
Abstract
Chimeric antigen receptor-modified T cells (CAR-T) have emerged as a new modality for cancer immunotherapy due to their potent efficacy against terminal cancers. CAR-Ts are reported to exert higher efficacy than monoclonal antibodies and antibody–drug conjugates, and act via mechanisms distinct from T cell receptor-engineered T cells. These cells are constructed by transducing genes encoding fusion proteins of cancer antigen-recognizing single-chain Fv linked to intracellular signaling domains of T cell receptors. CAR-Ts are classified as first-, second- and third-generation, depending on the intracellular signaling domain number of T cell receptors. This review covers the current status of CAR-T research, including basic proof-of-concept investigations at the cell and animal levels. Currently ongoing clinical trials of CAR-T worldwide are additionally discussed. Owing to the lack of existing approved products, several unresolved concerns remain with regard to safety, efficacy and manufacturing of CAR-T, as well as quality control issues. In particular, the cytokine release syndrome is the major side-effect impeding the successful development of CAR-T in clinical trials. Here, we have addressed the challenges and regulatory perspectives of CAR-T therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, immunotherapy has attracted considerable research attention as a new modality of cancer treatment. Among the newly developed cancer immunotherapy technologies, those using chimeric antigen receptor-modified T cells (CAR-T) have been of particular interest. Substantial progress has been made in the CAR-T-based cancer immunotherapy field following the initial generation of CAR-T in 1989 (Kershaw et al. 2013; 2014; Wang and Riviere 2015). Currently, dozens of CAR-T clinical trials are ongoing worldwide (Fig. 1). To construct CAR-T, T cells are transduced with genes encoding fusion proteins for cancer antigen-recognizing single chain Fv (scFv) linked to the intracellular signaling domain of T cell receptors.

Clinical trials of CAR-T worldwide
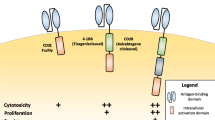
CAR-Ts are classified as first-, second- and third-generation, depending on the intracellular signaling domain numbers of T cell receptors (Fig. 2). First-generation CAR-T cells were designed to express scFv in the extracellular region and the signaling sequence of the T cell receptor intracellular domain with no co-stimulatory molecules. However, first-generation CAR-Ts were limited in their tumor cell-killing efficacy after specific recognition of tumor cells by antigens. To overcome these limitations, second- and third-generation CAR-Ts were designed to express co-stimulatory molecules in the intracellular domain (Fig. 2, Casucci and Bondanza 2011; Maus et al. 2014). Chimeric antigen receptor (CAR) gene cassettes for second-generation CAR-Ts encompass one co-stimulatory molecule, such as CD28 or 4-1BB. Third-generation CAR-Ts have been further developed to include two co-stimulatory molecules among CD27, CD28, 4-1BBand OX40.

Construction of CAR in each CAR-T generation
Manufacture and administration of CAR-T
CAR-Ts are manufactured using three consecutive steps (Lee et al. 2012; Wang and Rivière 2015; Levine 2015). The first step is to generate genetic constructs of CAR to encode tumor antigen-specific Fv linked to signaling sequences of T cell receptors. Next, T cells are transduced with CAR using viral, nonviral or physical methods. Retroviral or lentiviral vectors have been successfully employed as viral vectors for transduction of T cells with CAR. In other studies, T cells have been transduced with plasmid DNA (Huang et al. 2012; Kumaresan et al. 2014; Wang et al. 2014a, b) or RNA (Zhao et al. 2010) encoding CAR via electroporation. The third step is cultivation of CAR-T cells.
Several protocols have attempted to activate T cells for transduction, one of which is to use anti-CD3-antibodies and cytokines, such as interleukin-2. Various other T cell activation methods are currently under investigation (Fig. 3). Antigen-presenting cells expressing 4-1BBL and Fc receptor have additionally been employed to activate T cells. Another procedure involves the use of beads modified with anti-CD3 and anti-CD28 antibodies for T cell activation. The resulting CAR-T cells are derived from CD4 or CD8 T cells, expanded using cytokines, and administered to patients via intravenous infusion (Lee et al. 2012).

Activation of T cells and expansion of CAR-T
Advantages of CAR-T over existing cancer immunotherapy
CAR-Ts have attracted considerable research attention due to their potent efficacy against terminal cancers, relative to monoclonal antibodies and antibody–drug conjugates. Moreover, CAR-Ts act through different mechanisms from T cell receptor-engineered T cells (TCR-T). In TCR-T, TCRs recognize complexes of tumor antigens that are processed in APC cells and presented on APC cell surfaces with MHC class molecules. Unlike TCR-T, CAR-Ts do not require processing and presentation of tumor antigen-recognizing moieties with MHC molecules (Kershaw et al. 2014; Fig. 4). The differences between CAR-T and TCR-T are summarized in Table 1 (Gill and June 2015). Notably, the lack of MHC restrictions means that eligible patient groups for CAR-T are wider compared to those for TCR-T that requires the identification of patient MHC types.

Different cell surface structures between CAR-T and TCR-T
Current status of CAR-T studies
The utility of CAR-T for treatment of lymphoma and solid tumors has been examined in several studies, grouped as cell-level (Table 2; Fig. 5), animal-level (Table 2; Fig. 5), preclinical (Table 3; Fig. 5), and clinical trials (Table 4; Fig. 6). Current status summary of CAR-T under basic investigational stages shows that retroviral vectors are most actively used for introducing CAR genes into T cells. Although the use of viral vectors are dominating, nonviral approaches using cationic polymers or electroporations have been attempted (Fig. 5a). Hematological cancers such as lymphoma have been the major target of CAR-T in basic research stage (Fig. 5b). Second-generation CAR-Ts have been most widely investigated in basic cell-level and animal studies, with focus on hematological cancers and solid tumors (Fig. 5c). The application of CAR-T has been extended from cancer immunotherapy to treatment of autoimmune diseases, such as multiple sclerosis in Europe (Fransson et al. 2012). Another recent study reported on the effectiveness of CAR-T in treating fungal infection, suggesting a new field of CAR-T application (Kumaresan et al. 2014). Similar to the investigational stage research, preclinical trial studies used retroviral vectors and second generation CAR constructions in higher frequency than other viral vectors (Fig. 5d), and generations, respectively (Fig. 5f). Notably, the number of preclinical trials for hematological tumors were lower than brain cancers (Fig. 5e).

Current status of CAR-T therapeutics under investigational stages and preclinical trials. CAR-T therapeutics under investigational stages were analyzed by delivery vectors (a), target diseases (b), and generations (c). CAR-T therapeutics in preclinical trials were analyzed by delivery vectors (d), target diseases (e), and generations (f)

Current status of CAR-T therapeutics in clinical trials. CAR-T therapeutics in preclinical stages were analyzed by tumor antigens (a), target diseases (b), and generations (c)
For introduction of CAR-T genes into T cells, viral vectors have been predominantly used. The duration of CAR-T survival in vivo is reported to be longer than several months. Since month-long sustainable expression of CAR can evoke undesirable side-effects, the optimal length of expression time requires further investigation. To shorten the duration of CAR expression and minimize safety concerns, physical electroporation studies are underway. Introduction of plasmid DNA encoding CAR directly into the cytoplasm of T cells via electroporation may lead to an expected duration of CAR of several days, while avoiding side-effects of delivery vectors.
Currently, more than 80 CAR-T cases are in clinical trials worldwide (www.clinicaltrials.gov). No clinical trials are in phase 3 as yet, but the potential is high, given the number in phase 2 (Table 4). The majority of CAR-T clinical trials is being held in USA, and has also been initiated in Asia, China and Japan (Fig. 1). CD19, widely studied as a tumor antigen target of CAR-T, is overexpressed on the surfaces of leukemia cells of acute lymphocytic leukemia (ALL) patients. CD19 CAR-T therapy is reported to be effective in children with recurrent ALL after bone marrow transplantation (Lee et al. 2015). Among the products in clinical trials, CTL019 (Novartis, USA), CD19-targeted CAR-T (CD19-CAR-T) against ALL has been recognized as a “breakthrough therapy” by the Food and Drug Administration of USA and is in phase 2 development. Several other global pharmaceutical companies are developing CAR-T products in the pipeline. In clinical trials, CD19 has been most extensively used as target tumor antigens of hematological cancers (Fig. 6a). Other antigens in clinical trials include carcinoembryonic antigen (CEA), human epidermal growth factor receptor 2 (HER2), GD2, CD30, and CD20 (Fig. 6a). Hematological cancers have been predominantly studied in clinical phase (Fig. 6b). Until now, phase 2 trial are the most advanced stage for CAR-Ts (Fig. 6c).
Challenges
Limitations of each generation of CAR-T
Although CAR-T takes advantage of the immune response of T cell killing abnormal cells, tumor cell-killing effects are decreased in solid tumors. This reduced antitumor activity is attributable to the immunosuppressive microenvironment of tumor tissues, resulting in low penetration efficiencies of CAR-T into solid tumor tissues. Moreover, leukocytes in tumor tissues are known to secrete factors that lower T cell activity.
Second-generation CAR-Ts containing a co-stimulatory signaling domain can induce the expression of immunosuppressive receptors, such as T cell membrane protein-3, cytotoxic T lymphocyte associated antigen 4 (CTLA-4), and programmed death-1 (PD-1). To overcome suppression of CAR-T activity by PD-1, the effects of co-administration of anti-PD-1 antibody with CAR-T were recently examined (John et al. 2013). An ongoing clinical trial (NCT00586391) is exploring the effects of co-administration of CAR-T and ipilimumab, an anti-CTLA-4 antibody (Maher 2014).
Third-generation CAR-Ts with two co-stimulatory molecules, such as OX40 and 4-1BB, display enhanced activity in vivo. However, excessive stimulation of T cell activity by two co-stimulatory molecules may induce an abrupt increase in cytokine secretion, known as ‘cytokine release syndrome’ (CRS). The onset of this severe side- effects one of the biggest safety concerns that require addressing for further successful development of CAR-T.
Side-effects
The biggest hurdle in CAR-T clinical trials is severe side-effects, the most acute being CRS. The mortality list of patients undergoing clinical trials of CAR-T highlights the need for design of improved clinical protocols and regulatory decisions of investigational new drug development applications. Symptoms of CRS include high fever, joint pain, muscle pain, low blood pressure, and dyspnea, with death in a few cases. In 2014, clinical trials were temporarily held in Memorial Sloan Kettering Cancer Center after deaths of two patients within two weeks after infusion with CD19-CAR-T from Juno Therapeutics (USA). CRS has been determined as the main cause of death of patients in clinical trials to date.
Although CRS is the most common side-effect related to CAR-T therapy, the development of CRS has been considered to be correlated to the response to therapy. In previous studies, it has been observed that CAR-T responsive patients developed CRS, whereas non-responsive patients did not develop CRS (Maude et al. 2015). The severity of CRS has been reported to be rather correlated with tumor burden at injection time of CAR-T. Given the importance of CRS in clinical monitoring, the markers which can predict the severity of CRS need to be identified. Peak levels of cytokines such as interferon-γ have been found to be more elevated in severe CRS than mild CRS (Davila et al. 2014). Other studies proposed C-reactive protein as an indicator of severe CRS (Maude et al. 2014). However, the decisive biomarkers for CRS still remains to be studied.
To minimize these side-effects, it is crucial to select the appropriate group among enrolled patients and optimize the CAR-T dose in clinical protocol design. Currently, anti-interleukin 6-antibody or steroid drugs are co-administered with CAR-T to reduce CRS (Davila et al. 2014), although further studies are required in this respect. In addition, safety studies assessing whether CAR-T can induce autoimmunity or graft-versus-host disease are warranted. Several studies have examined the efficacy of co-administration of cytokine inhibitors with CAR-T or a suicide gene, with the aim of reducing CRS. A recent study (Grupp et al. 2013) reported that the co-administration of CAR-T with tocilizumab, an anti-interleukin-6 antibody, alleviates CRS.
The majority of CAR-Ts for clinical trials have been constructed using viral vectors for CAR gene transfection. Although these vectors are designed to be non-replicating, a long-term study (over a number of years) should be performed to monitor potential replicative ability. Profiling and standardization of cytokines after CAR-T administration is necessary. The deaths of two patients in 2014 have highlighted CRS as the most severe limitation in clinical trials of CAR-T. This excessive increase in cytokine secretion results from amplification of CAR-T cells in vivo and may be sufficiently fatal to cause death. To monitor and control CRS, identification of the specific roles of individual cytokines and types of relevant cytokines is required to predict the risk of CRS in clinical trials. Moreover, profiling of cytokines and CRS marker cytokines during clinical trials should be standardized.
In the case of CD19-CAR-T, anticancer effects are accompanied by a certain level of side effects. The possibility of segregating efficacy from side-effects of CAR-T products should be explored, with the aim of establishing an optimal regimen with minimization of CRS. Autoimmunity of CAR-T is another potential concern. Low doses of CAR-T and serial tumor cell-killing effects of single CAR-T cells may lower the possibility of autoimmunity. However, the artificial nature of CAR-T may increase the risk of autoimmunity. Thus, to guarantee safety, regulatory concerns regarding the autoimmunity issue should not be overlooked.
In addition to CRS, another concern is “on-target/off-tumor” side effects. The side effect is due to that majority of target antigens for CAR-Ts are existing on both tumor and normal tissues, showing overexpression on tumor cells (Kakarla and Gottschalk 2014). To minimize the “on-target/off-tumor” side effects, the discovery of new target molecule exclusively expressing on tumor tissues would be necessary. Another approach is to remove inappropriately activated CAR-Ts. A recent study reported that small molecule drug AP1903 could induce caspase 9 and apoptosis in transduced cells, killing only activated cells expressing high levels of CAR (Gargett and Brown 2014).
Regulatory perspective
CTL019, a CD19-CAR-T designed by Novartis (USA), is the first known compound in the CAR-T class that has entered phase 2 clinical trials. From the regulatory perspective, manufacturing and clinical trials are major concerns (Puri 2014). Production issues include consistency of CAR-T products, patient-dependent variations in T cell transfection efficiency, optimal T cell types for CAR transfection, and labeling of CAR-T. The major clinical trial concerns include potency and safety.
Pharmacokinetics and biodistribution of CAR-T
Pharmacokinetics and biodistribution experiments provide essential information for predicting the possible side-effects of CAR-T. A recent study reported that CAR-T is distributed to the bone marrow after intravenous administration and circulates in the blood up to 10 months post-injection (Ritchie et al. 2013). Further quantitative analyses of pharmacokinetics, tissue distribution and retention in the body are essential. The tumor antigen, HER-2, has been shown to be expressed in brain tissues and the mammary gland (Wang et al. 2010). Carcinoembryonic antigen, another tumor antigen, has been identified in colon or normal tissues (Schölzel et al. 2000).The nonexclusive expression patterns of tumor antigens in normal tissues increase the possibility of normal tissue damage upon administration of CAR-T targeting these tumor antigens. From this viewpoint, regulatory considerations associated with the distribution and pharmacokinetics of CAR-T should be addressed.
Efficacy of CAR-T
One concern with regard to the efficacy of CAR-T is the consideration of optimal T cell subsets. The majority of CAR-Ts in clinical trials introduce CAR genes after isolation of total T cells from patients. The issue of whether the T cell subtype affects the transfection efficiency and potency of CAR-Ts should be examined.
Efficacy evaluation methods for CAR-T should be further explored. Currently, tumor cell lysis induction and cytokine secretion capabilities of CAR-Ts are under examination. Establishment of a relevant method reflecting the efficacy of CAR-T may facilitate standardized evaluation of CAR-T products. Moreover, owing to in vivo amplification, it is necessary to assess the relationships among the ratio of CAR-T within total administered T cells, CAR copy numbers and efficacy.
Unlike chemical or protein drugs, CAR-T is a living drug that amplifies in the body after administration. This in vivo amplification property leads to an increase in the actual effective dose of CAR-T. The severity of symptoms and ages of patients may serve as factors affecting the in vivo amplification efficiency of CAR-T. The discrepancy between administration and working doses is a unique feature of CAR-T. Further analysis of the relationships between administered and working doses and therapeutic effects is warranted.
The specific T cell subtypes contributing to anticancer activity need to be identified. Currently, CAR-T is administered as mixtures of various T cell subsets. A recent study reported that increasing the frequency of CD8(+)CD45RA(+)CCR7(+) CAR-T cells, a subset closest to T cell memory stem cells, within total CAR-T enhances anticancer activity in an animal model (Xu et al. 2014).
Clinical trials of CAR-T
Several issues require clarification with regard to the optimal dose of CAR-T. As a result of in vivo amplification of intravenously infused CAR-T, the initial doses infused are not the same as the actual working cell number. Moreover, adjustment criteria of doses should be fixed between the weight and body surface of patients. Considering the amplification of CAR-Ts in bone marrow after infusion, we need to ascertain whether measurement of CAR-T numbers in the blood reflects the amplification extent in bone marrow.
Manufacture and quality control of CAR-T products
To validate consistency in CAR-T quality among batches, regulatory studies on chemistry, manufacturing and control (CMC) are essential. Analysis of quality control for each step of CAR-T production is important (Fig. 7). Quality control should be performed to maintain transfection efficiency of CAR among different batches, with parameters including the acceptable ranges of gene-modified CAR-T ratios among total T cells and quantification of copy numbers of the CAR gene per cell. For consistent production of CAR-T among batches, standardization of the stock of viral vectors is required to provide constant multiplicity of infection. Moreover, effects of patient age and medical treatment history on transfection of T cells with CAR-encoding vectors should be assessed.

Quality control flow chart for production of CAR-T
Viral vectors, such as retroviral or lentiviral vectors, are frequently used for introduction of CAR into T cells (Wang and Riviere 2015). Quality control of viral vectors for CAR gene delivery should be performed in terms of purity, safety, T cell transfection efficiency, and physicochemical characterization.
Moreover, for quality control, validation of CAR-T sterility is essential. Since CAR-T is manufactured ex vivo by isolation of T cells, introduction of CAR genes, amplification, microbial assays and the scope of microorganisms for CAR-T need to be established.
Production and distribution of CAR-T need to be standardized. After conceptual design and proof-of-concept studies, production techniques have mostly been transferred from the laboratory benches of academia to industry. The manufacturing processes of CAR-carrying viral vectors and CAR-T are complex and differ among developers. CAR-T products under clinical trials are generated and distributed to patients using different protocols. The production and distribution processes of gene-modified cells are sufficiently crucial to affect quality. Quality control and standardization of manufacturing and distribution processes should thus be performed under good practice principles.
Labeling of CAR-T products should be carefully assessed. Given the autologous nature of CAR-T in which patient T cells are transfected with CAR genes and infused back into the same patient, it is important to clarify labeling of CAR-T to minimize the fatal risk of potential administration to the wrong patients.
Conclusions
CAR-Ts have attracted considerable research attention as a novel and potent modality of cancer immunotherapy. Global pharmaceutical companies have started investing in CAR-Ts, with several products in the pipeline for approval. However, substantial regulatory issues for CAR-T need to be addressed. The living nature of CAR-T necessitates careful assessment of safety and efficacy issues. The ex vivo manufacturing process of CAR-T highlights the significance of validating sterility and extensive product quality control. Further focus on regulatory studies and establishment of regulatory science-based guidelines may expedite the development of effective CAR-T products for patient use.
References
Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y (2013) Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 24:717–727
Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z (2014) Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther 22:1018–1028
Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, Brouns SA, Spencer DM, Till BG, Jensen MC, Riddell SR, Press OW (2013) Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One 8:e82742
Casucci M, Bondanza A (2011) Suicide gene therapy to increase the safety of chimeric antigen receptor-redirected T lymphocytes. J Cancer 2:378–382
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, Rainusso N, Wu MF, Liu H, Kew Y, Grossman RG, Powell S, Lee D, Ahmed N, Gottschalk S (2013) T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther 21:629–637
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, He X, Devine SM, Zhang X, Caligiuri MA, Hofmeister CC, Yu J (2014) CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 28:917–927
Davies DM, Foster J, Van Der Stegen SJ, Parente-Pereira AC, Chiapero-Stanke L, Delinassios GJ, Burbridge SE, Kao V, Liu Z, Bosshard-Carter L, Van Schalkwyk MC, Box C, Eccles SA, Mather SJ, Wilkie S, Maher J (2012) Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med 18:565–576
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra25
Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, Loskog AS (2012) CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflam 9:112
Gargett T, Brown MP (2014) The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol 5:235
Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, Del Vecchio M, Creighton CJ, Ittmann M, Ferrone S, Dotti G (2014) T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res 20:962–971
Gill S, June CH (2015) Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263:68–89
Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G, Heslop HE, Gottschalk S, Wels WS, Baker ML, Ahmed N (2013) TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucl Acids 2:e105
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368:1509–1518
Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, Gao X, Guo L, Yvon E, Hicks J, Liu H, Dotti G, Metelitsa LS (2014) Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 124:2824–2833
Hu WX, Chen HP, Yu K, Shen LX, Wang SuSZ, Sui WJ, Shan DM, Li HZ (2012) Gene therapy of malignant solid tumors by targeting erbB2 receptors and by activating T cells. Cancer Biother Radiopharm 27:711–718
Huang G, Yu L, Cooper LJ, Hollomon M, Huls H, Kleinerman ES (2012) Genetically modified T cells targeting interleukin-11 receptor α-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res 72:271–281
Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR (2013) Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res 19:3153–3164
Inaguma Y, Akahori Y, Murayama Y, Shiraishi K, Tsuzuki-Iba S, Endoh A, Tsujikawa Demachi-Okamura A, Hiramatsu K, Saji H, Yamamoto Y, Yamamoto N, Nishimura Y, Takahashi T, Kuzushima K, Emi N, Akatsuka Y (2014) Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specific for minor histocompatibility antigen HA-1H. Gene Ther 21:575–584
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, Zeng T, Huang H, Zhang X, Sun W, Man-Yuen Sze D, Yi Q, Hou J (2014) Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mole Oncol 8:297–310
John LB, Kershaw MH, Darcy PK (2013) Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunol 2:e26286
Kakarla S, Gottschalk S (2014) CAR T cells for solid tumors: armed and ready to go? Cancer J 20:151–155
Kershaw MH, Westwood JA, Darcy PK (2013) Gene-engineered T cells for cancer therapy. Nat Rev Cancer 13:525–541
Kershaw MH, Westwood JA, Slaney CY, Darcy PK (2014) Clinical application of genetically modified T cells in cancer therapy. Clin Transl Immunol 3(5):e16
Kobayashi E, Kishi Ozawa T, Hamana H, Nakagawa H, Jin A, Lin Z, Muraguchi A (2014) A chimeric antigen receptor for TRAIL-receptor 1 induces apoptosis in various types of tumor cells. Biochem Biophys Res Com 453:798–803
Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, Sahin A, Carter BS, Brem H, Junghans RP, Sampath P (2012) Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res 18:5949–5960
Krebs S, Chow KK, Yi Z, Rodriguez-Cruz T, Hegde M, Gerken C, Ahmed N, Gottschalk S (2014) T cells redirected to interleukin-13Rα2 with interleukin-13 mutein-chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytothera 16:1121–1131
Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, Roszik J, Rabinovich B, Olivares S, Krishnamurthy J, Zhang L, Najjar AM, Huls MH, Lee DA, Champlin RE, Kontoyiannis DP, Cooper LJ (2014) Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci USA 111:10660–10665
Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, Powell DJ (2012) Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther 20:633–643
Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, Powell DJ (2013) Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res 1:43–53
Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA (2012) The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin Cancer Res 18:2780–2790
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL (2015) T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385:517–528
Levine BL (2015) Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther 22:79–84
Ma Q, Safar M, Holmes E, Wang Y, Boynton AL, Junghans RP (2014) Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate 61:12–25
Maher J (2014) Clinical immunotherapy of B-cell malignancy using CD19-targeted CAR T-cells. Curr Gene Ther 14:35–43
Mata M, Vera JF, Gerken C, Rooney CM, Miller T, Pfent C, Wang LL, Wilson-Robles HM, Gottschalk S (2014) Toward immunotherapy with redirected T cells in a large animal model: ex vivo activation, expansion, and genetic modification of canine T cells. J Immunother 37:407–415
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371:1507–1517
Maude SL, Teachey DT, Porter DL, Grupp SA (2015) CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125:4017–4023
Maus MV, Grupp SA, Porter DL, June CH (2014) Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123:2625–2635
Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, Zhang W, Fine HA, Rosenberg SA (2012) Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human Gene Ther 23:1043–1053
Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G (2014) Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res 74:5195–5205
Puri RK (2014) FDA Perspective on the regulation of TCR/CAR T-cell products. In: 12th CIMT (Cancer Immunotherapy) annual symposium
Rainusso N, Brawley VS, Ghazi A, Hicks MJ, Gottschalk S, Rosen JM, Ahmed N (2012) Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther 19:212–217
Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, Shin M, Wall DM, Hönemann D, Gambell P, Westerman DA, Haurat J, Westwood JA, Scott AM, Kravets L, Dickinson M, Trapani JA, Smyth MJ, Darcy PK, Kershaw MH, Prince HM (2013) Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther 21:2122–2129.HM
Saito S, Nakazawa Y, Sueki A, Matsuda K, Tanaka M, Yanagisawa R, Maeda Y, Sato Y, Okabe S, Inukai T, Sugita K, Wilson MH, Rooney CM, Koike K (2014) Anti-leukemic potency of piggyBac-mediated CD19-specific T cells against refractory Philadelphia chromosome-positive acute lymphoblastic leukemia. Cytother 6:1257–1269
Schölzel S, Zimmermann W, Schwarzkopf G, Grunert F, Rogaczewski B, Thompson J (2000) Carcinoembryonic antigen family members CEACAM6 and CEACAM7 are differentially expressed in normal tissues and oppositely deregulated in hyperplastic colorectal polyps and early adenomas. Am J Pathol 156:595–605
Schuberth PC, Jakka G, Jensen SM, Wadle A, Gautschi F, Haley D, Haile S, Mischo A, Held G, Thiel M, Tinguely M, Bifulco CB, Fox BA, Renner C, Petrausch U (2013) Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther 20:386–395
Shin JH, Park HB, Oh YM, Lim DP, Lee JE, Seo HH, Lee SJ, Eom HS, Kim IH, Lee SH, Choi K (2012) Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 119:5678–5687
Sun M, Shi H, Liu C, Liu J, Liu X, Sun Y (2014) Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res 16:R61
Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E (2012) Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res 18:6436–6445
Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovitch B, Lee DA, Champlin RE, Bonini C, Naldini L, Rebar EJ, Gregory PD, Holmes MC, Cooper LJ (2012) A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119:5697–5705
Tsukahara TN, Iwase N, Kawakami K, Iwasaki M, Yamamoto C, Ohmine K, Uchibori R, Teruya T, Ido H, Saga Y, Urabe M, Mizukami H, Kume A, Nakamura M, Brentjens R, Ozawa K (2015) The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther 22:209–215
Wang LX, Westwood JA, Moeller M, Duong CP, Wei WZ, Malaterre J, Trapani JA, Neeson P, Smyth MJ, Kershaw MH, Darcy PK (2010) Tumor ablation by gene-modified T cells in the absence of autoimmunity. Cancer Res 70:9591–9598
Wang X, Rivière I (2015) Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther 22:85–94
Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, Aguilar B, Ostberg JR, Riddell SR, Forman SJ, Jensen MC (2012) Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8 + central memory T cells manufactured at clinical scale. J Immunother 35:689–701
Wang C, Hu W, Shen L, Dou R, Zhao S, Shan D, Yu K, Huang R, Li H (2014a) Adoptive antitumor immunotherapy in vitro and in vivo using genetically activated erbB2-specific T cells. J Immunother 37:351–359
Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL, Bo J, Fan H, Zhang Y, Zhang YJ, Chen MX, Feng KC, Wang QS, Fu XB, Han WD (2014b) Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol 155:160–175
Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, Sakemura R, Goto T, Hanajiri R, Imahashi N, Shimada K, Tomita A, Kiyoi H, Nishida T, Naoe T, Murata M (2015) Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol 94:911–920
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, Liu H, Creighton CJ, Gee AP, Heslop HE, Rooney CM, Savoldo B, Dotti G (2014) Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123:3750–3759
Zhao YE, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, Albelda SM, June CH (2010) Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 70:9053–9061
Acknowledgments
This research was supported by a grant (15172MFDS163) from Ministry of Food and Drug Safety in 2015.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kim, MG., Kim, D., Suh, SK. et al. Current status and regulatory perspective of chimeric antigen receptor-modified T cell therapeutics. Arch. Pharm. Res. 39, 437–452 (2016). https://doi.org/10.1007/s12272-016-0719-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0719-7