
Abstract
Performance enhancement of the immune system can now be generated through ex vivo gene modification of T cells in order to redirect native specificity to target tumor antigens. This approach combines the specificity of antibody therapy, the expanded response of cellular therapy and the memory activity of vaccine therapy. Recent clinical trials of chimeric antigen receptor (CAR) T cells directed toward CD19 as a stand-alone therapy have shown sustained complete responses in patients with acute lymphoblastic leukemia and chronic lymphocytic leukemia. As these drug products are individually derived from a patient’s own cells, a different manufacturing approach is required for this kind of personalized therapy compared with conventional drugs. Key steps in the CAR T-cell manufacturing process include the selection and activation of isolated T cells, transduction of T cells to express CARs, ex vivo expansion of modified T cells and cryopreservation in infusible media. In this review, the steps involved in isolating, genetically modifying and scaling-out the CAR T cells for use in a clinical setting are described in the context of in-process and release testing and regulatory standards.
Similar content being viewed by others

Introduction
Cell-based therapies have been under investigation for a number of years in a variety of clinical applications, including tissue regeneration, immune reconstitution, vaccination, and generation of antigen-specific cells. More recently, a great deal of research has investigated the therapeutic potential of enhancing the immune response in cancer. A variety of therapeutic approaches, including adoptive T-cell therapy, have attempted to harness the cytotoxic potential of immune cells in targeting cancers. Following collection from the patient or a matched donor, T cells may be enriched and expanded ex vivo.1 Early studies attempted to source tumor-specific T cells from surgically excised tumor tissue. T cells could be expanded and tested for tumor specificity and significant clinical responses were observed in a subset of patients.1 In another early strategy, repeated ex vivo stimulation of peripheral blood mononuclear cells with antigen-presenting cells (APCs) expressing viral or tumor antigens was shown to generate a large number of antigen-specific cytotoxic T cells.2 The challenge in these early approaches was not only in their labor-intensive requirements and several weeks of culture, but in defining a consistent and potent T-cell product.
A more efficient path to augment the immune response to cancer can be found through gene transfer. T-cell receptors with high avidity to tumor antigens can be identified, cloned and transduced into patients’ T cells for subsequent reinfusion, thereby redirecting cytotoxic T-cell activity against select tumor antigens.3 Recent advances in gene transfer technologies, particularly with retroviral and lentiviral vectors, have ushered in an era of combining cell-based therapies with gene-based therapies, in which genetically engineered chimeric antigen receptors (CARs) or tumor-specific T-cell receptor genes are expressed in immune effector cells.4 CARs combine intracellular signaling domains with a single-chain variable fragment of an antibody (Ab) into a single chimeric protein.
The first CARs consisted of chimeric T-cell receptor with the antigen-binding site of an Ab.5 The use of ‘first-generation’ CARs consisted of Ab-binding domains and the CD3ζ signaling domain. In clinical trials in patients with various cancers, including lymphoma and ovarian cancer, these showed only modest efficacy, primarily owing to insufficient activation, expansion and persistence of the modified T cells.6, 7, 8, 9 Physiologically, T cells are activated via two signals: (1) engagement of the T-cell receptor with the presented antigen, and (2) engagement of costimulatory molecules such as CD28, 4-1BB, OX40 or CD40L.10 However, tumors do not generally express the ligands to activate the costimulatory molecules and the absence of costimulation leads to anergy and insufficient in vivo expansion. In fact, many tumors express ligands that engage negative costimulatory receptors. Addressing this limitation ‘second-generation’ CARs were developed that incorporated costimulatory domains, such as CD28 or 4-1BB (CD137), to enhance survival and increase the proliferation of the genetically engineered T cells, leading to increased antitumor activity.11, 12, 13, 14 In 2011, a team at the University of Pennsylvania used modified T cells employing a lentiviral vector to express a second-generation CAR; this CAR retargets genetically engineered T cells to CD19 and stimulates T-cell activation and proliferation. CD19 is present on B-cell leukemias and lymphomas as well as on healthy B cells, but not on hematopoietic stem cells or other tissues. An efficient method of activating and expanding T cells for this trial and subsequent trials is described in this article. The redirected T cells have produced lasting complete responses in clinical trials in most pediatric and adult patients with relapsed/refractory acute lymphoblastic leukemia and some adult patients with relapsed/refractory chronic lymphocytic leukemia.15, 16, 17
Considerations of CAR components
CARs are chimeric constructs composed of several domains derived from different proteins, namely: (1) an antigen recognition domain that is usually taken from an Ab, (2) a CD3ζ T-cell co-receptor signaling domain, and (3) a costimulatory domain required for T-cell activation during antigen presentation.11, 12, 13, 14 CD19 is currently the most actively investigated target in CAR T-cell therapy and has spurred the development of a number of CD19 CARs. CD19 CARs vary by type of costimulatory domain- none, CD28 or 4-1BB (CD137), source of anti-CD19 Ab, and gene delivery method (Table 1).9, 16, 18, 19, 20, 21, 22, 23 Although a number of CARs have proceeded to clinical trials, there are CAR T-cell properties, ranging from vector design to the manufacturing process, that may be further improved and require additional investigation.
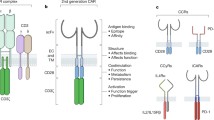
In general, the process of CAR T-cell manufacturing and delivery involves the following major steps (Figure 1): (1) leukapheresis: apheresis in which a patient’s T cells are harvested from peripheral blood; (2) T-cell activation: T cells are activated using Ab-coated beads that serve as artificial dendritic cells (DCs); (3) transduction or transfection: T cells are genetically transduced or transfected ex vivo with a construct encoding the anti-gene target chimeric antigen receptor; (4) expansion: gene-modified cells undergo further ex vivo expansion; (5) chemotherapy: the patient receives a preparative lymphodepleting regimen before T-cell infusion; (6) infusion: genetically engineered T cells are infused into the patient. In this review, we describe the procedure of isolating and manufacturing genetically engineered T cells for use in a clinical setting, taking into account reagents, in-process testing and regulatory considerations.

Overview of CAR T therapy in the clinic.Figure reprinted from Novartis. Copyright 2014 Novartis. Used with permission.
Isolating and manufacturing T cells for CAR T-cell therapy
Harvesting T cells—leukapheresis and T-cell selection
Apheresis (Greek for ‘to take away’) is the process in which whole blood is removed from an individual’s body and separated into components, one or more components are removed, and the remainder is returned to circulation.24 Overall, apheresis is a largely safe procedure for both healthy donors and patients. A recent retrospective analysis of 15 763 procedures found only 59 (0.37%) moderate to severe adverse events including dizziness or fainting episodes (0.12%), citrate toxicity (0.02%), a combination of dizziness/fainting and citrate toxicity (0.11%), vascular injuries (0.07%) and miscellaneous events (0.04%).25 Leukapheresis should take place before administering lymphodepleting chemotherapy as an absolute T-cell count below 200–300 will likely result in a poor T-cell collection.
Collection by leukapheresis will normally take place in the absence of hematopoietic stem cell mobilizing agents (that is, steady state). In settings where CAR T-cell therapy may take place in the setting of stem cell transplantation, or when a mobilized leukapheresis product has been previously collected and the patient is profoundly lymphopenic, the use of a mobilized product may be desired or necessary. The literature shows that there is conflicting evidence as to the effect of mobilizing agents on T-cell function. Reports show that granulocyte colony-stimulating factor (G-CSF) mobilization skews T cells toward a Th2 phenotype and impairs signaling through CD28.26, 27 Depletion of monocytes from the collected product may reduce the effect, but one study showed that there appeared to be a direct effect upon T cells.27 However, a recent study showed that functional virus-specific T cells could be isolated from G-CSF mobilized units.28 Interestingly, when T-cell function in plerixafor mobilized unit was compared with the T-cell function in G-CSF mobilized units, decreased CD62L expression and altered cytokine-associated gene expression were noted in G-CSF, but not plerixafor mobilized units.29 The potency of T cells isolated from mobilized apheresis units may therefore vary depending on the mobilization regimen, patient treatment history, method of T-cell isolation and stimulation.
The duration of the apheresis procedure depends on multiple factors, including the efficiency of apheresis machines, the types of target cells and their number in the blood, patient weight and number of cells desired.30 In order to stop the blood from clotting in the apheresis device, anti-coagulants such as citrates are mixed with blood as it is being pumped through the machine.31 An automated cell-washing device, such as the Haemonetics (Braintree, MA, USA) CellSaver, can be used to wash the apheresis product out of the collection buffer. Many research and stem cell laboratories still use the COBE 2991 Cell Processor (Terumo BCT, Lakewood, CO, USA), which is a batch-fed closed-system centrifuge that has been used in cell processing applications for >30 years. A number of laboratory devices may be used to separate or enrich the T-cell or lymphocyte fraction from the leukapheresis product. Counterflow centrifugal elutriation is used to separate cells according to size and density based on the cell’s sedimentation velocity, resulting in high purity, good recovery and excellent cell viability.32 Depletion of monocytes and isolation of the lymphocytes may be performed by directly loading the leukapheresis product into a Terumo Elutra Cell Separation System.33 Further separation of cell subsets within the lymphocyte fractions from an elutriated product can be performed via magnetic separation of cells labeled with nanoparticle-conjugated antibodies coated on cells. The Miltenyi CliniMACS Instrument (Bergisch Gladbach, Germany) is an electromechanical device that accepts a closed and sterile single-use tubing and column set, allowing CD4+, CD8+ or CD25+ T cells to be isolated or depleted from elutriated lymphocyte fractions or directly from the apheresis product. The CliniMACS and the cell-preparation bag are connected by a single-use tubing set and a separation column that contains the cell sample. Following a series of washing steps, the purified target cells are eluted and the enriched T cells are cultured (as generally described below). Importantly, the ClinicMACS has a Device Master File with the Food and Drug Administration. However, this system may result in longer processing time, depending on the frequency of the population and is associated with higher costs, particularly if sequential negative and positive selection is required, because every Ab-bead conjugate used for separation may cost several thousand dollars.
T-cell activation and transduction
Once T cells have been collected by apheresis and separated or enriched from the remainder of the cell population, T cells are activated, transduced and expanded before being reinfused into the patient. In vivo, DCs or B cells may present a variety of antigens and costimulatory molecules to activate T cells. DCs have been used in a variety of cell culture settings. However, autologous DCs or B cells present a number of laboratory and particularly clinical application difficulties, including patient inherent variability and logistics of maintaining separate culture systems. Therefore, DCs are not practical in a clinical setting, particularly with respect to CAR T therapy.34 An early method for the expansion of T cells was to co-culture them with irradiated allogeneic peripheral blood mononuclear cells as APCs, also known as feeder cells.35 To facilitate expansion to specific antigens, peptides are exogenously loaded onto the APCs. A method referred to as the ‘Rapid Expansion Protocol,’ uses OKT3, IL-2 and irradiated peripheral blood mononuclear cell feeders to generate a 500- to 2000-fold expansion of tumor-infiltrating lymphocytes within 14 days.36 However, there is difficulty scaling up with this technique, which can be cost prohibitive and time consuming owing to the Food and Drug Administration requirements for the validation and qualification of the allogeneic feeder cells.37
A more efficient and potent method of T-cell activation—and the one used in the CAR T therapy that is the furthest along in clinical development, CTL019—is via stimulation with anti-CD3/anti-CD28 monoclonal Ab (mAb) coated paramagnetic 4.5-μm diameter beads (Life Technologies, Carlsbad, CA, USA), which can also enable simultaneous positive selection and activation of T cells.38, 39 Both the antibodies are covalently linked to the same surface. The anti-CD3 mAb provides a strong proliferative signal through the T-Cell receptor complex and the anti-CD28 mAb provides a potent costimulatory signal. In this method, CD4+ T cells have shown strong evidence of activation that produces cytokines at levels 1 to 2 logs higher than anti-CD3 mAb plus IL-2, and high levels of proliferation38, 40Nanomatrix beads coated with anti-CD3 and anti-CD28 antibodies are also commercially available, though their use in clinical trials has not yet been reported.40 Concurrently with activation with 4.5-μm diameter beads, T cells are transduced with a viral vector containing the CAR transgene. A packaging line is used to generate the viral vector, that is able to transduce target cells and contains the transgene of interest.41, 42 The vector is incubated with the cells for several days and then washed out.
Considerations of CAR delivery systems
Different vectors have been used including lentiviral and retroviral vectors, as well as transfection of the Sleeping Beauty transposon system.22, 23, 43 Currently, viral transduction with either murine retroviral or lentiviral vectors is the most frequently used method of gene transfer for mammalian gene therapy because of the high efficiency of gene delivery.44 After transduction, the retrovirus (RNA is reverse transcribed) to produce viral DNA, which can then be integrated into the host DNA, allowing for stable long-term gene expression. However, retroviruses only efficiently transduce dividing cells. Furthermore, the site of integration for retroviruses appears to be nonrandom and there are data suggesting a preference for insertion into promoters of host genes, leading to aberrant gene expression and oncogenicity—though this was more readily observed in stem cells rather than T cells. Finally, gammaretroviral vectors are more susceptible to transgene silencing by host restriction factors.45
Lentiviruses require the presence of several regulatory genes for neutralizing host cell defenses, mitigating immune responses and regulating viral replication.46 As lentiviruses integrate into the host genome, there is the likely benefit of long-term, stable expression. Furthermore, lentiviruses theoretically have a lower risk of insertional mutagenesis than retroviral vectors because they usually integrate away from host promoters.46, 47 In a Phase 1/2 study evaluating the safety of lentivirus-modified CD4+ T cells infusion in early-stage human immunodeficiency virus-infected subjects, it was shown that there was no preferential survival or expansion of cells with integration sites near proto-oncogenes or tumor suppressor genes.48 The data to date suggest that it is extremely unlikely that the observed persistence of T cells genetically modified by lentiviruses is due to the dysregulation of the proliferative and survival pathways. Multiple generations of split-component lentiviral constructs have been developed to improve safety and increase transgene expression. In the case of CTL019, this generation of lentiviral construct separates viral genes onto three different plasmids: two packaging plasmids and an envelope plasmid; the fourth plasmid carries the transgene, namely the CAR.49 When ex vivo transduced cells are in culture for a period of time⩾4 days from the start of transduction retrovirally transduced or lentivirally transduced cells must be tested for replication competent virus as per the Food and Drug Administration guidance.50 To improve the efficacy of transgene integration into the modified T cell, the Sleeping Beauty transposon and transposase have also been investigated.22, 43 In this process, two DNA plasmids, one containing the transgene of interest and the other containing the transposase, are transfected using electroporation. Stably transfected cells can then be stimulated by irradiated antigen-presenting cell (aAPC).
Ex vivo culture systems
Various cell culture systems may be used for this process of activation, transduction and expansion. Traditional static cell culture systems are expensive, labor intensive and do not scale easily. Furthermore, and more importantly, open cell culture systems are not compatible with the standards for large scale clinical and commercial production. Therefore, bioreactor culture systems provide substantial advantages over traditional static cell culture.51 Currently, two systems, namely the WAVE Bioreactor (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) and the G-Rex Bioreactor (Wilson Wolf, New Brighton, MN, USA), are under investigation for their ability to scale out and commercialize cell therapies. The WAVE Bioreactor provides a sealed system, is scalable and disposable, and permits oxygen transfer and mixing via wave-induced agitation. This system has shown a preference for CD4+ T-cell growth; CD8+ T cells express a slightly less activated and differentiated phenotype.51 In the G-Rex Bioreactor system, cells grow on a gas-permeable membrane, providing a highly oxygenated environment. Although the static G-Rex Bioreactor flasks are compatible with standard tissue culture incubators and have shown comparable or even improved expansions compared with the WAVE Bioreactor system, producing large cell numbers currently requires many flasks.51 In our experience in CTL019 manufacturing, transduced T cells are expanded in WAVE Bioreactor bags.
After expansion in the bioreactor for a period of 9–11 days, the magnetic beads are removed and the cells are harvested, washed and cryopreserved in an infusible medium. At the time of harvest following ex vivo expansion, the volume of the cell culture may be as much as 5–10 liters and, therefore, several rounds of washing and concentrating the product are required. A cell processor, such as a Haemonetics CellSaver, which provides a closed and sterile system, can be used for the washing and concentration steps before formulation and cryopreservation. Recently, a closed-system formulation kit (CryoDoc system) has been developed and consists of closed-system tubing and manifolds without the need for a good manufacturing practices-grade clean room.52 After release testing for specified criteria, the cryopreserved, genetically modified T cells are shipped back to the treatment center where the cells are thawed and infused into the patient.
In-process and release testing considerations
Gene-modified cellular therapy products are regulated by the Food and Drug Administration Center for Biologics Evaluation and Research in the United States, or by parallel agencies in other regions, such as the European Medicines Agency in the European Union. Furthermore, cell-therapy products that are intended for either investigation or licensed therapeutic use in humans must be manufactured using current Good Manufacturing Practices (cGMP). Requirements for in-process and release testing for CAR T therapy contrast with those for most other cellular therapy products because additional testing related to the vector transduction and production must be performed. Owing to the time required to perform the testing that includes transduction efficiency, measuring residuals from vector production and testing for the absence of replicate competent virus, gene therapy products are cryopreserved. Testing can therefore be performed and repeated as necessary, which also can be beneficial. In-process testing may include the mycoplasma assay on preharvest cells, cell phenotype, and assays analyzing the decline or absence of contaminating tumor cells.
Testing is intended to certify (1) safety: using preclinical experiments and in-process or final product testing to ensure the removal of reagents that were used in the manufacturing process, (2) sterility: to ensure that the final product is free from contaminating microorganisms, (3) purity: to ensure the removal of any extraneous matter, (4) potency: to examine whether the therapeutic capability of the cell product will be as it was intended (this may not be confirmed until later phase 2 or phase 3 clinical trials), (5) identity: to establish and certify the product characteristics via the use of macroscopic or microscopic methods, specific cultural tests, or in vitro or in vivo immunological tests.
Laboratory techniques in development to increase manufacturing efficiency
Artificial APCs
Artificial aAPCs have been developed from K562 cells, a chronic myelogenous leukemia cell line that does not express the major histocompatibility complex or T-cell-related costimulatory ligands.53, 54, 55 These cells have been transduced with lentiviral vectors, resulting in the specific expression of stimulatory and costimulatory molecules for the activation and expansion of different subsets of T cells. In addition to expressing CD32 or CD64, the high-affinity Fc receptor that can bind anti-CD3 and anti-CD28 mAbs, K562 cells can be modified to express other molecules on their surface, such as 4-1BB or a wide variety of other costimulatory receptors. These aAPCs have have been shown to result in increased activation and expansion of T cells compared with the magnetic bead–based aAPC.55 K562 cells may also be engineered to express cytokines and have a history in clinical trials as tumor antigen vaccines.56, 57 Therefore, K562 cells may be an ideal cell scaffold on which the desired major histocompatibility molecules and costimulatory ligands can be expressed for the use of T-cell activation and expansion.
Antigen-independent aAPCs have been developed by genetically modifying cells to express receptors specific to a domain present on CARs.58, 59 These genetically modified cells are then used in co-culture as aAPCs to stimulate the specific expansion of CAR T cells, which may be of varied specificities as long as the conserved IgG4 extracellular domain is expressed. Antigen-dependent aAPCs are also under investigation.60 In this technique, peripheral blood mononuclear cells are genetically modified to express both a target tumor-associated antigen and a costimulatory molecule, most notably PSCA antigen, CD80 and 4-1BBL. Stimulation is achieved via co-culture after 10 days, and in one study, this technique was shown to enrich the frequency of CAR T cells from <40% to nearly 90%.60 When combined with the G-Rex Bioreactor culturing system, this antigen-dependent aAPC approach resulted in nearly >90-fold increase in T cells with only 1 liter of culture medium.60
Temporary CAR expression
The temporary expression of CARs in T cells has been shown in preclinical models via RNA electroporation.61, 62, 63 One of the benefits of the temporary expression of CARs in T cells is the possibility of examining their clinical safety in a self-limiting manner, particularly if there are concerns regarding the on-target, but off-tumor toxic effects. Furthermore, it is possible that multiple injections of messenger RNA CAR T cells may also be more economical than retroviral and lentiviral vectors when considering the initial evaluation of novel CARs. It is yet unknown whether multiple, repeated treatments with RNA-electroporated T cells will yield similar efficacy to that of stably expressed CARs in a clinical setting, but preclinical models seem promising,64, 65 as do early clinical data. Recently, it was reported that the adoptive transfer of messenger RNA CAR T cells targeting mesothelin displayed antitumor activity in two patients in ongoing clinical trials.66
Future of CAR T therapy manufacturing
The majority of current CAR T therapies use a process that begins with selecting for and isolating a population of collected cells enriched for the patient’s own T cells for genetic modification. Clearly, there are technical and financial challenges in manufacturing single-patient product lots. Thus, preclinical development has been done toward generating universal T-cell products in which ‘off-the-shelf’ CAR T-cell therapies from allogeneic donors could be used, coupled with a knockdown of histocompatibility antigen genes. Although further investigation is needed with respect to the preclinical work before reaching clinical trials and approval, the successful development of ‘off-the-shelf’ CAR T cells would likely lead to therapies that would be easier to manufacture.
The significant hurdles to ‘off-the-shelf’ CAR T cells are potency and, especially, persistence. Recently, several pharmaceutical and biotechnology companies have entered the field of CAR therapy development and it is likely that the existing infrastructure required for the manufacturing and distribution of CAR T cells will also reduce the cost and increase efficiency.67 Until such time, it is crucial to critically examine current manufacturing techniques, in-process and release testing standards, and efficacy in the clinic. Clearly a streamlined process is desirable, but more importantly may also lead to increased therapeutic efficacy in patients, particularly those with advanced malignancies.
Several challenges of CAR T-cell therapy have been identified and are currently under investigation, including the presence of host immunity against the transgene and/or vector, the risk of insertional mutagenesis, and ensuring the persistence of the transgene and/or transgenic target cells. In the clinic, immunotherapy for cancer treatment has shown some promising results, particularly the use of CAR T-cell therapy, which combines the specificity of gene therapy, the expanded physiological response of cellular therapy and the memory activity of vaccine therapy.
References
Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH . Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol 2014; 32: 189–225.
Ahmed N, Heslop HE, Mackall CL . T-cell-based therapies for malignancy and infection in childhood. Pediatr Clin North Am 2010; 57: 83–96.
Restifo NP, Dudley ME, Rosenberg SA . Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012; 12: 269–281.
Lipowska-Bhalla G, Gilham DE, Hawkins RE, Rothwell DG . Targeted immunotherapy of cancer with CAR T cells: achievements and challenges. Cancer Immunol Immunother 2012; 61: 953–962.
Gross G, Waks T, Eshhar Z . Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 1989; 86: 10024–10028.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006; 12: 6106–6115.
Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006; 24: e20–e22.
Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008; 112: 2261–2271.
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 2011; 121: 1822–1826.
Stuart RW, Racke MK . Targeting T cell costimulation in autoimmune disease. Expert Opin Ther Targets 2002; 6: 275–289.
Finney HM, Lawson AD, Bebbington CR, Weir AN . Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol 1998; 161: 2791–2797.
Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M . Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 2002; 20: 70–75.
Sadelain M, Riviere I, Brentjens R . Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer 2003; 3: 35–45.
Sadelain M, Brentjens R, Riviere I . The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3: 388–398.
Porter DL, Levine BL, Kalos M, Bagg A, June CH . Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365: 725–733.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3: 95ra73.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368: 1509–1518.
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18: 676–684.
Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother 2009; 32: 169–180.
Davila ML, Brentjens R . Chimeric antigen receptor therapy for chronic lymphocytic leukemia: what are the challenges? Hematol Oncol Clin North Am 2013; 27: 341–353.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119: 2709–2720.
Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, Yang G et al. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLoS One 2013; 8: e64138.
Singh H, Huls H, Kebriaei P, Cooper LJ . A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev 2014; 257: 181–190.
Smith JW . Apheresis techniques and cellular immunomodulation. Ther Apher 1997; 1: 203–206.
Yuan S, Ziman A, Smeltzer B, Lu Q, Goldfinger D . Moderate and severe adverse events associated with apheresis donations: incidences and risk factors. Transfusion 2010; 50: 478–486.
Tanaka J, Mielcarek M, Torok-Storb B . Impaired induction of the CD28-responsive complex in granulocyte colony-stimulating factor mobilized CD4 T cells. Blood 1998; 91: 347–352.
Sloand EM, Kim S, Maciejewski JP, Van Rhee F, Chaudhuri A, Barrett J et al. Pharmacologic doses of granulocyte colony-stimulating factor affect cytokine production by lymphocytes in vitro and in vivo. Blood 2000; 95: 2269–2274.
Beloki L, Ramirez N, Olavarria E, Samuel ER, Lowdell MW . Manufacturing of highly functional and specific T cells for adoptive immunotherapy against virus from granulocyte colony-stimulating factor-mobilized donors. Cytotherapy; 2014; 16: 1390–1408.
Lundqvist A, Smith AL, Takahashi Y, Wong S, Bahceci E, Cook L et al. Differences in the phenotype, cytokine gene expression profiles, and in vivo alloreactivity of T cells mobilized with plerixafor compared with G-CSF. J Immunol 2013; 191: 6241–6249.
Cottler-Fox MH, Lapidot T, Petit I, Kollet O, DiPersio JF, Link D et al. Stem cell mobilization. Hematology Am Soc Hematol Educ Program 2003; 2003: 419–437.
Lee G, Arepally GM . Anticoagulation techniques in apheresis: from heparin to citrate and beyond. J Clin Apher 2012; 27: 117–125.
Hoffman R, Benz E, Silberstein L, Heslop H, Weitz J, Anastasi J . Hematology: Basic Principles and Practice, 6th edn. Elsevier Saunders: Philadelphia, PA, USA, 2013.
Powell DJ Jr, Brennan AL, Zheng Z, Huynh H, Cotte J, Levine BL . Efficient clinical-scale enrichment of lymphocytes for use in adoptive immunotherapy using a modified counterflow centrifugal elutriation program. Cytotherapy 2009; 11: 923–935.
June CH . Principles of adoptive T cell cancer therapy. J Clin Invest 2007; 117: 1204–1212.
Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD . Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992; 257: 238–241.
Nguyen LT, Yen PH, Nie J, Liadis N, Ghazarian D, Al-Habeeb A et al. Expansion and characterization of human melanoma tumor-infiltrating lymphocytes (TILs). PLoS One 2010; 5: e13940.
Levine BL . T lymphocyte engineering ex vivo for cancer and infectious disease. Expert Opin Biol Ther 2008; 8: 475–489.
Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol 1997; 159: 5921–5930.
Hami LS, Green C, Leshinsky N, Markham E, Miller K, Craig S . GMP production and testing of Xcellerated T Cells for the treatment of patients with CLL. Cytotherapy 2004; 6: 554–562.
Casati A, Varghaei-Nahvi A, Feldman SA, Assenmacher M, Rosenberg SA, Dudley ME et al. Clinical-scale selection and viral transduction of human naive and central memory CD8+ T cells for adoptive cell therapy of cancer patients. Cancer Immunol Immunother 2013; 62: 1563–1573.
Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D . Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 1997; 15: 871–875.
Sakuma T, Barry MA, Ikeda Y . Lentiviral vectors: basic to translational. Biochem J 2012; 443: 603–618.
Huls MH, Figliola MJ, Dawson MJ, Olivares S, Kebriaei P, Shpall EJ et al. Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood. J Vis Exp 2013; 72: e50070.
Kay MA . State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet 2011; 12: 316–328.
Ellis J . Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther 2005; 16: 1241–1246.
Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M . Viral vectors: a look back and ahead on gene transfer technology. New Microbiol 2013; 36: 1–22.
Durand S, Cimarelli A . The inside out of lentiviral vectors. Viruses 2011; 3: 132–159.
Wang GP, Levine BL, Binder GK, Berry CC, Malani N, McGarrity G et al. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther 2009; 17: 844–850.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17: 1453–1464.
US Food and Drug Administration. Guidance for industry: supplemental guidance on testing for replication competent retrovirus in retroviral vector based gene therapy products and during follow-up of patients in clinical trials using retroviral vectors. November, 2006, http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/ucm072961.htm#vii accessed 21 December 2014.
Somerville RP, Dudley ME . Bioreactors get personal. Oncoimmunology 2012; 1: 1435–1437.
Hollyman D, McDonald D, Senior I, Thompson J, Deavall H, Turner G et al. The development and validation of the CryoDoc system; a novel method for the cryopreservation of cell therapy products without the use of a clean room. Transfusion 2012; 52 (Suppl S3: 182A).
Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol 2002; 20: 143–148.
Thomas AK, Maus MV, Shalaby WS, June CH, Riley JL . A cell-based artificial antigen-presenting cell coated with anti-CD3 and CD28 antibodies enables rapid expansion and long-term growth of CD4 T lymphocytes. Clin Immunol 2002; 105: 259–272.
Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther 2007; 15: 981–988.
Nemunaitis J, Jahan T, Ross H, Sterman D, Richards D, Fox B et al. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther 2006; 13: 555–562.
Borrello IM, Levitsky HI, Stock W, Sher D, Qin L, DeAngelo DJ et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood 2009; 114: 1736–1745.
Rushworth D, Jena B, Olivares S, Maiti S, Briggs N, Somanchi S et al. Universal artificial antigen presenting cells to selectively propagate T cells expressing chimeric antigen receptor independent of specificity. J Immunother 2014; 37: 204–213.
Numbenjapon T, Serrano LM, Chang WC, Forman SJ, Jensen MC, Cooper LJ . Antigen-independent and antigen-dependent methods to numerically expand CD19-specific CD8+ T cells. Exp Hematol 2007; 35: 1083–1090.
Bajgain P, Mucharla R, Anurathapan U, Lapteva N, Leen AM, Heslop HE et al. A novel approach to manufacture CAR-T cells for clinical applications. ASBMT BMT Tandem Meeting; February 13-17, 2013; Salt Lake City, UT (abstract 2276).
Riet T, Holzinger A, Dorrie J, Schaft N, Schuler G, Abken H . Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol Biol 2013; 969: 187–201.
Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma 2012; 53: 958–965.
Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther 2011; 22: 1575–1586.
Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 2010; 70: 9053–9061.
Barrett DM, Liu X, Jiang S, Jun e CH, Grupp SA, Zhao Y . Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 2013; 24: 717–727.
Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2: 112–120.
Levine BL, June CH . Perspective: assembly line immunotherapy. Nature 2013; 498: S17.
Acknowledgements
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. I thank Matthew Hoelzle, for his assistance with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Levine declares a financial interest due to intellectual property and patents in the field of cell and gene therapy. Conflict of interest is managed in accordance with University of Pennsylvania policy and oversight.
Rights and permissions
About this article

Cite this article
Levine, B. Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther 22, 79–84 (2015). https://doi.org/10.1038/cgt.2015.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2015.5
- Springer Nature America, Inc.
This article is cited by
-
CAR-modified immune cells as a rapidly evolving approach in the context of cancer immunotherapies
Medical Oncology (2023)
-
A digital platform for the design of patient-centric supply chains
Scientific Reports (2022)
-
Engineered red blood cells as an off-the-shelf allogeneic anti-tumor therapeutic
Nature Communications (2021)
-
Novel biomanufacturing platform for large-scale and high-quality human T cells production
Journal of Biological Engineering (2019)
-
Optimized DNA electroporation for primary human T cell engineering
BMC Biotechnology (2018)