
Abstract
Tumor growth depends on angiogenesis. The complex tissue environment surrounding tumor cells, which is composed of a variety of resident and infiltrating host cells, secreted factors and extracellular matrix proteins, influences tumor angiogenesis and progression. Moreover, the tumor microenvironment contributes to determining therapeutic responses and resistance to therapy. The ability to block tumor resistance is related to the understanding of the cellular and molecular pathways activated in the tumor microenvironment. Novel emerging targeted therapeutic strategies are based on the combination of different antitumor approaches with the aim of resolving refractory tumors and improving cancer treatment efficiency.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Inflammatory infiltrate
- Resistance to therapy
- Tumor angiogenesis
- Tumor microenvironment
- Tumor progression
14.1 Tumor Angiogenesis
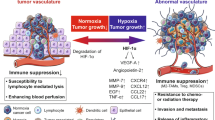
Healthy and pathologic tissue homeostasis requires an adequate supply of oxygen and nutrients that is connected to efficient development of the vascular system. Additionally, tumor cells to survive and proliferate need oxygen and nutrients and consequently the closeness to blood vessels. Angiogenesis is the physiological process through which new blood vessels form from pre-existing vessels (Carmeliet and Jain 2011). Generally, tumor development is an angiogenesis-dependent process, and the angiogenetic process depends on the temporal coordination of factors and related pathways needed for the establishment of stable channels to provide a supply to tumor cells (Weis and Cheresh 2011). It has been well established that during cancer progression, the interactions between tumor cells and inflammatory cells are closely associated with each other and with angiogenesis (Wang et al. 2019a).
The growth of solid tumor mass, its progression and the metastatic process, how it is widely described, are strongly influenced by angiogenesis (Folkman 1971). In 1966, Warren and collaborators implanted melanoma nodules in experimental animals and observed a rapid vessel sprout toward the mass, the formation of new capillaries, their penetration into the tumor, and the establishment of blood flow. This phenomenon was more evident during tumor growth than in inflammation processes (Warren and Shubik 1966). Research conducted by Folkman showed that without appropriate vascularization and therefore oxygen and nutrient supply, a tumor can grow limitedly to a size of a few millimeters and a cell content of approximately a few thousand cells (Folkman 1971; Nishida et al. 2006). Under these conditions, tumors induce a process recognized as an angiogenic switch in which tumor cells acquire angiogenic properties, leading to the transition from a quiescent to active endothelium and consequently the vascularization of the growing cell mass (Baeriswyl and Christofori 2009; Ribatti et al. 2007). In tumor murine models, this switch coincides with malignant transition of the growing mass and is needed for malignant tumor progression (Lin et al. 2006; Folkman et al. 1989). It became evident that some soluble factors released by the tumor induced the activation of angiogenesis. Folkman hypothesized that until the appropriate blood flow is created, the tumor mass stops its growth and enters a dormant state (Folkman et al. 1971). On this basis, in the last 50 years, research on mechanisms related to tumor angiogenesis has intensified to discover molecules usable as new targets in anticancer therapy. Tumor angiogenesis is a multiphasic process initiated directly by the tumor when it reaches a size that makes it hypoxic, which further leads to cancer development.
14.2 Tumor Microenvironment
It is well known that tumor cells develop in a complex tissue environment, the so-called tumor microenvironment (TME), which includes cancer cells, stromal cells, blood vessels, nerve fibers, extracellular matrix, and acellular components. The TME is involved in tumor initiation as well as during tumor progression and metastasis; furthermore, it also has important effects on therapeutic efficacy (Tamma et al. 2019a). It is believed that although cancer initiation is due to the acquisition of oncogenic mutations in cells, its progression depends on the surrounding cells that are recruited and subsequently release many cytokines and chemokines (Tysnes and Bjerkvig 2007). In 1863, Rudolf Virchow postulated the crosstalk between inflammation and cancer (Virchow 1989), and 20 years after Stephen Paget illustrated the “seed and soil” theory assuming that the choice of the target organ depends on the interactions between metastatic tumor cells (the “seed”) and their organ microenvironment (the “soil”) (Paget 1989). One hundred years later, Hanahan and Weinberg expanded from six to ten hallmarks of cancer and recognized the important role of the TME in cancer development (Hanahan and Weinberg 2011). The main cytokines and chemokines secreted by cells of the TME are involved in the regulation of angiogenesis, including proangiogenic factors, such as the vascular endothelial growth factor (VEGF) family, fibroblast growth factors (FGFs), platelet-derived growth factor (PDGF), angiopoietins (Ang), and hypoxia-inducible factor (HIF), and angiostatic factors, such as angiostatin, endostatin, platelet factor 4 (PF4), and thrombospondin-1 (TSP1) (Ucuzian et al. 2010).
14.3 Pro-Angiogenic Factors
VEGF
The human VEGF family includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF) originating from different genes (Melincovici et al. 2018). The VEGF family exerts its function by binding three transmembrane tyrosine kinase receptors (RTKs), VEGFR-1 (FLT1), VEGFR-2 (KDR, FLK1), and VEGFR-3 (FLT4). VEGFR-1 is expressed in monocytes, macrophages, hematopoietic stem cells, vascular smooth cells, and leukemic cells. VEGFR-2 is expressed in vascular endothelial cells, endothelial progenitor cells, and megakaryocytes, whereas VEGFR-3 is expressed in lymphatic endothelial cells. VEGFs can also interact with other proteins, integrins, cadherins, heparan sulfate proteoglycans, and with the coreceptors neuropilin-1 and -2 (NRP-1 and NRP-2), which enhance VEGFR-1 and VEGFR-2 action (Stuttfeld and Ballmer-Hofer 2009). VEGF-A is the main component of the VEGF family and is produced by endothelial and vascular smooth muscle cells, activated platelets, fibroblasts, lymphocytes, macrophages, and tumor cells. It is considered a crucial angiogenic stimulator involved in numerous pleiotropic effects, including the proliferation and inhibition of apoptosis of vascular endothelial cells (Ferrara and Davis-Smyth 1997; Gerber et al. 1998), permeability, chemotaxis and activation of monocytes and hematopoietic stem cells, and exerts neurotrophic and neuroprotective action (Storkebaum and Carmeliet 2004). Through alternative splicing, the VEGF-A transcript produces several isoforms with proangiogenic or antiangiogenic activities, including VEGF-A121, VEGF-A145, VEGF-A165, VEGF-A189, and VEGF-A206 (Yang et al. 2018a; Logue et al. 2016; Dehghani et al. 2018). VEGFA165 is the most important both quantitatively and qualitatively. VEGF-B is involved in pulmonary angiogenesis after chronic hypoxia and has been found in cardiac and skeletal muscle. VEGF-C and VEGF-D are important lymphangiogenesis regulators (Rauniyar et al. 2018; Stacker and Achen 2018). PlGF, discovered in the human placenta, is highly expressed in trophoblast cells (Hang et al. 2013) and has also been found in the thyroid, lungs, heart, and skeletal muscle (Maglione et al. 1991). It includes four different subtypes that bind VEGFR-1, and the PlGF isoform also binds NRP-1 and NRP-2. PlGF regulates the growth, migration, and survival of endothelial cells directly through VEGFR-1 or indirectly through VEGFR-2/VEGF-A-mediated activation or formation of a PlGF/VEGF-A heterodimer (Autiero et al. 2003).
Fibroblast Growth Factors (FGFs)
The human FGF family includes 22 members involved in the regulation of endothelial cell differentiation, proliferation, migration, survival, and vessel maturation (Yun et al. 2010). FGF-1 and FGF-2, the first known as acid FGF and the latter as basic FGF, mostly mediate the angiogenic response (Motomura et al. 2008). FGF receptors (FGFRs) belong to the RTK superfamily. Upon activation, they undergo dimerization and internalization and initiate large-scale tyrosine phosphorylation responses and signaling cascades activating the Ras/MAP-kinase pathway (Mathew et al. 2016).
Platelet-Derived Growth Factor (PDGF)
The PDGF family comprises four PDGF homodimers, namely, PDGF-AA, PDGF-BB, PDGF-CC, and PDGF-DD, and one heterodimer, PDGF-AB (Fredriksson et al. 2004). PDGF was originally isolated from platelets, but it has been expressed by numerous other cell types, including epithelial and endothelial cells. PDGF receptors (PDGFRs) belong to the family of RTKs and include PDGFRα and PDGFRβ, which are encoded by two different genes (Gao et al. 2018). These receptors are expressed by fibroblasts, pericytes, vascular smooth muscle cells, monocytes, macrophages, lymphocytes, and mast cells and stimulate their proliferation and motility. PDGFs participate in vascular development by acting on the proliferation and survival of vascular mural cells (Olson and Soriano 2011).
Angiopoietins (Angs)
The Ang protein family includes four members: Ang-1, Ang-2, Ang-3, and Ang-4 (Lee et al. 2004); the first two are the major members involved in vasculogenesis and vascular repair (Akwii et al. 2019). Angs bind to two receptors belonging to the family of RTKs named Tie1 and Tie2. Tie2 is expressed by endothelial and myeloid cells (Patan 1998). Tie1 is an orphan poorly characterized receptor that seems to be involved in the modulation of Ang/Tie-2 through the formation of heterodimers with Tie-2 (Eklund et al. 2017). Ang-1 is expressed by both mural cells and other nonvascular stromal and tumor cells. It is involved in the regulation of vessel stabilization during embryonic development, vessel remodeling, and maintenance of the normal vasculature (Brindle et al. 2006). Ang-2 is produced by the VEGF-stimulated endothelium, hypoxia, and shear stress, promoting blood vessel wall destabilization through competitive inhibition of Tie-2 and integrin activation. Furthermore, Ang-2 stimulates pericyte detachment, permeability, vascular regression, and lymphangiogenesis (Akwii et al. 2019).
Hypoxia-Inducible Factors (HIFs)
HIFs are DNA-binding transcription factors that associate with specific nuclear cofactors under hypoxia (Palazon et al. 2014). They are heterodimers that include both the constitutively expressed HIF-1β subunit and oxygen-regulated HIF-1α or HIF-2α subunit (Hu et al. 2003). In humans, HIF-1α is ubiquitously expressed, while HIF-2α, although it is expressed mainly in the endothelium, in hypoxic conditions, is also expressed in the kidney, pancreas, brain, liver, intestine, and myocardium. When cells are in a hypoxic environment, the hydroxylation process is inhibited, and HIF-α escapes proteasomal degradation, dimerizes with HIF-1β, and associates with transcriptional coactivators (Berra et al. 2001). The latter recognizes hypoxia-responsive genes, resulting in physiological adaptation to hypoxia. Other stimuli, such as nitric oxide and reactive oxygen species (ROS), can also activate HIFs (Wellman et al. 2004).
Many human cancers are characterized by increased levels of HIF, and its expression correlates with mortality (Zhong et al. 1999; Talks et al. 2000). Hypoxic conditions contribute to increased HIF activity, which translates into the regulation of genes involved in angiogenesis, cell survival, metabolism, invasion, and metastasis. In solid tumors, the rapid proliferation of cancer cells limits oxygen diffusion within the tumor, decreasing its concentrations under physiological conditions. This leads to increased expression and activity of HIF, contributing to tumor angiogenesis (Huang et al. 2017; Shi and Fang 2004).
14.4 Angiogenic Inhibitors
Angiostatin
Angiostatin is a 38 kDa internal fragment of plasminogen (Cao and Xue 2004; Ji et al. 1998). Angiostatin inhibits endothelial cell proliferation, migration, and tube formation (Pozzi et al. 2000) and induces apoptosis of endothelial cells (Ramirez-Moreno et al. 2020). Moreover, angiostatin inhibits the signaling induced by FGF-2 and VEGF in human microvascular endothelial cells (Redlitz et al. 1999) and inhibits primary tumor growth as well as angiogenesis-dependent growth of metastases (Dell’Eva et al. 2002).
Endostatin
Endostatin is an angiostatic 20 kDa internal type XVIII collagen fragment released by proteolytic activity (Wenzel et al. 2006). The hinge region of endostatin contains several proteolytic cleavage sites where matrix metalloproteinases (MMPs), cathepsins, and elastases induce its release and consequently the interaction with cell membrane receptors, including α5β1, αvβ3, and αvβ5 integrin receptors, on endothelial cells (Zatterstrom et al. 2000). Endostatin inhibits the mitogen-activated protein kinase pathway in endothelial cells, leading to the inhibition of angiogenesis (Wickstrom et al. 2005). Endostatin affects VEGF to VEGFR-2 binding and tyrosine phosphorylation (Jia et al. 2004) and inhibits the activities of matrix metalloproteinases-2, -9, and -13 (MMP-2, MMP-9, and MMP-13) (Kim et al. 2000).
Platelet Factor 4 (PF4)
PF4 is the most abundant chemokine member of the C-X-C family found in platelets and megakaryocytes. It exhibits antiangiogenic effects both in vivo and in vitro and directly interacts with VEGF-A165 (Hang et al. 2013; Maurer et al. 2006).
Thrombospondin-1 (TSP-1)
TSP-1 belongs to a family of extracellular matrix (ECM) glycoproteins. TSP-1, initially discovered in platelet granules, is also produced by endothelial cells, monocytes/macrophages, and smooth muscle cells. TSP-1 interacts with numerous ECM proteins, modulates extracellular protease levels, and activates transforming growth factor beta (TGF-β) (Lawler 2002). TSP-1 inhibits angiogenesis by inhibiting the growth, sprouting, and motility of endothelial cells. High concentrations of TSP-1 have the opposite effect, promoting angiogenesis (Lawler and Lawler 2012).
14.5 TME Infiltrating Cells
Macrophages
Tumor-associated macrophages (TAMs) are one of the major tumor-infiltrating innate immune cells and play an important role in the TME because they are involved in promoting tumor growth, invasion, metastasis, and therapeutic resistance (Chanmee et al. 2014). TAMs are described in two different polarization states: M1 CD68-positive and M2, CD-163 and CD-206-positive (Medbury et al. 2013). It is generally believed that M1 macrophages are involved in proinflammatory processes by migrating to inflamed tissues and targeting pathogens directly or activating cells of the adaptive immune system. It has been demonstrated that the M1 subpopulation has antitumor function because of its ability to kill tumor cells and recruit cytotoxic T lymphocytes to activate adaptive immune responses (Chanmee et al. 2014). The M2 subpopulation, on the other hand, has the functions of debris removal, angiogenesis stimulation, and tissue reconstruction and promotes tumorigenesis. They induce immune tolerance and attract T regulatory cells and Th2 T cells. It is believed that M2 TAMs have protumor activity because they stimulate angiogenesis and tumor growth (Jayasingam et al. 2019). Usually, TAM recruitment is correlated with the induction of angiogenic switching and is associated with a poor prognosis in most cancer types. Many cytokines and chemokines are secreted by vascular and perivascular cells, stromal cells, and cancer cells that recruit TAMs in the TME and include C-C motif ligand 2 (CCL2), CCL5, CCL7, Ang-2, colony-stimulating factor-1 (CSF1), VEGF, interleukin-33 (IL-33), semaphorin 3D (Sema 3D), endothelial monocyte-activating polypeptide-II (EMAP-II), endothelin (ET)-1 and 2, stromal cell-derived factor 1α (SDF1α/CXCL12), eotaxin, and oncostatin (Wang et al. 2019a).
TAMs can transdifferentiate into vessel-like structures by vasculogenic mimicry. In gliomas, the areas where vascular mimicry is found are characterized by high TAM infiltration and correlated with M2 density (Rong et al. 2016). The angiogenic factors secreted by TAMs include EGF-A, TGF-β, FGF-2, CCL18, Sema4D, adrenomedullin (ADM), and PlGF (Riabov et al. 2014). TAMs express the MCT1-lactate transporter. Furthermore, TAMs express VEGF-A when exposed to hypoxia or in the presence of lactate produced by tumor cells following aerobic or anaerobic glycolysis (Zhang et al. 2020). This effect is mediated by HIF1α, and lactate seems to lead to M2-like polarization of TAMs (Colegio et al. 2014). TAMs have been found to localize frequently in avascular and hypoxic areas of invasive carcinoma of the breast, where the expression of VEGF-A is upregulated (Lewis and Pollard 2006). Fra-1 and the IL-6/JAK/Stat3 signaling pathway in TAMs are involved in the secretion of proangiogenic factors (Choi et al. 2018). TAMs produce CCL18, which stimulates angiogenesis in synergy with VEGF-A (Lin et al. 2015). ADM is a potent vasodilator belonging to the calcitonin superfamily whose secretion by macrophages is upregulated by inflammatory factors and hypoxia. In melanoma, TAM-derived ADM induces angiogenesis in a paracrine manner via the endothelial nitric oxide synthase (eNOS) signaling pathway (Chen et al. 2011). MMP-9, which is highly expressed by M2 macrophages, triggers the angiogenic switch during carcinogenesis by the release of VEGF-A from the ECM in colorectal cancer (Deryugina and Quigley 2015; Yahaya et al. 2019).
Mast Cells (MCs)
MCs are involved in a large spectrum of biological processes, ranging from inflammation and immune modulation to angiogenesis, tissue repair, remodeling, and cancer (Welker et al. 2000). MC precursors complete their differentiation and maturation in target tissues under the control of local growth factors, including IL-9, IL-10, IL-3, IL-4, IL-33, CXCL12, nerve growth factor (NGF), and TGF-β (Hu et al. 2007). MCs are traditionally classified based on the production of tryptase and chymase, and resident MCs of various organs are characterized by the expression and release of peculiar factors related to their tissue-specific functions (Krystel-Whittemore et al. 2015). MCs can be recruited to the tumor microenvironment by tumor cell-released chemoattractants, including stem cell factor (SCF) or CCL-15 (Yu et al. 2018). In the TME, MCs release proangiogenic factors such as FGF2, VEGFA, tumor necrosis factor alpha (TNFα), and CXCL8 (Norrby 2002). Furthermore, they produce MMPs and chymase, and tryptase activates pro-MMPs (Kanbe et al. 1999; Johnson et al. 1998). The localization of MCs in the TME is determined by interactions of CCR2, CXCR2, and CXCR3 with their respective ligands CCL2, CXCL1, and CXCL10. In this way, MCs facilitate tumor angiogenesis and promote tumor invasiveness (Ramirez-Moreno et al. 2020; Komi and Redegeld 2020). On the other hand, numerous cytokines released by MCs contribute to inflammation, inhibiting tumor cell growth and inducing tumor cell apoptosis (Ribatti and Crivellato 2012). MC tryptase activates the Ang-1 pathway and induces endothelial cell proliferation in pancreatic cancer (Guo et al. 2016). MC inactivation delayed the angiogenic switch and malignant progression in early preneoplastic lesion experimental squamous epithelial, intestinal, and pancreatic islet cancer models (Maciel et al. 2015).
Neutrophils
Neutrophils release large amounts of soluble factors, including cytokines and chemokines, through which they recruit and activate other immune cells (Malech et al. 2014). Moreover, they are involved in chronic inflammation regulation and in various steps of tumor progression and angiogenesis, exerting both pro-(tumor-associated neutrophil, TAN-N2) and antitumor (TAN-N1) roles. Normal density neutrophils (NDNs) have been associated with cytotoxic antitumor activities, while immature low-density neutrophils (LDNs) exert immunosuppressive protumor activities (Cerecedo et al. 2021). The TME is infiltrated with CD66b+ neutrophils, and their number is correlated with poor clinical outcome (Carus et al. 2013). TGFβ reduces endothelial adhesiveness of neutrophils and neutrophil transmigration through the endothelium as well as the number of antitumor neutrophils in the TME (Granot 2019). In a Nod Scid mouse model of human prostate cancer, TANs are the major source of MMP-9 (Li et al. 2020a). In gliomas, the high TME infiltration of neutrophils was correlated with the tumor grade as well as resistance to anti-VEGF therapy (Liang et al. 2014). Neutrophils produce low amounts of tissue inhibitors of metalloproteinases-1 (TIMP-1), thus enhancing the angiogenic effect of MMP-9 (Wang et al. 2019b). In a RIP-Tag murine model, granulocyte-CSF (G-CSF) stimulates neutrophils to release the proangiogenic molecule Bv8, which is critical for VEGF-independent tumor angiogenesis (Bjornmalm et al. 2017). Resistance to anti-VEGF therapy in tumors has been correlated with the infiltration of neutrophils and associated with Bv8 neutrophil expression (Shojaei et al. 2008). On the other hand, neutrophils are also involved in the inhibition of angiogenesis through the release of antiangiogenic factors, such as affecting neutrophil migration toward CXCL1 and CXCL8 (Jeronimo et al. 2017).
Lymphocytes
The role of lymphocytes in tumor progression and angiogenesis remains to be further explored, and conflicting data about their function in the TME are emerging (Paijens et al. 2021). B cells are often present in the TME, and it is believed that they may contribute to tumor angiogenesis via STAT3 activation. STAT3 activation in cancer promotes tumor cell survival and proliferation, and a positive correlation has been established between its expression and VEGF release (Yang et al. 2013). It is thought that although only a subset of B cells infiltrating the tumor express STAT3, this might be enough to potentiate and maintain persistent STAT3 activation. Transplantation of STAT3-expressing B cells in tumor mouse models increased tumor growth and angiogenesis through the production of VEGF (Wang et al. 2019c). Another way by which B cells contribute to tumor angiogenesis is the antibody-mediated activation of Fcγ receptors on TAMs. This mechanism induces the secretion of IL-1, leading to the recruitment of myofibroblasts and promotion of tumor angiogenesis (Voronov et al. 2014). Tumor-infiltrating T cells play an important role in the antitumor response by the production of many cytokines, such as TNF-α, interferon gamma (IFN-γ), IL-2, IL-17, IL-22, and IL-36. TAMs inhibit CD8+ T-cell infiltration and antitumor function (de Ruiter et al. 2017; Lan et al. 2021). Regulatory T (Treg) cells are immunosuppressive cells that affect the specialization and function of antigen-presenting cells (APCs), decrease their interactions with T cells, and subsequently inhibit effector T-cell function (Maimela et al. 2019). In addition, Tregs suppress natural killer (NK) cell activities (Li et al. 2020b). Cytotoxic T cells in the TME release IL-2, IL-12, and IFN-γ, improving the cytotoxic functions of CD8+ T cells through the production of TNF-related apoptosis-inducing ligands (TRAILs), ROS, and perforin (Grossman et al. 2004). Tumor cells express coinhibitory receptors such as programmed death ligand-1 (PD-L1) and CD80 that interact with the inhibitory molecules programmed death-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) expressed by CD8+ T cells. These interactions may inhibit CD8+ T-cell activation and function (Cai et al. 2019). CD4+ and CD8+ T cells produce FGF-2 and heparin-binding epidermal-like growth factor (HB-EGF), which are both proangiogenic factors (Blotnick et al. 1994). On the other hand, T cells are also involved in the antiangiogenic response through TNFα, TGFβ, and IFNs. IFNs induce the expression of CXCL-9, CXCL-10, and CXCL-11 with angiostatic activities that can directly bind CXCR3 on endothelial cells (Blotnick et al. 1994; Beatty and Paterson 2001). NK cells are able to control tumor growth through their cytotoxic activity (Wu and Lanier 2003). Intratumor NK cells display phenotypic and/or functional alterations compared with peripheral NK cells depending on the influence of local factors and/or the interaction with other cell types of the TME (Larsen et al. 2014). The presence of TGF-β inhibits CD16, perforins, granzymes, and IFN-γ secretion, reverting NK cells to a proangiogenic phenotype characterized by the secretion of VEGF. Furthermore, the interaction between the immunoregulatory class I MHC molecule HLA-G and the KIR2DL4, ILT-4, and ILT-2 inhibitory NK cell receptors induces NK cells to acquire proangiogenic activities. Prostaglandin E2 (PGE2) is believed to contribute to the NK cell angiogenic switch (Bassani et al. 2019). Tumor-infiltrating NK cells express high levels of CD56, but low levels or none of CD16 produce several factors, such as VEGF, angiogenin, Ang-1, PIGF, CXCL8, and MMPs, which stimulate endothelial cell growth and angiogenesis (Bruno et al. 2018).
Cancer-Associated Fibroblasts (CAFs)
CAFs are able to interact with tumor cells and form a myofibroblastic microenvironment that supports tumor progression and angiogenesis via secretion of various growth factors, cytokines, chemokines, and the degradation of ECM (Liu et al. 2019). A significant percentage of CAFs may share endothelial markers such as PECAM/CD31, and this allows us to suppose that they originate from an endothelial subpopulation through endothelial-to-mesenchymal transition (Potenta et al. 2008). Regarding their influence on angiogenesis, several studies have shown that their secretome is rich in several cytokines with proangiogenic effects, including VEGF, CXCL-8, and FGFs (Linares et al. 2020). Furthermore, CAF release is able to form capillary-like structures through vasculogenic mimicry by TGF-β and SDF-1 paracrine action (Yang et al. 2016a). Moreover, SDF-1 recruits endothelial precursor cells (EPCs), which may transdifferentiate into endothelial cells and stimulate the formation of novel vasculature at the tumor-host cell interface (Orimo et al. 2005). CAFs express podoplanin, which promotes angiogenesis in breast cancer via upregulation of VEGF-C rather than VEGF-A or VEGF-D (Kubouchi et al. 2018). The galectin family of glycan-binding proteins displays important functions in cancer development and progression. In gastric cancer, CAF expression of Galectin-1 is upregulated, leading to enhanced VEGF expression. Under hypoxic conditions, G-protein-coupled estrogen receptor (GPER) downregulation in CAFs reduces VEGF expression (Ham et al. 2019). In human pancreatic adenocarcinoma, VEGF expression by CAFs may be regulated by fibroblast activation protein α (FAP α), which is involved in affecting the balance of pro- and anti-angiogenic mediators (Higashino et al. 2019).
14.6 TME Inflammatory Cells and Angiogenesis. Our Experience in the Study of Human Lymphomas
We studied the inflammatory cell infiltrate and its role in tumor angiogenesis in diffuse large B-cell lymphoma (DLBCL) by comparing activated B-cell-like (ABC) patients to germinal center B-cell-like (GCB) patients. We demonstrated that increased ABC expression of STAT3 was correlated with poor prognosis in DLBCL and was associated with higher M2 TAM (Fig. 14.1a, b) and CD8+ (Fig. 14.1c, d) cell infiltration into the TME, which, in turn, induced a strong angiogenic response in the ABC group (Tamma et al. 2020). Moreover, tumor vessels appeared lined by endothelial cells expressing both FVIII and STAT3 (Tamma et al. 2019b). Regarding the morphological distribution of the different TME cells in DLBCL, we established that cell patterns generated by CD4+, CD8+, CD68+, CD163+, and tryptase+ mast cell profiles have a higher uniformity index in the ABC, indicating a tendency of the cells to assume a more uniform distribution in the tissues in this more aggressive DLBCL subtype (Guidolin et al. 2021). Recently, Laddaga and coworkers suggested that the number of tumor infiltrating lymphocytes in the DLBCL TME is connected to a pre-existing antitumor immune response and then to an improved therapy response (Laddaga et al. 2021).

Immunohistochemical staining of CD163+ macrophages in ABC (a) and GCB (b) DLBCL samples; CD8+ T cells in ABC (c) and GCB (d) DLBCL samples; CD68+ macrophages in MALT lymphoma (e) and control (f) samples. Scale bar 60 mm
In a further study, we demonstrated that mucosa-associated lymphoid tissue (MALT)-type lymphoma and the tumor inflammatory TME included a high number of CD3+, CD4+ and CD8+ lymphocytes, CD68+ (Fig. 14.1e, f), CD163+ macrophages, and tryptase+ mast cells. Interestingly, CD8+ cell content positively correlated with both CD34+ vessels, remarking on the important role of these cells in tumor angiogenesis and with CD163+ TAMs. Moreover, tryptase+ mast cells correlated with CD4+ lymphocytes (Tamma et al. 2021).
14.7 Targeting Angiogenesis and Inflammatory Cells in TME
Chemotherapy associated with surgery and/or radiotherapy is the principal cancer therapy worldwide (Bjornmalm et al. 2017). The TME has been gradually recognized as a crucial contributor to cancer progression and drug resistance (Heinrich et al. 2012), so the study of the components of the TME was deepened to identify new therapeutic targets.
Targeting Angiogenesis
Bevacizumab was the first anti-VEGF antibody Food and Drug Administration (FDA) approved and actually used in different cancers, including metastatic colorectal cancer, lung cancer, kidney cancer, glioblastoma metastasis, and HER2-negative breast cancer, with response rates and durations highly variable (Jang et al. 2017). The addition of bevacizumab to chemotherapy has shown improvements in progression-free and overall survival with respect to chemotherapy alone (Jang et al. 2017; Yang et al. 2017). Another strategy consists of the inhibition of VEGF binding to its receptors by soluble decoy receptors (Holash et al. 2002). Aflibercept is a recombinant fusion protein containing portions of human VEGFR-1 and VEGFR-2 extracellular domains fused to the Fc portion of human immunoglobulin G1 able to bind with high-affinity VEGF and PlGF, inhibiting the activation of cognate VEGFRs (Holash et al. 2002). Experimental data about the use of aflibercept in cancer xenograft models demonstrated greater antitumor activity than bevacizumab (Chiron et al. 2014). Ramucirumab is a monoclonal anti-VEGFR-2 antibody used as monotherapy or in combination with paclitaxel for the treatment of advanced gastric or gastroesophageal junction adenocarcinoma, metastatic non-small cell lung cancer (NSCLC), and colorectal cancer (Singh and Parmar 2015; Aprile et al. 2014). Tyrosine kinase inhibitors (TKIs) are used for the inhibition of VEGFRs, PDGF-A and PDGF-BRs, and c-Kit (Hamberg et al. 2010; Wang et al. 2016). Among TKIs, pazopanib is commonly used for the treatment of advanced renal cell carcinoma and soft tissue sarcoma (Hamberg et al. 2010; Nakano et al. 2019) and sunitinib is used in metastatic renal cell carcinoma (Roma-Rodrigues et al. 2019). Sunitinib has more benefits than sorafenib as a first-line therapy, although sunitinib has higher toxicity than sorafenib (Deng et al. 2019). M-TOR inhibitors decrease endothelial cell proliferation through the mTOR/AP-1/VEGF pathway, among which everolimus (Wang et al. 2016). Patients treated with antiangiogenic agents have a reduced response to therapies for the acquisition of drug resistance. Two mechanisms of this resistance are the activation of alternative signaling pathways and the upregulation of alternative angiogenic factors and cytokines. Deepening these pathways would allow us to elaborate new treatments and the development of combination regimens with more durable clinical benefits (Philips and Atkins 2014). Anti-VEGF treatment in pancreatic cancer induces increased expression of FGF-1 and -2 and Ang-1 (Zhuang et al. 2010). In patients affected by colorectal cancer treated with bevacizumab, high levels of Ang-2 were detectable (Goede et al. 2010). In glioblastoma multiforme, anti-VEGFR therapy leads to increased levels of FGF-2 and SDF-1. Similar results have been found in lung cancer models resistant to angiogenesis inhibitors in which epidermal growth factor receptors (EGFRs) and FGFRs are overexpressed (Cascone et al. 2011). In colorectal and renal cancer patients treated with TKIs, increased levels of PIGF and VEGF were detectable (Motzer and Bukowski 2006). Vanucizumab, a bispecific anti-Ang-2/anti-VEGF-A antibody, revealed an acceptable safety profile and promising antitumor activity (Hidalgo et al. 2018). FGFR inhibitors restore the sensitivity to bevacizumab in tumor mouse models (Gyanchandani et al. 2013), but further research failed to determine the relevance of this association (Norden et al. 2015; Semrad et al. 2017). The VEGFR, FGFR, and PDGFR multiple receptor TKI lenvatinib showed promising effects in several tumors and should be considered for counteracting resistance to antiangiogenic agents (Suyama and Iwase 2018).
Anti-angiogenic therapies induce the production of cytokines, such as SDF1, IL-8, and G-CSF, involved in the recruitment of bone marrow-derived cells (BMDCs), which contributes negatively to the anti-angiogenic effect (Montemagno and Pages 2020). An increase in CD11b+ Gr1+ myeloid-derived suppressor cells (MDSCs) has been observed in tumors not sensitive to anti-VEGF-A treatment (Shojaei et al. 2007). Th-17 cells induce the expression of G-CSF by CAFs and consequently the recruitment of MDSCs (Shojaei et al. 2009). Hypoxia has been related to sunitinib resistance in glioblastoma and breast and metastatic renal cell carcinoma as a consequence of the increased recruitment of MDSCs to the tumor niche (Piao et al. 2012).
Vessel co-option is believed to be correlated with refractoriness to anti-VEGF drug treatment of colorectal cancer liver metastases (Frentzas et al. 2016) and has been observed following anti-VEGFR-2 inhibition in cerebral melanoma metastases (Frentzas et al. 2016). Moreover, vessel co-option has been evidenced in human breast cancer liver metastases, NSCLC, and lung metastases (Kuczynski et al. 2016). The blockade of both VEGF-A and ARP2/3, VEGFA and c-MET or VEGF-A and ZEB2 suppresses vessel co-option and tumor invasion (Sennino et al. 2012; Depner et al. 2016). Vasculogenic mimicry is deeply associated with poor patient survival (Sun et al. 2004). In ovarian cancer models, bevacizumab may induce the progression of metastatic disease, which would correlate with a hypoxic response and vasculogenic mimicry (Xu et al. 2012). Studies on the TME in everolimus-resistant renal carcinoma demonstrated that the antiangiogenic drug stimulates vasculogenic mimicry by differentiating tumor cells into endothelial-like cells (Serova et al. 2016). Moreover, everolimus induces triple-negative breast cancer invasion via vasculogenic mimicry; thus, its evaluation could be helpful in predicting the efficacy of antiangiogenic therapy in these patients (Sun et al. 2017).
Targeting TAMs
Targeting TAM-recruiting mediators, which include chemokines, complement components, CSF-1, and VEGF, is being studied (Liu et al. 2020). It has been reported that the inhibition of CSF1R in glioblastoma and cervical and breast cancer murine models induces a dramatic reduction in tumor volume and survival of mice (Pyonteck et al. 2013). This inhibition seems to reprogram TAMs by GM-CSF to induce their repolarization to an antitumoral state (Quail and Joyce 2013; DeNardo et al. 2011). The monoclonal antibody RG7155 in human patients led to a remarkable reduction in CSF-1R+ CD163+ macrophages in diffuse-type giant cell tumor patients (Ackermann et al. 2013). TAM reduction improves antiangiogenic treatments. Treatment with vascular-disrupting agents such as combretastatin-A4-phosphate has been reported to markedly increase its efficacy when TIE2+ TAM recruitment is blocked (Welford et al. 2011). The reduction in TAMs augmented the effects of sorafenib (Zhang et al. 2010). In addition, TAMs improved the antiangiogenic and antitumor effects of VEGF/VEGFR2 antibodies in subcutaneous tumor models (Priceman et al. 2010). TAMs limit the cytotoxic activity of CD8+ cytotoxic T cells during tumor progression, mainly in the M2 polarization state. Inhibiting TAM recruitment or blocking TAM polarization to the M2 phenotype may enhance T-cell-mediated antitumor responses and improve the efficacy of immunotherapies (Coussens et al. 2013). Moreover, some immunotherapies may also depend on the reprogramming of TAMs toward an M1 phenotype. One method used to reprogram TAMs is histidine-rich glycoprotein (HRG) treatment, which induces macrophage downregulation of PlGF and stimulates the normalization of blood vessels and the efficiency of chemotherapy in mouse tumor models (Rolny et al. 2011). Other strategies include the suppression of nuclear factor-kB signaling (Hagemann et al. 2008) or exposure to anti-IL-10R antibodies combined with the TLR9 ligand CpG (Guiducci et al. 2005).
Targeting TANs
Inhibition of the protumor functions of TANs (Hsu et al. 2020) may be combined with conventional or new anticancer therapies to improve the antitumor effects (Khan et al. 2020). CXCR2 inhibitors are also used in combination with other therapies in clinical evaluation in patients with different tumors (Li et al. 2019; Timaxian et al. 2021; Groth et al. 2021; Cabrero-de Las Heras and Martinez-Balibrea 2018). The neutrophil-derived enzyme elastase promotes tumor growth and invasiveness. The elastase inhibitor ONO-5046 reduced tumor growth in NSCLC (Houghton et al. 2010). Another approach has been to reprogram neutrophil function in the TME through the inhibition of TGFβ (Qin et al. 2020). The inhibition of angiotensin-converting enzyme and the angiotensin II type 1 receptor nicotinamide phosphoribosyltransferase (NAMPT) or CXCR4 is another approach to reprogram neutrophils to an antitumor state (Shrestha et al. 2016; Yang et al. 2018b).
Targeting CAFs
The protein FAP is considered a candidate for targeting CAFs because it is expressed in tumors but not in healthy tissues and is considered a predictor of poor survival (Liao et al. 2013). Nevertheless, both sibrotuzumab, an antibody targeting FAP, and inhibitors of FAP activity induced lower survival rates (Liu et al. 2019; Yang et al. 2016b). An IL-2 variant targeting FAP, RO6874281, is under investigation (Joshi 2020; Koustoulidou et al. 2021).
14.8 Concluding Remarks
Cytokines and chemokines secreted by cells of the TME are involved in the regulation of tumor angiogenesis based on the balance of pro- and antiangiogenic factors. Deepening the mechanisms underlying the crosstalk between the TME and tumor cells has allowed the discovery of numerous molecular-targeted drugs that control diverse elements of the TME. Different approaches varying from traditional and emerging inhibitors of angiogenic cytokines and their receptors to the modulation of TME cell activities and novel immune checkpoint inhibitors proved to be promising in tumor progression. Despite the promising results of these new therapeutic approaches, their efficacy is often limited by evasion, and resistance mechanisms have emerged. Overcoming resistance to antitumor therapies is a great challenge but might lead to the improvement of the clinical outcome of patients and, for this reason, currently constitutes a major focus of research.
References
Ackermann M, Tsuda A, Secomb TW, Mentzer SJ, Konerding MA (2013) Intussusceptive remodeling of vascular branch angles in chemically-induced murine colitis. Microvasc Res 87:75–82
Akwii RG, Sajib MS, Zahra FT, Mikelis CM (2019) Role of angiopoietin-2 in vascular physiology and pathophysiology. Cell 8(5):471
Aprile G, Rijavec E, Fontanella C, Rihawi K, Grossi F (2014) Ramucirumab: preclinical research and clinical development. Onco Targets Ther 7:1997–2006
Autiero M, Luttun A, Tjwa M, Carmeliet P (2003) Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J Thromb Haemost 1(7):1356–1370
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19(5):329–337
Bassani B, Baci D, Gallazzi M, Poggi A, Bruno A, Mortara L (2019) Natural killer cells as key players of tumor progression and angiogenesis: old and novel tools to divert their pro-tumor activities into potent anti-tumor effects. Cancers (Basel) 11(4):461
Beatty G, Paterson Y (2001) IFN-gamma-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-gamma. J Immunol 166(4):2276–2282
Berra E, Roux D, Richard DE, Pouyssegur J (2001) Hypoxia-inducible factor-1 alpha (HIF-1 alpha) escapes O(2)-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep 2(7):615–620
Bjornmalm M, Thurecht KJ, Michael M, Scott AM, Caruso F (2017) Bridging bio-Nano science and cancer nanomedicine. ACS Nano 11(10):9594–9613
Blotnick S, Peoples GE, Freeman MR, Eberlein TJ, Klagsbrun M (1994) T lymphocytes synthesize and export heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor, mitogens for vascular cells and fibroblasts: differential production and release by CD4+ and CD8+ T cells. Proc Natl Acad Sci U S A 91(8):2890–2894
Brindle NP, Saharinen P, Alitalo K (2006) Signaling and functions of angiopoietin-1 in vascular protection. Circ Res 98(8):1014–1023
Bruno A, Bassani B, D’Urso DG, Pitaku I, Cassinotti E, Pelosi G et al (2018) Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J 32(10):5365–5377
Cabrero-de Las Heras S, Martinez-Balibrea E (2018) CXC family of chemokines as prognostic or predictive biomarkers and possible drug targets in colorectal cancer. World J Gastroenterol 24(42):4738–4749
Cai J, Wang D, Zhang G, Guo X (2019) The role of PD-1/PD-L1 axis in Treg development and function: implications for cancer immunotherapy. Onco Targets Ther 12:8437–8445
Cao Y, Xue L (2004) Angiostatin. Semin Thromb Hemost 30(1):83–93
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307
Carus A, Ladekarl M, Hager H, Nedergaard BS, Donskov F (2013) Tumour-associated CD66b+ neutrophil count is an independent prognostic factor for recurrence in localised cervical cancer. Br J Cancer 108(10):2116–2122
Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB et al (2011) Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest 121(4):1313–1328
Cerecedo D, Martinez-Vieyra I, Lopez-Villegas EO, Hernandez-Cruz A, Loza-Huerta ADC (2021) Heterogeneity of neutrophils in arterial hypertension. Exp Cell Res 402(2):112577
Chanmee T, Ontong P, Konno K, Itano N (2014) Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 6(3):1670–1690
Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X et al (2011) Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res 17(23):7230–7239
Chiron M, Bagley RG, Pollard J, Mankoo PK, Henry C, Vincent L et al (2014) Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol Cancer Ther 13(6):1636–1644
Choi J, Gyamfi J, Jang H, Koo JS (2018) The role of tumor-associated macrophage in breast cancer biology. Histol Histopathol 33(2):133–145
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V et al (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513(7519):559–563
Coussens LM, Zitvogel L, Palucka AK (2013) Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339(6117):286–291
de Ruiter EJ, Ooft ML, Devriese LA, Willems SM (2017) The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: a systematic review and meta-analysis. Onco Targets Ther 6(11):e1356148
Dehghani S, Nosrati R, Yousefi M, Nezami A, Soltani F, Taghdisi SM et al (2018) Aptamer-based biosensors and nanosensors for the detection of vascular endothelial growth factor (VEGF): a review. Biosens Bioelectron 110:23–37
Dell’Eva R, Pfeffer U, Indraccolo S, Albini A, Noonan D (2002) Inhibition of tumor angiogenesis by angiostatin: from recombinant protein to gene therapy. Endothelium 9(1):3–10
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF et al (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1(1):54–67
Deng H, Liu W, He T, Hong Z, Yi F, Wei Y et al (2019) Comparative efficacy, safety, and costs of sorafenib vs. sunitinib as first-line therapy for metastatic renal cell carcinoma: a systematic review and meta-analysis. Front Oncol. 9:479
Depner C, Zum Buttel H, Bogurcu N, Cuesta AM, Aburto MR, Seidel S et al (2016) EphrinB2 repression through ZEB2 mediates tumour invasion and anti-angiogenic resistance. Nat Commun 7:12329
Deryugina EI, Quigley JP (2015) Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol 44-46:94–112
Eklund L, Kangas J, Saharinen P (2017) Angiopoietin-Tie signalling in the cardiovascular and lymphatic systems. Clin Sci (Lond) 131(1):87–103
Ferrara N, Davis-Smyth T (1997) The biology of vascular endothelial growth factor. Endocr Rev 18(1):4–25
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285(21):1182–1186
Folkman J, Merler E, Abernathy C, Williams G (1971) Isolation of a tumor factor responsible for angiogenesis. J Exp Med 133(2):275–288
Folkman J, Watson K, Ingber D, Hanahan D (1989) Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 339(6219):58–61
Fredriksson L, Li H, Eriksson U (2004) The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev 15(4):197–204
Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E et al (2016) Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med 22(11):1294–1302
Gao Z, Daquinag AC, Su F, Snyder B, Kolonin MG (2018) PDGFRalpha/PDGFRbeta signaling balance modulates progenitor cell differentiation into white and beige adipocytes. Development 145(1):dev155861
Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V et al (1998) Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273(46):30336–30343
Goede V, Coutelle O, Neuneier J, Reinacher-Schick A, Schnell R, Koslowsky TC et al (2010) Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer 103(9):1407–1414
Granot Z (2019) Neutrophils as a therapeutic target in cancer. Front Immunol 10:1710
Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ (2004) Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21(4):589–601
Groth C, Arpinati L, Shaul ME, Winkler N, Diester K, Gengenbacher N et al (2021) Blocking migration of polymorphonuclear myeloid-derived suppressor cells inhibits mouse melanoma progression. Cancers (Basel) 13(4):726
Guidolin D, Tamma R, Annese T, Tortorella C, Ingravallo G, Gaudio F et al (2021) Different spatial distribution of inflammatory cells in the tumor microenvironment of ABC and GBC subgroups of diffuse large B cell lymphoma. Clin Exp Med 21(4):573–578
Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP (2005) Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res 65(8):3437–3446
Guo X, Zhai L, Xue R, Shi J, Zeng Q, Gao C (2016) Mast cell Tryptase contributes to pancreatic cancer growth through promoting angiogenesis via activation of angiopoietin-1. Int J Mol Sci 17(6):834
Gyanchandani R, Ortega Alves MV, Myers JN, Kim S (2013) A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol Cancer Res 11(12):1585–1596
Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG et al (2008) “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 205(6):1261–1268
Ham IH, Lee D, Hur H (2019) Role of cancer-associated fibroblast in gastric cancer progression and resistance to treatments. J Oncol 2019:6270784
Hamberg P, Verweij J, Sleijfer S (2010) (Pre-)clinical pharmacology and activity of pazopanib, a novel multikinase angiogenesis inhibitor. Oncologist 15(6):539–547
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hang TC, Tedford NC, Reddy RJ, Rimchala T, Wells A, White FM et al (2013) Vascular endothelial growth factor (VEGF) and platelet (PF-4) factor 4 inputs modulate human microvascular endothelial signaling in a three-dimensional matrix migration context. Mol Cell Proteomics 12(12):3704–3718
Heinrich EL, Walser TC, Krysan K, Liclican EL, Grant JL, Rodriguez NL et al (2012) The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron 5(1):5–18
Hidalgo M, Martinez-Garcia M, Le Tourneau C, Massard C, Garralda E, Boni V et al (2018) First-in-human phase I study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin Cancer Res 24(7):1536–1545
Higashino N, Koma YI, Hosono M, Takase N, Okamoto M, Kodaira H et al (2019) Fibroblast activation protein-positive fibroblasts promote tumor progression through secretion of CCL2 and interleukin-6 in esophageal squamous cell carcinoma. Lab Investig 99(6):777–792
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M et al (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 99(17):11393–11398
Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE et al (2010) Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 16(2):219–223
Hsu BE, Shen Y, Siegel PM (2020) Neutrophils: orchestrators of the malignant phenotype. Front Immunol 11:1778
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003) Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23(24):9361–9374
Hu ZQ, Zhao WH, Shimamura T (2007) Regulation of mast cell development by inflammatory factors. Curr Med Chem 14(28):3044–3050
Huang Y, Lin D, Taniguchi CM (2017) Hypoxia inducible factor (HIF) in the tumor microenvironment: friend or foe? Sci China Life Sci 60(10):1114–1124
Jang HJ, Kim BJ, Kim JH, Kim HS (2017) The addition of bevacizumab in the first-line treatment for metastatic colorectal cancer: an updated meta-analysis of randomized trials. Oncotarget 8(42):73009–73016
Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES (2019) Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol 9:1512
Jeronimo A, Rodrigues G, Vilas-Boas F, Martins GG, Bagulho A, Real C (2017) Hydrogen peroxide regulates angiogenesis-related factors in tumor cells. Biochem Cell Biol 95(6):679–685
Ji WR, Castellino FJ, Chang Y, Deford ME, Gray H, Villarreal X et al (1998) Characterization of Kringle domains of angiostatin as antagonists of endothelial cell migration, an important process in angiogenesis. FASEB J 12(15):1731–1738
Jia YH, Dong XS, Wang XS (2004) Effects of endostatin on expression of vascular endothelial growth factor and its receptors and neovascularization in colonic carcinoma implanted in nude mice. World J Gastroenterol 10(22):3361–3364
Johnson JL, Jackson CL, Angelini GD, George SJ (1998) Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 18(11):1707–1715
Joshi S (2020) Targeting the tumor microenvironment in neuroblastoma: recent advances and future directions. Cancers (Basel) 12(8):2057
Kanbe N, Tanaka A, Kanbe M, Itakura A, Kurosawa M, Matsuda H (1999) Human mast cells produce matrix metalloproteinase 9. Eur J Immunol 29(8):2645–2649
Khan S, Mittal S, McGee K, Alfaro-Munoz KD, Majd N, Balasubramaniyan V et al (2020) Role of neutrophils and myeloid-derived suppressor cells in glioma progression and treatment resistance. Int J Mol Sci 21(6):1954
Kim YM, Jang JW, Lee OH, Yeon J, Choi EY, Kim KW et al (2000) Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res 60(19):5410–5413
Komi DEA, Redegeld FA (2020) Role of mast cells in shaping the tumor microenvironment. Clin Rev Allergy Immunol 58(3):313–325
Koustoulidou S, Hoorens MWH, Dalm SU, Mahajan S, Debets R, Seimbille Y et al (2021) Cancer-associated fibroblasts as players in cancer development and progression and their role in targeted radionuclide imaging and therapy. Cancers (Basel) 13(5):1100
Krystel-Whittemore M, Dileepan KN, Wood JG (2015) Mast cell: a multi-functional master cell. Front Immunol 6:620
Kubouchi Y, Yurugi Y, Wakahara M, Sakabe T, Haruki T, Nosaka K et al (2018) Podoplanin expression in cancer-associated fibroblasts predicts unfavourable prognosis in patients with pathological stage IA lung adenocarcinoma. Histopathology 72(3):490–499
Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S et al (2016) Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J Natl Cancer Inst 108(8):djw030
Laddaga FE, Ingravallo G, Mestice A, Tamma R, Perrone T, Maiorano E et al (2021) Correlation between circulating blood and microenvironment T lymphocytes in diffuse large B-cell lymphomas. J Clin Pathol. https://doi.org/10.1136/jclinpath-2020-207048
Lan HR, Du WL, Liu Y, Mao CS, Jin KT, Yang X (2021) Role of immune regulatory cells in breast cancer: foe or friend? Int Immunopharmacol 96:107627
Larsen SK, Gao Y, Basse PH (2014) NK cells in the tumor microenvironment. Crit Rev Oncog 19(1–2):91–105
Lawler J (2002) Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med 6(1):1–12
Lawler PR, Lawler J (2012) Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med 2(5):a006627
Lee HJ, Cho CH, Hwang SJ, Choi HH, Kim KT, Ahn SY et al (2004) Biological characterization of angiopoietin-3 and angiopoietin-4. FASEB J 18(11):1200–1208
Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res 66(2):605–612
Li Y, He Y, Butler W, Xu L, Chang Y, Lei K et al (2019) Targeting cellular heterogeneity with CXCR2 blockade for the treatment of therapy-resistant prostate cancer. Sci Transl Med 11(521):eaax0428
Li P, Lu M, Shi J, Hua L, Gong Z, Li Q et al (2020a) Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat Commun 11(1):4387
Li C, Jiang P, Wei S, Xu X, Wang J (2020b) Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer 19(1):116
Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N et al (2014) Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res 20(1):187–198
Liao Y, Ni Y, He R, Liu W, Du J (2013) Clinical implications of fibroblast activation protein-alpha in non-small cell lung cancer after curative resection: a new predictor for prognosis. J Cancer Res Clin Oncol 139(9):1523–1528
Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA et al (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66(23):11238–11246
Lin L, Chen YS, Yao YD, Chen JQ, Chen JN, Huang SY et al (2015) CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 6(33):34758–34773
Linares J, Marin-Jimenez JA, Badia-Ramentol J, Calon A (2020) Determinants and functions of CAFs secretome during cancer progression and therapy. Front Cell Dev Biol 8:621070
Liu T, Zhou L, Li D, Andl T, Zhang Y (2019) Cancer-associated fibroblasts build and secure the tumor microenvironment. Front Cell Dev Biol 7:60
Liu H, Yang Z, Lu W, Chen Z, Chen L, Han S et al (2020) Chemokines and chemokine receptors: a new strategy for breast cancer therapy. Cancer Med 9(11):3786–3799
Logue OC, McGowan JW, George EM, Bidwell GL 3rd. (2016) Therapeutic angiogenesis by vascular endothelial growth factor supplementation for treatment of renal disease. Curr Opin Nephrol Hypertens 25(5):404–409
Maciel TT, Moura IC, Hermine O (2015) The role of mast cells in cancers. F1000Prime Rep 7:09
Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG (1991) Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci U S A 88(20):9267–9271
Maimela NR, Liu S, Zhang Y (2019) Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J 17:1–13
Malech HL, Deleo FR, Quinn MT (2014) The role of neutrophils in the immune system: an overview. Methods Mol Biol 1124:3–10
Mathew G, Hannan A, Hertzler-Schaefer K, Wang F, Feng GS, Zhong J et al (2016) Targeting of Ras-mediated FGF signaling suppresses Pten-deficient skin tumor. Proc Natl Acad Sci U S A 113(46):13156–13161
Maurer AM, Zhou B, Han ZC (2006) Roles of platelet factor 4 in hematopoiesis and angiogenesis. Growth Factors 24(4):242–252
Medbury HJ, James V, Ngo J, Hitos K, Wang Y, Harris DC et al (2013) Differing association of macrophage subsets with atherosclerotic plaque stability. Int Angiol 32(1):74–84
Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M et al (2018) Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Romanian J Morphol Embryol 59(2):455–467
Montemagno C, Pages G (2020) Resistance to anti-angiogenic therapies: a mechanism depending on the time of exposure to the drugs. Front Cell Dev Biol 8:584
Motomura K, Hagiwara A, Komi-Kuramochi A, Hanyu Y, Honda E, Suzuki M et al (2008) An FGF1:FGF2 chimeric growth factor exhibits universal FGF receptor specificity, enhanced stability and augmented activity useful for epithelial proliferation and radioprotection. Biochim Biophys Acta 1780(12):1432–1440
Motzer RJ, Bukowski RM (2006) Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol 24(35):5601–5608
Nakano K, Funauchi Y, Hayakawa K, Tanizawa T, Ae K, Matsumoto S et al (2019) Relative dose intensity of induction-phase pazopanib treatment of soft tissue sarcoma: its relationship with prognoses of pazopanib responders. J Clin Med 8(1):60
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M (2006) Angiogenesis in cancer. Vasc Health Risk Manag 2(3):213–219
Norden AD, Schiff D, Ahluwalia MS, Lesser GJ, Nayak L, Lee EQ et al (2015) Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J Neuro-Oncol 121(2):297–302
Norrby K (2002) Mast cells and angiogenesis. APMIS 110(5):355–371
Olson LE, Soriano P (2011) PDGFRbeta signaling regulates mural cell plasticity and inhibits fat development. Dev Cell 20(6):815–826
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R et al (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121(3):335–348
Paget S (1989) The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 8(2):98–101
Paijens ST, Vledder A, de Bruyn M, Nijman HW (2021) Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol 18(4):842–859
Palazon A, Goldrath AW, Nizet V, Johnson RS (2014) HIF transcription factors, inflammation, and immunity. Immunity 41(4):518–528
Patan S (1998) TIE1 and TIE2 receptor tyrosine kinases inversely regulate embryonic angiogenesis by the mechanism of intussusceptive microvascular growth. Microvasc Res 56(1):1–21
Philips GK, Atkins MB (2014) New agents and new targets for renal cell carcinoma. Am Soc Clin Oncol Educ Book:e222–e227
Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV et al (2012) Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 14(11):1379–1392
Potenta S, Zeisberg E, Kalluri R (2008) The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 99(9):1375–1379
Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA (2000) Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A 97(5):2202–2207
Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL et al (2010) Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood 115(7):1461–1471
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF et al (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19(10):1264–1272
Qin F, Liu X, Chen J, Huang S, Wei W, Zou Y et al (2020) Anti-TGF-beta attenuates tumor growth via polarization of tumor associated neutrophils towards an anti-tumor phenotype in colorectal cancer. J Cancer 11(9):2580–2592
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437
Ramirez-Moreno IG, Ibarra-Sanchez A, Castillo-Arellano JI, Blank U, Gonzalez-Espinosa C (2020) Mast cells localize in hypoxic zones of tumors and secrete CCL-2 under hypoxia through activation of L-type calcium channels. J Immunol 204(4):1056–1068
Rauniyar K, Jha SK, Jeltsch M (2018) Biology of vascular endothelial growth factor C in the morphogenesis of lymphatic vessels. Front Bioeng Biotechnol 6:7
Redlitz A, Daum G, Sage EH (1999) Angiostatin diminishes activation of the mitogen-activated protein kinases ERK-1 and ERK-2 in human dermal microvascular endothelial cells. J Vasc Res 36(1):28–34
Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J (2014) Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol 5:75
Ribatti D, Crivellato E (2012) Mast cells, angiogenesis, and tumour growth. Biochim Biophys Acta 1822(1):2–8
Ribatti D, Nico B, Crivellato E, Roccaro AM, Vacca A (2007) The history of the angiogenic switch concept. Leukemia 21(1):44–52
Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C et al (2011) HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 19(1):31–44
Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR (2019) Targeting tumor microenvironment for cancer therapy. Int J Mol Sci 20(4):840
Rong X, Huang B, Qiu S, Li X, He L, Peng Y (2016) Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 7(51):83976–83986
Semrad TJ, Kim EJ, Tanaka MS, Sands J, Roberts C, Burich RA et al (2017) Phase II study of Dovitinib in patients progressing on anti-vascular endothelial growth factor therapy. Cancer Treat Res Commun 10:21–26
Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V et al (2012) Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov 2(3):270–287
Serova M, Tijeras-Raballand A, Dos Santos C, Martinet M, Neuzillet C, Lopez A et al (2016) Everolimus affects vasculogenic mimicry in renal carcinoma resistant to sunitinib. Oncotarget 7(25):38467–38486
Shi YH, Fang WG (2004) Hypoxia-inducible factor-1 in tumour angiogenesis. World J Gastroenterol 10(8):1082–1087
Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S et al (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol 25(8):911–920
Shojaei F, Singh M, Thompson JD, Ferrara N (2008) Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci U S A 105(7):2640–2645
Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M et al (2009) G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A 106(16):6742–6747
Shrestha S, Noh JM, Kim SY, Ham HY, Kim YJ, Yun YJ et al (2016) Angiotensin converting enzyme inhibitors and angiotensin II receptor antagonist attenuate tumor growth via polarization of neutrophils toward an antitumor phenotype. Onco Targets Ther 5(1):e1067744
Singh AD, Parmar S (2015) Ramucirumab (Cyramza): a breakthrough treatment for gastric cancer. P T 40(7):430–468
Stacker SA, Achen MG (2018) Emerging roles for VEGF-D in human disease. Biomol Ther 8(1):1
Storkebaum E, Carmeliet P (2004) VEGF: a critical player in neurodegeneration. J Clin Invest 113(1):14–18
Stuttfeld E, Ballmer-Hofer K (2009) Structure and function of VEGF receptors. IUBMB Life 61(9):915–922
Sun B, Zhang S, Zhao X, Zhang W, Hao X (2004) Vasculogenic mimicry is associated with poor survival in patients with mesothelial sarcomas and alveolar rhabdomyosarcomas. Int J Oncol 25(6):1609–1614
Sun H, Zhang D, Yao Z, Lin X, Liu J, Gu Q et al (2017) Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol Ther 18(4):205–213
Suyama K, Iwase H (2018) Lenvatinib: a promising molecular targeted agent for multiple cancers. Cancer Control 25(1):1073274818789361
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ et al (2000) The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 157(2):411–421
Tamma R, Annese T, Ruggieri S, Brunetti O, Longo V, Cascardi E et al (2019a) Inflammatory cells infiltrate and angiogenesis in locally advanced and metastatic cholangiocarcinoma. Eur J Clin Investig 49(5):e13087
Tamma R, Ingravallo G, Albano F, Gaudio F, Annese T, Ruggieri S et al (2019b) STAT-3 RNAscope determination in human diffuse large B-cell lymphoma. Transl Oncol 12(3):545–549
Tamma R, Ingravallo G, Gaudio F, Annese T, Albano F, Ruggieri S et al (2020) STAT3, tumor microenvironment, and microvessel density in diffuse large B cell lymphomas. Leuk Lymphoma 61(3):567–574
Tamma R, Ingravallo G, Annese T, Giorgis MDE, DI Giovanni F, Gaudio F et al (2021) Tumor cell microenvironment and microvessel density analysis in MALT type lymphoma. Anticancer Res 41(3):1291–1297
Timaxian C, Vogel CFA, Orcel C, Vetter D, Durochat C, Chinal C et al (2021) Pivotal role for Cxcr2 in regulating tumor-associated neutrophil in breast cancer. Cancers (Basel) 13(11):2584
Tysnes BB, Bjerkvig R (2007) Cancer initiation and progression: involvement of stem cells and the microenvironment. Biochim Biophys Acta 1775(2):283–297
Ucuzian AA, Gassman AA, East AT, Greisler HP (2010) Molecular mediators of angiogenesis. J Burn Care Res 31(1):158–175
Virchow R (1989) Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--Atheromatous affection of arteries. 1858. Nutr Rev 47(1):23–25
Voronov E, Carmi Y, Apte RN (2014) The role IL-1 in tumor-mediated angiogenesis. Front Physiol 5:114
Wang S, Lu J, You Q, Huang H, Chen Y, Liu K (2016) The mTOR/AP-1/VEGF signaling pathway regulates vascular endothelial cell growth. Oncotarget 7(33):53269–53276
Wang J, Li D, Cang H, Guo B (2019a) Crosstalk between cancer and immune cells: role of tumor-associated macrophages in the tumor microenvironment. Cancer Med 8(10):4709–4721
Wang X, Rojas-Quintero J, Wilder J, Tesfaigzi Y, Zhang D, Owen CA (2019b) Tissue inhibitor of metalloproteinase-1 promotes polymorphonuclear neutrophil (PMN) pericellular proteolysis by anchoring matrix metalloproteinase-8 and -9 to PMN surfaces. J Immunol 202(11):3267–3281
Wang SS, Liu W, Ly D, Xu H, Qu L, Zhang L (2019c) Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol Immunol 16(1):6–18
Warren BA, Shubik P (1966) The growth of the blood supply to melanoma transplants in the hamster cheek pouch. Lab Investig 15(2):464–478
Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17(11):1359–1370
Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F et al (2011) TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest 121(5):1969–1973
Welker P, Grabbe J, Zuberbier T, Guhl S, Henz BM (2000) Mast cell and myeloid marker expression during early in vitro mast cell differentiation from human peripheral blood mononuclear cells. J Invest Dermatol 114(1):44–50
Wellman TL, Jenkins J, Penar PL, Tranmer B, Zahr R, Lounsbury KM (2004) Nitric oxide and reactive oxygen species exert opposing effects on the stability of hypoxia-inducible factor-1alpha (HIF-1alpha) in explants of human pial arteries. FASEB J 18(2):379–381
Wenzel D, Schmidt A, Reimann K, Hescheler J, Pfitzer G, Bloch W et al (2006) Endostatin, the proteolytic fragment of collagen XVIII, induces vasorelaxation. Circ Res 98(9):1203–1211
Wickstrom SA, Alitalo K, Keski-Oja J (2005) Endostatin signaling and regulation of endothelial cell-matrix interactions. Adv Cancer Res 94:197–229
Wu J, Lanier LL (2003) Natural killer cells and cancer. Adv Cancer Res 90:127–156
Xu Y, Li Q, Li XY, Yang QY, Xu WW, Liu GL (2012) Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J Exp Clin Cancer Res 31:16
Yahaya MAF, Lila MAM, Ismail S, Zainol M, Afizan N (2019) Tumour-associated macrophages (TAMs) in colon cancer and how to reeducate them. J Immunol Res 2019:2368249
Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W et al (2013) B cells promote tumor progression via STAT3 regulated-angiogenesis. PLoS One 8(5):e64159
Yang J, Lu Y, Lin YY, Zheng ZY, Fang JH, He S et al (2016a) Vascular mimicry formation is promoted by paracrine TGF-beta and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett 383(1):18–27
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W et al (2016b) FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res 76(14):4124–4135
Yang SB, Gao KD, Jiang T, Cheng SJ, Li WB (2017) Bevacizumab combined with chemotherapy for glioblastoma: a meta-analysis of randomized controlled trials. Oncotarget 8(34):57337–57344
Yang JG, Wang LL, Ma DC (2018a) Effects of vascular endothelial growth factors and their receptors on megakaryocytes and platelets and related diseases. Br J Haematol 180(3):321–334
Yang J, Kumar A, Vilgelm AE, Chen SC, Ayers GD, Novitskiy SV et al (2018b) Loss of CXCR4 in myeloid cells enhances antitumor immunity and reduces melanoma growth through NK cell and FASL mechanisms. Cancer Immunol Res 6(10):1186–1198
Yu Y, Blokhuis B, Derks Y, Kumari S, Garssen J, Redegeld F (2018) Human mast cells promote colon cancer growth via bidirectional crosstalk: studies in 2D and 3D coculture models. Onco Targets Ther 7(11):e1504729
Yun YR, Won JE, Jeon E, Lee S, Kang W, Jo H et al (2010) Fibroblast growth factors: biology, function, and application for tissue regeneration. J Tissue Eng 2010:218142
Zatterstrom UK, Felbor U, Fukai N, Olsen BR (2000) Collagen XVIII/endostatin structure and functional role in angiogenesis. Cell Struct Funct 25(2):97–101
Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX et al (2010) Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 16(13):3420–3430
Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E et al (2020) Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab 31(6):1136–53 e7
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D et al (1999) Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59(22):5830–5835
Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S et al (2010) Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res 70(1):299–308
Acknowledgments
This work was supported by Associazione “Il Sorriso di Antonio,” Corato, Italy.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tamma, R., Annese, T., Ribatti, D. (2022). The Role of Inflammatory Cells in Tumor Angiogenesis. In: Kovalszky, I., Franchi, M., Alaniz, L.D. (eds) The Extracellular Matrix and the Tumor Microenvironment. Biology of Extracellular Matrix, vol 11. Springer, Cham. https://doi.org/10.1007/978-3-030-99708-3_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-99708-3_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-99707-6
Online ISBN: 978-3-030-99708-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)