
Abstract
Chimeric antigen receptor (CAR) T-cells are considered “living drugs” and offer a compelling alternative to conventional anticancer therapies. Briefly, T-cells are redirected, using gene engineering technology, toward a specific cancer cell surface target antigen via a synthetic chimeric antigen receptor (CAR) protein. CARs have a modular design comprising four main structures: an antigen-binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains for T-cell activation. A major challenge in the CAR T-cell manufacturing field is balancing product quality with scalability and cost-effectiveness, especially when transitioning from an academic clinical trial into a marketed product, to be implemented across many collection, manufacturing, and treatment sites. Achieving product consistency while circumnavigating the intrinsic variability associated with autologous products is an additional barrier. To overcome these limitations, a robust understanding of the product and its biological actions is crucial to establish a target product profile with a defined list of critical quality attributes to be assessed for each batch prior to product certification. Additional challenges arise as the field progresses, such as new safety considerations associated with the use of allogenic T-cells and genome editing tools. In this chapter, we will discuss the release and potency assays required for CAR T-cell manufacturing, covering their relevance, current challenges, and future perspectives.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Allogeneic CAR T-cells
- CAR T-cell cytotoxicity
- CAR T-cell manufacturing
- CAR T-cell potency
- CAR T-cell therapy
- Direct assays
- Indirect assays
- Purity criteria
- T-cell potency
- Vector copy number
8.1 Introduction
Chimeric antigen receptor (CAR) T-cells are considered “living drugs” and offer a compelling alternative to conventional anticancer therapies. Briefly, T-cells are redirected, using gene engineering technology, toward a specific cancer cell surface target antigen via a synthetic CAR protein. CARs have a modular design comprising four main structures: an antigen-binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains for T-cell activation (Fig. 8.1) [71, 81]. The antigen-binding domain is typically composed of a single-chain variable fragment, derived from a monoclonal antibody, providing specificity against the desired antigen.

Structure of chimeric antigen receptors (CAR). (a) The core structure of a CAR, highlighting its main components: the extracellular domain (responsible for antigen recognition), the transmembrane domain, and the intracellular domain (endodomain). The antigen-recognition domain is a single-chain fragment variant (scFV) generally composed of the variable light and heavy chain regions of an antigen-specific immunoglobulin separated by a flexible linker. This is linked to the transmembrane domain through the hinge. This spacers region generally supplies stability and flexibility for efficient CAR expression and activity, and it is often derived from the structure of immunoglobulins. The endodomain contains the intracellular motifs that enable downstream signaling proteins to be recruited and phosphorylated upon antigen binding for T-cell activation. Most CARs contain the intracellular domain of CD3ζ, which contains three immunoreceptor tyrosine-based activation motifs (ITAMs), as well as different co-stimulatory domains (e.g., CD28 and 41BB). (b) Evolution of the development of CARs from the first generation, which contained only ITAM motifs in the intracellular domain. Introduction of one (second generation) or more (third generation) co-stimulatory domains were crucial for the success of CAR T-cell therapies. New CARs are now under development to further improve efficacy by introduction of constitutive or inducible chemokines (e.g., IL-12) (fourth generation) or intracellular domains of cytokine receptors (fifth or next generation). (Image reproduced from Tokarew et al. [82] under the terms of the Creative Commons CC BY license (http://creativecommons.org/licenses/by/4.0/))
CD19-targeting is at the forefront of CAR T-cell technology development. This antigen is highly expressed across different types of B cell malignancies, but virtually absent outside the B-cell compartment and its expression is confined to the B cell development stages but lost upon terminal differentiation into plasma cells. These characteristics confer a high specificity and high coverage that is ideal for CAR T-cell therapy targets [93]. The unprecedented responses observed in clinical trials using CD19-targeting CAR T-cells have led to U.S. Food and Drug Administration (FDA) approvals for four different CAR T-cell products for relapsed/refractory (r/r) B-cell malignancies: YESCARTA™ (axicabtagene ciloleucel), KYMRIAH™ (tisagenlecleucel), TECARTUS™ (brexucabtagene autoleucel), and most recently, BREYANZI® (lisocabtagene maraleucel) [62].
The CAR T-cell field is rapidly evolving: a growing number of new targets and indications are under development, such as B-cell maturation antigen (BCMA) for multiple myeloma, CD30 for Hodgkin’s lymphoma, and CD20/CD22 for B-cell malignancies [94], with the first BCMA-targeting therapy, ABECMA (idecabtagene vicleucel), recently approved by the FDA [69]. CAR T-cells for application in solid tumor oncology are also the subject of intense investigation, posing additional challenges in overcoming the immunosuppressive tumor microenvironment, and low-expression/promiscuous target antigens. Despite this, encouraging results have been observed with EGFR, HER2, mesothelin, MUC1, and EpCAM CAR targeting for a broad range of indications [54].
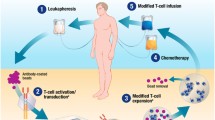
Manufacturing protocols for CAR T-cell products vary between products and institutions but are always governed by the principles of good manufacturing practice (GMP). Briefly, patient T-cells are harvested (using apheresis) followed by enrichment, activation, and transduction steps ex vivo, typically using a viral vector as a transgene delivery system. Transduced T-cells are expanded for 6–22 days ex vivo to obtain the target therapeutic dose and subsequently cryopreserved while awaiting completion of quality control testing, batch certification, and release to the patient [71].
A major challenge in the CAR T-cell manufacturing field is balancing product quality with scalability and cost-effectiveness, especially when transitioning from an academic clinical trial into a marketed product, to be implemented across many collection, manufacturing, and treatment sites. Achieving product consistency while circumnavigating the intrinsic variability associated with autologous products is an additional barrier. To overcome these limitations, a robust understanding of the product and its biological actions is crucial to establish a target product profile with a defined list of critical quality attributes to be assessed for each batch prior to product certification. Additional challenges arise as the field progresses, such as new safety considerations associated with the use of allogenic T-cells and genome-editing tools.
In this chapter, we will discuss the release and potency assays required for CAR T-cell manufacturing, covering their relevance, current challenges, and future perspectives.
8.2 Regulations and Requirements for Quality Control Testing and Batch Release
CAR T-cell therapies are considered advanced therapy medicinal products (ATMPs) in Europe, under the scope of the European Commission (EC) regulation 1394/2007 (as amended) and must be manufactured following the Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products (EudraLex, Volume 4, Part IV, 2017) [24]. In the United States, these therapies are regulated by the FDA Office of Tissues and Advanced Therapies of the Center for Biologics Evaluation and Research [53].
Both the FDA [53] and the European Medicines Agency (EMA) [16] have historically published guidelines for cell and gene therapy that are applicable, although not specific, to CAR T-cell products. The FDA’s “Considerations for the Development of Chimeric Antigen Receptor (CAR) T-Cell Therapies” was released on March 2022 and a revised version of the EMA Guideline on quality, nonclinical, and clinical aspects of medicinal products containing genetically modified cells came into effect in June 2021 and covers more details pertinent to CAR T-cell therapies [26].
Due to the rapidly evolving nature of the field, diversity of manufacturing practice, product complexity, and critical knowledge gaps concerning the biological action and the determinant features associated with clinical efficacy and safety of CAR T-cell therapies, it is challenging to establish harmonized and consolidated guidelines that apply to the entire industry. Furthermore, compendial testing methods are often not suitable for analysis of this type of product, so alternative assays should be validated.
In general, rigorous characterization studies throughout the earlier stages of development for each individual CAR T-cell product are essential to identify critical quality attributes, i.e., molecular and biological characteristics found to be necessary to ensure product safety and efficacy. These should cover the determinants of product safety, identity, purity, and potency that will form the requirements for final batch release (Table 8.1). A certificate that summarizes the test methods used, the corresponding test results, and the acceptable range must be provided for release of each batch. Specifications should be appropriate to the stage of product development and should be refined and tightened as product development progresses toward licensing. For Phase 1 trials, it is generally understood by the regulatory agencies that few specifications are finalized and that assays may still be under development. However, as a minimum, specifications and acceptance criteria for product safety and quantity (cell doses) should be defined and an appropriate testing plan for characterization defined. It is also generally accepted that validation of analytical procedures will not be complete at this stage. Nevertheless, methods should be appropriately controlled, specific, sensitive, and reproducible and, whenever possible, compendial methods should be used. Furthermore, safety-related assays should be qualified prior to initiation of clinical trials [26, 86]. The quality attributes, most commonly assessed for CAR T-cell product batch release, are discussed below.
8.3 Safety
Safety testing must be in place for all Phase 1 trials and usually includes assays to ensure products are free from microbial contamination, adventitious agents, and replication competent virus. These are outlined in detail below. Specifications with established acceptance criteria must be defined based on the quality attributes of each specific CAR T-cell product and details of manufacturing methods and transgene delivery strategy should be considered.
Due to the rapidly evolving nature of CAR technology and clinical trials, the risks associated with novel products must be accordingly mapped to define critical quality attributes and appropriate testing/assays to ensure product safety. Examples include fourth-generation CAR designs that combine direct tumor targeting with programmed cytokine secretion [13]; genome-editing tools that have the potential to induce (off target) double-stranded DNA breaks; and the immunological implications of off-the-shelf, allogeneic CAR T-cell products [41].
8.3.1 Sterility Assessment
In Europe and the United States, sterility testing of biopharmaceutical products is historically performed as defined by USP <71> [88] and Ph. Eur. 2.6.1 [15], with detection of microbial or fungal growth in test samples by turbidity assessment after 14 days of incubation. These time-consuming assays greatly increase the turnaround time of CAR T-cell products. Over recent years, automation and introduction of colorimetric and fluorescence-based CO2 measurements of metabolic activity (e.g., BacT/Alert 3D® and BD BACTEC™ systems) or adenosine triphosphate (ATP) detection by bioluminescence (Rapid Milliflex® Detection System) have increased the sensitivity of culture-based methods and permit faster detection of contamination when compared with standard methods. Further, the use of enriched aerobic or anaerobic media and incubation at 35–37 °C for a minimum of 7 days is an approach formally recognized by Ph. Eur. 2.6.27 [34].
The use of rapid and nonculture methods is also covered by the American legislation, under the Code of Federal Regulation (CFR) on “General Biological Product Standards” (21 CFR 610) [85]. Any alternative methods must be validated (as covered by Ph. Eur. 5.1.6) [15] and results must be demonstrated to be equal or superior to the compendial references. PCR-based approaches for bacteria and fungi detection through amplification of highly conserved sequences, such as the bacterial 16S rRNA, are currently under development [83]. Although optimization and comparability in sensitivity to the gold standard compendial tests are still to be determined, these methods are promising and have the potential to generate results within a few hours.
For products with a short shelf-life, product release prior to final sterility results can be accepted as part of a risk-based approach. The FDA mandate a combination of in-process sterility controls, a rapid detection test (such as Gram staining) and final sterility assessment based on 21 CFR 610 compliant methods, with a clear management strategy for positive results detected following product administration. This is not currently common practice for CAR T-cell therapies where products are cryopreserved prior to infusion. However, disease progression (and in some cases patient death) during the manufacturing period represents a significant challenge, affecting up to 13% of the patients in pivotal trials [36]. Shorter vein-to-vein times and the use of closed, automated manufacturing platforms (where the risks of in-process product contamination are considerably reduced) are of critical importance to the field.
8.3.2 Mycoplasma Detection
Mycoplasma contamination can arise from the use of cell culture reagents of animal origin, from the starting donor material, or the environment and personnel. Contrary to most bacterial contaminations, the presence of Mycoplasma does not always result in noticeable changes to cell culture turbidity or cell morphology and may go undetected for several cell passages. To address this risk, a compendial culture-based assay using indicator cell lines and multiple cell passages is described by USP <63> [88] and Ph. Eur. 2.6.7 [15], but this laborious and time-consuming testing method is not well suited for release testing of single-batch cell products. Indeed, the use of alternative detection methods such as PCR-based assays is supported by the FDA and EMA for use in the CAR T-cell therapy space [84].
8.3.3 Replication Competent Lentivirus (RCL) or Retrovirus (RCR)
Retroviral and lentiviral vectors are commonly used in CAR T-cell manufacture as efficient tools for delivery of transgene to target cells. Viral vector design has improved significantly over the last decade, with safety features designed to reduce the likelihood of generating replication competent viral vector during the manufacture process. However, exposure to replication-competent lentivirus (RCL) or retrovirus (RCR) remains a theoretical safety concern for patients treated with CAR T-cell therapies. Recombination events could lead to the generation of novel, replicating viruses during CAR T-cell manufacture or post-infusion, posing a risk of genotoxicity and malignant cell transformation. To mitigate for this, recommendations for RCL/RCR testing include assessment of all viral vector lots, manufactured cell products, and monitoring patients post-infusion. Assays for RCL/RCR detection in the viral vector batch rely on the use of permissive cell lines such as the PG4 cell line, which assumes a transformed phenotype in the presence of RCR, cultivated during multiple passages with the test material to support virus entry, amplification, and particle production [14]. The amplified material is then detected with a bespoke indicator assay, developed specifically for the vector under investigation.
For analysis of ex vivo transduced cells for batch release, PCR-based assays may be considered appropriate, particularly when time constraints are present. The use of alternative assays should be defined based on a risk assessment and should be validated, with an appropriate limit of detection. Recent guidelines from both European and American regulatory bodies have introduced flexibility to the requirement for RCL/RCR testing as part of final batch release. Indeed, RCR/RCL testing can be omitted once sufficient manufacturing and clinical experience is obtained to demonstrate that transduced cell products are consistently RCL/RCR-negative [87], or if the absence of RCL/RCR is demonstrated for each viral vector batch and generation of replicating virus during manufacture is ruled out by appropriate risk assessments [26]. Reassuringly, long-term safety data from multiple clinical trials using genetically modified cell products continues to accumulate, without evidence of RCR/RCL, indicating that any associated risks are low [36, 49, 51].
8.3.4 Vector Copy Number (VCN) per Transduced Cell
When cells are transduced with integrating vectors, the risk of insertional mutagenesis needs to be carefully considered. The risks are determined by several factors, including the insertion profile of the vector used, the vector design including the choice of enhancer and promoter sequences, the transgene product, and the vector copy number (VCN) per transduced cell.
Gammaretroviral vectors confer a risk of leukemogenesis due to their pattern of integration near transcription start sites and proto-oncogenes. This is also a potential (lesser) risk for lentiviral vectors [26]. Available clinical data suggest that newer generation vectors strongly reduce the risks of insertional mutagenesis [57], nevertheless as the total number of transduced cell infusions increases, the likelihood of infusing cells bearing at-risk insertions also increases.
Regulatory agencies require characterization of integration profile and integration sites to support marketing authorization applications. Analysis of VCN per transduced cell is a critical quality attribute determining product safety. Since VCN also has a direct impact on transgene expression, products must be carefully designed to achieve a balance between safety and efficacy. Less than five copies per transduced cell is usually considered a safe limit [99].
VCN assessment of CAR T-cell products and patient peripheral blood during follow-up is usually performed by quantitative PCR (qPCR), although a recommended, standardized assay is yet to be defined [40]. Methods employing single-cell level analysis such as droplet digital PCR (ddPCR) have the advantage of allowing detection of cell-to-cell variability in the distribution of vector copies rather than an average of the whole cell population, thus allowing identification of clones with a high number of integrations that could pose a higher risk [73].
8.3.5 Identity
Identity testing is required to identify a product and distinguish it from other products in the same facility. Most CAR T-cell therapies are patient-specific, autologous products and efficient traceability systems must be in place from apheresis to the final cell product, such that the correct product is infused to the correct patient.
For CAR T-cell products, identity assays include an assessment of specific cell populations such as CD3+/CD4+/CD8+ T-cells in addition to the intended genetic modification(s) by qPCR or flow cytometry. Transduction efficiency can be defined based on CAR expression or on the expression of marker genes, using polyclonal anti-mouse Fab reagents for CARs derived from murine scFv, anti-idiotype antibodies generated against specific binders or antigen-Fc detection reagents [40].
Immunophenotyping by flow cytometry is widely used in the clinical setting, but a lack of assay standardization remains. Promoting standardization is a priority for the field and efforts to address this include the definition of standard panels for evaluation of major immune cell subsets, the availability of internal controls, the development of automated analysis strategies, the definition of proficiency assessment programs, and the requirement for accreditation of flow facilities by external agencies such as the UK National External Quality Assessment Scheme (UK NEQAS) system. The EuroFlow consortium [63] and the Human ImmunoPhenotyping Consortium [27] are examples of initiatives to streamline and standardize immunophenotyping assays, so that data can be compared across different sites and studies. However, each CAR T-cell product has unique characteristics such that there may be a requirement to develop and validate new transduction efficiency assays for each new construct.
8.3.6 Purity
Purity is defined as the relative freedom from extraneous materials in the final product, excepting the drug substance and excipients. Purity criteria should be defined according to the nature and intended use of the cell product, the manufacturing method used, and the consistency of the production process. Assays to demonstrate product purity should be adequate to the phase of development and adjusted as data accumulates or whenever the manufacture process changes.
Process-related impurities may include media and supplements, growth factors and cytokines, antibiotics, activation and enrichment reagents, and vectors. These should be kept to a minimum in the final formulation. Risk assessment should consider the clearance of each substance throughout the manufacturing process and the risk to the patient upon infusion, setting quantitative limits for the final product as appropriate. An example of the CAR T-cell space is residual activation beads, generally quantified by microscopy [84, 90].
Viral vectors require particular consideration. Calculations based on initial vector volumes added alongside the reduction ratio achieved (defined by the vector half-life, inactivation steps, and final dilution) can help to ensure that free infectious vector particles in the final product are reduced to negligible concentrations.
There are significant technical challenges in demonstrating absence of infectious viral particles in the final product and this is acknowledged by EMA [26]. Residual infectious particle concentration can be roughly detected using permissible cell lines (HEK 293T cells), but for the purposes of environmental risk determination, theoretical calculations are generally accepted by the regulatory authorities. For lentiviral vectors, the Dutch Commission on Genetic Modification (COGEM) proposed a formula based on available experimental data that can estimate residual free infectious viral particles in the cell product. However, variability between vectors, products, and processes means that this should be used with caution [18].
Product-related impurities can include nontarget cells, unmodified target cells, and nonviable cells, which may be present after selection or enrichment. For CAR T-cell products, a minimum of 70% viability is recommended by the FDA [83]. Release criteria for CAR T-cell products often include % CD3+ T-cells, but a full characterization of final cell composition (including residual tumor burden) is desirable, especially when the manufacturing method does not include an enrichment step.
With regard to safety, evaluation of bacterial endotoxin level is mandatory. The FDA may require in vivo rabbit pyrogen tests for some licensed products. More often, the Limulus Amoebocyte Lysate (LAL) method is used, as defined by Ph. Eur. 2.6.14 [15] and USP <85> [88]. This test uses hemolymph extracted from the Limulus Polyphemus crab, which clots in the presence of bacterial endotoxins. The FDA recommends that the upper limit acceptance criterion for endotoxin should be set at 5 Endotoxin Units (EU)/kg body weight/hour for intravenous infusion. For intrathecal and/or intraocular administration, the recommendation is 0.2 EU/kg body weight/hour and 2.0 EU/dose/eye, respectively [86]. Although most CAR T-cell therapies are intravenously infused, local administration in the tumor or at the resection site are being evaluated for solid tumors [74].
Dimethyl sulfoxide (DMSO) is considered an excipient for cryopreserved cells rather than an impurity, but a safety limit for infusion is defined as 1 mL/kg/day, and this should be taken into account for high volume CAR T-cell products.
8.3.7 Quantity
To ensure consistent dosing throughout clinical investigation, specification of methods to measure dosing should also be defined at Phase 1. For CAR T-cell products, methods to determine absolute cell counts and flow cytometry assays for detection of CAR expression are usually used.
Image-based automated counting methods, such as the NucleoCounter® and Vi-CELL™ are useful for determination of total cell numbers and viability. Alongside automated hematological analyzers, these can be useful as quick tools for in-process controls. Precise assessment of final product dose is usually performed by flow cytometry, as this permits determination of CAR expression in viable CD3+ T-cells/other target cells. It also gives additional information on the expression of other proteins such as memory and exhaustion markers, which may be relevant features for potency assessment and allows enumeration of cells using counting beads.
The FDA recommends that assays to determine dose should be qualified prior to initiation of clinical studies and a detailed description of the qualification protocol submitted in the original Investigational New Drug (IND) application, along with data supporting the accuracy, reproducibility, sensitivity, and specificity of the method [86].
8.4 Potency
Potency assessment is an essential aspect of the quality control system to evaluate biological function of cellular products and to ensure batch-to-batch consistency. These assays should be defined according to the products’ mechanisms of action and critical attributes assessed by well-controlled investigations throughout the development stages and conducted with consistently manufactured products (Table 8.2). In the CAR T-cell arena, development of potency assays is challenging, due to the intrinsic batch-to-batch variability associated with the use of autologous cells.
Potency assessment for CAR T-cell batch release generally involves analysis of target-specific cytotoxicity, but this does not give insights into many aspects of their biological potential such as the ability to deliver long-term responses and persistence. It is unlikely that the immense complexity of these products can be captured by a single assay. This is discussed in more detail in Sect. 8.3.
8.4.1 Potency Assessment for CAR T-Cell Therapies
Potency assessment of CAR T-cells is a quantitative measurement of their biological activity and should ensure the quality and consistency of released batches [29]. These assays should be capable of identifying subpotent batches and used as a measure of drug product quality and consistency. Traditional approaches to potency testing are based on the development of in vivo and in vitro assays that measure the product’s mechanism of action (MoA). Assays should be developed, optimized, and validated to characterize product attributes/biological activity that reflects or predicts clinical outcome and that could be defined as a critical quality feature [7].
Potency assay development for cellular therapy products poses several challenges [67]. First, each drug product is manufactured using patient-specific starting material, such that there is limited QC material available for potency assessment(s). Second, autologous products can be highly variable, making it challenging to define and validate a consistent assay. Furthermore, CAR T-cells exert their action through multiple, complementary mechanisms and it is difficult to capture this complexity in a single, accurate assay. Potency testing can be time-consuming, and development of rapid assays should be the priority so as not to delay final batch certification, particularly for patients with rapidly progressive disease. For the reasons outlined, standardized potency assays for CAR T-cell products are not yet defined for widespread use.
Although potency testing is not a prerequisite for early-stage clinical studies and is only essential for product release from Phase III onward, implementation of potency assays in earlier phase clinical studies may facilitate the development of more sophisticated and well-defined assays for use in Phase III via continuous optimization.
8.5 Regulatory Aspects
According to American and EU pharmaceutical legislation, cellular therapeutic products (and therefore CAR T-cell products) require potency evaluation prior to market entry [25, 28]. Both regulators stress the complexity of potency assay development and adopt a flexible regulatory approach, albeit the FDA specifies certain requirements for potency testing, outlined in Table 8.2 [25].
The analytical method used for potency assessment should ideally be quantitative, with appropriate controls and standards. However, regulatory agencies acknowledge that quantitative methods are not always feasible and will accept semiquantitative assays in this setting.
Although not required at early stages, a progressive developmental approach to potency assays is suggested and acceptance criteria specifications set for Phases 1 and 2 should be adjusted throughout product development stages to reflect manufacturing and clinical experience. The presentation of early-stage results as “information only” is also valid.
Both the FDA and EMA accept the use of in vivo and in vitro functional biological assays for product characterization. Due to the time constraints for product release, both agencies recognize the usage of nonfunctional, surrogate assays if correlation has been previously established. Most importantly, accuracy, sensitivity, specific, and reproducibility of all types of assays should be established [29].
In some cases, assay development may require the generation of novel standards or procedures that are not yet covered in the guidelines. In which case, manufacturers are responsible for the assessment and development of suitable assays for their specific product.
8.6 Methods for Potency Assessment of CAR T-Cell Products
Potency assessment should be defined based on the proposed mode of action of the product and on how and why it is expected to give therapeutic benefit. This can be based on scientific literature around nonclinical studies (animal or in vitro), or preliminary clinical data from early-stage trials. If the MoA is not fully defined, an assay matrix approach can permit assessment of biological activity, but when the MoA is known, the assay should focus on that attribute. For simplicity, most CAR T-cell potency assays are designed to measure cytotoxic activity in vitro against target-expressing cell lines. However, this simplified assessment does not account for the complex factors that determine product efficacy and influence response in vivo, such as the interaction with other immune cells, the role of the tumor microenvironment, the effects of chronic activation, and other factors not yet defined (Fig. 8.2). Extended potency assays can be helpful in this regard and are essential during the development of novel therapeutics, to permit full comprehension of the product and to inform potential product improvements.

In vivo determinants of CAR T-cell therapy effectiveness to be considered for the design of potency assays. (A) The main feature associated with CAR T-cell function is target-directed cytotoxicity. (B) Target-directed cytotoxic function is directly dependent on antigen expression. Therefore, antigen escape, low antigen density, and heterogenous antigen expression are common concerns. (C) Robust in vivo expansion and (D) sustained persistence are key features related with long-lasting clinical responses and are often associated with the T stem cell-memory compartment (Tscm), defined as CD45RA−/CCR7+/CD62L+/CD95+ CAR T-cells, and the resistance to exhaustion and senescence upon prolonged antigen exposure. (E) Lack of immunogenicity is also determinant in therapy success. (F) Particularly in the case of solid tumors, the capacity to reach tumor site and bypass the physical barriers of the tumor stroma (migration/invasion) are critical. (G) CAR T-cells designed for such application must also be resistant to the hostile tumor microenvironment (TME), often hypoxic and acidic and (H) to the many immunosuppressive factors expressed by the tumor cells (such as PD-L1), secreted into the TME (such as TGFb), and the presence of suppressive immune cells, such as regulatory T (Treg)
In vivo assays are often central to product development, as in process controls or to evaluate the effect of manufacture process change [79], but even then, the lack of appropriate animal models, difficult standardization, technical complexity, and experimental duration limit their use [25]. Efforts have been made into the development, characterization, and standardization of xenograft mice models for anti-CD19 CAR T-cell therapies [1] and such comprehensive studies of tumor behavior and kinetics could be critical in widening the application of these models for the characterization of product potency, prediction of clinical outcomes, and particularly applicable in the field of solid tumors.
Simplified in vitro assays have the advantage of allowing a higher degree of standardization and are likely to remain the preferred choice for potency evaluation for batch release. For instance, the use of target-expressing cell line banks, although not fully representative of patient’s target tumor cells, provides a standardized model that allows a batch-to-batch comparison of product activity. As long as supported by efficiency data, simple in vitro assays are easier to qualify, allowing definition of a numeric acceptance criteria and providing invaluable comparative information on product quality. The most common in vitro methods for potency assessment are described and summarized in Fig. 8.3.

Summary of most commonly used strategies for CAR T-cell products potency assessment. Most potency assays currently used for batch release are associated with assessment of CAR T-cell effector function, either via directly cytolytic activity of target-expressing cells or via the use of surrogate markers that reflect T-cell activation and function upon exposure to targets. Other attributes can also be relevant in the determination of CAR T-cell activity. Characterization of an immunophenotypic profile that correlates with product efficacy is being sought. Proliferative capacity and, most recently, polyfunctionality profile, have also been demonstrated to correlate with responses and can be considered for potency assessment. Special considerations for the development of potency assays for solid tumors include the effect of the complex tumor microenvironment (TME) and CAR T-cell migration capacity and ability to reach tumor site
In the future, several assays may be required for full product characterization, but we acknowledge that more complex/advanced assays for product release may have an adverse impact on time to release and overall costs. As an example, CRISPR screening has recently emerged as a valuable tool for the identification of genes that are determinants for CAR T-cell function and clinical efficacy [91]. Although impractical for product release, such screening approaches, if implemented in the course of product development and characterization, have the potential to reveal critical quality attributes that can be used as biomarkers for an efficient product release assay.
8.6.1 Target-Directed Cytotoxic Activity
Cytotoxicity assays measure CAR T-cell tumor targeting with the use of methods such as flow cytometry, radioactive labeling, and impedance analysis. Direct assays aim to quantify effector activity and target cell lysis, whereas indirect assays measure a by-product of the effector–target interaction (e.g., measurement of cytokines).
8.6.2 Direct Assays
During cytotoxicity assay development, it is essential to optimize read-outs, incubation times, and effector to target cell ratios. Controls should be included to demonstrate antigen-specific cytotoxicity (e.g., antigen-negative targets) and to ensure that measured cytotoxicity is effector-specific (e.g., by culturing targets without effectors and with non-CAR T-cells to account for background signal).
It is essential to choose the most representative cell type(s) for the study. On occasion, primary patient-derived target cells can be used, but this adds complexity, as autologous target cell isolation and culture can be cumbersome, it can increase assay variability (and failure) between batches and can hinder inter-batch comparison [59]. Instead, CD19+ transduced cells or natively expressing CD19+ cell lines (e.g., Burkitt’s lymphoma derived Raji cells) are easier to cultivate and are routinely used as targets for CD19 CAR T-cell cytotoxicity assays [31, 75], with the accepted limitation that they do not fully replicate the variable and complex metabolic and genetic profile of autologous tumor cells. Careful evaluation and selection of a suitable surrogate target cell line, including features like antigen expression and resistance to lysing activity are critical to ensure correlation with in vivo effect and tangible relevance [19, 33].
Chromium (51Cr)-based cytotoxicity assays represent the gold standard for characterization of CAR T-cell cytolytic activity due to their high sensitivity. Target cells are labeled with radioactive 51Cr, which is released to the supernatant upon effector-mediated target cell lysis [10]. As an endpoint assay, 51Cr release is usually measured on a single short time point (usually 4 h), due to the spontaneous release of 51Cr from the cell over time impairing longer analysis. The need for target cell labellng in a radiation-restricted area, alongside the hazards and technical/equipment requirements associated with the use of radiation, as well as the lack of target lysis kinetic information obtained are the main drawbacks of this technique.
Cytotoxicity assays using alternative target labeling techniques (e.g., calcein, europium, bioluminescence) are now emerging as more user-friendly approaches, although sensitivity must be evaluated [49, 89]. For bioluminescence analysis, target cells are transduced with a luciferase reporter gene. As the added bioluminescent substrate (Luciferin) is only processed by live cells, direct quantification of live target cells, and thus quantification of cytotoxic activity is measured as a decrease in bioluminescent signal over time [44].
Cytotoxic assays based on quantification of cytosolic enzymes that are naturally present in the cell and whose enzymatic activity can be measured upon release from damaged cells (e.g. lactate dehydrogenase [21]) could be used for cytotoxicity measurement. The main limitation of this approach is that these enzymes are present in both effector and target cells. This impairs the ability to discern the relative contributions from individual cell populations to the final enzymatic read-out, leading to poor assay sensitivity.
Impedance-based assays allow label-free, real-time monitoring of specific effector-induced cytolysis, measured by the detachment of target cells from a treated surface [95]. This technique was first validated for assessment of NK cell-induced cytotoxicity [35]. Briefly, nonadherent effector cells are incubated together with adherent target cells following which cytolytic action leads to target cell detachment, loss of impedance, and the restoration of electric current flow, which correlates with cytolytic activity. These automated platforms (e.g., xCELLigence systems) permit real-time monitoring of target cell populations over extended periods, combining high-sensitivity analysis with minimal cell manipulation. Furthermore, these platforms are not limited to adherent tumor cell targets. Antibody-coated plates can be used to immobilize nonadherent cell targets such that impedance assays can be used in CAR T-cell potency assessment [11].
Flow cytometry-based cytotoxicity assays can be used to study cytolysis in heterogenous cell populations. Target and effector cells can be recognized in terms of size, granularity, and specific staining while evaluating target cell death using standard DNA intercalating agents (e.g., propidium iodide or 7-AAD). Detailed product characterization and target cell phenotyping for antigen expression and density can be conducted in parallel and the resulting profile(s) can be correlated with differential susceptibility to cytolysis [39, 97].
An alternative approach is “fluorometric assessment of T lymphocyte antigen-specific lysis” (FATAL), which is purported to be a sensitive and reliable alternative to the 51Cr assay [77]. Target cells are loaded with fluorescent dyes and cytotoxic activity detected by flow cytometry. This assay has the potential advantage of lower dye leakage, allowing longer incubation times in comparison to the 51Cr assay. The VITAL assay, based on the same principle, adds a further potential advantage, permitting differential labeling of distinct target cell populations and measurement of cytolysis against a range of targets simultaneously [38].
Overall, flow-based assays have much utility in the potency space, characterizing the dynamic relationship between target and effector cells [50, 64]. Current limitations include the need for individual sample data acquisition, increasing the time required for analysis. High-throughput multiparametric assays that allow workflow automatization and timely cytotoxicity evaluation are key to scalability and validity [9, 55].
8.6.3 Indirect Assays
Indirect assays aim to measure the by-product(s) of effector cell activation upon exposure to target cells and can be particularly useful where product availability is limited. Indirect assays measure cytokines and chemotoxins (e.g., IFN-γ, granzyme B) secreted upon effector cell activation [76, 78]. Both FDA-approved Tisagenlecleucel and Axicaptagene Ciloleucel products utilize IFN-γ secretion in response to CD19 expressing targets as part of a potency assessment for product release. Interestingly, Novartis reported that IFN-γ secretion varied greatly from batch to batch, complicating the correlation between limited potency assessment and clinical effect [30, 47].
IFN-γ detection via ELISA reflects cytokine release from the whole incubated cell population (not restricted to CAR T-cells), which can lead to an overestimation of cytokine secretion by CAR T-cells [20]. For a more specific read-out, flow cytometry assays can detect cytokines intracellularly and allow investigators to differentiate cytokine secretion between different cell types. However, a few drawbacks of this approach are the need for prolonged intracellular staining protocols and the requirement of blocking cellular secretory pathways. This assay, therefore, reflects cytokine production rather than cytokine release. Another method, the IFN-γ catch assay, utilizes a capture reagent that combines a pan-leukocyte CD45 binder and an IFN-γ binder, immobilizing the cytokine as released by each cell to its surface [22]. This overcomes the limitations outlined for ELISA and flow cytometric methods.
Some centers use indirect methods based on the correlation between T-cell degranulation and killing activity. Upon interaction with target cells, markers of T-cell activation and degranulation (e.g., CD107a) are expressed on the CAR T-cell surface and can be detected via flow cytometry. This technique is also compatible with extend phenotyping with the use of additional markers [2].
More recently, efforts have concentrated in the measurement of effector-released cytokines at a single-cell level via enzyme-linked immunospot assay (ELISPot). ELISPot requires only a low cell number for analysis, but the disadvantage is that it does not allow further immunophenotyping of the product and it is limited to the detection of only one or two enzymes [43]. Alternative approaches based on fluorophores (such as the FluoroSpot assay) could allow the accurate detection of multiple cytokines per cell [42]. Further refinement of this approach has led to the LysisPot platform that uses target cell lines expressing β-galactosidase, a nonsecreted enzyme that is released from the cells upon lysis. This method allows characterization at a single cell level of both the direct cytotoxic activity of the CAR T-cell product and cytokine (IFN-γ) release. Of note, this assay has demonstrated that not all cytokine-producing cells have cytolytic activity [4].
8.6.4 Immunophenotyping
Detailed immunophenotyping can inform CAR T-cell potency assessment, provided a correlation between specific phenotypes and product efficacy can be made [56]. Cell exhaustion and senescence are related to loss of function and disease relapse, such that expression of the phenotypic marker programmed cell death protein 1 (PD-1) (and others) could predict for functionality [32, 37]. Further, CD45RA and CD62L expression are used as markers of T-cell memory, which appears to correlate with product efficacy [56]. Immunophenotyping assays are quick and simple and allow analysis at single cell level, but results should be evaluated with caution, as these are surrogate markers of CAR T-cell functionality and results may vary significantly from patient to patient. To date, no precise immunophenotypic profile has been determined as a direct predictor of CAR T-cell function in a validated, quantitative assay.
8.6.5 Target-Induced Proliferation
Proliferation capacity upon target antigen recognition has been demonstrated to predict efficacy of CAR T-cell therapies in vivo [58, 70]. This feature could also be used as an alternative potency assay in vitro, using fluorescent markers such as carboxyfluorescein succinimidyl ester (CFSE). To date, the correlation between proliferation upon target antigen recognition in vitro and in vivo potency is still pending [14]. Cytotoxic activity is the main MoA of CAR T-cell therapies to reduce in tumor burden and as such these assays tend to be preferred for batch release assessment.
8.6.6 Polyfunctionality
Novel, high-throughput single-cell analysis platforms have the potential to revolutionize the field of CAR T-cell potency assessment. Several studies have positively correlated the presence of polyfunctional cells (cells that co-secrete multiple cytokines), with potent and durable immunity against certain infections [17, 48] and tumors [8]. Recently, highly polyfunctional CD19 CAR T-cell products were demonstrated to be associated with clinical responses in non-Hodgkin’s lymphoma (NHL) patients [72]. High-throughput platforms such as the IsoPlexis system, use barcode chip assays [6] that can accurately and simultaneously measure up to 16 cytokine/chemokines secreted by thousands of CAR T-cells at a single-cell level [96]. Implementation of such high-throughput assays in potency testing may permit a more thorough characterization of CAR T-cells at single cell level and improve prediction of clinical response.
8.7 Challenges and Potential Improvements for CAR T-Cell Potency Assays
In the future, assessment of potency will encompass in vitro assays designed to study cell behavior and activity in an environment that more closely mirrors what is found in vivo. Antigen-stress tests will assist in the investigation of maintained cytotoxic activity after several rounds of exposure to target, mirroring the chronic cell activation observed in vivo and providing a model to investigate mechanisms associated with CAR T-cell failure. Target cells expressing a continuum of antigen densities can also be used to investigate the correlation between antigen density and product cytotoxicity. Soluble factors or cytokines such us TGFβ are present in vivo and can influence the biological activity of CAR T-cells [19]. TGFβ challenge assays may help to quantitate this impact on CAR T-cell function.
Due to the inherent variability of autologous CAR T-cell product, assays selected for potency assessment for final product release should have appropriate acceptance criteria that consider inter-batch variability and should be defined prior to the commencement of pivotal clinical trials [25, 28] in order to accurately define potent versus non-potent products. In the solid tumor CAR T-cell space, potency assays may have additional requirements beyond those outlined here, such as measures of CAR T-cell migration capacity to remote and immunologically hostile tumor sites [45, 61].
8.8 Future Challenges and Directions for CAR T-Cell Product Release Testing
In recent years, the field of cell therapy has developed at unprecedented speed. New CAR designs, new manufacturing technologies, and new approaches to address the current limitations of CAR T-cell therapies continuously emerge, and researchers and regulatory agencies are faced with the challenges of developing new assays and guidelines to address additional unknowns and risks [46].
Although the evaluation of cytotoxicity against CD19-expressing cells is a relatively well-described measure of CD19 CAR T potency, the panorama can be complex in the case of not as well-characterized targets and more complex and heterogenous tumors. Particularly, in the field of solid tumors, more advanced in vitro antitumor efficacy assays are likely to be required, taking into account the differential expression levels of target antigens for definition of activation thresholds, the impact of prolonged antigen exposure and the effects of the immunomodulatory tumor microenvironment. Recent approaches to overcome these challenges include the establishment of cell libraries expressing different amounts of surface antigens using CRISPR/Cas9 knock out, reexpression, FACS sorting and single-cell cloning [52], and the development of ex vivo tumor-derived culture systems that can account for the environment-derived immunomodulation [80]. Conversely, in vitro assays to evaluate homing and tumor infiltration are challenging. The use and characterization of animal models [1], as well as the emergence of methods that combine the use of human tumor slices and real-time imaging [23] are likely to provide unvaluable insights into some of the key quality attributes associated with in vivo efficacy of CAR T-cell therapy in solid tumors.
On the other hand, one of the biggest developments in the CAR T-cell field is the move away from viral vectors and toward alternative gene delivery methods. Older methods such as the Transposon/Transposase platform relies upon DNA plasmids and mRNA transposase electro- or lipo-transfected into T-cells [60]. Several groups have shown the feasibility of generating CAR T-cells using the Sleeping Beauty system and minicircle vectors [51, 60]. Safety concerns with this technology include residual DNA plasmids and transposase (activity) in the final cell product and the potential risks of insertional mutagenesis and transposon remobilization. Release assays for this type of product would require an additional set of safety analyses and risk assessments to investigate the additional risk(s) posed to product recipients through use of this manufacture methodology. As an alternative to Transposon/Transposase technology, genome-editing tools such as transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR (Cas) tools allow specific modification of target genes, via disruption, correction, or replacement and have unlimited potential to improve CAR T-cell therapies [3]. In an attempt to minimize risks associated with insertional mutagenesis, targeting genes into genomic safe harbors is now possible [65].
Genome-editing tools have found favor in the development of third party or “universal” CAR T-cell therapeutics. TCRαβ/CD3 disruption has been demonstrated as a feasible approach to develop CAR T-cells products from mismatched donors, minimizing the risks of graft-versus-host disease [59, 65]. Several groups are combining TCRαβ/CD3 knockdown with additional strategies to prevent allogenic CAR T-cell rejection, which is another formidable challenge in the universal CAR T-cell space. Universal CAR therapies may overcome some of the limitations associated with autologous products such as poor-quality patient starting material, logistic and manufacturing challenges, disease progression prior to product availability, and batch-to-batch product variability. Alternative approaches using an endoplasmic reticulum retention signal to prevent CD3 surface expression have also been reported and these have the potential advantage of overcoming some of the limitations described above with genome-editing tools [68].
Genome-edited products require extensive characterization to demonstrate safety. Off-target effects are a major concern when using the CRISPR/Cas9 system, as these can lead to unintended mutagenesis and increase the risks of malignant cellular transformation [3, 12]. Although in silico methods are used to screen for potential off-target sites, they cannot precisely predict mutations that occur in vivo. EMA indicates that for genetically modified cells derived using genome-editing tools, in vitro assays for editing efficiency and off-target editing should be conducted [26]. However, development of sensitive and specific assays to detect off-target editing remains a challenge. Approaches like the T7 endonuclease 1 (T7E1) assay, deep sequencing and Chromatin Immunoprecipitation Sequencing (ChIP-seq) have been employed for detection of off-target editing. In silico prediction tools have also been developed [98], but many shortcomings are associated with these techniques. Indeed, off-target mutations with a frequency below 0.5% remain mostly undetected by current genome-wide analyses. Furthermore, targeting more than one gene for editing confers additional risk, as complex, multiplex gene editing can potentially lead to translocations induced by simultaneous double-stranded breaks at multiple loci. These have been reported to occur with a frequency as high as 7% in T-cells and have been detected by different methods, including cytogenetic analysis, qPCR, and droplet digital PCR [5, 66, 92]. Base-editing is a next-generation approach to CRISPR-Cas, which may overcome some of the risks described.
An additional risk posed by allogeneic CAR T-cells is the presence of alloreactive cells in the final product. This confers a risk of GvHD and should be addressed by efficient cell selection/depletion strategies, along with stringent purity criteria for product release. Alternative allogeneic cell sources such as NK cells or γδ T-cells might avoid the development of GvHD although challenges with rejection and persistence remain [68]. Characterization of these cell products and development of assays for identity, purity, potency, and so on will mirror, but will not be the same as those required for T-cell products.
In conclusion, as new developments increase the efficacy, applicability, and accessibility of CAR T-cell therapies, adoption of this technology for more widespread use in cancer therapy should become a reality. It is crucial for the field to develop a solid understanding of individual products and their biological activity so that critical quality attributes can be defined to ensure efficiency, consistency, and safety. In the coming years, as new data emerges from preclinical research and early clinical trials, researchers and regulatory agencies worldwide will face the challenge to keep pace with clinical development. There will be a need to generate new and harmonized guidelines to ensure patient safety and product quality to cover the diversity of emerging novel CAR T-cell therapies.
References
Ahmadbeigi N, Alatab S, Vasei M, Ranjbar A, Aghayan S, Khorsand A, Moradzadeh K, Darvishyan Z, Jamali M, Muhammadnejad S (2021) Characterization of a xenograft model for anti-CD19 CAR T cell studies. Clin Transl Oncol 23:2181. https://doi.org/10.1007/s12094-021-02626-5
Aktas E, Kucuksezer UC, Bilgic S, Erten G, Deniz G (2009) Relationship between CD107a expression and cytotoxic activity. Cell Immunol 254:149–154. https://doi.org/10.1016/j.cellimm.2008.08.007
Ashmore-Harris C, Fruhwirth GO (2020) The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin Transl Med 9:15. https://doi.org/10.1186/s40169-020-0268-z
Bordignon V, Cordiali-Fei P, Rinaldi M, Signori E, Cottarelli A, Zonfrillo M, Ensoli F, Rasi G, Fuggetta M (2012) Evaluation of antigen specific recognition and cell mediated cytotoxicity by a modified LysisPot assay in a rat colon carcinoma model. J Exp Clin Cancer Res 31:9. https://doi.org/10.1186/1756-9966-31-9
Bothmer A, Gareau KW, Abdulkerim HS, Buquicchio F, Cohen L, Viswanathan R, Zuris JA, Marco E, Fernandez CA, Myer VE, Cotta-Ramusino C (2020) Detection and modulation of DNA translocations during multi-gene genome editing in T cells. CRISPR J 3:177–187. https://doi.org/10.1089/crispr.2019.0074
Bowman N, Liu D, Paczkowski P, Chen J, Rossi J, Mackay S, Bot A, Zhou J (2020) Advanced cell mapping visualizations for single cell functional proteomics enabling patient stratification. Proteomics 20:1900270. https://doi.org/10.1002/pmic.201900270
Bravery CA, Carmen J, Fong T, Oprea W, Hoogendoorn KH, Woda J, Burger SR, Rowley JA, Bonyhadi ML, Van’t Hof W (2013) Potency assay development for cellular therapy products: an ISCT∗ review of the requirements and experiences in the industry. Cytotherapy 15:9–19.e9. https://doi.org/10.1016/j.jcyt.2012.10.008
Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, Novellis P, Veronesi G, Lugli E (2018) High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J Exp Med 215:2520–2535. https://doi.org/10.1084/jem.20180684
Brummelman J, Haftmann C, Núñez NG, Alvisi G, Mazza EMC, Becher B, Lugli E (2019) Development, application and computational analysis of high-dimensional fluorescent antibody panels for single-cell flow cytometry. Nat Protoc 14:1946–1969. https://doi.org/10.1038/s41596-019-0166-2
Brunner KT, Mauel J, Cerottini JC, Chapuis B (1968) Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 14:181–196
Cerignoli F, Abassi YA, Lamarche BJ, Guenther G, Santa Ana D, Guimet D, Zhang W, Zhang J, Xi B (2018) In vitro immunotherapy potency assays using real-time cell analysis. PLoS One 13:e0193498. https://doi.org/10.1371/journal.pone.0193498
Cheng Y, Tsai SQ (2018) Illuminating the genome-wide activity of genome editors for safe and effective therapeutics. Genome Biol 19:226. https://doi.org/10.1186/s13059-018-1610-2
Chmielewski M, Abken H (2020) TRUCKS, the fourth-generation CAR T cells: current developments and clinical translation. Adv Cell Gene Ther 3:e84. https://doi.org/10.1002/acg2.84
Clay TM, Hobeika AC, Mosca PJ, Lyerly HK, Morse MA (2001) Assays for monitoring cellular immune responses to active immunotherapy of cancer. Clin Cancer Res 75:1127–1135
Council of Europe (2018) European Pharmacopoeia
Dai X, Mei Y, Cai D, Han W (2019) Standardizing CAR-T therapy: getting it scaled up. Biotechnol Adv 37:239–245. https://doi.org/10.1016/j.biotechadv.2018.12.002
Darrah PA, Patel DT, De Luca PM, Lindsay RWB, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA (2007) Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med 13:843–850. https://doi.org/10.1038/nm1592
Dautzenberg IJC, Rabelink MJWE, Hoeben RC (2021) The stability of envelope-pseudotyped lentiviral vectors. Gene Ther 28:89–104. https://doi.org/10.1038/s41434-020-00193-y
de Charette M, Marabelle A, Houot R (2016) Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? Eur J Cancer 68:134–147. https://doi.org/10.1016/j.ejca.2016.09.010
de Wolf C, van de Bovenkamp M, Hoefnagel M (2018) Regulatory perspective on in vitro potency assays for human T cells used in anti-tumor immunotherapy. Cytotherapy 20:601–622. https://doi.org/10.1016/j.jcyt.2018.01.011
Decker T, Lohmann-Matthes ML (1988) A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods 115:61–69. https://doi.org/10.1016/0022-1759(88)90310-9
Desombere I, Meuleman P, Rigole H, Willems A, Irsch J, Leroux-Roels G (2004) The interferon gamma secretion assay: a reliable tool to study interferon gamma production at the single cell level. J Immunol Methods 286:167–185. https://doi.org/10.1016/j.jim.2004.01.001
Donnadieu E, Dupré L, Pinho LG, Cotta-de-Almeida V (2020) Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J Leukoc Biol 108:1067–1079. https://doi.org/10.1002/JLB.1MR0520-746R
EudraLex (2018) The rules governing medicinal products in the European Union. Volume 4 – Good manufacturing practice. European Comission. https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en
European Medicines Agency, Committee for Advanced Therapies (2016) Guideline on potency testing of cell based immunotherapy medicinal products for the treatment of cancer. EMA/CHMP/BWP/271475/2006 rev.1
European Medicines Agency, Committee for Advanced Therapies (2021) Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. EMA/CAT/GTWP/671639/2008 Rev 1
Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, Raddassi K, Devine L, Obermoser G, Pekalski ML, Pontikos N, Diaz A, Heck S, Villanova F, Terrazzini N, Kern F, Qian Y, Stanton R, Wang K, Brandes A, Ramey J, Aghaeepour N, Mosmann T, Scheuermann RH, Reed E, Palucka K, Pascual V, Blomberg BB, Nestle F, Nussenblatt RB, Brinkman RR, Gottardo R, Maecker H, McCoy JP (2016) Standardizing flow cytometry immunophenotyping analysis from the human immunophenotyping consortium. Sci Rep 6:20686. https://doi.org/10.1038/srep20686
Food and Drug Administration (2008) Guidance for FDA reviewers and sponsors: content and review of chemistry, manufacturing, and control (CMC) information for human somatic cell therapy investigational new drug applications (INDs). FDA-2008-D-0206. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/content-and-review-chemistry-manufacturing-and-control-cmc-information-human-somatic-celltherapy
Food and Drug Administration (2011) Guidance for industry: potency tests for cellular and gene therapy products. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. https://www.fda.gov/regulatory-information/search-fda-guidancedocuments/potency-tests-cellular-and-gene-therapy-products
Food and Drug Administration (2017) BLA 125646, tisagenlecleucel, Novartis Pharmaceuticals Corporation. U.S. Department of Health and Human Services, Food and Drug Administration, US Oncologic Drugs Advisory Committee Meeting. https://www.fda.gov/media/106081/download
Fousek K, Watanabe J, Joseph SK, George A, An X, Byrd TT, Morris JS, Luong A, Martínez-Paniagua MA, Sanber K, Navai SA, Gad AZ, Salsman VS, Mathew PR, Kim HN, Wagner DL, Brunetti L, Jang A, Baker ML, Varadarajan N, Hegde M, Kim Y-M, Heisterkamp N, Abdel-Azim H, Ahmed N (2021) CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia 35:75–89. https://doi.org/10.1038/s41375-020-0792-2
Fraietta JA, Lacey SF, Wilcox NS, Bedoya F, Chen F, Orlando E, Brogdon JL, Hwang W-T, Frey N, Young RM, Pequignot E, Ambrose DE, Levine BL, Bitter H, Porter DL, Xu J, June CH, Melenhorst JJ (2016) Biomarkers of response to anti-CD19 chimeric antigen receptor (CAR) T-cell therapy in patients with chronic lymphocytic leukemia. Blood 128:57–57. https://doi.org/10.1182/blood.V128.22.57.57
Friedl J, Stift A, Paolini P, Roth E, Steger GG, Mader R, Jakesz R, Gnant MFX (2000) Tumor antigen pulsed dendritic cells enhance the cytolytic activity of tumor infiltrating lymphocytes in human hepatocellular cancer. Cancer Biother Radiopharm 15:477–486. https://doi.org/10.1089/cbr.2000.15.477
Gebo JET, Lau AF (2020) Sterility testing for cellular therapies: what is the role of the clinical microbiology laboratory? J Clin Microbiol 58:e01492-19. https://doi.org/10.1128/JCM.01492-19
Glamann J, Hansen AJ (2006) Dynamic detection of natural killer cell-mediated cytotoxicity and cell adhesion by electrical impedance measurements. Assay Drug Dev Technol 4:555–563. https://doi.org/10.1089/adt.2006.4.555
Graham C, Jozwik A, Pepper A, Benjamin R (2018) Allogeneic CAR-T cells: more than ease of access? Cell 7:155. https://doi.org/10.3390/cells7100155
Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, Trebska-McGowan K, Wunderlich JR, Yang JC, Rosenberg SA (2016) Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 22:433–438. https://doi.org/10.1038/nm.4051
Hermans IF, Silk JD, Yang J, Palmowski MJ, Gileadi U, McCarthy C, Salio M, Ronchese F, Cerundolo V (2004) The VITAL assay: a versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J Immunol Methods 285:25–40. https://doi.org/10.1016/j.jim.2003.10.017
Höppner M, Luhm J, Schlenke P, Koritke P, Frohn C (2002) A flow-cytometry based cytotoxicity assay using stained effector cells in combination with native target cells. J Immunol Methods 267:157–163. https://doi.org/10.1016/S0022-1759(02)00167-9
Hu Y, Huang J (2020) The chimeric antigen receptor detection toolkit. Front Immunol 11:1770. https://doi.org/10.3389/fimmu.2020.01770
Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, Gao L, Wen Q, Zhong JF, Zhang C, Zhang X (2020) Recent advances in CAR-T cell engineering. J Hematol Oncol 13:86. https://doi.org/10.1186/s13045-020-00910-5
Jahnmatz P, Bengtsson T, Zuber B, Färnert A, Ahlborg N (2016) An antigen-specific, four-color, B-cell FluoroSpot assay utilizing tagged antigens for detection. J Immunol Methods 433:23–30. https://doi.org/10.1016/j.jim.2016.02.020
Janetzki S, Price L, Schroeder H, Britten CM, Welters MJP, Hoos A (2015) Guidelines for the automated evaluation of Elispot assays. Nat Protoc 10:1098–1115. https://doi.org/10.1038/nprot.2015.068
Karimi MA, Lee E, Bachmann MH, Salicioni AM, Behrens EM, Kambayashi T, Baldwin CL (2014) Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS One 9:e89357. https://doi.org/10.1371/journal.pone.0089357
Kiesgen S, Chicaybam L, Chintala NK, Adusumilli PS (2018) Chimeric antigen receptor (CAR) T-cell therapy for thoracic malignancies. J Thorac Oncol 13:16–26. https://doi.org/10.1016/j.jtho.2017.10.001
Kiesgen S, Messinger JC, Chintala NK, Tano Z, Adusumilli PS (2021) Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat Protoc 16:1331–1342. https://doi.org/10.1038/s41596-020-00467-0
Kite Pharma Incorporated (2017) Summary basis for regulatory action for BLA 125643 (YESCARTA™). U.S. Food and Drug Administration, Review Committee. https://www.fda.gov/media/108788/download
Lewinsohn DA, Lewinsohn DM, Scriba TJ (2017) Polyfunctional CD4+ T cells as targets for tuberculosis vaccination. Front Immunol 8:1262. https://doi.org/10.3389/fimmu.2017.01262
Lichtenfels R, Biddison WE, Schulz H, Vogt AB, Martin R (1994) CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T lymphocyte activity. J Immunol Methods 172:227–239. https://doi.org/10.1016/0022-1759(94)90110-4
Liu L, Chahroudi A, Silvestri G, Wernett ME, Kaiser WJ, Safrit JT, Komoriya A, Altman JD, Packard BZ, Feinberg MB (2002) Visualization and quantification of T cell-mediated cytotoxicity using cell-permeable fluorogenic caspase substrates. Nat Med 8:185–189. https://doi.org/10.1038/nm0202-185
Magnani CF, Tettamanti S, Alberti G, Pisani I, Biondi A, Serafini M, Gaipa G (2020) Transposon-based CAR T cells in acute leukemias: where are we going? Cell 9:1337. https://doi.org/10.3390/cells9061337
Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, Myklebust JH, Kadapakkam M, Weber EW, Tousley AM, Richards RM, Heitzeneder S, Nguyen SM, Wiebking V, Theruvath J, Lynn RC, Xu P, Dunn AR, Vale RD, Mackall CL (2020) Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov 10:702–723. https://doi.org/10.1158/2159-8290.CD-19-0945
Marks P (2019) The FDA’s regulatory framework for chimeric antigen receptor-T cell therapies. Clin Transl Sci 12:428–430. https://doi.org/10.1111/cts.12666
Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, Shomali N, Chartrand MS, Pathak Y, Jarahian M, Izadi S, Hassanzadeh A, Shirafkan N, Tahmasebi S, Khiavi FM (2021) CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther 12:81. https://doi.org/10.1186/s13287-020-02128-1
Martinez EM, Klebanoff SD, Secrest S, Romain G, Haile ST, Emtage PCR, Gilbert AE (2018) High-throughput flow cytometric method for the simultaneous measurement of CAR-T cell characterization and cytotoxicity against solid tumor cell lines. SLAS Discov Adv Sci Drug Discov 23:603–612. https://doi.org/10.1177/2472555218768745
McLellan AD, Ali Hosseini Rad SM (2019) Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol 97:664–674. https://doi.org/10.1111/imcb.12254
Milone MC, O’Doherty U (2018) Clinical use of lentiviral vectors. Leukemia 32:1529–1541. https://doi.org/10.1038/s41375-018-0106-0
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH (2009) Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17:1453–1464. https://doi.org/10.1038/mt.2009.83
Mitra A, Mishra L, Li S (2013) Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol 31:347–354. https://doi.org/10.1016/j.tibtech.2013.03.006
Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, Ivics Z, Hudecek M (2017) Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 31:186–194. https://doi.org/10.1038/leu.2016.180
Morello A, Sadelain M, Adusumilli PS (2016) Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 6:133–146. https://doi.org/10.1158/2159-8290.CD-15-0583
Mullard A (2021) FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov 20:166. https://doi.org/10.1038/d41573-021-00031-9
On behalf of the EuroFlow Consortium (EU-FP6, LSHB-CT-2006-018708), Kalina T, Flores-Montero J, van der Velden VHJ, Martin-Ayuso M, Böttcher S, Ritgen M, Almeida J, Lhermitte L, Asnafi V, Mendonça A, de Tute R, Cullen M, Sedek L, Vidriales MB, Pérez JJ, te Marvelde JG, Mejstrikova E, Hrusak O, Szczepański T, van Dongen JJM, Orfao A (2012) EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 26:1986–2010. https://doi.org/10.1038/leu.2012.122
Packard BZ, Komoriya A (2008) Intracellular protease activation in apoptosis and cell-mediated cytotoxicity characterized by cell-permeable fluorogenic protease substrates. Cell Res 18:238–247. https://doi.org/10.1038/cr.2008.17
Papapetrou EP, Schambach A (2016) Gene insertion into genomic safe harbors for human gene therapy. Mol Ther 24:678–684. https://doi.org/10.1038/mt.2016.38
Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, Ghorashian S, Pinner D, Ahsan G, Gilmour K, Lucchini G, Inglott S, Mifsud W, Chiesa R, Peggs KS, Chan L, Farzaneh F, Thrasher AJ, Vora A, Pule M, Veys P (2017) Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med 9:eaaj2013. https://doi.org/10.1126/scitranslmed.aaj2013
Quintarelli C, Locatelli F, Caruana I, De Angelis B (2016) Overcoming challenges in CAR T-cell product CGMP release. Mol Ther 24:845–846. https://doi.org/10.1038/mt.2016.72
Rafiq S, Hackett CS, Brentjens RJ (2020) Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 17:147–167. https://doi.org/10.1038/s41571-019-0297-y
Richter JR (2021) Multiple myeloma: “if you don’t stop to look around once in a while… you could miss it”. Cancer J 27:183–184. https://doi.org/10.1097/PPO.0000000000000527
Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Rosenberg SA (2004) Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol 173:7125–7130. https://doi.org/10.4049/jimmunol.173.12.7125
Roddie C, O’Reilly M, Dias Alves Pinto J, Vispute K, Lowdell M (2019) Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy 21:327–340. https://doi.org/10.1016/j.jcyt.2018.11.009
Rossi J, Paczkowski P, Shen Y-W, Morse K, Flynn B, Kaiser A, Ng C, Gallatin K, Cain T, Fan R, Mackay S, Heath JR, Rosenberg SA, Kochenderfer JN, Zhou J, Bot A (2018) Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132:804–814. https://doi.org/10.1182/blood-2018-01-828343
Santeramo I, Bagnati M, Harvey EJ, Hassan E, Surmacz-Cordle B, Marshall D, Di Cerbo V (2020) Vector copy distribution at a single-cell level enhances analytical characterization of gene-modified cell therapies. Mol Ther Methods Clin Dev 17:944–956. https://doi.org/10.1016/j.omtm.2020.04.016
Schaft N (2020) The landscape of CAR-T cell clinical trials against solid tumors—a comprehensive overview. Cancers 12:2567. https://doi.org/10.3390/cancers12092567
Schneider D, Xiong Y, Wu D, Nӧlle V, Schmitz S, Haso W, Kaiser A, Dropulic B, Orentas RJ (2017) A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer 5:42. https://doi.org/10.1186/s40425-017-0246-1
Shafer-Weaver K, Sayers T, Strobl S, Derby E, Ulderich T, Baseler M, Malyguine A (2003) The Granzyme B ELISPOT assay: an alternative to the 51Cr-release assay for monitoring cell-mediated cytotoxicity. J Transl Med 1:14
Sheehy ME, McDermott AB, Furlan SN, Klenerman P, Nixon DF (2001) A novel technique for the fluorometric assessment of T lymphocyte antigen specific lysis. J Immunol Methods 249:99–110. https://doi.org/10.1016/S0022-1759(00)00329-X
Streeck H, Frahm N, Walker BD (2009) The role of IFN-γ Elispot assay in HIV vaccine research. Nat Protoc 4:461–469. https://doi.org/10.1038/nprot.2009.7
Stroncek DF, Jin P, Wang E, Jett B (2007) Potency analysis of cellular therapies: the emerging role of molecular assays. J Transl Med 5:24. https://doi.org/10.1186/1479-5876-5-24
Tano Z, Kiesgen S, Chintala N, Dozier J, Messinger J, Tan KS, Adusumilli P (2018) MA06.06 An ex-vivo patient-derived, immunocompetent (PDI) culture system to evaluate immunotherapeutic agents’ anti-tumor efficacy. J Thorac Oncol 13:S376. https://doi.org/10.1016/j.jtho.2018.08.362
Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S (2019) Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer 120:26–37. https://doi.org/10.1038/s41416-018-0325-1
Tokarew N, Ogonek J, Endres S et al (2019) Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer 120:26–37. https://doi.org/10.1038/s41416-018-0325-1
Tokuno O, Hayakawa A, Yanai T, Mori T, Ohnuma K, Tani A, Minami H, Sugimoto T (2015) Sterility testing of stem cell products by broad-range bacterial 16S ribosomal DNA polymerase chain reaction. Lab Med 46:34–41. https://doi.org/10.1309/LMKT4P9FFI2BBSIU
Tyagarajan S, Spencer T, Smith J (2020) Optimizing CAR-T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev 16:136–144. https://doi.org/10.1016/j.omtm.2019.11.018
U.S. Department of Health and Human Services, Food and Drug Administration (2021) Code of Federal Regulations Title 21. Part 610 – General biological products standards. Rev. 2
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research (2020) Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drug applications (INDs)
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research (2020) Testing of retroviral vector-based human gene therapy products for replication competent retrovirus during product manufacture and patient follow-up; Guidance for Industry
United States Pharmacopeia and National Formulary (2018) USP 41-NF 36 U.S
von Zons P, Crowley-Nowick P, Friberg D, Bell M, Koldovsky U, Whiteside TL (1997) Comparison of europium and chromium release assays: cytotoxicity in healthy individuals and patients with cervical carcinoma. Clin Diagn Lab Immunol 4:202–207. https://doi.org/10.1128/CDLI.4.2.202-207.1997
Wang X, Rivière I (2016) Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics 3:16015. https://doi.org/10.1038/mto.2016.15
Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, Lv D, Starr R, Brito A, Wu Q, Kim LJY, Qiu Z, Lin P, Lorenzini MH, Badie B, Forman SJ, Xie Q, Brown CE, Rich JN (2021) CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov 11:1192–1211. https://doi.org/10.1158/2159-8290.CD-20-1243
Webber BR, Lonetree C, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, Lahr WS, Draper GM, Slipek NJ, Smeester BA, Lovendahl KN, McElroy AN, Gordon WR, Osborn MJ, Moriarity BS (2019) Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun 10:5222. https://doi.org/10.1038/s41467-019-13007-6
Wei J, Han X, Bo J, Han W (2019) Target selection for CAR-T therapy. J Hematol Oncol 12:62. https://doi.org/10.1186/s13045-019-0758-x
Wei J, Guo Y, Wang Y, Wu Z, Bo J, Zhang B, Zhu J, Han W (2020) Clinical development of CAR T cell therapy in China: 2020 update. Cell Mol Immunol 18:792. https://doi.org/10.1038/s41423-020-00555-x
Xi B, Berahovich R, Zhou H, Xu S, Wei Y, Guan J, Harto H, Guan J, Wu L, Santa Ana D, Cerignoil F, Lamarche B, Abassi YA, Golubovskaya V (2019) A real-time potency assay for chimeric antigen receptor T cells targeting solid and hematological cancer cells. J Vis Exp (153):59033. https://doi.org/10.3791/59033
Xue Q, Bettini E, Paczkowski P, Ng C, Kaiser A, McConnell T, Kodrasi O, Quigley MF, Heath J, Fan R, Mackay S, Dudley ME, Kassim SH, Zhou J (2017) Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J Immunother Cancer 5:85. https://doi.org/10.1186/s40425-017-0293-7
Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM (2010) New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev Vaccines 9:601–616. https://doi.org/10.1586/erv.10.49
Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H (2015) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4:e264. https://doi.org/10.1038/mtna.2015.37
Zhao Y, Stepto H, Schneider CK (2017) Development of the first World Health Organization lentiviral vector standard: toward the production control and standardization of lentivirus-based gene therapy products. Hum Gene Ther Methods 28:205–214. https://doi.org/10.1089/hgtb.2017.078
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Dias, J., Cadiñanos-Garai, A., Roddie, C. (2023). Release Assays and Potency Assays for CAR T-Cell Interventions. In: Burns, J.S. (eds) Potency Assays for Advanced Stem Cell Therapy Medicinal Products. Advances in Experimental Medicine and Biology, vol 1420. Springer, Cham. https://doi.org/10.1007/978-3-031-30040-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-031-30040-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-30039-4
Online ISBN: 978-3-031-30040-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)