrima syndrome or Allgrove Syndrome, also known as Triple-A syndrome, is a rare autosomal recessive disorder characterized by ACTH-resistant adrenal insufficiency, alacrima (absence of tear secretion), achalasia (dilatation of thoracic esophagus), autonomic dysfunction, and neurodegeneration. This syndrome is caused by mutations in the AAAS gene, which encodes the nuclear pore protein ALADIN with as yet unknown functions [38]. Adrenal insufficiency develops gradually within the first 10 years of life but sometimes becomes evident much later. It may present in some cases as an adrenal crisis with hypoglycemia and seizures.
3 Adrenal Disorders Associated with Alterations in the HPA Axis
3.1 Cushing Syndrome
Cushing syndrome can be caused by both benign and malignant lesions, such as glucocorticoid-producing adenomas, micro-nodular dysplasia, ACTH-independent macro-nodular hyperplasia or adenocarcinoma of the adrenal cortex [23, 33]. Patients with this syndrome appear with “Cushingoid features”, as described above; however, their clinical manifestations depend on the severity and duration of cortisol excess. Cushing syndrome caused by adenocarcinoma is characterized by large tumors of more than 5 cm in diameter, and are usually rapidly progressing, whereas adenomas are usually small (less than 3 cm) and have generally an insidious onset and chronic course [39]. First choice treatment for Cushing syndrome remains the surgical removal of the tumor. Laparoscopic surgery is currently the approach of choice for the treatment of Cushing syndrome caused by adenomas [40]. In case of failure of resection of the entire mass, usually adenocarcinomas, or those with metastatic lesions, mitotane is used for suppressing both tumor growth and production of steroids [41]. This compound inhibits the cholesterol side-chain cleavage (SCC) enzyme (the human p450 CYP, cholesterol desmolase, or 20, 22 desmolase) and the 11β-hydroxylase (p450 11β or CYP11B1) [41]. Adrenal enzyme inhibitors, such as ketoconazole, metyrapone and etomidate, are also used in clinical practice for patients with surgical failure who remain hypercortisolemic [32].
3.2 Adrenal Disorders That Result in Hypofunction of the HPA Axis
Adrenal insufficiency is a disorder first described by Thomas Addison in 1855, which is characterized by deficient production or action of glucocorticoids and/or mineralocorticoids and adrenal androgens [24]. Any pathologic condition associated with destruction of the adrenal cortex can cause primary adrenal insufficiency. The etiology of primary adrenal insufficiency has changed over time. Prior to 1920, the most common cause of primary adrenal insufficiency was tuberculosis, while since 1950, the majority of cases (80–90 %) have been ascribed to autoimmune adrenalitis, which can be isolated (40 %) or in the context of an autoimmune polyendocrinopathy syndrome (60 %) [24]. The clinical symptoms of adrenal insufficiency include weakness, fatigue, anorexia, abdominal pain, weight loss, orthostatic hypotension, salt craving, and characteristic hyperpigmentation of the skin. Regardless of etiology, adrenal insufficiency was an invariably fatal disorder, until the synthesis of cortisone by Kendall and Reichstein in 1949, and the introduction of substitution therapy with life-saving synthetic glucocorticoids. Today, patients with primary adrenal insufficiency are treated with hydrocortisone or cortisone acetate if hydrocortisone is not available, and fludrocortisone to prevent intravascular volume depletion, hyponatremia and hyperkalemia [24]. One of the most important aspects of the management of chronic primary adrenal insufficiency is patient and family education. Patients should understand the reason for life-long replacement therapy, the need to increase the dose of glucocorticoids during minor or major stress and to inject hydrocortisone or methylprednisolone in emergencies.
4 Other Disorders Associated with Changes in the HPA Axis
4.1 Generalized Glucocorticoid Resistance Syndrome (Chrousos Syndrome)
Primary generalized glucocorticoid resistance (Chrousos syndrome) is a rare, familial or sporadic condition, which is characterized by generalized partial end-organ insensitivity to glucocorticoids [42, 43]. Affected patients have compensatory elevation in circulating cortisol and ACTH concentrations, which maintain circadian rhythmicity and appropriate responsiveness to stressors, and resistance of the HPA axis to dexamethasone suppression, but no clinical manifestations suggestive of Cushing syndrome. The clinical spectrum of this condition is broad, ranging from completely asymptomatic cases, displaying biochemical alterations only, to severe forms of mineralocorticoid and/or androgen excess [44]. The molecular basis of Chrousos syndrome has been ascribed to inactivating mutations in the hGR gene [16, 42–45]. Most of these mutations are located in the GR LBD, which alter functions of this subdomain, such as ligand-binding, activation of transcription and nuclear translocation. So far only two mutations have been reported in the DBD of GR: R477H and V423A [46]. All reported pathologic mutations causing familial or sporadic Chrousos syndrome are depicted in Fig. 6.4. Recently, a heterozygous mutation causing glucocorticoid hypersensitivity, ie the mirror image of glucocorticoid resistance, was described [47]. In this patient, Cushingoid features such as obesity and metabolic syndrome were associated with low normal cortisol levels.

Location of the identified mutations in the hGR gene causing Chrousos syndrome. A mutation that causes glucocorticoid hypersensitivity is included in red color. NTD N-terminal domain, DBD DNA-binding domain, LBD ligand-binding domain, ATG translation initiation codon, TGA stop codon, 1-8, 9α, 9β Exons 1-8, 9α, 9β (Modified from Ref. [43])
4.2 hGR Polymorphisms
Inter-individual variations in tissue sensitivity to glucocorticoids have been described within the normal population and have been partly attributed to polymorphisms in the hGR gene [16]. A heterozygous polymorphism replacing aspartic acid to serine at amino acid 363 that mildly increases the transcriptional activity of the affected receptor in vitro is associated with glucocorticoid hypersensitivity, weakly correlating with the development of central obesity and, thus, influencing the metabolic profile and the longevity of humans in a negative fashion [48, 49].
The polymorphism in the GR gene that causes arginine to lysine replacement at amino acid 23 (ER22/23EK: GAG AGG to GAA AAG) is associated with relative glucocorticoid resistance by altering the expression levels of GR translational isoforms [50]. This polymorphism increases muscle mass in males and reduces waist to hip ratio in females [51], and is associated with greater insulin sensitivity, and lower total and low-density lipoprotein cholesterol levels [52], indicating that this polymorphism has a beneficial effect on longevity by reducing glucocorticoid action.
4.3 Ectopic Tumors Producing ACTH or CRH
Ectopic ACTH producing tumors account for ~15 % of ACTH-dependent Cushing syndrome [53]. Most of them are malignant and occur more frequently in men than in women. These tumors usually derive from neuroendocrine cells, but adenocarcinoma and squamous cell carcinoma may also cause the syndrome [53]. Approximately half of the cases are due to small cell carcinoma of the lung. Carcinoids originating from the thymus, bronchi and other organs, pancreatic islet tumors, pheochromocytomas and ovarian adenocarcinomas may also be associated with this syndrome [54]. Patients with this condition have elevated concentrations of cortisol and ACTH, while concentrations of other POMC-related peptides and bioactive molecules, such as other pituitary hormones, calcitonin, somatostatin are sometimes increased because of their concurrent ectopic production by the tumors [54]. Hypokalemia occurs in most cases with this syndrome. Manifestations are of sudden onset and their progression is usually rapid and sometimes not responsive to treatment.
4.4 The Cardiomyocyte as a Glucocorticoid Target Cell
In addition to common cardiovascular manifestations, such as hypertension and metabolic syndrome, often observed in several pathologic conditions associated with alterations in the activity of the HPA axis, accumulating evidence suggests that glucocorticoids exert direct effects on cardiomyocytes. Indeed, glucocorticoids stimulate cardiomyocyte contraction by influencing calcium homeostasis [55–57]. Moreover, mice overexpressing GR in their cardiomyocytes exhibit atrioventricular block [58]. Furthermore, glucocorticoids may cause cardiac hypertrophy, a condition that represents a severe cause of congestive heart failure in humans [59]. A recent in vitro study elucidated the molecular mechanisms underlying glucocorticoid-induced cardiac hypertrophy [60]. Addition of dexamethasone in primary cardiomyocytes and rat embryonic H9C2 cardiomyocytes resulted in up-regulation of cardiac hypertrophic molecules, such as β-myosin heavy chain, atrial natriuretic factor and α-actin, resulting in an increased cardiomyocyte size [60]. These glucocorticoid effects were mediated by the GR, as exposure of cells to the GR antagonist RU 486 or addition of GR shRNA inhibited dexamethasone-induced cardiomyocyte hypertrophy [60]. In addition to hypertrophic effects, this study demonstrated that glucocorticoids had strong anti-apoptotic actions on cardiomyocytes. Serum deprivation and addition of TNFα triggered the apoptotic process in cardiomyocytes, whereas treatment of these cells with dexamethasone caused significant up-regulation of the key anti-apoptotic molecule Bcl-xL and concurrent down-regulation of Gas2, a known target of caspases-3 and −7 [60]. Importantly, these anti-apoptotic glucocorticoid actions may prevent cardiomyocyte death in severe pathologic conditions, such as ischemia/reperfusion injury.
4.5 Beneficial Nongenomic Glucocorticoid Effects in Cardiac Ischemia/Reperfusion Injury
In the past, glucocorticoids were used in acute myocardial ischemia due to their cardioprotective effects. Indeed, the acute administration of glucocorticoids decreased myocardial ischemic injury in animal models [61, 62] and increased the critical short-term survival of patients after acute myocardial infarction [63]. However, synthetic glucocorticoids are not used in the current treatment of acute myocardial ischemia because of their severe side effects, such as cardiac rupture [64]. An interesting study shed light on the acute cardioprotective effects of glucocorticoids both in in vitro experiments and in cardiac ischemia /reperfusion injury animal models [65]. Acute administration of high doses of dexamethasone activated a GR-dependent signaling pathway, which induced the activation of PI3K leading to increased activity of endothelial nitric oxide synthase (eNOS). The resultant NO-mediated vasorelaxation ameliorated cardiovascular inflammation and significantly decreased the size of myocardial infarct in mice that underwent ischemia and reperfusion injury [65]. Interestingly, these cardioprotective effects of glucocorticoids were GRE-independent and nongenomic, since the dexamethasone-bound GR activated acutely the synthesis of NO through non-transcriptional mechanisms. The acute cardioprotective effects of glucocorticoids represent a strong evidence for the importance of nongenomic glucocorticoid actions, which are currently under intense investigation.
5 Conclusions
The HPA axis plays a fundamental role in the maintenance of basal and stress-related homeostasis; dysfunction of hypothalamic nuclei, pituitary or adrenal glands may cause symptoms and signs associated with glucocorticoid excess or deficiency. Accordingly, the aim of treatment is to correct both the primary cause and the associated hormonal abnormality. During the last several decades, despite the well-recognized significant progress in the clinical care of patients with disorders of the HPA axis, there are still numerous challenges regarding the diagnosis and treatment of these conditions. Indeed, the ubiquitous presence and actions of glucocorticoids, the rapid advances in our general knowledge of the human genome and our ever-increasing understanding of cell signaling allow us to view the actions and functions of the HPA axis and its involvement in human health and disease in a more integrated fashion.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- AP-1:
-
Activator protein 1
- AV:
-
Arginine vasopressin
- CBP:
-
p300/CREB-binding protein
- CLOCK:
-
Circadian locomotor output cycle kaput
- CNS:
-
Central nervous system
- CRH:
-
Corticotropin-releasing hormone
- DRIP/TRAP:
-
Vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein
- eNOS:
-
Endothelial nitric oxide synthase
- GR:
-
Glucocorticoid receptor
- GREs:
-
Glucocorticoid response elements
- HPA Axis:
-
hypothalamic-pituitary-adrenal axis
- NF-κB:
-
Nuclear factor κΒ
- NL:
-
Nuclear localization signal
- p/CAF:
-
p300/CBP-associated factor
- POMC:
-
Pro-opiomelanocortin
- PVN:
-
Paraventricular nucleus
- SCN:
-
Suprachiasmatic nucleus
- STAT:
-
Signal transducer and activator of transcription
References
Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5:374–81.
Chrousos GP, Gold PW. The concepts of stress and stress system disorders: overview of physical and behavioral homeostasis. JAMA. 1992;267:1244–52.
Charmandari E, Tsigos C, Chrousos GP. Endocrinology of the stress response. Annu Rev Physiol. 2005;67:259–84.
Chrousos GP, Kino T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress. 2007;10:213–9.
Kontopoulou TD, Marketos SG. Homeostasis. The ancient Greek origin of a modern scientific principle. Hormones. 2002;1:124–5.
Fink G. Stress: definition and history. In: Squire LR, editor. Encyclopedia of neuroscience, vol. 9. Oxford: Academic; 2009. p. 549–55.
Weinberger C, Hollenberg SM, Ong ES, Harmon JM, Brower ST, Cidlowski J, et al. Identification of human glucocorticoid receptor complementary DNA clones by epitope selection. Science. 1985;228(4700):740–2.
Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 2005;(304):e48.
Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–62.
Nader N, Chrousos GP, Kino T. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab. 2010;21:277–86.
Chrousos GP, Calabrese JR, Avgerinos P, Kling MA, Rubinow D, Oldfield EH, et al. Corticotropin releasing factor: basic studies and clinical applications. Prog Neuropsychopharmacol Biol Psychiatry. 1985;9:349–59.
Calogero AE, Bernardini R, Gold PW, Chrousos GP. Regulation of rat hypothalamic corticotropin-releasing hormone secretion in vitro: potential clinical implications. Adv Exp Med Biol. 1988;245:167–81.
Smith MA, Kling MA, Whitfield HJ, Brandt HA, Demitrack MA, Geracioti TD, Chrousos GP, Gold PW. Corticotropin-releasing hormone: from endocrinology to psychobiology. Horm Res. 1989;31:66–71.
Stevens A, White A. ACTH: cellular peptide hormone synthesis and secretory pathways. Results Probl Cell Differ. 2010;50:63–84.
Bornstein SR, Chrousos GP. Clinical review 104: Adrenocorticotropin (ACTH)- and non-ACTH-mediated regulation of the adrenal cortex: neural and immune inputs. J Clin Endocrinol Metab. 1999;84:1729–36.
Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75:1–12.
Kino Τ, Chrousos GP. Glucocorticoid and mineralocorticoid receptors and associated diseases. Essays Biochem. 2004;40:137–55.
Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor β, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95:2435–41.
Kino T, De Martino MU, Charmandari E, Mirani M, Chrousos GP. Tissue glucocorticoid resistance/hypersensitivity syndromes. J Steroid Biochem Mol Biol. 2003;85:457–67.
Charmandari E, Chrousos GP, Ichijo T, Bhattacharyya N, Vottero A, Souvatzoglou E, et al. The human glucocorticoid receptor (hGR) β isoform suppresses the transcriptional activity of hGRα by interfering with formation of active coactivator complexes. Mol Endocrinol. 2005;19:52–64.
Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. Molecular origins for the dominant negative function of human glucocorticoid receptor β. Mol Cell Biol. 2003;23:4319–30.
Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–42.
Chrousos GP. Glucocorticoid therapy. In: Felig P, Frohman LA, editors. Endocrinology & metabolism. 4th ed. New York: McGraw-Hill; 2001. p. 609–32.
Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet. 2014;383:2152–67.
de Lind van Wijngaarden RF, Otten BJ, Festen DA, Joosten KF, de Jong FH, Sweep FC, et al. High prevalence of central adrenal insufficiency in patients with Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93:1649–54.
Gold PW, Chrousos GP. The endocrinology of melancholic and atypical depression: relation to neurocircuitry and somatic consequences. Proc Assoc Am Physicians. 1999;111:22–34.
Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (2). N Engl J Med. 1988;319:413–20.
Kaye WH, Gwirtsman HE, George DT, Ebert MH, Jimerson DC, Tomai TP, et al. Elevated cerebrospinal fluid levels of immunoreactive corticotropin-releasing hormone in anorexia nervosa: relation to state of nutrition, adrenal function, and intensity of depression. J Clin Endocrinol Metab. 1987;64:203–8.
Nader N, Ng SS, Lambrou GI, Pervanidou P, Wang Y, Chrousos GP, et al. AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK. Mol Endocrinol. 2010;24:1748–64.
Kino T, Chrousos GP. Circadian CLOCK-mediated regulation of target-tissue sensitivity to glucocorticoids: implications for cardiometabolic diseases. Endocr Dev. 2011;20:116–26.
Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009;23:1572–83.
Veytsman I, Nieman L, Fojo T. Management of endocrine manifestations and the use of mitotane as a chemotherapeutic agent for adrenocortical carcinoma. J Clin Oncol. 2009;27:4619–29.
Orth DN, Kovacs WJ, DeBold CR. The adrenal cortex. In: Wilson JD, Foster DW, Kronenberg HM, Larsen R, editors. Textbook of endocrinology. 9th ed. Philadelphia: W.B. Saunders Co; 1998. p. 517–664.
Tsigos C, Chrousos GP. Differential diagnosis and management of Cushing’s syndrome. Annu Rev Med. 1996;47:443–61.
Aghi MK. Management of recurrent and refractory Cushing disease. Nat Clin Pract Endocrinol Metab. 2008;4:560–8.
Kelberman D, Dattani MT. Hypopituitarism oddities: congenital causes. Horm Res. 2007;68 Suppl 5:138–44.
Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–7.
Cronshaw JM, Matunis MJ. The nuclear pore complex: disease associations and functional correlations. Trends Endocrinol Metab. 2004;15:34–9.
Patalano A, Brancato V, Mantero F. Adrenocortical cancer treatment. Horm Res. 2009;71 Suppl 1:99–104.
Porterfield JR, Thompson GB, Young Jr WF, Chow JT, Fryrear RS, van Heerden JA, et al. Surgery for Cushing’s syndrome: an historical review and recent ten-year experience. World J Surg. 2008;32:659–77.
Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356:2372–80.
Charmandari E. Primary generalized glucocorticoid resistance and hypersensitivity. Horm Res Paediatr. 2011;76:145–55.
Charmandari E, Kino T. Chrousos syndrome: a seminal report, a phylogenetic enigma and the clinical implications of glucocorticoid signalling changes. Eur J Clin Invest. 2010;40:932–42.
Charmandari E. Primary generalized glucocorticoid resistance and hypersensitivity: the end-organ involvement in the stress response. Sci Signal. 2012;5(244):pt5.
Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93(5):1563–72.
Roberts ML, Kino T, Nicolaides NC, Hurt DE, Katsantoni E, Sertedaki A, et al. A novel point mutation in the DNA-binding domain (DBD) of the human glucocorticoid receptor causes primary generalized glucocorticoid resistance by disrupting the hydrophobic structure of its DBD. J Clin Endocrinol Metab. 2013;98:E790–5.
Charmandari E, Ichijo T, Jubiz W, Baid S, Zachman K, Chrousos GP, et al. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR-mediated gene expression. J Clin Endocrinol Metab. 2008;93:4963–8.
Huizenga NA, Koper JW, De Lange P, Pols HA, Stolk RP, Burger H, et al. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab. 1998;83:144–51.
Dobson MG, Redfern CP, Unwin N, Weaver JU. The N363S polymorphism of the glucocorticoid receptor: potential contribution to central obesity in men and lack of association with other risk factors for coronary heart disease and diabetes mellitus. J Clin Endocrinol Metab. 2001;86:2270–4.
Russcher H, van Rossum EF, de Jong FH, Brinkmann AO, Lamberts SW, Koper JW. Increased expression of the glucocorticoid receptor-A translational isoform as a result of the ER22/23EK polymorphism. Mol Endocrinol. 2005;19:1687–96.
van Rossum EF, Voorhoeve PG, te Velde SJ, Koper JW, Delemarre-van de Waal HA, Kemper HC, et al. The ER22/23EK polymorphism in the glucocorticoid receptor gene is associated with a beneficial body composition and muscle strength in young adults. J Clin Endocrinol Metab. 2004;89:4004–9.
van Rossum EF, Koper JW, Huizenga NA, Uitterlinden AG, Janssen JA, Brinkmann AO, et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes. 2002;51:3128–34.
Nieman LK, Ilias I. Evaluation and treatment of Cushing’s syndrome. Am J Med. 2005;118:1340–6.
Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newell-Price J, Reznek RH, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91:371–7.
Whitehurst Jr RM, Zhang M, Bhattacharjee A, Li M. Dexamethasone-induced hypertrophy in rat neonatal cardiac myocytes involves an elevated L-type Ca(2+)current. J Mol Cell Cardiol. 1999;31:1551–8.
Hadoke PW, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712.
Wang L, Feng ZP, Duff HJ. Glucocorticoid regulation of cardiac K+ currents and L-type Ca2+ current in neonatala mice. Circ Res. 1999;85:168–73.
Sainte-Marie Y, Nguyen Dinh Cat A, Perrier R, Mangin L, Soukaseum C, Peuchmaur M, et al. Conditional glucocorticoid receptor expression in the heart induces atrio-ventricular block. FASEB J. 2007;21:3133–41.
Lister K, Autelitano DJ, Jenkins A, Hannan RD, Sheppard KE. Cross talk between corticosteroids and alpha-adrenergic signaling augments cardiomyocyte hypertrophy: a possible role for SGK1. Cardiovasc Res. 2006;70:555–65.
Ren R, Oakley RH, Cruz-Topete D, Cidlowski JA. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology. 2012;153:5346–60.
Libby P, Maroko PR, Bloor CM, Sobel BE, Braunwald E. Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest. 1973;52:599–607.
Spath Jr JA, Lane DL, Lefer AM. Protective action of methylprednisolone on the myocardium during experimental myocardial ischemia in the cat. Circ Res. 1974;35:44–51.
Barzilai D, Plavnick J, Hazani A, Einath R, Kleinhaus N, et al. Use of hydrocortisone in the treatment of acute myocardial infarction. Summary of a clinical trial in 446 patients. Chest. 1972;61:488–91.
Hammerman H, Schoen FJ, Braunwald E, Kloner RA. Drug-induced expansion of infarct: morphologic and functional correlations. Circulation. 1984;69:611–7.
Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Nicolaides, N.C., Charmandari, E., Chrousos, G.P. (2015). The Hypothalamic-Pituitary-Adrenal Axis in Human Health and Disease. In: Cokkinos, D. (eds) Introduction to Translational Cardiovascular Research. Springer, Cham. https://doi.org/10.1007/978-3-319-08798-6_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-08798-6_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-08797-9
Online ISBN: 978-3-319-08798-6
eBook Packages: MedicineMedicine (R0)